 Xuebing Chen
Xuebing Chen Jiancong Lu
Jiancong Lu Xu Zhao†
Xu Zhao† Dongfang Qiao
Dongfang Qiao Xia Yue
Xia Yue
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 21 August 2019
Sec. Non-Neuronal Cells
Volume 13 - 2019 | https://doi.org/10.3389/fncel.2019.00366
Methamphetamine (MA) is a widely abused psychoactive drug that primarily damages the nervous system. However, the involvement of MA in the survival of microglia remains poorly understood. CCAAT-enhancer binding protein (C/EBP-β) is a transcription factor and an important regulator of cell apoptosis. Lipocalin2 (lcn2) is a known apoptosis inducer and is involved in many cell death processes. We hypothesized that C/EBP-β is involved in MA-induced lcn2-mediated microglial apoptosis. To test this hypothesis, we measured the protein expression of C/EBP-β after MA treatment and evaluated the effects of silencing C/EBP-β or lcn2 on MA-induced apoptosis in BV-2 cells and the mouse striatum after intrastriatal MA injection. MA exposure increased the expression of C/EBP-β and stimulated the lcn2-mediated modulation of apoptosis. Moreover, silencing the C/EBP-β-dependent lcn2 upregulation reversed the MA-induced microglial apoptosis. The in vivo relevance of these findings was confirmed in mouse models, which demonstrated that the microinjection of anti-C/EBP-β into the striatum ameliorated the MA-induced decrease survival of microglia. These findings provide a new insight regarding the specific contributions of C/EBP-β-lcn2 to microglial survival in the context of MA abuse.
HIGHLIGHTS
- Methamphetamine upregulated the expression of C/EBP-β in microglia.
- Methamphetamine increased the expression of lcn2 through C/EBP-β pathway.
- C/EBP-β is involved in Methamphetamine-induced lcn2-mediated microglial apoptosis.
Methamphetamine (MA), a widely abused psychoactive drug, has long been known to elevate the concentration of cytoplasmic dopamine through binding with the dopamine transporter and to ultimately induce neuronal death (Gouzoulis-Mayfrank et al., 2017; Li et al., 2017; Moszczynska and Callan, 2017). Microglial activation that is thought to participate in either protoxic or protective mechanisms in the MA-induced neurotoxicity (Coelho-Santos et al., 2012; Fernandes et al., 2016). Microglia, the resident macrophages of the central nervous system (CNS), play an important role in immune surveillance, and compromised microglial function is associated with various neurological pathologies (Streit et al., 1999). Accumulating evidence suggests that activated microglia have been associated with MA-induced neurotoxicity both in vitro and in vivo (Urrutia et al., 2013; Goncalves et al., 2017; Shaerzadeh et al., 2018). In addition, chronic MA abuse is also associated with microglial activation in the brains of MA-addicted humans (Sekine et al., 2008). Activated microglia eventually undergo apoptosis by a process known as activation-induced cell death (AICD) through the regulation of apoptotic proteins, including caspases and the BCL2 family of proteins (Zhang et al., 2016). Although MA is known to induce microglial activation, whether MA induces AICD in microglia and the molecular mechanisms in this process remain elusive.
CCAAT-enhancer binding protein (C/EBP-β) is a member of the C/EBP family of bZIP transcription factors that bind DNA as dimers. Through the alternative use of three translation initiation codons, three C/EBP isoforms (called LAP∗, LAP, and LIP) are translated from a single mRNA (Bradley et al., 2003). LAP∗ and LAP function as transcriptional activators, while the LIP function as a transcriptional repressor in a dominant negative fashion because of lacking a transactivation domain. Recent studies have reported that C/EBP-β participates in multiple cellular functions, such as cell death and survival (Zahnow, 2009; van der Krieken et al., 2015; Xu X. et al., 2018). Many studies have demonstrated that C/EBP-β is an important regulator of the pro-inflammatory program in microglia (Strohmeyer et al., 2014). Our recent study demonstrated that C/EBP-β mediated neuronal apoptosis through mitochondrial pathways (Xu X. et al., 2018). MA can induce microglial apoptosis (Zhang et al., 2016; Sharikova et al., 2018; Du et al., 2019). However, whether C/EBP-β is involved in MA-induced microglial apoptosis remains elusive.
Lipocalin2 (lcn2) is involved in cell death in different situations (Bi et al., 2013; Kim et al., 2016). The proapoptotic role of lcn2 is thought to be involved in mitochondrial pathways (Jin et al., 2011; Chien et al., 2012). The expression of lcn2 is regulated by a network of stimuli acting through different transduction pathways. Notably, C/EBPs are implicated in the transcriptional control of lcn2 (Jha et al., 2015). C/EBPs have transcription factor binding sites that influence lcn2 promoter activity (Cowland et al., 2003; Shen et al., 2006). The LPS-induced upregulation of C/EBP-β increased lcn2 expression in the lung and liver (Sunil et al., 2007). We hypothesized that C/EBP-β regulates lcn2 expression after MA exposure. In the current work, we investigated the role of C/EBP-β in MA-induced microglial death.
Male C57BL/6 mice (6–8 weeks old) were purchased from the Laboratory Animal Center of Southern Medical University (SMU), Guangzhou, China. Each mouse was housed singly at room temperature with a 12 h light/12 h dark cycle. Food and water were available ad libitum. Animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at SMU and followed the latest NIH Guidelines for the Care and Use of Laboratory Animals (National Research Council, 2011). The animals were habituated to the animal facility for 1 week before the experiments began. The mice were divided randomly into control group, acute group and subacute group experimental groups (n = 5–8/group). The acute group received two injections [15 mg/kg, intraperitoneal (i.p.), at 12-h intervals]. The subacute group received eight injections [15 mg/kg, intraperitoneal (i.p.), at 12-h intervals] of either saline vehicle or MA (>99% purity; National Institutes for Food and Drug Control, Guangzhou, China). This exposure paradigm was based on previous studies (Winek et al., 2001; Cadet et al., 2003; Krasnova and Cadet, 2009; Zhang et al., 2016). The animals were euthanized 48 h after the last injection. Brain samples were quickly removed, and the striatal tissues were dissected on ice and stored at −80°C until use.
The mouse microglia cell line (BV-2) was purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences. BV-2 cells were cultured in high glucose DMEM (Gibco, CA, United States) supplemented with 10% fetal bovine serum (FBS) in a humidified incubator (37°C, 5% CO2, 95% air). BV-2 cells were pretreated with a range of MA concentrations (0.25–1.5 mM) for 24 h to evaluate the corresponding apoptotic events. This concentration was chosen for subsequent experiments based on previous studies (Frank et al., 2016; Zhang et al., 2016).
BV-2 cells and the brain tissue from mice exposed to vehicle or MA were lysed in ice-cold RIPA buffer with protease inhibitor. Protein concentrations were determined with the BCA-100 Protein Quantitative Analysis Kit (Biocolors, Shanghai, China). Protein samples were separated by 10–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto 0.22 mm polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, United States). The membranes were blocked at room temperature for 2 h in 5% non-fat milk or for 1 h in 5% bovine serum albumin (BSA) (Biosharp, Guangzhou, China) and then incubated with specific primary antibodies overnight at 4°C, followed by incubation with horseradish peroxidase conjugated secondary antibodies for 1 h at room temperature. The primary antibodies were as follows: anti C/EBP-β (1:1000) and anti-lcn2 (1:500) from Abcam (Cambridge, United Kingdom) and anti-cleaved caspase3 (1:1000), anti-cleaved PARP (1:1000), anti-bax (1:1000), anti-bcl2 (1:1000), anti-cytochrome c (cyto-c, 1:1000), and anti-β-actin (1:1000) purchased from the Cell Signaling Technology (Danvers, MA, United States). The protein bands were developed with Super ECL Western Blotting Detection Reagents. The expression of the housekeeping gene β-actin was used as a reference control. The signal of band intensities was quantitated by a Gel-Pro analyzer (Media Cybernetics, Inc., Rockville, MD, United States).
Specific small interfering RNA (siRNA) sequences targeting C/EBP-β and lcn2 were synthesized by GenePharma (Shanghai, China) as follows: C/EBP-β siRNA: 5′-GCGACAAGAAGCTGACTTT-3′; and lcn2 siRNA: 5′-CCAAGCTGCCAGACTATAA-3′. The scrambled siRNA sequence was used as a control. siRNAs were dissolved in diethylpyrocarbonate (DEPC) water at a concentration of 20 μM. Cells were placed on a 6-well plate at a density of 5 × 105 cells/well. When cells reached 60% confluence, 20 μmol siRNA, and 5 μl of Lipofectamine 3000 Reagent (Invitrogen, Carlsbad, CA, United States) were added to Opti-MEM medium (Gibco BRL, Paisley, United Kingdom). The mixed solution was incubated for 15 min at room temperature. Then, cells were incubated with standard culture medium for 12–18 h and thereafter were exposed to 1.0 mM MA in non-serum medium for another 24 h.
BV-2 cells were seeded in confocal dishes with different treatments, and brain tissues were fixed with paraformaldehyde and permeabilized with 0.1% Triton-X. After blocking with 5% BSA, cells and tissue sections were incubated with rabbit C/EBP-β (1:500; Abcam) and lcn2 (1:100; Abcam) at 4°C. After washing and incubating with Alexa 488-conjugated mouse anti-rabbit IgG (1:500; Invitrogen), Alexa 555-conjugated goat anti-rabbit IgG (1:500; Invitrogen), and Alexa 594-conjugated goat anti-rabbit IgG (1:500; Invitrogen), the cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI). The secondary antibodies were purchased from the Thermo Life Technologies (CA, United States). The images were observed by confocal microscopy (Carl ZeissLSM 880 with Airyscan, Germany) using the same settings to improve the contrast.
BV-2 cells were seeded on confocal dishes overnight. Cells were pretreated with lipo/scrambled siRNA and then exposed to MA (1.0 mM) or vehicle for 24 h. Visualization of DNA fragmentation was performed using the fluorometric terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) system for apoptotic cells (KeyGen Biotech, China) according to the manufacturer’s instructions. Both TUNEL- and DAPI-positive cells were counted by Image J software. Data are calculated based on the total number of TUNEL-positive cells.
BV-2 cells were placed on a 6-well plate at a density of 3–5 × 105cells/ml. Cells were pretreated with lipo/scrambled siRNA and then exposed to MA (1.0 mM) or vehicle for 24 h. The cells were collected after washing twice with 4°C PBS and stained with Annexin V-FITC and propidium iodide (PI; 4A Biotech Co., Ltd., Nanjing, China) according to instructions of the Annexin V Apoptosis Detection Kit. The percentage of apoptotic cells was quantified with a flow cytometry system (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, United States). Either early apoptosis (Annexin V-FITC-positive, PI-negative) or late apoptosis (Annexin V-FITC-positive, PI-positive) was considered apoptotic cells.
The fluorescent dye JC-1 (KeyGen Biotech) was used to assess the change in mitochondrial membrane potential. Briefly, cells seeded into confocal dishes were incubated with a mixture of 1 μl of JC-1 staining fluid and 500 μl of incubation buffer in the dark at 37°C for 30 min, washed twice with the incubation buffer, and observed by confocal microscopy (Carl ZeissLSM 880 with Airyscan, Germany).
The shRNA synthesis and stereotaxic injection protocol were based on our recent studies (Huang et al., 2015; Li et al., 2017). In brief, the C/EBP-β shRNA sequence (5′-TTTAAGTGATTACTCAGGG-3′) was cloned into the pGC-LV vector and transfected into human embryonic kidney (HEK 293FT) cells with the pHelper 1.0 and 2.0 vectors. The LV-shC/EBP-β lentivirus was harvested (109 transducing units/ml). LV-GFP was used as a control virus. The C57 mice were randomly divided into four groups (n = 5–8/group): the LV-GFP group, LV-shC/EBP-β group, LV-GFP + MA group, and LV-shC/EBP-β + MA group. LV-shC/EBP-β lentivirus or LV-GFP lentivirus were bilaterally injected into the striatum [coordinates relative to bregma: anteroposterior (AP) +0.4 mm; mediolateral (ML) ±2 mm; and dorsoventral (DV) −3.5 mm] via a stereotaxic apparatus (Yuan et al., 2018). After 4 days, the animals were injected with saline vehicle or MA (15 mg/kg × 8 injections, i.p., at 12-h intervals) and were euthanized 48 h after the final injection. Brain samples were quickly removed and stored at −80°C until use.
All data were expressed as the mean ± standard deviation (SD) with at least three independent replicates. Statistical analysis was performed using one-way ANOVA followed by least significant difference (LSD) post hoc analysis and independent-samples t-test (as appropriate) with the statistical software SPSS version 20.0 (SPSS Inc., Chicago, IL, United States). P < 0.05 was considered statistically significant.
To address the role of the C/EBP-β isoform LAP after MA exposure, C57BL/6 mice were treated with MA or a control, and the expression of the C/EBP-β isoform LAP was detected. Western blot analysis showed that the expression of the C/EBP-β isoform LAP in the striatum of MA-treated mice was significantly increased in both the acute group and subacute group, and this change was at a greater extent after longer exposure (by 3.48-fold for subacute group and 3.07-fold for acute group, Figures 1A,B). MA-treated mice was significantly increased in the acute group while decreased in the subacute group (Supplementary Figures 1A,B). Therefore, the subacute group was selected for further studies. Immunofluorescence staining for the striatum (Figure 1C) showed C/EBP-β was significantly increased in the subacute group. Furthermore, BV-2 cells were treated with different concentrations of MA (0, 0.25, 0.5, 0.75, and 1.5 mM) for different durations (0, 1, 3, 6, 12, or 24 h), and C/EBP-β was determined by Western blot analysis. The results showed that the expression levels of the C/EBP-β isoform LAP, but not C/EBP-β LIP (data not shown), increased significantly in a dose- and time-dependent manner in MA-treated BV-2 cells (Figures 2A–D). The results from the immunofluorescence staining of BV-2 cells (Figure 2E) showed that MA exposure increased the expression of C/EBP-β. Collectively, these data suggested that MA exposure induces the expression of C/EBP-β in microglia both in vitro and in vivo.
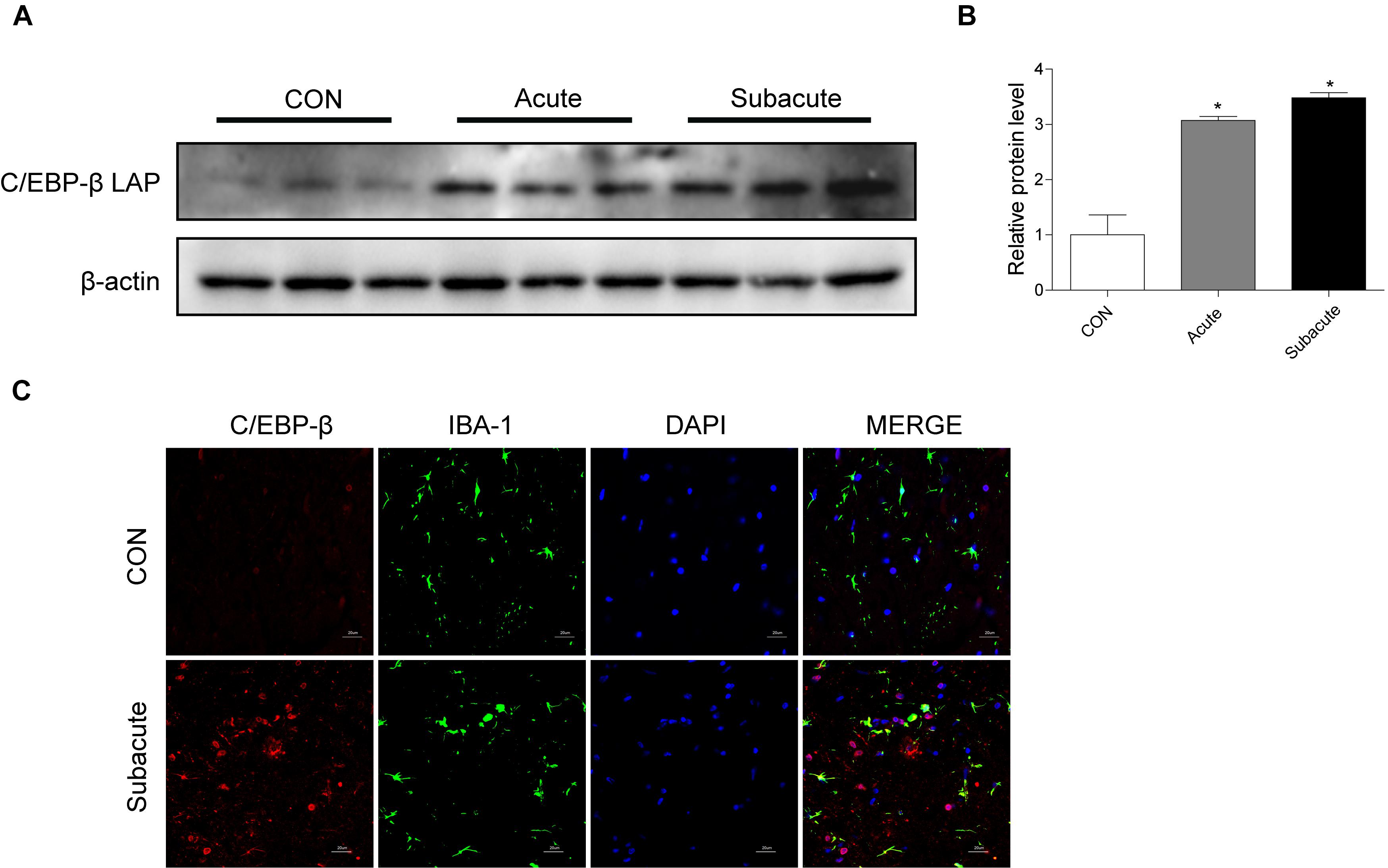
Figure 1. MA exposure increases C/EBP-β isoform LAP expression in vivo. Representative Western blots (A) and quantitative analyses (B) of C/EBP-β in the corpus striatum tissues of mice from the control group, acute group and subacute group. N = 5–8 animals/group. Fold induction relative to the vehicle-treated group (∗p < 0.05 vs. the vehicle-treated group using one-way ANOVA).β-actin was used as a loading control. All data are presented as the mean ± SD. (C) Representative immunofluorescence microscopy images of C/EBP-β (red) in the striatal microglia (green, IBA-1) of mice in the vehicle-treated group, subacute MA-treated group (scale bar = 20 μm). Nuclei were counterstained with DAPI (blue). C/EBP-β (red) merged with DAPI (blue) was pink. All data were expressed at least three independent replicates.
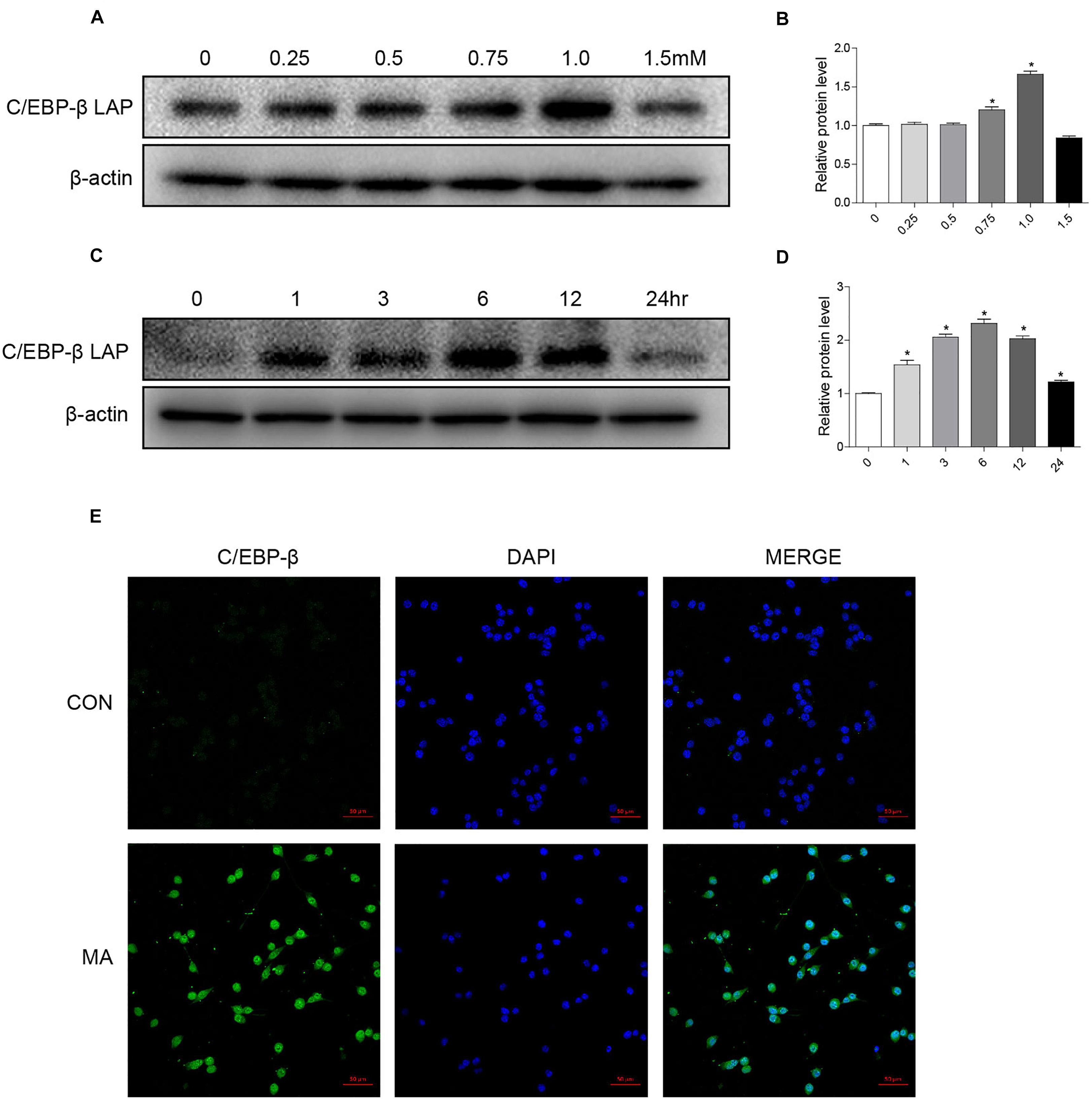
Figure 2. MA exposure increases C/EBP-β isoform LAP expression in vitro. Microglial BV-2 cells were treated with 0.25–1.5 mM MA for 24 h (A,B) and exposed to 1.0 mM MA for 1–24 h (C,D). Representative immunofluorescence microscopy images of C/EBP-β (green) in BV-2 cells in the vehicle-treated group and MA-treated group (E, scale bar = 50 μm). Nuclei were counterstained with DAPI (blue). All data were expressed at least three independent replicates.β-actin was used as a loading control. The fold induction relative to that in the vehicle-treated group is shown. All data are presented as the mean ± SD, ∗p < 0.05 vs. the vehicle-treated group using one-way ANOVA.
To assess the effect of MA exposure in microglial cell toxicity, BV-2 cells were treated with 0.25–1.5 mM MA for 24 h and 1.0 mM MA for 1–24 h. Then, Western blot analysis was performed to measure cleaved caspase3, cleaved PARP, the ratio of bax/bcl-2, and cyto-c expression levels, which were also significantly increased in a dose-dependent manner after MA exposure (Supplementary Figures 2A–C). As shown in Figures 3A–C, the apoptosis-related proteins were significantly increased in a time-dependent manner after MA exposure. This result was further confirmed by TUNEL staining (Figures 3D,E). MA treatment resulted in increasing the ratio of JC-1 monomers (green) to JC-1 aggregates (red), standing for the loss of mitochondrial membrane potential (MΨm) in BV-2 cells (Figures 3F,G). Together, these results indicated that the accumulation of MA might play a key role in microglial cell toxicity.
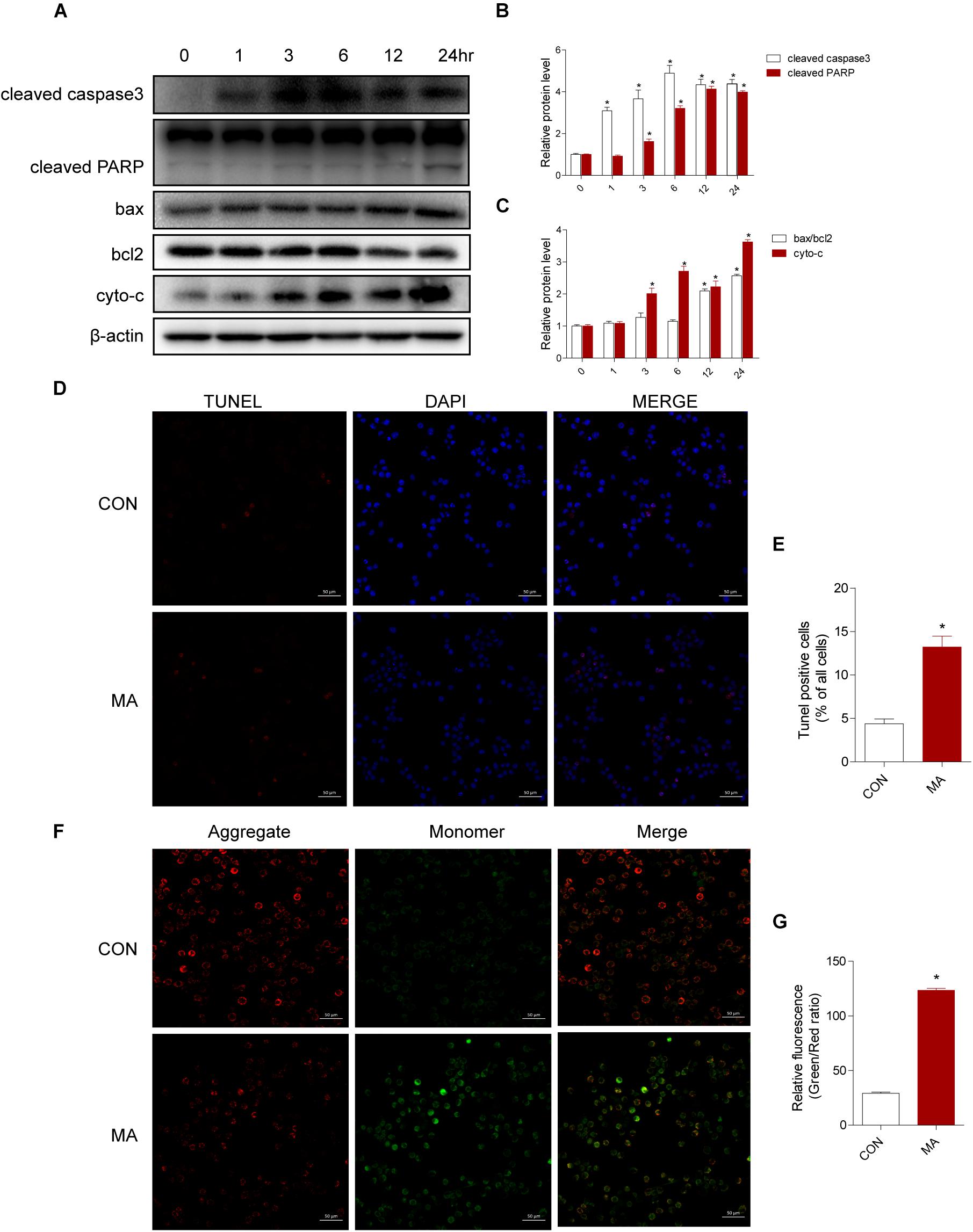
Figure 3. MA increases apoptosis in vitro. Microglial BV-2 cells were treated with 1.0 mM MA for 1–24 h (A–C). The protein expression levels of cleaved caspase3 and cleaved PARP, the ratio of bax/bcl2 and cytochrome c were determined with Western blot and quantitative analyses. Representative data from three independent experiments are shown. β-actin was used as a loading control. Apoptotic cells were subjected to TUNEL staining (D, scale bar = 50 μm). Nuclei were counterstained with DAPI (blue). Quantitative analysis of the percentage of apoptotic cells using a standard cell counting method with the TUNEL assay (E). Confocal microscopy analysis using JC-1 staining revealed that MA (1.0 mM, 24 h) challenge decreased the MΨm level in BV-2 cells (F, scale bar = 50 μm). Red stands for the JC-1 aggregate fluorescence from healthy mitochondria, and green exhibits cytosolic JC-1 monomers. The increased ratio of JC-1 monomers (green) to JC-1 aggregates (red) stands for the decreased MΨm. The fold induction relative to that in vehicle-treated cells is shown (G). Data are presented as the means ± SD with at least three independent replicates. ∗p < 0.05 vs. the saline vehicle-treated control group. Data in panels (B,C) were analyzed by one-way ANOVA. Data in panels (E,G) were analyzed by Student’s t-test.
We next evaluated the involvement of C/EBP-β in the MA-induced activation of the intrinsic apoptosis pathway. We used siRNAs targeting C/EBP-β to silence its expression and then examined the effects on MA-induced apoptosis in BV-2 cells. Western blot analysis showed that the MA-induced increases in apoptotic-related proteins were significantly blocked by siRNAs targeting C/EBP-β (Figures 4A–C). This result was further confirmed by flow cytometry (Figures 4D,E), the JC-1 assay (Figures 5A,B) and the TUNEL assay (Figures 5C,D). These results suggest that knockdown of C/EBP-β can effectively decrease MA-induced apoptosis in microglia.
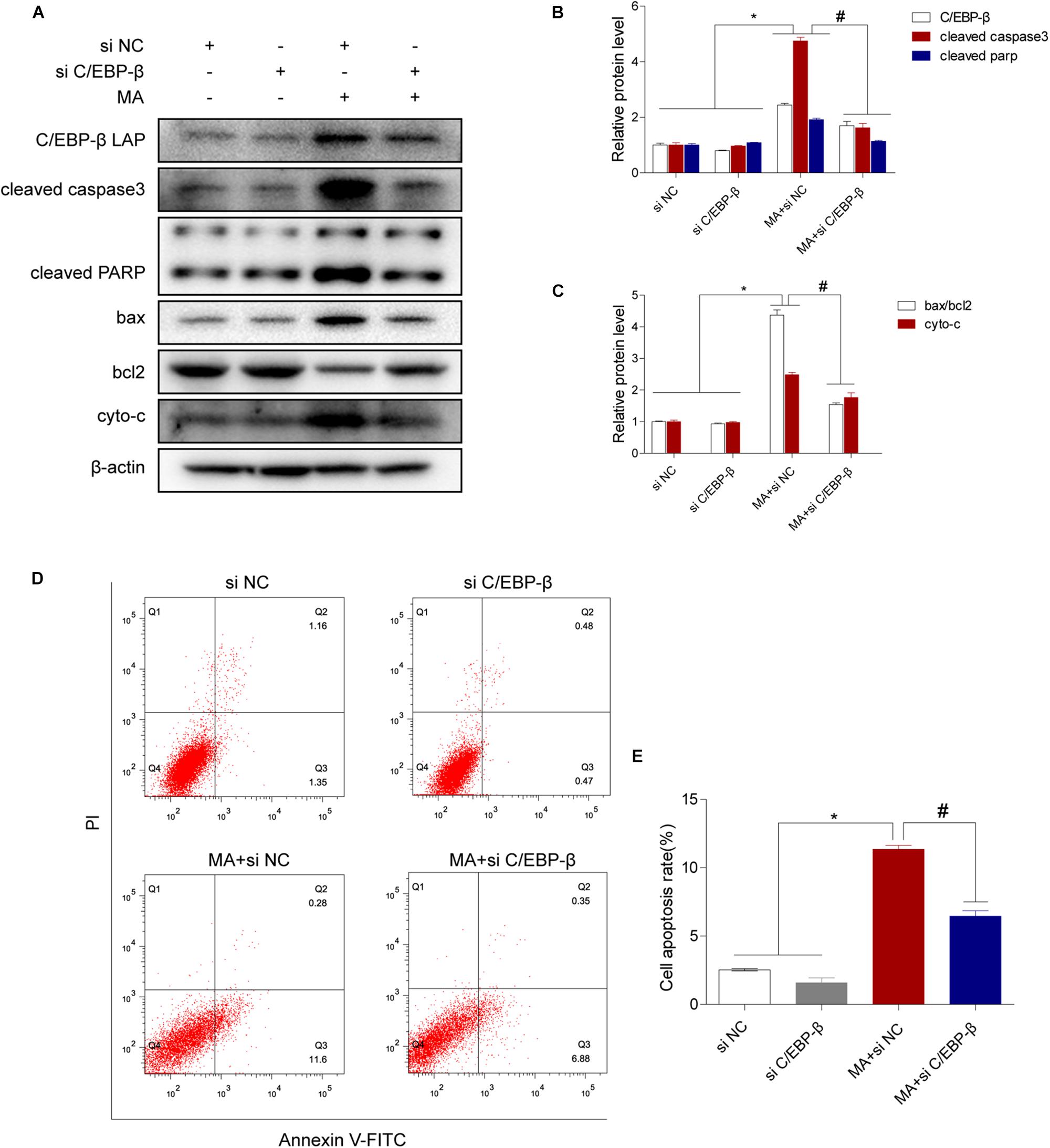
Figure 4. The C/EBP-β isoform LAP is responsible for MA-induced apoptosis in vitro. Representative Western blots (A) and quantitative analyses (B,C) of C/EBP-β, cleaved caspase3 and cleaved PARP, the ratio of bax/bcl2 and cytochrome c in BV-2 cells exposed to vehicle + control siRNA, 1.0 mM MA + control siRNA, vehicle + C/EBP-β siRNA and 1.0 mM MA + C/EBP-β siRNA. All data were expressed at least three independent replicates. β-actin was used as a loading control. Apoptotic cells were stained and analyzed with flow cytometry (D). The percentage of apoptosis is presented as the mean ± SD (E). ∗p < 0.05 vs. the saline vehicle-treated control group, #p < 0.05 vs. the scrambled + MA group (one-way ANOVA).
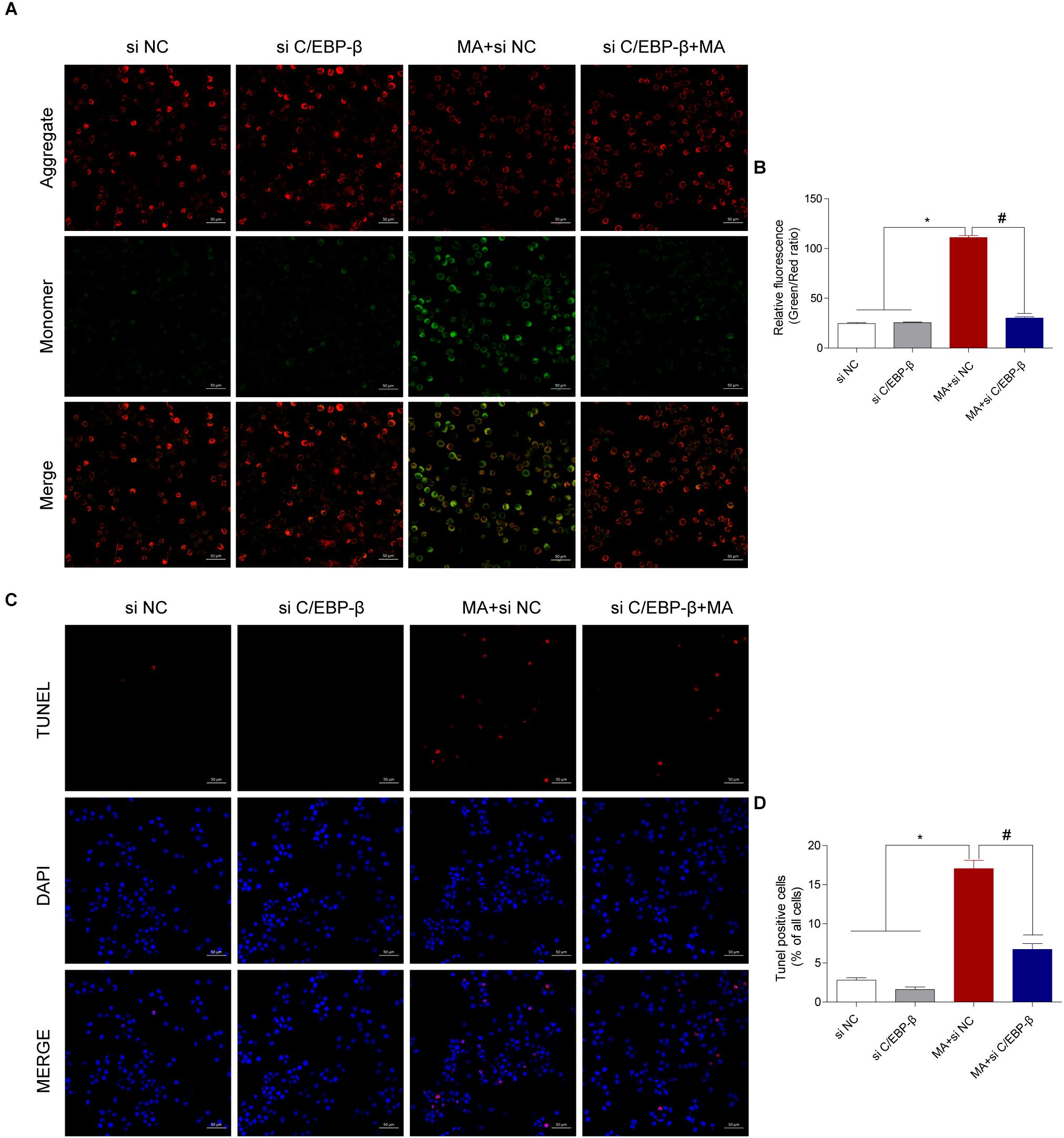
Figure 5. Silencing C/EBP-β decreases the cell apoptosis induced by MA exposure in vitro with the TUNEL assay and JC-1 staining. Representative images of the JC-1 staining (A, scale bar = 50 μm) and TUNEL assay (C, scale bar = 50 μm) in BV-2cells from different groups: vehicle + control siRNA, 1.0 mM MA + control siRNA, vehicle + C/EBP-β siRNA, and 1.0 mM MA + C/EBP-β siRNA for 24 h exposure. Bar graphs show the ratio of aggregated and monomeric JC-1 (B). The percentage of apoptosis is presented as the mean ± SD (D). ∗p < 0.05 vs. the saline vehicle-treated control group, #p < 0.05 vs. the scrambled + MA group (one-way ANOVA).
Therefore, we hypothesized that C/EBP-β may mediate MA-induced microglial apoptosis via upregulation of lcn2 expression. To investigate whether lcn2 is the target of C/EBP-β, Western blot and immunofluorescence staining were used, and the results showed that lcn2 was increased after MA exposure (Figures 6A–C). In addition, we observed that the MA-induced increases in lcn2 expression were significantly blocked after C/EBP-β knockdown in 1.0 mM MA-exposed BV-2 cells (Figures 6D,E), suggesting that lcn2 may be a downstream protein of C/EBP-β.
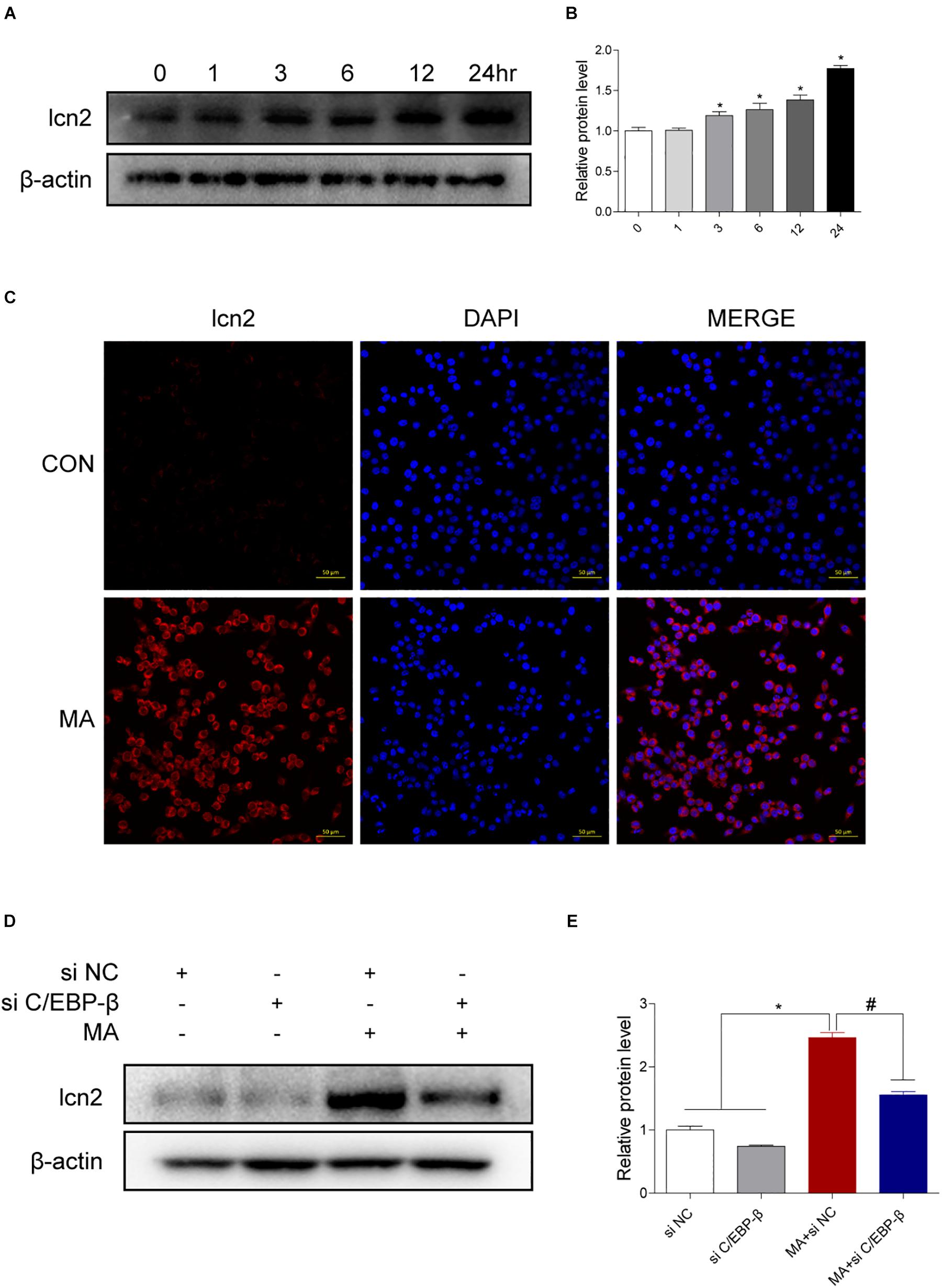
Figure 6. Lcn2 is a downstream target of C/EBP-β. (A,B) Representative Western blots of lcn2 expression in BV-2 cells exposed to MA (0–24 h). (C) Representative immunofluorescence microscopy images of lcn2 (red) in BV-2 cells in the vehicle-treated group and MA-treated group (scale bar = 50 μm). Nuclei were counterstained with DAPI (blue). Western blot results revealed that the MA-induced lcn2 increased expression could be blocked by preincubating with C/EBP-β siRNA (D,E). β-actin was used as a loading control. The fold induction relative to that in vehicle-treated cells is shown. Data are presented as the means ± SD. ∗p < 0.05 vs. the saline vehicle-treated control group, #p < 0.05 vs. the scrambled + MA group (one-way ANOVA). All data were expressed at least three independent replicates.
To test the hypothesis that the C/EBP-β/lcn2 axis mediates MA-induced microglial apoptosis, we used siRNAs targeting lcn2 to silence its expression and then determined the bax, bcl2, cleaved caspase3, cleaved PARP and cytochrome c expression levels in MA-exposed BV-2 cells. The results of Western blotting showed that the expression levels of lcn2 and apoptosis-related proteins were increased by MA and decreased following lcn2 knockdown except the protein of C/EBP-β (Figures 7A–D). These results were further confirmed by flow cytometry (Figures 7E,F), the JC-1 assay (Figures 8A,B) and the TUNEL staining (Figures 8C,D). These results suggested that knockdown of lcn2 can effectively decrease MA-induced apoptosis in microglia.
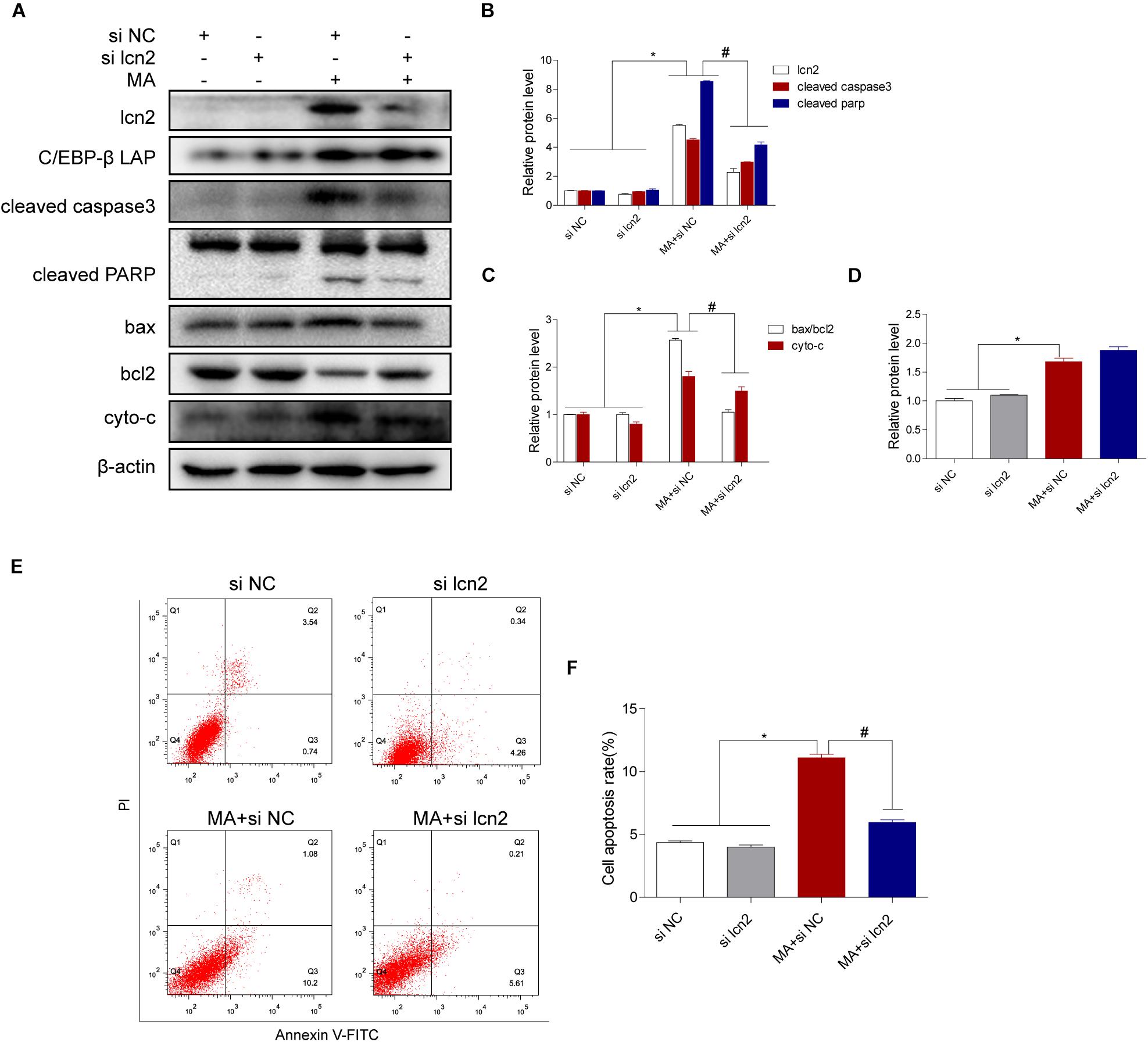
Figure 7. Lcn2 plays a critical role in C/EBP-β-mediated MA-induced apoptotic signaling pathways. Representative Western blots of lcn2, cleaved caspase3 and cleaved PARP, the ratio of bax/bcl2, cytochrome c (A–C, respectively) and C/EBP-β (D) in BV-2 cells exposed to vehicle + control siRNA, 1.0 mM METH + control siRNA, vehicle + lcn2 siRNA, and 1.0 mM METH + lcn2 siRNA. All data were expressed at least three independent replicates. β-actin was used as a loading control. Apoptotic cells were stained and analyzed with flow cytometry (E). The percentage of apoptosis is presented as the mean ± SD (F). ∗p < 0.05 vs. the saline vehicle-treated control group, #p < 0.05 vs. the scrambled + MA group (one-way ANOVA).
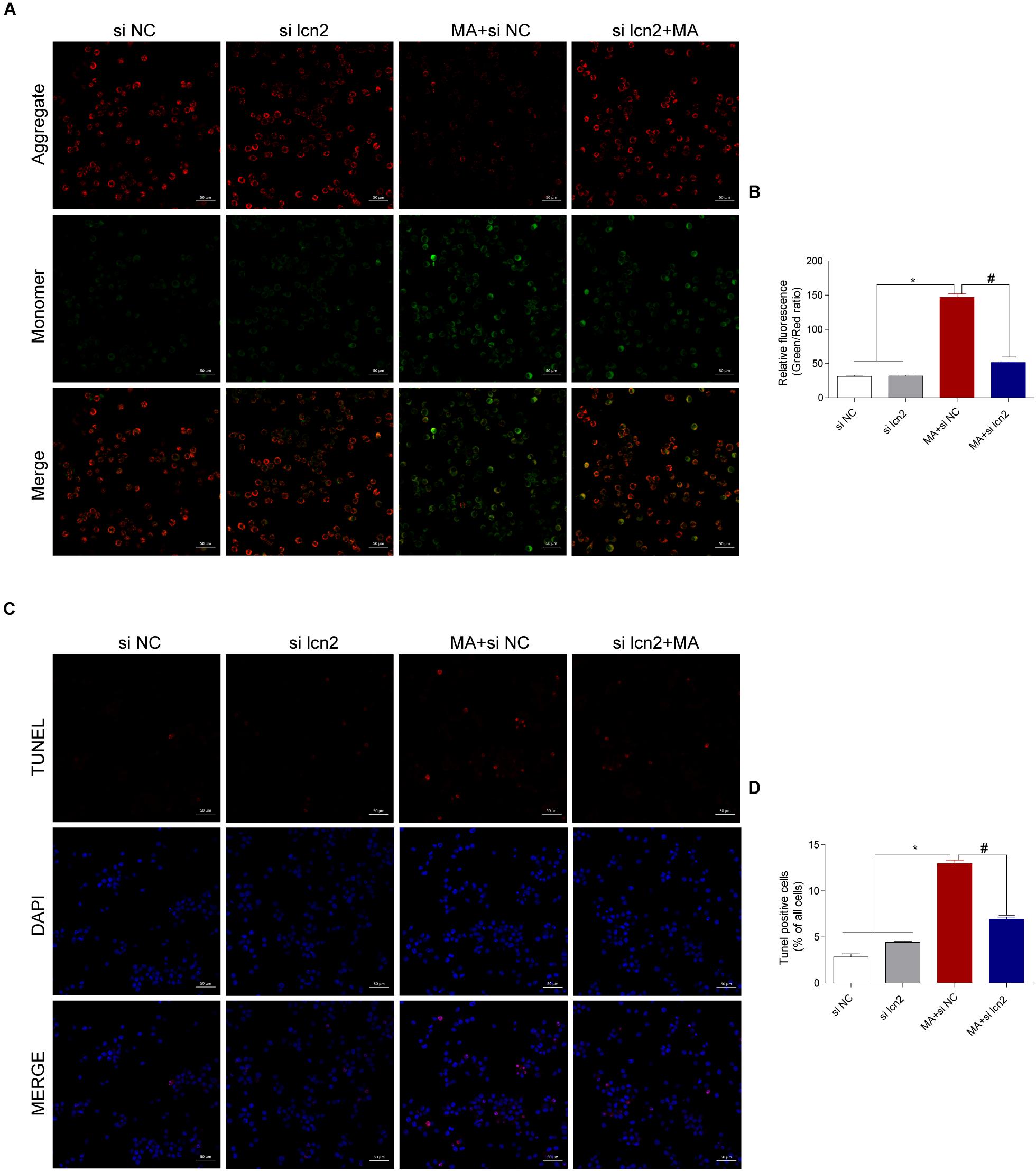
Figure 8. Silencing lcn2 decreases the cell apoptosis induced by MA exposure in vitro with the TUNEL assay and JC-1 staining. Representative images of the JC-1 staining (A, scale bar = 50 μm) and TUNEL assay (C, scale bar = 50 μm) in BV-2 cells from different groups: vehicle + control siRNA, 1.0 mM MA + control siRNA, vehicle + C/EBP-β siRNA and 1.0 mM MA + C/EBP-β siRNA after 24 h exposure. Bar graphs show the ratio of aggregated and monomeric JC-1 (B). The percentage of apoptosis is presented as the mean ± SD (D). ∗p < 0.05 vs. the saline vehicle-treated control group, #p < 0.05 vs. the scrambled + MA group (one-way ANOVA).
To confirm the role of C/EBP-β in MA-induced microglial apoptosis in vivo, lentivirus vector (LV)-GFP and LV-shC/EBP-β were injected into the striatum of mice via a stereotaxic apparatus to silence the C/EBP-β expression in the striatal region (n = 5–8/group). After 4 days of stereotaxic injection, the mice were treated with saline or MA. Western blot analyses showed that MA-increased C/EBP-β protein expression in the LV-GFP group was significantly blocked by treatment with LV-shC/EBP-β in the mouse striatum. The expression levels of Lcn2, ratio of bax/bcl2, cytochrome c, cleaved caspase3, and cleaved PARP had similar effects (Figures 9A–C).
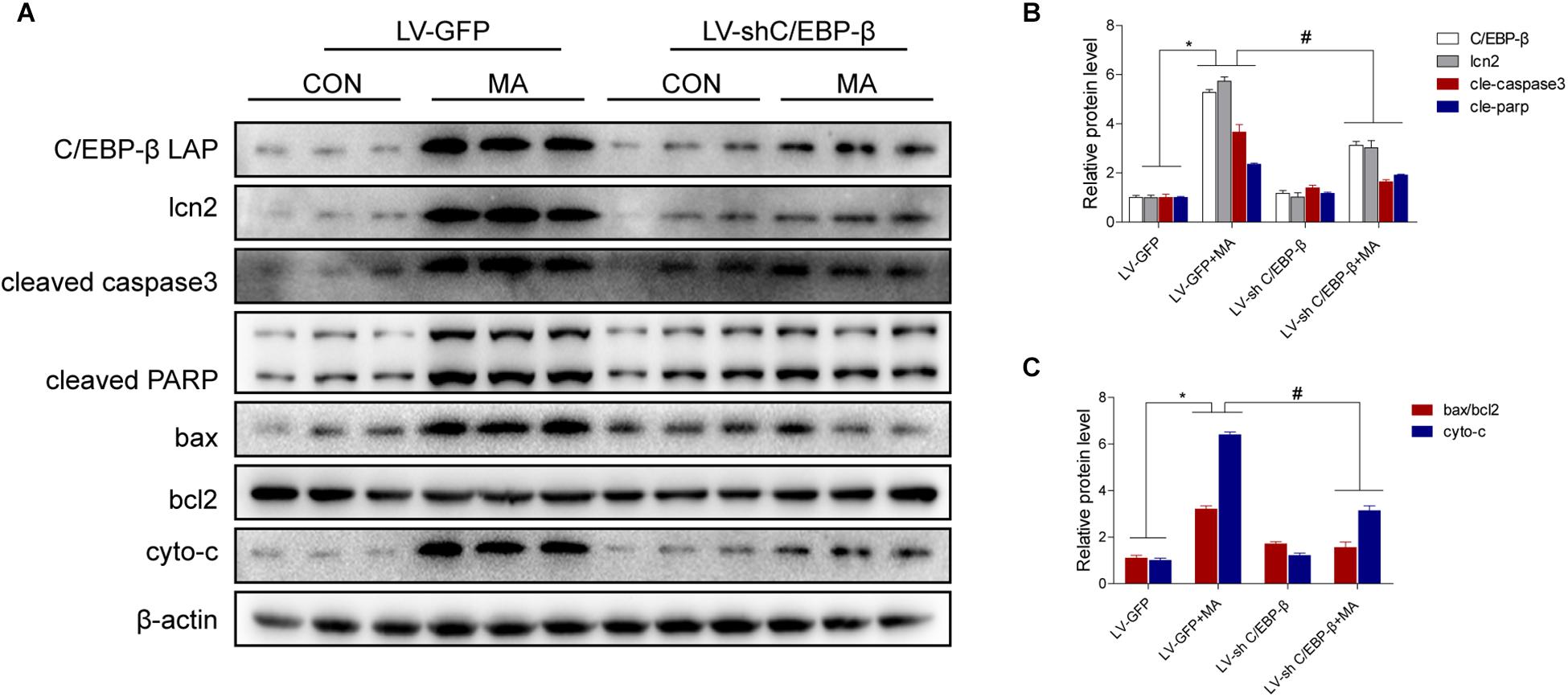
Figure 9. Silencing of C/EBP-β expression reduces MA-induced microglial apoptosis in vivo. LV-GFP and LV-shC/EBP-β lentiviruses were injected separately into the bilateral striatum of mice via a stereotaxic apparatus with saline or MA (15 mg/kg × 8 injections, at 12-h intervals, i.p.). Western blot and quantitative analyses were performed to determine the C/EBP-β, lcn2, cleaved caspase3 and PARP, ratio of bax/bcl2 and cytochrome c protein expression (A–C). All data were expressed at least three independent replicates. β-actin was used as a loading control. The fold induction relative to that in vehicle-treated cells is shown. Data are presented as the means ± SD.∗p < 0.05 vs. the saline vehicle-treated LV-GFP group, #P < 0.05 vs. the LV-GFP + MA group. Data were analyzed by one-way ANOVA.
In this study, we provided evidence that MA, as a recreational drug, induced apoptosis in the microglial cell line BV-2 and in mouse brain tissues. Specifically, we found that MA-induced microglial apoptosis was due to the activation of mitochondrial apoptosis, and this pathway was mediated by the C/EBP-β-lcn2 axis.
Microglia are the resident macrophages of the CNS and represent 5 to 15% of adult brain cells, with densities varying between distinct brain regions (Allen and Lyons, 2018; Thion et al., 2018). Following stimulation with external stimuli, microglia undergo proliferation and morphological changes and play a key role in CNS homeostasis by mediating a protective response (Thion et al., 2018). Microglia are also phagocytic and phagocytize harmful substances and senescent cells, including abnormal neurons. However, prolonged exposure to toxic stimuli such as MA or secreted cytokines can lead to prominent neuronal dysfunction and impair microglial function, even leading to cell death, thereby resulting in worsened damage within the CNS. MA exposure contributes to microglial activation and secretion of inflammatory factors to promote neuroinflammation (Robson et al., 2013; Urrutia et al., 2013) and even induces microglial death through the Mir-143-BBC signaling pathway (Zhang et al., 2016). In the current study, we found that C/EBP-β mediated MA-induced microglial mitochondrial apoptosis.
Recent studies have also shown an upregulation of microglial C/EBP-β upon pro-inflammatory stimulation (Dasgupta et al., 2003; Ejarque-Ortiz et al., 2007, 2010). MA as a widely abused psychoactive drug, induced microglial activation and secretion of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α (Wang et al., 2018). Pro-inflammatory cytokines IL-1β, IL-6, or TNF-α and LPS induced C/EBP-β upregulation in microglia through activation of the mitogen-activated protein kinases (MAPKs) signaling pathway (Ejarque-Ortiz et al., 2007). Furthermore, MA potentiates LPS stimulation of IL-1β production in microglia (Xu E. et al., 2018). We hypothesize that MA induced microglial reaction and secretion pro-inflammatory cytokines to involve in C/EBP-β expression. In addition, TLR4- HMGB1 involved in C/EBP-β expression in LPS-induced macrophage polarization (Gao et al., 2018). Our previous study has demonstrated that MA induced TLR4 upregulation in the microglia and astrocytes (Du et al., 2017). So the interaction of TLR4 with C/EBP-β in MA-induced microglia survival is a direction in the future research.
A number of studies have reported that C/EBP-β expressed in many tissues (Huber et al., 2012; Pulido-Salgado et al., 2015). C/EBP-β involved in neuronal death in our recent study (Xu X. et al., 2018) and others (Marshall et al., 2003; Grewal et al., 2009). LPS induces C/EBP-β upreglulation in astrocytes and microglia (Ejarque-Ortiz et al., 2007; Nakazato et al., 2015). The upregulation of microglial C/EBP-β in pro-inflammatory stimulation has been observed in the microglial cell lines BV-2 (Ejarque-Ortiz et al., 2010; Gresa-Arribas et al., 2010). In the present study, we found that C/EBP-β plays key roles in MA-induced microglial death, moreover, C/EBP-β is not only unique to microglia in the brain (Figure 1C). MA induced C/EBP-β upregulation in other non-identified cells possibly. Our previous study has found that MA induced C/EBP-β upregulation in neurons in the striatum (Xu X. et al., 2018). Further research are required to substantiate the C/EBP-β expression in diverse cell type after MA exposure. The interactions of microglia with neurons and other non-identified cells in MA-induced neurotoxicity are a direction in the future research. Knockdown of C/EBP-β with lentivirus can inhibit the apoptosis-related proteins bax, bcl2, cytochrome c, cleaved caspase3, and PARP and protect microglia from MA-induced toxicity in vivo and in vitro.
The function of lcn2 may be dependent on cell type because lcn2 can either stimulate or inhibit cell growth and differentiation in different cell types (Ferreira et al., 2015; Jha et al., 2015). Emerging evidences suggested that activated microglia in vivo may secrete lcn2 to facilitate morphological transformation and the secretion of cytokines (Lee et al., 2007, 2011; Naude et al., 2012). In microglia and astrocytes, the expression and secretion of lcn2 are increased under inflammatory conditions (Lee et al., 2009; Jang et al., 2013). Microglia- and astrocyte-derived lcn2 contributes to neuronal death during neurodegenerative conditions (Bi et al., 2013; Tong et al., 2013). Series reports indicated that lcn2 is involved in mitochondrial apoptosis and mediates cell apoptosis (Devireddy et al., 2005; El Karoui et al., 2016). Lee et al. (2007) reported that lcn2 makes microglia more vulnerable to apoptotic stimuli. We found that lcn2 is involved in MA-induced microglial apoptosis. In addition, C/EBP-β are implicated in the transcriptional control of lcn2 (Jha et al., 2015). Positions for putative binding sites of transcription factor of C/EBP-β was −190 and −170 nt in the lcn2 promoter were measured by the luciferase reporter assay (Fujino et al., 2006). Both the NF-κB and C/EBP-β sites in the Lcn2 promoter are important for IL-17-mediated transcription (Shen et al., 2006). In this study, C/EBP-β was shown to be a transcription factor involved in lcn2 after MA-induced microglial apoptosis.
BV-2 is a certainly popular cell line to study microgla, Some researchers study pathophysiological functions of microglia using BV-2 cells in numbers of studies, including MA exposure (Dasgupta et al., 2003; Guo et al., 2015; Zhang et al., 2016). However, BV-2 cells do not always reproduce well the phenotype of primary microglia or in vivo microglia (Butovsky et al., 2014). This is a limitation of our research. Further research are required to substantiate the present findings using C/EBP-β knockout animal models.
In summary, C/EBP-β upregulation might be a potential pathogenic mechanism leading to further disruption of the striatum induced by MA neurotoxicity. C/EBP-β is involved in MA-induced microglial apoptosis in vivo and in vitro. lcn2 is a downstream target of C/EBP-β-mediated MA-induced apoptosis. Knockdown of C/EBP-β decreased lcn2 expression to protect microglia from MA-induced apoptosis (Figure 10). These results suggested that MA-induced microglial apoptosis could be partly reversed by knocking down C/EBP-β. These findings provide a new insight regarding the specific contributions of C/EBP-β-lcn2 to microglial survival in the context of MA abuse.
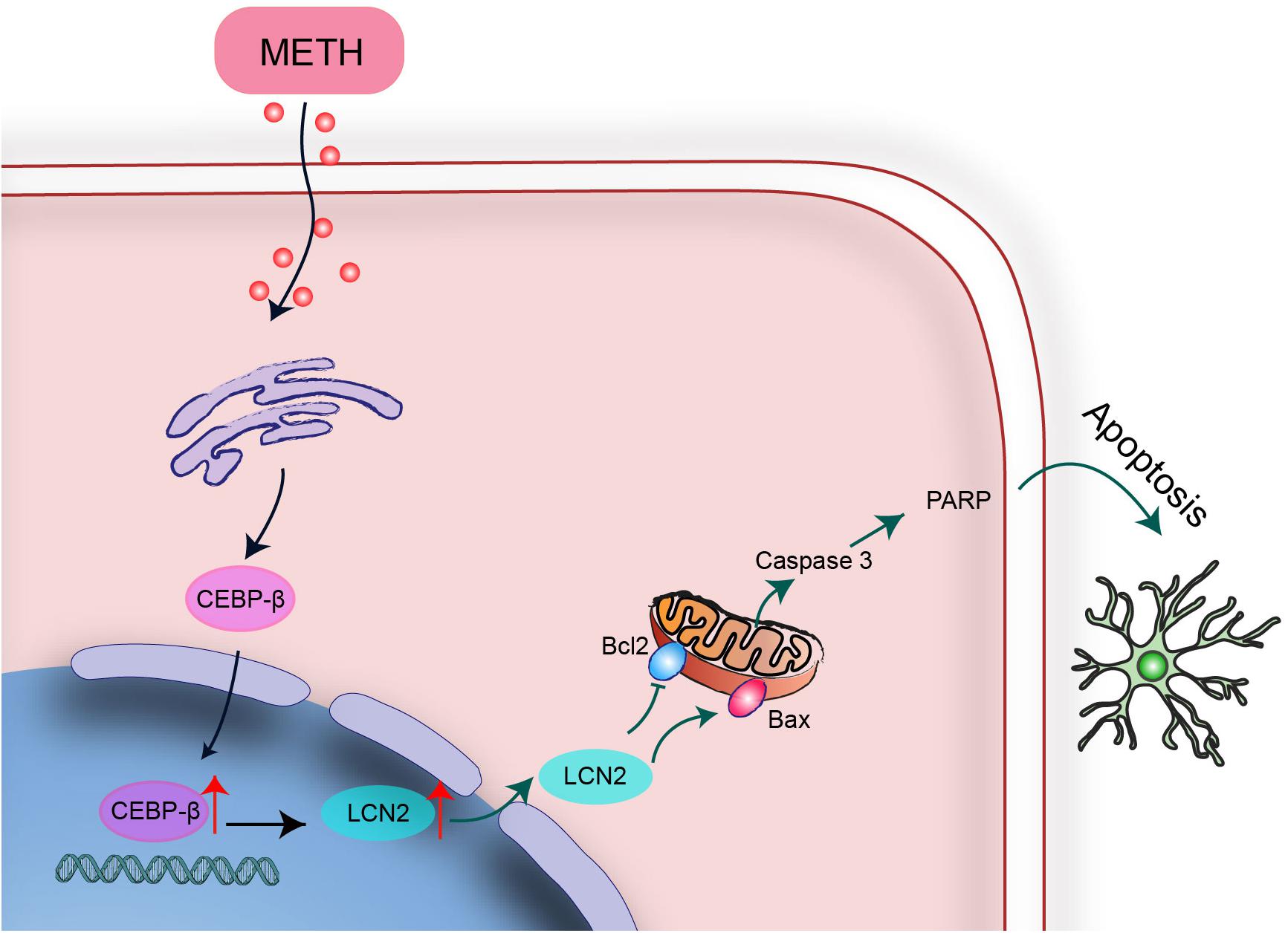
Figure 10. A schematic depicting the role of the C/EBP-β-lcn2 signaling pathway in MA-induced microglial apoptosis. A schematic depicting the role of the C/EBP-β/lcn2 signaling pathway in MA-induced microglial apoptosis via a mitochondrial-dependent apoptosis pathway. Briefly, C/EBP-β expression is induced after MA exposure, which upregulates the expression level of lcn2 by transactivating its promoter. Then, lcn2 triggers apoptosis through the mitochondrial pathway.
The authors declare that all data supporting the findings of this study are available within the article.
XC and XY conducted all the experiments with the help of JL, DQ, CC, and XZ. XC and HW designed the experiments. XC, HW, and XY analyzed and interpreted the results. HW and XC wrote the manuscript with the help of XY, JL, and XZ. All authors approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81430045 and 81871528 to HW), Guangzhou Sci-Tech Project (Grant No. 201804010307 to XY), and Natural Science Foundation of Guangdong Province (Grant No. 2018A030313876 to XY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank American Journal Experts for its linguistic assistance during the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00366/full#supplementary-material
AICD, activation-induced cell death; BSA, bovine serum albumin; C/EBP-β, CCAAT-enhancer binding protein; CNS, central nervous system; LSD, least significant difference; LPS, lipopolysaccharide; MA, methamphetamine; SD, standard deviation; siRNA, small interfering RNA; SMU, Southern Medical University.
Allen, N. J., and Lyons, D. A. (2018). Glia as architects of central nervous system formation and function. Science 362, 181–185. doi: 10.1126/science.aat0473
Bi, F., Huang, C., Tong, J., Qiu, G., Huang, B., Wu, Q., et al. (2013). Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. U.S.A. 110, 4069–4074. doi: 10.1073/pnas.1218497110
Bradley, M. N., Zhou, L., and Smale, S. T. (2003). C/EBP regulation in lipopolysaccharide-stimulated macrophages. Mol. Cell. Biol. 23, 4841–4858. doi: 10.1128/mcb.23.14.4841-4858.2003
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Cadet, J. L., Jayanthi, S., and Deng, X. (2003). Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J. 17, 1775–1788. doi: 10.1096/fj.03-0073rev
Chien, M. H., Ying, T. H., Yang, S. F., Yu, J. K., Hsu, C. W., Hsieh, S. C., et al. (2012). Lipocalin-2 induces apoptosis in human hepatocellular carcinoma cells through activation of mitochondria pathways. Cell Biochem. Biophys. 64, 177–186. doi: 10.1007/s12013-012-9370-1
Coelho-Santos, V., Gonçalves, J., Fontes-Ribeiro, C., and Silva, A. P. (2012). Prevention of methamphetamine-induced microglial cell death by TNF-α and IL-6 through activation of the JAK-STAT pathway. J. Neuroinflammation 9:103.
Cowland, J. B., Sorensen, O. E., Sehested, M., and Borregaard, N. (2003). Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1, but not by TNF-alpha. J. Immunol. 171, 6630–6639. doi: 10.4049/jimmunol.171.12.6630
Dasgupta, S., Jana, M., Liu, X., and Pahan, K. (2003). Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J. Biol. Chem. 278, 22424–22431. doi: 10.1074/jbc.M301789200
Devireddy, L. R., Gazin, C., Zhu, X., and Green, M. R. (2005). A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123, 1293–1305. doi: 10.1016/j.cell.2005.10.027
Du, L., Shen, K., Bai, Y., Chao, J., Hu, G., Zhang, Y., et al. (2019). Involvement of NLRP3 inflammasome in methamphetamine-induced microglial activation through miR-143/PUMA axis. Toxicol. Lett. 301, 53–63. doi: 10.1016/j.toxlet.2018.10.020
Du, S.-H., Qiao, D.-F., Chen, C.-X., Chen, S., Liu, C., Lin, Z., et al. (2017). Toll-Like receptor 4 mediates methamphetamine-induced neuroinflammation through caspase-11 signaling pathway in astrocytes. Front. Mol. Neurosci. 10:409. doi: 10.3389/fnmol.2017.00409
Ejarque-Ortiz, A., Gresa-Arribas, N., Straccia, M., Mancera, P., Sola, C., Tusell, J. M., et al. (2010). CCAAT/enhancer binding protein delta in microglial activation. J. Neurosci. Res. 88, 1113–1123. doi: 10.1002/jnr.22272
Ejarque-Ortiz, A., Medina, M. G., Tusell, J. M., Perez-Gonzalez, A. P., Serratosa, J., and Saura, J. (2007). Upregulation of CCAAT/enhancer binding protein beta in activated astrocytes and microglia. Glia 55, 178–188. doi: 10.1002/glia.20446
El Karoui, K., Viau, A., Dellis, O., Bagattin, A., Nguyen, C., Baron, W., et al. (2016). Endoplasmic reticulum stress drives proteinuria-induced kidney lesions via lipocalin 2. Nat. Commun 7:10330. doi: 10.1038/ncomms10330
Fernandes, N. C., Sriram, U., Gofman, L., Cenna, J. M., Ramirez, S. H., and Potula, R. (2016). Methamphetamine alters microglial immune function through P2X7R signaling. J Neuroinflammation 13:91. doi: 10.1186/s12974-016-0553-3
Ferreira, A. C., Da Mesquita, S., Sousa, J. C., Correia-Neves, M., Sousa, N., Palha, J. A., et al. (2015). From the periphery to the brain: Lipocalin-2, a friend or foe? Prog. Neurobiol. 131, 120–136. doi: 10.1016/j.pneurobio.2015.06.005
Frank, M. G., Adhikary, S., Sobesky, J. L., Weber, M. D., Watkins, L. R., and Maier, S. F. (2016). The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 51, 99–108. doi: 10.1016/j.bbi.2015.08.001
Fujino, R. S., Tanaka, K., Morimatsu, M., Tamura, K., Kogo, H., and Hara, T. (2006). Spermatogonial cell-mediated activation of an IkappaBzeta-independent nuclear factor-kappaB pathway in Sertoli cells induces transcription of the lipocalin-2 gene. Mol. Endocrinol. 20, 904–915. doi: 10.1210/me.2005-2423
Gao, S., Wang, Y., Li, D., Guo, Y., Zhu, M., Xu, S., et al. (2018). TanshinoneIIA alleviates inflammatory response and directs macrophage polarization in lipopolysaccharide-stimulated RAW264.7 cells. Inflammation 42, 264–275. doi: 10.1007/s10753-018-0891-7
Goncalves, J., Leitao, R. A., Higuera-Matas, A., Amparo Assis, M., Coria, S. M., Fontes-Ribeiro, C., et al. (2017). Extended-access methamphetamine self-administration elicits neuroinflammatory response along with blood-brain barrier breakdown. Brain Behav. Immun. 62, 306–317. doi: 10.1016/j.bbi.2017.02.017
Gouzoulis-Mayfrank, E., Hartel-Petri, R., Hamdorf, W., Havemann-Reinecke, U., Muhlig, S., and Wodarz, N. (2017). Methamphetamine-related disorders. Dtsch. Arztebl. Int. 114, 455–461. doi: 10.3238/arztebl.2017.0455
Gresa-Arribas, N., Serratosa, J., Saura, J., and Sola, C. (2010). Inhibition of CCAAT/enhancer binding protein delta expression by chrysin in microglial cells results in anti-inflammatory and neuroprotective effects. J. Neurochem. 115, 526–536. doi: 10.1111/j.1471-4159.2010.06952.x
Grewal, S., Defamie, N., Zhang, X., De Gois, S., Shawki, A., Mackenzie, B., et al. (2009). SNAT2 amino acid transporter is regulated by amino acids of the SLC6 gamma-aminobutyric acid transporter subfamily in neocortical neurons and may play no role in delivering glutamine for glutamatergic transmission. J. Biol. Chem. 284, 11224–11236. doi: 10.1074/jbc.M806470200
Guo, M. L., Liao, K., Periyasamy, P., Yang, L., Cai, Y., Callen, S. E., et al. (2015). Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 11, 995–1009. doi: 10.1080/15548627.2015.1052205
Huang, W., Xie, W. B., Qiao, D., Qiu, P., Huang, E., Li, B., et al. (2015). Caspase-11 plays an essential role in methamphetamine-induced dopaminergic neuron apoptosis. Toxicol. Sci. 145, 68–79. doi: 10.1093/toxsci/kfv014
Huber, R., Pietsch, D., Panterodt, T., and Brand, K. (2012). Regulation of C/EBPbeta and resulting functions in cells of the monocytic lineage. Cell Signal. 24, 1287–1296. doi: 10.1016/j.cellsig.2012.02.007
Jang, E., Kim, J.-H., Lee, S., Kim, J.-H., Seo, J.-W., Jin, M., et al. (2013). Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 191, 5204–5219. doi: 10.4049/jimmunol.1301637
Jha, M. K., Lee, S., Park, D. H., Kook, H., Park, K.-G., Lee, I.-K., et al. (2015). Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci. Biobehav. Rev. 49, 135–156. doi: 10.1016/j.neubiorev.2014.12.006
Jin, D., Zhang, Y., and Chen, X. (2011). Lipocalin 2 deficiency inhibits cell proliferation, autophagy, and mitochondrial biogenesis in mouse embryonic cells. Mol. Cell. Biochem. 351, 165–172. doi: 10.1007/s11010-011-0724-6
Kim, B.-W., Jeong, K. H., Kim, J.-H., Jin, M., Kim, J.-H., Lee, M.-G., et al. (2016). Pathogenic upregulation of glial lipocalin-2 in the parkinsonian dopaminergic system. J. Neurosci. 36, 5608–5622. doi: 10.1523/jneurosci.4261-15.2016
Krasnova, I. N., and Cadet, J. L. (2009). Methamphetamine toxicity and messengers of death. Brain Res. Rev. 60, 379–407. doi: 10.1016/j.brainresrev.2009.03.002
Lee, S., Kim, J. H., Kim, J. H., Seo, J. W., Han, H. S., Lee, W. H., et al. (2011). Lipocalin-2 Is a chemokine inducer in the central nervous system: role of chemokine ligand 10 (CXCL10) in lipocalin-2-induced cell migration. J. Biol. Chem. 286, 43855–43870. doi: 10.1074/jbc.M111.299248
Lee, S., Lee, J., Kim, S., Park, J. Y., Lee, W. H., Mori, K., et al. (2007). A Dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J. Immunol. 179, 3231–3241. doi: 10.4049/jimmunol.179.5.3231
Lee, S., Park, J.-Y., Lee, W.-H., Kim, H., Park, C., Mori, K., et al. (2009). Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J. Neurosci. 29, 234–249. doi: 10.1523/jneurosci.5273-08.2009
Li, B., Chen, R., Chen, L., Qiu, P., Ai, X., Huang, E., et al. (2017). Effects of DDIT4 in methamphetamine-induced autophagy and apoptosis in dopaminergic neurons. Mol. Neurobiol. 54, 1642–1660. doi: 10.1007/s12035-015-9637-9
Marshall, J., Dolan, B. M., Garcia, E. P., Sathe, S., Tang, X., Mao, Z., et al. (2003). Calcium channel and NMDA receptor activities differentially regulate nuclear C/EBPβ levels to control neuronal survival. Neuron 39, 625–639. doi: 10.1016/s0896-6273(03)00496-3
Moszczynska, A., and Callan, S. P. (2017). Molecular, behavioral and physiological consequences methamphetamine neurotoxicity: implications for treatment. J. Pharmacol. Exp. Ther. 362, 474–488. doi: 10.1124/jpet.116.238501
Nakazato, R., Takarada, T., Ikeno, S., Nakamura, S., Kutsukake, T., Hinoi, E., et al. (2015). Upregulation of runt-related transcription factor-2 through CCAAT enhancer binding protein-beta signaling pathway in microglial BV-2 cells exposed to ATP. J. Cell Physiol. 230, 2510–2521. doi: 10.1002/jcp.24988
National Research Council (2011). Guide for the Care and Use of Laboratory Animals, 8th Edn, Washington, DC: National Academies Press, 963–965.
Naude, P. J. W., Csaba, N., Eiden, L. E., Ait-Ali, D., van der Heide, R., Engelborghs, S., et al. (2012). Lipocalin 2: novel component of proinflammatory signaling in Alzheimer’s disease. Faseb J. 26, 2811–2823. doi: 10.1096/fj.11-202457
Pulido-Salgado, M., Vidal-Taboada, J. M., and Saura, J. (2015). C/EBPbeta and C/EBPdelta transcription factors: basic biology and roles in the CNS. Prog. Neurobiol. 132, 1–33. doi: 10.1016/j.pneurobio.2015.06.003
Robson, M. J., Turner, R. C., Naser, Z. J., McCurdy, C. R., Huber, J. D., and Matsumoto, R. R. (2013). SN79, a sigma receptor ligand, blocks methamphetamine-induced microglial activation and cytokine upregulation. Exp. Neurol. 247, 134–142. doi: 10.1016/j.expneurol.2013.04.009
Sekine, Y., Ouchi, Y., Sugihara, G., Takei, N., Yoshikawa, E., Nakamura, K., et al. (2008). Methamphetamine causes microglial activation in the brains of human abusers. J. Neurosci. 28, 5756–5761. doi: 10.1523/JNEUROSCI.1179-08.2008
Shaerzadeh, F., Streit, W. J., Heysieattalab, S., and Khoshbouei, H. (2018). Methamphetamine neurotoxicity, microglia, and neuroinflammation. J. Neuroinflammation 15:341. doi: 10.1186/s12974-018-1385-0
Sharikova, A. V., Quaye, E., Park, J. Y., Maloney, M. C., Desta, H., Thiyagarajan, R., et al. (2018). Methamphetamine induces apoptosis of microglia via the intrinsic mitochondrial-dependent pathway. J. Neuroimmune Pharmacol. 13, 396–411. doi: 10.1007/s11481-018-9787-4
Shen, F., Hu, Z., Goswami, J., and Gaffen, S. L. (2006). Identification of common transcriptional regulatory elements in interleukin-17 target genes. J. Biol. Chem. 281, 24138–24148. doi: 10.1074/jbc.M604597200
Streit, W. J., Walter, S. A., and Pennell, N. A. (1999). Reactive microgliosis. Prog. Neurobiol. 57, 563–581. doi: 10.1016/s0301-0082(98)00069-0
Strohmeyer, R., Shelton, J., Lougheed, C., and Breitkopf, T. (2014). CCAAT-enhancer binding protein-beta expression and elevation in Alzheimer’s disease and microglial cell cultures. PLoS One 9:e86617. doi: 10.1371/journal.pone.0086617
Sunil, V. R., Patel, K. J., Nilsen-Hamilton, M., Heck, D. E., Laskin, J. D., and Laskin, D. L. (2007). Acute endotoxemia is associated with upregulation of lipocalin 24p3/Lcn2 in lung and liver. Exp. Mol. Pathol. 83, 177–187. doi: 10.1016/j.yexmp.2007.03.004
Thion, M. S., Ginhoux, F., and Garel, S. (2018). Microglia and early brain development: an intimate journey. Science 362, 185–189. doi: 10.1126/science.aat0474
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., Liu, X., et al. (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. Embo J. 32, 1917–1926. doi: 10.1038/emboj.2013.122
Urrutia, A., Granado, N., Gutierrez-Lopez, M. D., Moratalla, R., O’Shea, E., and Colado, M. I. (2013). The JNK inhibitor, SP600125, potentiates the glial response and cell death induced by methamphetamine in the mouse striatum. Int. J. Neuropsychopharmacol. 17, 235–246. doi: 10.1017/s1461145713000850
van der Krieken, S. E., Popeijus, H. E., Mensink, R. P., and Plat, J. (2015). CCAAT/enhancer binding protein beta in relation to ER stress, inflammation, and metabolic disturbances. Biomed. Res. Int. 2015:324815. doi: 10.1155/2015/324815
Wang, B., Chen, T., Wang, J., Jia, Y., Ren, H., Wu, F., et al. (2018). Methamphetamine modulates the production of interleukin-6 and tumor necrosis factor-alpha via the cAMP/PKA/CREB signaling pathway in lipopolysaccharide-activated microglia. Int. Immunopharmacol. 56, 168–178. doi: 10.1016/j.intimp.2018.01.024
Winek, C. L., Wahba, W. W., Winek, C. L., and Balzer, T. W. (2001). Drug and chemical blood-level data 2001. Forensic Sci. Int. 122, 107–123. doi: 10.1016/s0379-0738(01)00483-2
Xu, E., Liu, J., Liu, H., Wang, X., and Xiong, H. (2018). Inflammasome activation by methamphetamine potentiates lipopolysaccharide stimulation of IL-1β production in microglia. J. Neuroimmune Pharmacol. 13, 237–253. doi: 10.1007/s11481-018-9780-y
Xu, X., Huang, E., Luo, B., Cai, D., Zhao, X., Luo, Q., et al. (2018). Methamphetamine exposure triggers apoptosis and autophagy in neuronal cells by activating the C/EBPbeta-related signaling pathway. FASEB J. doi: 10.1096/fj.201701460RRR [Epub ahead of print].
Yuan, Y. Q., Wang, Y. L., Yuan, B. S., Yuan, X., Hou, X. O., Bian, J. S., et al. (2018). Impaired CBS-H2S signaling axis contributes to MPTP-induced neurodegeneration in a mouse model of Parkinson’s disease. Brain Behav. Immun. 67, 77–90. doi: 10.1016/j.bbi.2017.07.159
Zahnow, C. A. (2009). CCAAT/enhancer-binding protein beta: its role in breast cancer and associations with receptor tyrosine kinases. Expert. Rev. Mol. Med. 11:e12. doi: 10.1017/S1462399409001033
Keywords: methamphetamine, C/EBP-β, lipocalin2, microglia, apoptosis
Citation: Chen X, Lu J, Zhao X, Chen C, Qiao D, Wang H and Yue X (2019) Role of C/EBP-β in Methamphetamine-Mediated Microglial Apoptosis. Front. Cell. Neurosci. 13:366. doi: 10.3389/fncel.2019.00366
Received: 30 April 2019; Accepted: 29 July 2019;
Published: 21 August 2019.
Edited by:
Arturo Ortega, Center for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV), MexicoReviewed by:
Esther López-Bayghen, Center for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV), MexicoCopyright © 2019 Chen, Lu, Zhao, Chen, Qiao, Wang and Yue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijun Wang, aGp3YW5nQHNtdS5lZHUuY24=; Xia Yue, eWlsdXlhbmdndWFuZ0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.