 Marta Pellegatta
Marta Pellegatta Carla Taveggia
Carla Taveggia- Division of Neuroscience and INSPE at IRCCS San Raffaele Scientific Institute, Milan, Italy
After damage, axons in the peripheral nervous system (PNS) regenerate and regrow following a process termed Wallerian degeneration, but the regenerative process is often incomplete and usually the system does not reach full recovery. Key steps to the creation of a permissive environment for axonal regrowth are the trans-differentiation of Schwann cells and the remodeling of the extracellular matrix (ECM). In this review article, we will discuss how proteases and secretases promote effective regeneration and remyelination. We will detail how they control neuregulin-1 (NRG-1) activity at the post-translational level, as the concerted action of alpha, beta and gamma secretases cooperates to balance activating and inhibitory signals necessary for physiological myelination and remyelination. In addition, we will discuss the role of other proteases in nerve repair, among which A Disintegrin And Metalloproteinases (ADAMs) and gamma-secretases substrates. Moreover, we will present how matrix metalloproteinases (MMPs) and proteases of the blood coagulation cascade participate in forming newly synthetized myelin and in regulating axonal regeneration. Overall, we will highlight how a deeper comprehension of secretases and proteases mechanism of action in Wallerian degeneration might be useful to develop new therapies with the potential of readily and efficiently improve the regenerative process.
Peripheral Nerve Regeneration
In the peripheral nervous system (PNS), nerves can be damaged following various events such as traumatic injuries, metabolic alterations and genetic peripheral neuropathies. All these events significantly impact the quality of life of both patients and caregivers. Regardless of the nature of the triggering cause, after injury both Schwann cells and neurons are compromised as their reciprocal interdependence determines that damaging one cell type will eventually influence the other. Unlike the central nervous system, the PNS possesses the intrinsic capability to regenerate, as axons can regrow over long distances to reach their final target and Schwann cells are able to remyelinate them. Nonetheless, the regenerative process often remains incomplete, due to the inability of the repair machinery to maintain an extended growth promoting response (Gaudet et al., 2011).
After traumatic injury, PNS axons can regrow and remyelinate following a process termed Wallerian degeneration. The cascade of events leading to the axon regrow and the re-establishment of a functional myelinated axo-glia unit has been originally described by August Waller as the result of nerve axotomy (Waller and Owen, 1850), and might partially differ from those observed after crush injury (Stoll et al., 2002; Gaudet et al., 2011; Rotshenker, 2011). In this review article, we will report the role of molecules and modifiers of the extracellular matrix (ECM) in the process of regeneration and remyelination. Since these molecules have been investigated in both models of crush injury and axotomy we will generally refer to their role in peripheral nerve injury.
During early Wallerian degeneration, axons degenerate and Schwann cells adopt a specific response to injury: the repair program (Gerdts et al., 2016). Specifically, denervated Schwann cells lose their myelin sheath and downregulate the expression of myelin-associated genes, among which Egr2/Krox20, MPZ, MBP, MAG and periaxin. In addition, they start to proliferate and upregulate molecules characteristic of their immature stage such as L1, NCAM, p75NTR and GFAP (Jessen and Mirsky, 2008). However, after injury, Schwann cells do not simply revert their phenotype to an earlier developmental stage, but undergo a proper “trans-differentiation” phase in which they acquire unique characteristics, becoming true “repair cells” (Arthur-Farraj et al., 2012; Jessen et al., 2015). In particular, they upregulate neurotrophic factors supporting axon survival and elongation, a necessary step towards regeneration (Fontana et al., 2012; Brushart et al., 2013). In addition, they specifically activate a massive myelin-breakdown and play an early pivotal role in removing myelin debris, which represents a barrier to the regrowth of axons distal to the injury site (Brown et al., 1994; Perry et al., 1995; Hirata and Kawabuchi, 2002; Gomez-Sanchez et al., 2015; Jang et al., 2016). During the first week after injury, repair Schwann cells also produce cytokines that induce macrophages’ activation and recruitment from the bloodstream (Bolin et al., 1995; Kurek et al., 1996; Shamash et al., 2002; Tofaris et al., 2002; Roberts et al., 2017). In the following stages, indeed, macrophages assume a primary role in debris removal and growth factors production, supporting axons regeneration and Schwann cells migration through the distal nerve stump (Gaudet et al., 2011; Gerdts et al., 2016). Within this complex pro-regenerative environment, repair Schwann cells adopt an elongated bipolar morphology and align to form regenerative tracks from the injury site to the nerve target, called “Bungner bands” (Gomez-Sanchez et al., 2017). These structures generate along the basal lamina that previously surrounded the Schwann cell-axon unit in intact nerves and support injured axons regrowth through the distal stump to reach their final target.
Axo-glial communication is essential for nerve repair: not only Schwann cells create a permissive environment for axons regrowth, but also axon-derived signals promote differentiation of repair cells (Fawcett and Keynes, 1990; Stassart et al., 2018). The re-establishment of the axo-glial unit during later phases of the regenerative process allows Schwann cells to re-differentiate and remyelinate the newly generated axon and to re-establish rapid saltatory conduction of the nerve impulse. Nonetheless, remyelinated axons have shorter internodes and thinner myelin sheaths as compared to uninjured nerves (Fancy et al., 2011; Stassart et al., 2018).
Given the inefficiency in the regenerative process, there is an evident need to envisage more effective therapeutic strategies allowing complete recovery. Recent advances in biomaterial and biomedical engineering have provided new strategies to improve regenerative capabilities in injured nerves with synthetic conduits (Tajdaran et al., 2018). These scaffolds support the reconstruction of long nerve gaps, as they allow the application of exogenous agents with neurotrophic properties. Although not pertinent to this review, nerve-scaffolding techniques represent a strategic line of research in nerve regeneration.
Finally, axonal loss and repeated attempts of nerve regeneration are key features also of several peripheral neuropathies, like Charcot Marie Tooth hereditary neuropathies (Stassart et al., 2018). Thus, the development of more adequate treatments to promote nerve repair could similarly benefit these pathologies for which there is no effective therapy.
Neuregulin-1
Neuregulins are transmembrane proteins belonging to the family of epidermal growth factor (EGF)-like growth factors (Falls, 2003). They signal by binding to the tyrosine kinase receptors of the ErbB family expressed on Schwann cells (Yarden and Sliwkowski, 2001), thereby activating different intracellular signal transduction pathways in glial cells (Yang et al., 2004; Gu et al., 2005). Neuregulin-1 (NRG-1) is the most important gene among the members of this family and it is also the best characterized. The Nrg1 gene encodes for several isoforms, all containing an EGF-like domain, which is required to activate the ErbB receptors. All NRG1 isoforms differ mainly for the composition of their N-terminal end, on the base of which they have been classified into six different groups (Garratt et al., 2000; Buonanno and Fischbach, 2001; Mei and Nave, 2014). Distinct NRG1 isoforms use specific promoters to drive their expression in a tight spatio-temporal regulated pattern. Indeed, they are implicated in different cellular function, like migration, proliferation, morphogenesis and control of cell size (Buonanno and Fischbach, 2001; Esper et al., 2006; Britsch, 2007; Mei and Nave, 2014).
Among the various types of Neuregulins, we will focus on NRG1 type I and type III, which are the main isoforms thus far investigated in PNS development and regeneration. NRG1 type I is a single-pass membrane protein and presents an Immunoglobulin-like domain at the N-terminus, while NRG1 type III, in addition to the transmembrane domain, possesses a cysteine-rich domain in the N-terminal portion that further anchors the protein to the plasma membrane (Ho et al., 1995).
In the PNS, NRG1 type III is essentially expressed on axonal membranes (Figure 1). During nerve formation, NRG1 type III is required for Schwann cells proliferation and differentiation (Nave and Salzer, 2006). Indeed, in mutant mice lacking NRG1 type III expression, Schwann cells are absent, and this causes a severe cell death of dorsal root ganglia (DRG) and motor neurons (Wolpowitz et al., 2000). During development, NRG1 type III forces Schwann cells to choose between myelination and non-myelination (Taveggia et al., 2005). Of note, axonal levels of NRG1 type III also determine the thickness of the myelin sheath: mice overexpressing axonal NRG1 type III are hypermyelinated, whereas mice haploinsufficient for NRG1 type III are hypomyelinated (Michailov et al., 2004; Taveggia et al., 2005). Binding of NRG1 to their cognate ErbB2/ErbB3 receptors, activates multiple signal transduction pathways in Schwann cells, including PI3K-AKT1, calcineurin and MAPK signaling pathways (Taveggia et al., 2005, 2010; Kao et al., 2009; Pereira et al., 2012). Of note, NRG1 type III activity is regulated by the extracellular cleavage of secretases (Willem, 2016). While the beta secretase β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) activates NRG1 type III, enhancing myelination (Hu et al., 2006; Willem et al., 2006), cleavage of NRG1 type III by the α-secretase ADAM17 inhibits myelination (La Marca et al., 2011; Bolino et al., 2016). Accordingly, transgenic mice lacking BACE1 are hypomyelinated (Hu et al., 2006; Willem et al., 2006) and mice depleted of neuronal ADAM17 are hypermyelinated, with a phenotype that remarkably resembles NRG1 type III overexpressing mice (Michailov et al., 2004;La Marca et al., 2011).
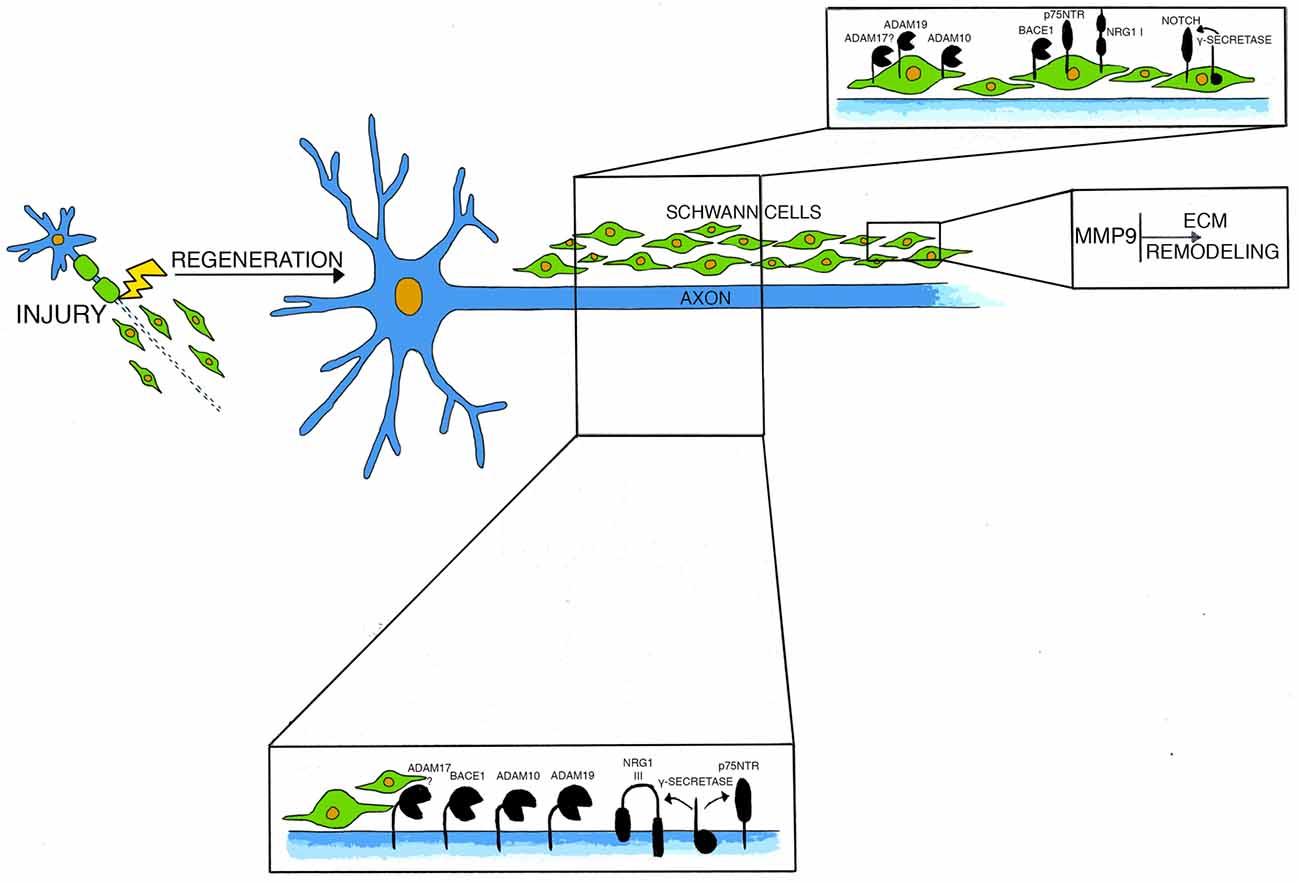
Figure 1. This model depicts molecules and secretases mainly involved in axonal regeneration after injury and their cellular localization.
Following traumatic nerve injury, the expression of several components of the NRG1 signaling machinery is dys-regulated (Birchmeier and Bennett, 2016; Morano et al., 2018). For example, ErbB2/ErbB3 receptors expression is increased after damage and their activation status is enhanced (Cohen et al., 1992; Carroll et al., 1997; Kwon et al., 1997; Guertin et al., 2005), although the effective role of ErbB receptors in peripheral nerve injury is partly controversial (Atanasoski et al., 2006). Moreover, neuronal NRG1 type III expression decreases as soon as the Schwann cells-axon unit is destroyed but it is subsequently re-expressed during late phases of nerve regeneration, when axons re-innervate their target (Bermingham-Mcdonogh et al., 1997; Stassart et al., 2013).
Axonal NRG1 plays an important role in peripheral nerve repair, which, under many aspects, is different from that accomplished during nerve development (Birchmeier and Bennett, 2016). After an injury insult, axonal NRG1 is a driving force for regeneration and it controls the expression of genes involved in peripheral nerve injury (El Soury et al., 2018). Its juxtacrine interactions with Schwann cells, in fact, prompt both remyelination and normal re-innervation of neuromuscular junctions (Fricker et al., 2011). Accordingly, remyelination in mice lacking NRG1 expression in a subset of motor and sensory neurons is severely impaired (Fricker et al., 2011). Unexpectedly, these defects are transient, and axons in mutant nerves are efficiently remyelinated during late nerve regeneration (Fricker et al., 2013). NRG1 therefore is an important factor in regulating nerve repair following injury but, unlike development, it does not control the myelinating fate of axons.
While the above-mentioned studies suggest that axonal NRG1 is dispensable for long term remyelination, it has also been reported that neuronal overexpression of NRG1 type I and type III isoforms facilitates nerve repair and improves remyelination after injury (Stassart et al., 2013). Although these findings seem contradictory, it is possible that gain and loss of function approaches are targeting different mechanisms. Lack of NRG1 after injury might in fact be compensated with time by other molecules, whereas its overexpression could promote Schwann cells re-differentiation and favor nerve regeneration through a distinct process.
Unlike NRG1 type III, NRG1 type I expression is potently induced in Schwann cells early after nerve damage, and although dispensable for developmental myelination, it is critical for remyelination (Figure 1; Loeb et al., 1999; Falls, 2003; Stassart et al., 2013). Indeed, mutant mice specifically lacking NRG1 type I expression in Schwann cells poorly remyelinate after injury, suggesting that Schwann cells require autocrine NRG1 type I signaling to support re-differentiation and repair (Stassart et al., 2013).
Interestingly, administration of exogenous NRG1 type III and NRG1 type I promotes neurite outgrowth and nerve regeneration (Mahanthappa et al., 1996; Cai et al., 2004). Moreover, modulation of NRG1 activity in animal models of CMT1A (Fledrich et al., 2014), CMT1B (Scapin et al., 2018) and Congenital Hypomyelinating Neuropathy (Belin et al., 2018), can ameliorate the peripheral hypomyelination observed in these diseases, prevent axonal loss and promote nerve functional recovery, the latter all features observed also after nerve injury. Nonetheless, it has also been recently reported that NRG1 is already highly expressed in nerves of CMT1A pre-clinical models, thus indicating that delivery of exogenous NRG1 should be carefully evaluated (Fornasari et al., 2018).
Collectively these studies suggest that after damage, Schwann cells might have developed new strategies to accomplish many of their cellular functions. Thus, a better understanding of the molecular mechanism underlying their extraordinary plasticity remains crucial towards the development of powerful therapies leading to complete regeneration.
BACE1
The beta secretase BACE1 is an aspartyl protease mostly involved in processing type I transmembrane proteins, which results in the generation of a cleaved, membrane-bound portion of the target molecule and in the release of a soluble fragment. BACE1 has been well characterized in the pathogenesis of Alzheimer’s disease, since it is one of the main enzymes responsible for the processing of the amyloid precursor protein and for the generation of amyloid beta plaques, a pathological hallmark of the disease (Sinha and Lieberburg, 1999; Vassar et al., 1999; Yan et al., 1999; Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001).
Besides the amyloid precursor protein, BACE1 cleaves a variety of substrates (Hemming et al., 2009). Among them, in the nervous system, there are voltage-gated sodium channels (Wong et al., 2005; Kim et al., 2007, 2011; Gersbacher et al., 2010), the potassium channel proteins KCNE1 and KCNE2 (Hitt et al., 2012; Sachse et al., 2013), the adhesion molecules L1 and close homolog of L1 (CHL1; Hitt et al., 2012; Kuhn et al., 2012; Zhou et al., 2012), contactin-2 (Kuhn et al., 2012) and Seizure-related gene 6 protein (Sez 6; Kuhn et al., 2012). Processing of these substrates by BACE1 perturbs several brain functions, as these proteins are involved in different tasks. Indeed, BACE1 is crucial to generate axonal connections (Hitt et al., 2012), regulate axonal guidance (Kuhn et al., 2012; Zhou et al., 2012) and modulate neurite outgrowth and neural connectivity (Kuhn et al., 2012). Surprisingly, despite being implicated in processing many substrates, constitutive ablation of BACE1 in mice results in viable and fertile animals, with only moderate behavioral abnormalities (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001; Laird et al., 2005; Savonenko et al., 2008).
In the PNS, BACE1 is mainly expressed in motor neurons and in DRG sensory neurons (Willem et al., 2006) and its expression is highly similar to that of NRG1 type III. Nonetheless, BACE1 is present at very low levels also in Schwann cells in normal nerves and at higher levels in damaged nerves (Hu et al., 2015; Figure 1). Constitutive deletion of BACE1 results in severe myelination defects during development (Hu et al., 2006; Willem et al., 2006; Velanac et al., 2012). Interestingly, NRG1 cleavage is impaired in BACE1 null mice (Hu et al., 2006; Willem et al., 2006), and the hypomyelinating phenotype resembles that observed in NRG1 type III haploinsufficient mice (Michailov et al., 2004; Taveggia et al., 2005). Accordingly, in peripheral nerves, BACE1 directly cleaves axonal NRG1 type III, generating a membrane-tethered portion of the molecule that exposes the EGF-like domain allowing the activation of the ErbB2/ErbB3 receptors complex on Schwann cells, thereby promoting myelination (Hu et al., 2008; Velanac et al., 2012).
BACE1 is also implicated in peripheral nerve injury; specifically, in the absence of BACE1 remyelination is impaired. Accordingly, injured nerves of BACE1 null mice have thinner myelin sheath and increased number of un-myelinated axons (Hu et al., 2008). These defects have been ascribed to aberrant processing of axonal NRG1 type III, which impairs PI3K/AKT1 signaling pathway activity (Hu et al., 2008). More recently, also the contribution of glial BACE1, i.e., expressed in Schwann cells, has been investigated in nerve remyelination (Hu et al., 2015). Nerve graft experiments suggest that both axonal and Schwann cell-derived BACE1 activity are decisive for remyelination (Hu et al., 2015). Interestingly, since both BACE1 and NRG1 type I are induced in Schwann cells after injury (Hu et al., 2008, 2015; Stassart et al., 2013), it is likely that glial BACE1 contributes to nerve repair by promoting NRG1 type I cleavage (Hu et al., 2015), though this has never been formally demonstrated.
Other studies reported an opposite role for BACE1 in PNS remyelination. By analyzing early regenerative events, Farah et al. (2011) observed that BACE1 knockout mice present enhanced myelin and axonal debris clearance, an increased number of regenerated fibers and precocious re-innervation of neuromuscular junctions. These phenomena are likely due to neuronal BACE1, as overexpression of BACE1 in neurons determines a marked decrease in peripheral axons regeneration capacity (Tallon et al., 2017). These data suggest that neuronal BACE1 could act as a negative regulator of axonal regeneration in early events following injury, contradicting previous studies. To reconcile these conflicting results and to better clarify the role of BACE1 in nerve regeneration and remyelination, further studies in conditional knockout mice allowing ablation in defined temporal windows are necessary. Moreover, since BACE1 processes many substrates, which are expressed in different cell types and at specific stages of the repair process, the study of its cell autonomous role could help in defining the effector(s) through which BACE1 regulates specific stages of axonal regeneration and remyelination.
Since BACE1 has a prominent role in the generation of amyloid beta plaques during AD pathogenesis, a lot of effort has been devoted in developing small molecules that could inhibit the enzyme’s activity (Coimbra et al., 2018), however only one study has tested their efficacy in a model of nerve crush injury. A 7 days treatment with a BACE1 inhibitor starting immediately after damage increases regeneration and enhances myelin debris clearance (Farah et al., 2011), supporting the hypothesis that BACE1 inhibits early axonal regeneration (Farah et al., 2011; Tallon et al., 2017). Although the possibility of using BACE1 inhibitors to boost axonal regeneration is potentially promising, it is essential to better define their application, given the contradictory role of BACE1 after injury (Hu et al., 2008, 2015). Thus, further studies are required to exactly determine the efficacy of BACE1 inhibitor(s) in the resolution of PNS injury. Particularly relevant would be to define the exact time frame in which to administer these compounds to promote axonal growth without altering remyelination.
A Disintegrin and Metalloproteinases
A Disintegrin And Metalloproteinases (ADAMs) are membrane-anchored metalloproteinases involved in the proteolytic processing of several transmembrane proteins and in the consequent regulation of the cleaved substrates’ activity (Schlöndorff and Blobel, 1999). ADAM proteases have both proteolytic and disintegrin characteristics (Schlöndorff and Blobel, 1999). They present a conserved structure composed by distinct domains (Schlöndorff and Blobel, 1999; Primakoff and Myles, 2000) and have been implicated in the release of several molecules among which EGF receptor ligands, tumor necrosis factor family members, neuregulins and other transmembrane proteins (Black and White, 1998; Blobel, 2000). ADAM proteins participate in many biological functions such as cell migration, cell membrane fusion and cytokines and growth factors shedding. Moreover, members of this family cooperate in muscle development, fertilization, neurogenesis, angiogenesis and cell fate determination (Schlöndorff and Blobel, 1999; Primakoff and Myles, 2000; Horiuchi et al., 2003; Seals and Courtneidge, 2003). Pathologies like inflammation-related disorders and cancer also implicate ADAM proteins (Seals and Courtneidge, 2003; Blobel, 2005).
It is thus not surprising that ADAM secretases have a role also in PNS myelination. In particular, the α-secretase ADAM17 possesses an intrinsic proteolytic activity, which inhibits NRG1 type III and the process of myelination during development. Thus, only a fine balance between activating and inhibitory signals coming from the activity of BACE1 and ADAM17 could physiologically regulate the correct formation of the myelin sheath (La Marca et al., 2011).
Not only ADAM17, but also ADAM10 and ADAM19, play important roles in PNS functioning (Figure 1). In particular, ADAM10 is highly expressed in Schwann-cells DRG neuronal co-cultures during myelination and its chemical inhibition impairs axonal outgrowth in vitro (Jangouk et al., 2009). Nonetheless, in vivo studies have shown that ADAM10 is dispensable for developmental myelination. In fact, both overexpression of a dominant negative form of the protein (Freese et al., 2009) as well as its conditional inactivation in a transgenic model (Meyer zu Horste et al., 2015) do not alter myelination. Interestingly, ADAM10 promotes axonal sprouting, as its deficiency in motor neurons impairs the extent of axons growth in vitro (Meyer zu Horste et al., 2015). Moreover, this secretase is critical for axonal outgrowth during peripheral nerve regeneration as deletion of ADAM10 in motor neurons reduces the axonal density of small caliber fibers after crush injury. Thus, ADAM10 seems mainly implicated in the specific control of post-traumatic outgrowth of axons (Meyer zu Horste et al., 2015).
ADAM19 is highly expressed in developing PNS, and its expression pattern resembles that of NRG1 (Kurisaki et al., 1998; Shirakabe et al., 2001); both proteins are co-expressed in DRG and in motor neurons (Marchionni et al., 1993; Shirakabe et al., 2001). Of note, ADAM19 can cleave membrane-anchored NRG1 proteins in cultured neurons (Kurisaki et al., 1998; Shirakabe et al., 2001; Wakatsuki et al., 2004). In vivo studies have shown that while ADAM19 is dispensable for PNS development, remyelination after nerve injury is impaired in its absence (Wakatsuki et al., 2009). Further, after injury, ADAM19 regulates the re-differentiation of Schwann cells from a pro-myelinating stage to a fully myelinating one by activating the AKT pathway (Wakatsuki et al., 2009).
Although ADAM10 and ADAM19 participate in the progression of nerve regeneration, nerve repair is eventually achieved in both mutants. It is thus possible that compensatory mechanisms intervene to favor remyelination, likely carried out by other secretases. Since ADAM17 is fundamental in nerve development and NRG1 processing, it would be important to assess the role of this secretase during nerve regeneration.
Gamma Secretases
Gamma secretases (γ-secretases) are a multi-subunit protease complex responsible for the intramembrane cleavage of transmembrane proteins, mostly known for the generation of the β-amyloid peptide in Alzheimer’s disease (De Strooper et al., 1998; Wolfe et al., 1999). Beside the amyloid precursor protein, γ-secretases process several type I transmembrane proteins, among which the adhesion molecules N-cadherin and E-cadherin (Struhl and Adachi, 2000; Golde and Eckman, 2003), the neurotrophin receptor p75 (p75NTR; Jung et al., 2003), the β2 subunit of voltage-gated sodium channel (Kim et al., 2005), the protein receptor Notch (De Strooper et al., 1999) and Neuregulins (Dejaegere et al., 2008; Trimarco et al., 2014). Although no direct functional role of the γ-secretase complex has been described in peripheral nerve regeneration, some of its substrates are relevant for such process, in particular Notch and p75NTR.
Notch activity is regulated by a classical regulated intramembrane proteolysis (RIP) mechanism (Figure 1). After binding to its substrates, Notch is first cleaved in its extracellular domain (Pan and Rubin, 1997; Brou et al., 2000; Hartmann et al., 2002; van Tetering et al., 2009); this cleavage induces a subsequent process in the transmembrane domain, which is mediated by the γ-secretase complex (De Strooper et al., 1999; Struhl and Greenwald, 1999). Notch intracellular domain then translocates in the cell nucleus, where it regulates the transcription of genes important for cell development and differentiation (Kopan and Goate, 2000; Kopan and Ilagan, 2004; Louvi and Artavanis-Tsakonas, 2006). In peripheral nerves, Notch negatively regulates developmental myelination (Woodhoo et al., 2009). After injury, Notch is rapidly upregulated in peripheral nerves and, in a mouse model in which Notch is conditionally ablated in Schwann cells, the rate of Schwann cells dedifferentiation after axotomy is reduced (Woodhoo et al., 2009). This impairment relies on Notch intracellular domain, as a similar defect is found also in mice deficient in Schwann cells RBPJ (Recombination signal Binding Protein for Immunoglobulin Kappa J region), the key transcriptional mediator of canonical Notch intracellular signaling (Woodhoo et al., 2009).
p75NTR regulates a variety of cellular function in the nervous system, from programmed cell death, axonal growth and degeneration, to cell proliferation, myelination and synaptic plasticity (Kraemer et al., 2014; Meeker and Williams, 2015). This wide range of biological effects is due to the multiplicity of ligands and co-receptors which partner with p75NTR to regulate its signaling. In particular, p75NTR promotes cells survival through association with TRK receptors (Hempstead et al., 1991; Meeker and Williams, 2015; Pathak et al., 2018), inhibits axonal regeneration by interacting with Nogo receptor and Lingo (Wang et al., 2002; Wong et al., 2002; Mi et al., 2004) and induces apoptosis via high affinity binding with Sortilin (Friedman, 2000; Beattie et al., 2002; Nykjaer et al., 2004). Similarly to Notch, also p75NTR is regulated by RIP: upon ligand binding, the extracellular domain of the receptor is first cleaved by ADAM17 (Weskamp et al., 2004) and then processed by the γ-secretase complex, thereby releasing a small intracellular domain (Zampieri et al., 2005). p75NTR is highly expressed in Schwann cells and neurons during development, and its expression levels decline with age (Cosgaya et al., 2002). Both nerve crush injury and axotomy induce a strong upregulation of p75NTR in neurons and Schwann cells in distal nerve segments (Ernfors et al., 1989; Koliatsos et al., 1991; Saika et al., 1991; Syroid et al., 2000; Petratos et al., 2003; Figure 1). The role of p75NTR after injury is complex and partially contradictory (for a comprehensive review see Meeker and Williams, 2015), nonetheless, grafting of Schwann cells deficient for p75NTR impairs motor neuron survival after injury (Tomita et al., 2007). Moreover, remyelination is hampered in mice lacking p75NTR and nerve repair is delayed (Song et al., 2006; Chen et al., 2016). Thus, p75NTR likely has a pivotal role in supporting neuronal survival, nerve regeneration and remyelination after injury.
In addition to the above-described roles, p75NTR also regulates fibrinolysis. Upon nerve injury, fibrin accumulates in the nerves to promote axon degeneration and Schwann cells dedifferentiation. However, to achieve efficient regeneration, fibrin has to be degraded in a process called fibrinolysis (Akassoglou et al., 2002). In injured nerves, accumulation of p75NTR in Schwann cells inhibits fibrinolysis thus maintaining Schwann cells in a un-differentiated state (Akassoglou et al., 2002; Sachs et al., 2007). During nerve regeneration, fibrin is degraded, p75NTR is downregulated and Schwann cells can re-differentiate to initiate remyelination (Akassoglou et al., 2000; Sachs et al., 2007; Figure 2). Thus, the biological role of p75NTR in axonal regeneration and remyelination is complex and probably depends on the relative contribution of its function as a regulator of cell death/survival and as an inhibitor of fibrin degradation.
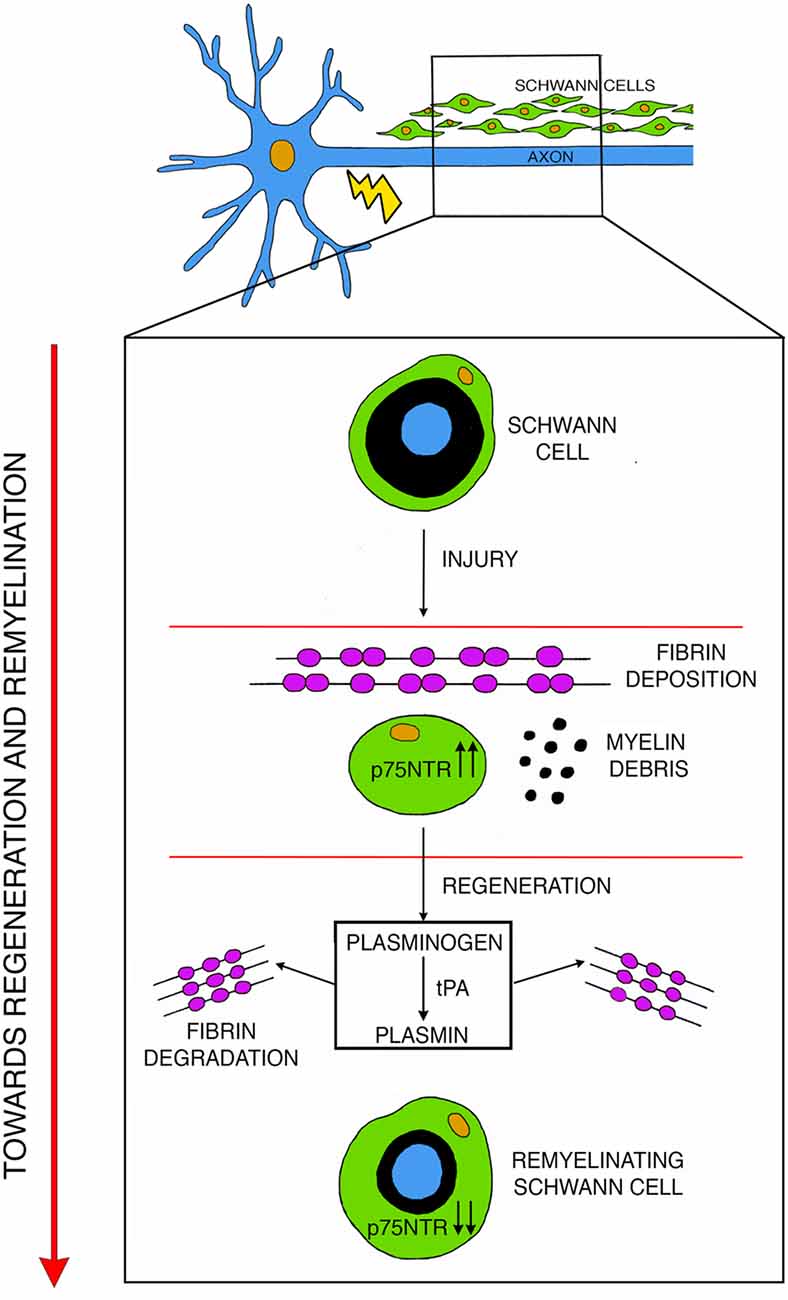
Figure 2. After injury, fibrin deposits inside the nerve while Schwann cells undergo de-differentiation and upregulate p75NTR. During active phases of regeneration p75NTR expression is diminished in remyelinating Schwann cells, and the induction of the plasminogen and of the tissue plasminogen activator (tPA)/plasmin cascade leads to fibrin degradation thus promoting remyelination. Modified with permission from Akassoglou et al. (2002).
Of note, the γ-secretase complex processes also axonal NRG1 type III (Bao et al., 2003, 2004). As already described, the extracellular processing of NRG1 type III prompts a forward signaling to initiate myelination (Hu et al., 2006; Willem et al., 2006; La Marca et al., 2011). The γ-secretase intracellular cleavage of axonal NRG1 type III, instead, generates a NRG1-ICD intracellular fragment, which activates the transcription of the lipocalin-type prostaglandin-D synthase (L-Pgds) gene (Trimarco et al., 2014). L-PGDS further induces a pro-myelinating pathway in Schwann cells controlling formation and maintenance of the myelin sheath (Trimarco et al., 2014). Given the importance of NRG1 in nerve regeneration, additional studies to clarify the contribution of NRG1-ICD and L-PGDS after injury could contribute to the understanding of the processes involved in nerve repair.
Matrix Metalloproteinases
The matrix metalloproteinases (MMPs) are a family of zinc-dependent enzymes, which can degrade all components of the ECM (McCawley and Matrisian, 2001). At least 24 members, including collagenases, gelatinases, matrilysins, stromelysins and membrane-type MMPs belong to the MMPs family (Nagase et al., 2006). Proteolytic MMPs activity is necessary to regulate expression levels and function of ECM components and cell surface signaling receptors (Page-McCaw et al., 2007).
Among the various MMPs, MMP-2 and MMP-9 have been implicated in nerve development, and after injury (Platt et al., 2003). During development, MMP-2 and MMP-9 process dystroglycan to facilitate the formation of the dystroglycan receptor complex, a laminin receptor required for the correct formation of apposition and Cajal bands along myelinated fibers (Court et al., 2011).
Upon axotomy, MMP-9 mRNA levels are rapidly upregulated starting few hours after injury and peak at day 3 (La Fleur et al., 1996; Siebert et al., 2001; Hughes et al., 2002; Platt et al., 2003) while its activity increases already 12 h after axotomy, peaks around day 2 and returns to basal levels after 4 days (Siebert et al., 2001). In a model of crush nerve injury, MMP-9 localizes in myelinating Schwann cells, immune cells and endothelial cells (La Fleur et al., 1996; Shubayev and Myers, 2000; Hughes et al., 2002; Figure 1). During nerve regeneration, MMP-9 negatively controls Schwan cells proliferation (Chattopadhyay et al., 2007). Accordingly, nerves of mice lacking MMP-9 have increased numbers of post-mitotic Schwann cells, together with shorter internodes and nodal abnormalities (Kim et al., 2012). In vivo studies in mutants lacking MMP-9 indicate that MMP-9 activity during axonal degeneration is crucial to promote myelin sheath collapse and to prompt the blood-nerve-barrier breakdown allowing the influx of immune cells into the nerve (Shubayev and Myers, 2000; Chattopadhyay et al., 2007; Kobayashi et al., 2008). More recent studies have implicated also MMP-9, together with MMP-7, in regulating claudin proteins expression at the tight junction in nerves after crush injury, suggesting that high levels of MMPs might alter nerve physical barriers (Wang et al., 2018). Further, MMP-9 stimulates the rearrangement of the basal lamina (La Fleur et al., 1996; Hartung and Kieseier, 2000; Platt et al., 2003; Shubayev et al., 2006; Chattopadhyay et al., 2007; Kobayashi et al., 2008).
Interestingly, together with MMP-9, also its endogenous inhibitor TIMP-1 is upregulated in nerves early after injury (Kim et al., 2012); thus, it has been proposed that a fine balance between MMP-9 and TIMP-1 activity might regulate Schwann cells mitogenesis and maturation and control the molecular and structural assembly of myelin domains in remyelinated fibers to prompt a correct nerve repair (Kim et al., 2012). Thus, during early phases of axonal degeneration, a physiological inhibition of MMP-9 activity might be precisely coupled with its activation to coordinate an effective nerve repair.
Plasminogen Activators and Related Proteases
The plasminogen activators (PAs), namely tissue PA (tPA) and urokinase PA (uPA), are key enzymes of the blood coagulation cascade implicated in several biological processes, including vascular and tissue remodeling, tumor development and progression (Mekkawy et al., 2014) and nervous system pathophysiology (Deryugina and Quigley, 2012; Thiebaut et al., 2018). These secreted proteases cleave the proenzyme plasminogen to turn it into its active form, plasmin (Collen, 1999; Rijken and Lijnen, 2009). Plasmin is a serine protease involved in processing a vast spectrum of molecules (Liotta et al., 1981), but its main substrate is fibrin (Bugge et al., 1996). Plasmin, in fact, degrades fibrin clots allowing scar resolution (Weisel and Litvinov, 2017). In addition, plasmin can activate other proteins, like MMPs (Keski-Oja et al., 1992; Murphy and Docherty, 1992; Zou et al., 2006) and growth factors (Saksela and Rifkin, 1990; Brauer and Yee, 1993; Munger et al., 1997). In the PNS, PAs activity is elevated during axonal outgrowth. PAs are secreted by primary neurons and Schwann cells in culture (Krystosek and Seeds, 1981, 1984) and their activity is localized in growth cones (Krystosek and Seeds, 1984). Moreover, the PA system is induced in vivo in embryonic DRG and motor neurons during axonal outgrowth (Seeds et al., 1992; Sumi et al., 1992). Interestingly, the PA system is potently induced also after peripheral nerve injury in mice (Siconolfi and Seeds, 2001a,b). Regeneration of peripheral nerves relies on the ability of axons to regrow in a very complex environment at the injury site, made of a structurally altered ECM, myelin and axon debris, surrounding tissues and infiltrating inflammatory cells. These adverse conditions represent an obstacle for axonal growth, thus regenerating neurites secrete PAs to dissolve cell-cell and cell-matrix adhesion. Accordingly, tPA and uPA mRNAs are promptly upregulated early after peripheral nerve injury in Schwann cells and neurons and their activity increases at the injury site up to 7 days post-crush (Akassoglou et al., 2000; Siconolfi and Seeds, 2001a; Figure 2). Moreover, recovery in knockout mice deficient for tPA, uPA and Plasminogen is significantly delayed after nerve injury, indicating that they are necessary for timely regeneration of peripheral nerves and the re-establishment of peripheral sensitivity (Siconolfi and Seeds, 2001b). Plasmin is therefore protective for nerve injury (Akassoglou et al., 2000) and this effect is linked to the proteolytic activity plasmin exerts on fibrin (Figure 2). Accordingly, the vast axonal damage observed in tPA and plasminogen knockout nerves correlates with excessive accumulation of fibrin. Interestingly, genetic and pharmacological depletion of fibrinogen, the fibrin molecular precursor, enhances regeneration in these mutants (Akassoglou et al., 2000). Thus, fibrin deposition exacerbates axonal injury and the physiological induction of the tPA/plasmin cascade after injury prompts fibrin removal and stimulates repair (Figure 2). Of note, the important role of this cascade has been described also in humans, as there is a direct correlation between the extent of fibrinolytic activity and the degree of regeneration in patients with peripheral neuropathies (Previtali et al., 2008; Rivellini et al., 2012).
In addition to the PA system, thrombin, another key enzyme of the blood coagulation system, and its inhibitor Nexin-1, are potently induced after sciatic nerve injury. In the blood coagulation cascade, thrombin converts fibrinogen into fibrin, its insoluble derivative (Weisel and Litvinov, 2017). Nexin-1, instead, a member of the large serpin superfamily, directly inhibits thrombin proteolytic activity (Cavanaugh et al., 1990; Gurwitz and Cunningham, 1990). In injured sciatic nerves, thrombin levels are elevated immediately after damage and reach their peak 1 day post-injury, declining after one week (Smirnova et al., 1996; Gera et al., 2016). On the contrary, upregulation of Nexin-1 in injured nerves occurs 6–9 days after thrombin induction (Meier et al., 1989; Smirnova et al., 1996) and is required to neutralize excessive thrombin proteolytic activity.
Collectively, these studies indicate that the proteases of the blood coagulation system and their regulators are essential to stimulate recovery in injured PNS and a precise balance between activation and inhibition of their proteolytic activity is required to correctly modulate nerve repair.
Concluding Remarks
In the present review article, we presented a selected range of molecules and pathways involved in injured PNS. We specifically focused on extra cellular matrix and tissue modifiers and on growth factors’ processing. Several studies have demonstrated how crucial is the role of these enzymes in tissue remodeling in response to damage. Nonetheless, despite the extensive studies conducted thus far, additional work is required to better define both the cell autonomy and the time frame in which these molecules and pathways are mostly active. We thus think that clarifying these aspects could be beneficial to define new therapeutic strategies that, together with the development of conduit devices, could efficiently improve the regenerative process and prompt full functional recovery.
Author Contributions
MP and CT searched PubMed for articles published up until August 2018. Both MP and CT discussed the topics, decided the outline of the review and wrote the manuscript.
Funding
The studies from our laboratory are supported by grants from the Italian Ministry of Health (Ministero della Salute) RF PE 2013-02355206 (CT), Italy; Fondazione Telethon award number GPP 14040 and GGP 15012 (CT), Italy; the National Institutes of Health (NIH) NS099102-02, USA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Maria Grazia Forese for extensive discussions and suggestions. We apologize to colleagues whose relevant work could not be cited because of space limitations.
References
Akassoglou, K., Kombrinck, K. W., Degen, J. L., and Strickland, S. (2000). Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J. Cell Biol. 149, 1157–1166. doi: 10.1083/jcb.149.5.1157
Akassoglou, K., Yu, W. M., Akpinar, P., and Strickland, S. (2002). Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron 33, 861–875. doi: 10.1016/s0896-6273(02)00617-7
Arthur-Farraj, P. J., Latouche, M., Wilton, D. K., Quintes, S., Chabrol, E., Banerjee, A., et al. (2012). c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647. doi: 10.1016/j.neuron.2012.06.021
Atanasoski, S., Scherer, S. S., Sirkowski, E., Leone, D., Garratt, A. N., Birchmeier, C., et al. (2006). ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. J. Neurosci. 26, 2124–2131. doi: 10.1523/JNEUROSCI.4594-05.2006
Bao, J., Lin, H., Ouyang, Y., Lei, D., Osman, A., Kim, T. W., et al. (2004). Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat. Neurosci. 7, 1250–1258. doi: 10.1038/nn1342
Bao, J., Wolpowitz, D., Role, L. W., and Talmage, D. A. (2003). Back signaling by the Nrg-1 intracellular domain. J. Cell Biol. 161, 1133–1141. doi: 10.1083/jcb.200212085
Beattie, M. S., Harrington, A. W., Lee, R., Kim, J. Y., Boyce, S. L., Longo, F. M., et al. (2002). ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36, 375–386. doi: 10.1016/s0896-6273(02)01005-x
Belin, S., Ornaghi, F., Shackleford, G., Wang, J., Scapin, C., Lopez-Anido, C., et al. (2018). Neuregulin 1 type III improves peripheral nerve myelination in a mouse model of congenital hypomyelinating neuropathy. Hum. Mol. Genet. doi: 10.1093/hmg/ddy420 [Epub ahead of print].
Bermingham-Mcdonogh, O., Xu, Y. T., Marchionni, M. A., and Scherer, S. S. (1997). Neuregulin expression in PNS neurons: isoforms and regulation by target interactions. Mol. Cell. Neurosci. 10, 184–195. doi: 10.1006/mcne.1997.0654
Birchmeier, C., and Bennett, D. L. (2016). Neuregulin/ErbB signaling in developmental myelin formation and nerve repair. Curr. Top. Dev. Biol. 116, 45–64. doi: 10.1016/bs.ctdb.2015.11.009
Black, R. A., and White, J. M. (1998). ADAMs: focus on the protease domain. Curr. Opin. Cell Biol. 10, 654–659. doi: 10.1016/s0955-0674(98)80042-2
Blobel, C. P. (2000). Remarkable roles of proteolysis on and beyond the cell surface. Curr. Opin. Cell Biol. 12, 606–612. doi: 10.1016/s0955-0674(00)00139-3
Blobel, C. P. (2005). ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43. doi: 10.1038/nrm1548
Bolin, L. M., Verity, A. N., Silver, J. E., Shooter, E. M., and Abrams, J. S. (1995). Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J. Neurochem. 64, 850–858. doi: 10.1046/j.1471-4159.1995.64020850.x
Bolino, A., Piguet, F., Alberizzi, V., Pellegatta, M., Rivellini, C., Guerrero-Valero, M., et al. (2016). Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol. Med. 8, 1438–1454. doi: 10.15252/emmm.201606349
Brauer, P. R., and Yee, J. A. (1993). Cranial neural crest cells synthesize and secrete a latent form of transforming growth factor β that can be activated by neural crest cell proteolysis. Dev. Biol. 155, 281–285. doi: 10.1006/dbio.1993.1026
Britsch, S. (2007). The neuregulin-I/ErbB signaling system in development and disease. Adv. Anat. Embryol. Cell Biol. 190, 1–65. doi: 10.1007/978-3-540-37107-6
Brou, C., Logeat, F., Gupta, N., Bessia, C., Lebail, O., Doedens, J. R., et al. (2000). A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol. Cell 5, 207–216. doi: 10.1016/S1097-2765(00)80417-7
Brown, M. C., Perry, V. H., Hunt, S. P., and Lapper, S. R. (1994). Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration. Eur. J. Neurosci. 6, 420–428. doi: 10.1111/j.1460-9568.1994.tb00285.x
Brushart, T. M., Aspalter, M., Griffin, J. W., Redett, R., Hameed, H., Zhou, C., et al. (2013). Schwann cell phenotype is regulated by axon modality and central-peripheral location and persists in vitro. Exp. Neurol. 247, 272–281. doi: 10.1016/j.expneurol.2013.05.007
Bugge, T. H., Kombrinck, K. W., Flick, M. J., Daugherty, C. C., Danton, M. J., and Degen, J. L. (1996). Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 87, 709–719. doi: 10.1016/s0092-8674(00)81390-2
Buonanno, A., and Fischbach, G. D. (2001). Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr. Opin. Neurobiol. 11, 287–296. doi: 10.1016/s0959-4388(00)00210-5
Cai, J., Peng, X., Nelson, K. D., Eberhart, R., and Smith, G. M. (2004). Synergistic improvements in cell and axonal migration across sciatic nerve lesion gaps using bioresorbable filaments and heregulin-β1. J. Biomed. Mater. Res. A 69, 247–258. doi: 10.1002/jbm.a.20119
Cai, H., Wang, Y., Mccarthy, D., Wen, H., Borchelt, D. R., Price, D. L., et al. (2001). BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 4, 233–234. doi: 10.1038/85064
Carroll, S. L., Miller, M. L., Frohnert, P. W., Kim, S. S., and Corbett, J. A. (1997). Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J. Neurosci. 17, 1642–1659. doi: 10.1523/JNEUROSCI.17-05-01642.1997
Cavanaugh, K. P., Gurwitz, D., Cunningham, D. D., and Bradshaw, R. A. (1990). Reciprocal modulation of astrocyte stellation by thrombin and protease nexin-1. J. Neurochem. 54, 1735–1743. doi: 10.1111/j.1471-4159.1990.tb01228.x
Chattopadhyay, S., Myers, R. R., Janes, J., and Shubayev, V. (2007). Cytokine regulation of MMP-9 in peripheral glia: implications for pathological processes and pain in injured nerve. Brain Behav. Immun. 21, 561–568. doi: 10.1016/j.bbi.2006.10.015
Chen, X., Zhang, J., Wang, X., and Bi, J. (2016). Functional recovery from sciatic nerve crush injury is delayed because of increased distal atrophy in mice lacking the p75 receptor. Neuroreport 27, 940–947. doi: 10.1097/WNR.0000000000000635
Cohen, J. A., Yachnis, A. T., Arai, M., Davis, J. G., and Scherer, S. S. (1992). Expression of the neu proto-oncogene by Schwann cells during peripheral nerve development and Wallerian degeneration. J. Neurosci. Res. 31, 622–634. doi: 10.1002/jnr.490310406
Coimbra, J. R. M., Marques, D. F. F., Baptista, S. J., Pereira, C. M. F., Moreira, P. I., Dinis, T. C. P., et al. (2018). Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 6:178. doi: 10.3389/fchem.2018.00178
Collen, D. (1999). The plasminogen (fibrinolytic) system. Thromb. Haemost. 82, 259–270. doi: 10.1055/s-0037-1615841
Cosgaya, J. M., Chan, J. R., and Shooter, E. M. (2002). The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298, 1245–1248. doi: 10.1126/science.1076595
Court, F. A., Zambroni, D., Pavoni, E., Colombelli, C., Baragli, C., Figlia, G., et al. (2011). MMP2-9 cleavage of dystroglycan alters the size and molecular composition of Schwann cell domains. J. Neurosci. 31, 12208–12217. doi: 10.1523/JNEUROSCI.0141-11.2011
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., et al. (1999). A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. doi: 10.1038/19083
De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., et al. (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390. doi: 10.1038/34910
Dejaegere, T., Serneels, L., Schäfer, M. K., Van Biervliet, J., Horre, K., Depboylu, C., et al. (2008). Deficiency of Aph1B/C-γ-secretase disturbs Nrg1 cleavage and sensorimotor gating that can be reversed with antipsychotic treatment. Proc. Natl. Acad. Sci. U S A 105, 9775–9780. doi: 10.1073/pnas.0800507105
Deryugina, E. I., and Quigley, J. P. (2012). Cell surface remodeling by plasmin: a new function for an old enzyme. J. Biomed. Biotechnol. 2012:564259. doi: 10.1155/2012/564259
El Soury, M., Fornasari, B. E., Morano, M., Grazio, E., Ronchi, G., Incarnato, D., et al. (2018). Soluble neuregulin1 down-regulates myelination genes in schwann cells. Front. Mol. Neurosci. 11:157. doi: 10.3389/fnmol.2018.00157
Ernfors, P., Henschen, A., Olson, L., and Persson, H. (1989). Expression of nerve growth factor receptor mRNA is developmentally regulated and increased after axotomy in rat spinal cord motoneurons. Neuron 2, 1605–1613. doi: 10.1016/0896-6273(89)90049-4
Esper, R. M., Pankonin, M. S., and Loeb, J. A. (2006). Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res. Rev. 51, 161–175. doi: 10.1016/j.brainresrev.2005.11.006
Falls, D. (2003). Neuregulins: functions, forms and signaling strategies. Exp. Cell Res. 284, 14–30. doi: 10.1016/s0014-4827(02)00102-7
Fancy, S. P., Chan, J. R., Baranzini, S. E., Franklin, R. J., and Rowitch, D. H. (2011). Myelin regeneration: a recapitulation of development? Annu. Rev. Neurosci. 34, 21–43. doi: 10.1146/annurev-neuro-061010-113629
Farah, M. H., Pan, B. H., Hoffman, P. N., Ferraris, D., Tsukamoto, T., Nguyen, T., et al. (2011). Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J. Neurosci. 31, 5744–5754. doi: 10.1523/JNEUROSCI.6810-10.2011
Fawcett, J. W., and Keynes, R. J. (1990). Peripheral nerve regeneration. Annu. Rev. Neurosci. 13, 43–60. doi: 10.1146/annurev.ne.13.030190.000355
Fledrich, R., Stassart, R. M., Klink, A., Rasch, L. M., Prukop, T., Haag, L., et al. (2014). Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat. Med. 20, 1055–1061. doi: 10.1038/nm.3664
Fontana, X., Hristova, M., Da Costa, C., Patodia, S., Thei, L., Makwana, M., et al. (2012). c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 198, 127–141. doi: 10.1083/jcb.201205025
Fornasari, B. E., Ronchi, G., Pascal, D., Visigalli, D., Capodivento, G., Nobbio, L., et al. (2018). Soluble Neuregulin1 is strongly up-regulated in the rat model of Charcot-Marie-Tooth 1A disease. Exp. Biol. Med. 243, 370–374. doi: 10.1177/1535370218754492
Freese, C., Garratt, A. N., Fahrenholz, F., and Endres, K. (2009). The effects of α-secretase ADAM10 on the proteolysis of neuregulin-1. FEBS J. 276, 1568–1580. doi: 10.1111/j.1742-4658.2009.06889.x
Fricker, F. R., Antunes-Martins, A., Galino, J., Paramsothy, R., La Russa, F., Perkins, J., et al. (2013). Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain 136, 2279–2297. doi: 10.1093/brain/awt148
Fricker, F. R., Lago, N., Balarajah, S., Tsantoulas, C., Tanna, S., Zhu, N., et al. (2011). Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J. Neurosci. 31, 3225–3233. doi: 10.1523/JNEUROSCI.2568-10.2011
Friedman, W. J. (2000). Neurotrophins induce death of hippocampal neurons via the p75 receptor. J. Neurosci. 20, 6340–6346. doi: 10.1523/JNEUROSCI.20-17-06340.2000
Garratt, A. N., Britsch, S., and Birchmeier, C. (2000). Neuregulin, a factor with many functions in the life of a schwann cell. Bioessays 22, 987–996. doi: 10.1002/1521-1878(200011)22:11<987::aid-bies5>3.3.co;2-x
Gaudet, A. D., Popovich, P. G., and Ramer, M. S. (2011). Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflammation 8:110. doi: 10.1186/1742-2094-8-110
Gera, O., Shavit-Stein, E., Bushi, D., Harnof, S., Shimon, M. B., Weiss, R., et al. (2016). Thrombin and protein C pathway in peripheral nerve Schwann cells. Neuroscience 339, 587–598. doi: 10.1016/j.neuroscience.2016.10.034
Gerdts, J., Summers, D. W., Milbrandt, J., and Diantonio, A. (2016). Axon self-destruction: new links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. doi: 10.1016/j.neuron.2015.12.023
Gersbacher, M. T., Kim, D. Y., Bhattacharyya, R., and Kovacs, D. M. (2010). Identification of BACE1 cleavage sites in human voltage-gated sodium channel β2 subunit. Mol. Neurodegener. 5:61. doi: 10.1186/1750-1326-5-61
Golde, T. E., and Eckman, C. B. (2003). Physiologic and pathologic events mediated by intramembranous and juxtamembranous proteolysis. Sci. STKE 2003:RE4. doi: 10.1126/stke.2003.172.re4
Gomez-Sanchez, J. A., Carty, L., Iruarrizaga-Lejarreta, M., Palomo-Irigoyen, M., Varela-Rey, M., Griffith, M., et al. (2015). Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 210, 153–168. doi: 10.1083/jcb.201503019
Gomez-Sanchez, J. A., Pilch, K. S., van der Lans, M., Fazal, S. V., Benito, C., Wagstaff, L. J., et al. (2017). After nerve injury, lineage tracing shows that myelin and remak schwann cells elongate extensively and branch to form repair schwann cells, which shorten radically on remyelination. J. Neurosci. 37, 9086–9099. doi: 10.1523/JNEUROSCI.1453-17.2017
Gu, Z., Jiang, Q., Fu, A. K., Ip, N. Y., and Yan, Z. (2005). Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J. Neurosci. 25, 4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005
Guertin, A. D., Zhang, D. P., Mak, K. S., Alberta, J. A., and Kim, H. A. (2005). Microanatomy of axon/glial signaling during Wallerian degeneration. J. Neurosci. 25, 3478–3487. doi: 10.1523/JNEUROSCI.3766-04.2005
Gurwitz, D., and Cunningham, D. D. (1990). Neurite outgrowth activity of protease nexin-1 on neuroblastoma cells requires thrombin inhibition. J. Cell. Physiol. 142, 155–162. doi: 10.1002/jcp.1041420119
Hartmann, D., de Strooper, B., Serneels, L., Craessaerts, K., Herreman, A., Annaert, W., et al. (2002). The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for α-secretase activity in fibroblasts. Hum. Mol. Genet. 11, 2615–2624. doi: 10.1093/hmg/11.21.2615
Hartung, H. P., and Kieseier, B. C. (2000). The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J. Neuroimmunol. 107, 140–147. doi: 10.1016/s0165-5728(00)00225-3
Hemming, M. L., Elias, J. E., Gygi, S. P., and Selkoe, D. J. (2009). Identification of β-secretase (BACE1) substrates using quantitative proteomics. PLoS One 4:e8477. doi: 10.1371/journal.pone.0008477
Hempstead, B. L., Martin-Zanca, D., Kaplan, D. R., Parada, L. F., and Chao, M. V. (1991). High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350, 678–683. doi: 10.1038/350678a0
Hirata, K., and Kawabuchi, M. (2002). Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc. Res. Tech. 57, 541–547. doi: 10.1002/jemt.10108
Hitt, B., Riordan, S. M., Kukreja, L., Eimer, W. A., Rajapaksha, T. W., and Vassar, R. (2012). β-site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J. Biol. Chem. 287, 38408–38425. doi: 10.1074/jbc.M112.415505
Ho, W. H., Armanini, M. P., Nuijens, A., Phillips, H. S., and Osheroff, P. L. (1995). Sensory and motor neuron-derived factor. A novel heregulin variant highly expressed in sensory and motor neurons. J. Biol. Chem. 270, 14523–14532. doi: 10.1074/jbc.270.24.14523
Horiuchi, K., Weskamp, G., Lum, L., Hammes, H. P., Cai, H., Brodie, T. A., et al. (2003). Potential role for ADAM15 in pathological neovascularization in mice. Mol. Cell. Biol. 23, 5614–5624. doi: 10.1128/mcb.23.16.5614-5624.2003
Hu, X., He, W., Diaconu, C., Tang, X., Kidd, G. J., Macklin, W. B., et al. (2008). Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 22, 2970–2980. doi: 10.1096/fj.08-106666
Hu, X., Hicks, C. W., He, W., Wong, P., Macklin, W. B., Trapp, B. D., et al. (2006). Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 9, 1520–1525. doi: 10.1038/nn1797
Hu, X., Hu, J., Dai, L., Trapp, B., and Yan, R. (2015). Axonal and Schwann cell BACE1 is equally required for remyelination of peripheral nerves. J. Neurosci. 35, 3806–3814. doi: 10.1523/JNEUROSCI.5207-14.2015
Hughes, P. M., Wells, G. M., Perry, V. H., Brown, M. C., and Miller, K. M. (2002). Comparison of matrix metalloproteinase expression during Wallerian degeneration in the central and peripheral nervous systems. Neuroscience 113, 273–287. doi: 10.1016/s0306-4522(02)00183-5
Jang, S. Y., Shin, Y. K., Park, S. Y., Park, J. Y., Lee, H. J., Yoo, Y. H., et al. (2016). Autophagic myelin destruction by Schwann cells during Wallerian degeneration and segmental demyelination. Glia 64, 730–742. doi: 10.1002/glia.22957
Jangouk, P., Dehmel, T., Meyer Zu Hörste, G., Ludwig, A., Lehmann, H. C., and Kieseier, B. C. (2009). Involvement of ADAM10 in axonal outgrowth and myelination of the peripheral nerve. Glia 57, 1765–1774. doi: 10.1002/glia.20889
Jessen, K. R., and Mirsky, R. (2008). Negative regulation of myelination: relevance for development, injury and demyelinating disease. Glia 56, 1552–1565. doi: 10.1002/glia.20761
Jessen, K. R., Mirsky, R., and Lloyd, A. C. (2015). Schwann cells: development and role in nerve repair. Cold Spring Harb. Perspect. Biol. 7:a020487. doi: 10.1101/cshperspect.a020487
Jung, K. M., Tan, S., Landman, N., Petrova, K., Murray, S., Lewis, R., et al. (2003). Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J. Biol. Chem. 278, 42161–42169. doi: 10.1074/jbc.M306028200
Kao, S. C., Wu, H., Xie, J., Chang, C. P., Ranish, J. A., Graef, I. A., et al. (2009). Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science 323, 651–654. doi: 10.1126/science.1166562
Keski-Oja, J., Lohi, J., Tuuttila, A., Tryggvason, K., and Vartio, T. (1992). Proteolytic processing of the 72,000-Da type IV collagenase by urokinase plasminogen activator. Exp. Cell Res. 202, 471–476. doi: 10.1016/0014-4827(92)90101-d
Kim, D. Y., Carey, B. W., Wang, H., Ingano, L. A. M., Binshtok, A. M., Wertz, M. H., et al. (2007). BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 9, 755–764. doi: 10.1038/ncb1602
Kim, D. Y., Gersbacher, M. T., Inquimbert, P., and Kovacs, D. M. (2011). Reduced sodium channel Nav1.1 levels in BACE1-null mice. J. Biol. Chem. 286, 8106–8116. doi: 10.1074/jbc.M110.134692
Kim, D. Y., Ingano, L. A., Carey, B. W., Pettingell, W. H., and Kovacs, D. M. (2005). Presenilin/γ-secretase-mediated cleavage of the voltage-gated sodium channel β2-subunit regulates cell adhesion and migration. J. Biol. Chem. 280, 23251–23261. doi: 10.1074/jbc.M412938200
Kim, Y., Remacle, A. G., Chernov, A. V., Liu, H., Shubayev, I., Lai, C., et al. (2012). The MMP-9/TIMP-1 axis controls the status of differentiation and function of myelin-forming Schwann cells in nerve regeneration. PLoS One 7:e33664. doi: 10.1371/journal.pone.0033664
Kobayashi, H., Chattopadhyay, S., Kato, K., Dolkas, J., Kikuchi, S., Myers, R. R., et al. (2008). MMPs initiate Schwann cell-mediated MBP degradation and mechanical nociception after nerve damage. Mol. Cell. Neurosci. 39, 619–627. doi: 10.1016/j.mcn.2008.08.008
Koliatsos, V. E., Crawford, T. O., and Price, D. L. (1991). Axotomy induces nerve growth factor receptor immunoreactivity in spinal motor neurons. Brain Res. 549, 297–304. doi: 10.1016/0006-8993(91)90471-7
Kopan, R., and Goate, A. (2000). A common enzyme connects notch signaling and Alzheimer’s disease. Genes Dev. 14, 2799–2806. doi: 10.1101/gad.836900
Kopan, R., and Ilagan, M. X. (2004). γ-secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 5, 499–504. doi: 10.1038/nrm1406
Kraemer, B. R., Yoon, S. O., and Carter, B. D. (2014). The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb. Exp. Pharmacol. 220, 121–164. doi: 10.1007/978-3-642-45106-5_6
Krystosek, A., and Seeds, N. W. (1981). Plasminogen activator release at the neuronal growth cone. Science 213, 1532–1534. doi: 10.1126/science.7197054
Krystosek, A., and Seeds, N. W. (1984). Peripheral neurons and Schwann cells secrete plasminogen activator. J. Cell Biol. 98, 773–776. doi: 10.1083/jcb.98.2.773
Kuhn, P. H., Koroniak, K., Hogl, S., Colombo, A., Zeitschel, U., Willem, M., et al. (2012). Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 31, 3157–3168. doi: 10.1038/emboj.2012.173
Kurek, J. B., Austin, L., Cheema, S. S., Bartlett, P. F., and Murphy, M. (1996). Up-regulation of leukaemia inhibitory factor and interleukin-6 in transected sciatic nerve and muscle following denervation. Neuromuscul. Disord. 6, 105–114. doi: 10.1016/0960-8966(95)00029-1
Kurisaki, T., Masuda, A., Osumi, N., Nabeshima, Y., and Fujisawa-Sehara, A. (1998). Spatially- and temporally-restricted expression of meltrin α (ADAM12) and β (ADAM19) in mouse embryo. Mech. Dev. 73, 211–215. doi: 10.1016/s0925-4773(98)00043-4
Kwon, Y. K., Bhattacharyya, A., Alberta, J. A., Giannobile, W. V., Cheon, K., Stiles, C. D., et al. (1997). Activation of ErbB2 during wallerian degeneration of sciatic nerve. J. Neurosci. 17, 8293–8299. doi: 10.1523/jneurosci.17-21-08293.1997
La Fleur, M., Underwood, J. L., Rappolee, D. A., and Werb, Z. (1996). Basement membrane and repair of injury to peripheral nerve: defining a potential role for macrophages, matrix metalloproteinases, and tissue inhibitor of metalloproteinases-1. J. Exp. Med. 184, 2311–2326. doi: 10.1084/jem.184.6.2311
La Marca, R., Cerri, F., Horiuchi, K., Bachi, A., Feltri, M. L., Wrabetz, L., et al. (2011). TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci. 14, 857–865. doi: 10.1038/nn.2849
Laird, F. M., Cai, H., Savonenko, A. V., Farah, M. H., He, K., Melnikova, T., et al. (2005). BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 25, 11693–11709. doi: 10.1523/jneurosci.2766-05.2005
Liotta, L. A., Goldfarb, R. H., Brundage, R., Siegal, G. P., Terranova, V., and Garbisa, S. (1981). Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res. 41, 4629–4636.
Loeb, J. A., Khurana, T. S., Robbins, J. T., Yee, A. G., and Fischbach, G. D. (1999). Expression patterns of transmembrane and released forms of neuregulin during spinal cord and neuromuscular synapse development. Development 126, 781–791.
Louvi, A., and Artavanis-Tsakonas, S. (2006). Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 7, 93–102. doi: 10.1038/nrn1847
Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., et al. (2001). Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 4, 231–232. doi: 10.1038/85059
Mahanthappa, N. K., Anton, E. S., and Matthew, W. D. (1996). Glial growth factor 2, a soluble neuregulin, directly increases Schwann cell motility and indirectly promotes neurite outgrowth. J. Neurosci. 16, 4673–4683. doi: 10.1523/jneurosci.16-15-04673.1996
Marchionni, M. A., Goodearl, A. D., Chen, M. S., Bermingham-Mcdonogh, O., Kirk, C., Hendricks, M., et al. (1993). Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature 362, 312–318. doi: 10.1038/362312a0
McCawley, L. J., and Matrisian, L. M. (2001). Matrix metalloproteinases: they’re not just for matrix anymore! Curr. Opin. Cell Biol. 13, 534–540. doi: 10.1016/s0955-0674(00)00248-9
Meeker, R. B., and Williams, K. S. (2015). The p75 neurotrophin receptor: at the crossroad of neural repair and death. Neural Regen. Res. 10, 721–725. doi: 10.4103/1673-5374.156967
Mei, L., and Nave, K. A. (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. doi: 10.1016/j.neuron.2014.06.007
Meier, R., Spreyer, P., Ortmann, R., Harel, A., and Monard, D. (1989). Induction of glia-derived nexin after lesion of a peripheral nerve. Nature 342, 548–550. doi: 10.1038/342548a0
Mekkawy, A. H., Pourgholami, M. H., and Morris, D. L. (2014). Involvement of urokinase-type plasminogen activator system in cancer: an overview. Med. Res. Rev. 34, 918–956. doi: 10.1002/med.21308
Meyer zu Horste, G., Derksen, A., Stassart, R., Szepanowski, F., Thanos, M., Stettner, M., et al. (2015). Neuronal ADAM10 promotes outgrowth of small-caliber myelinated axons in the peripheral nervous system. J. Neuropathol. Exp. Neurol. 74, 1077–1085. doi: 10.1097/nen.0000000000000253
Mi, S., Lee, X., Shao, Z., Thill, G., Ji, B., Relton, J., et al. (2004). LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 7, 221–228. doi: 10.1038/nn1188
Michailov, G. V., Sereda, M. W., Brinkmann, B. G., Fischer, T. M., Haug, B., Birchmeier, C., et al. (2004). Axonal neuregulin-1 regulates myelin sheath thickness. Science 304, 700–703. doi: 10.1126/science.1095862
Morano, M., Ronchi, G., Nicolo, V., Fornasari, B. E., Crosio, A., Perroteau, I., et al. (2018). Modulation of the Neuregulin 1/ErbB system after skeletal muscle denervation and reinnervation. Sci. Rep. 8:5047. doi: 10.1038/s41598-018-23454-8
Munger, J. S., Harpel, J. G., Gleizes, P.-E., Mazzieri, R., Nunes, I., and Rifkin, D. B. (1997). Latent transforming growth factor-β: structural features and mechanisms of activation. Kidney Int. 51, 1376–1382. doi: 10.1038/ki.1997.188
Murphy, G., and Docherty, A. J. (1992). The matrix metalloproteinases and their inhibitors. Am. J. Respir. Cell Mol. Biol. 7, 120–125. doi: 10.1165/ajrcmb/7.2.120
Nagase, H., Visse, R., and Murphy, G. (2006). Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 69, 562–573. doi: 10.1016/j.cardiores.2005.12.002
Nave, K. A., and Salzer, J. L. (2006). Axonal regulation of myelination by neuregulin 1. Curr. Opin. Neurobiol. 16, 492–500. doi: 10.1016/j.conb.2006.08.008
Nykjaer, A., Lee, R., Teng, K. K., Jansen, P., Madsen, P., Nielsen, M. S., et al. (2004). Sortilin is essential for proNGF-induced neuronal cell death. Nature 427, 843–848. doi: 10.1038/nature02319
Page-McCaw, A., Ewald, A. J., and Werb, Z. (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233. doi: 10.1038/nrm2125
Pan, D., and Rubin, G. M. (1997). Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 90, 271–280. doi: 10.1016/s0092-8674(00)80335-9
Pathak, A., Stanley, E. M., Hickman, F. E., Wallace, N., Brewer, B., Li, D., et al. (2018). Retrograde degenerative signaling mediated by the p75 neurotrophin receptor requires p150(Glued) deacetylation by axonal HDAC1. Dev. Cell 46, 376.e7–387.e7. doi: 10.1016/j.devcel.2018.07.001
Pereira, J. A., Lebrun-Julien, F., and Suter, U. (2012). Molecular mechanisms regulating myelination in the peripheral nervous system. Trends Neurosci. 35, 123–134. doi: 10.1016/j.tins.2011.11.006
Perry, V. H., Tsao, J. W., Fearn, S., and Brown, M. C. (1995). Radiation-induced reductions in macrophage recruitment have only slight effects on myelin degeneration in sectioned peripheral nerves of mice. Eur. J. Neurosci. 7, 271–280. doi: 10.1111/j.1460-9568.1995.tb01063.x
Petratos, S., Butzkueven, H., Shipham, K., Cooper, H., Bucci, T., Reid, K., et al. (2003). Schwann cell apoptosis in the postnatal axotomized sciatic nerve is mediated via NGF through the low-affinity neurotrophin receptor. J. Neuropathol. Exp. Neurol. 62, 398–411. doi: 10.1093/jnen/62.4.398
Platt, C. I., Krekoski, C. A., Ward, R. V., Edwards, D. R., and Gavrilovic, J. (2003). Extracellular matrix and matrix metalloproteinases in sciatic nerve. J. Neurosci. Res. 74, 417–429. doi: 10.1002/jnr.10783
Previtali, S. C., Malaguti, M. C., Riva, N., Scarlato, M., Dacci, P., Dina, G., et al. (2008). The extracellular matrix affects axonal regeneration in peripheral neuropathies. Neurology 71, 322–331. doi: 10.1212/01.wnl.0000319736.43628.04
Primakoff, P., and Myles, D. G. (2000). The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet. 16, 83–87. doi: 10.1016/s0168-9525(99)01926-5
Rijken, D. C., and Lijnen, H. R. (2009). New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 7, 4–13. doi: 10.1111/j.1538-7836.2008.03220.x
Rivellini, C., Dina, G., Porrello, E., Cerri, F., Scarlato, M., Domi, T., et al. (2012). Urokinase plasminogen receptor and the fibrinolytic complex play a role in nerve repair after nerve crush in mice and in human neuropathies. PLoS One 7:e32059. doi: 10.1371/journal.pone.0032059
Roberds, S. L., Anderson, J., Basi, G., Bienkowski, M. J., Branstetter, D. G., Chen, K. S., et al. (2001). BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 10, 1317–1324. doi: 10.1093/hmg/10.12.1317
Roberts, S. L., Dun, X. P., Doddrell, R. D. S., Mindos, T., Drake, L. K., Onaitis, M. W., et al. (2017). Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development 144, 3114–3125. doi: 10.1242/dev.150656
Rotshenker, S. (2011). Wallerian degeneration: the innate-immune response to traumatic nerve injury. J. Neuroinflammation 8:109. doi: 10.1186/1742-2094-8-109
Sachs, B. D., Baillie, G. S., McCall, J. R., Passino, M. A., Schachtrup, C., Wallace, D. A., et al. (2007). p75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J. Cell Biol. 177, 1119–1132. doi: 10.1083/jcb.200701040
Sachse, C. C., Kim, Y. H., Agsten, M., Huth, T., Alzheimer, C., Kovacs, D. M., et al. (2013). BACE1 and presenilin/γ-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J. 27, 2458–2467. doi: 10.1096/fj.12-214056
Saika, T., Senba, E., Noguchi, K., Sato, M., Yoshida, S., Kubo, T., et al. (1991). Effects of nerve crush and transection on mRNA levels for nerve growth factor receptor in the rat facial motoneurons. Mol. Brain Res. 9, 157–160. doi: 10.1016/0169-328x(91)90142-k
Saksela, O., and Rifkin, D. B. (1990). Release of basic fibroblast growth factor-heparan sulfate complexes from endothelial cells by plasminogen activator-mediated proteolytic activity. J. Cell Biol. 110, 767–775. doi: 10.1083/jcb.110.3.767
Savonenko, A. V., Melnikova, T., Laird, F. M., Stewart, K. A., Price, D. L., and Wong, P. C. (2008). Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. U S A 105, 5585–5590. doi: 10.1073/pnas.0710373105
Scapin, C., Ferri, C., Pettinato, E., Zambroni, D., Bianchi, F., Del Carro, U., et al. (2018). Enhanced axonal neuregulin-1 type-III signaling ameliorates neurophysiology and hypomyelination in a charcot-marie-tooth type 1B mouse model. Hum. Mol. Genet. doi: 10.1093/hmg/ddy411 [Epub ahead of print].
Schlöndorff, J., and Blobel, C. P. (1999). Metalloprotease-disintegrins: modular proteins capable of promoting cell-cell interactions and triggering signals by protein-ectodomain shedding. J. Cell Sci. 112, 3603–3617.
Seals, D. F., and Courtneidge, S. A. (2003). The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30. doi: 10.1101/gad.1039703
Seeds, N. W., Verrall, S., Friedman, G., Hayden, S., Gadotti, D., Haffke, S., et al. (1992). Plasminogen activators and plasminogen activator inhibitors in neural development. Ann. N Y Acad. Sci. 667, 32–40. doi: 10.1111/j.1749-6632.1992.tb51592.x
Shamash, S., Reichert, F., and Rotshenker, S. (2002). The cytokine network of Wallerian degeneration: tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J. Neurosci. 22, 3052–3060. doi: 10.1523/JNEUROSCI.22-08-03052.2002
Shirakabe, K., Wakatsuki, S., Kurisaki, T., and Fujisawa-Sehara, A. (2001). Roles of Meltrin β /ADAM19 in the processing of neuregulin. J. Biol. Chem. 276, 9352–9358. doi: 10.1074/jbc.m007913200
Shubayev, V. I., Angert, M., Dolkas, J., Campana, W. M., Palenscar, K., and Myers, R. R. (2006). TNFα-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol. Cell. Neurosci. 31, 407–415. doi: 10.1016/j.mcn.2005.10.011
Shubayev, V. I., and Myers, R. R. (2000). Upregulation and interaction of TNFα and gelatinases A and B in painful peripheral nerve injury. Brain Res. 855, 83–89. doi: 10.1016/s0006-8993(99)02321-5
Siconolfi, L. B., and Seeds, N. W. (2001a). Induction of the plasminogen activator system accompanies peripheral nerve regeneration after sciatic nerve crush. J. Neurosci. 21, 4336–4347. doi: 10.1523/JNEUROSCI.21-12-04336.2001
Siconolfi, L. B., and Seeds, N. W. (2001b). Mice lacking tPA, uPA, or plasminogen genes showed delayed functional recovery after sciatic nerve crush. J. Neurosci. 21, 4348–4355. doi: 10.1523/JNEUROSCI.21-12-04348.2001
Siebert, H., Dippel, N., Mader, M., Weber, F., and Brück, W. (2001). Matrix metalloproteinase expression and inhibition after sciatic nerve axotomy. J. Neuropathol. Exp. Neurol. 60, 85–93. doi: 10.1093/jnen/60.1.85
Sinha, S., and Lieberburg, I. (1999). Cellular mechanisms of β-amyloid production and secretion. Proc. Natl. Acad. Sci. U S A 96, 11049–11053. doi: 10.1073/pnas.96.20.11049
Smirnova, I. V., Ma, J. Y., Citron, B. A., Ratzlaff, K. T., Gregory, E. J., Akaaboune, M., et al. (1996). Neural thrombin and protease nexin I kinetics after murine peripheral nerve injury. J. Neurochem. 67, 2188–2199. doi: 10.1046/j.1471-4159.1996.67052188.x
Song, X. Y., Zhou, F. H., Zhong, J. H., Wu, L. L., and Zhou, X. F. (2006). Knockout of p75(NTR) impairs re-myelination of injured sciatic nerve in mice. J. Neurochem. 96, 833–842. doi: 10.1111/j.1471-4159.2005.03564.x
Stassart, R. M., Fledrich, R., Velanac, V., Brinkmann, B. G., Schwab, M. H., Meijer, D., et al. (2013). A role for Schwann cell-derived neuregulin-1 in remyelination. Nat. Neurosci. 16, 48–54. doi: 10.1038/nn.3281
Stassart, R. M., Möbius, W., Nave, K. A., and Edgar, J. M. (2018). The axon-myelin unit in development and degenerative disease. Front. Neurosci. 12:467. doi: 10.3389/fnins.2018.00467
Stoll, G., Jander, S., and Myers, R. R. (2002). Degeneration and regeneration of the peripheral nervous system: from augustus waller’s observations to neuroinflammation. J. Peripher. Nerv. Syst. 7, 13–27. doi: 10.1046/j.1529-8027.2002.02002.x
Struhl, G., and Adachi, A. (2000). Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 6, 625–636. doi: 10.1016/s1097-2765(00)00061-7
Struhl, G., and Greenwald, I. (1999). Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 398, 522–525. doi: 10.1038/19091
Sumi, Y., Dent, M. A., Owen, D. E., Seeley, P. J., and Morris, R. J. (1992). The expression of tissue and urokinase-type plasminogen activators in neural development suggests different modes of proteolytic involvement in neuronal growth. Development 116, 625–637.
Syroid, D. E., Maycox, P. J., Soilu-Hanninen, M., Petratos, S., Bucci, T., Burrola, P., et al. (2000). Induction of postnatal schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J. Neurosci. 20, 5741–5747. doi: 10.1523/JNEUROSCI.20-15-05741.2000
Tajdaran, K., Chan, K., Gordon, T., and Borschel, G. H. (2018). Matrices, scaffolds, and carriers for protein and molecule delivery in peripheral nerve regeneration. Exp. Neurol. doi: 10.1016/j.expneurol.2018.08.014 [Epub ahead of print].
Tallon, C., Rockenstein, E., Masliah, E., and Farah, M. H. (2017). Increased BACE1 activity inhibits peripheral nerve regeneration after injury. Neurobiol. Dis. 106, 147–157. doi: 10.1016/j.nbd.2017.07.003
Taveggia, C., Feltri, M. L., and Wrabetz, L. (2010). Signals to promote myelin formation and repair. Nat. Rev. Neurol. 6, 276–287. doi: 10.1038/nrneurol.2010.37
Taveggia, C., Zanazzi, G., Petrylak, A., Yano, H., Rosenbluth, J., Einheber, S., et al. (2005). Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47, 681–694. doi: 10.1016/j.neuron.2005.08.017
Thiebaut, A. M., Gauberti, M., Ali, C., Martinez De Lizarrondo, S., Vivien, D., Yepes, M., et al. (2018). The role of plasminogen activators in stroke treatment: fibrinolysis and beyond. Lancet Neurol. 17, 1121–1132. doi: 10.1016/S1474-4422(18)30323-5
Tofaris, G. K., Patterson, P. H., Jessen, K. R., and Mirsky, R. (2002). Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J. Neurosci. 22, 6696–6703. doi: 10.1523/JNEUROSCI.22-15-06696.2002
Tomita, K., Kubo, T., Matsuda, K., Fujiwara, T., Yano, K., Winograd, J. M., et al. (2007). The neurotrophin receptor p75NTR in Schwann cells is implicated in remyelination and motor recovery after peripheral nerve injury. Glia 55, 1199–1208. doi: 10.1002/glia.20533
Trimarco, A., Forese, M. G., Alfieri, V., Lucente, A., Brambilla, P., Dina, G., et al. (2014). Prostaglandin D2 synthase/GPR44: a signaling axis in PNS myelination. Nat. Neurosci. 17, 1682–1692. doi: 10.1038/nn.3857
van Tetering, G., van Diest, P., Verlaan, I., van der Wall, E., Kopan, R., and Vooijs, M. (2009). Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 284, 31018–31027. doi: 10.1074/jbc.M109.006775
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Velanac, V., Unterbarnscheidt, T., Hinrichs, W., Gummert, M. N., Fischer, T. M., Rossner, M. J., et al. (2012). Bace1 processing of NRG1 type III produces a myelin-inducing signal but is not essential for the stimulation of myelination. Glia 60, 203–217. doi: 10.1002/glia.21255
Wakatsuki, S., Kurisaki, T., and Sehara-Fujisawa, A. (2004). Lipid rafts identified as locations of ectodomain shedding mediated by Meltrin β/ADAM19. J. Neurochem. 89, 119–123. doi: 10.1046/j.1471-4159.2003.02303.x
Wakatsuki, S., Yumoto, N., Komatsu, K., Araki, T., and Sehara-Fujisawa, A. (2009). Roles of meltrin-β/ADAM19 in progression of Schwann cell differentiation and myelination during sciatic nerve regeneration. J. Biol. Chem. 284, 2957–2966. doi: 10.1074/jbc.m803191200
Waller, V. A., and Owen, R. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lond. 140, 423–429. doi: 10.1098/rstl.1850.0021
Wang, K. C., Kim, J. A., Sivasankaran, R., Segal, R., and He, Z. (2002). P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420, 74–78. doi: 10.1038/nature01176
Wang, X., Miao, Y., Ni, J., Wang, Y., Qian, T., Yu, J., et al. (2018). Peripheral nerve injury induces dynamic changes of tight junction components. Front. Physiol. 9:1519. doi: 10.3389/fphys.2018.01519
Weisel, J. W., and Litvinov, R. I. (2017). Fibrin formation, structure and properties. Subcell. Biochem. 82, 405–456. doi: 10.1007/978-3-319-49674-0_13
Weskamp, G., Schlondorff, J., Lum, L., Becherer, J. D., Kim, T. W., Saftig, P., et al. (2004). Evidence for a critical role of the tumor necrosis factor α convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J. Biol. Chem. 279, 4241–4249. doi: 10.1074/jbc.M307974200
Willem, M. (2016). Proteolytic processing of Neuregulin-1. Brain Res. Bull. 126, 178–182. doi: 10.1016/j.brainresbull.2016.07.003
Willem, M., Garratt, A. N., Novak, B., Citron, M., Kaufmann, S., Rittger, A., et al. (2006). Control of peripheral nerve myelination by the β-secretase BACE1. Science 314, 664–666. doi: 10.1126/science.1132341
Wolfe, M. S., De Los Angeles, J., Miller, D. D., Xia, W., and Selkoe, D. J. (1999). Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer’s disease. Biochemistry 38, 11223–11230. doi: 10.1021/bi991080q
Wolpowitz, D., Mason, T. B., Dietrich, P., Mendelsohn, M., Talmage, D. A., and Role, L. W. (2000). Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron 25, 79–91. doi: 10.1016/s0896-6273(00)80873-9
Wong, S. T., Henley, J. R., Kanning, K. C., Huang, K. H., Bothwell, M., and Poo, M. M. (2002). A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat. Neurosci. 5, 1302–1308. doi: 10.1038/nn975
Wong, H. K., Sakurai, T., Oyama, F., Kaneko, K., Wada, K., Miyazaki, H., et al. (2005). β Subunits of voltage-gated sodium channels are novel substrates of β-site amyloid precursor protein-cleaving enzyme (BACE1) and γ-secretase. J. Biol. Chem. 280, 23009–23017. doi: 10.1074/jbc.m414648200
Woodhoo, A., Alonso, M. B., Droggiti, A., Turmaine, M., D’Antonio, M., Parkinson, D. B., et al. (2009). Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat. Neurosci. 12, 839–847. doi: 10.1038/nn.2323
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature 402, 533–537. doi: 10.1038/990107
Yang, X. L., Xiong, W. C., and Mei, L. (2004). Lipid rafts in neuregulin signaling at synapses. Life Sci. 75, 2495–2504. doi: 10.1016/j.lfs.2004.04.036
Yarden, Y., and Sliwkowski, M. X. (2001). Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137. doi: 10.1038/35052073
Zampieri, N., Xu, C. F., Neubert, T. A., and Chao, M. V. (2005). Cleavage of p75 neurotrophin receptor by α-secretase and γ-secretase requires specific receptor domains. J. Biol. Chem. 280, 14563–14571. doi: 10.1074/jbc.M412957200
Zhou, L., Barão, S., Laga, M., Bockstael, K., Borgers, M., Gijsen, H., et al. (2012). The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J. Biol. Chem. 287, 25927–25940. doi: 10.1074/jbc.M112.377465
Keywords: wallerian degeneration, remyelination, secretases, matrix metalloproteinases, fibrinolysis
Citation: Pellegatta M and Taveggia C (2019) The Complex Work of Proteases and Secretases in Wallerian Degeneration: Beyond Neuregulin-1. Front. Cell. Neurosci. 13:93. doi: 10.3389/fncel.2019.00093
Received: 24 October 2018; Accepted: 26 February 2019;
Published: 20 March 2019.
Edited by:
Giovanna Gambarotta, University of Turin, ItalyReviewed by:
Douglas William Zochodne, University of Alberta, CanadaPetr Dubový, Masaryk University, Czechia
Copyright © 2019 Pellegatta and Taveggia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Pellegatta, bWFpbHRvOnBlbGxlZ2F0dGEubWFydGFAaHNyLml0
Carla Taveggia, dGF2ZWdnaWEuY2FybGFAaHNyLml0