 Keren Asraf1†
Keren Asraf1† Ron N. Apte
Ron N. Apte Sigal Fleisher-Berkovich
Sigal Fleisher-Berkovich
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Neurosci. , 01 May 2018
Sec. Non-Neuronal Cells
Volume 12 - 2018 | https://doi.org/10.3389/fncel.2018.00116
The renin-angiotensin system (RAS) is an important peripheral system involved in homeostasis modulation, with angiotensin II (Ang II) serving as the main effector hormone. The main enzyme involved in Ang II formation is angiotensin-converting enzyme (ACE). ACE inhibitors (ACEIs) such as captopril (Cap) are predominantly used for the management of hypertension. All of the components of the RAS have also been identified in brain. Centrally located hormones such as Ang II can induce glial inflammation. Moreover, in Alzheimer’s disease (AD) models, where glial inflammation occurs and is thought to contribute to the propagation of the disease, increased levels of Ang II and ACE have been detected. Interestingly, ACE overexpression in monocytes, migrating to the brain was shown to prevent AD cognitive decline. However, the specific effects of captopril on glial inflammation and AD remain obscure. In the present study, we investigated the effect of captopril, given at a wide concentration range, on inflammatory mediators released by lipopolysaccharide (LPS)-treated glia. In the current study, both primary glial cells and the BV2 microglial cell line were used. Captopril decreased LPS-induced nitric oxide (NO) release from primary mixed glial cells as well as regulating inducible NO synthase (iNOS) expression, NO, tumor necrosis factor-α (TNF-α) and induced interleukin-10 (IL-10) production by BV2 microglia. We further obtained data regarding intranasal effects of captopril on cortical amyloid β (Aβ) and CD11b expression in 5XFAD cortex over three different time periods. Interestingly, we noted decreases in Aβ burden in captopril-treated mice over time which was paralleled by increased microglial activation. These results thus shed light on the neuroprotective role of captopril in AD which might be related to modulation of microglial activation.
Alzheimer’s disease (AD) is a progressive neurodegenerative disease considered as the most common type of dementia worldwide (Stansley et al., 2012; Kettenmann et al., 2013). It is well accepted that glial-mediated inflammation contributes to the progression of the disease (Griffin, 2006; Tejera and Heneka, 2016). The AD brain is characterized by activated microglia located in close vicinity to extra-cellular cerebral depositions of amyloid β (Aβ) aggregates and intra-cellular tau-associated neurofibrillary tangles (NFTs; Heneka et al., 2014). As activated microglia are responsible for brain homeostasis, they mediate the innate immune response in the central nervous system (CNS; Tejera and Heneka, 2016). Microglia assume a variety of functions, ranging from the release of inflammatory mediators to phagocytosis (Mandrekar-Colucci and Landreth, 2010; Tejera and Heneka, 2016). Thus, microglial reactions to pathological conditions may result in a detrimental inflammatory responses leading to neurodegeneration (Griffin, 2006; Heneka et al., 2014). For example, glial cytokine production plays crucial roles in the chronic and self-sustained inflammatory cycles seen in AD, subsequently leading to neuronal dysfunction (Griffin, 2006; Glass et al., 2010). High levels of pro-inflammatory cytokines, such as interleukin-1β (IL1β) and tumor necrosis factor-α (TNF-α), were observed in the cerebrospinal fluid (CSF) and brains of AD patients (Tarkowski et al., 1999; López González et al., 2016). Reactive oxygen species (ROS), nitric oxide (NO) and elevated levels of inducible nitric oxide synthase (iNOS) enzyme, originating from resident CNS glial cells, are also observed during AD (Heneka et al., 2014). It is well established that excessive amounts of NO in the brain can shift its role from physiological neuromodulator to neurotoxic factor (Jonnala and Buccafusco, 2001). Moreover, peroxynitrite was shown to enhance Aβ peptide aggregation, leading to amyloid plaque formation via nitration of Aβ peptide residues (Kummer et al., 2011). A direct interaction between Aβ proteins and TNF-a type 1 receptor (TNFR1) was reported to stimulate inflammatory cascades leading to neuronal apoptosis (Li et al., 2004).
Over the last 25 years, anti-inflammatory agents were suggested for blocking the complement system activation in AD, induced by Aβ peptides (Breitner et al., 1995; McGeer et al., 1996). Epidemiological studies have shown various degrees of benefit from prolonged consumption of NSAIDs on the onset of AD and symptomatic severity (McGeer et al., 1996). Other prospective randomized controlled trials in adults with normal cognition or mild cognitive impairment indicated no convincing evidence for the efficacy of pharmacologic intervention with NSAIDs in reducing the risk for dementia or improving cognition (Fink et al., 2018). Interestingly, recent studies suggest a lag time of 10 and possibly 20 years as opportunity for treating AD patients with anti-inflammatory drugs prior to clinical diagnosis in order to ameliorate or prevent the disease (McGeer et al., 2016).
There is evidence showing that the brain renin angiotensin system (RAS) is associated with the development of neurodegenerative diseases via a process involving peripheral and central inflammation (Saavedra, 2012, 2016). The classical RAS can be described as a hormone system which mediates blood pressure and body fluid metabolism regulation through the main effector peptide, angiotensin II (Ang II; Skrbic and Igic, 2009). Widespread neuronal injury following glial activation by Ang II, unregulated inflammation, oxidative stress and Aβ production have been reported (Zhang et al., 2010; Zhu et al., 2011; Wang et al., 2014; Faraco et al., 2016; Torika et al., 2017).
Active Ang II is produced upon cleavage of the angiotensinogen precursor protein (Wright et al., 2008). The main enzyme involved in Ang II formation from the non-active peptide angiotensin I is angiotensin-converting enzyme (ACE). ACE was identified with other RAS components in the brain (McKinley et al., 2003; Wright et al., 2008). Moreover, elevated levels of brain ACE have been observed during AD progression (Arregui et al., 1982; Barnes et al., 1991; Savaskan et al., 2001). ACE inhibitors (ACEIs) were shown to reduce glia-induced inflammation (Hou et al., 2008; Dong et al., 2011). Although ACEIs are widely prescribed for the treatment of cardiovascular disorders, diabetes and metabolic syndrome, limited clinical studies investigated the anti-inflammatory effects of these agents in humans (Zanchetti and Elmfeldt, 2006; Savoia and Schiffrin, 2007; Kaur et al., 2015). Recent clinical studies conducted by de Oliveira et al. (2014) with perindopril and captopril found beneficial effects for such pharmacological treatment in terms of cognitive decline in late onset-AD patients. Moreover, treatment with captopril resulted in reduced amyloidogenic processing of the amyloid precursor protein (APP) and ROS levels in the hippocampus of Tg2576 AD mice (AbdAlla et al., 2013).
In the present study, we investigated the effects of captopril, a potent ACEI, administered across a wide range of concentrations, on inflammatory mediators released by lipopolysaccharide (LPS)-induced glia. Both primary glial cells and the BV2 microglial cell line were used in these studies. Targeting microglia with LPS is a well-known model for understanding the interplay between infection and neuroinflammation associated with microglial activation in brain neurodegenerative diseases (Banks and Robinson, 2010). It is well established that LPS-induced acute systemic inflammation, via stimulation of toll-like receptors 4 (TLR4) expressed on innate immune cells, can lead to lasting changes in neuroimmunomodulation and behavior (Saavedra, 2012). Actually, in the CNS, all cell types express TLRs, however, microglia express the whole repertoire and TLR4 selectively (Pardon, 2015). Interestingly, Ang II, LPS and Aβ peptides share a common mechanism for microglial activation which involves the activation of TLR (Buchanan et al., 2010; Pardon, 2015; Winklewski et al., 2016). Moreover, we considered the time-dependent effects of intranasally administrated captopril on AD-associated pathological features, gliosis and Aβ aggregation, in the brains of five familial AD mice (5XFAD). The 5XFAD mice co-express mutations in the APP and presenilin 1 (PS1) genes, which in time lead to early expression of AD-associated brain pathological features (Oakley et al., 2006). In addition to Aβ lesions and gliosis that begin to develop at 8 weeks of age in the brains of these mice, this model is one of few AD mouse models that also display cholinergic neuronal loss in different brain regions as the mice age (Yan et al., 2018).
BV2 murine microglial cell line was provided by Professor Rosario Donato (Dep. of Experimental Medicine and Biochemical Sciences, University of Perugia, Italy). Cells were maintained at humified atmosphere of and 37°C and 5% CO2 in RPMI-1640 medium with 10% fetal calf serum (FCS), penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively) and 4 Mm of L-glutamine. For experiments, cells were cultured in 24- and 6-wells plates at a density of 3 × 105 and 1 × 106 cells per well, respectively. Following over-night incubation, serum free medium (SFM) was added for 4 h and additional 24 h incubation with SFM containing 10 mM HEPES, 0.1% bovine serum albumin (BSA) and drug treatments was performed.
Rat primary mixed glial cells cultures of astrocytes and microglia were obtained from the whole brain of neonatal (0–24 h age) Wistar rats, according to previous protocols (Brenner et al., 1992; Torika et al., 2016). Briefly, cells were harvested following meninges removal and mesh on a nylon sieves of 60 μm pore size and seeded in poly-l-lysine- coated- 24-well plates at a concentration of 1 × 106 cells per well. Cells were mentioned in high glucose DMEM medium supplemented with 10% FCS, penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively), 0.2 mM L-glutamine and 100 U/ml insulin. Cells were grown at humified atmosphere of and 37°C and 5% CO2 for 21 days, medium was replaced twice a week. For experiments, SFM was added for 4 h and replace with supplemented SFM with 10 mM HEPES, 0.1% BSA and drug treatments for 24 h.
Culture treatments included LPS from Escherichia coli, captopril and actinomycin D, all purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell Proliferation Kit (XTT; Biological Industries, Kibbutz Beit-Haemek, Israel) was used for determination of cells viability, according to the manufacturer’s instructions. The spectrophotometric analysis of the total formazan content was performed by using a microplate reader (model 680, Bio-Rad, Hercules, CA, USA), absorbance measured at 450 nm as previously described (Kwiecińska et al., 2012).
NO production was determined by measuring the nitrite content in the supernatant of the cell culture as described previously (Zhu et al., 2010). An equal volume (100 μl) of supernatants and griess reagent (Sigma–Aldrich, St. Louis, MO, USA) were incubated for 15 min, at room temperature and light avoided atmosphere. The spectrophotometric analysis of the total nitrite content was performed by using a microplate reader (model 680, Bio-Rad, Hercules, CA, USA), absorbance measured at 540 nm. The nitrite concentration was determined using sodium nitrite as a standard (0–50 μM). Nirite levels were normalized to cell count.
Supernatants TNF-α and IL-10 levels in the medium were assayed using ELISA kits (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions.
Whole cell lysates were obtained using lysis buffer containing protease and phosphatase cocktail (Stratech Scientific LTD., UK). Samples were separated on 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes and blocked using 4% BSA. Overnight incubation at 4°C with specific rabbit anti-iNOS antibody (1:1000, Cayman Chemicals, Ann Arbor, MI, USA) was performed. Then, membranes were incubated with donkey anti-rabbit antibody (1:10,000, GE Healthcare, Buckinghamshire, UK) for 90 min at room temperature. The bands were visualized using enhanced chemiluminescence (ECL) solution (according to the manufacturer’s instructions) and exposure to X-ray film (Fuji medical X-ray film, FujiFilm). Computerized image analysis system (EZ Quant-Gel 2.2, EZQuant Biology Software Solutions Ltd., Israel) was used for bands analysis. Protein load was normalized by β-actin protein level measurements using membrane exposure to mouse anti-β-actin antibody (1:4000, Sigma-Aldrich) and horseradish peroxidase-conjugated goat anti-mouse antibody (1:20,000, Jackson ImmunoReaserch Inc., West Grove, PA, USA).
The five familial AD mouse model (5XFAD) was used for animal experiments. 5XFAD mice express total of five familial AD (FAD) mutations, three mutations in the human APP695 gene (Swedish K670N, M671L; Florida I716V and London V717I) and two mutations in the human presenilin-1 gene (PSEN-1; M146L, L286V). C57BL/6 wild type (WT) mice (Harlan, Jerusalem, Israel) were reproduced with hemizygous 5XFAD mice. DNA tail polymerase chain reaction (PCR) for 3 weeks-old neonatal mice was used for detection of the human APP gene and dividing mice into WT or 5XFAD groups. Cages temperature and humidity conditions were set to 22 ± 2°C and 65 ± 5%, respectively. Mice were kept in 12 h light/dark cycle and available food/water supply conditions. For experiments, mice of both genders were randomly divided into three groups: (1) the control group included WT mice that were treated with 5 mg/kg/day of captopril (WT+Cap; n = 11 mice; 6 females/5 males); (2) 5XFAD mice that were treated with 5 mg/kg/day of captopril (5XFAD+Cap; n = 11; 5 females/6 males); and (3) 5XFAD mice that were treated with the vehicle (saline; 5XFAD+saline; n = 11; 6 females/5 males). Intranasal administration (2 μl drop to each nostril) of the solutions started when mice reached 8 weeks-old and lasted for 8 weeks. Surgical and experimental procedures were approved by the Institutional Animal Care and Use Committee of Ben-Gurion University of the Negev (Beer Sheeba, Israel; approval number: IL-30-08-2011-15, IL-55-09-2016).
Cardiac perfusion was performed in ketamine/Xylazine Hydrochloride anesthetized mice as previously described (Torika et al., 2016; Asraf et al., 2017). Brains were then removed and the two separated hemispheres were incubated in cold 4% paraformaldehyde (PFA) solution (4°C, overnight). Hemispheres were transferred into 30% sucrose solution for 48 h followed by −80°C freezing in molds filled with tissue adhesive (O.C.T compound Tissue-Tek, Torrance, CA, USA). Brain tissues were sliced into 40 μm thick sagittal sections by cryostat (Leica, Germany) and then rinsed in 0.05% PBS/Tween 20 solution followed by another rinsing in 0.5% PBS/Triton X-100 solution. Primary antibody diluting buffer (GBI Labs, Bothell, WA, USA) was used for blockage of non-specific binding. Immunostaining for Aβ and CD11b proteins was performed using 2 h incubation with rabbit anti-human Aβ antibody (1:250, gift from Prof. Alon Monsonego, the Shraga Segal Department of Microbiology and Immunology, faculty of Health Sciences and the National institute of Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer-Sheeba, Israel) and rat anti-mouse/human CD11b antibody (1:25, Biolegend) followed by incubation with the corresponding secondary antibodies, Cy3-conjugated donkey anti-rabbit IgG (1:1000, Jackson ImmunoResearch Laboratories, USA) and Alexa flour 488-conjugated goat anti-rat IgG (1:250, Jackson ImmunoResearch Laboratories, USA), respectively. Mounting medium with DAPI (Vector Labs, USA) was used for cells nuclei staining. Confocal images at a 1024 × 1024-pixel resolution with ×10 objective were obtained using the Olympus FluoView FV1000 confocal microscope (Olympus, Hamburg, Germany).
The threshold function in ImageJ software (version 1.40C, NIH, Bethesda, MD, USA) was used for quantification of the area stained for Aβ and CD11b proteins. Five cortical sections of each mouse were analyzed for the indicated proteins. The fluorescence intensity measured for the WT mice group was used as the baseline intensity.
Results are presented as the mean ± SEM. The statistical differences between the experimental groups were assessed by one-way analysis of variance (ANOVA) followed by post hoc multiple comparison test (Tukey–Kramer Multiple Comparison Test). Statistical significance was considered at p < 0.05.
We first investigated the possible cytotoxic effect of a 24 h captopril treatment on the BV2 microglial cell line using the XTT assay (Figure 1: F(5,18) = 123, p < 0.0001). The effect of captopril was compared to that of the already known cytotoxic drug actinomycin D. As indicated in Figure 1, while 0.25 μg/ml actinomycin D reduced cell viability by 95%, as compared to non-treated cells (control), captopril (0.1–3 mM) did not show any cytotoxic effect.

Figure 1. Captopril treatment does not affect BV2 microglial cell viability. BV2 cells were incubated in 96-well plates in the presence or absence of captopril (Cap; 0.1, 0.3, 1 or 3 mM) or actinomycin D (Actino; 0.25 μg/ml) for 24 h. XTT solution was added and viability was assessed using an enzyme-linked immunosorbent assay (ELISA) reader. Results are presented as means ± SEM of two independent experiments (n = 8). One-way ANOVA and a Tukey–Kramer multiple comparison test were used to determine statistical significance. ***p < 0.001 vs. control (non-treated cells).
We investigated the effect of captopril on NO production levels by BV2 cells treated with two different LPS doses (7 and 100 ng/ml). As shown in Figure 2A, a significant increase in NO production was observed following 7 ng/ml LPS treatment of BV2 microglia, when compared to control non-treated cells. Treatment of BV2 cells with 7 ng/ml LPS and low doses of captopril (0.3 and 1 mM) resulted in 50% and 45% increased NO production, respectively, as compared to cells induced with LPS alone (Figure 2A: F(5,96) = 160.8, p < 0.0001). Higher dose of captopril (3 mM) reduced LPS-induced NO levels in the BV2 microglial cell line by more than 40% (Figure 2A). Low captopril doses of 0.3 and 1 mM also increased NO production levels by 16% and 19%, respectively, when compared to 100 ng/ml LPS alone (Figure 2B: F(5,90) = 107.8, p < 0.0001). As shown in Figure 2C, captopril increased basal NO production by 3-fold, compared with control (Figure 2C: F(4,132) = 4.488, p < 0.0001).
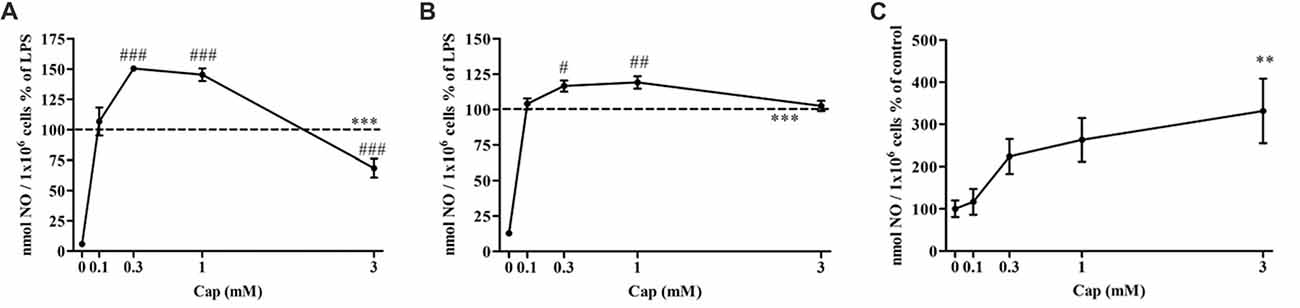
Figure 2. Captopril treatment differentially modulates lipopolysaccharide (LPS)-induced nitric oxide (NO) release from BV2 microglia cells. BV2 microglia were incubated in 24-well plates in the presence of 7 ng/ml (A) or 100 ng/ml (B) LPS alone or with captopril (Cap; 0.1, 0.3, 1 or 3 mM) for 24 h. 0 mM captopril represent control sample, non-treated cells. Thereafter, supernatants were analyzed for nitrite levels using the Griess reaction. Nitrite levels were normalized to cell counts. Dashed lines represent the LPS value (normalized as 100%). (C) NO levels measured in non-stimulated BV2 cells treated with captopril (Cap; 0.1, 0.3, 1 or 3 mM) for 24 h, presented as % of control (0 mM Cap). Results are presented as means ± SEM of three-four independent experiments ((A) n = 30, (B,C) n = 24). Statistical significance was determined using one-way ANOVA and a Tukey–Kramer multiple comparison test. **p < 0.01, ***p < 0.001 vs. control non-treated cells (0 mM Cap); #p < 0.05 vs. LPS; ##p < 0.01 vs. LPS; ###p < 0.001 vs. LPS.
The effect of captopril on the secretion of pro-inflammatory TNF-α, and anti-inflammatory IL-10 from BV2 microglial cells was assessed (Figure 3). TNF-α levels were significantly increased by more than 97% in 7 ng/ml LPS-treated BV2 microglial cells, as compared to controls (Figure 3A: F(5,283) = 202.4, p < 0.0001). Incubation with low doses of captopril (0.1 and 0.3 mM) did not affect LPS-induced TNF-α production levels. When administered at a 1 mM concentration, captopril elicited a 32% increase over that observed following treatment with LPS alone (Figure 3A). By contrast, captopril provided at 3 mM abrogated the effect of LPS on TNF-α production by about 50% (Figure 3A). Basal TNF-α levels were significantly reduced upon captopril treatment (Figure 3B: F(4,100) = 38.78, p < 0.0001). 7 ng/ml LPS treatment significantly increased the IL-10 production levels when compared to controls (Figure 3C: F(5,63) = 236.7, p < 0.0001). Treatment with lower doses of captopril (0.1 and 0.3 mM) reduced IL-10 release by LPS-treated cells (Figure 3B). However, a robust increase in IL-10 production was observed following treatment with higher doses of captopril (1 and 3 mM). Captopril alone significantly affected IL-10 production levels in non-stimulated BV2 cells (Figure 3D: F(4,57) = 35.9, p < 0.0001). While low doses of 0.1 and 0.3 mM captopril reduced IL-10 production by approximately 23%, compared to control cells, 3 mM captopril increased its production by 59% (Figure 3D).
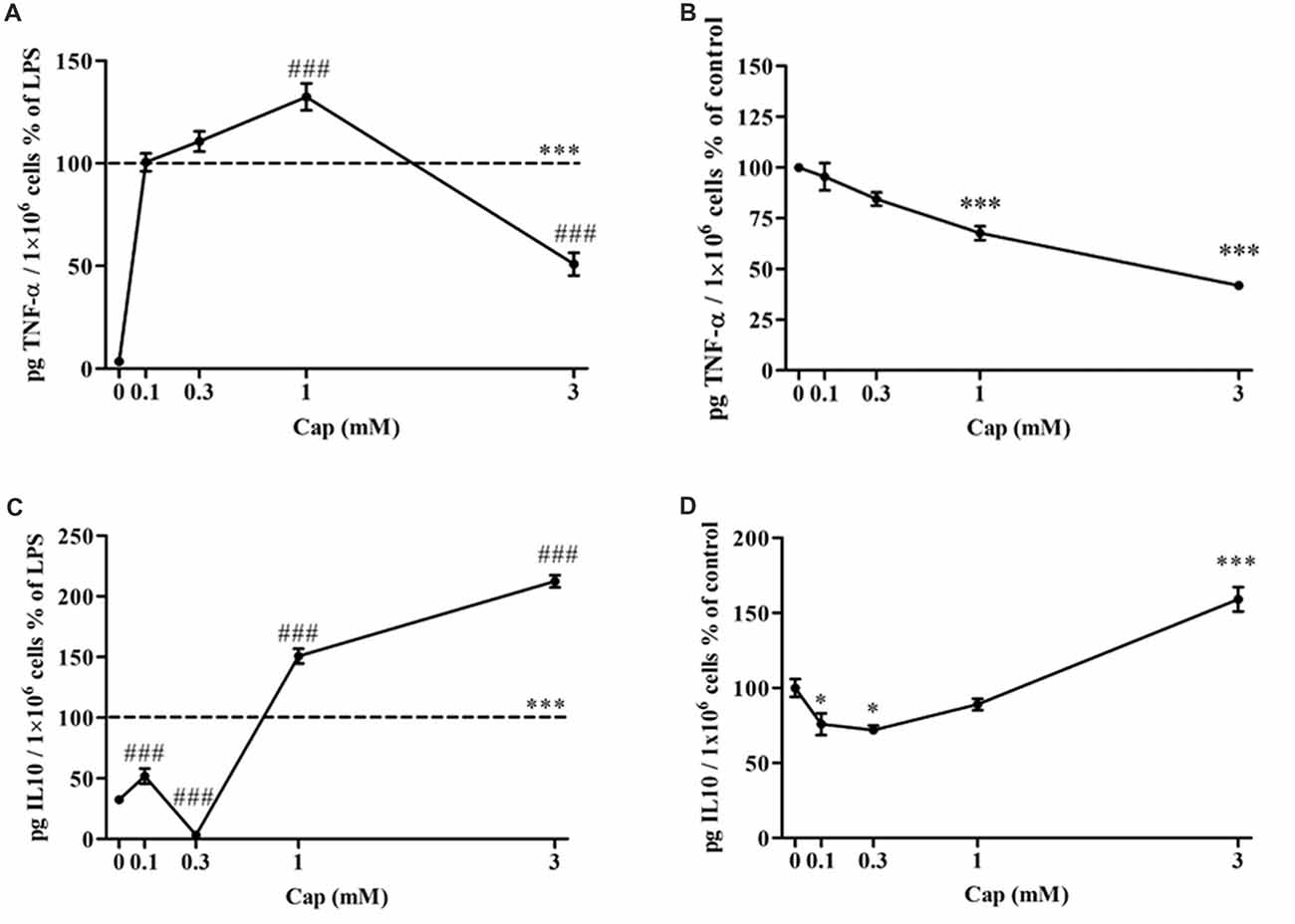
Figure 3. Captopril serves a dually regulates TNF-α and IL-10 production by LPS-treated BV2 microglial cells. BV2 microglial cells were incubated for 24 h with 7 ng/ml LPS alone or in the presence of captopril (Cap; 0.1, 0.3, 1 or 3 mM) (A,C). Captopril (0 mM) represent control sample, non-treated cells. Supernatants were analyzed for TNF-α (A,B) and IL-10 (C,D) levels using an ELISA kits and normalized to cell counts. Dashed lines represent the LPS value (normalized as 100%). TNF-α (B) and IL-10 (D) levels measured in non-stimulated BV2 cells treated with captopril (Cap; 0.1, 0.3, 1 or 3 mM) for 24 h, presented as % of control (0 mM Captopril). Results are presented as means ± SEM of two-four independent experiments ((A) n = 30, (B) n = 24, (C) n = 32, (D) n = 30). Statistical significance was determined using one-way ANOVA and a Tukey–Kramer multiple comparison test. *p < 0.05 vs. control non-treated cells; ***p < 0.001 vs. control non-treated cells; ###p < 0.001 vs. LPS.
The effect of captopril on NO production levels by primary mixed glial cells was investigated in 100 ng/ml and 0.5 μg/ml LPS-treated cells. Treatment with 100 ng/ml (Figure 4A) and 0.5 μg/ml (Figure 4B) LPS resulted in robust induction of NO production levels. 3 mM captopril treatment significantly reduced the NO production levels by approximately 50% compared to 100 ng/ml LPS-treated cells (Figure 4A: F(5, 51) = 14.55, p < 0.0001). While lower captopril doses (0.1 and 0.3 mM) reduced the LPS (0.5 μg/ml)-induced NO production by 11% and 24%, respectively, higher doses of the inhibitor (1 mM and 3 mM) resulted in 40% and 50% reduction of NO production by LPS-treated primary mixed glial cells, respectively (Figure 4B: F(5,205) = 293.7, p < 0.0001). Moreover, 30% reduction in TNF-α production levels were observed following 3 mM captopril treatment of 0.5 μg/ml LPS-treated primary mixed glial cells (Figure 4C: F(5,92) = 101.6, p < 0.0001).
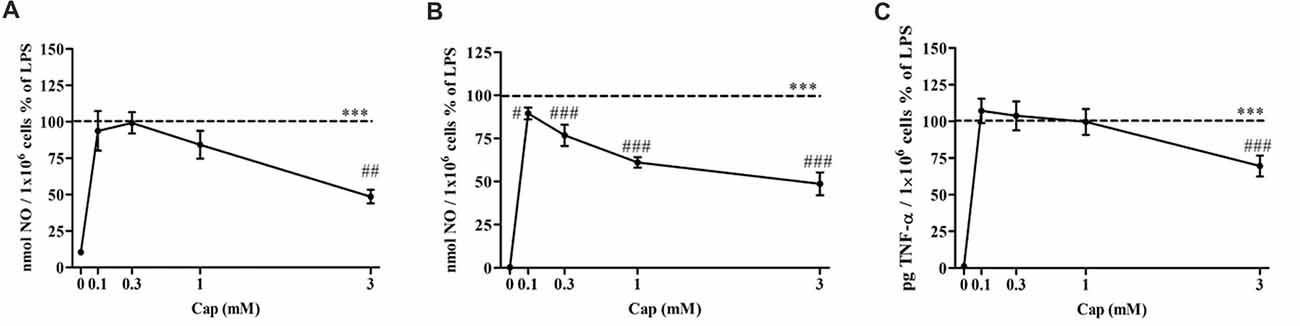
Figure 4. Captopril attenuates NO and TNF-α production levels in LPS-treated primary mixed glial cells. Primary neonatal mixed glial cells were incubated in 24-well plates in the presence of 100 ng/ml (A) or 0.5 μg/ml (B,C) LPS alone or with captopril (Cap; 0.1, 0.3, 1 or 3 mM) for 24 h. Captopril (0 mM) represent control sample, non-treated cells. Supernatants were analyzed for nitrite and TNF-α levels and normalized to cell counts. Dashed lines represent the LPS value (normalized as 100%). Results are presented as means ± SEM of two-three independent experiments ((A) n = 24, (B) n = 36, (C) n = 24). Statistical significance was determined using one-way ANOVA and a Tukey–Kramer multiple comparison test. ***p < 0.001 vs. control non-treated cells (0 mM Cap); #p < 0.05 vs. LPS; ##p < 0.01 vs. LPS; ###p < 0.001 vs. LPS.
We previously showed a 50% reduction in iNOS expression upon treatment with higher concentrations of captopril in LPS-treated BV2 cells, as compared to cells treated with LPS (7 ng/ml) alone for 24 h (Torika et al., 2016). In contrast, iNOS expression was amplified 2-fold by adding 1 mM captopril over the level of enzyme expression in cells treated solely with LPS (Figure 5: F(3,15) = 96.22, p < 0.0001).
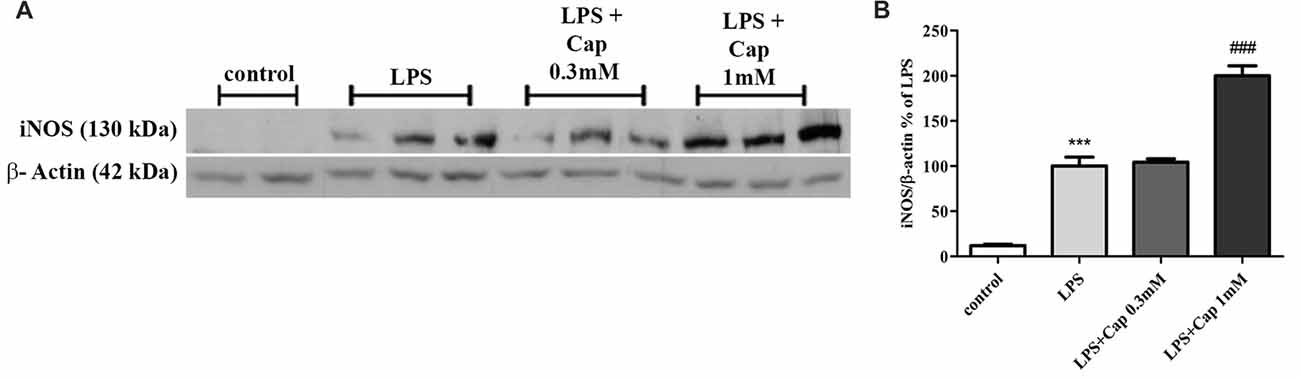
Figure 5. Captopril (at low concentrations) increases LPS-induced inducible NO synthase (iNOS) expression in BV2 microglial cell line. Cells were incubated for 24 h in the presence or absence of LPS (7 ng/ml) alone or with captopril (Cap; 0.1, 0.3, 1 or 3 mM). Thereafter, whole cell lysates were obtained and proteins were separated by SDS-PAGE. Levels of iNOS protein (130 kDa) were determined relative to β-actin (42 kDa) levels by Western analysis using target-specific primary antibodies. Representative blots (A) are shown. Results are presented in graph (B) as means ± SEM of two independent experiments (n = 18). One-way ANOVA and a Tukey–Kramer multiple comparison test were used to determine statistical significance. ***p < 0.001 vs. control non-treated cells; ###p < 0.001 vs. LPS.
The effects of captopril, given at a clinically relevant dose via intranasal administration procedure, on CD11b expression and amyloid burden in 5XFAD mice cortex were studied, as was brain immunohistochemistry (Figure 6). Cortical sections of 4 month-old WT mice showed low CD11b expression (Figures 6C,D), with no Aβ formation (Figures 6A,B). By contrast, cortical section of age-matched 5XFAD mice exhibited increased levels of Aβ plaques (Figures 6A,B) and the CD11b marker (Figures 6C,D), when compared to WT-treated mice. Intranasal administration of 5 mg/kg/day captopril significantly reduced the areas stained for CD11b (Figures 6C,D: F(2,43) = 515.8, p < 0.0001) and Aβ (Figures 6A,B: F(2,43) = 272.3, p < 0.0001) proteins in the cortex of 4 month-old 5XFAD mice, when compared to saline-treated 5XFAD mice brain sections.
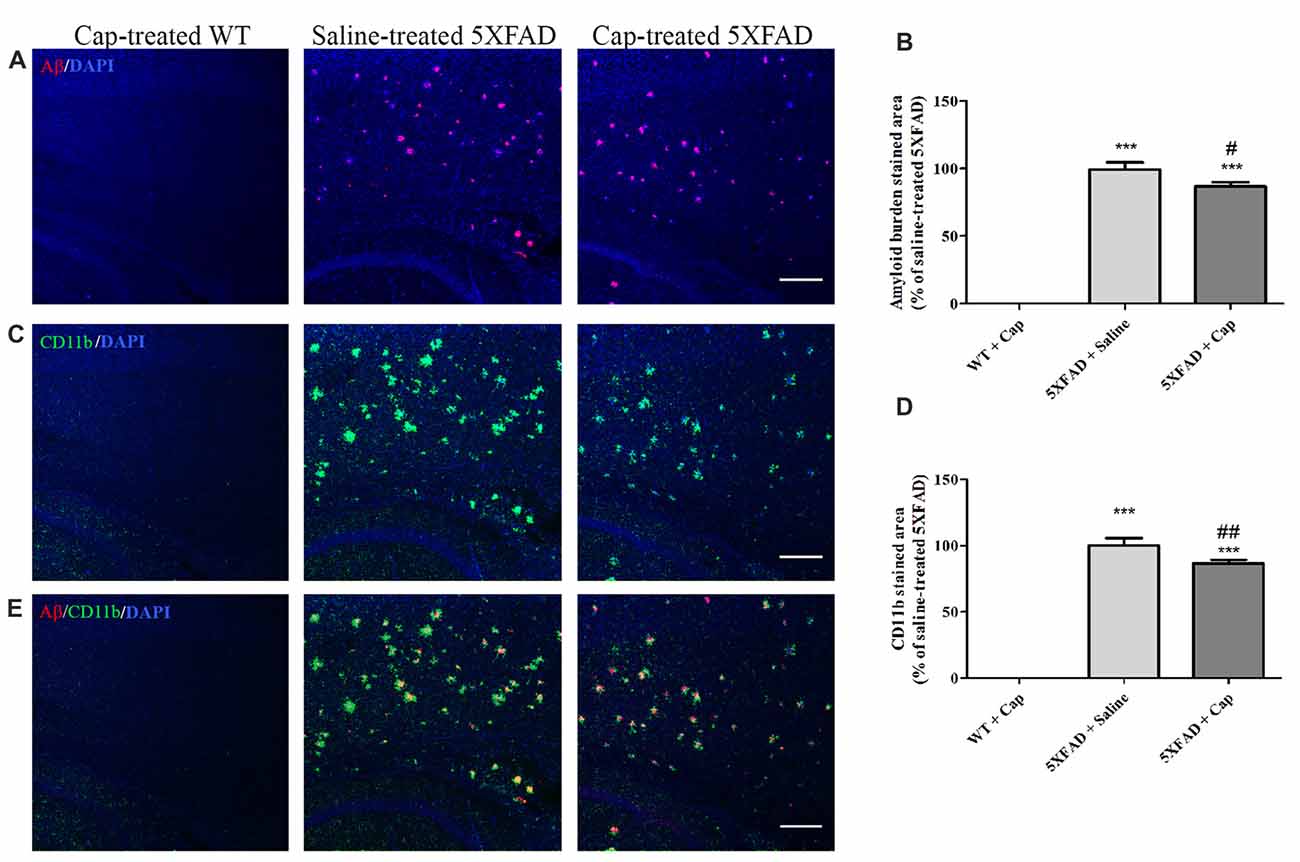
Figure 6. Intranasal administration of captopril reduces amyloid burden and CD11b expression in the cortex of 5XFAD mice. Eight week-old wild type (WT) or 5XFAD mice were treated intranasally with either saline or captopril (cap; 5 mg/kg/day) for 2 months. At the end of treatment, the mice were anesthetized and cardiac perfusion with cold PBS was performed. Brains were fixed in 4% paraformaldehyde (PFA) and 30% sucrose solutions. Then, 40 μm-thick brain sagittal sections were stained for Aβ (red) and CD11b (green) proteins using target-specific antibodies and counterstained with mounting solution containing DAPI (blue). Representative cortical layers from the three mice groups are presented. The experiment included 11 mice per group (n = 33). The calculated average sums of Aβ- (A,B) and CD11b-stained (C,D) cortical areas are represented as mean percentage ± SEM of the stained area in the saline-treated group in five repeats. Merged images of anti-Aβ and anti-CD11b staining are presented (E). One-way ANOVA and a Tukey–Kramer multiple comparison test were used to determine statistical significance. The scale bar is 200 μm. ***p < 0.001 vs. WT+cap; #p < 0.05 vs. 5XFAD+saline; ##p < 0.01 vs. 5XFAD+saline.
We compared the effects of intranasal exposure of mice to captopril (5 mg/kg/day) for different periods of time on gliosis and amyloid burden expression in the cortical areas of 5XFAD mice (Table 1). As indicated in Table 1, the decrease of Aβ burden in captopril-treated mice over time (125.5%–75.1%) was paralleled by increased microglial CD11b expression (67.8%–101%).

Table 1. Overview of the time-dependent effects of intranasal captopril on gliosis and Aβ expression in 5XFAD mice cortical areas.
Although microglia comprise only 10% of the CNS cell population, much of the innate immune response in the CNS is mediated by these cells (DiSabato et al., 2016). The microglial inflammatory response can be mimicked by the use of LPS endotoxin, which triggers microglia to secrete a wide variety of inflammatory cytokines (Pardon, 2015).
In this study, a robust inflammatory response by BV2 microglial cells was observed following LPS treatment, and resulted in the enhanced release of TNF-α, and NO, as well as elevated levels of iNOS expression (Figures 2–5). The present study also provides evidence for the first time that ACE inhibition by captopril serves a dual role in microglia-mediated neuroinflammation.
Dual regulation of neuroinflammation was also observed by us with kinins. Stimulation of the bradykinin 2 receptor (BK2R) enhanced glial inflammation in a manner that was blocked by BK2R antagonist. By contrast, a BK 1 receptor (BK1R) agonist attenuated the glial inflammatory response (Levant et al., 2006). This may partially explain the dual effects of captopril shown in the present study. In addition to intervening in Ang II metabolism, ACE can also metabolize bradykinin (BK) to form a non-active peptide (Camargo et al., 2012; Igic and Skrbic, 2014). It is well established that BK has high affinity to BK2Rs, while B1Rs are specialized for responding to BK metabolites (Moreau et al., 2005). ACE inhibition interferes with BK breakdown and prolongs its half-life (Igic and Skrbic, 2014). Based on the above, it is assumed that different captopril doses lead to differential BK1R/BK2R activation balance.
Previously, Bhat et al. (2016) showed the anti-inflammatory effects of perindopril (1 nM–1 μM), a centrally active ACEI, in LPS-treated glial cell culture. RAS intervention by perindopril ameliorated astrocytic and glial activation and reduced the production of TNF-α and oxidative stress markers and in parallel, elevated IL-10 levels (Bhat et al., 2016). Furthermore, 1 mM captopril was reported to suppress the production of the pro-inflammatory cytokine interleukin 12 (IL-12) by human peripheral blood mononuclear cells (Constantinescu et al., 1998).
In contrast to the proposal that captopril acts as an anti-inflammatory, some data argues that this agent mediates the opposing effect at certain concentrations. For instance, Coelho dos Santos et al. (2010) showed that captopril induced inflammation in human monocytes and peripheral mononuclear cells. The authors also suggested that captopril increased the monocyte infection involved in Chagas disease by induction of interleukin 17 (IL-17) and inhibition of IL-10 production (Coelho dos Santos et al., 2010). Goel et al. (2015) studied the effects of orally administered perindopril (0.1 mg/kg) on inflammatory and oxidative stress features in spontaneous hypertensive rats (SHRs) brain. In their study, the already high levels of TNF-α, iNOS, nitrite and ROS observed in the brains of SHRs were further exaggerated following intracervical LPS administration but decreased in response to perindopril treatment (Goel et al., 2015). Similar anti- inflammatory effects and suppression of pro-inflammatory mediators were also observed in LPS-injected rats treated with captopril (1–100 mg/kg) via inhibition of NF-κB pathways (Ilieva et al., 2006; Muñoz et al., 2006).
Captopril dually regulated both NO and TNF-α secretion from BV2 cells (Figures 2, 3). An inhibitory effect of captopril on both NO and TNF-α was observed in primary mixed glial cultures comprising both microglia and astrocytes (Figure 4). The reciprocal interactions between microglia and astrocytes may be particularly important for the distinct effects observed with captopril in mixed glial cultures, as compared with microglial cell lines. Differential sensitivity of cells to captopril may be due to the presence of different types or quantities of endopeptidases or to different densities of BK and/or Ang II receptors in these cells. Different LPS concentrations were used to induce inflammation in both cell types. The dual effects of captopril, on “inflammatory molecules” release, probably do not depend on inflammation grade (Figures 2, 4).
In addition to in vitro anti-inflammatory properties of captopril, we demonstrated reduced amyloid burden and macrophage/microglia accumulation in the cortex of 5XFAD mice following a 2-month-long intranasal captopril treatment, when compared to age-matched saline-treated 5XFAD mice (Figure 6). Although ACE is believed to convert neurotoxic Aβ42 peptides into a shorter form of Aβ40, thought to be less toxic in AD, the manner in which ACE inhibition affects amyloid peptide forms in AD is not yet well understood (Eckman et al., 2006; Zou et al., 2007, 2013; Regenold et al., 2017). It was previously reported that a 6-month-long captopril treatment of AD mice reduced markers of amyloidogenic processing of full-length APP and resulted in slower hippocampal Aβ accumulation (AbdAlla et al., 2013). Up-regulation of APP and tau hyper-phosphorylation mediated by captopril were also reported in a recent study by the same research group (AbdAlla et al., 2015). By contrast, captopril was shown to promote Aβ42 deposits in an AD mouse model and in cell culture studies (Hemming and Selkoe, 2005; Zou et al., 2007). Other AD studies suggested that ACEIs do not alter brain Aβ levels (Hemming et al., 2007; Dong et al., 2011; Wharton et al., 2012). As the present study showed that a 2-month-long intranasal captopril treatment reduced the Aβ burden and gliosis in the 5XFAD mouse cortex, we assume that the intranasal delivery procedure employed promotes an additive beneficial effect over systemic ACEI administration. Intranasal delivery has been suggested to enhance therapeutic delivery to the brain and allow direct entry to the CNS with minimal systemic exposure (Dhuria et al., 2010). However, the mechanism by which intranasal captopril administration ameliorates the amyloid burden in the cortex of 5XFAD mice should be further examined. This effect could be mediated by mechanisms which involve elevated clearance of Aβ via phagocytic microglial cells (Doens and Fernández, 2014), variations in Aβ-degrading enzyme expression (Nalivaeva et al., 2012) or changes in the generation of Aβ peptides followed by lowered brain inflammation (Griffin, 2006).
Table 1 summarizes what we have observed (Torika et al., 2016) with respect to the effects of intranasal captopril treatment on cortical Aβ and CD11b expression in the brain of 5XFAD mice over three different time periods. Interestingly, our findings show decreased burden in captopril-treated mice over time which was paralleled by increased microglial activation. In AD patient brain, the amyloid burden is accompanied by a clustering of activated microglia around the amyloid plaques. Reduced Aβ depositions, alongside microglial activation and enhanced phagocytic ability by angiotensin-related drugs, was shown to potentially improve cognitive performance in AD mice (Tsukuda et al., 2009; Shindo et al., 2012; Torika et al., 2017). It is worth noting that reduced amyloid burden can also involve other mechanisms which are not necessarily related to changes in microglial activity. Further studies are required to conclude whether intranasally administered captopril alters Aβ-degrading enzyme expression or influences other mechanisms involved in Aβ production.
KA, NT and SF-B designed the experiments. KA, RNA and NT performed cell culture experiments. KA performed in vivo experiments and analyzed the data. SF-B secured funds for this work. NT and SF-B wrote the manuscript. All authors read the manuscript and approved its final content.
This work was supported by the Israel Science Foundation (grant no. 101/11-16).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Prof. Abraham Danon for careful reading of the manuscript and useful advice.
AbdAlla, S., El Hakim, A., Abdelbaset, A., Elfaramawy, Y., and Quitterer, U. (2015). Inhibition of ACE retards tau hyperphosphorylation and signs of neuronal degeneration in aged rats subjected to chronic mild stress. Biomed. Res. Int. 2015:917156. doi: 10.1155/2015/917156
AbdAlla, S., Langer, A., Fu, X., and Quitterer, U. (2013). ACE inhibition with captopril retards the development of signs of neurodegeneration in an animal model of Alzheimer’s disease. Int. J. Mol. Sci. 14, 16917–16942. doi: 10.3390/ijms140816917
Arregui, A., Perry, E. K., Rossor, M., and Tomlinson, B. E. (1982). Angiotensin converting enzyme in Alzheimer’s disease increased activity in caudate nucleus and cortical areas. J. Neurochem. 38, 1490–1492. doi: 10.1111/j.1471-4159.1982.tb07930.x
Asraf, K., Torika, N., Danon, A., and Fleisher-Berkovich, S. (2017). Involvement of the bradykinin b1 receptor in microglial activation: in vitro and in vivo studies. Front. Endocrinol. 8:82. doi: 10.3389/fendo.2017.00082
Banks, W. A., and Robinson, S. M. (2010). Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav. Immun. 24, 102–109. doi: 10.1016/j.bbi.2009.09.001
Barnes, N. M., Cheng, C. H., Costall, B., Naylor, R. J., Williams, T. J., and Wischik, C. M. (1991). Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer’s disease. Eur. J. Pharmacol. 200, 289–292. doi: 10.1016/0014-2999(91)90584-d
Bhat, S. A., Goel, R., Shukla, R., and Hanif, K. (2016). Angiotensin receptor blockade modulates NFkappaB and STAT3 signaling and inhibits glial activation and neuroinflammation better than angiotensin-converting enzyme inhibition. Mol. Neurobiol. 53, 6950–6967. doi: 10.1007/s12035-015-9584-5
Breitner, J. C., Welsh, K. A., Helms, M. J., Gaskell, P. C., Gau, B. A., Roses, A. D., et al. (1995). Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol. Aging 16, 523–530. doi: 10.1016/0197-4580(95)00049-k
Brenner, T., Boneh, A., Shohami, E., Abramsky, O., and Weidenfeld, J. (1992). Glucocorticoid regulation of eicosanoid production by glial cells under basal and stimulated conditions. J. Neuroimmunol. 40, 273–279. doi: 10.1016/0165-5728(92)90143-9
Buchanan, M. M., Hutchinson, M., Watkins, L. R., and Yin, H. (2010). Toll-like receptor 4 in CNS pathologies. J. Neurochem. 114, 13–27. doi: 10.1111/j.1471-4159.2010.06736.x
Camargo, A. C., Ianzer, D., Guerreiro, J. R., and Serrano, S. M. (2012). Bradykinin-potentiating peptides: beyond captopril. Toxicon 59, 516–523. doi: 10.1016/j.toxicon.2011.07.013
Coelho dos Santos, J. S., Menezes, C. A., Villani, F. N., Magalhães, L. M., Scharfstein, J., Gollob, K. J., et al. (2010). Captopril increases the intensity of monocyte infection by Trypanosoma cruzi and induces human T helper type 17 cells. Clin. Exp. Immunol. 162, 528–536. doi: 10.1111/j.1365-2249.2010.04270.x
Constantinescu, C. S., Goodman, D. B., and Ventura, E. S. (1998). Captopril and lisinopril suppress production of interleukin-12 by human peripheral blood mononuclear cells. Immunol. Lett. 62, 25–31. doi: 10.1016/s0165-2478(98)00025-x
de Oliveira, F. F., Bertolucci, P. H., Chen, E. S., and Smith, M. C. (2014). Brain-penetrating angiotensin-converting enzyme inhibitors and cognitive change in patients with dementia due to Alzheimer’s disease. J. Alzheimers Dis. 42, S321–S324. doi: 10.3233/JAD-132189
Dhuria, S. V., Hanson, L. R., and Frey, W. H. II. (2010). Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J. Pharm. Sci. 99, 1654–1673. doi: 10.1002/jps.21924
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the devil is in the details. J. Neurochem. 139, 136–153. doi: 10.1111/jnc.13607
Doens, D., and Fernández, P. L. (2014). Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflammation 11:48. doi: 10.1186/1742-2094-11-48
Dong, Y. F., Kataoka, K., Tokutomi, Y., Nako, H., Nakamura, T., Toyama, K., et al. (2011). Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J. 25, 2911–2920. doi: 10.1096/fj.11-182873
Eckman, E. A., Adams, S. K., Troendle, F. J., Stodola, B. A., Kahn, M. A., Fauq, A. H., et al. (2006). Regulation of steady-state β-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem. 281, 30471–30478. doi: 10.1074/jbc.M605827200
Faraco, G., Park, L., Zhou, P., Luo, W., Paul, S. M., Anrather, J., et al. (2016). Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J. Cereb. Blood Flow Metab. 36, 241–252. doi: 10.1038/jcbfm.2015.79
Fink, H. A., Jutkowitz, E., McCarten, J. R., Hemmy, L. S., Butler, M., Davila, H., et al. (2018). Pharmacologic interventions to prevent cognitive decline, mild cognitive impairment, and clinical Alzheimer-type dementia: a systematic review. Ann. Intern. Med. 168, 39–51. doi: 10.7326/M17-1529
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Goel, R., Bhat, S. A., Rajasekar, N., Hanif, K., Nath, C., and Shukla, R. (2015). Hypertension exacerbates predisposition to neurodegeneration and memory impairment in the presence of a neuroinflammatory stimulus: protection by angiotensin converting enzyme inhibition. Pharmacol. Biochem. Behav. 133, 132–145. doi: 10.1016/j.pbb.2015.04.002
Griffin, W. S. (2006). Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 83, 470s–474s. doi: 10.1093/ajcn/83.2.470S
Hemming, M. L., and Selkoe, D. J. (2005). Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J. Biol. Chem. 280, 37644–37650. doi: 10.1074/jbc.M508460200
Hemming, M. L., Selkoe, D. J., and Farris, W. (2007). Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid β-protein metabolism in mouse models of Alzheimer disease. Neurobiol. Dis. 26, 273–281. doi: 10.1016/j.nbd.2007.01.004
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Hou, D. R., Wang, Y., Zhou, L., Chen, K., Tian, Y., Song, Z., et al. (2008). Altered angiotensin-converting enzyme and its effects on the brain in a rat model of Alzheimer disease. Chin. Med. J. 121, 2320–2323.
Igic, R., and Skrbic, R. (2014). The renin-angiotensin system and its blockers. Srp. Arh. Celok. Lek. 142, 756–763. doi: 10.2298/SARH1412756I
Ilieva, I., Ohgami, K., Jin, X. H., Suzuki, Y., Shiratori, K., Yoshida, K., et al. (2006). Captopril suppresses inflammation in endotoxin-induced uveitis in rats. Exp. Eye Res. 83, 651–657. doi: 10.1016/j.exer.2006.03.005
Jonnala, R. R., and Buccafusco, J. J. (2001). Inhibition of nerve growth factor signaling by peroxynitrite. J. Neurosci. Res. 63, 27–34. doi: 10.1002/1097-4547(20010101)63:1<27::aid-jnr4>3.3.co;2-r
Kaur, P., Muthuraman, A., and Kaur, M. (2015). The implications of angiotensin-converting enzymes and their modulators in neurodegenerative disorders: current and future perspectives. ACS Chem. Neurosci. 6, 508–521. doi: 10.1021/cn500363g
Kettenmann, H., Kirchhoff, F., and Verkhratsky, A. (2013). Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. doi: 10.1016/j.neuron.2012.12.023
Kummer, M. P., Hermes, M., Delekarte, A., Hammerschmidt, T., Kumar, S., Terwel, D., et al. (2011). Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 71, 833–844. doi: 10.1016/j.neuron.2011.07.001
Kwiecińska, P., Taubøll, E., and Gregoraszczuk, E. L. (2012). Effects of valproic acid and levetiracetam on viability and cell cycle regulatory genes expression in the OVCAR-3 cell line. Pharmacol. Rep. 64, 157–165. doi: 10.1016/s1734-1140(12)70742-9
Levant, A., Levy, E., Argaman, M., and Fleisher-Berkovich, S. (2006). Kinins and neuroinflammation: dual effect on prostaglandin synthesis. Eur. J. Pharmacol. 546, 197–200. doi: 10.1016/j.ejphar.2006.06.074
Li, R., Yang, L., Lindholm, K., Konishi, Y., Yue, X., Hampel, H., et al. (2004). Tumor necrosis factor death receptor signaling cascade is required for amyloid-β protein-induced neuron death. J. Neurosci. 24, 1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004
López González, I., Garcia-Esparcia, P., Llorens, F., and Ferrer, I. (2016). Genetic and transcriptomic profiles of inflammation in neurodegenerative diseases: Alzheimer, parkinson, creutzfeldt-jakob and tauopathies. Int. J. Mol. Sci. 17:206. doi: 10.3390/ijms17020206
Mandrekar-Colucci, S., and Landreth, G. E. (2010). Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 9, 156–167. doi: 10.2174/187152710791012071
McGeer, P. L., Rogers, J., and McGeer, E. G. (2016). Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J. Alzheimers Dis. 54, 853–857. doi: 10.3233/jad-160488
McGeer, P. L., Schulzer, M., and McGeer, E. G. (1996). Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 47, 425–432. doi: 10.1212/wnl.47.2.425
McKinley, M. J., Albiston, A. L., Allen, A. M., Mathai, M. L., May, C. N., McAllen, R. M., et al. (2003). The brain renin-angiotensin system: location and physiological roles. Int. J. Biochem. Cell Biol. 35, 901–918. doi: 10.1016/S1357-2725(02)00306-0
Moreau, M. E., Garbacki, N., Molinaro, G., Brown, N. J., Marceau, F., and Adam, A. (2005). The kallikrein-kinin system: current and future pharmacological targets. J. Pharmacol. Sci. 99, 6–38. doi: 10.1254/jphs.srj05001x
Muñoz, A., Rey, P., Guerra, M. J., Mendez-Alvarez, E., Soto-Otero, R., and Labandeira-Garcia, J. L. (2006). Reduction of dopaminergic degeneration and oxidative stress by inhibition of angiotensin converting enzyme in a MPTP model of parkinsonism. Neuropharmacology 51, 112–120. doi: 10.1016/j.neuropharm.2006.03.004
Nalivaeva, N. N., Beckett, C., Belyaev, N. D., and Turner, A. J. (2012). Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 120, 167–185. doi: 10.1111/j.1471-4159.2011.07510.x
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Pardon, M.-C. (2015). Lipopolysaccharide hyporesponsiveness: protective or damaging response to the brain? Rom. J. Morphol. Embryol. 56, 903–913.
Regenold, W. T., Blumenthal, J. B., Loreck, D. J., Mordecai, K. L., Scarinzi, G., Doddi, S. R., et al. (2017). Elevated plasma Aβ42 in cognitively impaired individuals taking ACE inhibitor antihypertensives. Am. J. Alzheimers Dis. Other Demen. 32, 347–352. doi: 10.1177/1533317517707288
Saavedra, J. M. (2012). Angiotensin II AT1 receptor blockers as treatments for inflammatory brain disorders. Clin. Sci. 123, 567–590. doi: 10.1042/CS20120078
Saavedra, J. M. (2016). Evidence to consider angiotensin II receptor blockers for the treatment of early Alzheimer’s disease. Cell. Mol. Neurobiol. 36, 259–279. doi: 10.1007/s10571-015-0327-y
Savaskan, E., Hock, C., Olivieri, G., Bruttel, S., Rosenberg, C., Hulette, C., et al. (2001). Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol. Aging 22, 541–546. doi: 10.1016/s0197-4580(00)00259-1
Savoia, C., and Schiffrin, E. L. (2007). Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin. Sci. 112, 375–384. doi: 10.1042/cs20060247
Shindo, T., Takasaki, K., Uchida, K., Onimura, R., Kubota, K., Uchida, N., et al. (2012). Ameliorative effects of telmisartan on the inflammatory response and impaired spatial memory in a rat model of Alzheimer’s disease incorporating additional cerebrovascular disease factors. Biol. Pharm. Bull. 35, 2141–2147. doi: 10.1248/bpb.b12-00387
Skrbic, R., and Igic, R. (2009). Seven decades of angiotensin (1939–2009). Peptides 30, 1945–1950. doi: 10.1016/j.peptides.2009.07.003
Stansley, B., Post, J., and Hensley, K. (2012). A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflammation 9:115. doi: 10.1186/1742-2094-9-115
Tarkowski, E., Blennow, K., Wallin, A., and Tarkowski, A. (1999). Intracerebral production of tumor necrosis factor-α, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol. 19, 223–230. doi: 10.1023/A:1020568013953
Tejera, D., and Heneka, M. T. (2016). Microglia in Alzheimer’s disease: the good, the bad and the ugly. Curr. Alzheimer Res. 13, 370–380. doi: 10.2174/1567205013666151116125012
Torika, N., Asraf, K., Cohen, H., and Fleisher-Berkovich, S. (2017). Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav. Immun. 64, 80–90. doi: 10.1016/j.bbi.2017.04.001
Torika, N., Asraf, K., Roasso, E., Danon, A., and Fleisher-Berkovich, S. (2016). Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: possible implications for Alzheimer’s disease. J. Neuroimmune Pharmacol. 11, 774–785. doi: 10.1007/s11481-016-9703-8
Tsukuda, K., Mogi, M., Iwanami, J., Min, L. J., Sakata, A., Jing, F., et al. (2009). Cognitive deficit in amyloid-β-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-γ activation. Hypertension 54, 782–787. doi: 10.1161/HYPERTENSIONAHA.109.136879
Wang, J., Pang, T., Hafko, R., Benicky, J., Sanchez-Lemus, E., and Saavedra, J. M. (2014). Telmisartan ameliorates glutamate-induced neurotoxicity: roles of AT1 receptor blockade and PPARγ activation. Neuropharmacology 79, 249–261. doi: 10.1016/j.neuropharm.2013.11.022
Wharton, W., Stein, J. H., Korcarz, C., Sachs, J., Olson, S. R., Zetterberg, H., et al. (2012). The effects of ramipril in individuals at risk for Alzheimer’s disease: results of a pilot clinical trial. J. Alzheimers Dis. 32, 147–156. doi: 10.3233/JAD-2012-120763
Winklewski, P. J., Radkowski, M., and Demkow, U. (2016). Neuroinflammatory mechanisms of hypertension: potential therapeutic implications. Curr. Opin. Nephrol. Hypertens. 25, 410–416. doi: 10.1097/MNH.0000000000000250
Wright, J. W., Yamamoto, B. J., and Harding, J. W. (2008). Angiotensin receptor subtype mediated physiologies and behaviors: new discoveries and clinical targets. Prog. Neurobiol. 84, 157–181. doi: 10.1016/j.pneurobio.2007.10.009
Yan, H., Pang, P., Chen, W., Zhu, H., Henok, K. A., Li, H., et al. (2018). The lesion analysis of cholinergic neurons in 5XFAD mouse model in the three-dimensional level of whole brain. Mol. Neurobiol. 55, 4115–4125. doi: 10.1007/s12035-017-0621-4
Zanchetti, A., and Elmfeldt, D. (2006). Findings and implications of the Study on COgnition and Prognosis in the Elderly (SCOPE)—a review. Blood Press 15, 71–79. doi: 10.1080/08037050600771583
Zhang, M., Mao, Y., Ramirez, S. H., Tuma, R. F., and Chabrashvili, T. (2010). Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 171, 852–858. doi: 10.1016/j.neuroscience.2010.09.029
Zhu, X. Y., Liu, S. J., Liu, Y. J., Wang, S., and Ni, X. (2010). Glucocorticoids suppress cystathionine γ-lyase expression and H2S production in lipopolysaccharide-treated macrophages. Cell. Mol. Life Sci. 67, 1119–1132. doi: 10.1007/s00018-009-0250-9
Zhu, D., Shi, J., Zhang, Y., Wang, B., Liu, W., Chen, Z., et al. (2011). Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLoS One 6:0016037. doi: 10.1371/journal.pone.0016037
Zou, K., Liu, J., Watanabe, A., Hiraga, S., Liu, S., Tanabe, C., et al. (2013). Aβ43 is the earliest-depositing Aβ species in APP transgenic mouse brain and is converted to Aβ41 by two active domains of ACE. Am. J. Pathol. 182, 2322–2331. doi: 10.1016/j.ajpath.2013.01.053
Keywords: Alzheimer’s disease, angiotensin II, angiotensin-converting enzyme, captopril, glial inflammation
Citation: Asraf K, Torika N, Apte RN and Fleisher-Berkovich S (2018) Microglial Activation Is Modulated by Captopril: in Vitro and in Vivo Studies. Front. Cell. Neurosci. 12:116. doi: 10.3389/fncel.2018.00116
Received: 21 December 2017; Accepted: 12 April 2018;
Published: 01 May 2018.
Edited by:
Elena García-Martín, Universidad de Extremadura, SpainReviewed by:
Regina Pekelmann Markus, Universidade de São Paulo, BrazilCopyright © 2018 Asraf, Torika, Apte and Fleisher-Berkovich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigal Fleisher-Berkovich, ZmxlaXNoZXJAYmd1LmFjLmls
† These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.