 Priyamvada M. Pitale
Priyamvada M. Pitale Oleg Gorbatyuk
Oleg Gorbatyuk Marina Gorbatyuk
Marina Gorbatyuk- 1Department of Optometry and Vision Science, School of Optometry, University of Alabama at Birmingham, Birmingham, AL, United States
- 2Department of Neurology, Center for Neurodegeneration and Experimental Therapeutics, University of Alabama at Birmingham, Birmingham, AL, United States
Activation of the endoplasmic reticulum (ER) stress and ER stress response, also known as the unfolded protein response (UPR), is common to various degenerative disorders. Therefore, signaling components of the UPR are currently emerging as potential targets for intervention and treatment of human diseases. One UPR signaling member, activating transcription factor 4 (ATF4), has been found up-regulated in many pathological conditions, pointing to therapeutic potential in targeting its expression. In cells, ATF4 governs multiple signaling pathways, including autophagy, oxidative stress, inflammation, and translation, suggesting a multifaceted role of ATF4 in the progression of various pathologies. However, ATF4 has been shown to trigger both pro-survival and pro-death pathways, and this, perhaps, can explain the contradictory opinions in current literature regarding targeting ATF4 for clinical application. In this review, we summarized recent published studies from our labs and others that focus on the therapeutic potential of the strategy controlling ATF4 expression in different retinal and neurodegenerative disorders.
Introduction
Mammalian cells activate the unfolded protein response (UPR) in response to various internal and external cellular stimuli that disturb the balance in the endoplasmic reticulum (ER). These stimuli include, but are not limited to, accumulation of misfolded proteins, glucose deprivation (Roth et al., 2010; Csala et al., 2012), calcium dysfunction (Krebs et al., 2011; Shinde et al., 2016), hypoxia (Zheng et al., 2012), inflammation (Wang et al., 2016), redox potential changes (Hagiwara and Nagata, 2012), and mechanical stress (Husa et al., 2013). The normal physiological process involves activation of a signaling cascade, mediated by dissociation of the stress sensors protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme I (IRE-1), and activating transcription factor 6 (ATF 6) from glucose regulated protein 78 (Grp78), a key regulator of the UPR (Patil and Walter, 2001). This activation results in adjustment of the ER capacity to assist protein in the folding, activation of the ER-associated degradation (ERAD) pathway for degradation of abnormal proteins, and re-establishment of cellular homeostasis. However, a stimulus of long-lasting duration may cause a chronic ER stress. In this case, the cell experiences a “pathological” UPR that triggers programmed cell death, usually through sustained activation of the PERK UPR arm and pro-apoptotic components of cellular signaling (Figure 1; Woehlbier and Hetz, 2011). Detection of the physiological UPR could be challenging in cells due to the dynamics of downstream molecular events and quick equilibration of cellular homeostasis. By contrast, pathological UPR could be traced by accumulation of the UPR hallmark proteins and histological observation of consequent cell death (Hiramatsu et al., 2011). Recognition of a molecular switch operating between cell life and death decisions is perhaps a cornerstone of contemporary cell physiology and potentially will be a focus of future investigations.
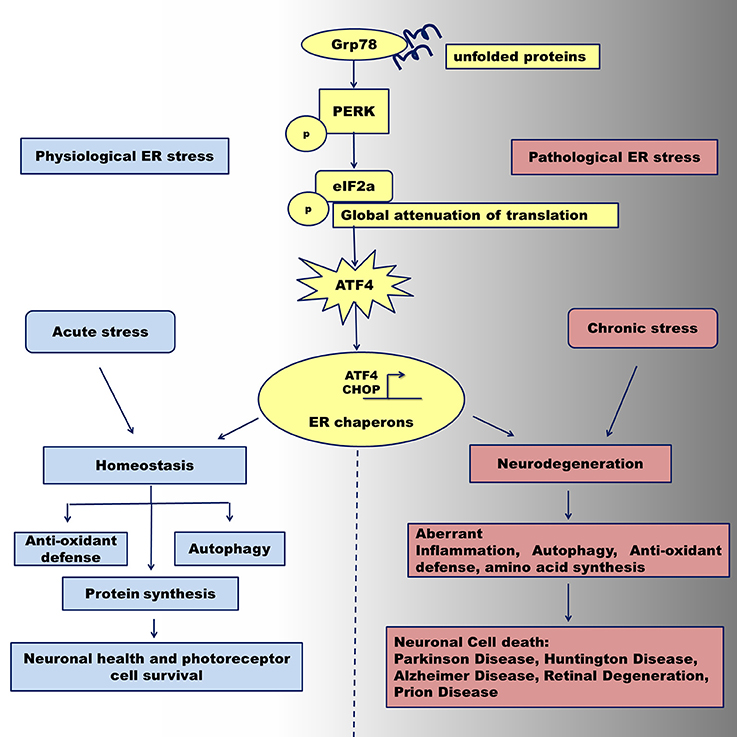
Figure 1. Activation of the PERK UPR signaling under physiological and pathological ER stress conditions. Abnormally folded proteins within the ER are sensed by GRP78 chaperone that in turn activates PERK pathway program. Under acute ER stress, the PERK kinase activates downstream mediator, phosphorylated eIF2a leading to up-regulation of ATF4. The latest together with CHOP protein activates transcription of ER stress chaperones that can regulate the transition from adaptation and neuronal cell survival via temporal translation inhibition leading to reestablishing of cellular homeostasis. After prolonged pathological ER stress, ATF4 can regulate the transition from adaptation/survival in neuronal cells to a pro-apoptotic phase via provoking aberrant autophagy signaling, translational program, and inflammatory response. Sustained malfunction of these cellular signaling may contribute to neuronal cells death through activation of apoptosis and leads to neurodegeneration seen in Parkinson disease, Alzheimer Disease, Huntington Diseases, Prion Disease and Retinal Degeneration.
At the molecular level, a cell under chronic ER stress continues to activate the PERK signaling pathway (Woehlbier and Hetz, 2011). Following transduction of information regarding the protein capacity of the ER, the IRE1 pathway is turned off (Woehlbier and Hetz, 2011). At the same time, the activation of the PERK downstream mediator activating transcription factor 4 (ATF4) induces the expression of a transcriptional factor C/EBP Homologous Protein (CHOP), which is, in turn, responsible for the activation of apoptosis through, Bcl-2-like protein 11 (BIM), p53 upregulated modulator of apoptosis (PUMA), and Noxa up-regulation (Huang et al., 2015). Therefore, the expression, activation, and duration of ATF4 activity could be an important parameter that determines cell fate. In this review, we summarized the latest discoveries on the role of the ATF4 protein in various retinal and neurodegenerative diseases.
ATF4: Expression and Post-Translational Modification
ATF4 is a 351 amino acid cAMP-response element binding protein that belongs to the cAMP response element-binding protein (CREB)-2 family of proteins (Vattem and Wek, 2004). The protein is structured into several domains that are essential for ATF4 function and degradation. The N-terminal of ATF4 contains a p300/CBP associated factor (PCAF) domain for p300 interaction, a leucine zing finger region II for the binding of neuronal cell death putative kinase, and a prolyl-4 hydroxylase domain (Hai and Curran, 1991; Karin and Smeal, 1992; Chérasse et al., 2007; Chan et al., 2013). The leucine zipper is essential for the dimerization of two DNA binding regions and for recognition of 5′-TGACGTCA-3′ DNA sequence within promoters(Vincent and Struhl, 1992). Two other domains, the oxygen dependent degradation (ODDD) domain and βTrCP motif, are involved in ATF4 degradation.
At the C-terminal, ATF4 contains the basic/leucine zipper domain (bZIP domain) that serves as a DNA binding sequence for interaction with abraxas brother 1 (Abro1), CCAAT/enhancer binding protein β (C/EBPβ), factor-inhibiting ATF4-mediated transcription (FIAT), AT-rich sequence-binding protein 2 (SATB2), death-associated protein Ser/Thr kinase 3 (DAPK3), and nuclear factor (erythroid- derived 2)-like 2 (Nrf2) (He et al., 2001). The ATF4 mRNA is transcribed ubiquitously at low levels, but its protein expression depends on various stress conditions, such as hypoxia, anoxia, and lack of nutrition, as well as glucose deprivation. The mouse ATF4 mRNA contains two upstream open reading frames (uORFs): the uORF1 and uORF2 located at 5′ of coding sequence. ATF4 expression involves a differential contribution of each upstream ORF(Vattem and Wek, 2004).
The uORF1 is a positive acting element that facilitates ribosome scanning (Wek et al., 2006), whereas the uORF2 acts as an inhibitory element that blocks the ATF4 expression that promotes ribosome scanning and re-initiation at the ATF4 coding sequence. As a part of a translational complex, the eukaryotic initiation factor 2 alpha (eIF2a) forms a ternary structure with GTP and Met-tRNA to bind the 40S ribosomal subunit and initiate translation (Kilberg et al., 2009). Under non-stress conditions, when eIF2a-GTP is plentiful due to the absence of stress, the complex binds a ribosome, GTP is hydrolyzed, and eIF2a-GDP is released from the complex for further association with eIF2b, an eIF2a GDP-GTP exchanger. This results in re-initiation of downstream ribosome scanning at the next uORF2 (Vattem and Wek, 2004; Wek et al., 2006). After translation of uORF2, ribosomes dissociate from the ATF4 mRNA. This leads to a reduction in expression of the ATF4-coding region and a weak ATF4 expression (Vattem and Wek, 2004). Under stress conditions, eIF2a is phosphorylated by four different kinases [PERK, protein kinase R(PKR), general control nonderepressible 2 (GCN2), and heme-regulated eIF2α kinase (HRI) kinases; (Vattem and Wek, 2004; Wek et al., 2006)], and this phosphorylation inhibits eIF2b-mediated eIF2a GDP-GTP exchange and represses global protein synthesis (Vattem and Wek, 2004). Meanwhile, eIF2a-GTP levels are lowered, and this deficit causes further ribosome scanning to be performed more carefully through negative-acting ORF2 positions at the initiation of the ATF4-coding region (Vattem and Wek, 2004). Therefore, translation of ATF4 is significantly enhanced in response to cellular stress.
In addition to the bZip domain, the ATF4 protein contains multiple sites for post-translational modification, including phosphorylation sites and sites for ubiquitination, SUMOylation, and acetylation. Protein kinase A (Karpinski et al., 1992), ribosomal S6 kinase 2 (Li et al., 2014), protein kinase CK2 (Manni et al., 2012), and RET tyrosine kinase (Bagheri-Yarmand et al., 2015) are all known to phosphorylate ATF4. At many positions, phosphorylation leads to ATF4 degradation (Lassot et al., 2001). ATF4 degradation also relies on a phosphorylation-dependent interaction with the SCF (betaTrCP) ubiquitin ligase (Lassot et al., 2001). Along with phosphorylation, another mechanism that controls the ATF4 activity is the binding of p300, NAD-dependent deacetylase sirtuin-1 (SIRT1), and FIAT. Histone acetyltransferase p300 binds to the N-terminal of ATF4, thereby stabilizing and enhancing ATF4 transcriptional activity (Lassot et al., 2005). Conversely, FIAT protein blocks the ATF4 activity by binding to a leucine zipper motif and preventing ATF4 from further associations. This motif is directly responsible for ATF4 inhibition when FIAT is over-expressed in cells (St-Arnaud and Elchaarani, 2007; Yu et al., 2009; St-Arnaud et al., 2010). In addition, NAD+-dependent deacetylase SIRT1 also downregulates ATF4 synthesis during proteasome inhibition in cells (Woo et al., 2013).
ATF4 Activates Pro-Survival and Pro-Death Signaling During Neurodegeneration
The activity of ATF4 is associated with its cellular localization. Under conditions of ER stress, ATF4 migrates to the nucleus, using the nucleus targeting KKLKK signal (amino acids 280 to 284) located within the basic region of the ATF4 gene. In the nucleus, ATF4 binds targeted DNAs and regulates their transcription (Han et al., 2013). The C/EBP homologous protein (CHOP, also known as GADD153) is one of the targeted genes. Together with ATF4, this protein regulates the expression of various cellular genes (Han et al., 2013). The list of shared targets includes 218 proteins. CHOP and ATF4 regulate multiple cellular processes, including the cellular response to ER stress (chaperones), protein biosynthesis, translation, and amino-tRNA synthetase activity. In addition, ATF4 controls the expression of 254 genes independently of CHOP (Han et al., 2013). These genes mostly govern cellular metabolism, including amino acid biosynthesis and transporter activity.
Multiple studies have identified ATF4 downstream targeted genes, but very few in vivo experiments have investigated up- and down-regulation of ATF4 in the brain or in eye tissues. We recently over-expressed ATF4 in the mouse retinal and rat dopamine nigral neurons (Bhootada et al., 2016; Gully et al., 2016). To do this, we leveraged the ability of the adeno-associated viruses with serotype 5 to deliver ATF4 cDNA, and we efficiently transduced the cDNA in photoreceptors of the retina or dopaminergic neurons of the substantia nigra pars compacta (SNc). We revealed that a 2.3-fold increase in ATF4 triggers retinal degeneration in wild type mice and mimics conditions with ATF4 over-expression in transgenic T17M rhodopsin retinas. Analysis of ERG recordings demonstrated a significant reduction in the functional tests in both types of retina (Bhootada et al., 2016). The functional loss in the retina was associated with a higher rate of photoreceptor cell death in mutant retinas over-expressing ATF4. Therefore, both the wild type and the mutant retinas experienced activation of apoptotic pathway.
We also conducted a second study, in which we investigated rats with a 3.2-fold increase in ATF4 in the SNc. These rats showed a loss of TH-positive cells when compared to control rats given AAV5-GFP injections (Gully et al., 2016). Cell loss was associated with a drop in the dopamine level and an increased activation of caspase-3/7. Taken together, our data on retinal and nigral neurons have demonstrated that over-expression of ATF4 results in progressive neurodegeneration. This discovery led to the next intriguing question regarding the cause of death of the neuronal cells over-expressing ATF4. We detected activation of caspase 3/7 in both studies, but we have not yet identified a direct molecular path that leads to neuronal cell death. The ATF4 and the ATF4 → CHOP signaling together can modulate the expression of 575 genes, which could eventually lead to neuronal death in these tissues (Han et al., 2013). Therefore, conducting further experiments with ATF4 titration and CHOP knockdown in these cells would be interesting in order to obtain a precise answer to this question.
ATF4 and Retinal Degenerative Diseases
Retinal degeneration is a progressive deterioration of retinal cells that eventually leads to their demise. The group of retinal degenerative disorders includes inherited, trauma-associated, diabetic, and age- related retinal degenerations. The cellular mechanism of retinopathies is complex and is often linked to multiple molecular markers of autophagy, oxidative stress, and inflammation. The UPR PERK → p- eIF2a → ATF4 pathway seems to play a significant role in photoreceptor deterioration in mice expressing aberrant proteins. Thus, we and other investigators have recently proposed that the UPR contributes markedly to retinal pathogenesis under various degenerative conditions (Gorbatyuk and Gorbatyuk, 2013; Zhang et al., 2014; Hiramatsu et al., 2015; Karthikeyan et al., 2017). Some investigators have suggested that “ER stress is a general upstream mechanism for neurodegeneration” and that “targeting ER stress molecules is a promising therapeutic strategy for neuroprotection (Huang et al., 2017).”
Research conducted over the past 5 years has identified ATF4 as one of the cellular markers that is elevated by various stress conditions in the retina. We have demonstrated that mice expressing mutant and truncated rhodopsins show dramatically elevated ATF4 levels (Kunte et al., 2012; Rana et al., 2014; Bhootada et al., 2016), while other investigators have detected ATF4 upregulation in different models of inherited retinal degeneration (Comitato et al., 2016; Lobo et al., 2016; Ooe et al., 2017). ATF4 regulates inflammatory signaling by governing the expression of multiple inflammatory genes. For example, in wild type mice, we found that ATF4 over-expression significantly increased the production of pro-inflammatory IL-1β (Rana et al., 2014). Huang et al. reported that ATF4 is a novel regulator of monocyte chemoattractant protein-1 (MCP-1) (Huang et al., 2015). These cytokines are all overproduced in the mouse retina during retinal degeneration. Therefore, not surprisingly, ATF4 downregulation in the T17M rhodopsin retina diminishes the ER stress response and prevents of loss of retinal function and photoreceptor cell death (Bhootada et al., 2016).
In degenerating retinas, enhanced ATF4 expression could be provoked by excessive light. Kuse et al. reported that blue light triggers photoreceptor cell death through activation of an ER stress response and ATF4 over-production (Kuse et al., 2014). This group subsequently demonstrated that light-induced S- opsin aggregation could be responsible for the activation of ATF4 (Ooe et al., 2017). Taken together, these findings would imply that mislocalization of opsin in the photoreceptors, on its own, is capable of triggering the ER stress response through activation of ATF4 signaling.
ATF4 plays a critical role in the progression of diabetic retinopathy (DR). Studies of proliferative DR have proposed ATF4 as one of the bio-markers of DR and suggest that it may represent a new therapeutic target for proliferative DR (Wang et al., 2017). Support for this proposal comes from a mouse model of type 1 diabetes with an activated ER stress response that inhibits ATF4 activity; these mice show a marked attenuation of the high–glucose-induced production of Intercellular Adhesion Molecule (iCAM), Tumor necrosis factor (TNFa), and vascular endothelial growth factor (VEGF) and a significant overall amelioration of retinal inflammation (Chen et al., 2012). Similarly, ATF4 over-expression triggers an inflammatory response in endothelial cells through activation of the Signal transducer and activator of transcription 3 (STAT3) pathway (Chen et al., 2012). This could explain why T17M rhodopsin mice with inherited retinal degeneration experience a preservation of retinal function and photoreceptor cell death when their retina is deficient in TNFa, a downstream ATF4 target (Rana et al., 2017).
Another hallmark of proliferative DR, and of the wet form of age-related macular degeneration, is angiogenesis. Our study of the hypoxic retina demonstrated that ATF4 deficiency could alleviate hypoxia-driven neovascularization in mice (Wang et al., 2013). Other investigators have also confirmed that ATF4 is a powerful promoter of angiogenesis (Liu et al., 2015; Chen et al., 2017). Taken together, these studies emphasize the need for further studies to determine the role of ATF4 in various neovascularization conditions.
ATF4 and CNS Neurodegenerative Diseases
Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and prion disease, are characterized by the slow and progressive loss of neural cells and subsequent dysfunction of the nervous system. Accumulation of misfolded proteins has been proposed as a common link in many neurodegenerative disorders (Li et al., 2013; Roussel et al., 2013; Scheper and Hoozemans, 2015; Remondelli and Renna, 2017). Thus, a study of neurodegeneration in a mouse model of AD revealed an activation of ATF4-mediated intra-axonal translation and identified ATF4 as a mediator of the spread of AD neurodegeneration (Baleriola et al., 2014). The AD brain (Ohno, 2014) and the brain of this AD-like mouse model (Jankowsky et al., 2004) both showed significant upregulation of the protein level of ATF4. This is perhaps associated with the fact that ATF4 upregulation “may not only act as the downstream effector of Aβ but also as an upstream initiator for the memory deficit and pathological hallmarks in AD (Wei et al., 2015).” An association is possible between ATF4 elevation and increased phosphorylation of tau, through Glycogen Synthase Kinase 3 (GSK-3) and Protein phosphatase 1 (PP1) kinases, and this could give rise to neuronal damage.
Sbodio et al., in their studies on the pathogenesis of HD, have demonstrated that dysfunction of ATF4, as a master regulator of amino acid homeostasis in cells, significantly contributes to molecular neurodegeneration. They postulated a disruption of cysteine biosynthesis in cells with polyglutamine repeats due to low activity of the cystathionine γ-lyase enzyme, which is due, in turn, to low ATF4 activity (Sbodio et al., 2016). They proposed that, under conditions where the oxidative stress persists or exceeds a certain level in a cell, ATF4 loses its ability to restore cellular responses and homeostasis. This implies that patients with HD might benefit from restoration of ATF4 activity that regulates a diverse spectrum of amino acid metabolism.
The cellular mechanism of PD pathogenesis involves the aggregation of α-synuclein (αS) in the neurons in the form of Lewy bodies and substantial loss of nigral dopaminergic neurons in the SNc. Studies in a PD transgenic mouse model expressing αS120 (Bellucci et al., 2011) and in patient brain biopsies (Sun et al., 2013) have reported elevated levels of ATF4 in the SNc. For example, Sun et al. demonstrated that 50% of all studied human PD brain sections contained nearly 80% neuromelanin-positive neurons that showed strong ATF4 immunostaining (Sun et al., 2013). The remaining 50% of the sections showed ATF4 staining compatible with that observed in control brains. The authors proposed that the mean duration of the disease significantly affected ATF4 expression and that a longer PD progression gave rise to a higher ATF4 expression.
The role of ATF4 has been further explored by Imai et al. and Bouman et al., who proposed that the PD-like progression in mice could involve the binding of activated ATF4 to the E3 ligase parkin promoter (Imai et al., 2000, 2001) and the mediation of parkin expression in response to ER and mitochondrial stress (Bouman et al., 2011). The authors suggest that increased expression of parkin is beneficial in the context of protection of cells from ER stress and mitochondrial damage, so that ATF4 may play a cytoprotective role during PD development. Notably, in our previous work, we showed the association between experimental PD progression and ATF4 protein level (Gorbatyuk et al., 2012). We found that recombinant adeno-associated virus r(AAV)- mediated overexpression of human αS results in a seven-fold induction of the ATF4 protein. Our study of α-synucleopathy progression in rats demonstrated that the elevation of ATF4 occurring during PD progression is associated with a loss of TH positive cells, a reduced dopamine level, and an increased behavior deficit (Gorbatyuk et al., 2012). We therefore hypothesized that elevation of ATF4 could have a deleterious role in the brain in patients with PD. To this end, we used AAV to overexpress ATF4 in the SNc of the rat and compare human αS over-expression. We found that the loss of TH positive cells and the reduction in dopamine levels were greater in the SNc of rats over-expressing ATF4 than in those over-expressing αS. Moreover, the animals overexpressing ATF4 demonstrated substantial behavior deficits when compared to a control group (Gorbatyuk et al., 2012).
ATF4 and Aging
Age affects the human body by altering proteostasis and enzymatic activity, thereby modulating multiple cellular pathways (Kaushik and Cuervo, 2015). Unfortunately, wide-ranging studies have mainly reported ATF4 over-expression under neurodegenerative conditions, which are prominent in aged tissues and organs. By contrast, ATF4 expression in healthy aged tissues has not received proper attention, with a few exceptions. Our study of aged retinas demonstrated a higher ATF4 expression in retinas from 24-month-old rats than from 4-month-old rats (Lenox et al., 2015). This increase was associated with elevated levels of GADD34 and CHOP proteins. In addition, Rantes/CCL5 expression was enhanced in aged retinas as well. The latest findings for CCL5 support the proposed ATF4 regulation of Rantes secretion through its interaction with the c- Jun molecule of the toll-like receptor 4 (TLR4)- signaling pathway (Zhang et al., 2013). In a further study of naive aged rats, we also demonstrated that 24-month-old rats show significant two-fold elevation of nigral ATF4 levels, which are associated with increases in endogenous α-syn in males and declines in GRP78 in both male and female aged brains (Salganik et al., 2015).
ATF4 has recently been proposed as a potential mediator of age-related muscle weakness and atrophy, as ATF4 downregulation by ursolic acid and tomatidine treatment significantly reduced age- related deficits in skeletal muscle strength, quality, and mass (Ebert et al., 2015). Other studies on age-related muscular dystrophy have revealed that increases in ATF4 expression alone are sufficient to induce fiber atrophy (Ebert et al., 2010). Therefore, ATF4 downregulation could be a potential therapeutic target for restoring skeletal muscle deficit in the elderly.
Conclusions
In this review, we have presented the latest findings concerning ATF4 involvement in retinal and neurodegenerative disorders. An analysis of the existing literature indicated contradictory opinions on the role of ATF4 in triggering the pro-survival or pro-death trend during developing experimental pathologies. The discrepancy could be due to the specificity of the activated molecular signaling, the stages of neurodegeneration, and the environmental conditions. Moreover, it is noteworthy to mention that the downstream targets and cellular signaling, including autophagy, oxidative stress, inflammation, and translation regulated by ATF4 are quite diverse. However, we believe that the major discrepancy between these studies' results lies in the use of in vivo and in vitro systems. Importantly, the therapy we are seeking has to define the potential of ATF4 to regulate the progression of neurodegeneration during chronic ER stress, which apparently is difficult, if not impossible, to reproduce in cell-culture disease models. This is also indicated by the fact that most, if not all, published works that have studied the consequences of direct modulation of ATF4 in animal disease models indicate that restriction of the excessive protein production can significantly diminish retinal and neurodegenerative disorders (Sidrauski et al., 2013; Halliday et al., 2015; Rozpedek et al., 2015; Wei et al., 2015; Bhootada et al., 2016; Gully et al., 2016). Therefore, future experiments are necessary to investigate whether downregulation of ATF4 would be a feasible therapeutic strategy for varied neurodegenerative pathologies.
Author Contributions
PP prepared figures, reviewed the literature and prepared the draft. OG reviewed the literature and wrote the review. MG reviewed the literature and wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the M. J. Fox Foundation grant (to OG) and the National Eye Institute EI R01EY020905 (to MG).
References
Bagheri-Yarmand, R., Sinha, K. M., Gururaj, A. E., Ahmed, Z., Rizvi, Y. Q., Huang, S. C., et al. (2015). A novel dual kinase function of the RET proto-oncogene negatively regulates activating transcription factor 4-mediated apoptosis. J. Biol. Chem. 290, 11749–11761. doi: 10.1074/jbc.M114.619833
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Bellucci, A., Navarria, L., Zaltieri, M., Falarti, E., Bodei, S., Sigala, S., et al. (2011). Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson's disease. J. Neurochem. 116, 588–605. doi: 10.1111/j.1471-4159.2010.07143.x
Bhootada, Y., Kotla, P., Zolotukhin, S., Gorbatyuk, O., Bebok, Z., Athar, M., et al. (2016). Limited ATF4 expression in degenerating retinas with ongoing ER stress promotes photoreceptor survival in a mouse model of autosomal dominant retinitis pigmentosa. PLoS ONE 11:e0154779. doi: 10.1371/journal.pone.0154779
Bouman, L., Schlierf, A., Lutz, A. K., Shan, J., Deinlein, A., Kast, J., et al. (2011). Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 18, 769–782. doi: 10.1038/cdd.2010.142
Chan, C. P., Kok, K. H., Tang, H. M., Wong, C. M., and Jin, D. Y. (2013). Internal ribosome entry site-mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim. Biophys. Acta 1833, 2165–2175. doi: 10.1016/j.bbamcr.2013.05.002
Chen, D., Fan, Z., Rauh, M., Buchfelder, M., Eyupoglu, I. Y., Savaskan, N., et al. (2017). ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 36, 5593–5608. doi: 10.1038/onc.2017.146
Chen, Y., Wang, J. J., Li, J., Hosoya, K. I., Ratan, R., Townes, T., et al. (2012). Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia 55, 2533–2545. doi: 10.1007/s00125-012-2594-1
Chérasse, Y., Maurin, A. C., Chaveroux, C., Jousse, C., Carraro, V., Parry, L., et al. (2007). The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 35, 5954–5965. doi: 10.1093/nar/gkm642
Comitato, A., Di Salvo, M. T., Turchiano, G., Montanari, M., Sakami, S., Palczewski, K., et al. (2016). Dominant and recessive mutations in rhodopsin activate different cell death pathways. Hum. Mol. Genet. 25, 2801–2812. doi: 10.1093/hmg/ddw137
Csala, M., Kereszturi, É., Mandl, J., and Bánhegyi, G. (2012). The endoplasmic reticulum as the extracellular space inside the cell: role in protein folding and glycosylation. Antioxid. Redox Signal. 16, 1100–1108. doi: 10.1089/ars.2011.4227
Ebert, S. M., Dyle, M. C., Bullard, S. A., Dierdorff, J. M., Murry, D. J., Fox, D. K., et al. (2015). Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)-dependent pathway to age-related skeletal muscle weakness and atrophy. J. Biol. Chem. 290, 25497–25511. doi: 10.1074/jbc.M115.681445
Ebert, S. M., Monteys, A. M., Fox, D. K., Bongers, K. S., Shields, B. E., Malmberg, S. E., et al. (2010). The transcription factor ATF4 promotes skeletal myofiber atrophy during fasting. Mol. Endocrinol. 24, 790–799. doi: 10.1210/me.2009-0345
Gorbatyuk, M. S., Shabashvili, A., Chen, W., Meyers, C., Sullivan, L. F., Salganik, M., et al. (2012). Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 20, 1327–1337. doi: 10.1038/mt.2012.28
Gorbatyuk, M., and Gorbatyuk, O. (2013). Review: retinal degeneration: focus on the unfolded protein response. Mol. Vis. 19, 1985–1998.
Gully, J. C., Sergeyev, V. G., Bhootada, Y., Mendez-Gomez, H., Meyers, C., Zolotukhin, S., et al. (2016). Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson's disease. Neurosci. Lett. 627, 36–41. doi: 10.1016/j.neulet.2016.05.039
Hagiwara, M., and Nagata, K. (2012). Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation. Antioxid. Redox Signal. 16, 1119–1128. doi: 10.1089/ars.2011.4495
Hai, T., and Curran, T. (1991). Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. U.S.A. 88, 3720–3724
Halliday, M., Radford, H., Sekine, Y., Moreno, J., Verity, N., le Quesne, J., et al. (2015). Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6:e1672. doi: 10.1038/cddis.2015.49
Han, J., Back, S. H., Hur, J., Lin, Y. H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. doi: 10.1038/ncb2738
He, C. H., Gong, P., Hu, B., Stewart, D., Choi, M. E., Choi, A. M., et al. (2001). Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 276, 20858–20865. doi: 10.1074/jbc.M101198200
Hiramatsu, N., Chiang, W. C., Kurt, T. D., Sigurdson, C. J., and Lin, J. H. (2015). Multiple mechanisms of unfolded protein response-induced cell death. Am. J. Pathol. 185, 1800–1808. doi: 10.1016/j.ajpath.2015.03.009
Hiramatsu, N., Joseph, V. T., and Lin, J. H. (2011). Monitoring and manipulating mammalian unfolded protein response. Meth. Enzymol. 491, 183–198. doi: 10.1016/B978-0-12-385928-0.00011-0
Huang, H., Jing, G., Wang, J. J., Sheibani, N., and Zhang, S. X. (2015). ATF4 is a novel regulator of MCP-1 in microvascular endothelial cells. J. Inflamm. (Lond). 12:31. doi: 10.1186/s12950-015-0076-1
Huang, H., Miao, L., Liang, F., Liu, X., Xu, L., Teng, X., et al. (2017). Neuroprotection by eIF2alpha-CHOP inhibition and XBP-1 activation in EAE/optic neuritiss. Cell Death Dis. 8:e2936. doi: 10.1038/cddis.2017.329
Husa, M., Petursson, F., Lotz, M., Terkeltaub, R., and Liu-Bryan, R. (2013). C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthr. Res. Ther. 15:R218. doi: 10.1186/ar4415
Imai, Y., Soda, M., and Takahashi, R. (2000). Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275, 35661–35664. doi: 10.1074/jbc.C000447200.
Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y., Takahashi, R., et al. (2001). An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105, 891–902. doi: 10.1016/S0092-8674(01)00407-X
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170. doi: 10.1093/hmg/ddh019
Karin, M., and Smeal, T. (1992). Control of transcription factors by signal transduction pathways: the beginning of the end. Trends Biochem. Sci. 17, 418–422
Karpinski, B. A., Morle, G. D., Huggenvik, J., Uhler, M. D., and Leiden, J. M. (1992). Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc. Natl. Acad. Sci. U.S.A. 89, 4820–4824
Karthikeyan, B., Harini, L., Krishnakumar, V., Kannan, V. R., Sundar, K., and Kathiresan, T. (2017). Insights on the involvement of (-)-epigallocatechin gallate in ER stress-mediated apoptosis in age-related macular degeneration. Apoptosis 22, 72–85. doi: 10.1007/s10495-016-1318-2
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Kilberg, M. S., Shan, J., and Su, N. (2009). ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 20, 436–443. doi: 10.1016/j.tem.2009.05.008
Krebs, J., Groenendyk, J., and Michalak, M. (2011). Ca2+-signaling, alternative splicing and endoplasmic reticulum stress responses. Neurochem. Res. 36, 1198–1211. doi: 10.1007/s11064-011-0431-4
Kunte, M. M., Choudhury, S., Manheim, J. F., Shinde, V. M., Miura, M., Chiodo, V. A., et al. (2012). ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 53, 3792–3800. doi: 10.1167/iovs.11-9235
Kuse, Y., Ogawa, K., Tsuruma, K., Shimazawa, M., and Hara, H. (2014). Damage of photoreceptor-derived cells in culture induced by light emitting diode-derived blue light. Sci. Rep. 4:5223. doi: 10.1038/srep05223
Lassot, I., Estrabaud, E., Emiliani, S., Benkirane, M., Benarous, R., and Margottin-Goguet, F. (2005). p300 modulates ATF4 stability and transcriptional activity independently of its acetyltransferase domain. J. Biol. Chem. 280, 41537–41545. doi: 10.1074/jbc.M505294200
Lassot, I., Ségéral, E., Berlioz-Torrent, C., Durand, H., Groussin, L., Hai, T., et al. (2001). ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol. Cell. Biol. 21, 2192–2202. doi: 10.1128/MCB.21.6.2192-2202.2001
Lenox, A. R., Bhootada, Y., Gorbatyuk, O., Fullard, R., and Gorbatyuk, M. (2015). Unfolded protein response is activated in aged retinas. Neurosci. Lett. 609, 30–35. doi: 10.1016/j.neulet.2015.10.019.
Li, J., O, W., Li, W., Jiang, Z. G., and Ghanbari, H. A. (2013). Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 14, 24438–24475. doi: 10.3390/ijms141224438
Li, T. F., Yukata, K., Yin, G., Sheu, T., Maruyama, T., Jonason, J. H., et al. (2014). BMP-2 induces ATF4 phosphorylation in chondrocytes through a COX-2/PGE2 dependent signaling pathway. Osteoarthr. Cartil. 22, 481–489. doi: 10.1016/j.joca.2013.12.020
Liu, C., Li, Z., Wang, L., Tong, L., He, N., Chen, Y., et al. (2015). Activating transcription factor 4 promotes angiogenesis of breast cancer through enhanced macrophage recruitment. Biomed Res. Int. 2015:974615. doi: 10.1155/2015/974615
Lobo, G. P., Au, A., Kiser, P. D., and Hagstrom, S. A. (2016). Involvement of Endoplasmic Reticulum Stress in TULP1 induced retinal degeneration. PLoS ONE 11:e0151806. doi: 10.1371/journal.pone.0151806
Manni, S., Brancalion, A., Tubi, L. Q., Colpo, A., Pavan, L., Cabrelle, A., et al. (2012). Protein kinase CK2 protects multiple myeloma cells from ER stress-induced apoptosis and from the cytotoxic effect of HSP90 inhibition through regulation of the unfolded protein response. Clin. Cancer Res. 18, 1888–1900. doi: 10.1158/1078-0432.CCR-11-1789
Ohno, M. (2014). Roles of eIF2alpha kinases in the pathogenesis of Alzheimer's disease. Front. Mol. Neurosci. 7:22. doi: 10.3389/fnmol.2014.00022
Ooe, E., Tsuruma, K., Kuse, Y., Kobayashi, S., Shimazawa, M., and Hara, H. (2017). The involvement of ATF4 and S-opsin in retinal photoreceptor cell damage induced by blue LED light. Mol. Vis. 23, 52–59.
Patil, C., and Walter, P. (2001). Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 13, 349–355. doi: 10.1016/S0955-0674(00)00219-2
Rana, T., Kotla, P., Fullard, R., and Gorbatyuk, M. (2017). TNFa knockdown in the retina promotes cone survival in a mouse model of autosomal dominant retinitis pigmentosa. Biochim. Biophys. Acta 1863, 92–102. doi: 10.1016/j.bbadis.2016.10.008
Rana, T., Shinde, V. M., Starr, C. R., Kruglov, A. A., Boitet, E. R., Kotla, P., et al. (2014). An activated unfolded protein response promotes retinal degeneration and triggers an inflammatory response in the mouse retina. Cell Death Dis. 5:e1578. doi: 10.1038/cddis.2014.539
Remondelli, P., and Renna, M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10:187. doi: 10.3389/fnmol.2017.00187
Roth, J., Zuber, C., Park, S., Jang, I., Lee, Y., Kysela, K. G., et al. (2010). Protein N-glycosylation, protein folding, and protein quality control. Mol. Cells 30, 497–506. doi: 10.1007/s10059-010-0159-z
Roussel, B. D., Kruppa, A. J., Miranda, E., Crowther, D. C., Lomas, D. A., and Marciniak, S. J. (2013). Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 12, 105–118. doi: 10.1016/S1474-4422(12)70238-7
Rozpedek, W., Markiewicz, L., Diehl, J. A., Pytel, D., and Majsterek, I. (2015). Unfolded protein response and PERK kinase as a new therapeutic target in the pathogenesis of Alzheimer's disease. Curr. Med. Chem. 22, 3169–3184. doi: 10.2174/0929867322666150818104254
Salganik, M., Sergeyev, V. G., Shinde, V., Meyers, C. A., Gorbatyuk, M. S., Lin, J. H., et al. (2015). The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (alpha-syn) toxicity to rat nigral neurons. Neurobiol. Aging 36, 2213–2223. doi: 10.1016/j.neurobiolaging.2015.02.018
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2016). Transcriptional control of amino acid homeostasis is disrupted in Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 113, 8843–8848. doi: 10.1073/pnas.1608264113
Scheper, W., and Hoozemans, J. J. (2015). The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. doi: 10.1007/s00401-015-1462-8
Shinde, V., Kotla, P., Strang, C., and Gorbatyuk, M. (2016). Unfolded protein response-induced dysregulation of calcium homeostasis promotes retinal degeneration in rat models of autosomal dominant retinitis pigmentosa. Cell Death Dis. 7:e2085. doi: 10.1038/cddis.2015.325
Sidrauski, C., Acosta-Alvear, D., Khoutorsky, A., Vedantham, P., Hearn, B. R., Li, H., et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2:e00498. doi: 10.7554/eLife.00498
St-Arnaud, R., and Elchaarani, B. (2007). Identification of additional dimerization partners of FIAT, the factor inhibiting ATF4-mediated transcription. Ann. N.Y. Acad. Sci. 1116, 208–215. doi: 10.1196/annals.1402.028
St-Arnaud, R., Mandic, V., and Elchaarani, B. (2010). FIAT, the factor-inhibiting ATF4-mediated transcription, also represses the transcriptional activity of the bZIP factor FRA-1. Ann. N.Y. Acad. Sci. 1192, 338–343. doi: 10.1111/j.1749-6632.2009.05211.x
Sun, X., Liu, J., Crary, J. F., Malagelada, C., Sulzer, D., Greene, L. A., et al. (2013). ATF4 protects against neuronal death in cellular Parkinson's disease models by maintaining levels of parkin. J. Neurosci. 33, 2398–2407. doi: 10.1523/JNEUROSCI.2292-12.2013
Vattem, K. M., and Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11269–11274. doi: 10.1073/pnas.0400541101
Vincent, A. C., and Struhl, K. (1992). ACR1, a yeast ATF/CREB repressor. Mol. Cell. Biol. 12, 5394–5405.
Wang, X., Wang, G., Kunte, M., Shinde, V., and Gorbatyuk, M. (2013). Modulation of angiogenesis by genetic manipulation of ATF4 in mouse model of oxygen-induced retinopathy [corrected]. Invest. Ophthalmol. Vis. Sci. 54, 5995–6002. doi: 10.1167/iovs.13-12117
Wang, Y., Gao, S., Zhu, Y., and Shen, X. (2017). Elevated activating transcription factor 4 and glucose-regulated 78 Kda protein levels correlate with inflammatory cytokines in the aqueous humor and vitreous of proliferative diabetic retinopathy. Curr. Eye Res. 42, 1–7. doi: 10.1080/02713683.2017.1297998
Wang, Y., Gong, J., Zeng, H., Liu, R., Jin, B., Chen, L., et al. (2016). Lipopolysaccharide activates the unfolded protein response in human periodontal ligament fibroblasts. J. Periodontol. 87, e75–e81. doi: 10.1902/jop.2015.150413
Wei, N., Zhu, L. Q., and Liu, D. (2015). ATF4: a novel potential therapeutic target for Alzheimer's disease. Mol. Neurobiol. 52, 1765–1770. doi: 10.1007/s12035-014-8970-8
Wek, R. C., Jiang, H. Y., and Anthony, T. G. (2006). Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11. doi: 10.1042/BST20060007
Woehlbier, U., and Hetz, C. (2011). Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem. Sci. 36, 329–337. doi: 10.1016/j.tibs.2011.03.001
Woo, S. R., Park, J. E., Kim, Y. H., Ju, Y. J., Shin, H. J., Joo, H. Y., et al. (2013). SIRT1 suppresses activating transcription factor 4 (ATF4) expression in response to proteasome inhibition. J. Microbiol. Biotechnol. 23, 1785–1790. doi: 10.4014/jmb.1309.09027
Yu, V. W., El-Hoss, J., and St-Arnaud, R. (2009). FIAT inhibition increases osteoblast activity by modulating Atf4-dependent functions. J. Cell. Biochem. 106, 186–192. doi: 10.1002/jcb.21995
Zhang, C., Bai, N., Chang, A., Zhang, Z., Yin, J., Shen, W., et al. (2013). ATF4 is directly recruited by TLR4 signaling and positively regulates TLR4-trigged cytokine production in human monocytes. Cell. Mol. Immunol. 10, 84–94. doi: 10.1038/cmi.2012.57
Zhang, S. X., Sanders, E., Fliesler, S. J., and Wang, J. J. (2014). Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 125, 30–40. doi: 10.1016/j.exer.2014.04.015
Keywords: activating transcription factor 4, ER stress response, unfolded protein response (UPR), neurodegenerative diseases, retinal diseases, neurons, photoreceptor cells, vertebrate
Citation: Pitale PM, Gorbatyuk O and Gorbatyuk M (2017) Neurodegeneration: Keeping ATF4 on a Tight Leash. Front. Cell. Neurosci. 11:410. doi: 10.3389/fncel.2017.00410
Received: 20 September 2017; Accepted: 05 December 2017;
Published: 15 December 2017.
Edited by:
Davide Cervia, Università degli Studi della Tuscia, ItalyCopyright © 2017 Pitale, Gorbatyuk and Gorbatyuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oleg Gorbatyuk, b2xlZ2dvckB1YWIuZWR1
Marina Gorbatyuk, bWdvcnRrQHVhYi5lZHU=