 Jessica Di Re
Jessica Di Re Paul A. Wadsworth
Paul A. Wadsworth Fernanda Laezza
Fernanda Laezza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 19 April 2017
Sec. Cellular Neuropathology
Volume 11 - 2017 | https://doi.org/10.3389/fncel.2017.00103
This article is part of the Research Topic Biology of Brain Disorders View all 43 articles
The finely tuned regulation of neuronal firing relies on the integrity of ion channel macromolecular complexes. Minimal disturbances of these tightly regulated networks can lead to persistent maladaptive plasticity of brain circuitry. The intracellular fibroblast growth factor 14 (FGF14) belongs to the nexus of proteins interacting with voltage-gated Na+ (Nav) channels at the axonal initial segment. Through isoform-specific interactions with the intracellular C-terminal tail of neuronal Nav channels (Nav1.1, Nav1.2, Nav1.6), FGF14 controls channel gating, axonal targeting and phosphorylation in neurons effecting excitability. FGF14 has been also involved in synaptic transmission, plasticity and neurogenesis in the cortico-mesolimbic circuit with cognitive and affective behavioral outcomes. In translational studies, interest in FGF14 continues to rise with a growing list of associative links to diseases of the cognitive and affective domains such as neurodegeneration, depression, anxiety, addictive behaviors and recently schizophrenia, suggesting its role as a converging node in the etiology of complex brain disorders. Yet, a full understanding of FGF14 function in neurons is far from being complete and likely to involve other functions unrelated to the direct regulation of Nav channels. The goal of this Mini Review article is to provide a summary of studies on the emerging role of FGF14 in complex brain disorders.
In 2014, nearly one in four adults in the United States was diagnosed with a mental illness (National Institutes of Mental Health1). Treatment for many of these illnesses is hampered by limited efficacy of the medications and patient noncompliance due to intolerable side effects. To address this need, the National Institutes of Mental Health has launched an initiative to research these illnesses from all levels, ranging from genomic to behavioral. Dubbed the Research Domain Criteria (RDoC), this initiative proposes to complement top-down understanding of these diseases, beginning with human behavior, with bottom-up research by understanding the molecular and cellular causes of these disorders.
Because many neuropsychiatric disorders are associated with maladaptive plasticity and excitability, one area of importance within neurons is the axon initial segment (AIS), which serves as the action potential initiation site (Palmer and Stuart, 2006). This highly complex subcellular region contains a nexus of scaffolding and regulatory proteins that ensure proper targeting, clustering and function of the ion channels underlying the action potential (Ogawa and Rasband, 2008; Hsu et al., 2014). Neuropsychiatric disorders have been associated with many of these proteins, including ankyrin-G, α-spectrins, β-spectrins, neurofascin, contactin and intracellular fibroblast growth factors (iFGFs; Hsu et al., 2014). One such protein, fibroblast growth factor 14 (FGF14, also known as fibroblast homologous factor 4 or FHF4), is an iFGF that binds to voltage-gated Na+ (Nav) channels and promotes their localization to the proximal region of the axon, providing the fine-tuned regulation necessary for normal functioning (Lou et al., 2005; Laezza et al., 2007, 2009; Goetz et al., 2009; Wang et al., 2011, 2012; Wildburger et al., 2015; Ali et al., 2016; Bosch et al., 2016; Hsu et al., 2016; Pablo et al., 2016). Loss of functional FGF14 may change the biophysical properties of Nav channels or alter their localization to the AIS, leading to changes in neuronal excitability (Goldfarb et al., 2007; Shakkottai et al., 2009; Bosch et al., 2015; Hsu et al., 2016). Recent findings also show that FGF14 regulates the function of voltage-gated K+ and Ca2+ channels, however none of these interactions are direct, and therefore might represent a different type of regulation from what has been described for Nav channels (Yan et al., 2013; Pablo and Pitt, 2017).
Initially cloned on the basis of sequence similarity with other FGF members, FGF14 was first associated to a human disease with the F145S mutation causing spinocerebellar ataxia 27 (SCA27), a naturally occurring complex neurodegenerative disorder characterized by onset of ataxia in early adulthood and deficits in cognition, memory and behavior (Smallwood et al., 1996; van Swieten et al., 2003; Brusse et al., 2006). Genetic deletion of FGF14 in mice recapitulates some of these symptoms at the behavior and circuitry level (Wang et al., 2002; Wozniak et al., 2007).
Since then, FGF14 has been indicated by several linkage and genome wide association studies (GWAS) to be a putative risk factor for other neuropsychiatric diseases including depression, addiction and schizophrenia, as well as neurodegenerative diseases, such as Alzheimer’s Disease (Detera-Wadleigh et al., 1999; Park et al., 2004; Mulle et al., 2005; Need et al., 2009; Johnson et al., 2011; Verbeek et al., 2012; Singh and Rajeswari, 2015; Yang et al., 2015). These recent associations clearly indicate that the role of this gene in the CNS is yet to be fully understood. In the next paragraphs, we will summarize some of the most recent studies on FGF14 in animal models and human tissue.
While iFGFs share a conserved core β-trefoil region with other FGFs their functions and distributions are distinct from canonical FGFs (Itoh and Ornitz, 2008). Canonical FGFs are normally secreted to activate FGF receptors on the cell surface, however iFGFs lack a secretory sequence, fail to activate or antagonize FGF receptors and are primarily found in the cytoplasm, nucleus or the AIS (Smallwood et al., 1996; Olsen et al., 2003; Ornitz and Itoh, 2015; Pablo et al., 2016). Initial discoveries using yeast-two-hybrid screening identified FGF12 and FGF13 as direct interactors of Nav channels (Liu et al., 2003; Wittmack et al., 2004; Rush et al., 2006) and subsequent studies resulted in similar discoveries for the two isoforms of FGF14 (Lou et al., 2005; Laezza et al., 2007, 2009), aligned in the illustration of Figure 1A. To date, the evidence for direct interaction and functional modulation of Nav channels by FGF14 ranges from crystal structure to biochemical in cell assays to animal models and includes recent identification of critical amino acid residues at the FGF14: Nav1.6 channel complex illustrated in Figure 1B (Lou et al., 2005; Laezza et al., 2007, 2009; Goetz et al., 2009; Ali et al., 2014, 2016; Hsu et al., 2016; Pablo et al., 2016).
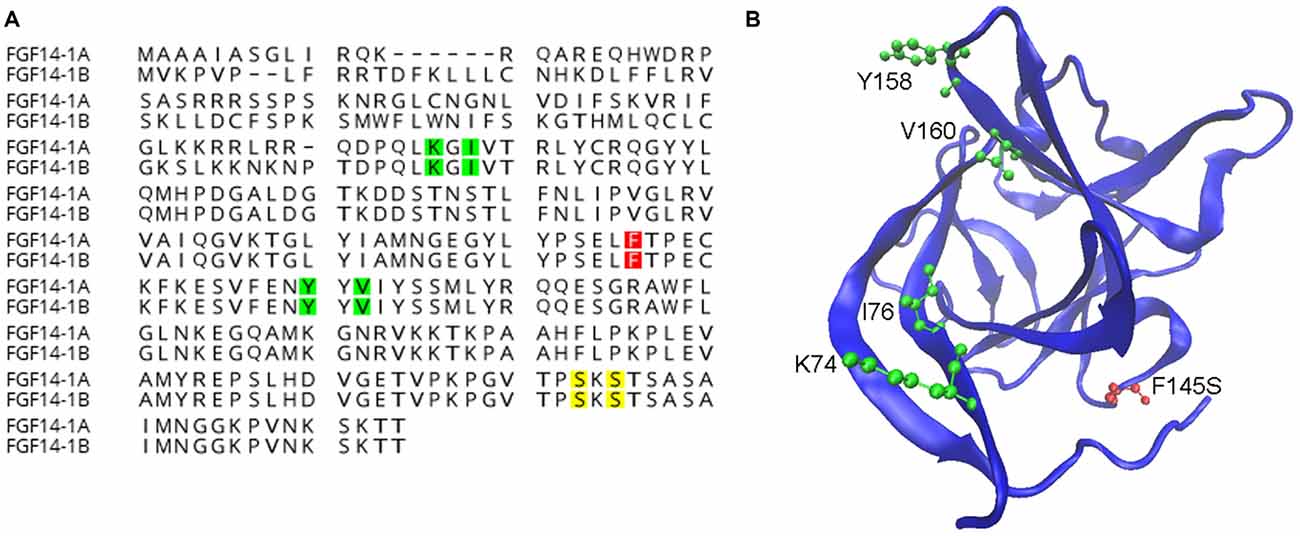
Figure 1. Known functions of amino acids in fibroblast 14 (FGF14). (A) Alignment of wild-type FGF14-1a and FGF14-1b protein sequences with highlighted residues representing surface hot-spots for FGF14:Nav1.6 interactions (green, Ali et al., 2016), casein kinase II (CK-2) phosphorylation sites (yellow, Hsu et al., 2016), and the spinocerebellar ataxia 27 (SCA27) F145S mutation site (red, Laezza et al., 2007). (B) An FGF14 homology model generated using the FGF13:Nav1.5 crystal structure (Protein Data Bank ID: 4DCK) as template, visualized using the visual molecular dynamic (VMD) software package (Humphrey et al., 1996). Surface hot-spots important for protein:protein interactions (green) and the SCA27 F145S mutation (red) are shown.
The N-terminus of FGF14 is alternatively spliced into two isoforms: FGF14-1a and FGF14-1b. FGF14-1a shares sequence homology with FGF12-1a and FGF13-1a, while the amino terminus of FGF14-1b contains a unique 69 amino-acid sequence and is the more prevalent isoform in the CNS (Munoz-Sanjuan et al., 2000). Importantly, the interaction of FGF14 in cells that heterologously express individual Nav isoforms shows that FGF14 is unique, as it results in very distinct isoform-specific Na+ current phenotypes that are dictated by the two FGF14 splice variants (Lou et al., 2005; Laezza et al., 2009; Ali et al., 2016). In primary hippocampal neurons, overexpression of FGF14-1b increases Na+ current density, causes a hyperpolarizing shift in the voltage-dependence of activation and a depolarizing shift in the voltage-dependence of inactivation, while the F145S, SCA27 dominant negative loss-of-function mutation causes opposite phenotypes, possibly by disrupting the function of wild-type FGF14 (Laezza et al., 2007). Neurons from Fgf14−/− mice exhibit impaired excitability in both the hippocampus and cerebellum (Goldfarb et al., 2007; Shakkottai et al., 2009; Bosch et al., 2015; Hsu et al., 2016). The effect of FGF14 on Nav channels and excitability is summarized in Table 1.
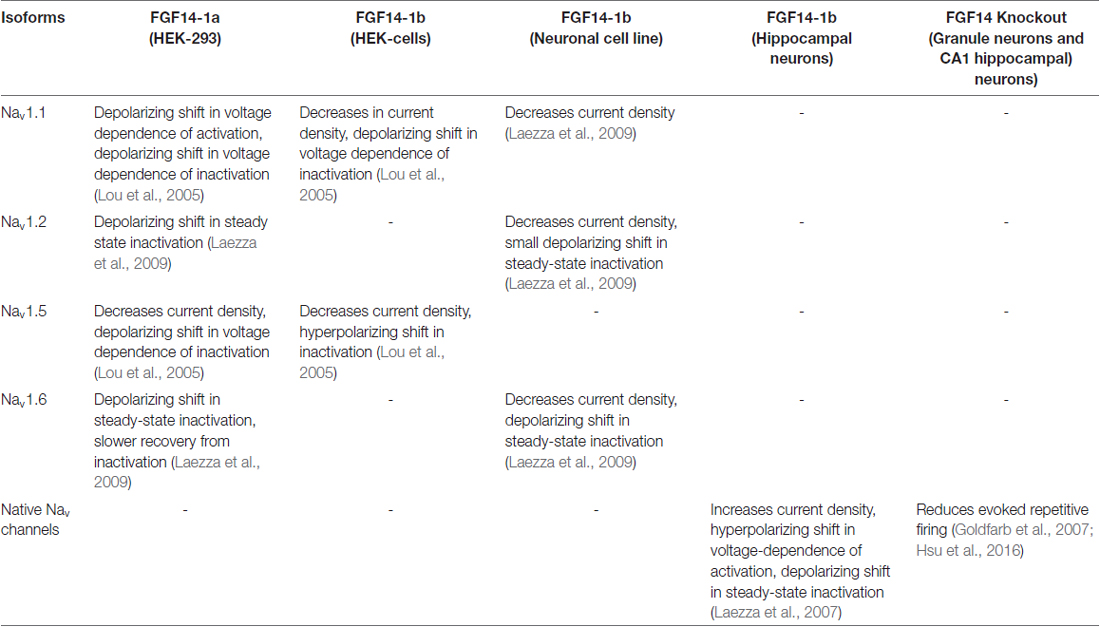
Table 1. Effect of splice variants in heterologous systems and knockout animals.
Recent studies have added a new dimension to FGF14, showing that its interaction with the Nav channel is controlled by selective kinases (Shavkunov et al., 2012, 2013; Hsu et al., 2015, 2016). Initial studies using the luciferase complementation assay demonstrated that the FGF14:Nav1.6 complex formation is controlled by glycogen synthase kinase 3 (GSK; Shavkunov et al., 2013) and more recently by the GSK3 priming kinase, casein kinase II (CK2) which phosphorylates FGF14 at S228 and S230 (Hsu et al., 2016; Figure 1B). Inhibition of either GSK3 or CK2 is sufficient to disrupt the FGF14:Nav channel complex formation with consequences for targeting of the two proteins to the AIS and for intrinsic excitability (Shavkunov et al., 2013; Hsu et al., 2016). It is possible that phosphorylation at these kinase specific sites that confers functional specificity to FGF14 contributing to regulation of other ion channels (i.e., voltage-gated K+ and Ca2+ channels).
More is known about the specific phosphorylation of FGF14 by GSK3 and CK2, however other kinases have been shown to affect FGF14:Nav1.6 interactions (Shavkunov et al., 2012; Hsu et al., 2015). Importantly, many kinases involved in tyrosine receptor kinase signaling are implicated in this regulation, including the mitogen activated protein kinase (MAPK), C-Jun N-terminal kinase (JNK; Hsu et al., 2015). JNK signaling is disrupted in insulin resistance associated with type-II diabetes and Alzheimer’s disease (Najem et al., 2016). Functional enrichment of single nucleotide polymorphisms (SNPs) in patients with type-II diabetes and Alzheimer’s disease shows that FGF14 is significantly overrepresented in these two diseases because of its phosphorylation by JNK (Hao et al., 2015). Changes in the mRNA expression of MAPK/JNK signaling proteins, including FGF14, are also significantly overrepresented in early-onset Alzheimer’s disease patients (Antonell et al., 2013). Taken together, these results indicate that the interaction between JNK and FGF14 might be an important area for future research in Alzheimer’s disease.
Numerous are the reports of the effect of genetic deletion of Fgf14 on synaptic transmission. Studies in the cerebellum of Fgf14−/− mice revealed decreased excitatory transmission from granule cells to Purkinje cells (parallel fibers, PF), a phenotype that is accompanied by reduced AMPA receptor-mediated excitatory postsynaptic currents and decreased expression of vesicular glutamate transporter 1, a specific presynaptic marker at PF-Purkinje neuron synapses (Tempia et al., 2015). Presynaptic changes in neurotransmitter release have been also reported at the Schaffer’s collaterals to CA1 synapses where deletion of Fgf14 results in reduction in the ready-releasable pool of presynaptic glutamate and diminished expression of synaptobrevin, synaptophysin, syntaxin I (Xiao et al., 2007). Other changes in presynaptic function have been reported at inhibitory GABAergic terminals onto CA1 pyramidal cells of Fgf14−/− mice, which exhibit reduced expression of glutamic acid decarboxylase 67 (GAD67) and vesicular GABA transporter (vGAT), presumably deriving from fast-spiking parvalbumin (PV) interneurons synapses (Alshammari T. K. et al., 2016). Additional studies in the same animal model identified selective loss of PV interneurons, reduced γ frequency oscillations and deficits in working memory (Alshammari T. K. et al., 2016). Collectively, these results recapitulate some endophenotypes of schizophrenia and are supported by human studies finding significant reduction and co-variation of FGF14, PV, vGAT and GAD67 in post-mortem samples from schizophrenic patients compared to healthy control individuals (Alshammari T. K. et al., 2016). Whether all these changes at presynaptic glutamatergic and GABAergic terminals result from neuroadaptive responses to impaired firing or represent disruption of a separate function of the FGF14 protein remains to be determined. However, the evidence for genetic links between vGAT, GAD67 and FGF14 might argue for a “separate function” hypothesis of FGF14 that results from a control at the gene level.
Studies have also supported a role of FGF14 in synaptic plasticity in the hippocampus. Fgf14−/− mice show impaired long-term potentiation (LTP) at the Schaffer’s collaterals to CA1 synapses, which is accompanied by decreased expression of synaptic vesicles docked at the active zone, and fewer miniature excitatory postsynaptic currents in primary hippocampal neurons (Xiao et al., 2007). Short-term plasticity is also impaired at these Fgf14−/− terminals, at which repetitive stimuli causes significant synaptic fatigue, consistent with impaired presynaptic function (Xiao et al., 2007).
Adult neurogenesis, or the proliferation, differentiation and integration of new neurons into existing brain circuitry has become an area of research interest in part due to its implication in the cognitive pathophysiology of several neuropsychiatric disorders, including Alzheimer’s disease, depression and schizophrenia (Ming and Song, 2005; Taupin, 2005, 2008; Reif et al., 2007; Johnson et al., 2009; Sun et al., 2011; Jun et al., 2012; Walton et al., 2012; Ouchi et al., 2013). It was recently found that FGF14 is required for the maturation of progenitor cells in the dentate gyrus of the hippocampus. Fgf14−/− mice show impaired transition from late immature neuronal progenitor cells to mature neurons, which is accompanied by reduced paired-pulse facilitation at the perforant path to granule neurons in the dentate gyrus (Alshammari M. A. et al., 2016). Overall, deletion of FGF14 results in an immature dentate gyrus, an endophenotype that corroborates a link between the gene and schizophrenia (Hagihara et al., 2013).
As many neuropsychiatric disorders are heterogeneous and complex, GWAS have become an important tool for sorting relevant genetic information from large patient populations. Numerous GWAS have reported SNPs in FGF14 in the context of neuropsychiatric disorders (Table 2). Although all these SNPs are in the FGF14 intronic region and thus their role on the protein expression and function are unclear, they might provide guidance for future investigations. A Brazilian pilot study on early onset/familial schizophrenia found a link between early-onset schizophrenia and FGF14 (Gadelha et al., 2012). GWAS in German cohort found an association between FGF14 and schizophrenia, which is corroborated by a linkage study of familial schizophrenia in Canadian families of Celtic or German descent (Brzustowicz et al., 1999; Need et al., 2009). Additionally, SNPs in FGF14 have been associated with dependence on alcohol and illegal substances in humans, and a fine-mapping study found several SNPs to be associated with major depressive disorder in a study of Dutch twins (Drgon et al., 2011; Johnson et al., 2011; Verbeek et al., 2012). Furthermore, an FGF14 SNP is associated with volumetric changes in the entorhinal cortex in AD patients (Yang et al., 2015). Overall, genetic variations in FGF14 are linked to the pathophysiology of several neuropsychiatric disorders, a promising area for further research that is supported by studies in Fgf14−/− preclinical models (Alshammari T. K. et al., 2016; Alshammari M. A. et al., 2016).
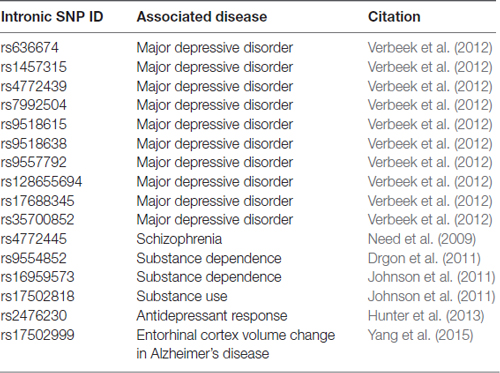
Table 2. Single nucleotide polymorphisms (SNPs) in FGF14 introns associated with neuropsychiatric disease.
FGF14 plays a role in all fundamental properties of neurons: intrinsic firing, synaptic transmission of excitatory and inhibitory neurons and plasticity, while deletion of the gene leads to disruptive motor and cognitive behaviors. The role of FGF14 in humans is yet to be fully understood, but the emerging technologies for genome sequencing and protein characterization will provide potential opportunities for identifying new disease signatures associated with FGF14.
All of the authors have contributed substantially to the work. JDR and FL contributed to writing and editing the manuscript. PAW created the image and legend for Figure 1.
This research was funded by National Institute of Mental Health: grant no. 1R01MH111107-01A1, R01 MH095995-A1 and the National Institutes of Health: grant no 5T32AG051131-02.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ali, S. R., Singh, A. K., and Laezza, F. (2016). Identification of amino acid residues in fibroblast growth factor 14 (FGF14) required for structure-function interactions with voltage-gated sodium channel Nav1.6. J. Biol. Chem. 291, 11268–11284. doi: 10.1074/jbc.m115.703868
Ali, S., Shavkunov, A., Panova, N., Stoilova-McPhie, S., and Laezza, F. (2014). Modulation of the FGF14:FGF14 homodimer interaction through short peptide fragments. CNS. Neurol. Disord. Drug. Targets 13, 1559–15570. doi: 10.2174/1871527313666141126103309
Alshammari, T. K., Alshammari, M. A., Nenov, M. N., Hoxha, E., Cambiaghi, M., Marcinno, A., et al. (2016). Genetic deletion of fibroblast growth factor 14 recapitulates phenotypic alterations underlying cognitive impairment associated with schizophrenia. Transl. Psychiatry 6:e806. doi: 10.1038/tp.2016.66
Alshammari, M. A., Alshammari, T. K., Nenov, M. N., Scala, F., and Laezza, F. (2016). Fibroblast growth factor 14 modulates the neurogenesis of granule neurons in the adult dentate gyrus. Mol. Neurobiol. 53, 7254–7270. doi: 10.1007/s12035-015-9568-5
Antonell, A., Lladó, A., Altirriba, J., Botta-Orfila, T., Balasa, M., Fernández, M., et al. (2013). A preliminary study of the whole-genome expression profile of sporadic and monogenic early-onset alzheimer’s disease. Neurobiol. Aging 34, 1772–1778. doi: 10.1016/j.neurobiolaging.2012.12.026
Bosch, M. K., Carrasquillo, Y., Ransdell, J. L., Kanakamedala, A., Ornitz, D. M., and Nerbonne, J. M. (2015). Intracellular FGF14 (iFGF14) is required for spontaneous and evoked firing in cerebellar purkinje neurons and for motor coordination and balance. J. Neurosci. 35, 6752–6769. doi: 10.1523/JNEUROSCI.2663-14.2015
Bosch, M. K., Nerbonne, J. M., Townsend, R. R., Miyazaki, H., Nukina, N., Ornitz, D. M., et al. (2016). Proteomic analysis of native cerebellar iFGF14 complexes. Channels (Austin) 10, 297–312. doi: 10.1080/19336950.2016.1153203
Brusse, E., de Koning, I., Maat-Kievit, A., Oostra, B. A., Heutink, P., and van Swieten, J. C. (2006). Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov. Disord. 21, 396–401. doi: 10.1002/mds.20708
Brzustowicz, L. M., Honer, W. G., Chow, E. W., Little, D., Hogan, J., Hodgkinson, K., et al. (1999). Linkage of familial schizophrenia to chromosome 13q32. Am. J. Hum. Genet. 65, 1096–1103. doi: 10.1086/302579
Detera-Wadleigh, S. D., Badner, J. A., Berrettini, W. H., Yoshikawa, T., Goldin, L. R., Turner, G., et al. (1999). A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p11.2. Proc. Natl. Acad. Sci. U S A 96, 5604–5609. doi: 10.1073/pnas.96.10.5604
Drgon, T., Johnson, C. A., Nino, M., Drgonova, J., Walther, D. M., and Uhl, G. R. (2011). “Replicated” genome wide association for dependence on illegal substances: genomic regions identified by overlapping clusters of nominally positive SNPs. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 125–138. doi: 10.1002/ajmg.b.31143
Gadelha, A., Ota, V. K., Cano, J. P., Melaragno, M. I., Smith, M. A., de Jesus Mari, J., et al. (2012). Linkage replication for chromosomal region 13q32 in schizophrenia: evidence from a Brazilian pilot study on early onset schizophrenia families. PLoS One 7:e52262. doi: 10.1371/journal.pone.0052262
Goetz, R., Dover, K., Laezza, F., Shtraizent, N., Huang, X., Tchetchik, D., et al. (2009). Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J. Biol. Chem. 284, 17883–17896. doi: 10.1074/jbc.m109.001842
Goldfarb, M., Schoorlemmer, J., Williams, A., Diwakar, S., Wang, Q., Huang, X., et al. (2007). Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron 55, 449–463. doi: 10.1016/j.neuron.2007.07.006
Hagihara, H., Takao, K., Walton, N. M., Matsumoto, M., and Miyakawa, T. (2013). Immature dentate gyrus: an endophenotype of neuropsychiatric disorders. Neural Plast. 2013:318596. doi: 10.1155/2013/318596
Hao, K., Di Narzo, A. F., Ho, L., Luo, W., Li, S., Chen, R., et al. (2015). Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol. Aspects Med. 43–44, 66–76. doi: 10.1016/j.mam.2015.06.006
Hsu, W. C., Nenov, M. N., Shavkunov, A., Panova, N., Zhan, M., and Laezza, F. (2015). Identifying a kinase network regulating FGF14:Nav1.6 complex assembly using split-luciferase complementation. PLoS One 10:e0117246. doi: 10.1371/journal.pone.0117246
Hsu, W.-C. J., Nilsson, C. L., and Laezza, F. (2014). Role of the axonal initial segment in psychiatric disorders: function, dysfunction, and intervention. Front. Psychiatry 5:109. doi: 10.3389/fpsyt.2014.00109
Hsu, W.-C. J., Scala, F., Nenov, M. N., Wildburger, N. C., Elferink, H., Singh, A. K., et al. (2016). CK2 activity is required for the interaction of FGF14 with voltage-gated sodium channels and neuronal excitability. FASEB J. 30, 2171–2186. doi: 10.1096/fj.201500161
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Hunter, A. M., Leuchter, A. F., Power, R. A., Muthén, B., McGrath, P. J., Lewis, C. M., et al. (2013). A genome-wide association study of a sustained pattern of antidepressant response. J. Psychiatr. Res. 47, 1157–1165. doi: 10.1016/j.jpsychires.2013.05.002
Itoh, N., and Ornitz, D. M. (2008). Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 237, 18–27. doi: 10.1002/dvdy.21388
Johnson, M. A., Ables, J. L., and Eisch, A. J. (2009). Cell-intrinsic signals that regulate adult neurogenesis in vivo: insights from inducible approaches. BMB Rep. 42, 245–259. doi: 10.5483/bmbrep.2009.42.5.245
Johnson, C., Drgon, T., Walther, D., and Uhl, G. R. (2011). Genomic regions identified by overlapping clusters of nominally-positive SNPs from genome-wide studies of alcohol and illegal substance dependence. PLoS One 6:e19210. doi: 10.1371/journal.pone.0019210
Jun, H., Mohammed Qasim Hussaini, S., Rigby, M. J., and Jang, M. H. (2012). Functional role of adult hippocampal neurogenesis as a therapeutic strategy for mental disorders. Neural Plast. 2012:854285. doi: 10.1155/2012/854285
Laezza, F., Gerber, B. R., Lou, J. Y., Kozel, M. A., Hartman, H., Craig, A. M., et al. (2007). The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J. Neurosci. 27, 12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007
Laezza, F., Lampert, A., Kozel, M. A., Gerber, B. R., Rush, A. M., Nerbonne, J. M., et al. (2009). FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol. Cell. Neurosci. 42, 90–101. doi: 10.1016/j.mcn.2009.05.007
Liu, C. J., Dib-Hajj, S. D., Renganathan, M., Cummins, T. R., and Waxman, S. G. (2003). Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 278, 1029–1036. doi: 10.1074/jbc.M207074200
Lou, J. Y., Laezza, F., Gerber, B. R., Xiao, M., Yamada, K. A., Hartmann, H., et al. (2005). Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J. Physiol. 569, 179–193. doi: 10.1113/jphysiol.2005.097220
Ming, G. L., and Song, H. (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250. doi: 10.1146/annurev.neuro.28.051804.101459
Mulle, J. G., McDonough, J. A., Chowdari, K. V., Nimgaonkar, V., and Chakravarti, A. (2005). Evidence for linkage to chromosome 13q32 in an independent sample of schizophrenia families. Mol. Psychiatry 10, 429–431. doi: 10.1038/sj.mp.4001639
Munoz-Sanjuan, I., Smallwood, P. M., and Nathans, J. (2000). Isoform diversity among fibroblast growth factor homologous factors is generated by alternative promoter usage and differential splicing. J. Biol. Chem. 275, 2589–2597. doi: 10.1074/jbc.275.4.2589
Najem, D., Bamji-Mirza, M., Yang, Z., and Zhang, Z. (2016). Aβ-induced insulin resistance and the effects of insulin on the cholesterol synthesis pathway and Aβ secretion in neural cells. Neurosci. Bull. 32, 227–238. doi: 10.1007/s12264-016-0034-9
Need, A. C., Ge, D., Weale, M. E., Maia, J., Feng, S., Heinzen, E. L., et al. (2009). A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 5:e1000373. doi: 10.1371/journal.pgen.1000373
Ogawa, Y., and Rasband, M. N. (2008). The functional organization and assembly of the axon initial segment. Curr. Opin. Neurobiol. 18, 307–313. doi: 10.1016/j.conb.2008.08.008
Olsen, S. K., Garbi, M., Zampieri, N., Eliseenkova, A. V., Ornitz, D. M., Goldfarb, M., et al. (2003). Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 278, 34226–34236. doi: 10.1074/jbc.M303183200
Ornitz, D. M., and Itoh, N. (2015). The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266. doi: 10.1002/wdev.176
Ouchi, Y., Banno, Y., Shimizu, Y., Ando, S., Hasegawa, H., Adachi, K., et al. (2013). Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J. Neurosci. 33, 9408–9419. doi: 10.1523/JNEUROSCI.2700-12.2013
Pablo, J. L., and Pitt, G. S. (2017). FGF14 is a regulator of KCNQ2/3 channels. Proc. Natl. Acad. Sci. U S A 114, 154–159. doi: 10.1073/pnas.1610158114
Pablo, J. L., Wang, C., Presby, M. M., and Pitt, G. S. (2016). Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proc. Natl. Acad. Sci. U S A 113, E2665–E2674. doi: 10.1073/pnas.1521194113
Palmer, L. M., and Stuart, G. J. (2006). Site of action potential initiation in layer 5 pyramidal neurons. J. Neurosci. 26, 1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006
Park, N., Juo, S. H., Cheng, R., Liu, J., Loth, J. E., Lilliston, B., et al. (2004). Linkage analysis of psychosis in bipolar pedigrees suggests novel putative loci for bipolar disorder and shared susceptibility with schizophrenia. Mol. Psychiatry 9, 1091–1099. doi: 10.1038/sj.mp.4001541
Reif, A., Schmitt, A., Fritzen, S., and Lesch, K. P. (2007). Neurogenesis and schizophrenia: dividing neurons in a divided mind? Eur. Arch. Psychiatry Clin. Neurosci. 257, 290–299. doi: 10.1007/s00406-007-0733-3
Rush, A. M., Wittmack, E. K., Tyrrell, L., Black, J. A., Dib-Hajj, S. D., and Waxman, S. G. (2006). Differential modulation of sodium channel Nav1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. Eur. J. Neurosci. 23, 2551–2562. doi: 10.1111/j.1460-9568.2006.04789.x
Shakkottai, V. G., Xiao, M., Xu, L., Wong, M., Nerbonne, J. M., Ornitz, D. M., et al. (2009). FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol. Dis. 33, 81–88. doi: 10.1016/j.nbd.2008.09.019
Shavkunov, A., Panova, N., Prasai, A., Veselenak, R., Bourne, N., Stoilova-McPhie, S., et al. (2012). Bioluminescence methodology for the detection of protein-protein interactions within the voltage-gated sodium channel macromolecular complex. Assay Drug Dev. Technol. 10, 148–160. doi: 10.1089/adt.2011.413
Shavkunov, A. S., Wildburger, N. C., Nenov, M. N., James, T. F., Buzhdygan, T. P., Panova-Elektronova, N. I., et al. (2013). The fibroblast growth factor 14· Voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3). J. Biol. Chem. 288, 19370–19385. doi: 10.1074/jbc.M112.445924
Singh, H. N., and Rajeswari, M. R. (2015). Role of long purine stretches in controlling the expression of genes associated with neurological disorders. Gene 572, 175–183. doi: 10.1016/j.gene.2015.07.007
Smallwood, P. M., Munoz-Sanjuan, I., Tong, P., Macke, J. P., Hendry, S. H., Gilbert, D. J., et al. (1996). Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc. Natl. Acad. Sci. U S A 93, 9850–9857. doi: 10.1073/pnas.93.18.9850
Sun, J., Sun, J., Ming, G.-L., and Song, H. (2011). Epigenetic regulation of neurogenesis in the adult mammalian brain. Eur. J. Neurosci. 33, 1087–1093. doi: 10.1111/j.1460-9568.2011.07607.x
Taupin, P. (2005). Adult neurogenesis in the mammalian central nervous system: functionality and potential clinical interest. Med. Sci. Monit. 11, RA247–RA252.
Taupin, P. (2008). Adult neurogenesis pharmacology in neurological diseases and disorders. Expert Rev. Neurother. 8, 311–320. doi: 10.1586/14737175.8.2.311
Tempia, F., Hoxha, E., Negro, G., Alshammari, M. A., Alshammari, T. K., Panova-Elektronova, N., et al. (2015). Parallel fiber to Purkinje cell synaptic impairment in a mouse model of spinocerebellar ataxia type 27. Front. Cell. Neurosci. 9:205. doi: 10.3389/fncel.2015.00205
van Swieten, J. C., Brusse, E., de Graaf, B. M., Krieger, E., van de Graaf, R., de Koning, I., et al. (2003). A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am. J. Hum. Genet. 72, 191–199. doi: 10.1086/345488
Verbeek, E. C., Bakker, I. M., Bevova, M. R., Bochdanovits, Z., Rizzu, P., Sondervan, D., et al. (2012). A fine-mapping study of 7 top scoring genes from a GWAS for major depressive disorder. PLoS One 7:e37384. doi: 10.1371/journal.pone.0037384
Walton, N. M., Zhou, Y., Kogan, J. H., Shin, R., Webster, M., Gross, A. K., et al. (2012). Detection of an immature dentate gyrus feature in human schizophrenia/bipolar patients. Transl. Psychiatry 2:e135. doi: 10.1038/tp.2012.56
Wang, Q., Bardgett, M. E., Wong, M., Wozniak, D. F., Lou, J., McNeil, B. D., et al. (2002). Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron 35, 25–38. doi: 10.1016/s0896-6273(02)00744-4
Wang, C., Chung, B. C., Yan, H., Lee, S. Y., and Pitt, G. S. (2012). Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 20, 1167–1176. doi: 10.1016/j.str.2012.05.001
Wang, C., Wang, C., Hoch, E. G., and Pitt, G. S. (2011). Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors, and voltage-gated sodium channels. J. Biol. Chem. 286, 24253–24563. doi: 10.1074/jbc.M111.245803
Wildburger, N. C., Ali, S. R., Hsu, W. C., Shavkunov, A. S., Nenov, M. N., Lichti, C. F., et al. (2015). Quantitative proteomics reveals protein-protein interactions with fibroblast growth factor 12 as a component of the voltage-gated sodium channel 1.2 (nav1.2) macromolecular complex in Mammalian brain. Mol. Cell. Proteomics 14, 1288–1300. doi: 10.1074/mcp.M114.040055
Wittmack, E. K., Rush, A. M., Craner, M. J., Goldfarb, M., Waxman, S. G., and Dib-Hajj, S. D. (2004). Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J. Neurosci. 24, 6765–6775. doi: 10.1523/JNEUROSCI.1628-04.2004
Wozniak, D. F., Xiao, M., Xu, L., Yamada, K. A., and Ornitz, D. M. (2007). Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol. Dis. 26, 14–26. doi: 10.1016/j.nbd.2006.11.014
Xiao, M., Xu, L., Laezza, F., Yamada, K., Feng, S., and Ornitz, D. M. (2007). Impaired hippocampal synaptic transmission and plasticity in mice lacking fibroblast growth factor 14. Mol. Cell. Neurosci. 34, 366–377. doi: 10.1016/j.mcn.2006.11.020
Yan, H., Pablo, J. L., and Pitt, G. S. (2013). FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Rep. 4, 66–75. doi: 10.1016/j.celrep.2013.06.012
Keywords: neuronal excitability, intracellular signaling, protein-protein interactions, biological psychiatry
Citation: Di Re J, Wadsworth PA and Laezza F (2017) Intracellular Fibroblast Growth Factor 14: Emerging Risk Factor for Brain Disorders. Front. Cell. Neurosci. 11:103. doi: 10.3389/fncel.2017.00103
Received: 21 December 2016; Accepted: 28 March 2017;
Published: 19 April 2017.
Edited by:
Daniela Tropea, Trinity College, Dublin, IrelandReviewed by:
Alexander Dityatev, German Center for Neurodegenerative Diseases (DZNE), GermanyCopyright © 2017 Di Re, Wadsworth and Laezza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernanda Laezza, ZmVsYWV6emFAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.