 Chang-Qing Li1
Chang-Qing Li1 Qi Wang
Qi Wang Qing-Ping Zeng
Qing-Ping Zeng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Cell. Neurosci. , 23 November 2016
Sec. Non-Neuronal Cells
Volume 10 - 2016 | https://doi.org/10.3389/fncel.2016.00269
Alzheimer's disease (AD), a neurodegenerative condition, is characterized by deficient synaptic plasticity, dramatic neuronal dysfunction, and massive neuronal loss. Apart from familial or early-onset AD (5–10%), most AD cases are non-familial or late-onset/sporadic (90–95%; Ballard et al., 2011) with a complicated etiology. Some competing theories have been suggested regarding the cause of AD, such as the amyloid hypothesis (Hardy and Allsop, 1991) and tau hypothesis (Mudher and Lovestone, 2002), but minimal data on initial triggers are available despite intensive explorations over recent decades.
We summarized the published evidence into an opinion that deciphers how the multifaceted adverse environmental factors drive the onset and development of AD. Etiological drivers can be categorized as biotic stressors and abiotic stressors, with the latter category divided into physical stressors and chemical stressors. Ultimately, biotic/abiotic stressors can be integrated into reactive oxygen species (ROS)/oxidative stressors and reactive nitrogen species (RNS)/nitrosative stressors that impact the transition of neurons from dysfunction to death (Barone et al., 2011a,b; Butterfield et al., 2014).
Our opinion on biotic/abiotic stress-triggered AD links the various stressors to the genesis and progression of AD through a neuroinflammatory signaling cascade, which initiates nuclear factor κB (NF-κB) and induces pro-inflammatory cytokines that evoke potent ROS/RNS burst for neuronal/glial killing. To trigger AD, biotic stressors convey the external biological signals via lipopolysaccharide (LPS)-toll-like receptor 4 (TLR4), LPS-receptor of advanced glycation end products (RAGE), and amyloid β peptide (Aβ)/senile plaques (SP)-RAGE interactions (Yan et al., 1996; Yamamoto et al., 2011). Alternatively, abiotic stressors transduce the external non-biological signals via AGEs-RAGE, high-mobility group protein B1 (HMGB1)-RAGE, and Aβ/SP-RAGE interactions (Mazarati et al., 2011; Horst et al., 2016). Specifically, hypothermia, as well as anesthesia and aging that induce hypothermia, can execute a neurotoxic role to kill neurons and glia via neurofibrillary tangles (NFTs) derived from hyperphosphorylated Tau (p-Tau) (Carrettiero et al., 2015; Figure 1).
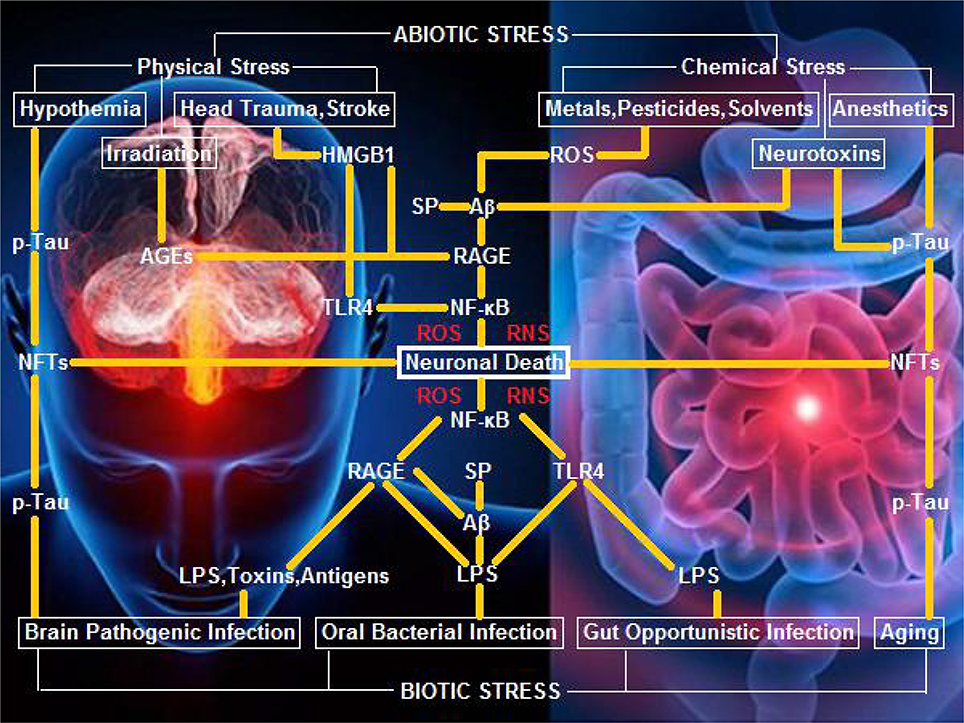
Figure 1. A hypothetical schematic of biotic/abiotic stress-triggered AD. Biotic stress from brain, oral, or gut infection can activate NF-κB-primed neuroinflammatory cascades, elicit ROS/RNS burst, and kill neurons and glia via LPS-TLR4/RAGE and Aβ/SP-RAGE interactions and subsequent signaling. Abiotic stress encompassing physical stress (e.g., head trauma, stroke, or irradiation) and chemical stress (e.g., metals, pesticides, solvents, or neurotoxins) can also activate NF-κB-primed neuroinflammatory cascades, elicit ROS/RNS burst, and kill neurons and glia via AGEs-RAGE, HMGB1-RAGE/TLR4, and Aβ/SP-RAGE interactions and downstream signaling. Hypothermia, anesthetics, and aging, can exert a neurotoxic effect upon exposure of neurons and glia to NFTs (the background figure was adopted from the website https://zhidao.baidu.com/daily/view?id=5979).
Mounting evidence supports that LPS and interferon γ (IFN-γ) activate microglia to induce a pro-inflammatory neurotoxic M1 phenotype, whereas interleukin 4 (IL-4), IL-10, IL-13, and transforming growth factor β (TGF-β) activate microglia to give rise to an anti-inflammatory neuroprotective M2 phenotype (Tang and Le, 2016). Interestingly, we found that electric acupuncture can mimic mechanical wounding to firstly deteriorate LPS-induced AD-like brain pathogenesis, but secondly ameliorate the progressive neurodegeneration in a wounding-healing manner, suggesting a putative conversion from M1 microglia to M2 microglia (He, 2016).
Biotic stressors refer to any potential infectious pathogens or opportunistic infectious microbes, including Chlamydophila pneumoniae (Balin et al., 1998), Helicobacter pylori (Kountouras et al., 2012), Toxoplasma gondii (Prandota, 2014), human immunodeficiency virus (HIV; Borjabad and Volsky, 2012), and human cytomegalovirus (HCMV; Lurain et al., 2013). An international team recently urged that cerebral pathogenic infections by herpes simplex virus type 1 (HSV-1), C. pneumoniae, spirochetes, and fungi be considered as candidate AD initiators (Itzhaki et al., 2016). Similarly, extracerebral infectious pathogens were also considered as AD triggers; for example, oral pathogenic infections by the periodontal bacteria Porphyromonas gingivalis and Actinomyces naeslundii were identified as high-risk factors driving development toward AD (Noble et al., 2014; Singhrao et al., 2015). A recent study on gut microbiota dysbiosis indicated that intestinal microbiome alterations are related to the malfunctional motor phenotypes, suggesting the overgrowth of intestinal commensal microbes (i.e., opportunistic infection) acting as a neurodegenerative driver (Scheperjans et al., 2015).
Sulfate-reducing bacteria (SRB), such as the Gram-positive Firmicutes and Gram-negative Proteobacteria, colonize 50% of human guts (Stewart et al., 2006). Among which Desulfovibrio piger was shown as the most common SRB in a surveyed cohort of healthy US adults (Scanlan et al., 2009). Chondroitin sulfate, a daily dietary nutrient available from livestock and poultry products, can increase the abundance of sulfatase-free D. piger upon reducing sulfate released from sulfatase-secreting Bacteroides thetaiotaomicron (Rey et al., 2013), thereby raising the possibility of B. thetaiotaomicron degrading mucin in the gut. Red meat containing heme can also nourish the mucin-degrading bacteria (e.g., Akkermansia muciniphila; Ijssennagger et al., 2015). These observations predisposed that gut dysbiosis may lead to the thinned mucosal layers and permeable colon linings, which boost LPS leakage from the gut and entry into the blood stream (Qin et al., 2012).
Factors that link the leaky gut and serum LPS to neurodegenerative diseases include: the plasma level of LPS in patients with neurodegenerative disease is three times higher than in healthy persons (Zhang et al., 2009); and intraperitoneal injections of LPS into mice cause a prolonged elevation hippocampal Aβ levels and lead to cognitive deficits (Kahn et al., 2012). To this end, intranasal LPS infusion was successfully used to establish a neurodegenerative model in rodents (He et al., 2013). According to a recent introduction by Scheperjans (2016) on the relevance of gut microbiota to Aβ deposition, germ-free APPSWE/PS1ΔE9 mice show mitigated amyloidosis in the brain compared with conventional APPSWE/PS1ΔE9 mice. While colonization of germ-free APPSWE/PS1ΔE9 mice with harvested gut microbiota from conventional APPSWE/PS1ΔE9 mice aggravates cerebral amyloidosis, colonization with gut microbiota from wild-type mice fails to increase cerebral Aβ levels.
Evidence supporting a possible infectious origin of AD is also derived from the sequencing-classified single nucleotide polymorphism (SNP) in apolipoprotein E gene (APOE), which is involved in modulating the immune response and infectious susceptibility (Verghese et al., 2011). Genome-wide association studies have revealed that several immune system components including virus receptor genes serve as risk factors for AD (Licastro et al., 2011). For example, cholesterol 25-hydroxylase (CH25H), catalyzing the generation of 25-hydroxycholesterol (25OHC) and inducing the enhancement of innate antiviral immunity, is selectively upregulated by virus infection (Blanc et al., 2013; Liu et al., 2013).
An epidemiological study has associated an increased risk of AD with a medical history of traumatic head injury (Webster et al., 2015). Moreover, brain inflammation seems a common consequence of mechanical insults such as trauma and stroke (Fiebich et al., 2014). Trauma can significantly increase expression of the alarmin HMBG1 (Horst et al., 2016), which in turn activates an inflammatory cascade by stimulating multiple receptors including RAGE and TLR4 (Mazarati et al., 2011). A recent study showed that AD-like model mice, on a diet enriched in AGEs due to irradiation, exhibit significant memory dysfunction, accompanied with the hippocampal deposition of insoluble Aβ42 fragment and AGEs (Lubitz et al., 2016). This latter finding was consistent with the notion that Aβ can activate microglia and induce neurotoxicity by RAGE binding (Yan et al., 1996).
Many naturally occurring and synthesized chemicals such as heavy metals, pesticides, bactericides, and solvents are ROS generators, and therefore are potential initiators of AD (Chin-Chan et al., 2015). A recent study showed that magnetite from air pollution might be an important risk factor for AD; particularly, those magnetite pollutant particles that are <200 nm in diameter can enter the brain directly via the olfactory bulb (Maher et al., 2016). Cyanobacteria or blue-green algae residing in the gut may produce the neurotoxin β-N-methylamino-L-alanine (BMAA), which was implicated in the development of AD (Banack et al., 2010; Brenner, 2013). Chronic dietary exposure to BMAA was identified as a causal factor of neurodegeneration in the Chamorros villagers on the Pacific island of Guam, and vervets (Chlorocebus sabaeus) fed with BMAA-dosed fruit were observed to develop neurodegenerative diseases exhibiting Aβ and NFTs (Cox et al., 2016).
It was highlighted that aggregation of p-Tau into NFTs or even development of tauopathies seems an essential consequence of hypothermia as well as anesthetic-induced hypothermia (Planel et al., 2007; Carrettiero et al., 2015). Due to reduced peripheral vasoconstriction, mitigated heat production, and other reasons, the core body temperature of healthy individuals over 60 years of age is 0.4°C lower than adults aged 20–60 years, suggesting that aging should facilitate p-Tau formation by inducing cerebral hypothermia. It was suggested that tau phosphorylation at later stages is mostly a consequence of hypothermia although hyperphosphorylation at early stages may be due to the deregulation of JNK and PP2A (El-Khoury et al., 2016).
In contrast to the conventional assertion of a causative role of Aβ in AD pathogenesis, the peptide was surprisingly recognized as an antimicrobial peptide (AMP) with potent activity against pathogenic infections (Soscia et al., 2010). Aβ has been confirmed to protect mouse, nematode, and cell culture models of AD from fungal and bacterial infections because propagating fibrils mediate the agglutination and eventual entrapment of pathogens. Indeed, bacterial infection by Salmonella typhimurium in the brains of transgenic AD mice results in accelerated Aβ deposition, which can co-localize with invading bacteria (Kumar et al., 2016). It was recently reported that a long-term antibiotic treatment regime inducing a prolonged change of gut microbiota decreases Aβ deposition in the APPSWE/PS1ΔE9 mouse AD model. In the observation, soluble Aβ levels were elevated, plaque-localized glial reactivity attenuated, and microglial morphology altered, suggesting a diversity of gut microbiota regulating host innate immunity, and impacting amyloidosis (Minter et al., 2016).
Aβ was also found to possess antiviral activity against HSV-1 and influenza A (White et al., 2014; Bourgade et al., 2015, 2016). Interestingly, another AMP, β-defensin 1, has similarly shown overproduction in AD patients (Williams et al., 2013). An SNP in human CH25H governs both AD susceptibility and Aβ deposition, implying Aβ induction may be a 25OHC target, and also providing a potential mechanistic link between pathogenic infection and Aβ accumulation (Papassotiropoulos et al., 2005; Lathe et al., 2014).
Why Aβ progressively deposits remains largely unknown, but S-nitrosylation of cysteine residues in Aβ-degrading enzymes might be relevant, and nitric oxide (NO) involved. The impact from NO-mediated nitrosative stress was found to prompt the S-nitrosylation of insulin-degrading enzyme (IDE) and dynamin-related protein 1 (Drp1) responsible for Aβ degradation, thus inhibiting Aβ catabolism and hyperactivating mitochondrial fission machinery. The raised Aβ levels and compromised mitochondrial bioenergetics were shown to result in dysfunctional synaptic plasticity and synapse loss in cortical and hippocampal neurons (Akhtar et al., 2016).
Interventions against AD involving eradicating Aβ from brain tissues hold promise in avoiding microglial activation, immune attack, and neuron killing. It was shown that aducanumab, a human monoclonal antibody that selectively targets the aggregated Aβ, enters the brain, binds parenchymal Aβ, and reduces Aβ in a transgenic mouse AD model, and that aducanumab even reduces brain Aβ in patients with prodromal AD after 1 year of monthly intravenous infusions (Sevigny et al., 2016).
Alternatively, prohibition of Aβ formation by impeding the cleavage of APP might also prevent AD. An ongoing human trial is assessing the therapeutic value of the β-secretase inhibitor solanezumab (Sheridan, 2015) although a clinical trial with the γ-secretase inhibitor semagacestat failed just 1 year ago (De Strooper, 2014). The preliminary data indicated that solanezumab can decrease cognitive decline in mild AD by about 30% in a clinical study recruiting 440 subjects (Reardon, 2015).
Considering Aβ as a pathogenic hallmark of AD, it is anticipated that treatments by monoclonal antibodies to remove Aβ or block APP cleavage would justify optimism and show progress in clinical trials. However, Aβ is unlikely an initiator, and more likely a mediator of AD, so Aβ-targeted interventions should not be an eventual solution to attenuating progressive aggravation toward AD. Once infectious agents have been verified as the primordial etiological cues leading to AD, the more practical medications treating AD should at least include, for example, anti-infection agents such as minocycline (El-Shimy et al., 2015; Budni et al., 2016), anti-inflammation agents such as anhydroexfoliamycin (Leirós et al., 2015) or rapamycin (Siman et al., 2015), and anti-oxidation agents such as allicin (Zhu et al., 2015). With similar importance, modulation of gut microbiota from dysbiosis to homeostasis for the early-phase prophylaxis of AD through personalized diet and prebiotic/probiotic supplementation should also be addressed (Hu et al., 2016).
QPZ wrote the manuscript. CQL, QZ, and QW critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Shi-Jie Zhang, Jiang He, Tao Liao, and Yan-Ping Chen for their assistance in manuscript preparation. This work was supported by the National Natural Science Foundation of China (NSFC, No. 81273620 to QPZ and No. 81673861 to CQL).
Akhtar, M. W., Sanz Blasco, S., Donatabadi, N., Parker, J., Chon, K., Lee, M. S., et al. (2016). Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat. Commun. 7:10242. doi: 10.1038/ncomms10242
Balin, B. J., Gerard, H. C., Arking, E. J., Appelt, D. M., Branigan, P. J., Abrams, J. T., et al. (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med. Microbiol. Immunol. 187, 23–42.
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer's disease. Lancet 377, 1019–1031. doi: 10.1016/S0140-6736(10)61349-9
Banack, S. A., Caller, T. A., and Stommel, E. W. (2010). The cynobacteria derived toxin beta-N-methylamino-L-alanine and amyotrophic lateral sclerosis. Toxins (Basel) 2, 2837–2850. doi: 10.3390/toxins2122837
Barone, E., Cenini, G., Di Domenico, F., Martin, S., Sultana, R., Mancuso, C., et al. (2011a). Long-term high-dose atorvastatin decreases brain oxidative and nitrosative stress in a preclinical model of Alzheimer disease: a novel mechanism of action. Pharmacol. Res. 63, 172–180. doi: 10.1016/j.phrs.2010.12.007
Barone, E., Di Domenico, F., Cenini, G., Sultana, R., Coccia, R., Preziosi, P., et al. (2011b). Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer's disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 25, 623–633. doi: 10.3233/JAD-2011-110092
Blanc, M., Hsieh, W. Y., Robertson, K. A., Kropp, K. A., Forster, T., Shui, G., et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. doi: 10.1016/j.immuni.2012.11.004
Borjabad, A., and Volsky, D. J. (2012). Common transcriptional signatures in brain tissue from patients with HIV-associated neurocognitive disorders, Alzheimer's disease, and multiple sclerosis. J. Neuroimmune Pharmacol. 7, 914–726. doi: 10.1007/s11481-012-9409-5
Bourgade, K., Garneau, H., Giroux, G., Le Page, Y., Bocti, C., Dupuis, G., et al. (2015). β-amyloid peptides display protective activity against the human Alzheimer's disease-associated herpes simplex virus-1. Biogerontology 16, 85–98. doi: 10.1007/s10522-014-9538-8
Bourgade, K., Le Page, A., Bocti, C., Witkowski, J. M., Dupuis, G., Frost, E. H., et al. (2016). Protective effect of amyloid-beta peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimers. Dis. 50, 1227–1241. doi: 10.3233/JAD-150652
Brenner, S. R. (2013). Blue-green algae or Cyanobacteria in the intestinal micro-flora may produce neurotoxins such as beta-N-methylamino-L-alanine (BMAA) which may be related to development of amyotrophic lateral sclerosis, Alzheimer's disease, and Parkinson-dementia-complex in humans and equine motor neuron disease in horses. Med. Hypotheses 80, 103. doi: 10.1016/j.mehy.2012.10.010
Budni, J., Garcez, M. L., de Medeiros, J., Cassaro, E., Santos-Bellettini, T., Mina, F., et al. (2016). The anti-inflammatory role of minocycline in Alzheimer's disease. Curr. Alzheimer Res. 13, 1319–1329. doi: 10.2174/1567205013666160819124206
Butterfield, D. A., Di Domenico, F., and Barone, E. (2014). Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochim. Biophys. Acta 1842, 1693–1706. doi: 10.1016/j.bbadis.2014.06.010
Carrettiero, D. C., Santiago, F. E., Motzko-Soares, A. C., and Almeida, M. C. (2015). Temperature and toxic Tau in Alzheimer's disease: new insights. Temperature (Austin) 2, 491–498. doi: 10.1080/23328940.2015.1096438
Chin-Chan, M., Navarro-Yepes, J., and Quintanilla-Vega, B. (2015). Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 9:124. doi: 10.3389/fncel.2015.00124
Cox, P. A., Davis, D. A., Mash, D. C., Metcalf, J. S., and Banack, S. A. (2016). Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc. Biol. Sci. 283:20152397. doi: 10.1098/rspb.2015.2397
De Strooper, B. (2014). Lessons from a failed γ-secretase Alzheimer trial. Cell 159, 721–726. doi: 10.1016/j.cell.2014.10.016
El-Khoury, N. B., Gratuze, M., Petry, F., Papon, M. A., Julien, C., Marcouiller, F., et al. (2016). Hypothermia mediates age-dependent increase of tau phosphorylation in db/db mice. Neurobiol. Dis. 88, 55–65. doi: 10.1016/j.nbd.2016.01.005
El-Shimy, I. A., Heikal, O. A., and Hamdi, N. (2015). Minocycline attenuates Aβ oligomers-induced pro-inflammatory phenotype in primary microglia while enhancing Aβ fibrils phagocytosis. Neurosci. Lett. 609, 36–41. doi: 10.1016/j.neulet.2015.10.024
Fiebich, B. L., Akter, S., and Akundi, R. S. (2014). The two-hit hypothesis for neuroinflammation: role of exogenous ATP in modulating inflammation in the brain. Front. Cell. Neurosci. 8:260. doi: 10.3389/fncel.2014.00260
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388. doi: 10.1016/0165-6147(91)90609-V
He, J. (2016). Electroacupuncture Ameliorates Collagen-Induced Alzheimer's Disease-like Brain Pathogenesis in Mice. Dissertation/master's thesis, Guangzhou University of Chinese Medicine, China.
He, Q., Yu, W., Wu, J., Chen, C., Lou, Z., Zhang, Q., et al. (2013). Intranasal LPS-mediated Parkinson's model challenges the pathogenesis of nasal cavity and environmental toxins. PLoS ONE 8:e78418. doi: 10.1371/journal.pone.0078418
Horst, K., Eschbach, D., Pfeifer, R., Relja, B., Sassen, M., Steinfeldt, T., et al. (2016). Long-term effects of induced hypothermia on local and systemic inflammation - Results from a porcine long-term trauma model. PLoS ONE 11:e0154788. doi: 10.1371/journal.pone.0154788
Hu, X., Wang, T., and Jin, F. (2016). Alzheimer's disease and gut microbiota. Sci. China Life Sci. 59, 1006–1023. doi: 10.1007/s11427-016-5083-9
Ijssennagger, N., Belzer, C., Hooiveld, G. J., Dekker, J., van Mil, S. W., Müller, M., et al. (2015). Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. U.S.A. 112, 10038–10043. doi: 10.1073/pnas.1507645112
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Bearer, E. L., Braak, H., et al. (2016). Microbes and Alzheimer's disease. J. Alzheimers Dis. 51, 979–984. doi: 10.3233/JAD-160152
Kahn, M. S., Kranjac, D., Alonzo, C. A., Haase, J. F., Cedillos, R. O., McLinden, K. A., et al. (2012). Prolonged elevation in hippocampal A beta and cognitive deficits following repeated endotoxin exposure in the mouse. Behav. Brain Res. 229, 176–184. doi: 10.1016/j.bbr.2012.01.010
Kountouras, J., Bozik, M., Zavos, C., Gavalas, E., Giartza-Taxidou, E., Venizelos, I., et al. (2012). A potential impact of chronic Helicobacter pylori infection on Alzheimer's disease pathobiology and course. Neurobiol. Aging 33, e3–e4. doi: 10.1016/j.neurobiolaging.2012.01.003
Kumar, D. K., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci. Transl. Med. 8, 340ra72. doi: 10.1126/scitranslmed.aaf1059
Lathe, R., Sapronova, S., and Kotelevtsev, Y. (2014). Atherosclerosis and Alzheimer-diseases with a common cause? Inflammation, oxysterols, vasculature. BMC Geriatr. 14:36. doi: 10.1186/1471-2318-14-36
Leirós, M., Alonso, E., Rateb, M. E., Ebel, R., Jaspars, M., Alfonso, A., et al. (2015). The streptomyces metabolite anhydroexfoliamycin ameliorates hallmarks of Alzheimer's disease in vitro and in vivo. Neuroscience 305, 26–35. doi: 10.1016/j.neuroscience.2015.07.082
Licastro, F., Carbone, I., Ianni, M., and Porcellini, E. (2011). Gene signature in Alzheimer's disease and environmental factors: the virus chronicle. J. Alzheimers Dis. 27, 809–817. doi: 10.3233/JAD-2011-110755
Liu, S. Y., Aliyari, R., Chikere, K., Li, G., Marsden, M. D., Smith, J. K., et al. (2013). Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. doi: 10.1016/j.immuni.2012.11.005
Lubitz, I., Ricny, J., Atrakchi-Baranes, D., Shemesh, C., and Kravitz, E. (2016). High dietary advanced glycation end products are associated with poorer spatial learning and accelerated Aβ deposition in an Alzheimer mouse model. Aging Cell 15, 309–316. doi: 10.1111/acel.12436
Lurain, N. S., Hanson, B. A., Martinson, J., Leurgans, S. E., Landay, A. L., Bennett, D. A., et al. (2013). Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J. Infect. Dis. 208, 564–572. doi: 10.1093/infdis/jit210
Maher, B. A., Ahmed, I. A. M., Karloukovski, V., MacLaren, D. A., Foulds, P. G., Allsop, D., et al. (2016). Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. U.S.A. 113, 10797–10801. doi: 10.1073/pnas.1605941113
Mazarati, A., Maroso, M., Iori, V., Vezzani, A., and Carli, M. (2011). High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and Receptor for Advanced Glycation End Products. Exp. Neurol. 232, 143–148. doi: 10.1016/j.expneurol.2011.08.012
Minter, M. R., Zhang, C., Leone, V., Ringus, D., Zhang, X., Oyler-Castrillo, P., et al. (2016). Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6:30028. doi: 10.1038/srep30028
Mudher, A., and Lovestone, S. (2002). Alzheimer's disease-do tauists and baptists finally shake hands? Trends Neurosci. 25, 22–26. doi: 10.1016/S0166-2236(00)02031-2
Noble, J. M., Scarmeas, N., Celenti, R. S., Elkind, M. S. V., Wright, C. B., Schupf, N., et al. (2014). Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer's disease. PLoS ONE 9:e114959. doi: 10.1371/journal.pone.0114959
Papassotiropoulos, A., Lambert, J. C., Wavrant-De Vrieze, F., Wollmer, M. A., von der Kammer, H., and Streffer, J. R. (2005). Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer's disease. Neurodegener. Dis. 2, 233–241. doi: 10.1159/000090362
Planel, E., Richter, K. E., Nolan, C. E., Finley, J. E., Liu, L., Wen, Y., et al. (2007). Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J. Neurosci. 27, 3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007
Prandota, J. (2014). Possible link between Toxoplasma gondii and the anosmia associated with neurodegenerative diseases. Am. J. Alzheimers Dis. Other Demen. 29, 205–214. doi: 10.1177/1533317513517049
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Reardon, S. (2015). Antibody drugs for Alzheimer's show glimmers of promise. Nature 523, 509–510. doi: 10.1038/nature.2015.18031
Rey, F. E., Gonzalez, M. D., Cheng, J., Wu, M., Ahern, P. P., and Gordon, J. I. (2013). Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. U.S.A. 110, 13582–13587. doi: 10.1073/pnas.1312524110
Scanlan, P. D., Shanahan, F., and Marchesi, J. R. (2009). Culture-independent analysis of desulfovibrios in the human distal colon of healthy, colorectal cancer and polypectomized individuals. FEMS Microbiol. Ecol. 69, 213–221. doi: 10.1111/j.1574-6941.2009.00709.x
Scheperjans, F. (2016). Can microbiota research change our understanding of neurodegenerative diseases? Neurodegener. Dis. Manage. 6, 81–85. doi: 10.2217/nmt-2015-0012
Scheperjans, F., Aho, V., Pereira, P. A., Koskinen, K., Paulin, L., Pekkonen, E., et al. (2015). Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov. Disord. 30, 350–358. doi: 10.1002/mds.26069
Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56. doi: 10.1038/nature19323
Sheridan, C. (2015). Pivotal trials for β-secretase inhibitors in Alzheimer's. Nat. Biotechnol. 33, 115–116. doi: 10.1038/nbt0215-115
Siman, R., Cocca, R., and Dong, Y. (2015). The mTOR inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage Alzheimer-type tauopathy. PLoS ONE 10:e0142340. doi: 10.1371/journal.pone.0142340
Singhrao, S. K., Harding, A., Poole, S., Kesavalu, L., and Crean, S. (2015). Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer's disease. Mediat. Inflamm. 2015:137357. doi: 10.1155/2015/137357
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 5:e9505. doi: 10.1371/journal.pone.0009505
Stewart, J. A., Chadwick, V. S., and Murray, A. (2006). Carriage, quantification, and predominance of methanogens and sulfate-reducing bacteria in faecal samples. Lett. Appl. Microbiol. 43, 58–63. doi: 10.1111/j.1472-765X.2006.01906.x
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Verghese, P. B., Castellano, J. M., and Holtzman, D. M. (2011). Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 10, 241–252. doi: 10.1016/S1474-4422(10)70325-2
Webster, S. J., Van Eldik, L. J., Watterson, D. M., and Bachstetter, A. D. (2015). Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J. Neurosci. 35, 6554–6569. doi: 10.1523/JNEUROSCI.0291-15.2015
White, M. R., Kandel, R., Tripathi, S., Condon, D., Qi, L., Taubenberger, J., et al. (2014). Alzheimer's associated beta-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS ONE 9:e101364. doi: 10.1371/journal.pone.0101364
Williams, W. M., Torres, S., Siedlak, S. L., Castellani, R. J., Perry, G., Smith, M. A., et al. (2013). Antimicrobial peptide beta-defensin-1 expression is upregulated in Alzheimer's brain. J. Neuroinflammation 10:127. doi: 10.1186/1742-2094-10-127
Yamamoto, Y., Harashima, A., Saito, H., Tsuneyama, K., Munesue, S., Motoyoshi, S., et al. (2011). Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J. Immunol. 186, 3248–3257. doi: 10.4049/jimmunol.1002253
Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., et al. (1996). RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 382, 685–691.
Zhang, R., Miller, R. G., Gascon, R., Champion, S., Katz, J., Lancero, M., et al. (2009). Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 206, 121–124. doi: 10.1016/j.jneuroim.2008.09.017β
Keywords: Alzheimer's disease (AD), biotic stress, abiotic stress, gut microbiota, opportunistic infection, lipopolysaccharide (LPS), reactive oxygen species (ROS), reactive nitrogen species (RNS)
Citation: Li C-Q, Zheng Q, Wang Q and Zeng Q-P (2016) Biotic/Abiotic Stress-Driven Alzheimer's Disease. Front. Cell. Neurosci. 10:269. doi: 10.3389/fncel.2016.00269
Received: 23 May 2016; Accepted: 07 November 2016;
Published: 23 November 2016.
Edited by:
Egidio D'Angelo, University of Pavia, ItalyReviewed by:
Fabio Blandini, Fondazione Instituto Neurologico Nazionale Casimiro Mondino (IRCCS), ItalyCopyright © 2016 Li, Zheng, Wang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Wang, d2FuZ3FpQGd6dWNtLmVkdS5jbg==
Qing-Ping Zeng, cXB6ZW5nQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.