 Yu-Chih Lin
Yu-Chih Lin Jeannine A. Frei
Jeannine A. Frei Michaela B. C. Kilander
Michaela B. C. Kilander Wenjuan Shen
Wenjuan Shen Gene J. Blatt
Gene J. Blatt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci. , 17 November 2016
Sec. Cellular Neuropathology
Volume 10 - 2016 | https://doi.org/10.3389/fncel.2016.00263
This article is part of the Research Topic Synaptic Diseases: From Biology to Potential Therapy View all 32 articles
Autism spectrum disorder (ASD) comprises a range of neurological conditions that affect individuals’ ability to communicate and interact with others. People with ASD often exhibit marked qualitative difficulties in social interaction, communication, and behavior. Alterations in neurite arborization and dendritic spine morphology, including size, shape, and number, are hallmarks of almost all neurological conditions, including ASD. As experimental evidence emerges in recent years, it becomes clear that although there is broad heterogeneity of identified autism risk genes, many of them converge into similar cellular pathways, including those regulating neurite outgrowth, synapse formation and spine stability, and synaptic plasticity. These mechanisms together regulate the structural stability of neurons and are vulnerable targets in ASD. In this review, we discuss the current understanding of those autism risk genes that affect the structural connectivity of neurons. We sub-categorize them into (1) cytoskeletal regulators, e.g., motors and small RhoGTPase regulators; (2) adhesion molecules, e.g., cadherins, NCAM, and neurexin superfamily; (3) cell surface receptors, e.g., glutamatergic receptors and receptor tyrosine kinases; (4) signaling molecules, e.g., protein kinases and phosphatases; and (5) synaptic proteins, e.g., vesicle and scaffolding proteins. Although the roles of some of these genes in maintaining neuronal structural stability are well studied, how mutations contribute to the autism phenotype is still largely unknown. Investigating whether and how the neuronal structure and function are affected when these genes are mutated will provide insights toward developing effective interventions aimed at improving the lives of people with autism and their families.
Autism spectrum disorder (ASD) is a neurodevelopmental clinical condition currently diagnosed based on the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria reflecting symptoms, possibly of varying severity, in social interaction, communication and behavior (American Psychiatric Association, 2013; Lord and Jones, 2013). ASD occurs in 1:68 individuals in the United States (Baio, 2014) and complex genetic interactions appear responsible for a high degree of heterogeneity of the clinical symptoms in ASD. Individuals with ASD often co-express other comorbidities including epilepsy which often complicates diagnosis and treatment. Alterations in neuronal structures in different brain regions have been reported in ASD individuals, including increased dendritic spine density in cortical pyramidal neurons (Hutsler and Zhang, 2010; Tang et al., 2014) as well as stunting of dendritic branching in the hippocampus (Raymond et al., 1996; Bauman and Kemper, 2005). In addition, subcortical band heterotopia, representing alterations in cell migration has also been found in a child with ASD (Beaudoin et al., 2007). These brain regions are often characterized with neuroanatomical irregularities in ASD (Donovan and Basson, 2016). The defective regulation for structural stability of neurons may be one of the underlying mechanisms that contribute to the anatomical changes in ASD.
Autism spectrum disorder is typically diagnosed during the first 3 years of life, a period of extensive neurite formation, synaptogenesis and refinement (Huttenlocher and Dabholkar, 1997; Zoghbi and Bear, 2012; Stamou et al., 2013; McGee et al., 2014). Indeed, brain imaging studies from individuals with ASD and anatomical measurements of neuronal structure in post-mortem tissues exhibit differences in neuronal connectivity derived from the disruption of neurite outgrowth, synapse formation and stabilization (Raymond et al., 1996; Hutsler and Zhang, 2010; Penzes et al., 2011). Studies of human induced pluripotent stem cells (iPSCs) derived from people with ASD also have identified defects of neuronal structure (Habela et al., 2015; Nestor et al., 2015). Genome-wide association studies on individuals with ASD and their families revealed several risk genes that may be the common molecular targets in autism (Bucan et al., 2009; Glessner et al., 2009; Hussman et al., 2011; O’Roak et al., 2011, 2012a; Buxbaum et al., 2012, 2014; Sanders et al., 2012; Shi et al., 2013; Stamou et al., 2013; Yu et al., 2013; An et al., 2014; Brett et al., 2014; Cukier et al., 2014; De Rubeis et al., 2014; Iossifov et al., 2014; McGee et al., 2014; Pinto et al., 2014; Ronemus et al., 2014; Toma et al., 2014; Yuen et al., 2015). Animal studies of these genes further identify several specific cellular pathways during brain development that are vulnerable in ASD, including the disruption of neurite outgrowth, dendritic spine formation, and synaptic function (Figure 1) (Walsh et al., 2008; Bourgeron, 2009; Hussman et al., 2011; Penzes et al., 2011; Zoghbi and Bear, 2012; Ebert and Greenberg, 2013; Stamou et al., 2013; Bernardinelli et al., 2014; De Rubeis et al., 2014; Pinto et al., 2014; Phillips and Pozzo-Miller, 2015). Differences in environment as well as the presence of multiple gene mutations occurring in the same individual with autism complicate studies of the relationship between each gene and the phenotype observed. However, because similar cellular pathways (e.g., neurite outgrowth) are altered in different affected individuals, we can potentially develop therapeutic interventions to help mitigate the autism phenotypes.
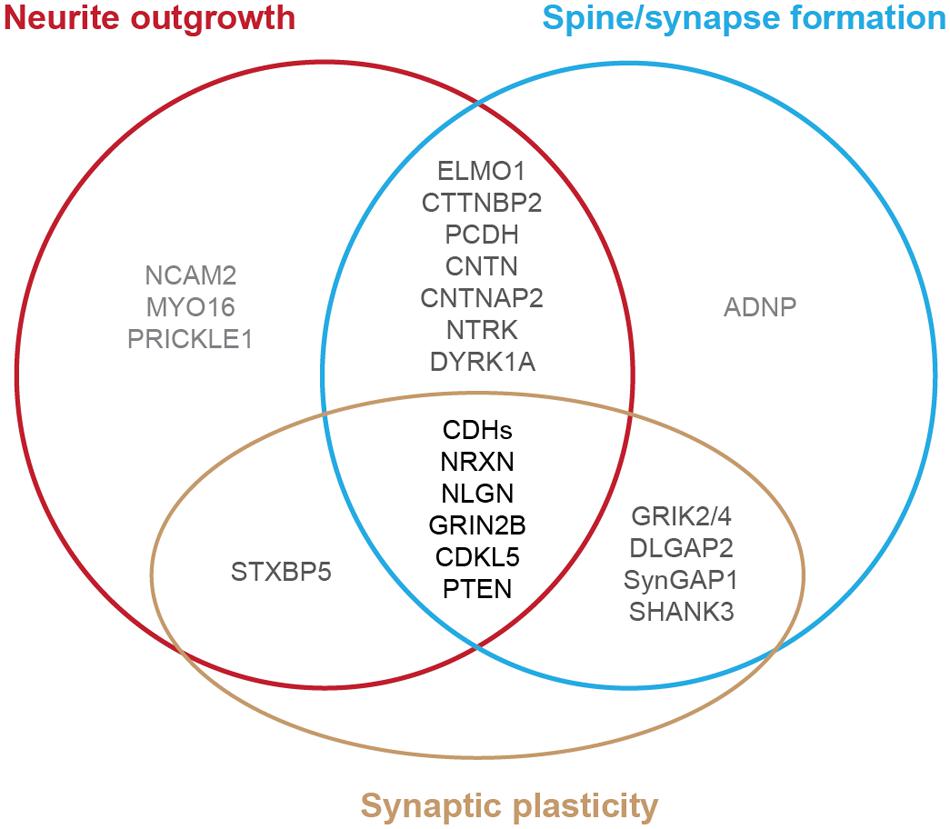
FIGURE 1. Diagram of autism-risk genes implicated in regulating the structural stability of neurons. Each circle represents a cellular pathway to regulate the structural stability of neurons, including neurite outgrowth (red), dendritic spine or synapse formation (blue), and synaptic plasticity (gold). Experimental evidence shows that many autism-risk genes regulate at least one cellular pathway to maintain the integrity of neuronal structures. Genes that regulate only one pathway are labeled in light gray. Genes that regulate two pathways are labeled in dark gray. Genes that regulate three pathways are labeled in black. The summaries of autism-risk genes that affect each cellular pathway can be found in Tables 1–3.
During development, neurite outgrowth and synapse formation are dynamic processes and their maturation is mutually dependent on proper guidance. Neurites initially exhibit frequent branch additions and retractions. Once dendrite arbors are established, productive synapse formation later in life and the accompanying activation of post-synaptic signaling machinery promotes arbor stability (Dailey and Smith, 1996; Wu and Cline, 1998; Rajan et al., 1999; Wong et al., 2000; Cline, 2001; Niell et al., 2004). Conversely, a loss of synaptic inputs leads to dendritic loss (Jones and Thomas, 1962; Matthews and Powell, 1962; Coleman and Riesen, 1968; Sfakianos et al., 2007). This reciprocal regulation contributes to the refinement of dendrites and synapses as the neurons mature (Wu et al., 1999; Trachtenberg et al., 2002; Holtmaat et al., 2005; Koleske, 2013). Thus, maintaining the structural stability of neurons and synapses is critical for proper brain function. Alterations in these processes likely underlie the disruption of normal dendrite and dendritic spine structure in neurological disorders, including neurodevelopmental conditions, psychiatric disorders, and neurodegenerative diseases (Fiala et al., 2002; Lin and Koleske 2010; Penzes et al., 2011; Kulkarni and Firestein, 2012; Zoghbi and Bear, 2012; Koleske, 2013; Bernardinelli et al., 2014).
It is well-accepted that ASD is not a monogenetic disorder, instead, it is often a neurological condition resulted from multiple mutations of several different genes. Although knockout, knockin, or transgenic approaches of autism-risk genes in animal models have demonstrated some of the autistic-like behaviors (Kazdoba et al., 2016), the limitation of the number of genes being manipulated in animals makes it difficult to recapitulate the human condition experimentally. Furthermore, ASD is a common comorbid condition in individuals with other neurodevelopmental disorders. The similar representation of the symptoms but different contribution of genetic mutations often complicates the diagnosis and the treatment. The complex profile of gene mutations makes it difficult to call a gene “the autism gene.” However, the list of autism-risk genes provides us a direction to understand the potentially vulnerable pathways in neurons that may be therapeutic targets to develop more efficient interventions for ASD. Indeed, in addition to the structural stability of neurons, several cellular pathways including transcriptional regulation (De Rubeis et al., 2014; Sanders, 2015), excitatory/inhibitory (E/I) balance (Blatt et al., 2001; Hussman, 2001; Rubenstein and Merzenich, 2003; Gao and Penzes, 2015; Nelson and Valakh, 2015), cerebellar development (Wang et al., 2014; Hampson and Blatt, 2015), and autoregulatory feedback loops (Mullins et al., 2016) have been proposed to be vulnerable in autism. In this review, we focus on recent identified autism-risk genes that have been shown to regulate neuronal structures and circuit formation, including aspects of neurite outgrowth (Table 1), synapse formation and spine stability (Table 2), and synaptic plasticity (Table 3). We will discuss the known biological function of those individual autism-risk genes in neurons and how they converge into common pathways. We have categorized these genes into cytoskeletal regulators, adhesion molecules, cell surface receptors, signaling molecules, as well as synaptic proteins (Figure 2). In addition, we include genes causing syndromic disorders in the discussion to highlight the importance of maintaining the neuronal structures for proper brain function.
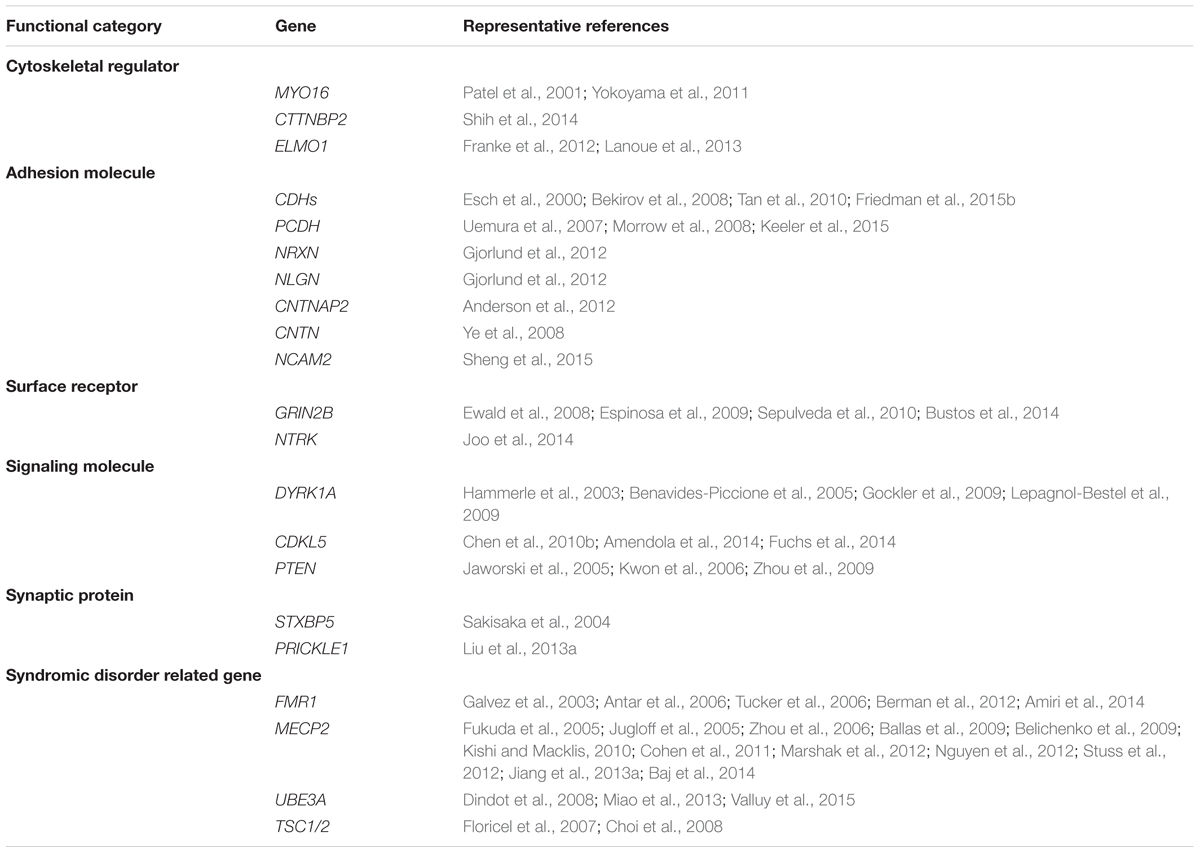
TABLE 1. Summary of autism-associated genes that regulate neurite outgrowth.
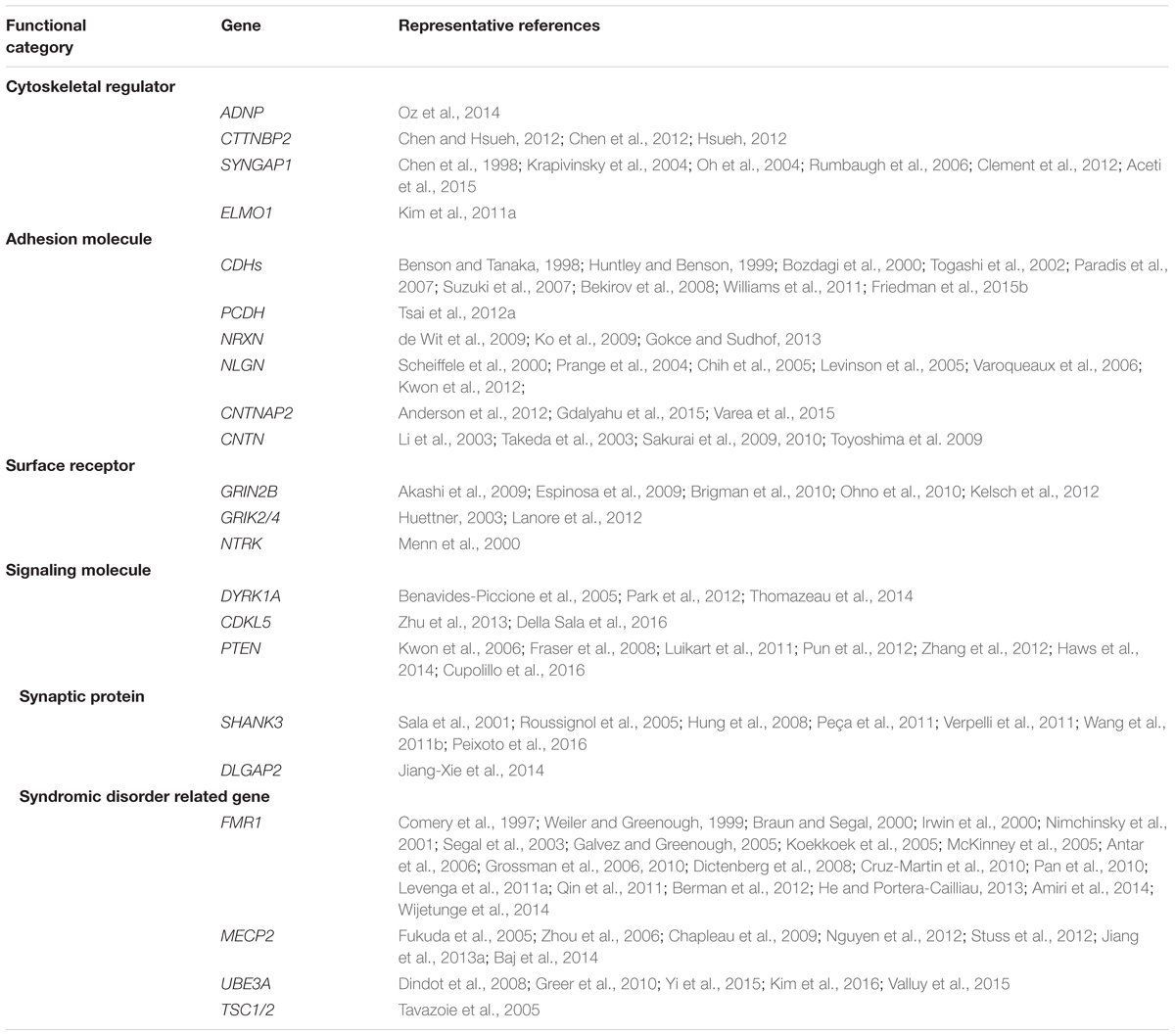
TABLE 2. Summary of autism-associated genes that regulate synapse/spine formation.
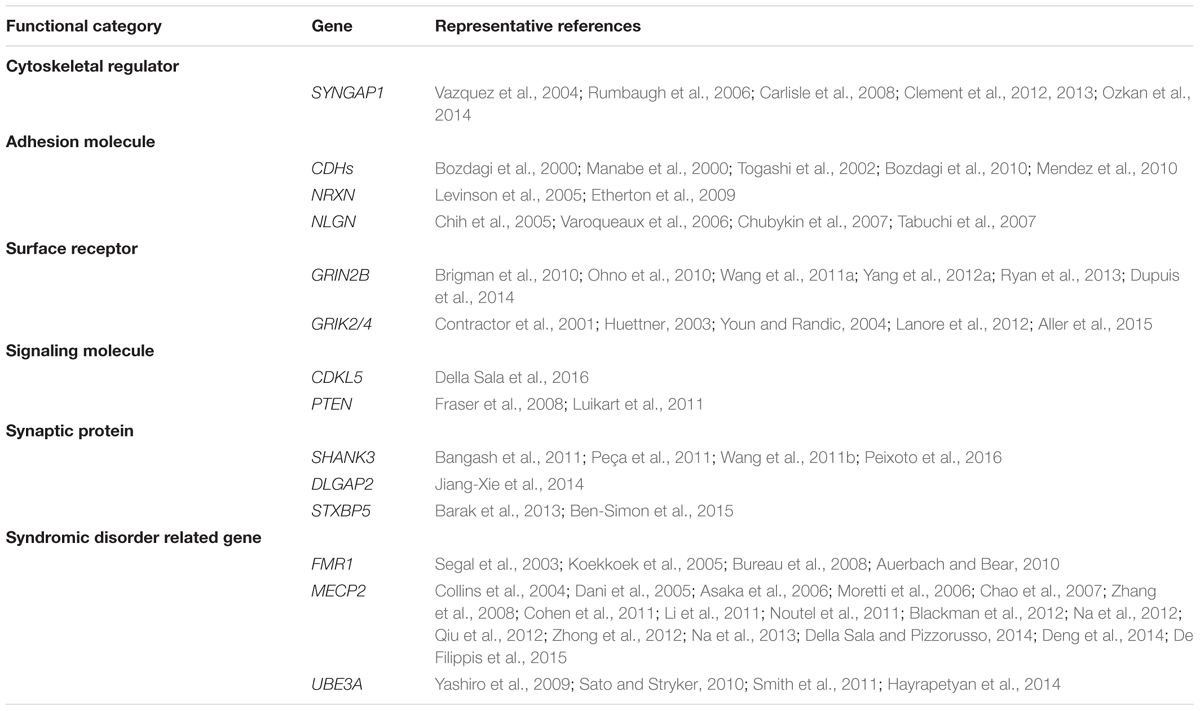
TABLE 3. Summary of autism-associated genes that regulate synaptic plasticity.
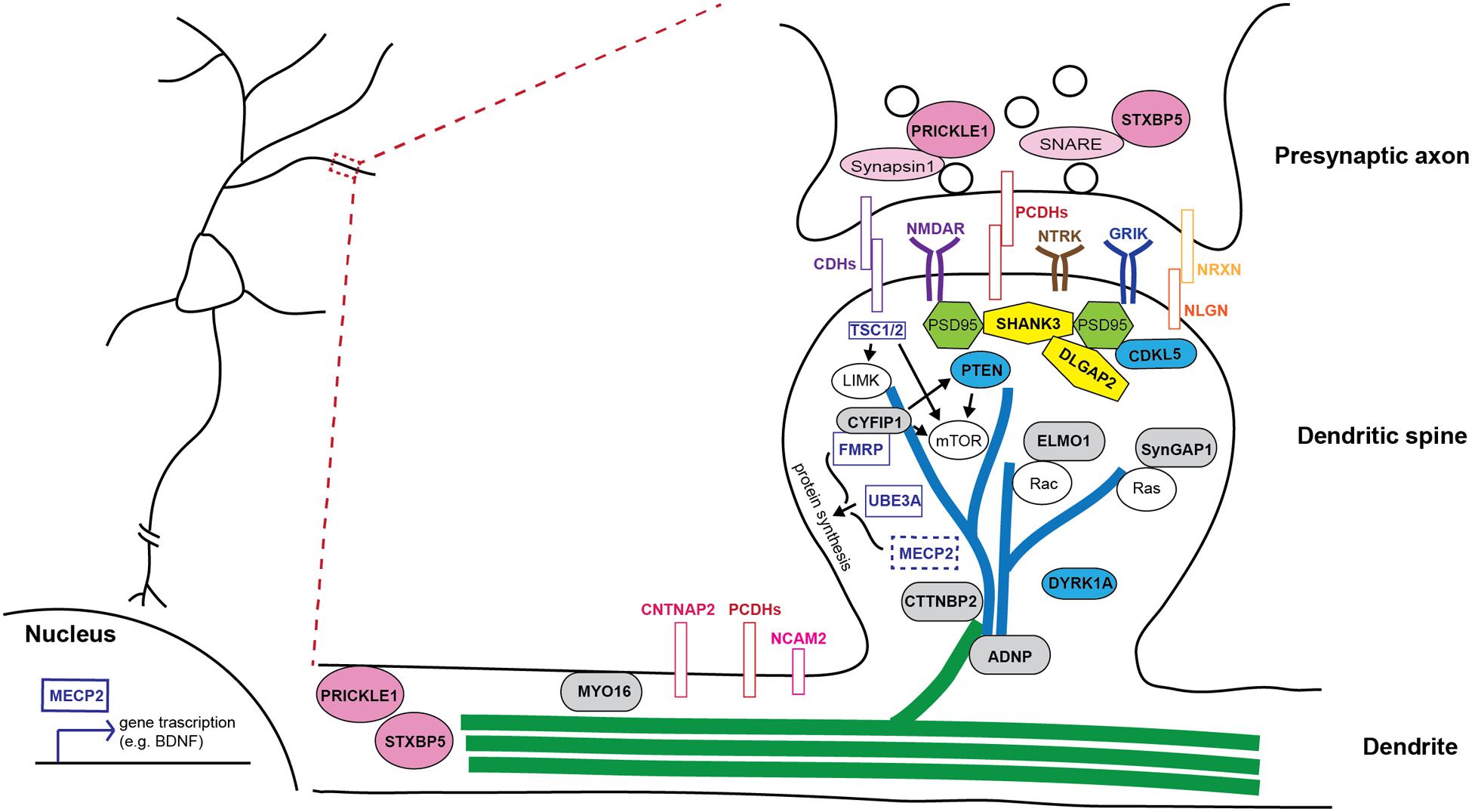
FIGURE 2. Schematic illustration of how autism-risk genes regulate neuronal structure and their sites of action. An illustration of a dendritic segment containing a dendrite and a dendritic spine is enlarged from the box region on the left and shown on the right. Microtubules (green) and actin filaments (blue) are two major cytoskeletons found in dendrites and dendritic spines, respectively. Autism-risk genes (in bold font) are categorized by their main function and color coded accordingly. (1) Cytoskeletal proteins (gray rounded rectangular box): MYO16, CTTNBP2, and ADNP, directly regulate actin and microtubule function to control dendritic spine and neurite stability. ELMO1 and SYNGAP1 regulate actin dynamics to control spine stability via small RhoGTPases. (2) Adhesion molecules (colored rectangular box): Cadherins (CDHs), protocadherins (PCDHs), and neurexin (NRXN)-neuroligin (NLGN) complex, as well as surface receptors, NTRK, GRIK, and NMDAR, act at synapses to regulate synaptic function. NCAM2 and CNTNAP2, also have functions in regulating neurite outgrowth. (3) Signaling molecules (blue ellipse shape): CDKL5, DYRK1A, and PTEN regulate several signaling pathways to maintain the stability of dendritic structures. (4) Scaffolding proteins (yellow polygon): SHANK3 and DLGAP2, locate at post-synaptic density and tightly associate with PSD95 and other signaling molecules to regulate spine stability and synaptic plasticity. (5) Synaptic proteins (pink ellipse shape): STXBP5 and PRICKLE1 not only regulate synaptic vesicle release, but also play a role in regulating neurite outgrowth. (6) Syndromic molecules (clear rectangular box): FMRP and UBE3A regulate the structural stability of neurons via the regulation of protein synthesis or binding with other molecules in dendritic spines. MECP2 mainly functions in the nucleus and regulates transcription of many genes to in turn affecting the structural stability of neurons. TSC1/2 regulates the mTOR pathway and cytoskeletal machinery to maintain dendritic stability.
The actin and microtubule cytoskeletons are the major components of dendritic spine and neurite structure, respectively (Hoogenraad and Akhmanova, 2010; Hotulainen and Hoogenraad, 2010; Dent et al., 2011; Shirao and Gonzalez-Billault, 2013). Precise regulation of these actin and microtubule networks is thus central to guide the proper development, plasticity, and long-term stability of these structures (Matus, 2000; Matus et al., 2000; Luo, 2002; Lin and Webb, 2009; Korobova and Svitkina, 2010; Lin and Koleske, 2010; Svitkina et al., 2010; Dent et al., 2011; Nicholson et al., 2012; Penzes and Cahill, 2012; Penzes and Rafalovich, 2012; Saneyoshi and Hayashi, 2012).
Myosins are motors that utilize ATPase activity to provide motility of actin or cargo transport on actin filaments (Pollard and Korn, 1973; Oliver et al., 1999; Tyska and Warshaw, 2002). Several myosin isoforms play central roles in regulating neurite outgrowth, as well as dendritic spine structural plasticity (Wylie et al., 1998; Wylie and Chantler, 2003; Ryu et al., 2006; Hammer and Wagner, 2013; Kneussel and Wagner, 2013; Yoshii et al., 2013; Koskinen et al., 2014; Ultanir et al., 2014). Among all isoforms, MYO16 (Myr8 or NYAP3) was recently implicated in ASD (Wang et al., 2009; Connolly et al., 2013; Kenny et al., 2014; Roberts et al., 2014; Liu et al., 2015b). MYO16 is expressed predominantly in the cortex and cerebellum. Levels and phosphorylation of MYO16 protein peak during early developmental stages, consistent with a role in regulating neuronal migration and neurite extension (Patel et al., 2001; Yokoyama et al., 2011). In addition to binding directly to filamentous- (F-)actin, MYO16 also physically interacts with PI3K and WAVE complex to regulate stress fiber remodeling in fibroblasts as well as the adhesion-dependent neurite outgrowth in neurons (Yokoyama et al., 2011).
CTTNBP2 encodes cortactin-binding protein 2 that interacts with cortactin, a nucleation-promoting factor of actin (Ohoka and Takai, 1998). CTTNBP2 is highly expressed in dendritic spines where it locally interacts with cortactin, striatin, a calcium binding protein, and PP2A, a serine/threonine protein phosphatase 2A, to control the formation and the maintenance of dendritic spines (Chen et al., 2012; Chen and Hsueh, 2012; Hsueh, 2012). In addition, oligomerization of CTTNBP2 induces microtubule bundling to promote dendrite arborization (Shih et al., 2014). Several mutations of CTTNBP2 have been reported in ASD cases, further indicating the importance of neurite outgrowth and dendritic spine formation for proper brain function (Cheung et al., 2001; Iossifov et al., 2012; De Rubeis et al., 2014).
Activity-dependent neuroprotective protein (ADNP) is a homeobox-containing protein secreted from glia and neurons (Bassan et al., 1999; Zamostiano et al., 2001; Furman et al., 2004; Mandel et al., 2008; Nakamachi et al., 2008). Through its interaction with the chromatin remodeling complex SWI/SNF, ADNP regulates hundreds of genes to modulate brain function (Pinhasov et al., 2003; Mandel and Gozes, 2007; Mandel et al., 2007). In addition to its traditional role in regulating transcription, ADNP has also been suggested to have function in regulating dendritic spines through interactions with microtubule end binding proteins (Oz et al., 2014). Mutations in or the alteration of the protein expression of ADNP have been associated with several neurological disorders, including schizophrenia and Alzheimer’s Disease (Vulih-Shultzman et al., 2007; Fernandez-Montesinos et al., 2010; Dresner et al., 2011; Yang et al., 2012b). Intriguingly, the association of mutations in ADNP and ASD is further emphasizing that the cytoskeletal integrity of neurons is vulnerable in ASD (O’Roak et al., 2012a,b; Ben-David and Shifman, 2013; De Rubeis et al., 2014; Helsmoortel et al., 2014; Vandeweyer et al., 2014; D’Gama et al., 2015).
Small RhoGTPases including Rho, Rac, and Cdc42 are central cytoskeletal regulators that control cell motility and morphology (Govek et al., 2005; Newey et al., 2005; Lin and Koleske, 2010; Tolias et al., 2011). Genetic mutations or dysregulation of the small RhoGTPase regulators, including guanine-exchange factors (GEFs) and GTPase-activating proteins (GAPs), have been implicated in several neurological conditions, including ASD (Newey et al., 2005; Lin and Koleske, 2010; Antoine-Bertrand et al., 2011; Stankiewicz and Linseman, 2014). Here, we will highlight those that regulate the morphological stability of neurons.
Engulfment and cell motility 1 (ELMO1) was first identified in a complex with a RacGEF, DOCK180, to activate Rac1 activity, which is essential for cell migration and phagocytosis (Gumienny et al., 2001; Brugnera et al., 2002; Grimsley et al., 2004). In hippocampal neurons, ELMO1 and DOCK180 colocalize at synaptic sites and together are required for spine formation (Kim et al., 2011a). Loss of Elmo1 shows a reduction in spine number but increased filopodia, suggesting a role in formation and/or maintenance of mature spines (Kim et al., 2011a). In addition, ELMO1 has been shown to regulate axonal and dendritic branching via Rac1 activation in response to different upstream signals (Franke et al., 2012; Lanoue et al., 2013).
SYNGAP1 encodes a synaptic-specific Ras/Rap GAP that associates with PSD-95 and specifically localizes to synaptic sites (Kim et al., 1998; Krapivinsky et al., 2004). The role of SYNGAP1 in regulating spine morphology and synaptic function has been well described. In response to CaMKII phosphorylation, SYNGAP1 directly regulates Ras/Rap activity to modulate MAPK signaling to maintain the stability of dendritic spines (Chen et al., 1998; Krapivinsky et al., 2004; Oh et al., 2004; Rumbaugh et al., 2006). Overexpression of SYNGAP1 decreases AMPAR-mediated currents and surface expression (Rumbaugh et al., 2006; Wang et al., 2013). Deletion or reduction of SYNGAP1 results in an elevated synaptic strength and an increase of mushroom spines (Vazquez et al., 2004; Rumbaugh et al., 2006; Carlisle et al., 2008). Mice with Syngap1 haploinsufficiency show accelerated maturation of dendritic spines followed by disruptions of synaptic transmission and cognitive function (Clement et al., 2012, 2013; Ozkan et al., 2014; Aceti et al., 2015). Coincidentally, haploinsufficiency in SYNGAP1 has been found in individuals with autism, intellectual disability, and a specific form of epilepsy (Berryer et al., 2013). Several other de novo mutations of SYNGAP1 also have been identified in different cases of ASD (Pinto et al., 2010; Cook, 2011; Hamdan et al., 2011; Berryer et al., 2013; Willsey et al., 2013; Brett et al., 2014; De Rubeis et al., 2014; O’Roak et al., 2014).
Since the actin and microtubule cytoskeletons are the major components of neuronal processes, it is not surprising that manipulating the cytoskeletal machinery dramatically affects neuronal structures. A small imbalance of cytoskeletal dynamics will create a huge impact on the structural stability of neurons, which in turn alters the formation of neuronal circuitry. Interestingly, most autism-associated cytoskeletal regulators control neurite outgrowth and synapse/spine formation thereby affecting the structural stability of neurons. These two processes are also the initial steps to establish correct neuronal connections during development. Failure to regulate these processes properly may result in significantly altered wiring of brain circuitries that is often found in ASD. The next research focus should investigate early in development to connect the dysregulatory effects of mutations in cytoskeletal genes.
Cell adhesion molecules (CAMs) play crucial roles in many aspects of neural circuit formation and, thus, it comes as no surprise that these molecules are found as top hits in lists of autism risk genes (Betancur et al., 2009; Pinto et al., 2010; Hussman et al., 2011; Chen et al., 2014b). Here, we discuss the current understanding of how CAMs that belong to the cadherin-, the neurexin/neuroligin- and the immunoglobulin-superfamily regulate neuronal stability.
The genetic association of cadherins with autism strongly supports their central roles in the development of the nervous system including synaptogenesis, dendrite arborization and dendritic spine regulation (Arikkath and Reichardt, 2008; Suzuki and Takeichi, 2008; Basu et al., 2015; Friedman et al., 2015a; Seong et al., 2015). The cadherin superfamily is comprised of more than a hundred different genes, subdivided into several classes including classical cadherins, protocadherins and atypical cadherins (Hulpiau and van Roy, 2009; Hirano and Takeichi, 2012). Several copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) are found in classical cadherins: CDH2. CDH5. CDH8. CDH9, CDH10, CDH11, and CDH13 (Wang et al., 2010; Chapman et al., 2011; Hussman et al., 2011; Pagnamenta et al., 2011; Sanders et al., 2011; O’Roak et al., 2012b; Prandini et al., 2012; Connolly et al., 2013; Walker and Scherer, 2013; Crepel et al., 2014; Krumm et al., 2015); non-clustered protocadherins: PCDH9, PCDH10, and PCDH19 (Morrow et al., 2008; Depienne et al., 2009; Camacho et al., 2012; O’Roak et al., 2012b; Prasad et al., 2012; Girirajan et al., 2013; van Harssel et al., 2013); and an atypical cadherin, FAT1 (Hussman et al., 2011; Neale et al., 2012; Cukier et al., 2014; Kenny et al., 2014). In general, the extracellular domain of cadherins contains five cadherin repeats/EC motifs that mediate Ca2+-dependent homophilic adhesion (Tanihara et al., 1994). However, heterophilic interactions between different sub-classes of classical cadherins create more combinations of interaction and function (Shimoyama et al., 2000). The cytosolic tail binds to catenins leading to the anchoring of the cadherin-catenin complex to the cytoskeleton (via β-catenin and α-catenin) and the clustering of cadherins in the plasma membrane (via p120-catenin) (Yap et al., 1998; Nelson, 2008; McCrea and Gu, 2010). The cadherin superfamily contains numerous cadherin members which impact neuronal structure and function from early neurite extension to the maintenance of mature synaptic networks (Basu et al., 2015; Friedman et al., 2015a). However, the pathways underlying disrupted cadherin signaling still requires further investigation. Here, we focus our discussion on the autism-associated cadherins shown to regulate neurite outgrowth and synapse morphogenesis.
N-cadherin, also known as cadherin 2 (CDH2), is the best studied classical cadherin. N-cadherin functions throughout the development of the nervous system, including neurite outgrowth, axon guidance, synaptogenesis and synaptic plasticity (Takeichi and Abe, 2005; Arikkath and Reichardt, 2008; Hirano and Takeichi, 2012; Friedman et al., 2015a). N-cadherin promotes dendritic outgrowth during development and is also required for activity-dependent dendrite expansion (Esch et al., 2000; Tan et al., 2010). N-cadherin is also required for the establishment of initial contacts between axons and filopodia followed by clustering at contact points to stabilize early synapses (Benson and Tanaka, 1998; Huntley and Benson, 1999; Togashi et al., 2002). Blocking N-cadherin adhesion in hippocampal neurons perturbs synapse formation and abolishes long-term potentiation (LTP)-induced stabilization of dendritic spines (Togashi et al., 2002; Mendez et al., 2010). Neural activity increases N-cadherin protein levels and dimerization leading to increased synapse number (Bozdagi et al., 2000). In mature synapses, N-cadherin is required for the persistence of dendritic spine enlargement and LTP (Bozdagi et al., 2000, 2010). Together with N-cadherin, CDH8 regulates the development of the hippocampal mossy fiber pathway (Bekirov et al., 2008). CDH8 also mediates assembly and maturation of corticostriatal synapses (Bekirov et al., 2008; Friedman et al., 2015b), whereas CDH9-mediated adhesion is involved in the formation and differentiation of dentate gyrus synapses on CA3 cells where it regulates synapse density, presynaptic bouton complexity and postsynaptic morphology (Williams et al., 2011). In contrast to CDH8 and CDH9, an RNAi screen for molecules required for synapse development identified CDH11 and CDH13 as positive regulators of glutamatergic synapse development (Paradis et al., 2007). Interestingly, Cdh11-deficient mice revealed enhanced LTP in the CA1 region of the hippocampus and mice show reduced fear- or anxiety-related behavior suggesting that CDH11 might restrict synaptic plasticity and efficacy (Manabe et al., 2000).
Protocadherins are the largest subgroup within the cadherin superfamily and are further subtyped into clustered (α-, β- and γ-PCDH) and non-clustered protocadherins (δ1- and δ2-PCDH) (Frank and Kemler, 2002). They share a similar structure to classical cadherins, but with six to seven cadherin domains/EC motifs. However, the cytosolic tails of protocadherins and cadherins do not show significant homology suggesting that they likely engage distinct intracellular signaling pathways. Protocadherins are highly expressed in the nervous system and localize to synapses. Based on their spatial and temporal expression pattern in the brain and on recent reports, protocadherins have roles in dendritic development and synaptic connections (Hirano et al., 1999; Frank and Kemler, 2002; Kim et al., 2007, 2011b; Keeler et al., 2015). For example, PCDH10 expression is regulated by neuronal activity and its function is crucial for forebrain axon outgrowth and the proper patterning of thalamocorticial projections (Uemura et al., 2007; Morrow et al., 2008). In addition, PCDH10 mediates synapse elimination by promoting proteasomal degradation of PSD-95 (Tsai et al., 2012a).
FAT atypical cadherin 1 (FAT1) belongs to the atypical cadherin family and consists of a huge extracellular domain comprising 34 cadherin domains/EC motifs (Tanoue and Takeichi, 2005; Sadeqzadeh et al., 2014). FAT1 expression is enriched during embryonic neurodevelopment and severe nervous system defects are found in FAT1-deficient mice (Ciani et al., 2003; Sadeqzadeh et al., 2014). At the cellular level, FAT1 localizes to cell–cell contacts as well as to the leading edge of lamellipodia and tips of filopodia to regulate cell polarity, cell migration, and cell-cell adhesion (Moeller et al., 2004; Tanoue and Takeichi, 2004). These functions are likely mediated through intracellular signaling via Ena/VASP proteins to regulate actin assembly and dynamics (Moeller et al., 2004; Tanoue and Takeichi, 2004). Other intracellular binding partners of FAT1 include the classical cadherin binding partner β-catenin as well as the synaptic scaffolding molecules Homer-1 and 3 (Hou et al., 2006; Schreiner et al., 2006).
Neurexins are encoded by three genes (NRXN1-3) while the neuroligin family consists of four isoforms in mice (Nlgn1-4) and five isoforms in humans (NLGN1-4X and 4Y) (Südhof, 2008). Neurexins localize to presynaptic terminals and form heterophilic interactions with neuroligins, which are localized to the postsynaptic compartment (Ichtchenko et al., 1995, 1996; Nguyen and Sudhof, 1997). Presynaptic neurexins link synaptic adhesion with the synaptic vesicle release machinery via binding to PDZ-domain containing proteins (Hata et al., 1996; Butz et al., 1998; Dean et al., 2003; Missler et al., 2003). At the postsynaptic site, the neurexin-neuroligin complex induces clustering of scaffolding proteins, such as PSD-95, and recruits NMDA- and AMPA-receptors (Irie et al., 1997; Chih et al., 2005; Nam and Chen, 2005; Heine et al., 2008; Barrow et al., 2009; Mondin et al., 2011). Distinct neurexin-neuroligin complexes play discrete roles during synaptogenesis with neuroligin-1 regulating excitatory synapse formation and maturation, while neuroligin-2 and 3 mediate inhibitory synapse formation (Song et al., 1999; Scheiffele et al., 2000; Prange et al., 2004; Varoqueaux et al., 2004, 2006; Chih et al., 2005; Levinson et al., 2005; Barrow et al., 2009; Kwon et al., 2012; Gokce and Sudhof, 2013). Intriguingly, neurexin-1 can also bind to leucine-rich repeat transmembrane protein 2 (LRRTM2) and promote synapse formation (de Wit et al., 2009; Ko et al., 2009; Gokce and Sudhof, 2013). In addition to their synaptic roles, interaction of neurexin-1 and neuroligin-1 regulates neurite outgrowth (Gjorlund et al., 2012). Several mutations and CNVs in NRXN1-3 have been found to be associated with ASD with the prevalence highest for mutations in NRXN1 (Feng et al., 2006; Autism Genome Project Consortium et al., 2007; Kim et al., 2008; Yan et al., 2008; Ching et al., 2010; Pinto et al., 2010; Wisniowiecka-Kowalnik et al., 2010; Gauthier et al., 2011; Voineskos et al., 2011; Camacho-Garcia et al., 2012; Duong et al., 2012; Iossifov et al., 2012; Kong et al., 2012; Liu et al., 2012; Prasad et al., 2012; Schaaf et al., 2012; Vaags et al., 2012; Bena et al., 2013; Dabell et al., 2013; Girirajan et al., 2013; Jiang et al., 2013b; Koshimizu et al., 2013; Walker and Scherer, 2013; Cukier et al., 2014; De Rubeis et al., 2014; Egger et al., 2014; Imitola et al., 2014; Vinas-Jornet et al., 2014; Tammimies et al., 2015). Similarly, NLGN1-4 genes have been implicated in the pathogenesis of ASD with NLGN3 and 4 being the most prevalent (Jamain et al., 2003; Laumonnier et al., 2004; Ylisaukko-oja et al., 2005; Lawson-Yuen et al., 2008; Glessner et al., 2009; Yu et al., 2011, 2013; Leblond et al., 2012; O’Roak et al., 2012b; Steinberg et al., 2012; Yanagi et al., 2012; Girirajan et al., 2013; Jiang et al., 2013b; Iossifov et al., 2014; Kenny et al., 2014; Li et al., 2014a; Krumm et al., 2015; Sanders et al., 2015; Yuen et al., 2015). However, several reports also indicate the negative association of NLGN3 and 4 with autism (Vincent et al., 2004; Gauthier et al., 2005; Blasi et al., 2006; Wermter et al., 2008; Avdjieva-Tzavella et al., 2012; Liu et al., 2013b; Xu et al., 2014). Further investigation is required to clarify this controversy.
Mutations in another member of the neurexin superfamily, contactin-associated protein-like 2 (CNTNAP2/CASPR2), have also been identified in individuals with autism (Alarcón et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Vernes et al., 2008; Li et al., 2010; Petrin et al., 2010; Poot et al., 2010; Nord et al., 2011; O’Roak et al., 2011; Peñagarikano et al., 2011; Whitehouse et al., 2011; Anney et al., 2012; Prasad et al., 2012; Girirajan et al., 2013; Sampath et al., 2013; Egger et al., 2014; Poot, 2014; Chiocchetti et al., 2015). CNTNAP2 is required for dendrite arborization and dendritic spine development and maintenance (Anderson et al., 2012; Gdalyahu et al., 2015). Mice deficient for Cntnap2 show defects in spine stabilization and synaptic function resulting in several core ASD-like behaviors such as deficits in communication and social interaction, as well as repetitive behaviors (Peñagarikano et al., 2011; Gdalyahu et al., 2015; Varea et al., 2015).
The immunoglobulin superfamily of CAMs (IgSF-CAMs), including contactin, L1CAM, NCAM or SynCAM, make up a third large group of trans-synaptic CAMs. IgSF-CAMs have been implicated in various processes during neural circuit formation, from neurite outgrowth and axonal navigation to synapse formation and plasticity (Rougon and Hobert, 2003).
The contactin (CNTN) subfamily consists of six members (CNTN1-6), each of which contain six Ig-like and four fibronectin III-like domains that are linked to the cell membrane via a glycosylphosphatidylinositol (GPI)-anchoring domain (Shimoda and Watanabe, 2009). While CNTN1 and 2 have been extensively studied in the context of neurite outgrowth, fasciculation, and axon guidance, less is known about the function of CNTN3-6 (Karagogeos, 2003; Shimoda and Watanabe, 2009; Mohebiany et al., 2014). However, CNTN3-6 have been implicated as risk genes in ASD (Fernandez et al., 2004; Christian et al., 2008; Morrow et al., 2008; Glessner et al., 2009; Roohi et al., 2009; Cottrell et al., 2011; Hussman et al., 2011; van Daalen et al., 2011; Leblond et al., 2012; Prasad et al., 2012; Vaags et al., 2012; Cukier et al., 2014; Kashevarova et al., 2014; Nava et al., 2014; Poot, 2014; Hu et al., 2015; Liu et al., 2015a). CNTN4 is strongly expressed in a subset of olfactory sensory neurons where it guides proper targeting of axon terminals to the corresponding glomeruli for the formation of olfactory circuits (Kaneko-Goto et al., 2008). The Cntn5 knockout mice display reduced fiber density and glutamatergic synapses in the auditory brainstem (Li et al., 2003; Toyoshima et al., 2009). CNTN6 is highly expressed in the postnatal cerebellum and plays an important role in the formation of synapses between parallel fibers and Purkinje cells (Takeda et al., 2003; Sakurai et al., 2009). Similarly, CNTN6 regulates the formation of glutamatergic synapses in the hippocampus and the orientation of apical dendrites of layer V pyramidal neurons in the visual cortex (Ye et al., 2008; Sakurai et al., 2010).
Neural cell adhesion molecule 2 (NCAM2) belongs to the NCAM family and is a paralog of NCAM1. Similar to other members of the Ig-superfamily, NCAMs contain five Ig- and two FN3-domains in the extracellular region and are differentially spliced to produce both transmembrane and GPI-anchored variants (Winther et al., 2012). NCAM2 is predominantly expressed in the brain and required for the formation and maintenance of axonal and dendritic compartmentalization in the olfactory glomeruli (Walz et al., 2006; Borisovska et al., 2011). In addition, NCAM2 regulates filopodia formation and neurite branching of cortical neurons via a CaMKII-dependent signaling pathway (Sheng et al., 2015). SNP and chromosomal deletion including NCAM2 has been reported in individuals with autism (Haldeman-Englert et al., 2010; Hussman et al., 2011; Petit et al., 2015).
Adhesion molecules are a huge group of proteins that display many similarities in molecular structure and in signaling property. Depending on the cellular localization, functions of adhesion molecules range from neurite outgrowth and synapse/spine formation, to neuronal plasticity, further highlighting their importance in regulating the structural stability of neurons. However, whether these molecules function to compensate each other or are developmentally regulated is still not clear. The interesting question is whether the temporal and spatial expression patterns of these autism-associated adhesion molecules correlate with the affected developmental time frame and affected brain regions in ASD.
Establishment of synaptic connections and modification of their strength and stability is intimately related to the receptor populations in the plasma membranes of pre- and postsynaptic cell compartments. Thus, several ASD risk genes code for cell surface receptor proteins including the ionotropic glutamate receptors (iGluR) and the receptor tyrosine kinases (RTK).
Glutamate-mediated ionotropic signaling occurs via activation of the glutamate-gated ion channel family which is divided into three subfamilies; the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), the N-methyl-D-aspartate receptors (NMDARs) and the kainate-type receptors (KARs) (Collingridge et al., 2009). The main function of these receptors focuses on the regulation of synaptic activity and plasticity, which in turn affect the structural stability of neurons. In particular, the genetic association of the GRIN2B gene, which encodes the GluN2B subunit of NMDARs, and the GRIK2/4 genes that encodes the GluK2 and 4 subunits of KARs with autism has been established (Jamain et al., 2002; Shuang et al., 2004; Holt et al., 2010; Myers et al., 2011; O’Roak et al., 2011, 2012a; Tarabeux et al., 2011; de Ligt et al., 2012; Griswold et al., 2012; Prasad et al., 2012; Talkowski et al., 2012; Yoo et al., 2012; Dimassi et al., 2013; De Rubeis et al., 2014; Kenny et al., 2014; Li et al., 2014a; Namjou et al., 2014; Poot, 2014; Aller et al., 2015; Pan et al., 2015).
NMDARs are composed of an obligatory GluN1 subunit and one or more GluN2 (GluN2A-GluN2D) subunits with the majority of the composition being GluN1/2A/2B (Buller et al., 1994; Petralia et al., 1994; Luo et al., 1997). The composition of GluN2 subunits are developmentally regulated and critically determine the synaptic properties (Laurie and Seeburg, 1994; Sheng et al., 1994; Li et al., 1998). The GluN2B subunit expresses early during development gradually being replaced by GluN2A indicating its role in the formation of neuronal circuitry (Sheng et al., 1994; Li et al., 1998; Bustos et al., 2014). Overexpression or knockdown of GluN2B alters dendrite arborization in neurons both in vivo and in vitro (Ewald et al., 2008; Espinosa et al., 2009; Sepulveda et al., 2010; Bustos et al., 2014). GluN2B is also required for the formation of dendritic spines, maturation of synapses, and the proper molecular compositions of several postsynaptic proteins (Akashi et al., 2009; Espinosa et al., 2009; Brigman et al., 2010; Kelsch et al., 2012). In turn, GluN2B is crucial for maintaining proper synaptic plasticity (Brigman et al., 2010; Ohno et al., 2010; Wang et al., 2011a; Yang et al., 2012a; Ryan et al., 2013; Dupuis et al., 2014). GRIN2B, an autism-risk gene, further suggests that pathways involved in early circuitry formation may be vulnerable targets in autism. Selective inhibition of GluN2B function has been shown to restore dendritic spine loss and associated behavior alterations in several experimental conditions providing insights to the potential therapeutic targets to correct some ASD phenotypes (Chen et al., 2011; Iafrati et al., 2014; Gupta et al., 2015).
Kainate-type receptors regulate axonal filopodia motility of hippocampal mossy fibers in response to neuronal stimulation during synaptogenesis (Tashiro et al., 2003). KAR subunits, in particular GluK2, interacts with structural elements of the synapse; such as the PSD-95 and SAP-102 scaffolding molecules, as well as the N-cadherin and β-catenin adhesion molecules (Carta et al., 2014; Pahl et al., 2014), indicating that Grik genes are involved in processes that regulate synapse architecture and stability. Indeed, GluK2 regulates hippocampal synapse maturation and stability (Huettner, 2003; Contractor et al., 2011; Lanore et al., 2012; Lerma and Marques, 2013). Animals with deficient GluK2 proteins exhibit a delay in the postnatal maturation of synaptic contacts between MF-CA3 in the hippocampus, suggesting that the expression of the GluK2 is important for the establishment of normal morphology and function of synaptic networks in the hippocampus (Contractor et al., 2001; Lanore et al., 2012). Expression of the GluK4 is mainly restricted to mossy fiber synapses in the hippocampal CA3 region where it co-assembles with GluK2 in functional pre- and postsynaptic GluK2/4 receptor complexes (Darstein et al., 2003). Mice with forebrain GluK4 overexpression exhibit altered synaptic transmission and display several autistic-like behaviors including social impairment, enhanced anxiety, and depressive states, coinciding with the finding of GRIK4 duplications in individuals with ASD (Griswold et al., 2012; Aller et al., 2015). Even though the phenotypes resulting from Grik gene dysfunction in mice are in the same general categories with symptoms of ASD, further investigation about the molecular consequences of impairments in GluK proteins in ASD is required for developing future therapeutic interventions.
Tyrosine receptor kinases (Trks) mediate neurotrophic growth factor-induced signaling via dimerization and trans-autophosphorylation of Tyr residues on the intracellular domains of the receptor and subsequent activation of intracellular signaling pathways (Deinhardt and Chao, 2014). This results in a number of neurogenic events, such as synaptic plasticity, maturation and stability, dendritic and axonal growth and differentiation as well as cell survival and maintenance (Martinez et al., 1998; Deinhardt and Chao, 2014). The Trk family consists of three proteins; TrkA, B and C, which are expressed by the neurotrophic tyrosine receptor kinase genes (NTRK. 1. 2 and 3, respectively. Each Trk receptor interacts selectively with a different neurotrophin resulting in preferential interaction pairs: TrkA is activated by NGF, TrkB by BDNF, and TrkC by NT-3 (Deinhardt and Chao, 2014). Considering its well-documented function in synaptophysiology (Minichiello, 2009), it would be reasonable to suspect a correlation between genetic variations in NTRK2 and ASD. However, to date, only one study has reported a weak association between NTRK2 mutations and ASD (Correia et al., 2010), while other studies were unable to confirm that link (Chakrabarti et al., 2009). Alternatively, a growing body of evidence generated from genetic evaluation of ASD risk genes has identified NTRK3, the gene coding for TrkC, as a plausible candidate in autism (Chakrabarti et al., 2009; Hussman et al., 2011; Vardarajan et al., 2013).
In the mammalian brain, TrkC (as well as other neurotrophic receptors) is present both as full length catalytically active receptor, as well as a splice variant that lacks the Tyr kinase domain and is catalytically inactive (Ichinose and Snider, 2000). Interestingly, knockout of the non-catalytic TrkC isoform in mice yields a more severe phenotype than does the depletion of the kinase-active receptor, indicating that TrkC has important functions beyond the ability to convey classical RTK signaling (Faux et al., 2007; Deinhardt and Chao, 2014). Indeed, recent studies have begun to elucidate the function assigned to non-catalytic isoforms by demonstrating a role for TrkC in synaptic adhesion complexes (Takahashi and Craig, 2013). Postsynaptic TrkC interacts across the presynaptic cleft with protein tyrosine phosphatase (PTP) σ to form an adhesion complex crucial for development and stability of excitatory, but not inhibitory, synapses (Takahashi et al., 2011; Coles et al., 2014). Formation of this adhesion complex is enhanced by the presence of the TrkC ligand, NT-3, which facilitates glutamatergic presynaptic assembly and function (Ammendrup-Johnsen et al., 2015). NT-3 binding to kinase domain-truncated TrkC isoforms has also been shown to induce cytoskeletal changes via recruitment of the scaffold protein tamalin, leading to activation of Arf6 and induction of Rac1-GTP (Esteban et al., 2006). Interestingly, the expression of non-catalytic TrkC relative to the kinase active isoform is upregulated during the second and third postnatal weeks, the most intense period of synaptogenesis, indicating that expression of the different Ntrk3 gene products is temporally associated with synapse formation (Valenzuela et al., 1993; Menn et al., 2000). Recent studies also found that NT-3-TrkC signaling between presynaptic granule neurons and postsynaptic Purkinje cells controls dendrite morphogenesis in cerebellum (Joo et al., 2014). Although no studies have evaluated the ratio of non-catalytic to catalytic TrkC receptors in individuals with ASD, it might be speculated that certain genetic variants could cause imbalances in the expression patterns of NTRK3 isoforms.
Surface receptors respond to extracellular signals such as neurotransmitters and trophic factors to activate downstream signaling pathways to diversify the cellular responses. Each receptor may have a unique signaling pathway associated with it and therefore, the mutations on selective receptors provide us with clues about which signaling pathways may be more susceptible to perturbations in ASD. Thus, identifying the downstream effectors and signaling pathways that are affected by these autism-associated receptor mutants should be an important direction of future investigation.
The dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1a (DYRK1A) is one of the isoforms in DYRK family and is a human homolog of the Drosophila kinase minibrain (MNB) (Shindoh et al., 1996). DYRK1A was first described as a cadidate gene for intellectual disability in Down syndrome because of its location on the “Down syndrome critical region” of chromosome 21 (van Bon et al., 1993; Shindoh et al., 1996; Hammerle et al., 2003). Interestingly, recent genetic analyses suggest that DYRK1A is also a risk gene in ASD (Iossifov et al., 2012; O’Roak et al., 2012a,b; Chen et al., 2014a; Krumm et al., 2014; Redin et al., 2014; Bronicki et al., 2015; van Bon et al., 2016). Expression of Dyrk1a in mouse brain is limited to early developmental periods and can promote neurite formation (Okui et al., 1999; Hammerle et al., 2003; Gockler et al., 2009). In addition, DYRK1A phosphorylates N-WASP, a cytoskeletal protein, to inhibit spine formation in primary hippocampal neurons (Park et al., 2012). Pyramidal neurons in Dyrk1a+/- mouse cortex have reduced dendritic branches and dendritic spine density, which potentially causes the reduced brain size in these mice (Fotaki et al., 2002; Benavides-Piccione et al., 2005). On the other hand, overexpression of DYRK1A in mice causes increased spine density in cortical pyramidal neurons, and these animals show prefrontal deficits including significant impairment of spatial learning and cognitive flexbitiliy (Altafaj et al., 2001; Thomazeau et al., 2014). However, overexpressing DYRK1A in primary cortical mouse neurons significantly reduces dendrite complexity through disruption of REST/NRSF levels and REST/NRSF-SWI/SNF chromatin remodeling complex (Lepagnol-Bestel et al., 2009).
The Cyclin-dependent kinase-like 5 (CDKL5) is a serine/threonine kinase, also known as serine/threonine kinase 9 (STK9). Mutations of CDKL5 have been associated with several X-linked neurodevelopmental disorders, as well as ASD (Weaving et al., 2004; Scala et al., 2005; Archer et al., 2006; Russo et al., 2009; Sprovieri et al., 2009; Schaaf et al., 2011; Bahi-Buisson and Bienvenu, 2012; Bartnik et al., 2012; Maortua et al., 2012; Carvill et al., 2013; Epi et al., 2013; Piton et al., 2013; Zhao et al., 2014; Codina-Sola et al., 2015; Szafranski et al., 2015). Expression of CDKL5 is enriched in the brain and increases gradually following development (Lin et al., 2005; Rusconi et al., 2008). In addition to a catalytic domain, CDKL5 contains nuclear localization and export signals and can shuttle between the cytoplasm and nucleus. In the nucleus, CDKL5 phosphorylates methyl-CpG-binding protein 2 (MECP2), a causative gene for Rett syndrome (see below), providing a suggestive molecular mechanism associated with the condition (Mari et al., 2005). In the cytosol, CDKL5 postively regulates neurite outgrowth and dendritic arborization via binding with Rac1 (Chen et al., 2010b). With this broad influence on neuronal function, Cdkl5 null mice have several defects ranging from neuronal survival, dendritie maturation, spine stability, synaptic plasticity, and behaviors (Amendola et al., 2014; Fuchs et al., 2014; Della Sala et al., 2016). Treatment of Cdkl5 null mice with insulin-like growth factor 1 (IGF-1) or the glycogen synthase kinase 3β (GSK3β) inhibitor can rescue these defective phenotypes (Della Sala et al., 2016; Fuchs et al., 2015). Furthermore, CDKL5 has been shown to bind to palmitoylated-PSD-95 and this interaction is important for synaptic targeting of CDKL5 and spine formation (Zhu et al., 2013).
Phosphatase and tensin homolog (PTEN) is a dual-specificity lipid/protein tyrosine phosphatase that negatively regulates the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of the rapamycin (mTOR) pathway to control cellular function (Maehama and Dixon, 1998, 1999; Stambolic et al., 1998; Vazquez and Sellers, 2000; Downes et al., 2001; Leslie and Downes, 2002; Hoeffer and Klann, 2010). PTEN was first identified as a tumor suppressor (Li et al., 1997) but later also found to be associated with neurodevelopmental conditions such as epilepsy, macrocephaly, and autism (Goffin et al., 2001; Butler et al., 2005; Buxbaum et al., 2007; Orrico et al., 2009; Varga et al., 2009; McBride et al., 2010; Redfern et al., 2010; Stein et al., 2010; Schaaf et al., 2011; O’Roak et al., 2012b; Busa et al., 2013; De Rubeis et al., 2014; Hobert et al., 2014; Marchese et al., 2014; Vanderver et al., 2014; Codina-Sola et al., 2015; D’Gama et al., 2015; Johnston and Raines, 2015; Krumm et al., 2015; Spinelli et al., 2015; Tammimies et al., 2015; Cupolillo et al., 2016; Schwerd et al., 2016; Tilot et al., 2016). PTEN expression in the brain is positively correlated with the developmental stages of neuronal dendrite formation and synaptogenesis suggesting a role in regulating neuronal function (Perandones et al., 2004). PTEN plays a critical role in regulating the stability of dendritic spines and synaptic activity. PTEN overexpression in hippocampal CA1 pyramidal neurons results in a decrease in spine density (Zhang et al., 2012). Deleting PTEN in cortical and hippocampal neurons causes loss of neuronal polarity and general neuronal hypertrophy, including increases in dendrite arborization and spine density (Jaworski et al., 2005; Kwon et al., 2006; Fraser et al., 2008; Zhou et al., 2009). Selective deletion of PTEN in dentate granule neurons results in increased spine density and synaptic activity, as well as increased mossy fiber sprouting (Luikart et al., 2011; Pun et al., 2012). Knockdown of PTEN in basaolateral amygdala and dentate gyrus, however, does not affect spine density, but spine morphology is dramatically altered with an increase of mature mushroom-shaped spines (Haws et al., 2014). The Pten knockout in cerebellum also results in significant alterations in neuronal morphology of Purkinje cells including swelling of dendrites, and an increase of axonal bouton and dendritic spine size (Cupolillo et al., 2016). The impact of PTEN on spine stability is dependent on the phosphorylation status of its serine/threonin residues and the PDZ-binding motif in its C-terminus (Zhang et al., 2012). The phenotypes observed following PTEN deletion result from hyperactivation of the PI3K/AKT/mTOR pathway and inhibition of this molecular pathway is sufficient to rescue these phenotypes (Jaworski et al., 2005; Kwon et al., 2006; Zhou et al., 2009; Pun et al., 2012).
The identification of the vulnerable intracellular signaling pathways will aid us in the pursuit to find new therapeutic drug targets in patients with ASD. Interestingly, a variety of gene mutations result in disruption of the mechanistic pathways that these signaling molecules participate in. Therefore, several of the experimental pharmacological agents currently proposed as possible treatment strategies for autistic phenotypes are targeting these signaling molecules (see “Perspectives”).
Synaptic signaling processes are key to proper neural function. Some pivotal components of synapses are postsynaptic scaffolding proteins, which cluster neurotransmitter receptors, cell adhesion proteins, ion channels and cytoskeletal molecules to a confined postsynaptic region (Kim and Sheng, 2004; Sheng and Hoogenraad, 2007). Dysfunction in scaffolding proteins often has a huge impact on neuronal function, including neuronal morphology and synaptic plasticity (Ting et al., 2012). Emerging evidence has recently linked ASD with mutations of several genes encoding scaffolding proteins as described below.
SHANK genes encode three large scaffolding proteins, SHANK1-3, that contain ankyrin repeats, the SH3 domain, the PDZ domain, the proline-rich domain, and the SAM domain (Naisbitt et al., 1999; Sheng and Kim, 2000; Baron et al., 2006). These multiple putative protein interaction domains enable shank proteins to function as a bridge linking inotropic glutamate receptors, PSD-95, SAPAPS (Naisbitt et al., 1999), Homers (Tu et al., 1999; Hayashi et al., 2009) and the cytoskeleton (Böckers et al., 2001; Qualmann et al., 2004). Altered function of all three SHANK genes have been implicated in autism, with SHANK3 showing the highest prevalence (Moessner et al., 2007; Gauthier et al., 2009; Awadalla et al., 2010; Berkel et al., 2010, 2012; Pinto et al., 2010; Schaaf et al., 2011; Waga et al., 2011; Leblond et al., 2012, 2014; Prasad et al., 2012; Sanders et al., 2012; Sato et al., 2012; Boccuto et al., 2013; Koshimizu et al., 2013; Coe et al., 2014; De Rubeis et al., 2014; Guilmatre et al., 2014; Li et al., 2014a; Cochoy et al., 2015; Krumm et al., 2015; Nemirovsky et al., 2015; Yuen et al., 2015). In general, SHANK2 and 3 promote dendritic spine formation, whereas SHANK1 promotes dendritic spine head size enlargement (Sala et al., 2001; Roussignol et al., 2005; Hung et al., 2008; Verpelli et al., 2011). SHANK3 also binds to a synaptic scaffold, Densin-180, to inhibit the Densin-180-induced dendrite arborization (Quitsch et al., 2005). Disruption of the SHANK3 gene is associated with the 22q13.3 deletion syndrome, characterized by severe expressive-language delay and mild cognitive challenges (Bonaglia et al., 2001). Several genetically manipulated Shank3 mutant mouse models were developed to study the role of SHANK3 mutations in ASD (Bangash et al., 2011; Peça et al., 2011; Wang et al., 2011b; Jiang and Ehlers, 2013; Peixoto et al., 2016). Neurons in these mice have morphological alterations in dendritic spines resulting in LTP deficiency and defects at striatal synapses. Furthermore, these animals display several behavioral deficits including abnormal vocalization, dyadic social interaction, and compulsive-repetitive behaviors. Intriguingly, re-expressing Shank3 in adulthood is sufficient to restore parts of the autism-related phenotypes in mice (Mei et al., 2016). This study sheds light on the application of gene therapy for individuals with SHANK3 mutations. Mechanistically, several actin regulators including Abp1, cortactin, cofilin, and Rac1, have altered expression or activity associated with Shank3 deficiency or autism-related mutations that contribute to dendritic spine reduction and synaptic dystrophy (Haeckel et al., 2008; Durand et al., 2012; Duffney et al., 2015).
Disks large-associated protein 2 (DLGAP2), also known as synapse-associated protein 90/postsynaptic density-95-associated proteins (SAPAP2), is a postsynaptic adapter protein in mammalian brains (Kindler et al., 2004). DLGAP2 directly interacts with DLG4 (also known as PSD-95) and Shank proteins to form the Dlg4-Dlgap-Shanks complex important for maintaining the PSD structure (Kim et al., 1997; Takeuchi et al., 1997; Boeckers et al., 1999). The Dlgap2-/- mice have reduced spine density in the orbitofrontal cortex accompanied with downregulation of synaptic proteins, Homer1 and αCaMKII, as well as receptors, NR1 and GluR1, and exhibit exacerbated aggressive behaviors (Jiang-Xie et al., 2014). Molecular and genetic studies have demonstrated that alterations in DLGAP2 are involved the pathophysiology of various psychiatric conditions, including schizophrenia, Alzheimer’s disease, post-tramautic syndrome, and pediatric obsessive-compulsive disorder (Chertkow-Deutsher et al., 2010; Wu et al., 2013; Li et al., 2014b; Chaudhry et al., 2015). Rare de novo CNVs, deletions, and duplications of DLGAP2 have been reported in individuals with ASD, but how mutations of DLGAP2 contribute to autism is still largely unknown (Marshall et al., 2008; Ozgen et al., 2009; Chien et al., 2010; Pinto et al., 2010; Cukier et al., 2014).
Neurotransmitter release is regulated by the cycling of synaptic vesicles at the axonal terminal. The regulation of synaptic vesicles contains several steps and requires precise interaction of several specialized proteins, including SNARE complex for membrane fusion and syntaxin for vesicle docking. STXBP5 encodes a syntaxin-binding protein, tomosyn that negatively regulates neurotransmitter release by forming a syntaxin-SNAP25-tomosyn complex (Fujita et al., 1998; Sakisaka et al., 2004; Yizhar et al., 2004; Yamamoto et al., 2009, 2010; Bielopolski et al., 2014). Neuron-specific tomosyn deletion in mouse hippocampal dentate gyrus impairs spatial learning and memory, whereas tomosyn knockdown in dentate gyrus decreases synaptic plasticity of mossy fibers (Barak et al., 2013; Ben-Simon et al., 2015). Tomosyn also regulates SNARE complexes via ROCK phosphorylation of syntaxin-1 to control neurite outgrowth (Sakisaka et al., 2004). Recent genetic studies have identified the association of STXBP5 and ASD (Davis et al., 2009; Cukier et al., 2014; De Rubeis et al., 2014).
PRICKLE1 encodes PRICKLE1 protein, which has been traditionally thought to regulate the Wnt/beta-catenin signaling pathway to control epithelial planar cell polarity and cell migration during neural tube formation (Heitzler et al., 1993; Carreira-Barbosa et al., 2003; Veeman et al., 2003; Jenny et al., 2005). Intriguingly, the Prickle1+/- mice exhibit autism-like behaviors, which may result from disrupted interaction with synapsin, a regulator of neurotransmitter release, suggesting that PRICKLE1 plays a critical role in synaptic vesicle regulation (Paemka et al., 2013). In addition, knockdown of PRICKLE1 in mice results in reduced axonal and dendrite formation in hippocampal neurons (Liu et al., 2013a). More recently, variants of PRICKLE1 have been found in individuals with autism (Cukier et al., 2014; Toma et al., 2014).
Synaptic scaffolds are crucial not only to maintain the structural stability of dendritic spines and synapses but also to link the signaling molecules and receptors to efficiently act in response to certain extracellular stimuli. Mutations in these molecules may disrupt several different signaling pathways and result in wide range of cellular defects, which sometimes are not limited to ASD. In addition, couple autism-associated genes that have been shown to regulate synaptic vesicles also play roles in neurite outgrowth or synaptic plasticity thereby regulating the structural stability of neurons. An interesting direction of investigation is whether the regulation of synaptic vesicles represents one of the key vulnerable cellular pathway that contributes to the alteration of neuronal structures in ASD.
Several autism-related neurodevelopmental disorders, such as Fragile X, Rett, Angelman syndromes (AS), and tuberous sclerosis are caused by a highly penetrable mutation of a single gene, e.g., FMR1 in Fragile X (Verkerk et al., 1991; Gedeon et al., 1992), MECP2 in Rett (Amir et al., 1999), UBE3A in AS (Kishino et al., 1997), and TSC1/2 in tuberous sclerosis complex (TSC; Povey et al., 1994). In recent DMS-5 criteria, however, ASD condition has been separated out from these single gene related disorders. Interestingly, these molecules all have a major function in regulating gene expression or protein synthesis, which in turn widely affects the structural stability of neurons. Because of the comorbidity between ASD and these single gene related disorders, we also review the current understanding of these genes and discuss how alterations of these genes may impair the structural integrity of neurons.
FMRP is a RNA-binding protein encoded by FMR1 gene (Ashley et al., 1993; Siomi et al., 1993). Mutations of the FMR1 gene in humans result in CGG repeat polymorphisms or in the deletion of FMRP protein contributing to Fragile X syndrome, the most common inherited form of intellectual disability (Verkerk et al., 1991; Gedeon et al., 1992). Interestingly, people with Fragile X syndrome often exhibit autistic behaviors and mutations of FMR1 genes are also found in several cases of ASD (Vincent et al., 1996; Feinstein and Reiss, 1998; Rogers et al., 2001; Hatton et al., 2006; Chaste et al., 2012; Yu et al., 2013). The absence of FMRP is associated with widespread morphological changes of dendrites and dendritic spines in different brain regions, including cortex (Comery et al., 1997; Weiler and Greenough, 1999; Irwin et al., 2000; Greenough et al., 2001; Nimchinsky et al., 2001; Galvez et al., 2003; Galvez and Greenough, 2005; McKinney et al., 2005; Bureau et al., 2008; Cruz-Martin et al., 2010; Pan et al., 2010; Qin et al., 2011; Berman et al., 2012; Amiri et al., 2014; Wijetunge et al., 2014), hippocampus (Braun and Segal, 2000; Segal et al., 2003; Antar et al., 2006; Grossman et al., 2010; Levenga et al., 2011a; Amiri et al., 2014), and cerebellum (Koekkoek et al., 2005).
Upon mGluR-activation, the local translation of Fmr1 in dendrite and dendritic spines is crucial for maintaining dendritic structure (Antar et al., 2004; Bear et al., 2004; Tucker et al., 2006; Osterweil et al., 2010; Pop et al., 2014). The hyperactivation of mGluR5 signaling, as well as the neuronal and behavioral deficits resulting from FMRP deficiency can be corrected by application of a mGluR5 antagonists (Bear et al., 2004; Bear, 2005; Tucker et al., 2006; Dolen et al., 2007; Price et al., 2007; Wilson and Cox, 2007; Auerbach and Bear, 2010; Osterweil et al., 2010; Levenga et al., 2011b; Michalon et al., 2012, 2014; Ronesi et al., 2012; Lohith et al., 2013; Pop et al., 2014). The activation of FMRP subsequently regulates the local synthesis of several other synaptic proteins, including AMPAR, CaMKII, and PSD95, which in turn modulates the activity-dependent dynamics and plasticity of dendritic spines (Muddashetty et al., 2007; Nakamoto et al., 2007; Kao et al., 2010; Ifrim et al., 2015). In addition, the Rac-PAK pathway is upregulated and coincides with the disruptive dendritic phenotype in the absence of FMRP (Lee et al., 2003; Hayashi et al., 2007; Chen et al., 2010a; Bongmba et al., 2011). A rare deletion and several variants of cytoplasmic FMR1 interacting protein 1 (CYFIP1) are also found in cases of ASD further suggesting genes involved in the FMRP signaling pathway are prevalent risk factors (van der Zwaag et al., 2010; Leblond et al., 2012; Toma et al., 2014; Waltes et al., 2014; Wang et al., 2015). Interestingly, CYFIP1 mediates FMRP-dependent protein translation to regulate the dendritic complexity and the stability of dendritic spines (Napoli et al., 2008; De Rubeis et al., 2013; Pathania et al., 2014). Overexpression of CYFIP1 results in an increase of dendritic branching and dendritic spine density (Oguro-Ando et al., 2015). Thus, the understanding of the molecular mechanisms regulated by FMRP provides insights into how mutations of FMR1 in ASD may affect neuronal function and contribute to autistic behaviors.
Methyl-CpG binding protein 2 (MECP2) is a transcriptional factor that has multiple functions in gene regulation (Meehan et al., 1989; Nan et al., 1997, 1998; Young et al., 2005; Chahrour et al., 2008; Cheng et al., 2014). Mutations of MECP2 gene contribute to 90% of cases with Rett syndrome, which is a severe developmental disorder exhibiting autistic phenotypes (Amir et al., 1999; Shahbazian and Zoghbi, 2001; Van den Veyver and Zoghbi, 2001). Interestingly, the MECP2 duplication syndrome also exhibit phenotypes that resemble those with ASD (Ramocki et al., 2009; Peters et al., 2013; Lombardi et al., 2015). Mutations and CNVs of MECP2, including duplication of the gene, have been identified in people with ASD without the diagnoses of either Rett or MECP2 duplication syndromes (Carney et al., 2003; Zappella et al., 2003; Shibayama et al., 2004; Swanberg et al., 2009; Campos et al., 2011; Schaaf et al., 2011; Cukier et al., 2012; Hanchard et al., 2012; Lim et al., 2013; Yu et al., 2013). Most studies on MECP2 function focus on the understanding of the etiology of Rett syndrome and the MECP2 duplication syndrome. Upregulation or downregulation of MECP2 dramatically alters the dendritic and axonal architecture of neurons and significantly disrupts the connectivity of neuronal networks (Fukuda et al., 2005; Jugloff et al., 2005; Ballas et al., 2009; Belichenko et al., 2009; Chapleau et al., 2009; Kishi and Macklis, 2010; Cohen et al., 2011; Marshak et al., 2012; Nguyen et al., 2012; Stuss et al., 2012; Jiang et al., 2013a; Baj et al., 2014). Overexpression of BDNF appears to restore the dendritic defects in Mecp2-null condition suggesting a molecular mechanism regulated by MECP2 to maintain the structural stability of neurons (Zhou et al., 2006; Larimore et al., 2009; Gao et al., 2015). Synaptic plasticity is also regulated by the expression or the phosphorylation of MECP2 (Collins et al., 2004; Dani et al., 2005; Asaka et al., 2006; Moretti et al., 2006; Chao et al., 2007; Zhang et al., 2008; Li et al., 2011; Noutel et al., 2011; Blackman et al., 2012; Na et al., 2012, 2013; Qiu et al., 2012; Zhong et al., 2012; Della Sala and Pizzorusso, 2014; Deng et al., 2014; De Filippis et al., 2015). Whether a similar mechanism to Rett or MECP2 duplication syndromes is altered in ASD individuals with MECP2 mutations needs to be further evaluated.
UBE3A gene is a paternally imprinted gene located at human chromosome 15 and encodes a member of the E3 ubiquitin ligase proteins (Huibregtse et al., 1993; Albrecht et al., 1997). Because UBE3A is selectively imprinted in mature neurons, epigenetic regulation of UBE3A has been associated with several neurodevelopmental disorders (Albrecht et al., 1997; LaSalle et al., 2015). Mutations resulting in loss-of-function in the maternally expressed copy of UBE3A causes AS, a severe developmental disorder characterized by delayed development, intellectual disability, severe speech impairment, and ataxia (Kishino et al., 1997). Maternal duplication of UBE3A results in Dup15q syndrome, a developmental disorder that has many similarities with AS but also exhibits several autistic traits (Cook et al., 1997; Wang et al., 2008; Hogart et al., 2010; Smith et al., 2011; Urraca et al., 2013; Al Ageeli et al., 2014; Germain et al., 2014). Coincidently, several genome-wide studies from individuals with autism identify UBE3A as an autism-risk gene (Nurmi et al., 2001; Glessner et al., 2009; Schaaf et al., 2011; Kelleher et al., 2012; Carvill et al., 2013; Iossifov et al., 2014; Yuen et al., 2015). In addition to its function of ubiquitin ligase to catalyze the protein degradation step, UBE3A also can act as a transcriptional coactivator for the nuclear hormone receptor superfamily of transcription factors (Nawaz et al., 1999). UBE3A localizes both in the nucleus and cytosol, including dendrite and pre- and post-synaptic compartments in neurons to regulate dendrite and dendritic spine morphology (Dindot et al., 2008; Valluy et al., 2015). Although maternal deletion of Ube3a does not affect dendrite arborization in mouse brains, knockdown of UBE3A in cultured neurons results in defects of dendrite polarization in pyramidal neurons (Dindot et al., 2008; Miao et al., 2013). Maternal-deficiency of Ube3a in mouse brain, however, shows defects in dendritic spine development in the cortex, hippocampus, and cerebellum (Dindot et al., 2008; Kim et al., 2016). Furthermore, several neuronal substrates for UBE3A have been identified, including Arc (Greer et al., 2010), the Rho-GEF Pbl/ECT2 (Reiter et al., 2006), Ephexin5 (Margolis et al., 2010), and TSC2 (Zheng et al., 2008). Their regulation by UBE3A provides molecular mechanisms to explain how synaptic integrity is maintained and how alteration of this interaction contributes in part to neuronal phenotypes in neurodevelopmental disorders. In addition, a recent study demonstrates that a PKA phosphorylation-defective mutation on UBE3A found in an individual with autism resulted in an increase of dendritic spine density (Yi et al., 2015).
TSC1 and TSC2 genes encode protein harmartin and tuberin, respectively, and they bind and function together (European Chromosome 16 Tuberous Sclerosis Consortium, 1993; van Slegtenhorst et al., 1997; van Slegtenhorst et al., 1998). Mutations of TSC1 or TSC2 genes cause an autosomal dominant disorder TSC, which is characterized by hamartomas in various organs (Povey et al., 1994; Crino et al., 2006). Some patients with TSC exhibit several neurological problems including autism (Smalley, 1998; Bolton, 2004). Similarly, mutations of TSC1 or TSC2 are also found in several ASD cases (Smalley, 1998; Serajee et al., 2003; Schaaf et al., 2011; Kelleher et al., 2012; O’Roak et al., 2012b; Koshimizu et al., 2013; Brett et al., 2014; Toma et al., 2014; Krumm et al., 2015). TSC1/2 act upstream as to suppress the mammalian target of rapamycin (mTOR) pathway and mTOR inhibitors have been promising therapeutic agents to ameliorate some symptoms in TSC (Gao et al., 2002; Meikle et al., 2008; Curatolo et al., 2016). One of the mTOR inhibitors, rapamycin, has also been shown to correct the autistic-like synaptic pruning deficits in Tsc2+/- mice (Tang et al., 2014). In addition to the mTOR pathway being the key downstream target for TSC1/2, LIMK-cofilin pathway is altered upon deletion of TSC1 or TSC2 and enlargement of somas and dendritic spines are observed (Tavazoie et al., 2005). The Tsc2 mutations result in a loss of Purkinje cells and Tsc1 mutant mice have increased dendritic spine density, which correlates with cerebellar dysfunction and several autistic-like behaviors in animals (Tsai et al., 2012b; Reith et al., 2013). TSC1/2 also negatively regulates neurite and axonal outgrowth (Floricel et al., 2007; Choi et al., 2008). It has yet to be determined whether TSC1/2 mutations in ASD correlate with perturbations of neuronal structures.
Diagnosis of ASD cases has risen dramatically in recent years. The increased awareness of the symptoms and the broader definition of the spectrum may be major contributing factors for the rising number of ASD cases. Thus, there is an increased interest on understanding the etiologies of ASD. It is widely accepted that the genetic component plays a major role in ASD, however, except for the direct inheritance of some syndromic conditions, it is difficult to identify risk factors for autism. It is possible due to the low sample size and the high heterogeneity of genetic variances to have sufficient statistical power to make conclusive correlations (Geschwind and State, 2015). Among those autism-risk genes identified to date, some of the autism associations are due to de novo mutations, and some are familial variants (Table 4). Whether the inheritance pattern exhibits a risk factor is still not clear, however, the diverse gene mutations found in different individuals with autism suggest that instead of focusing on the genes per se, identifying the vulnerable pathways that these genes regulate may provide better clues toward understanding the contributing cellular and molecular changes that reserve in the autism phenotypes. The cellular defects resulting from different combinations of gene mutations contribute to the diverse phenotypes observed in autism. The heterogeneity of symptoms in ASD further complicates the diagnosis and treatment. However, understanding how autism-associated genes function in the regulation of key cellular pathways will provide insights to how therapeutic intervention can be more targeted and efficient to treat affected individuals.

TABLE 4. Inheritance pattern of autism-risk genes that regulate the structural stability of neurons.
Neuroanatomical studies of individuals with autism suggest a common disruption of neuronal structures with a decrease of dendrite arborization but an increase of dendritic spine density in select brain regions (Raymond et al., 1996; Bauman and Kemper, 2005; Hutsler and Zhang, 2010; Tang et al., 2014). This feature is distinct from other neurodevelopmental disorders, such as Rett or Fragile X syndromes, where the dendrite arbors and dendritic spine density are both downregulated (Kulkarni and Firestein, 2012). Intriguingly, dendrite arborization completes prior to dendritic spine formation during development. Although it has been proposed that the pruning mechanism of dendritic spines is defective in ASD (Frith, 2003), it is also plausible that the increase of dendritic spine density may be a compensation to re-establish the sufficient quantity of connections with fewer dendrite arbors. However, the precise spatial arborization of dendrites is critical for correct pre- and post-synaptic innervation when establishing the brain circuitry. The local increase of dendritic spine density may not be sufficient to compensate the effect from the loss of dendrite arbors, and may instead result in abnormal synaptic activity to disrupt normal neuronal function. This further emphasizes the importance of the establishment of structural integrity for neurons in order to provide proper brain function. In addition, the mechanisms of action of many current pharmacological agents for treating ASD affect normal neuronal function including the structural stability of neurons. With the early onset of ASD, the treatment often occurs at a very young age when the brain is still undergoing the period of development and maturation. As these pharmacological treatments may be beneficial to ameliorate some symptoms in ASD, the general brain development of these individuals may also be affected (Penagarikano, 2015). Thus, more precise circuitry-specific therapeutic intervention is needed to reduce the unwanted effect to the developing brain. Understanding the genetic and cellular pathways affected in ASD should provide more selective candidates for developing targeted intervention.
Although the list of autism-associated genes that regulate neuronal structures is extensive, it stands out that several genes actually function in the same signaling pathways (Table 5). For example, the mGluR5 pathway is disrupted when Shank3 (Verpelli et al., 2011), Fmr1 (Giuffrida et al., 2005; Dolen et al., 2007; Price et al., 2007; Wilson and Cox, 2007; Hays et al., 2011; Ronesi et al., 2012), Ube3a (Pignatelli et al., 2014), or Mecp2 (Zhong et al., 2012) gene is altered. Application of mGluR5 antagonists has been shown to be promising to restore some phenotypes experimentally in neurons or animals with Fmr1 mutants (Price et al., 2007; Wilson and Cox, 2007; Levenga et al., 2011b; Michalon et al., 2012, 2014; Ronesi et al., 2012; Pop et al., 2014). Whether targeting mGluR5 pathway can be clinically effective for ASD with mutations beyond FMR1 will require further investigation. The mTOR pathway is defective when Pten (Jaworski et al., 2005; Kwon et al., 2006; Zhou et al., 2009; Pun et al., 2012), Tsc1/2 (Gao et al., 2002; Meikle et al., 2008; Curatolo et al., 2016), or Mecp2 (Ricciardi et al., 2011) gene is mutated. However, the mTOR pathway is upregulated in mice carrying defective gene products of Pten. Tsc1/2, or Fmr1, but downregulated in Mecp2-null mice. The mTOR inhibitor, rapamycin, has been shown to be effective to rescue some phenotypes caused by these mutations and has been used as a therapeutic agent to treat some of the autism symptoms (Meikle et al., 2008; Ehninger and Silva, 2011; Curatolo et al., 2016). The alteration of IGF-1/GSK3β pathway is implicated in Pten (Kwon et al., 2006), Cdk5l (Fuchs et al., 2014) or Mecp2 (Itoh et al., 2007) mutant animals. Inhibition of GSK3β or application of IGF-1 can rescue the dendritic phenotype in Cdk5l and Mecp2 mutant mice (Tropea et al., 2009; Fuchs et al., 2015; Della Sala et al., 2016). The mTOR pathway and the GSK3β pathway can be further linked together as they are both regulated by AKT. In addition, Rac1 activity is altered when Elmo1 (Grimsley et al., 2004), Cdk5l (Chen et al., 2010b), Shank3 (Duffney et al., 2015), or Fmr1 (Lee et al., 2003; Chen et al., 2010a; Bongmba et al., 2011) is mutated suggesting its crucial role to maintain the stability of neurons. The potential therapeutic approach targeting the Rac1 pathway to rescue the neuronal and behavioral phenotype in mutant animals is actively under investigation (Hayashi et al., 2007; Duffney et al., 2015).

TABLE 5. The common signaling pathways that are altered by mutations of autism-risk genes.
To date, several animal studies have tried to model the behavior phenotypes in autism, however, there remains a debate as to whether rodents can sufficiently recapitulate the complexities of the condition in human. The heterogeneity of genetic components also make it difficult to establish reliable animal models to describe the cellular and molecular mechanistic alterations in specific pathways. However, the studies on rodents can suggest which brain circuitry should be the area of interest for the corresponding behavior. iPSCs derived from ASD individuals appear to be an attractive model systems that allow researchers to directly investigate the interaction between the genetic contribution and the autism-relevant phenotypes. However, what is lacking in this system is a physiological relevant environment to correlate the behavior and the cellular phenotype. A recent emerging genetic editing technique, CRISPR/Cas9 (Jinek et al., 2012), is a powerful tool to study the mechanistic questions and identify the potential therapeutic interventions. Unlike the traditional knock-in or knock-out technique, CRISPR/Cas9 can introduce genomic editing of several genes at once. Using CRISPR/Cas9 in iPSCs can potentially determine the genetic contribution to the cellular phenotypes and provide a mechanism to correct them. However, there is still room for improvement of the efficiency and precision before this technique can be reliably used in clinical applications. Combining animal studies, iPSC models, and gene editing techniques, it is now possible to perform more comprehensive translational research in order to better understand the etiologies of ASD and design more efficient and effective therapeutic interventions.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
This work is supported by Hussman Foundation grant HIAS15003 to Y-CL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Anthony Koleske from Yale University and Dr. John Hussman for critical evaluation of the manuscript.
Aceti, M., Creson, T. K., Vaissiere, T., Rojas, C., Huang, W. C., Wang, Y. X., et al. (2015). Syngap1 haploinsufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol. Psychiatry 77, 805–815. doi: 10.1016/j.biopsych.2014.08.001
Akashi, K., Kakizaki, T., Kamiya, H., Fukaya, M., Yamasaki, M., Abe, M., et al. (2009). NMDA receptor GluN2B (GluR epsilon 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J. Neurosci. 29, 10869–10882. doi: 10.1523/JNEUROSCI.5531-08.2009
Al Ageeli, E., Drunat, S., Delanoe, C., Perrin, L., Baumann, C., Capri, Y., et al. (2014). Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases. Eur. J. Med. Genet. 57, 5–14. doi: 10.1016/j.ejmg.2013.10.008
Alarcón, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Albrecht, U., Sutcliffe, J. S., Cattanach, B. M., Beechey, C. V., Armstrong, D., Eichele, G., et al. (1997). Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 17, 75–78. doi: 10.1038/ng0997-75
Aller, M. I., Pecoraro, V., Paternain, A. V., Canals, S., and Lerma, J. (2015). Increased dosage of high-affinity kainate receptor gene grik4 Alters synaptic transmission and reproduces autism spectrum disorders features. J. Neurosci. 35, 13619–13628. doi: 10.1523/JNEUROSCI.2217-15.2015
Altafaj, X., Dierssen, M., Baamonde, C., Marti, E., Visa, J., Guimera, J., et al. (2001). Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum. Mol. Genet. 10, 1915–1923. doi: 10.1093/hmg/10.18.1915
Amendola, E., Zhan, Y., Mattucci, C., Castroflorio, E., Calcagno, E., Fuchs, C., et al. (2014). Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS ONE 9:e91613. doi: 10.1371/journal.pone.0091613
American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Arlington, VA: American Psychiatric Publishing.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Amiri, A., Sanchez-Ortiz, E., Cho, W., Birnbaum, S. G., Xu, J., McKay, R. M., et al. (2014). Analysis of FMR1 deletion in a subpopulation of post-mitotic neurons in mouse cortex and hippocampus. Autism Res. 7, 60–71. doi: 10.1002/aur.1342
Ammendrup-Johnsen, I., Naito, Y., Craig, A. M., and Takahashi, H. (2015). Neurotrophin-3 enhances the synaptic organizing function of TrkC-Protein tyrosine phosphatase sigma in rat hippocampal neurons. J. Neurosci. 35, 12425–12431. doi: 10.1523/JNEUROSCI.1330-15.2015
An, J. Y., Cristino, A. S., Zhao, Q., Edson, J., Williams, S. M., Ravine, D., et al. (2014). Towards a molecular characterization of autism spectrum disorders: an exome sequencing and systems approach. Transl. Psychiatry 4, 1–9. doi: 10.1038/tp.2014.38
Anderson, G. R., Galfin, T., Xu, W., Aoto, J., Malenka, R. C., and Sudhof, T. C. (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. U.S.A. 109, 18120–18125. doi: 10.1073/pnas.1216398109
Anney, R., Klei, L., Pinto, D., Almeida, J., Bacchelli, E., Baird, G., et al. (2012). Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Hum. Mol. Genet. 21, 4781–4792. doi: 10.1093/hmg/dds301
Antar, L. N., Afroz, R., Dictenberg, J. B., Carroll, R. C., and Bassell, G. J. (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 24, 2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004
Antar, L. N., Li, C., Zhang, H., Carroll, R. C., and Bassell, G. J. (2006). Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 32, 37–48. doi: 10.1016/j.mcn.2006.02.001
Antoine-Bertrand, J., Villemure, J. F., and Lamarche-Vane, N. (2011). Implication of rho GTPases in neurodegenerative diseases. Curr. Drug Targets 12, 1202–1215. doi: 10.2174/138945011795906543
Archer, H. L., Evans, J., Edwards, S., Colley, J., Newbury-Ecob, R., O’Callaghan, F., et al. (2006). CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J. Med. Genet. 43, 729–734. doi: 10.1136/jmg.2006.041467
Arikkath, J., and Reichardt, L. F. (2008). Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 31, 487–494. doi: 10.1016/j.tins.2008.07.001
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164. doi: 10.1016/j.ajhg.2007.09.015
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227. doi: 10.1016/j.nbd.2005.07.005
Ashley, C. T. Jr., Wilkinson, K. D., Reines, D., and Warren, S. T. (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. doi: 10.1126/science.7692601
Auerbach, B. D., and Bear, M. F. (2010). Loss of the fragile X mental retardation protein decouples metabotropic glutamate receptor dependent priming of long-term potentiation from protein synthesis. J. Neurophysiol. 104, 1047–1051. doi: 10.1152/jn.00449.2010
Autism Genome Project Consortium, Szatmari, P., Paterson, A. D., Zwaigenbaum, L., Roberts, W., Brian, J., et al. (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328. doi: 10.1038/ng1985
Avdjieva-Tzavella, D. M., Todorov, T. P., Todorova, A. P., Kirov, A. V., Hadjidekova, S. P., Rukova, B. B., et al. (2012). Analysis of the genes encoding neuroligins NLGN3 and NLGN4 in Bulgarian patients with autism. Genet. Couns. 23, 505–511.
Awadalla, P., Gauthier, J., Myers, R. A., Casals, F., Hamdan, F. F., Griffing, A. R., et al. (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet. 87, 316–324. doi: 10.1016/j.ajhg.2010.07.019
Bahi-Buisson, N., and Bienvenu, T. (2012). CDKL5-related disorders: from clinical description to molecular genetics. Mol. Syndromol. 2, 137–152.
Baio, J. (2014). Prevalence of autism spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 sites, united states, 2010. Surveill. Summ. 63, 1–21.
Baj, G., Patrizio, A., Montalbano, A., Sciancalepore, M., and Tongiorgi, E. (2014). Developmental and maintenance defects in Rett syndrome neurons identified by a new mouse staging system in vitro. Front. Cell. Neurosci. 8:18. doi: 10.3389/fncel.2014.00018
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 145, 758–772. doi: 10.1016/j.cell.2011.03.052
Barak, B., Okun, E., Ben-Simon, Y., Lavi, A., Shapira, R., Madar, R., et al. (2013). Neuron-specific expression of tomosyn1 in the mouse hippocampal dentate gyrus impairs spatial learning and memory. Neuromol. Med. 15, 351–363. doi: 10.1007/s12017-013-8223-4
Baron, M. K., Boeckers, T. M., Vaida, B., Faham, S., Gingery, M., Sawaya, M. R., et al. (2006). An architectural framework that may lie at the core of the postsynaptic density. Science 311, 531–535. doi: 10.1126/science.1118995
Barrow, S. L., Constable, J. R., Clark, E., El-Sabeawy, F., McAllister, A. K., and Washbourne, P. (2009). Neuroligin1: a cell adhesion molecule that recruits PSD-95 and NMDA receptors by distinct mechanisms during synaptogenesis. Neural Dev. 4:17. doi: 10.1186/1749-8104-4-17
Bartnik, M., Szczepanik, E., Derwinska, K., Wisniowiecka-Kowalnik, B., Gambin, T., Sykulski, M., et al. (2012). Application of array comparative genomic hybridization in 102 patients with epilepsy and additional neurodevelopmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 760–771. doi: 10.1002/ajmg.b.32081
Bassan, M., Zamostiano, R., Davidson, A., Pinhasov, A., Giladi, E., Perl, O., et al. (1999). Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J. Neurochem. 72, 1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x
Basu, R., Taylor, M. R., and Williams, M. E. (2015). The classic cadherins in synaptic specificity. Cell Adh. Migr. 9, 193–201. doi: 10.1080/19336918.2014.1000072
Bauman, M. L., and Kemper, T. L. (2005). Neuroanatomic observations of the brain in autism: a review and future directions. Int. J. Dev. Neurosci. 23, 183–187. doi: 10.1016/j.ijdevneu.2004.09.006
Bear, M. F. (2005). Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav. 4, 393–398. doi: 10.1111/j.1601-183X.2005.00135.x
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Beaudoin, B. S., Hill, J. M., and Ming, S. X. (2007). Brief report: the impact of subcortical band heterotopia and associated complications on the neuropsychological functioning of a 13-year-old child. J. Autism. Dev. Disord. 37, 983–992. doi: 10.1007/s10803-006-0236-5
Bekirov, I. H., Nagy, V., Svoronos, A., Huntley, G. W., and Benson, D. L. (2008). Cadherin-8 and N-cadherin differentially regulate pre- and postsynaptic development of the hippocampal mossy fiber pathway. Hippocampus 18, 349–363. doi: 10.1002/hipo.20395
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., et al. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258. doi: 10.1002/cne.22009
Bena, F., Bruno, D. L., Eriksson, M., van Ravenswaaij-Arts, C., Stark, Z., Dijkhuizen, T., et al. (2013). Molecular and clinical characterization of 25 individuals with exonic deletions of NRXN1 and comprehensive review of the literature. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 388–403. doi: 10.1002/ajmg.b.32148
Benavides-Piccione, R., Dierssen, M., Ballesteros-Yanez, I., Martinez de Lagran, M., Arbones, M. L., Fotaki, V., et al. (2005). Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/- mouse. Neurobiol. Dis. 20, 115–122. doi: 10.1016/j.nbd.2005.02.004
Ben-David, E., and Shifman, S. (2013). Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol. Psychiatry 18, 1054–1056. doi: 10.1038/mp.2012.148
Ben-Simon, Y., Rodenas-Ruano, A., Alvina, K., Lam, A. D., Stuenkel, E. L., Castillo, P. E., et al. (2015). A combined optogenetic-knockdown strategy reveals a major role of tomosyn in mossy fiber synaptic plasticity. Cell Rep. 12, 396–404. doi: 10.1016/j.celrep.2015.06.037
Benson, D. L., and Tanaka, H. (1998). N-cadherin redistribution during synaptogenesis in hippocampal neurons. J. Neurosci. 18, 6892–6904.
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Berkel, S., Tang, W., Trevino, M., Vogt, M., Obenhaus, H. A., Gass, P., et al. (2012). Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum. Mol. Genet. 21, 344–357. doi: 10.1093/hmg/ddr470
Berman, R. F., Murray, K. D., Arque, G., Hunsaker, M. R., and Wenzel, H. J. (2012). Abnormal dendrite and spine morphology in primary visual cortex in the CGG knock-in mouse model of the fragile X premutation. Epilepsia 53(Suppl. 1), 150–160. doi: 10.1111/j.1528-1167.2012.03486.x
Bernardinelli, Y., Nikonenko, I., and Muller, D. (2014). Structural plasticity: mechanisms and contribution to developmental psychiatric disorders. Front. Neuroanat. 8:123. doi: 10.3389/fnana.2014.00123
Berryer, M. H., Hamdan, F. F., Klitten, L. L., Moller, R. S., Carmant, L., Schwartzentruber, J., et al. (2013). Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 34, 385–394. doi: 10.1002/humu.22248
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412. doi: 10.1016/j.tins.2009.04.003
Bielopolski, N., Lam, A. D., Bar-On, D., Sauer, M., Stuenkel, E. L., and Ashery, U. (2014). Differential interaction of tomosyn with syntaxin and SNAP25 depends on domains in the WD40 beta-propeller core and determines its inhibitory activity. J. Biol. Chem. 289, 17087–17099. doi: 10.1074/jbc.M113.515296
Blackman, M. P., Djukic, B., Nelson, S. B., and Turrigiano, G. G. (2012). A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J. Neurosci. 32, 13529–13536. doi: 10.1523/JNEUROSCI.3077-12.2012
Blasi, F., Bacchelli, E., Pesaresi, G., Carone, S., Bailey, A. J., Maestrini, E., et al. (2006). Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 220–221. doi: 10.1002/ajmg.b.30287
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., and Bauman, M. L. (2001). Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J. Autism. Dev. Disord. 31, 537–543. doi: 10.1023/A:1013238809666
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Böckers, T. M., Mameza, M. G., Kreutz, M. R., Bockmann, J., Weise, C., Buck, F., et al. (2001). Synaptic scaffolding proteins in rat brain. Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein alpha-fodrin. J. Biol. Chem. 276, 40104–40112. doi: 10.1074/jbc.M102454200
Boeckers, T. M., Winter, C., Smalla, K. H., Kreutz, M. R., Bockmann, J., Seidenbecher, C., et al. (1999). Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem. Biophys. Res. Commun. 264, 247–252. doi: 10.1006/bbrc.1999.1489
Bolton, P. F. (2004). Neuroepileptic correlates of autistic symptomatology in tuberous sclerosis. Ment. Retard. Dev. Disabil. Res. Rev. 10, 126–131. doi: 10.1002/mrdd.20024
Bonaglia, M. C., Giorda, R., Borgatti, R., Felisari, G., Gagliardi, C., Selicorni, A., et al. (2001). Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 69, 261–268. doi: 10.1086/321293
Bongmba, O. Y., Martinez, L. A., Elhardt, M. E., Butler, K., and Tejada-Simon, M. V. (2011). Modulation of dendritic spines and synaptic function by Rac1: a possible link to Fragile X syndrome pathology. Brain Res. 1399, 79–95. doi: 10.1016/j.brainres.2011.05.020
Borisovska, M., McGinley, M. J., Bensen, A., and Westbrook, G. L. (2011). Loss of olfactory cell adhesion molecule reduces the synchrony of mitral cell activity in olfactory glomeruli. J. Physiol. 589(Pt 8), 1927–1941. doi: 10.1113/jphysiol.2011.206276
Bourgeron, T. (2009). A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. doi: 10.1016/j.conb.2009.06.003
Bozdagi, O., Shan, W., Tanaka, H., Benson, D. L., and Huntley, G. W. (2000). Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28, 245–259. doi: 10.1016/S0896-6273(00)00100-8
Bozdagi, O., Wang, X. B., Nikitczuk, J. S., Anderson, T. R., Bloss, E. B., Radice, G. L., et al. (2010). Persistence of coordinated long-term potentiation and dendritic spine enlargement at mature hippocampal CA1 synapses requires N-cadherin. J. Neurosci. 30, 9984–9989. doi: 10.1523/JNEUROSCI.1223-10.2010
Braun, K., and Segal, M. (2000). FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb. Cortex 10, 1045–1052. doi: 10.1093/cercor/10.10.1045
Brett, M., McPherson, J., Zang, Z. J., Lai, A., Tan, E. S., Ng, I., et al. (2014). Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS ONE 9:e93409. doi: 10.1371/journal.pone.0093409
Brigman, J. L., Wright, T., Talani, G., Prasad-Mulcare, S., Jinde, S., Seabold, G. K., et al. (2010). Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J. Neurosci. 30, 4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010
Bronicki, L. M., Redin, C., Drunat, S., Piton, A., Lyons, M., Passemard, S., et al. (2015). Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. Eur. J. Hum. Genet. 23, 1482–1487. doi: 10.1038/ejhg.2015.29
Brugnera, E., Haney, L., Grimsley, C., Lu, M., Walk, S. F., Tosello-Trampont, A. C., et al. (2002). Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 4, 574–582. doi: 10.1038/ncb824
Bucan, M., Abrahams, B. S., Wang, K., Glessner, J. T., Herman, E. I., Sonnenblick, L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5:e1000536. doi: 10.1371/journal.pgen.1000536
Buller, A. L., Larson, H. C., Schneider, B. E., Beaton, J. A., Morrisett, R. A., and Monaghan, D. T. (1994). The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J. Neurosci. 14, 5471–5484.
Bureau, I., Shepherd, G. M., and Svoboda, K. (2008). Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J. Neurosci. 28, 5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008
Busa, T., Chabrol, B., Perret, O., Longy, M., and Philip, N. (2013). Novel PTEN germline mutation in a family with mild phenotype: difficulties in genetic counseling. Gene 512, 194–197. doi: 10.1016/j.gene.2012.09.134
Bustos, F. J., Varela-Nallar, L., Campos, M., Henriquez, B., Phillips, M., Opazo, C., et al. (2014). PSD95 suppresses dendritic arbor development in mature hippocampal neurons by occluding the clustering of NR2B-NMDA receptors. PLoS ONE 9:e94037. doi: 10.1371/journal.pone.0094037
Butler, M. G., Dasouki, M. J., Zhou, X. P., Talebizadeh, Z., Brown, M., Takahashi, T. N., et al. (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 42, 318–321. doi: 10.1136/jmg.2004.024646
Butz, S., Okamoto, M., and Südhof, T. C. (1998). A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94, 773–782. doi: 10.1016/S0092-8674(00)81736-5
Buxbaum, J. D., Bolshakova, N., Brownfeld, J. M., Anney, R. J., Bender, P., Bernier, R., et al. (2014). The autism simplex collection: an international, expertly phenotyped autism sample for genetic and phenotypic analyses. Mol Autism 5:34. doi: 10.1186/2040-2392-5-34
Buxbaum, J. D., Cai, G., Chaste, P., Nygren, G., Goldsmith, J., Reichert, J., et al. (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 484–491. doi: 10.1002/ajmg.b.30493
Buxbaum, J. D., Daly, M. J., Devlin, B., Lehner, T., Roeder, K., State, M. W., et al. (2012). The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 76, 1052–1056. doi: 10.1016/j.neuron.2012.12.008
Camacho, A., Simon, R., Sanz, R., Vinuela, A., Martinez-Salio, A., and Mateos, F. (2012). Cognitive and behavioral profile in females with epilepsy with PDCH19 mutation: two novel mutations and review of the literature. Epilepsy Behav. 24, 134–137. doi: 10.1016/j.yebeh.2012.02.023
Camacho-Garcia, R. J., Planelles, M. I., Margalef, M., Pecero, M. L., Martinez-Leal, R., Aguilera, F., et al. (2012). Mutations affecting synaptic levels of neurexin-1beta in autism and mental retardation. Neurobiol. Dis. 47, 135–143. doi: 10.1016/j.nbd.2012.03.031
Campos, M. Jr., Pestana, C. P., dos Santos, A. V., Ponchel, F., Churchman, S., Abdalla-Carvalho, C. B., et al. (2011). A MECP2 missense mutation within the MBD domain in a Brazilian male with autistic disorder. Brain Dev. 33, 807–809. doi: 10.1016/j.braindev.2011.04.015
Carlisle, H. J., Manzerra, P., Marcora, E., and Kennedy, M. B. (2008). SynGAP regulates steady-state and activity-dependent phosphorylation of cofilin. J. Neurosci. 28, 13673–13683. doi: 10.1523/JNEUROSCI.4695-08.2008
Carney, R. M., Wolpert, C. M., Ravan, S. A., Shahbazian, M., Ashley-Koch, A., Cuccaro, M. L., et al. (2003). Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr. Neurol. 28, 205–211. doi: 10.1016/S0887-8994(02)00624-0
Carreira-Barbosa, F., Concha, M. L., Takeuchi, M., Ueno, N., Wilson, S. W., and Tada, M. (2003). Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development 130, 4037–4046. doi: 10.1242/dev.00567
Carta, M., Fievre, S., Gorlewicz, A., and Mulle, C. (2014). Kainate receptors in the hippocampus. Eur. J. Neurosci. 39, 1835–1844. doi: 10.1111/ejn.12590
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O’Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi: 10.1038/ng.2646
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chakrabarti, B., Dudbridge, F., Kent, L., Wheelwright, S., Hill-Cawthorne, G., Allison, C., et al. (2009). Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2, 157–177. doi: 10.1002/aur.80
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., et al. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233. doi: 10.1016/j.nbd.2009.05.001
Chapman, N. H., Estes, A., Munson, J., Bernier, R., Webb, S. J., Rothstein, J. H., et al. (2011). Genome-scan for IQ discrepancy in autism: evidence for loci on chromosomes 10 and 16. Hum. Genet. 129, 59–70. doi: 10.1007/s00439-010-0899-z
Chaste, P., Betancur, C., Gerard-Blanluet, M., Bargiacchi, A., Kuzbari, S., Drunat, S., et al. (2012). High-functioning autism spectrum disorder and fragile X syndrome: report of two affected sisters. Mol. Autism 3:5. doi: 10.1186/2040-2392-3-5
Chaudhry, M., Wang, X., Bamne, M. N., Hasnain, S., Demirci, F. Y., Lopez, O. L., et al. (2015). Genetic variation in imprinted genes is associated with risk of late-onset Alzheimer’s disease. J. Alzheimers. Dis. 44, 989–994. doi: 10.3233/JAD-142106
Chen, B. S., Thomas, E. V., Sanz-Clemente, A., and Roche, K. W. (2011). NMDA receptor-dependent regulation of dendritic spine morphology by SAP102 splice variants. J. Neurosci. 31, 89–96. doi: 10.1523/JNEUROSCI.1034-10.2011
Chen, C. K., Bregere, C., Paluch, J., Lu, J. F., Dickman, D. K., and Chang, K. T. (2014a). Activity-dependent facilitation of Synaptojanin and synaptic vesicle recycling by the Minibrain kinase. Nat. Commun. 5:4246. doi: 10.1038/ncomms5246
Chen, H. J., Rojas-Soto, M., Oguni, A., and Kennedy, M. B. (1998). A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20, 895–904. doi: 10.1016/S0896-6273(00)80471-7
Chen, J., Yu, S., Fu, Y., and Li, X. (2014b). Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 8:276. doi: 10.3389/fncel.2014.00276
Chen, L. Y., Rex, C. S., Babayan, A. H., Kramar, E. A., Lynch, G., Gall, C. M., et al. (2010a). Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J. Neurosci. 30, 10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010
Chen, Q., Zhu, Y. C., Yu, J., Miao, S., Zheng, J., Xu, L., et al. (2010b). CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 30, 12777–12786. doi: 10.1523/JNEUROSCI.1102-10.2010
Chen, Y. K., Chen, C. Y., Hu, H. T., and Hsueh, Y. P. (2012). CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis and synaptic distribution of the striatin-PP2A complex. Mol. Biol. Cell 23, 4383–4392. doi: 10.1091/mbc.E12-05-0365
Chen, Y. K., and Hsueh, Y. P. (2012). Cortactin-binding protein 2 modulates the mobility of cortactin and regulates dendritic spine formation and maintenance. J. Neurosci. 32, 1043–1055. doi: 10.1523/JNEUROSCI.4405-11.2012
Cheng, T. L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W. H., Xu, W., et al. (2014). MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 28, 547–560. doi: 10.1016/j.devcel.2014.01.032
Chertkow-Deutsher, Y., Cohen, H., Klein, E., and Ben-Shachar, D. (2010). DNA methylation in vulnerability to post-traumatic stress in rats: evidence for the role of the post-synaptic density protein Dlgap2. Int. J. Neuropsychopharmacol. 13, 347–359. doi: 10.1017/S146114570999071X
Cheung, J., Petek, E., Nakabayashi, K., Tsui, L. C., Vincent, J. B., and Scherer, S. W. (2001). Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics 78, 7–11. doi: 10.1006/geno.2001.6651
Chien, W. H., Gau, S. S., Wu, Y. Y., Huang, Y. S., Fang, J. S., Chen, Y. J., et al. (2010). Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin. Genet. 78, 449–456. doi: 10.1111/j.1399-0004.2010.01395.x
Chih, B., Engelman, H., and Scheiffele, P. (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328. doi: 10.1126/science.1107470
Ching, M. S., Shen, Y., Tan, W. H., Jeste, S. S., Morrow, E. M., Chen, X., et al. (2010). Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 937–947. doi: 10.1002/ajmg.b.31063
Chiocchetti, A. G., Kopp, M., Waltes, R., Haslinger, D., Duketis, E., Jarczok, T. A., et al. (2015). Variants of the CNTNAP2 5’ promoter as risk factors for autism spectrum disorders: a genetic and functional approach. Mol. Psychiatry 20, 839–849. doi: 10.1038/mp.2014.103
Choi, Y. J., Di Nardo, A., Kramvis, I., Meikle, L., Kwiatkowski, D. J., Sahin, M., et al. (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev. 22, 2485–2495. doi: 10.1101/gad.1685008
Christian, S. L., Brune, C. W., Sudi, J., Kumar, R. A., Liu, S., Karamohamed, S., et al. (2008). Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol. Psychiatry 63, 1111–1117. doi: 10.1016/j.biopsych.2008.01.009
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R., et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Ciani, L., Patel, A., Allen, N. D., and ffrench-Constant, C. (2003). Mice lacking the giant protocadherin mFAT1 exhibit renal slit junction abnormalities and a partially penetrant cyclopia and anophthalmia phenotype. Mol. Cell. Biol. 23, 3575–3582. doi: 10.1128/MCB.23.10.3575-3582.2003
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723. doi: 10.1016/j.cell.2012.08.045
Clement, J. P., Ozkan, E. D., Aceti, M., Miller, C. A., and Rumbaugh, G. (2013). SYNGAP1 links the maturation rate of excitatory synapses to the duration of critical-period synaptic plasticity. J. Neurosci. 33, 10447–10452. doi: 10.1523/JNEUROSCI.0765-13.2013
Cline, H. T. (2001). Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 11, 118–126. doi: 10.1016/S0959-4388(00)00182-3
Cochoy, D. M., Kolevzon, A., Kajiwara, Y., Schoen, M., Pascual-Lucas, M., Lurie, S., et al. (2015). Phenotypic and functional analysis of SHANK3 stop mutations identified in individuals with ASD and/or ID. Mol. Autism 6:23. doi: 10.1186/s13229-015-0020-5
Codina-Sola, M., Rodriguez-Santiago, B., Homs, A., Santoyo, J., Rigau, M., Aznar-Lain, G., et al. (2015). Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol. Autism 6:21. doi: 10.1186/s13229-015-0017-0
Coe, B. P., Witherspoon, K., Rosenfeld, J. A., van Bon, B. W., Vulto-van Silfhout, A. T., Bosco, P., et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 46, 1063–1071. doi: 10.1038/ng.3092
Cohen, S., Gabel, H. W., Hemberg, M., Hutchinson, A. N., Sadacca, L. A., Ebert, D. H., et al. (2011). Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85. doi: 10.1016/j.neuron.2011.08.022
Coleman, P. D., and Riesen, A. H. (1968). Evironmental effects on cortical dendritic fields. I. Rearing in the dark. J. Anat. 102(Pt 3), 363–374.
Coles, C. H., Mitakidis, N., Zhang, P., Elegheert, J., Lu, W., Stoker, A. W., et al. (2014). Structural basis for extracellular cis and trans RPTPsigma signal competition in synaptogenesis. Nat. Commun. 5:5209. doi: 10.1038/ncomms6209
Collingridge, G. L., Olsen, R. W., Peters, J., and Spedding, M. (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology 56, 2–5. doi: 10.1016/j.neuropharm.2008.06.063
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., et al. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689. doi: 10.1093/hmg/ddh282
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. U.S.A. 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Connolly, J. J., Glessner, J. T., and Hakonarson, H. (2013). A genome-wide association study of autism incorporating autism diagnostic interview-revised, autism diagnostic observation schedule, and social responsiveness scale. Child Dev. 84, 17–33. doi: 10.1111/j.1467-8624.2012.01838.x
Contractor, A., Mulle, C., and Swanson, G. T. (2011). Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 34, 154–163. doi: 10.1016/j.tins.2010.12.002
Contractor, A., Swanson, G., and Heinemann, S. F. (2001). Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron 29, 209–216. doi: 10.1016/S0896-6273(01)00191-X
Cook, E. H. Jr. (2011). De novo autosomal dominant mutation in SYNGAP1. Autism Res. 4, 155–156. doi: 10.1002/aur.198
Cook, E. H. Jr., Lindgren, V., Leventhal, B. L., Courchesne, R., Lincoln, A., Shulman, C., et al. (1997). Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 60, 928–934.
Correia, C. T., Coutinho, A. M., Sequeira, A. F., Sousa, I. G., Lourenco Venda, L., Almeida, J. P., et al. (2010). Increased BDNF levels and NTRK2 gene association suggest a disruption of BDNF/TrkB signaling in autism. Genes Brain Behav. 9, 841–848. doi: 10.1111/j.1601-183X.2010.00627.x
Cottrell, C. E., Bir, N., Varga, E., Alvarez, C. E., Bouyain, S., Zernzach, R., et al. (2011). Contactin 4 as an autism susceptibility locus. Autism Res. 4, 189–199. doi: 10.1002/aur.184
Crepel, A., De Wolf, V., Brison, N., Ceulemans, B., Walleghem, D., Peuteman, G., et al. (2014). Association of CDH11 with non-syndromic ASD. Am. J. Med. Genet. B Neuropsychiatr. Genet. 165B, 391–398. doi: 10.1002/ajmg.b.32243
Crino, P. B., Nathanson, K. L., and Henske, E. P. (2006). The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356. doi: 10.1056/NEJMra055323
Cruz-Martin, A., Crespo, M., and Portera-Cailliau, C. (2010). Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci. 30, 7793–7803. doi: 10.1523/JNEUROSCI.0577-10.2010
Cukier, H. N., Dueker, N. D., Slifer, S. H., Lee, J. M., Whitehead, P. L., Lalanne, E., et al. (2014). Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neurosychiatric disorders. Mol. Autism 5:1. doi: 10.1186/2040-2392-5-1
Cukier, H. N., Lee, J. M., Ma, D., Young, J. I., Mayo, V., Butler, B. L., et al. (2012). The expanding role of MBD genes in autism: identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res. 5, 385–397. doi: 10.1002/aur.1251
Cupolillo, D., Hoxha, E., Faralli, A., De Luca, A., Rossi, F., Tempia, F., et al. (2016). Autistic-like traits and cerebellar dysfunction in purkinje cell PTEN knock-out mice. Neuropsychopharmacology 41, 1457–1466. doi: 10.1038/npp.2015.339
Curatolo, P., Bjornvold, M., Dill, P. E., Ferreira, J. C., Feucht, M., Hertzberg, C., et al. (2016). The role of mTOR inhibitors in the treatment of patients with tuberous sclerosis complex: evidence-based and expert opinions. Drugs 76, 551–565. doi: 10.1007/s40265-016-0552-9
Dabell, M. P., Rosenfeld, J. A., Bader, P., Escobar, L. F., El-Khechen, D., Vallee, S. E., et al. (2013). Investigation of NRXN1 deletions: clinical and molecular characterization. Am. J. Med. Genet. A 161A, 717–731. doi: 10.1002/ajmg.a.35780
Dailey, M. E., and Smith, S. J. (1996). The dynamics of dendritic structure in developing hippocampal slices. J. Neurosci. 16, 2983–2994.
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565. doi: 10.1073/pnas.0506071102
Darstein, M., Petralia, R. S., Swanson, G. T., Wenthold, R. J., and Heinemann, S. F. (2003). Distribution of kainate receptor subunits at hippocampal mossy fiber synapses. J. Neurosci. 23, 8013–8019.
Davis, L. K., Meyer, K. J., Rudd, D. S., Librant, A. L., Epping, E. A., Sheffield, V. C., et al. (2009). Novel copy number variants in children with autism and additional developmental anomalies. J. Neurodev. Disord. 1, 292–301. doi: 10.1007/s11689-009-9013-z
De Filippis, B., Valenti, D., Chiodi, V., Ferrante, A., de Bari, L., Fiorentini, C., et al. (2015). Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 25, 889–901. doi: 10.1016/j.euroneuro.2015.03.012
de Ligt, J., Willemsen, M. H., van Bon, B. W., Kleefstra, T., Yntema, H. G., Kroes, T., et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. doi: 10.1056/NEJMoa1206524
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
De Rubeis, S., Pasciuto, E., Li, K. W., Fernandez, E., Di Marino, D., Buzzi, A., et al. (2013). CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79, 1169–1182. doi: 10.1016/j.neuron.2013.06.039
de Wit, J., Sylwestrak, E., O’Sullivan, M. L., Otto, S., Tiglio, K., Savas, J. N., et al. (2009). LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron 64, 799–806. doi: 10.1016/j.neuron.2009.12.019
Dean, C., Scholl, F. G., Choih, J., DeMaria, S., Berger, J., Isacoff, E., et al. (2003). Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 6, 708–716. doi: 10.1038/nn1074
Deinhardt, K., and Chao, M. V. (2014). Trk receptors. Handb. Exp. Pharmacol. 220, 103–119. doi: 10.1007/978-3-642-45106-5_5
Della Sala, G., and Pizzorusso, T. (2014). Synaptic plasticity and signaling in Rett syndrome. Dev. Neurobiol. 74, 178–196. doi: 10.1002/dneu.22114
Della Sala, G., Putignano, E., Chelini, G., Melani, R., Calcagno, E., Michele Ratto, G., et al. (2016). Dendritic spine instability in a mouse model of CDKL5 disorder is rescued by insulin-like growth factor 1. Biol. Psychiatry 80, 302–311. doi: 10.1016/j.biopsych.2015.08.028
Deng, J. V., Wan, Y., Wang, X., Cohen, S., Wetsel, W. C., Greenberg, M. E., et al. (2014). MeCP2 phosphorylation limits psychostimulant-induced behavioral and neuronal plasticity. J. Neurosci. 34, 4519–4527. doi: 10.1523/JNEUROSCI.2821-13.2014
Dent, E. W., Merriam, E. B., and Hu, X. (2011). The dynamic cytoskeleton: backbone of dendritic spine plasticity. Curr. Opin. Neurobiol. 21, 175–181. doi: 10.1016/j.conb.2010.08.013
Depienne, C., Bouteiller, D., Keren, B., Cheuret, E., Poirier, K., Trouillard, O., et al. (2009). Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 5:e1000381. doi: 10.1371/journal.pgen.1000381
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. doi: 10.1016/j.devcel.2008.04.003
D’Gama, A. M., Pochareddy, S., Li, M., Jamuar, S. S., Reiff, R. E., Lam, A. T., et al. (2015). Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88, 910–917. doi: 10.1016/j.neuron.2015.11.009
Dimassi, S., Andrieux, J., Labalme, A., Lesca, G., Cordier, M. P., Boute, O., et al. (2013). Interstitial 12p13.1 deletion involving GRIN2B in three patients with intellectual disability. Am. J. Med. Genet. A 161A, 2564–2569. doi: 10.1002/ajmg.a.36079
Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B., and Beaudet, A. L. (2008). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118. doi: 10.1093/hmg/ddm288
Dolen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Donovan, A. P., and Basson, M. A. (2016). The neuroanatomy of autism - a developmental perspective. J. Anat. doi: 10.1111/joa.12542 [Epub ahead of print].
Downes, C. P., Bennett, D., McConnachie, G., Leslie, N. R., Pass, I., MacPhee, C., et al. (2001). Antagonism of PI 3-kinase-dependent signalling pathways by the tumour suppressor protein, PTEN. Biochem. Soc. Trans. 29(Pt 6), 846–851. doi: 10.1042/bst0290846
Dresner, E., Agam, G., and Gozes, I. (2011). Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: deregulation in schizophrenia. Eur. Neuropsychopharmacol. 21, 355–361. doi: 10.1016/j.euroneuro.2010.06.004
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Duong, L., Klitten, L. L., Moller, R. S., Ingason, A., Jakobsen, K. D., Skjodt, C., et al. (2012). Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 354–358. doi: 10.1002/ajmg.b.32036
Dupuis, J. P., Ladepeche, L., Seth, H., Bard, L., Varela, J., Mikasova, L., et al. (2014). Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO J. 33, 842–861. doi: 10.1002/embj.201386356
Durand, C. M., Perroy, J., Loll, F., Perrais, D., Fagni, L., Bourgeron, T., et al. (2012). SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 17, 71–84. doi: 10.1038/mp.2011.57
Ebert, D. H., and Greenberg, M. E. (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. doi: 10.1038/nature11860
Egger, G., Roetzer, K. M., Noor, A., Lionel, A. C., Mahmood, H., Schwarzbraun, T., et al. (2014). Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 15, 117–127. doi: 10.1007/s10048-014-0394-0
Ehninger, D., and Silva, A. J. (2011). Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 17, 78–87. doi: 10.1016/j.molmed.2010.10.002
Epi, K. C., Epilepsy Phenome/Genome Project, Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Esch, T., Lemmon, V., and Banker, G. (2000). Differential effects of NgCAM and N-cadherin on the development of axons and dendrites by cultured hippocampal neurons. J. Neurocytol. 29, 215–223. doi: 10.1023/A:1026515426303
Espinosa, J. S., Wheeler, D. G., Tsien, R. W., and Luo, L. (2009). Uncoupling dendrite growth and patterning: single-cell knockout analysis of NMDA receptor 2B. Neuron 62, 205–217. doi: 10.1016/j.neuron.2009.03.006
Esteban, P. F., Yoon, H. Y., Becker, J., Dorsey, S. G., Caprari, P., Palko, M. E., et al. (2006). A kinase-deficient TrkC receptor isoform activates Arf6-Rac1 signaling through the scaffold protein tamalin. J. Cell Biol. 173, 291–299. doi: 10.1083/jcb.200512013
Etherton, M. R., Blaiss, C. A., Powell, C. M., and Südhof, T. C. (2009). Mouse neurexin-1α deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. U.S.A. 106, 17998–18003. doi: 10.1073/pnas.0910297106
European Chromosome 16 Tuberous Sclerosis Consortium (1993). Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315. doi: 10.1016/0092-8674(93)90618-Z
Ewald, R. C., Van Keuren-Jensen, K. R., Aizenman, C. D., and Cline, H. T. (2008). Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J. Neurosci. 28, 850–861. doi: 10.1523/JNEUROSCI.5078-07.2008
Faux, C., Hawadle, M., Nixon, J., Wallace, A., Lee, S., Murray, S., et al. (2007). PTPsigma binds and dephosphorylates neurotrophin receptors and can suppress NGF-dependent neurite outgrowth from sensory neurons. Biochim. Biophys. Acta 1773, 1689–1700. doi: 10.1016/j.bbamcr.2007.06.008
Feinstein, C., and Reiss, A. L. (1998). Autism: the point of view from fragile X studies. J. Autism. Dev. Disord. 28, 393–405. doi: 10.1023/A:1026000404855
Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., et al. (2006). High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 409, 10–13. doi: 10.1016/j.neulet.2006.08.017
Fernandez, T., Morgan, T., Davis, N., Klin, A., Morris, A., Farhi, A., et al. (2004). Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 74, 1286–1293. doi: 10.1086/421474
Fernandez-Montesinos, R., Torres, M., Baglietto-Vargas, D., Gutierrez, A., Gozes, I., Vitorica, J., et al. (2010). Activity-dependent neuroprotective protein (ADNP) expression in the amyloid precursor protein/presenilin 1 mouse model of Alzheimer’s disease. J. Mol. Neurosci. 41, 114–120. doi: 10.1007/s12031-009-9300-x
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54. doi: 10.1016/S0165-0173(02)00158-3
Floricel, F., Higaki, K., Maki, H., Nanba, E., Ninomiya, H., and Ohno, K. (2007). Antisense suppression of TSC1 gene product, hamartin, enhances neurite outgrowth in NGF-treated PC12h cells. Brain Dev. 29, 502–509. doi: 10.1016/j.braindev.2007.01.007
Fotaki, V., Dierssen, M., Alcantara, S., Martinez, S., Marti, E., Casas, C., et al. (2002). Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 22, 6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002
Frank, M., and Kemler, R. (2002). Protocadherins. Curr. Opin. Cell Biol. 14, 557–562. doi: 10.1016/S0955-0674(02)00365-4
Franke, K., Otto, W., Johannes, S., Baumgart, J., Nitsch, R., and Schumacher, S. (2012). miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J. 31, 2908–2921. doi: 10.1038/emboj.2012.130
Fraser, M. M., Bayazitov, I. T., Zakharenko, S. S., and Baker, S. J. (2008). Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151, 476–488. doi: 10.1016/j.neuroscience.2007.10.048
Friedman, L. G., Benson, D. L., and Huntley, G. W. (2015a). Cadherin-based transsynaptic networks in establishing and modifying neural connectivity. Curr. Top. Dev. Biol. 112, 415–465. doi: 10.1016/bs.ctdb.2014.11.025
Friedman, L. G., Riemslagh, F. W., Sullivan, J. M., Mesias, R., Williams, F. M., Huntley, G. W., et al. (2015b). Cadherin-8 expression, synaptic localization, and molecular control of neuronal form in prefrontal corticostriatal circuits. J. Comp. Neurol. 523, 75–92. doi: 10.1002/cne.23666
Frith, C. (2003). What do imaging studies tell us about the neural basis of autism? Novartis Found. Symp. 251, 149–166. doi: 10.1002/0470869380.ch10 discussion 166-176, 281-197.
Fuchs, C., Rimondini, R., Viggiano, R., Trazzi, S., De Franceschi, M., Bartesaghi, R., et al. (2015). Inhibition of GSK3beta rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 82, 298–310. doi: 10.1016/j.nbd.2015.06.018
Fuchs, C., Trazzi, S., Torricella, R., Viggiano, R., De Franceschi, M., Amendola, E., et al. (2014). Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 70, 53–68. doi: 10.1016/j.nbd.2014.06.006
Fujita, Y., Shirataki, H., Sakisaka, T., Asakura, T., Ohya, T., Kotani, H., et al. (1998). Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron 20, 905–915. doi: 10.1016/S0896-6273(00)80472-9
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544. doi: 10.1093/jnen/64.6.537
Furman, S., Steingart, R. A., Mandel, S., Hauser, J. M., Brenneman, D. E., and Gozes, I. (2004). Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 1, 193–199. doi: 10.1017/S1740925X05000013
Galvez, R., Gopal, A. R., and Greenough, W. T. (2003). Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 971, 83–89. doi: 10.1016/S0006-8993(03)02363-1
Galvez, R., and Greenough, W. T. (2005). Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am. J. Med. Genet. A 135, 155–160. doi: 10.1002/ajmg.a.30709
Gao, R., and Penzes, P. (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167. doi: 10.2174/1566524015666150303003028
Gao, X., Zhang, Y., Arrazola, P., Hino, O., Kobayashi, T., Yeung, R. S., et al. (2002). Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 4, 699–704. doi: 10.1038/ncb847
Gao, Y., Su, J., Guo, W., Polich, E. D., Magyar, D. P., Xing, Y., et al. (2015). Inhibition of miR-15a promotes BDNF expression and rescues dendritic maturation deficits in MeCP2-Deficient neurons. Stem Cells 33, 1618–1629. doi: 10.1002/stem.1950
Gauthier, J., Bonnel, A., St-Onge, J., Karemera, L., Laurent, S., Mottron, L., et al. (2005). NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 132B, 74–75. doi: 10.1002/ajmg.b.30066
Gauthier, J., Siddiqui, T. J., Huashan, P., Yokomaku, D., Hamdan, F. F., Champagne, N., et al. (2011). Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 130, 563–573. doi: 10.1007/s00439-011-0975-z
Gauthier, J., Spiegelman, D., Piton, A., Lafreniere, R. G., Laurent, S., St-Onge, J., et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 421–424. doi: 10.1002/ajmg.b.30822
Gdalyahu, A., Lazaro, M., Penagarikano, O., Golshani, P., Trachtenberg, J. T., and Gescwind, D. H. (2015). The autism related protein contactin-associated protein-like 2 (CNTNAP2) stabilizes new spines: an in vivo mouse study. PLoS ONE 10:e0125633. doi: 10.1371/journal.pone.0125633
Gedeon, A. K., Baker, E., Robinson, H., Partington, M. W., Gross, B., Manca, A., et al. (1992). Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat. Genet. 1, 341–344. doi: 10.1038/ng0892-341
Germain, N. D., Chen, P. F., Plocik, A. M., Glatt-Deeley, H., Brown, J., Fink, J. J., et al. (2014). Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol. Autism 5:44. doi: 10.1186/2040-2392-5-44
Geschwind, D. H., and State, M. W. (2015). Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 14, 1109–1120. doi: 10.1016/S1474-4422(15)00044-7
Girirajan, S., Dennis, M. Y., Baker, C., Malig, M., Coe, B. P., Campbell, C. D., et al. (2013). Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 92, 221–237. doi: 10.1016/j.ajhg.2012.12.016
Giuffrida, R., Musumeci, S., D’Antoni, S., Bonaccorso, C. M., Giuffrida-Stella, A. M., Oostra, B. A., et al. (2005). A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci. 25, 8908–8916. doi: 10.1523/JNEUROSCI.0932-05.2005
Gjorlund, M. D., Nielsen, J., Pankratova, S., Li, S., Korshunova, I., Bock, E., et al. (2012). Neuroligin-1 induces neurite outgrowth through interaction with neurexin-1beta and activation of fibroblast growth factor receptor-1. FASEB J. 26, 4174–4186. doi: 10.1096/fj.11-202242
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Gockler, N., Jofre, G., Papadopoulos, C., Soppa, U., Tejedor, F. J., and Becker, W. (2009). Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 276, 6324–6337. doi: 10.1111/j.1742-4658.2009.07346.x
Goffin, A., Hoefsloot, L. H., Bosgoed, E., Swillen, A., and Fryns, J. P. (2001). PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 105, 521–524. doi: 10.1002/ajmg.1477
Gokce, O., and Sudhof, T. C. (2013). Membrane-tethered monomeric neurexin LNS-domain triggers synapse formation. J. Neurosci. 33, 14617–14628. doi: 10.1523/JNEUROSCI.1232-13.2013
Govek, E. E., Newey, S. E., and Van Aelst, L. (2005). The role of the Rho GTPases in neuronal development. Genes Dev. 19, 1–49. doi: 10.1101/gad.1256405
Greenough, W. T., Klintsova, A. Y., Irwin, S. A., Galvez, R., Bates, K. E., and Weiler, I. J. (2001). Synaptic regulation of protein synthesis and the fragile X protein. Proc. Natl. Acad. Sci. U.S.A. 98, 7101–7106. doi: 10.1073/pnas.141145998
Greer, P. L., Hanayama, R., Bloodgood, B. L., Mardinly, A. R., Lipton, D. M., Flavell, S. W., et al. (2010). The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716. doi: 10.1016/j.cell.2010.01.026
Grimsley, C. M., Kinchen, J. M., Tosello-Trampont, A. C., Brugnera, E., Haney, L. B., Lu, M., et al. (2004). Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J. Biol. Chem. 279, 6087–6097. doi: 10.1074/jbc.M307087200
Griswold, A. J., Ma, D., Cukier, H. N., Nations, L. D., Schmidt, M. A., Chung, R. H., et al. (2012). Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 21, 3513–3523. doi: 10.1093/hmg/dds164
Grossman, A. W., Aldridge, G. M., Lee, K. J., Zeman, M. K., Jun, C. S., Azam, H. S., et al. (2010). Developmental characteristics of dendritic spines in the dentate gyrus of Fmr1 knockout mice. Brain Res. 1355, 221–227. doi: 10.1016/j.brainres.2010.07.090
Grossman, A. W., Elisseou, N. M., McKinney, B. C., and Greenough, W. T. (2006). Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 1084, 158–164. doi: 10.1016/j.brainres.2006.02.044
Guilmatre, A., Huguet, G., Delorme, R., and Bourgeron, T. (2014). The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 74, 113–122. doi: 10.1002/dneu.22128
Gumienny, T. L., Brugnera, E., Tosello-Trampont, A. C., Kinchen, J. M., Haney, L. B., Nishiwaki, K., et al. (2001). CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107, 27–41. doi: 10.1016/S0092-8674(01)00520-7
Gupta, S. C., Yadav, R., Pavuluri, R., Morley, B. J., Stairs, D. J., and Dravid, S. M. (2015). Essential role of GluD1 in dendritic spine development and GluN2B to GluN2A NMDAR subunit switch in the cortex and hippocampus reveals ability of GluN2B inhibition in correcting hyperconnectivity. Neuropharmacology 93, 274–284. doi: 10.1016/j.neuropharm.2015.02.013
Habela, C. W., Song, H., and Ming, G. L. (2015). Modeling synaptogenesis in schizophrenia and autism using human iPSC derived neurons. Mol. Cell. Neurosci. 73, 52–62. doi: 10.1016/j.mcn.2015.12.002
Haeckel, A., Ahuja, R., Gundelfinger, E. D., Qualmann, B., and Kessels, M. M. (2008). The actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head and synapse formation. J. Neurosci. 28, 10031–10044. doi: 10.1523/JNEUROSCI.0336-08.2008
Haldeman-Englert, C. R., Chapman, K. A., Kruger, H., Geiger, E. A., McDonald-McGinn, D. M., Rappaport, E., et al. (2010). A de novo 8.8-Mb deletion of 21q21.1-q21.3 in an autistic male with a complex rearrangement involving chromosomes 6, 10, and 21. Am. J. Med. Genet. A 152A, 196–202. doi: 10.1002/ajmg.a.33176
Hamdan, F. F., Daoud, H., Piton, A., Gauthier, J., Dobrzeniecka, S., Krebs, M. O., et al. (2011). De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol. Psychiatry 69, 898–901. doi: 10.1016/j.biopsych.2010.11.015
Hammer, J. A. III, and Wagner, W. (2013). Functions of class V myosins in neurons. J. Biol. Chem. 288, 28428–28434. doi: 10.1074/jbc.R113.514497
Hammerle, B., Carnicero, A., Elizalde, C., Ceron, J., Martinez, S., and Tejedor, F. J. (2003). Expression patterns and subcellular localization of the Down syndrome candidate protein MNB/DYRK1A suggest a role in late neuronal differentiation. Eur. J. Neurosci. 17, 2277–2286. doi: 10.1046/j.1460-9568.2003.02665.x
Hampson, D. R., and Blatt, G. J. (2015). Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 9:420. doi: 10.3389/fnins.2015.00420
Hanchard, N. A., Carvalho, C. M., Bader, P., Thome, A., Omo-Griffith, L., del Gaudio, D., et al. (2012). A partial MECP2 duplication in a mildly affected adult male: a putative role for the 3′ untranslated region in the MECP2 duplication phenotype. BMC Med. Genet. 13:71. doi: 10.1186/1471-2350-13-71
Hata, Y., Butz, S., and Südhof, T. C. (1996). CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 16, 2488–2494.
Hatton, D. D., Sideris, J., Skinner, M., Mankowski, J., Bailey, D. B. Jr., Roberts, J., et al. (2006). Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. A 140A, 1804–1813. doi: 10.1002/ajmg.a.31286
Haws, M. E., Jaramillo, T. C., Espinosa, F., Widman, A. J., Stuber, G. D., Sparta, D. R., et al. (2014). PTEN knockdown alters dendritic spine/protrusion morphology, not density. J. Comp. Neurol. 522, 1171–1190. doi: 10.1002/cne.23488
Hayashi, M. K., Tang, C., Verpelli, C., Narayanan, R., Stearns, M. H., Xu, R. M., et al. (2009). The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 137, 159–171. doi: 10.1016/j.cell.2009.01.050
Hayashi, M. L., Rao, B. S., Seo, J. S., Choi, H. S., Dolan, B. M., Choi, S. Y., et al. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11489–11494. doi: 10.1073/pnas.0705003104
Hayrapetyan, V., Castro, S., Sukharnikova, T., Yu, C., Cao, X., Jiang, Y. H., et al. (2014). Region-specific impairments in striatal synaptic transmission and impaired instrumental learning in a mouse model of Angelman syndrome. Eur. J. Neurosci. 39, 1018–1025. doi: 10.1111/ejn.12442
Hays, S. A., Huber, K. M., and Gibson, J. R. (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci. 31, 14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011
He, C. X., and Portera-Cailliau, C. (2013). The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128. doi: 10.1016/j.neuroscience.2012.03.049
Heine, M., Thoumine, O., Mondin, M., Tessier, B., Giannone, G., and Choquet, D. (2008). Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc. Natl. Acad. Sci. U.S.A. 105, 20947–20952. doi: 10.1073/pnas.0804007106
Heitzler, P., Coulson, D., Saenz-Robles, M. T., Ashburner, M., Roote, J., Simpson, P., et al. (1993). Genetic and cytogenetic analysis of the 43A-E region containing the segment polarity gene costa and the cellular polarity genes prickle and spiny-legs in Drosophila melanogaster. Genetics 135, 105–115.
Helsmoortel, C., Vulto-van Silfhout, A. T., Coe, B. P., Vandeweyer, G., Rooms, L., van den Ende, J., et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46, 380–384. doi: 10.1038/ng.2899
Hirano, S., and Takeichi, M. (2012). Cadherins in brain morphogenesis and wiring. Physiol. Rev. 92, 597–634. doi: 10.1152/physrev.00014.2011
Hirano, S., Yan, Q., and Suzuki, S. T. (1999). Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J. Neurosci. 19, 995–1005.
Hobert, J. A., Embacher, R., Mester, J. L., Frazier, T. W. II, and Eng, C. (2014). Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur. J. Hum. Genet. 22, 273–276. doi: 10.1038/ejhg.2013.114
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. doi: 10.1016/j.tins.2009.11.003
Hogart, A., Wu, D., LaSalle, J. M., and Schanen, N. C. (2010). The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 38, 181–191. doi: 10.1016/j.nbd.2008.08.011
Holt, R., Barnby, G., Maestrini, E., Bacchelli, E., Brocklebank, D., Sousa, I., et al. (2010). Linkage and candidate gene studies of autism spectrum disorders in European populations. Eur. J. Hum. Genet. 18, 1013–1019. doi: 10.1038/ejhg.2010.69
Holtmaat, A. J., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W., et al. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. doi: 10.1016/j.neuron.2005.01.003
Hoogenraad, C. C., and Akhmanova, A. (2010). Dendritic spine plasticity: new regulatory roles of dynamic microtubules. Neuroscientist 16, 650–661. doi: 10.1177/1073858410386357
Hotulainen, P., and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629. doi: 10.1083/jcb.201003008
Hou, R., Liu, L., Anees, S., Hiroyasu, S., and Sibinga, N. E. (2006). The Fat1 cadherin integrates vascular smooth muscle cell growth and migration signals. J. Cell Biol. 173, 417–429. doi: 10.1083/jcb.200508121
Hsueh, Y. P. (2012). Neuron-specific regulation on F-actin cytoskeletons: the role of CTTNBP2 in dendritic spinogenesis and maintenance. Commun. Integr. Biol. 5, 334–336. doi: 10.4161/cib.20364
Hu, J., Liao, J., Sathanoori, M., Kochmar, S., Sebastian, J., Yatsenko, S. A., et al. (2015). CNTN6 copy number variations in 14 patients: a possible candidate gene for neurodevelopmental and neuropsychiatric disorders. J Neurodev. Disord. 7:26. doi: 10.1186/s11689-015-9122-9
Huettner, J. E. (2003). Kainate receptors and synaptic transmission. Prog. Neurobiol. 70, 387–407. doi: 10.1016/S0301-0082(03)00122-9
Huibregtse, J. M., Scheffner, M., and Howley, P. M. (1993). Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 13, 775–784. doi: 10.1128/MCB.13.8.4918
Hulpiau, P., and van Roy, F. (2009). Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol. 41, 349–369. doi: 10.1016/j.biocel.2008.09.027
Hung, A. Y., Futai, K., Sala, C., Valtschanoff, J. G., Ryu, J., Woodworth, M. A., et al. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708. doi: 10.1523/JNEUROSCI.3032-07.2008
Huntley, G. W., and Benson, D. L. (1999). Neural (N)-cadherin at developing thalamocortical synapses provides an adhesion mechanism for the formation of somatopically organized connections. J. Comp. Neurol. 407, 453–471. doi: 10.1002/(SICI)1096-9861(19990517)407:4<453::AID-CNE1>3.3.CO;2-W
Hussman, J. P. (2001). Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J. Autism. Dev. Disord. 31, 247–248. doi: 10.1023/A:1010715619091
Hussman, J. P., Chung, R. H., Griswold, A. J., Jaworski, J. M., Salyakina, D., Ma, D., et al. (2011). A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol. Autism 2:1. doi: 10.1186/2040-2392-2-1
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Huttenlocher, P. R., and Dabholkar, A. S. (1997). Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 387, 167–178. doi: 10.1002/(SICI)1096-9861(19971020)387:2<167::AID-CNE1>3.0.CO;2-Z
Iafrati, J., Orejarena, M. J., Lassalle, O., Bouamrane, L., Gonzalez-Campo, C., and Chavis, P. (2014). Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 19, 417–426. doi: 10.1038/mp.2013.66
Ichinose, T., and Snider, W. D. (2000). Differential effects of TrkC isoforms on sensory axon outgrowth. J. Neurosci. Res. 59, 365–371. doi: 10.1002/(SICI)1097-4547(20000201)59:3<365::AID-JNR10>3.0.CO;2-I
Ichtchenko, K., Hata, Y., Nguyen, T., Ullrich, B., Missler, M., Moomaw, C., et al. (1995). Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81, 435–443. doi: 10.1016/0092-8674(95)90396-8
Ichtchenko, K., Nguyen, T., and Sudhof, T. C. (1996). Structures, alternative splicing, and neurexin binding of multiple neuroligins. J. Biol. Chem. 271, 2676–2682. doi: 10.1074/jbc.271.5.2676
Ifrim, M. F., Williams, K. R., and Bassell, G. J. (2015). Single-molecule imaging of PSD-95 mRNA translation in dendrites and its dysregulation in a mouse model of fragile X syndrome. J. Neurosci. 35, 7116–7130. doi: 10.1523/JNEUROSCI.2802-14.2015
Imitola, J., Walleigh, D., Anderson, C. E., Jethva, R., Carvalho, K. S., Legido, A., et al. (2014). Fraternal twins with autism, severe cognitive deficit, and epilepsy: diagnostic role of chromosomal microarray analysis. Semin. Pediatr. Neurol. 21, 167–171. doi: 10.1016/j.spen.2014.04.027
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Irie, M., Hata, Y., Takeuchi, M., Ichtchenko, K., Toyoda, A., Hirao, K., et al. (1997). Binding of neuroligins to PSD-95. Science 277, 1511–1515. doi: 10.1126/science.277.5331.1511
Irwin, S. A., Galvez, R., and Greenough, W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044. doi: 10.1093/cercor/10.10.1038
Itoh, M., Ide, S., Takashima, S., Kudo, S., Nomura, Y., Segawa, M., et al. (2007). Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 66, 117–123. doi: 10.1097/nen.0b013e3180302078
Jamain, S., Betancur, C., Quach, H., Philippe, A., Fellous, M., Giros, B., et al. (2002). Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 7, 302–310. doi: 10.1038/sj.mp.4000979
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jaworski, J., Spangler, S., Seeburg, D. P., Hoogenraad, C. C., and Sheng, M. (2005). Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005
Jenny, A., Reynolds-Kenneally, J., Das, G., Burnett, M., and Mlodzik, M. (2005). Diego and Prickle regulate Frizzled planar cell polarity signalling by competing for Dishevelled binding. Nat. Cell Biol. 7, 691–697. doi: 10.1038/ncb1271
Jiang, M., Ash, R. T., Baker, S. A., Suter, B., Ferguson, A., Park, J., et al. (2013a). Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J. Neurosci. 33, 19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013
Jiang, Y. H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Jiang, Y. H., Yuen, R. K., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013b). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263. doi: 10.1016/j.ajhg.2013.06.012
Jiang-Xie, L. F., Liao, H. M., Chen, C. H., Chen, Y. T., Ho, S. Y., Lu, D. H., et al. (2014). Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Mol. Autism 5:32.
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Johnston, S. B., and Raines, R. T. (2015). Conformational stability and catalytic activity of PTEN variants linked to cancers and autism spectrum disorders. Biochemistry 54, 1576–1582. doi: 10.1021/acs.biochem.5b00028
Jones, W. H., and Thomas, D. B. (1962). Changes in the dendritic organization of neurons in the cerebral cortex following deafferentation. J. Anat. 96, 375–381.
Joo, W., Hippenmeyer, S., and Luo, L. (2014). Neurodevelopment. Dendrite morphogenesis depends on relative levels of NT-3/TrkC signaling. Science 346, 626–629. doi: 10.1126/science.1258996
Jugloff, D. G., Jung, B. P., Purushotham, D., Logan, R., and Eubanks, J. H. (2005). Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol. Dis. 19, 18–27. doi: 10.1016/j.nbd.2004.11.002
Kaneko-Goto, T., Yoshihara, S., Miyazaki, H., and Yoshihara, Y. (2008). BIG-2 mediates olfactory axon convergence to target glomeruli. Neuron 57, 834–846. doi: 10.1016/j.neuron.2008.01.023
Kao, D. I., Aldridge, G. M., Weiler, I. J., and Greenough, W. T. (2010). Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc. Natl. Acad. Sci. U.S.A. 107, 15601–15606. doi: 10.1073/pnas.1010564107
Karagogeos, D. (2003). Neural GPI-anchored cell adhesion molecules. Front. Biosci. 8, s1304–s1320. doi: 10.2741/1214
Kashevarova, A. A., Nazarenko, L. P., Schultz-Pedersen, S., Skryabin, N. A., Salyukova, O. A., Chechetkina, N. N., et al. (2014). Single gene microdeletions and microduplication of 3p26.3 in three unrelated families: CNTN6 as a new candidate gene for intellectual disability. Mol. Cytogenet. 7:97. doi: 10.1186/s13039-014-0097-0
Kazdoba, T. M., Leach, P. T., and Crawley, J. N. (2016). Behavioral phenotypes of genetic mouse models of autism. Genes Brain Behav. 15, 7–26. doi: 10.1111/gbb.12256
Keeler, A. B., Molumby, M. J., and Weiner, J. A. (2015). Protocadherins branch out: multiple roles in dendrite development. Cell Adh. Migr. 9, 214–226. doi: 10.1080/19336918.2014.1000069
Kelleher, R. J. III, Geigenmuller, U., Hovhannisyan, H., Trautman, E., Pinard, R., Rathmell, B., et al. (2012). High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS ONE 7:e35003. doi: 10.1371/journal.pone.0035003
Kelsch, W., Li, Z., Eliava, M., Goengrich, C., and Monyer, H. (2012). GluN2B-containing NMDA receptors promote wiring of adult-born neurons into olfactory bulb circuits. J. Neurosci. 32, 12603–12611. doi: 10.1523/JNEUROSCI.1459-12.2012
Kenny, E. M., Cormican, P., Furlong, S., Heron, E., Kenny, G., Fahey, C., et al. (2014). Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 19, 872–879. doi: 10.1038/mp.2013.127
Kim, E., Naisbitt, S., Hsueh, Y. P., Rao, A., Rothschild, A., Craig, A. M., et al. (1997). GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J. Cell Biol. 136, 669–678. doi: 10.1083/jcb.136.3.669
Kim, E., and Sheng, M. (2004). PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5, 771–781. doi: 10.1038/nrn1517
Kim, H., Kunz, P. A., Mooney, R., Philpot, B. D., and Smith, S. L. (2016). Maternal loss of Ube3a impairs experience-driven dendritic spine maintenance in the developing visual cortex. J. Neurosci. 36, 4888–4894. doi: 10.1523/JNEUROSCI.4204-15.2016
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
Kim, J. H., Liao, D., Lau, L. F., and Huganir, R. L. (1998). SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20, 683–691. doi: 10.1016/S0896-6273(00)81008-9
Kim, J. Y., Oh, M. H., Bernard, L. P., Macara, I. G., and Zhang, H. (2011a). The RhoG/ELMO1/Dock180 signaling module is required for spine morphogenesis in hippocampal neurons. J. Biol. Chem. 286, 37615–37624. doi: 10.1074/jbc.M111.268029
Kim, S. Y., Chung, H. S., Sun, W., and Kim, H. (2007). Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 147, 996–1021. doi: 10.1016/j.neuroscience.2007.03.052
Kim, S. Y., Yasuda, S., Tanaka, H., Yamagata, K., and Kim, H. (2011b). Non-clustered protocadherin. Cell. Adh. Migr. 5, 97–105. doi: 10.4161/cam.5.2.14374
Kindler, S., Rehbein, M., Classen, B., Richter, D., and Bockers, T. M. (2004). Distinct spatiotemporal expression of SAPAP transcripts in the developing rat brain: a novel dendritically localized mRNA. Brain Res. Mol. Brain Res. 126, 14–21. doi: 10.1016/j.molbrainres.2004.03.014
Kishi, N., and Macklis, J. D. (2010). MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp. Neurol. 222, 51–58. doi: 10.1016/j.expneurol.2009.12.007
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Kneussel, M., and Wagner, W. (2013). Myosin motors at neuronal synapses: drivers of membrane transport and actin dynamics. Nat. Rev. Neurosci. 14, 233–247. doi: 10.1038/nrn3445
Ko, J., Fuccillo, M. V., Malenka, R. C., and Sudhof, T. C. (2009). LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron 64, 791–798. doi: 10.1016/j.neuron.2009.12.012
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., et al. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352. doi: 10.1016/j.neuron.2005.07.005
Koleske, A. J. (2013). Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 14, 536–550. doi: 10.1038/nrn3486
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. doi: 10.1038/nature11396
Korobova, F., and Svitkina, T. (2010). Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell 21, 165–176. doi: 10.1091/mbc.E09-07-0596
Koshimizu, E., Miyatake, S., Okamoto, N., Nakashima, M., Tsurusaki, Y., Miyake, N., et al. (2013). Performance comparison of bench-top next generation sequencers using microdroplet PCR-based enrichment for targeted sequencing in patients with autism spectrum disorder. PLoS ONE 8:e74167. doi: 10.1371/journal.pone.0074167
Koskinen, M., Bertling, E., Hotulainen, R., Tanhuanpaa, K., and Hotulainen, P. (2014). Myosin IIb controls actin dynamics underlying the dendritic spine maturation. Mol. Cell. Neurosci. 61, 56–64. doi: 10.1016/j.mcn.2014.05.008
Krapivinsky, G., Medina, I., Krapivinsky, L., Gapon, S., and Clapham, D. E. (2004). SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron 43, 563–574. doi: 10.1016/j.neuron.2004.08.003
Krumm, N., O’Roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi: 10.1016/j.tins.2013.11.005
Krumm, N., Turner, T. N., Baker, C., Vives, L., Mohajeri, K., Witherspoon, K., et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588. doi: 10.1038/ng.3303
Kulkarni, V. A., and Firestein, B. L. (2012). The dendritic tree and brain disorders. Mol. Cell. Neurosci. 50, 10–20. doi: 10.1016/j.mcn.2012.03.005
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Kwon, H. B., Kozorovitskiy, Y., Oh, W. J., Peixoto, R. T., Akhtar, N., Saulnier, J. L., et al. (2012). Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat. Neurosci. 15, 1667–1674. doi: 10.1038/nn.3256
Lanore, F., Labrousse, V. F., Szabo, Z., Normand, E., Blanchet, C., and Mulle, C. (2012). Deficits in morphofunctional maturation of hippocampal mossy fiber synapses in a mouse model of intellectual disability. J. Neurosci. 32, 17882–17893. doi: 10.1523/JNEUROSCI.2049-12.2012
Lanoue, V., Usardi, A., Sigoillot, S. M., Talleur, M., Iyer, K., Mariani, J., et al. (2013). The adhesion-GPCR BAI3, a gene linked to psychiatric disorders, regulates dendrite morphogenesis in neurons. Mol. Psychiatry 18, 943–950. doi: 10.1038/mp.2013.46
Larimore, J. L., Chapleau, C. A., Kudo, S., Theibert, A., Percy, A. K., and Pozzo-Miller, L. (2009). Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol. Dis. 34, 199–211. doi: 10.1016/j.nbd.2008.12.011
LaSalle, J. M., Reiter, L. T., and Chamberlain, S. J. (2015). Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 7, 1213–1228. doi: 10.2217/epi.15.70
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi: 10.1086/382137
Laurie, D. J., and Seeburg, P. H. (1994). Ligand affinities at recombinant N-methyl-D-aspartate receptors depend on subunit composition. Eur. J. Pharmacol 268, 335–345. doi: 10.1016/0922-4106(94)90058-2
Lawson-Yuen, A., Saldivar, J. S., Sommer, S., and Picker, J. (2008). Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur. J. Hum. Genet. 16, 614–618. doi: 10.1038/sj.ejhg.5202006
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. doi: 10.1371/journal.pgen.1004580
Lee, A., Li, W., Xu, K., Bogert, B. A., Su, K., and Gao, F. B. (2003). Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 130, 5543–5552. doi: 10.1242/dev.00792
Lepagnol-Bestel, A. M., Zvara, A., Maussion, G., Quignon, F., Ngimbous, B., Ramoz, N., et al. (2009). DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum. Mol. Genet. 18, 1405–1414. doi: 10.1093/hmg/ddp047
Lerma, J., and Marques, J. M. (2013). Kainate receptors in health and disease. Neuron 80, 292–311. doi: 10.1016/j.neuron.2013.09.045
Leslie, N. R., and Downes, C. P. (2002). PTEN: the down side of PI 3-kinase signalling. Cell. Signal. 14, 285–295. doi: 10.1016/S0898-6568(01)00234-0
Levenga, J., de Vrij, F. M., Buijsen, R. A., Li, T., Nieuwenhuizen, I. M., Pop, A., et al. (2011a). Subregion-specific dendritic spine abnormalities in the hippocampus of Fmr1 KO mice. Neurobiol. Learn. Mem. 95, 467–472. doi: 10.1016/j.nlm.2011.02.009
Levenga, J., Hayashi, S., de Vrij, F. M., Koekkoek, S. K., van der Linde, H. C., Nieuwenhuizen, I., et al. (2011b). AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis. 42, 311–317. doi: 10.1016/j.nbd.2011.01.022
Levinson, J. N., Chéry, N., Huang, K., Wong, T. P., Gerrow, K., Kang, R., et al. (2005). Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J. Biol. Chem. 280, 17312–17319. doi: 10.1074/jbc.M413812200
Li, H., Takeda, Y., Niki, H., Ogawa, J., Kobayashi, S., Kai, N., et al. (2003). Aberrant responses to acoustic stimuli in mice deficient for neural recognition molecule NB-2. Eur. J. Neurosci. 17, 929–936. doi: 10.1046/j.1460-9568.2003.02514.x
Li, H., Zhong, X., Chau, K. F., Williams, E. C., and Chang, Q. (2011). Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat. Neurosci. 14, 1001–1008. doi: 10.1038/nn.2866
Li, J., Shi, M., Ma, Z., Zhao, S., Euskirchen, G., Ziskin, J., et al. (2014a). Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol. Syst. Biol. 10, 774. doi: 10.15252/msb.20145487
Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S. I., et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. doi: 10.1126/science.275.5308.1943
Li, J. H., Wang, Y. H., Wolfe, B. B., Krueger, K. E., Corsi, L., Stocca, G., et al. (1998). Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur. J. Neurosci. 10, 1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x
Li, J. M., Lu, C. L., Cheng, M. C., Luu, S. U., Hsu, S. H., Hu, T. M., et al. (2014b). Role of the DLGAP2 gene encoding the SAP90/PSD-95-associated protein 2 in schizophrenia. PLoS ONE 9:e85373. doi: 10.1371/journal.pone.0085373
Li, X., Hu, Z., He, Y., Xiong, Z., Long, Z., Peng, Y., et al. (2010). Association analysis of CNTNAP2 polymorphisms with autism in the Chinese Han population. Psychiatr. Genet. 20, 113–117. doi: 10.1097/YPG.0b013e32833a216f
Lim, E. T., Raychaudhuri, S., Sanders, S. J., Stevens, C., Sabo, A., MacArthur, D. G., et al. (2013). Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 77, 235–242. doi: 10.1016/j.neuron.2012.12.029
Lin, C., Franco, B., and Rosner, M. R. (2005). CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum. Mol. Genet. 14, 3775–3786. doi: 10.1093/hmg/ddi391
Lin, W. H., and Webb, D. J. (2009). Actin and actin-binding proteins: masters of dendritic spine formation. Morphol. Funct. Open Neurosci. J. 3, 54–66. doi: 10.2174/1874082000903020054
Lin, Y. C., and Koleske, A. J. (2010). Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 33, 349–378. doi: 10.1146/annurev-neuro-060909-153204
Liu, C., Lin, C., Whitaker, D. T., Bakeri, H., Bulgakov, O. V., Liu, P., et al. (2013a). Prickle1 is expressed in distinct cell populations of the central nervous system and contributes to neuronal morphogenesis. Hum. Mol. Genet. 22, 2234–2246. doi: 10.1093/hmg/ddt075
Liu, Y., Du, Y., Liu, W., Yang, C., Liu, Y., Wang, H., et al. (2013b). Lack of association between NLGN3, NLGN4, SHANK2 and SHANK3 gene variants and autism spectrum disorder in a Chinese population. PLoS ONE 8:e56639. doi: 10.1371/journal.pone.0056639
Liu, X., Shimada, T., Otowa, T., Wu, Y. Y., Kawamura, Y., Tochigi, M., et al. (2015a). Genome-wide association study of autism spectrum disorder in the east asian populations. Autism Res. 9, 340–349. doi: 10.1002/aur.1536
Liu, Y., Hu, Z., Xun, G., Peng, Y., Lu, L., Xu, X., et al. (2012). Mutation analysis of the NRXN1 gene in a Chinese autism cohort. J. Psychiatr. Res. 46, 630–634. doi: 10.1016/j.jpsychires.2011.10.015
Liu, Y. F., Sowell, S. M., Luo, Y., Chaubey, A., Cameron, R. S., Kim, H. G., et al. (2015b). Autism and intellectual disability-associated KIRREL3 interacts with neuronal proteins MAP1B and MYO16 with potential roles in neurodevelopment. PLoS ONE 10:e0123106. doi: 10.1371/journal.pone.0123106
Lohith, T. G., Osterweil, E. K., Fujita, M., Jenko, K. J., Bear, M. F., and Innis, R. B. (2013). Is metabotropic glutamate receptor 5 upregulated in prefrontal cortex in fragile X syndrome? Mol. Autism 4:15. doi: 10.1186/2040-2392-4-15
Lombardi, L. M., Baker, S. A., and Zoghbi, H. Y. (2015). MECP2 disorders: from the clinic to mice and back. J. Clin. Invest. 125, 2914–2923. doi: 10.1172/JCI78167
Lord, C., and Jones, R. M. (2013). New strategies and findings for behavioral interventions in autism spectrum disorders. Ann. N. Y. Acad. Sci. 1304, 70–76. doi: 10.1111/nyas.12311
Luikart, B. W., Schnell, E., Washburn, E. K., Bensen, A. L., Tovar, K. R., and Westbrook, G. L. (2011). Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J. Neurosci. 31, 4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011
Luo, J., Wang, Y., Yasuda, R. P., Dunah, A. W., and Wolfe, B. B. (1997). The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol. Pharmacol. 51, 79–86.
Luo, L. (2002). Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu. Rev. Cell Dev. Biol. 18, 601–635. doi: 10.1146/annurev.cellbio.18.031802.150501
Maehama, T., and Dixon, J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378. doi: 10.1074/jbc.273.22.13375
Maehama, T., and Dixon, J. E. (1999). PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 9, 125–128. doi: 10.1016/S0962-8924(99)01519-6
Manabe, T., Togashi, H., Uchida, N., Suzuki, S. C., Hayakawa, Y., Yamamoto, M., et al. (2000). Loss of cadherin-11 adhesion receptor enhances plastic changes in hippocampal synapses and modifies behavioral responses. Mol. Cell. Neurosci. 15, 534–546. doi: 10.1006/mcne.2000.0849
Mandel, S., and Gozes, I. (2007). Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J. Biol. Chem. 282, 34448–34456. doi: 10.1074/jbc.M704756200
Mandel, S., Rechavi, G., and Gozes, I. (2007). Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev. Biol. 303, 814–824. doi: 10.1016/j.ydbio.2006.11.039
Mandel, S., Spivak-Pohis, I., and Gozes, I. (2008). ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J. Mol. Neurosci. 35, 127–141. doi: 10.1007/s12031-007-9013-y
Maortua, H., Martinez-Bouzas, C., Calvo, M. T., Domingo, M. R., Ramos, F., Garcia-Ribes, A., et al. (2012). CDKL5 gene status in female patients with epilepsy and Rett-like features: two new mutations in the catalytic domain. BMC Med. Genet. 13:68. doi: 10.1186/1471-2350-13-68
Marchese, M., Conti, V., Valvo, G., Moro, F., Muratori, F., Tancredi, R., et al. (2014). Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med. Genet. 15:26. doi: 10.1186/1471-2350-15-26
Margolis, S. S., Salogiannis, J., Lipton, D. M., Mandel-Brehm, C., Wills, Z. P., Mardinly, A. R., et al. (2010). EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 143, 442–455. doi: 10.1016/j.cell.2010.09.038
Mari, F., Azimonti, S., Bertani, I., Bolognese, F., Colombo, E., Caselli, R., et al. (2005). CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 14, 1935–1946. doi: 10.1093/hmg/ddi198
Marshak, S., Meynard, M. M., De Vries, Y. A., Kidane, A. H., and Cohen-Cory, S. (2012). Cell-autonomous alterations in dendritic arbor morphology and connectivity induced by overexpression of MeCP2 in Xenopus central neurons in vivo. PLoS ONE 7:e33153. doi: 10.1371/journal.pone.0033153
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
Martinez, A., Alcantara, S., Borrell, V., Del Rio, J. A., Blasi, J., Otal, R., et al. (1998). TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J. Neurosci. 18, 7336–7350.
Matthews, M. R., and Powell, T. P. (1962). Some observations on transneuronal cell degeneration in the olfactory bulb of the rabbit. J. Anat. 96, 89–102.
Matus, A. (2000). Actin-based plasticity in dendritic spines. Science 290, 754–758. doi: 10.1126/science.290.5492.754
Matus, A., Brinkhaus, H., and Wagner, U. (2000). Actin dynamics in dendritic spines: a form of regulated plasticity at excitatory synapses. Hippocampus 10, 555–560. doi: 10.1002/1098-1063(2000)10:5<555::AID-HIPO5>3.0.CO;2-Z
McBride, K. L., Varga, E. A., Pastore, M. T., Prior, T. W., Manickam, K., Atkin, J. F., et al. (2010). Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res 3, 137–141. doi: 10.1002/aur.132
McCrea, P. D., and Gu, D. (2010). The catenin family at a glance. J. Cell Sci. 123(Pt 5), 637–642. doi: 10.1242/jcs.039842
McGee, A., Li, G., Lu, Z., and Qiu, S. (2014). Convergent synaptic and circuit substrates underlying autism genetic risks. Front. Biol. 9, 137–150. doi: 10.1007/s11515-014-1298-y
McKinney, B. C., Grossman, A. W., Elisseou, N. M., and Greenough, W. T. (2005). Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136B, 98–102. doi: 10.1002/ajmg.b.30183
Meehan, R. R., Lewis, J. D., McKay, S., Kleiner, E. L., and Bird, A. P. (1989). Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58, 499–507. doi: 10.1016/0092-8674(89)90430-3
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Meikle, L., Pollizzi, K., Egnor, A., Kramvis, I., Lane, H., Sahin, M., et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 28, 5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008
Mendez, P., De Roo, M., Poglia, L., Klauser, P., and Muller, D. (2010). N-cadherin mediates plasticity-induced long-term spine stabilization. J. Cell Biol. 189, 589–600. doi: 10.1083/jcb.201003007
Menn, B., Timsit, S., Represa, A., Mateos, S., Calothy, G., and Lamballe, F. (2000). Spatiotemporal expression of noncatalytic TrkC NC2 isoform during early and late CNS neurogenesis: a comparative study with TrkC catalytic and p75NTR receptors. Eur. J. Neurosci. 12, 3211–3223. doi: 10.1046/j.1460-9568.2000.00215.x
Miao, S., Chen, R., Ye, J., Tan, G. H., Li, S., Zhang, J., et al. (2013). The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci. 33, 327–333. doi: 10.1523/JNEUROSCI.2509-12.2013
Michalon, A., Bruns, A., Risterucci, C., Honer, M., Ballard, T. M., Ozmen, L., et al. (2014). Chronic metabotropic glutamate receptor 5 inhibition corrects local alterations of brain activity and improves cognitive performance in fragile X mice. Biol. Psychiatry 75, 189–197. doi: 10.1016/j.biopsych.2013.05.038
Michalon, A., Sidorov, M., Ballard, T. M., Ozmen, L., Spooren, W., Wettstein, J. G., et al. (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. doi: 10.1016/j.neuron.2012.03.009
Minichiello, L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860. doi: 10.1038/nrn2738
Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R. E., Gottmann, K., et al. (2003). Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948. doi: 10.1038/nature01755
Moeller, M. J., Soofi, A., Braun, G. S., Li, X., Watzl, C., Kriz, W., et al. (2004). Protocadherin FAT1 binds Ena/VASP proteins and is necessary for actin dynamics and cell polarization. EMBO J. 23, 3769–3779. doi: 10.1038/sj.emboj.7600380
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297. doi: 10.1086/522590
Mohebiany, A. N., Harroch, S., and Bouyain, S. (2014). New insights into the roles of the contactin cell adhesion molecules in neural development. Adv. Neurobiol. 8, 165–194. doi: 10.1007/978-1-4614-8090-7_8
Mondin, M., Labrousse, V., Hosy, E., Heine, M., Tessier, B., Levet, F., et al. (2011). Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J. Neurosci. 31, 13500–13515. doi: 10.1523/JNEUROSCI.6439-10.2011
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., et al. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327. doi: 10.1523/JNEUROSCI.2623-05.2006
Morrow, E. M., Yoo, S. Y., Flavell, S. W., Kim, T. K., Lin, Y., Hill, R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi: 10.1126/science.1157657
Muddashetty, R. S., Kelic, S., Gross, C., Xu, M., and Bassell, G. J. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007
Mullins, C., Fishell, G., and Tsien, R. W. (2016). Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89, 1131–1156. doi: 10.1016/j.neuron.2016.02.017
Myers, R. A., Casals, F., Gauthier, J., Hamdan, F. F., Keebler, J., Boyko, A. R., et al. (2011). A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet. 7:e1001318. doi: 10.1371/journal.pgen.1001318
Na, E. S., Nelson, E. D., Adachi, M., Autry, A. E., Mahgoub, M. A., Kavalali, E. T., et al. (2012). A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 32, 3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012
Na, E. S., Nelson, E. D., Kavalali, E. T., and Monteggia, L. M. (2013). The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology 38, 212–219. doi: 10.1038/npp.2012.116
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/S0896-6273(00)80809-0
Nakamachi, T., Ohtaki, H., Yofu, S., Dohi, K., Watanabe, J., Hayashi, D., et al. (2008). Pituitary adenylate cyclase-activating polypeptide (PACAP) type 1 receptor (PAC1R) co-localizes with activity-dependent neuroprotective protein (ADNP) in the mouse brains. Regul. Pept. 145, 88–95. doi: 10.1016/j.regpep.2007.09.025
Nakamoto, M., Nalavadi, V., Epstein, M. P., Narayanan, U., Bassell, G. J., and Warren, S. T. (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 15537–15542. doi: 10.1073/pnas.0707484104
Nam, C. I., and Chen, L. (2005). Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc. Natl. Acad. Sci. U.S.A. 102, 6137–6142. doi: 10.1073/pnas.0502038102
Namjou, B., Marsolo, K., Caroll, R. J., Denny, J. C., Ritchie, M. D., Verma, S. S., et al. (2014). Phenome-wide association study (PheWAS) in EMR-linked pediatric cohorts, genetically links PLCL1 to speech language development and IL5-IL13 to Eosinophilic Esophagitis. Front. Genet. 5:401. doi: 10.3389/fgene.2014.00401
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/S0092-8674(00)81887-5
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., et al. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054. doi: 10.1016/j.cell.2008.07.031
Nava, C., Keren, B., Mignot, C., Rastetter, A., Chantot-Bastaraud, S., Faudet, A., et al. (2014). Prospective diagnostic analysis of copy number variants using SNP microarrays in individuals with autism spectrum disorders. Eur. J. Hum. Genet. 22, 71–78. doi: 10.1038/ejhg.2013.88
Nawaz, Z., Lonard, D. M., Smith, C. L., Lev-Lehman, E., Tsai, S. Y., Tsai, M. J., et al. (1999). The angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 19, 1182–1189. doi: 10.1128/MCB.19.2.1182
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Nelson, S. B., and Valakh, V. (2015). Excitatory/Inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Nelson, W. J. (2008). Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem. Soc. Trans. 36(Pt 2), 149–155. doi: 10.1042/BST0360149
Nemirovsky, S. I., Cordoba, M., Zaiat, J. J., Completa, S. P., Vega, P. A., Gonzalez-Moron, D., et al. (2015). Whole genome sequencing reveals a de novo SHANK3 mutation in familial autism spectrum disorder. PLoS ONE 10:e0116358. doi: 10.1371/journal.pone.0116358
Nestor, M. W., Phillips, A. W., Artimovich, E., Nestor, J. E., Hussman, J. P., and Blatt, G. J. (2015). Human inducible pluripotent stem cells and autism spectrum disorder: emerging technologies. Autism Res. 9, 513–535. doi: 10.1002/aur.1570
Newey, S. E., Velamoor, V., Govek, E. E., and Van Aelst, L. (2005). Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 64, 58–74. doi: 10.1002/neu.20153
Nguyen, M. V., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Nguyen, T., and Sudhof, T. C. (1997). Binding properties of neuroligin 1 and neurexin 1beta reveal function as heterophilic cell adhesion molecules. J. Biol. Chem. 272, 26032–26039. doi: 10.1074/jbc.272.41.26032
Nicholson, D. A., Cahill, M. E., Tulisiak, C. T., Geinisman, Y., and Penzes, P. (2012). Spatially restricted actin-regulatory signaling contributes to synapse morphology. J. Neurochem. 121, 852–860. doi: 10.1111/j.1471-4159.2012.07743.x
Niell, C. M., Meyer, M. P., and Smith, S. J. (2004). In vivo imaging of synapse formation on a growing dendritic arbor. Nat. Neurosci. 7, 254–260. doi: 10.1038/nn1191
Nimchinsky, E. A., Oberlander, A. M., and Svoboda, K. (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146.
Nord, A. S., Roeb, W., Dickel, D. E., Walsh, T., Kusenda, M., O’Connor, K. L., et al. (2011). Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 19, 727–731. doi: 10.1038/ejhg.2011.24
Noutel, J., Hong, Y. K., Leu, B., Kang, E., and Chen, C. (2011). Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron 70, 35–42. doi: 10.1016/j.neuron.2011.03.001
Nurmi, E. L., Bradford, Y., Chen, Y., Hall, J., Arnone, B., Gardiner, M. B., et al. (2001). Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 77, 105–113. doi: 10.1006/geno.2001.6617
Oguro-Ando, A., Rosensweig, C., Herman, E., Nishimura, Y., Werling, D., Bill, B. R., et al. (2015). Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol. Psychiatry 20, 1069–1078. doi: 10.1038/mp.2014.124
Oh, J. S., Manzerra, P., and Kennedy, M. B. (2004). Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 279, 17980–17988. doi: 10.1074/jbc.M314109200
Ohno, T., Maeda, H., Murabe, N., Kamiyama, T., Yoshioka, N., Mishina, M., et al. (2010). Specific involvement of postsynaptic GluN2B-containing NMDA receptors in the developmental elimination of corticospinal synapses. Proc. Natl. Acad. Sci. U.S.A. 107, 15252–15257. doi: 10.1073/pnas.0906551107
Ohoka, Y., and Takai, Y. (1998). Isolation and characterization of cortactin isoforms and a novel cortactin-binding protein, CBP90. Genes Cells 3, 603–612. doi: 10.1046/j.1365-2443.1998.00216.x
Okui, M., Ide, T., Morita, K., Funakoshi, E., Ito, F., Ogita, K., et al. (1999). High-level expression of the Mnb/Dyrk1A gene in brain and heart during rat early development. Genomics 62, 165–171. doi: 10.1006/geno.1999.5998
Oliver, T. N., Berg, J. S., and Cheney, R. E. (1999). Tails of unconventional myosins. Cell Mol. Life. Sci. 56, 243–257. doi: 10.1007/s000180050426
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
O’Roak, B. J., Stessman, H. A., Boyle, E. A., Witherspoon, K. T., Martin, B., Lee, C., et al. (2014). Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 5:5595. doi: 10.1038/ncomms6595
O’Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Orrico, A., Galli, L., Buoni, S., Orsi, A., Vonella, G., and Sorrentino, V. (2009). Novel PTEN mutations in neurodevelopmental disorders and macrocephaly. Clin. Genet. 75, 195–198. doi: 10.1111/j.1399-0004.2008.01074.x
Osterweil, E. K., Krueger, D. D., Reinhold, K., and Bear, M. F. (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 30, 15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010
Oz, S., Kapitansky, O., Ivashco-Pachima, Y., Malishkevich, A., Giladi, E., Skalka, N., et al. (2014). The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 19, 1115–1124. doi: 10.1038/mp.2014.97
Ozgen, H. M., van Daalen, E., Bolton, P. F., Maloney, V. K., Huang, S., Cresswell, L., et al. (2009). Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin. Genet. 76, 348–356. doi: 10.1111/j.1399-0004.2009.01254.x
Ozkan, E. D., Creson, T. K., Kramar, E. A., Rojas, C., Seese, R. R., Babyan, A. H., et al. (2014). Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 82, 1317–1333. doi: 10.1016/j.neuron.2014.05.015
Paemka, L., Mahajan, V. B., Skeie, J. M., Sowers, L. P., Ehaideb, S. N., Gonzalez-Alegre, P., et al. (2013). PRICKLE1 interaction with SYNAPSIN I reveals a role in autism spectrum disorders. PLoS ONE 8:e80737. doi: 10.1371/journal.pone.0080737
Pagnamenta, A. T., Khan, H., Walker, S., Gerrelli, D., Wing, K., Bonaglia, M. C., et al. (2011). Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J. Med. Genet. 48, 48–54. doi: 10.1136/jmg.2010.079426
Pahl, S., Tapken, D., Haering, S. C., and Hollmann, M. (2014). Trafficking of kainate receptors. Membranes 4, 565–595. doi: 10.3390/membranes4030565
Pan, F., Aldridge, G. M., Greenough, W. T., and Gan, W. B. (2010). Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 17768–17773. doi: 10.1073/pnas.1012496107
Pan, Y., Chen, J., Guo, H., Ou, J., Peng, Y., Liu, Q., et al. (2015). Association of genetic variants of GRIN2B with autism. Sci. Rep. 5:8296. doi: 10.1038/srep08296
Paradis, S., Harrar, D. B., Lin, Y., Koon, A. C., Hauser, J. L., Griffith, E. C., et al. (2007). An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron 53, 217–232. doi: 10.1016/j.neuron.2006.12.012
Park, J., Sung, J. Y., Park, J., Song, W. J., Chang, S., and Chung, K. C. (2012). Dyrk1A negatively regulates the actin cytoskeleton through threonine phosphorylation of N-WASP. J. Cell Sci. 125(Pt 1), 67–80. doi: 10.1242/jcs.086124
Patel, K. G., Liu, C., Cameron, P. L., and Cameron, R. S. (2001). Myr 8, a novel unconventional myosin expressed during brain development associates with the protein phosphatase catalytic subunits 1alpha and 1gamma1. J. Neurosci. 21, 7954–7968.
Pathania, M., Davenport, E. C., Muir, J., Sheehan, D. F., Lopez-Domenech, G., and Kittler, J. T. (2014). The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl. Psychiatry 4:e374. doi: 10.1038/tp.2014.16
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peixoto, R. T., Wang, W., Croney, D. M., Kozorovitskiy, Y., and Sabatini, B. L. (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B mice. Nat. Neurosci. 19, 716–724. doi: 10.1038/nn.4260
Penagarikano, O. (2015). New therapeutic options for autism spectrum disorder: experimental evidences. Exp. Neurobiol. 24, 301–311. doi: 10.5607/en.2015.24.4.301
Peñagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Penzes, P., and Cahill, M. E. (2012). Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton (Hoboken) 69, 426–441. doi: 10.1002/cm.21015
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Penzes, P., and Rafalovich, I. (2012). Regulation of the actin cytoskeleton in dendritic spines. Adv. Exp. Med. Biol. 970, 81–95. doi: 10.1007/978-3-7091-0932-8_4
Perandones, C., Costanzo, R. V., Kowaljow, V., Pivetta, O. H., Carminatti, H., and Radrizzani, M. (2004). Correlation between synaptogenesis and the PTEN phosphatase expression in dendrites during postnatal brain development. Brain Res. Mol. Brain Res. 128, 8–19. doi: 10.1016/j.molbrainres.2004.05.021
Peters, S. U., Hundley, R. J., Wilson, A. K., Warren, Z., Vehorn, A., Carvalho, C. M., et al. (2013). The behavioral phenotype in MECP2 duplication syndrome: a comparison with idiopathic autism. Autism Res. 6, 42–50. doi: 10.1002/aur.1262
Petit, F., Plessis, G., Decamp, M., Cuisset, J. M., Blyth, M., Pendlebury, M., et al. (2015). 21q21 deletion involving NCAM2: report of 3 cases with neurodevelopmental disorders. Eur. J. Med. Genet. 58, 44–46. doi: 10.1016/j.ejmg.2014.11.004
Petralia, R. S., Wang, Y. X., and Wenthold, R. J. (1994). The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J. Neurosci. 14, 6102–6120.
Petrin, A. L., Giacheti, C. M., Maximino, L. P., Abramides, D. V., Zanchetta, S., Rossi, N. F., et al. (2010). Identification of a microdeletion at the 7q33-q35 disrupting the CNTNAP2 gene in a Brazilian stuttering case. Am. J. Med. Genet. A 152A, 3164–3172. doi: 10.1002/ajmg.a.33749
Phillips, M., and Pozzo-Miller, L. (2015). Dendritic spine dysgenesis in autism related disorders. Neurosci. Lett. 601, 30–40. doi: 10.1016/j.neulet.2015.01.011
Pignatelli, M., Piccinin, S., Molinaro, G., Di Menna, L., Riozzi, B., Cannella, M., et al. (2014). Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling angelman syndrome. J. Neurosci. 34, 4558–4566. doi: 10.1523/JNEUROSCI.1846-13.2014
Pinhasov, A., Mandel, S., Torchinsky, A., Giladi, E., Pittel, Z., Goldsweig, A. M., et al. (2003). Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res. Dev. Brain Res. 144, 83–90. doi: 10.1016/S0165-3806(03)00162-7
Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694. doi: 10.1016/j.ajhg.2014.03.018
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Piton, A., Jouan, L., Rochefort, D., Dobrzeniecka, S., Lachapelle, K., Dion, P. A., et al. (2013). Analysis of the effects of rare variants on splicing identifies alterations in GABAA receptor genes in autism spectrum disorder individuals. Eur. J. Hum. Genet. 21, 749–756. doi: 10.1038/ejhg.2012.243
Pollard, T. D., and Korn, E. D. (1973). Acanthamoeba myosin. I. Isolation from Acanthamoeba castellanii of an enzyme similar to muscle myosin. J. Biol. Chem. 248, 4682–4690.
Poot, M. (2014). A candidate gene association study further corroborates involvement of contactin genes in autism. Mol. Syndromol. 5, 229–235. doi: 10.1159/000362891
Poot, M., Beyer, V., Schwaab, I., Damatova, N., Van’t Slot, R., Prothero, J., et al. (2010). Disruption of CNTNAP2 and additional structural genome changes in a boy with speech delay and autism spectrum disorder. Neurogenetics 11, 81–89. doi: 10.1007/s10048-009-0205-1
Pop, A. S., Levenga, J., de Esch, C. E., Buijsen, R. A., Nieuwenhuizen, I. M., Li, T., et al. (2014). Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology 231, 1227–1235. doi: 10.1007/s00213-012-2947-y
Povey, S., Burley, M. W., Attwood, J., Benham, F., Hunt, D., Jeremiah, S. J., et al. (1994). Two loci for tuberous sclerosis: one on 9q34 and one on 16p13. Ann. Hum. Genet. 58(Pt 2), 107–127. doi: 10.1111/j.1469-1809.1994.tb01881.x
Prandini, P., Pasquali, A., Malerba, G., Marostica, A., Zusi, C., Xumerle, L., et al. (2012). The association of rs4307059 and rs35678 markers with autism spectrum disorders is replicated in Italian families. Psychiatr. Genet. 22, 177–181. doi: 10.1097/YPG.0b013e32835185c9
Prange, O., Wong, T. P., Gerrow, K., Wang, Y. T., and El-Husseini, A. (2004). A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. U.S.A. 101, 13915–13920. doi: 10.1073/pnas.0405939101
Prasad, A., Merico, D., Thiruvahindrapuram, B., Wei, J., Lionel, A. C., Sato, D., et al. (2012). A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 2, 1665–1685. doi: 10.1534/g3.112.004689
Price, T. J., Rashid, M. H., Millecamps, M., Sanoja, R., Entrena, J. M., and Cervero, F. (2007). Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J. Neurosci. 27, 13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007
Pun, R. Y., Rolle, I. J., Lasarge, C. L., Hosford, B. E., Rosen, J. M., Uhl, J. D., et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. doi: 10.1016/j.neuron.2012.08.002
Qin, M., Entezam, A., Usdin, K., Huang, T., Liu, Z. H., Hoffman, G. E., et al. (2011). A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol. Dis. 42, 85–98. doi: 10.1016/j.nbd.2011.01.008
Qiu, Z., Sylwestrak, E. L., Lieberman, D. N., Zhang, Y., Liu, X. Y., and Ghosh, A. (2012). The Rett syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci. 32, 989–994. doi: 10.1523/JNEUROSCI.0175-11.2012
Qualmann, B., Boeckers, T. M., Jeromin, M., Gundelfinger, E. D., and Kessels, M. M. (2004). Linkage of the actin cytoskeleton to the postsynaptic density via direct interactions of Abp1 with the ProSAP/Shank family. J. Neurosci. 24, 2481–2495. doi: 10.1523/JNEUROSCI.5479-03.2004
Quitsch, A., Berhorster, K., Liew, C. W., Richter, D., and Kreienkamp, H. J. (2005). Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J. Neurosci. 25, 479–487. doi: 10.1523/JNEUROSCI.2699-04.2005
Rajan, I., Witte, S., and Cline, H. T. (1999). NMDA receptor activity stabilizes presynaptic retinotectal axons and postsynaptic optic tectal cell dendrites in vivo. J. Neurobiol. 38, 357–368. doi: 10.1002/(SICI)1097-4695(19990215)38:3<357::AID-NEU5>3.0.CO;2-#
Ramocki, M. B., Peters, S. U., Tavyev, Y. J., Zhang, F., Carvalho, C. M., Schaaf, C. P., et al. (2009). Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 66, 771–782. doi: 10.1002/ana.21715
Raymond, G. V., Bauman, M. L., and Kemper, T. L. (1996). Hippocampus in autism: a Golgi analysis. Acta Neuropathol. 91, 117–119. doi: 10.1007/s004010050401
Redfern, R. E., Daou, M. C., Li, L., Munson, M., Gericke, A., and Ross, A. H. (2010). A mutant form of PTEN linked to autism. Protein Sci 19, 1948–1956. doi: 10.1002/pro.483
Redin, C., Gerard, B., Lauer, J., Herenger, Y., Muller, J., Quartier, A., et al. (2014). Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 51, 724–736. doi: 10.1136/jmedgenet-2014-102554
Reiter, L. T., Seagroves, T. N., Bowers, M., and Bier, E. (2006). Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum. Mol. Genet. 15, 2825–2835. doi: 10.1093/hmg/ddl225
Reith, R. M., McKenna, J., Wu, H., Hashmi, S. S., Cho, S. H., Dash, P. K., et al. (2013). Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 51, 93–103. doi: 10.1016/j.nbd.2012.10.014
Ricciardi, S., Boggio, E. M., Grosso, S., Lonetti, G., Forlani, G., Stefanelli, G., et al. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 20, 1182–1196. doi: 10.1093/hmg/ddq563
Roberts, J. L., Hovanes, K., Dasouki, M., Manzardo, A. M., and Butler, M. G. (2014). Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 535, 70–78. doi: 10.1016/j.gene.2013.10.020
Rogers, S. J., Wehner, D. E., and Hagerman, R. (2001). The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 22, 409–417. doi: 10.1097/00004703-200112000-00008
Ronemus, M., Iossifov, I., Levy, D., and Wigler, M. (2014). The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 15, 133–141. doi: 10.1038/nrg3585
Ronesi, J. A., Collins, K. A., Hays, S. A., Tsai, N. P., Guo, W., Birnbaum, S. G., et al. (2012). Disrupted homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci. 431–440, S431. doi: 10.1038/nn.3033
Roohi, J., Montagna, C., Tegay, D. H., Palmer, L. E., DeVincent, C., Pomeroy, J. C., et al. (2009). Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 46, 176–182. doi: 10.1136/jmg.2008.057505
Rougon, G., and Hobert, O. (2003). New insights into the diversity and function of neuronal immunoglobulin superfamily molecules. Annu. Rev. Neurosci. 26, 207–238. doi: 10.1146/annurev.neuro.26.041002.131014
Roussignol, G., Ango, F., Romorini, S., Tu, J. C., Sala, C., Worley, P. F., et al. (2005). Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 25, 3560–3570. doi: 10.1523/JNEUROSCI.4354-04.2005
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183X.2003.00037.x
Rumbaugh, G., Adams, J. P., Kim, J. H., and Huganir, R. L. (2006). SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 4344–4351. doi: 10.1073/pnas.0600084103
Rusconi, L., Salvatoni, L., Giudici, L., Bertani, I., Kilstrup-Nielsen, C., Broccoli, V., et al. (2008). CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 283, 30101–30111. doi: 10.1074/jbc.M804613200
Russo, S., Marchi, M., Cogliati, F., Bonati, M. T., Pintaudi, M., Veneselli, E., et al. (2009). Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 10, 241–250. doi: 10.1007/s10048-009-0177-1
Ryan, T. J., Kopanitsa, M. V., Indersmitten, T., Nithianantharajah, J., Afinowi, N. O., Pettit, C., et al. (2013). Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior. Nat. Neurosci. 16, 25–32. doi: 10.1038/nn.3277
Ryu, J., Liu, L., Wong, T. P., Wu, D. C., Burette, A., Weinberg, R., et al. (2006). A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron 49, 175–182. doi: 10.1016/j.neuron.2005.12.017
Sadeqzadeh, E., de Bock, C. E., and Thorne, R. F. (2014). Sleeping giants: emerging roles for the fat cadherins in health and disease. Med Res. Rev. 34, 190–221. doi: 10.1002/med.21286
Sakisaka, T., Baba, T., Tanaka, S., Izumi, G., Yasumi, M., and Takai, Y. (2004). Regulation of SNAREs by tomosyn and ROCK: implication in extension and retraction of neurites. J. Cell Biol. 166, 17–25. doi: 10.1083/jcb.200405002
Sakurai, K., Toyoshima, M., Takeda, Y., Shimoda, Y., and Watanabe, K. (2010). Synaptic formation in subsets of glutamatergic terminals in the mouse hippocampal formation is affected by a deficiency in the neural cell recognition molecule NB-3. Neurosci. Lett. 473, 102–106. doi: 10.1016/j.neulet.2010.02.027
Sakurai, K., Toyoshima, M., Ueda, H., Matsubara, K., Takeda, Y., Karagogeos, D., et al. (2009). Contribution of the neural cell recognition molecule NB-3 to synapse formation between parallel fibers and Purkinje cells in mouse. Dev. Neurobiol. 69, 811–824. doi: 10.1002/dneu.20742
Sala, C., Piech, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/S0896-6273(01)00339-7
Sampath, S., Bhat, S., Gupta, S., O’Connor, A., West, A. B., Arking, D. E., et al. (2013). Defining the contribution of CNTNAP2 to autism susceptibility. PLoS ONE 8:e77906. doi: 10.1371/journal.pone.0077906
Sanders, S. J. (2015). First glimpses of the neurobiology of autism spectrum disorder. Curr. Opin. Genet. Dev. 33, 80–92. doi: 10.1016/j.gde.2015.10.002
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. doi: 10.1016/j.neuron.2015.09.016
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Saneyoshi, T., and Hayashi, Y. (2012). The Ca2+ and Rho GTPase signaling pathways underlying activity-dependent actin remodeling at dendritic spines. Cytoskeleton 69, 545–554. doi: 10.1002/cm.21037
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Sato, M., and Stryker, M. P. (2010). Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. U.S.A. 107, 5611–5616. doi: 10.1073/pnas.1001281107
Scala, E., Ariani, F., Mari, F., Caselli, R., Pescucci, C., Longo, I., et al. (2005). CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 42, 103–107. doi: 10.1136/jmg.2004.026237
Schaaf, C. P., Boone, P. M., Sampath, S., Williams, C., Bader, P. I., Mueller, J. M., et al. (2012). Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur. J. Hum. Genet. 20, 1240–1247. doi: 10.1038/ejhg.2012.95
Schaaf, C. P., Sabo, A., Sakai, Y., Crosby, J., Muzny, D., Hawes, A., et al. (2011). Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 20, 3366–3375. doi: 10.1093/hmg/ddr243
Scheiffele, P., Fan, J., Choih, J., Fetter, R., and Serafini, T. (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. doi: 10.1016/S0092-8674(00)80877-6
Schreiner, D., Müller, K., and Hofer, H. W. (2006). The intracellular domain of the human protocadherin hFat1 interacts with Homer signalling scaffolding proteins. FEBS Lett. 580, 5295–5300. doi: 10.1016/j.febslet.2006.08.079
Schwerd, T., Khaled, A. V., Schurmann, M., Chen, H., Handel, N., Reis, A., et al. (2016). A recessive form of extreme macrocephaly and mild intellectual disability complements the spectrum of PTEN hamartoma tumour syndrome. Eur. J. Hum. Genet. 24, 889–894. doi: 10.1038/ejhg.2015.209
Segal, M., Kreher, U., Greenberger, V., and Braun, K. (2003). Is fragile X mental retardation protein involved in activity-induced plasticity of dendritic spines? Brain Res. 972, 9–15. doi: 10.1016/S0006-8993(03)02410-7
Seong, E., Yuan, L., and Arikkath, J. (2015). Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh. Migr. 9, 202–213. doi: 10.4161/19336918.2014.994919
Sepulveda, F. J., Bustos, F. J., Inostroza, E., Zuniga, F. A., Neve, R. L., Montecino, M., et al. (2010). Differential roles of NMDA receptor subtypes NR2A and NR2B in dendritic branch development and requirement of RasGRF1. J. Neurophysiol. 103, 1758–1770. doi: 10.1152/jn.00823.2009
Serajee, F. J., Nabi, R., Zhong, H., and Mahbubul Huq, A. H. (2003). Association of INPP1, PIK3CG, and TSC2 gene variants with autistic disorder: implications for phosphatidylinositol signalling in autism. J. Med. Genet. 40:e119. doi: 10.1136/jmg.40.11.e119
Sfakianos, M. K., Eisman, A., Gourley, S. L., Bradley, W. D., Scheetz, A. J., Settleman, J., et al. (2007). Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J. Neurosci. 27, 10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007
Shahbazian, M. D., and Zoghbi, H. Y. (2001). Molecular genetics of Rett syndrome and clinical spectrum of MECP2 mutations. Curr. Opin. Neurol. 14, 171–176. doi: 10.1097/00019052-200104000-00006
Sheng, L., Leshchyns’ka, I., and Sytnyk, V. (2015). Neural cell adhesion molecule 2 promotes the formation of filopodia and neurite branching by inducing submembrane increases in Ca2+ levels. J. Neurosci. 35, 1739–1752. doi: 10.1523/JNEUROSCI.1714-14.2015
Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N., and Jan, L. Y. (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. doi: 10.1038/368144a0
Sheng, M., and Hoogenraad, C. C. (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76, 823–847. doi: 10.1146/annurev.biochem.76.060805.160029
Sheng, M., and Kim, E. (2000). The shank family of scaffold proteins. J. Cell Sci. 113(Pt 11), 1851–1856.
Shi, L., Zhang, X., Golhar, R., Otieno, F. G., He, M., Hou, C., et al. (2013). Whole-genome sequencing in an autism multiplex family. Mol. Autism 4:8. doi: 10.1186/2040-2392-4-8
Shibayama, A., Cook, E. H. Jr., Feng, J., Glanzmann, C., Yan, J., Craddock, N., et al. (2004). MECP2 structural and 3’-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 128B, 50–53. doi: 10.1002/ajmg.b.30016
Shih, P. Y., Lee, S. P., Chen, Y. K., and Hsueh, Y. P. (2014). Cortactin-binding protein 2 increases microtubule stability and regulates dendritic arborization. J. Cell Sci. 127(Pt 16), 3521–3534. doi: 10.1242/jcs.149476
Shimoda, Y., and Watanabe, K. (2009). Contactins: emerging key roles in the development and function of the nervous system. Cell. Adh. Migr. 3, 64–70. doi: 10.4161/cam.3.1.7764
Shimoyama, Y., Tsujimoto, G., Kitajima, M., and Natori, M. (2000). Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. Biochem. J. 349(Pt 1), 159–167. doi: 10.1042/bj3490159
Shindoh, N., Kudoh, J., Maeda, H., Yamaki, A., Minoshima, S., Shimizu, Y., et al. (1996). Cloning of a human homolog of the Drosophila minibrain/rat Dyrk gene from “the Down syndrome critical region” of chromosome 21. Biochem. Biophys. Res. Commun. 225, 92–99. doi: 10.1006/bbrc.1996.1135
Shirao, T., and Gonzalez-Billault, C. (2013). Actin filaments and microtubules in dendritic spines. J. Neurochem. 126, 155–164. doi: 10.1111/jnc.12313
Shuang, M., Liu, J., Jia, M. X., Yang, J. Z., Wu, S. P., Gong, X. H., et al. (2004). Family-based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am. J. Med. Genet. B Neuropsychiatr. Genet. 131B, 48–50. doi: 10.1002/ajmg.b.30025
Siomi, H., Siomi, M. C., Nussbaum, R. L., and Dreyfuss, G. (1993). The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298. doi: 10.1016/0092-8674(93)90420-U
Smalley, S. L. (1998). Autism and tuberous sclerosis. J. Autism. Dev. Disord. 28, 407–414. doi: 10.1023/A:1026052421693
Smith, S. E., Zhou, Y. D., Zhang, G., Jin, Z., Stoppel, D. C., and Anderson, M. P. (2011). Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3:103ra197. doi: 10.1126/scitranslmed.3002627
Song, J. Y., Ichtchenko, K., Südhof, T. C., and Brose, N. (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. U.S.A. 96, 1100–1105. doi: 10.1073/pnas.96.3.1100
Spinelli, L., Black, F. M., Berg, J. N., Eickholt, B. J., and Leslie, N. R. (2015). Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J. Med. Genet. 52, 128–134. doi: 10.1136/jmedgenet-2014-102803
Sprovieri, T., Conforti, F. L., Fiumara, A., Mazzei, R., Ungaro, C., Citrigno, L., et al. (2009). A novel mutation in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene associated with a severe Rett phenotype. Am. J. Med. Genet. A 149A, 722–725. doi: 10.1002/ajmg.a.32711
Stambolic, V., Suzuki, A., de la Pompa, J. L., Brothers, G. M., Mirtsos, C., Sasaki, T., et al. (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39. doi: 10.1016/S0092-8674(00)81780-8
Stamou, M., Streifel, K. M., Goines, P. E., and Lein, P. J. (2013). Neuronal connectivity as a convergent target of gene x environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol. Teratol. 36, 3–16. doi: 10.1016/j.ntt.2012.12.001
Stankiewicz, T. R., and Linseman, D. A. (2014). Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell Neurosci. 8:314. doi: 10.3389/fncel.2014.00314
Stein, M. T., Elias, E. R., Saenz, M., Pickler, L., and Reynolds, A. (2010). Autistic spectrum disorder in a 9-year-old girl with macrocephaly. J. Dev. Behav. Pediatr. 31, 632–634. doi: 10.1097/DBP.0b013e3181ef422a
Steinberg, K. M., Ramachandran, D., Patel, V. C., Shetty, A. C., Cutler, D. J., and Zwick, M. E. (2012). Identification of rare X-linked neuroligin variants by massively parallel sequencing in males with autism spectrum disorder. Mol. Autism 3:8. doi: 10.1186/2040-2392-3-8
Stuss, D. P., Boyd, J. D., Levin, D. B., and Delaney, K. R. (2012). MeCP2 mutation results in compartment-specific reductions in dendritic branching and spine density in layer 5 motor cortical neurons of YFP-H mice. PLoS ONE 7:e31896. doi: 10.1371/journal.pone.0031896
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Suzuki, S. C., Furue, H., Koga, K., Jiang, N., Nohmi, M., Shimazaki, Y., et al. (2007). Cadherin-8 is required for the first relay synapses to receive functional inputs from primary sensory afferents for cold sensation. J. Neurosci. 27, 3466–3476. doi: 10.1523/JNEUROSCI.0243-07.2007
Suzuki, S. C., and Takeichi, M. (2008). Cadherins in neuronal morphogenesis and function. Dev. Growth. Differ. 50(Suppl. 1), S119–S130. doi: 10.1111/j.1440-169X.2008.01002.x
Svitkina, T., Lin, W. H., Webb, D. J., Yasuda, R., Wayman, G. A., Van Aelst, L., et al. (2010). Regulation of the postsynaptic cytoskeleton: roles in development, plasticity, and disorders. J. Neurosci. 30, 14937–14942. doi: 10.1523/JNEUROSCI.4276-10.2010
Swanberg, S. E., Nagarajan, R. P., Peddada, S., Yasui, D. H., and LaSalle, J. M. (2009). Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum. Mol. Genet. 18, 525–534. doi: 10.1093/hmg/ddn380
Szafranski, P., Golla, S., Jin, W., Fang, P., Hixson, P., Matalon, R., et al. (2015). Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur. J. Hum. Genet. 23, 915–921. doi: 10.1038/ejhg.2014.217
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Takahashi, H., Arstikaitis, P., Prasad, T., Bartlett, T. E., Wang, Y. T., Murphy, T. H., et al. (2011). Postsynaptic TrkC and presynaptic PTPsigma function as a bidirectional excitatory synaptic organizing complex. Neuron 69, 287–303. doi: 10.1016/j.neuron.2010.12.024
Takahashi, H., and Craig, A. M. (2013). Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 36, 522–534. doi: 10.1016/j.tins.2013.06.002
Takeda, Y., Akasaka, K., Lee, S., Kobayashi, S., Kawano, H., Murayama, S., et al. (2003). Impaired motor coordination in mice lacking neural recognition molecule NB-3 of the contactin/F3 subgroup. J. Neurobiol. 56, 252–265. doi: 10.1002/neu.10222
Takeichi, M., and Abe, K. (2005). Synaptic contact dynamics controlled by cadherin and catenins. Trends Cell Biol 15, 216–221. doi: 10.1016/j.tcb.2005.02.002
Takeuchi, M., Hata, Y., Hirao, K., Toyoda, A., Irie, M., and Takai, Y. (1997). SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J. Biol. Chem. 272, 11943–11951. doi: 10.1074/jbc.272.18.11943
Talkowski, M. E., Rosenfeld, J. A., Blumenthal, I., Pillalamarri, V., Chiang, C., Heilbut, A., et al. (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537. doi: 10.1016/j.cell.2012.03.028
Tammimies, K., Marshall, C. R., Walker, S., Kaur, G., Thiruvahindrapuram, B., Lionel, A. C., et al. (2015). Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903. doi: 10.1001/jama.2015.10078
Tan, Z. J., Peng, Y., Song, H. L., Zheng, J. J., and Yu, X. (2010). N-cadherin-dependent neuron-neuron interaction is required for the maintenance of activity-induced dendrite growth. Proc. Natl. Acad. Sci. U.S.A. 107, 9873–9878. doi: 10.1073/pnas.1003480107
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tanihara, H., Sano, K., Heimark, R. L., St John, T., and Suzuki, S. (1994). Cloning of five human cadherins clarifies characteristic features of cadherin extracellular domain and provides further evidence for two structurally different types of cadherin. Cell Adh. Commun. 2, 15–26. doi: 10.3109/15419069409014199
Tanoue, T., and Takeichi, M. (2004). Mammalian Fat1 cadherin regulates actin dynamics and cell-cell contact. J. Cell Biol. 165, 517–528. doi: 10.1083/jcb.200403006
Tanoue, T., and Takeichi, M. (2005). New insights into Fat cadherins. J. Cell Sci. 118(Pt 11), 2347–2353. doi: 10.1242/jcs.02398
Tarabeux, J., Kebir, O., Gauthier, J., Hamdan, F. F., Xiong, L., Piton, A., et al. (2011). Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 1:e55. doi: 10.1038/tp.2011.52
Tashiro, A., Dunaevsky, A., Blazeski, R., Mason, C. A., and Yuste, R. (2003). Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: a two-step model of synaptogenesis. Neuron 38, 773–784. doi: 10.1016/S0896-6273(03)00299-X
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
Thomazeau, A., Lassalle, O., Iafrati, J., Souchet, B., Guedj, F., Janel, N., et al. (2014). Prefrontal deficits in a murine model overexpressing the down syndrome candidate gene dyrk1a. J. Neurosci. 34, 1138–1147. doi: 10.1523/JNEUROSCI.2852-13.2014
Tilot, A. K., Bebek, G., Niazi, F., Altemus, J. B., Romigh, T., Frazier, T. W., et al. (2016). Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Mol. Psychiatry 21, 118–125. doi: 10.1038/mp.2015.17
Ting, J. T., Peça, J., and Feng, G. (2012). Functional consequences of mutations in postsynaptic scaffolding proteins and relevance to psychiatric disorders. Annu. Rev. Neurosci. 35, 49–71. doi: 10.1146/annurev-neuro-062111-150442
Togashi, H., Abe, K., Mizoguchi, A., Takaoka, K., Chisaka, O., and Takeichi, M. (2002). Cadherin regulates dendritic spine morphogenesis. Neuron 35, 77–89. doi: 10.1016/S0896-6273(02)00748-1
Tolias, K. F., Duman, J. G., and Um, K. (2011). Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog. Neurobiol. 94, 133–148. doi: 10.1016/j.pneurobio.2011.04.011
Toma, C., Torrico, B., Hervas, A., Valdes-Mas, R., Tristan-Noguero, A., Padillo, V., et al. (2014). Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 19, 784–790. doi: 10.1038/mp.2013.106
Toyoshima, M., Sakurai, K., Shimazaki, K., Takeda, Y., Shimoda, Y., and Watanabe, K. (2009). Deficiency of neural recognition molecule NB-2 affects the development of glutamatergic auditory pathways from the ventral cochlear nucleus to the superior olivary complex in mouse. Dev. Biol. 336, 192–200. doi: 10.1016/j.ydbio.2009.09.043
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., et al. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794. doi: 10.1038/nature01273
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034. doi: 10.1073/pnas.0812394106
Tsai, N. P., Wilkerson, J. R., Guo, W., Maksimova, M. A., DeMartino, G. N., Cowan, C. W., et al. (2012a). Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594. doi: 10.1016/j.cell.2012.11.040
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012b). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/S0896-6273(00)80810-7
Tucker, B., Richards, R. I., and Lardelli, M. (2006). Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 15, 3446–3458. doi: 10.1093/hmg/ddl422
Tyska, M. J., and Warshaw, D. M. (2002). The myosin power stroke. Cell Motil. Cytoskeleton 51, 1–15. doi: 10.1002/cm.10014
Uemura, M., Nakao, S., Suzuki, S. T., Takeichi, M., and Hirano, S. (2007). OL-Protocadherin is essential for growth of striatal axons and thalamocortical projections. Nat. Neurosci. 10, 1151–1159. doi: 10.1038/nn1960
Ultanir, S. K., Yadav, S., Hertz, N. T., Oses-Prieto, J. A., Claxton, S., Burlingame, A. L., et al. (2014). MST3 kinase phosphorylates TAO1/2 to enable Myosin Va function in promoting spine synapse development. Neuron 84, 968–982. doi: 10.1016/j.neuron.2014.10.025
Urraca, N., Cleary, J., Brewer, V., Pivnick, E. K., McVicar, K., Thibert, R. L., et al. (2013). The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 6, 268–279. doi: 10.1002/aur.1284
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S., et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141. doi: 10.1016/j.ajhg.2011.11.025
Valenzuela, D. M., Maisonpierre, P. C., Glass, D. J., Rojas, E., Nunez, L., Kong, Y., et al. (1993). Alternative forms of rat TrkC with different functional capabilities. Neuron 10, 963–974. doi: 10.1016/0896-6273(93)90211-9
Valluy, J., Bicker, S., Aksoy-Aksel, A., Lackinger, M., Sumer, S., Fiore, R., et al. (2015). A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci. 18, 666–673. doi: 10.1038/nn.3996
van Bon, B. W., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2016). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132. doi: 10.1038/mp.2015.5
van Bon, B. W. M., Coe, B. P., de Vries, B. B. A., and Eichler, E. E. (1993). “DYRK1A-Related Intellectual Disability Syndrome,” in GeneReviews(R), eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H. Bean, et al. (Seattle, WA: University of Washington).
van Daalen, E., Kemner, C., Verbeek, N. E., van der Zwaag, B., Dijkhuizen, T., Rump, P., et al. (2011). Social responsiveness scale-aided analysis of the clinical impact of copy number variations in autism. Neurogenetics 12, 315–323. doi: 10.1007/s10048-011-0297-2
Van den Veyver, I. B., and Zoghbi, H. Y. (2001). Mutations in the gene encoding methyl-CpG-binding protein 2 cause Rett syndrome. Brain Dev. 23(Suppl. 1), S147–S151. doi: 10.1016/S0387-7604(01)00376-X
van der Zwaag, B., Staal, W. G., Hochstenbach, R., Poot, M., Spierenburg, H. A., de Jonge, M. V., et al. (2010). A co-segregating microduplication of chromosome 15q11.2 pinpoints two risk genes for autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 960–966. doi: 10.1002/ajmg.b.31055
van Harssel, J. J., Weckhuysen, S., van Kempen, M. J., Hardies, K., Verbeek, N. E., de Kovel, C. G., et al. (2013). Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders. Neurogenetics 14, 23–34. doi: 10.1007/s10048-013-0353-1
van Slegtenhorst, M., de Hoogt, R., Hermans, C., Nellist, M., Janssen, B., Verhoef, S., et al. (1997). Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. doi: 10.1126/science.277.5327.805
van Slegtenhorst, M., Nellist, M., Nagelkerken, B., Cheadle, J., Snell, R., van den Ouweland, A., et al. (1998). Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7, 1053–1057. doi: 10.1093/hmg/7.6.1053
Vanderver, A., Tonduti, D., Kahn, I., Schmidt, J., Medne, L., Vento, J., et al. (2014). Characteristic brain magnetic resonance imaging pattern in patients with macrocephaly and PTEN mutations. Am. J. Med. Genet. A 164A, 627–633. doi: 10.1002/ajmg.a.36309
Vandeweyer, G., Helsmoortel, C., Van Dijck, A., Vulto-van Silfhout, A. T., Coe, B. P., Bernier, R., et al. (2014). The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 315–326. doi: 10.1002/ajmg.c.31413
Vardarajan, B. N., Eran, A., Jung, J. Y., Kunkel, L. M., and Wall, D. P. (2013). Haplotype structure enables prioritization of common markers and candidate genes in autism spectrum disorder. Transl. Psychiatry 3:e262. doi: 10.1038/tp.2013.38
Varea, O., Martin-de-Saavedra, M. D., Kopeikina, K. J., Schurmann, B., Fleming, H. J., Fawcett-Patel, J. M., et al. (2015). Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc. Natl. Acad. Sci. U.S.A. 112, 6176–6181. doi: 10.1073/pnas.1423205112
Varga, E. A., Pastore, M., Prior, T., Herman, G. E., and McBride, K. L. (2009). The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 11, 111–117. doi: 10.1097/GIM.0b013e31818fd762
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., et al. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. doi: 10.1016/j.neuron.2006.09.003
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 83, 449–456. doi: 10.1078/0171-9335-00410
Vazquez, F., and Sellers, W. R. (2000). The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling. Biochim. Biophys. Acta 1470, M21–M35.
Vazquez, L. E., Chen, H. J., Sokolova, I., Knuesel, I., and Kennedy, M. B. (2004). SynGAP regulates spine formation. J. Neurosci. 24, 8862–8872. doi: 10.1523/JNEUROSCI.3213-04.2004
Veeman, M. T., Slusarski, D. C., Kaykas, A., Louie, S. H., and Moon, R. T. (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 13, 680–685. doi: 10.1016/S0960-9822(03)00240-9
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359, 2337–2345. doi: 10.1056/NEJMoa0802828
Verpelli, C., Dvoretskova, E., Vicidomini, C., Rossi, F., Chiappalone, M., Schoen, M., et al. (2011). Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 286, 34839–34850. doi: 10.1074/jbc.M111.258384
Vinas-Jornet, M., Esteba-Castillo, S., Gabau, E., Ribas-Vidal, N., Baena, N., San, J., et al. (2014). A common cognitive, psychiatric, and dysmorphic phenotype in carriers of NRXN1 deletion. Mol. Genet. Genomic Med. 2, 512–521. doi: 10.1002/mgg3.105
Vincent, J. B., Kolozsvari, D., Roberts, W. S., Bolton, P. F., Gurling, H. M., and Scherer, S. W. (2004). Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am. J. Med. Genet. B Neuropsychiatr. Genet. 129B, 82–84. doi: 10.1002/ajmg.b.30069
Vincent, J. B., Konecki, D. S., Munstermann, E., Bolton, P., Poustka, A., Poustka, F., et al. (1996). Point mutation analysis of the FMR-1 gene in autism. Mol. Psychiatry 1, 227–231.
Voineskos, A. N., Lett, T. A., Lerch, J. P., Tiwari, A. K., Ameis, S. H., Rajji, T. K., et al. (2011). Neurexin-1 and frontal lobe white matter: an overlapping intermediate phenotype for schizophrenia and autism spectrum disorders. PLoS ONE 6:e20982. doi: 10.1371/journal.pone.0020982
Vulih-Shultzman, I., Pinhasov, A., Mandel, S., Grigoriadis, N., Touloumi, O., Pittel, Z., et al. (2007). Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther. 323, 438–449. doi: 10.1124/jpet.107.129551
Waga, C., Okamoto, N., Ondo, Y., Fukumura-Kato, R., Goto, Y., Kohsaka, S., et al. (2011). Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr. Genet. 21, 208–211. doi: 10.1097/YPG.0b013e328341e069
Walker, S., and Scherer, S. W. (2013). Identification of candidate intergenic risk loci in autism spectrum disorder. BMC Genomics 14:499. doi: 10.1186/1471-2164-14-499
Walsh, C. A., Morrow, E. M., and Rubenstein, J. L. (2008). Autism and brain development. Cell 135, 396–400. doi: 10.1016/j.cell.2008.10.015
Waltes, R., Duketis, E., Knapp, M., Anney, R. J., Huguet, G., Schlitt, S., et al. (2014). Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum. Genet. 133, 781–792. doi: 10.1007/s00439-013-1416-y
Walz, A., Mombaerts, P., Greer, C. A., and Treloar, H. B. (2006). Disrupted compartmental organization of axons and dendrites within olfactory glomeruli of mice deficient in the olfactory cell adhesion molecule, OCAM. Mol. Cell. Neurosci. 32, 1–14. doi: 10.1016/j.mcn.2006.01.013
Wang, C. C., Held, R. G., and Hall, B. J. (2013). SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS ONE 8:e83941. doi: 10.1371/journal.pone.0083941
Wang, C. C., Held, R. G., Chang, S. C., Yang, L., Delpire, E., Ghosh, A., et al. (2011a). A critical role for GluN2B-containing NMDA receptors in cortical development and function. Neuron 72, 789–805. doi: 10.1016/j.neuron.2011.09.023
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011b). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wang, J., Tao, Y., Song, F., Sun, Y., Ott, J., and Saffen, D. (2015). Common regulatory variants of CYFIP1 contribute to susceptibility for autism spectrum disorder (ASD) and Classical Autism. Ann. Hum. Genet. 79, 329–340. doi: 10.1111/ahg.12121
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Wang, K. S., Liu, X. F., and Aragam, N. (2010). A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 124, 192–199. doi: 10.1016/j.schres.2010.09.002
Wang, N. J., Parokonny, A. S., Thatcher, K. N., Driscoll, J., Malone, B. M., Dorrani, N., et al. (2008). Multiple forms of atypical rearrangements generating supernumerary derivative chromosome 15. BMC Genet. 9:2. doi: 10.1186/1471-2156-9-2
Wang, S. S., Kloth, A. D., and Badura, A. (2014). The cerebellum, sensitive periods, and autism. Neuron 83, 518–532. doi: 10.1016/j.neuron.2014.07.016
Weaving, L. S., Christodoulou, J., Williamson, S. L., Friend, K. L., McKenzie, O. L., Archer, H., et al. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 75, 1079–1093. doi: 10.1086/426462
Weiler, I. J., and Greenough, W. T. (1999). Synaptic synthesis of the Fragile X protein: possible involvement in synapse maturation and elimination. Am. J. Med. Genet. 83, 248–252. doi: 10.1002/(SICI)1096-8628(19990402)83:4<248::AID-AJMG3>3.0.CO;2-1
Wermter, A. K., Kamp-Becker, I., Strauch, K., Schulte-Korne, G., and Remschmidt, H. (2008). No evidence for involvement of genetic variants in the X-linked neuroligin genes NLGN3 and NLGN4X in probands with autism spectrum disorder on high functioning level. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 535–537. doi: 10.1002/ajmg.b.30618
Whitehouse, A. J., Bishop, D. V., Ang, Q. W., Pennell, C. E., and Fisher, S. E. (2011). CNTNAP2 variants affect early language development in the general population. Genes Brain Behav. 10, 451–456. doi: 10.1111/j.1601-183X.2011.00684.x
Wijetunge, L. S., Angibaud, J., Frick, A., Kind, P. C., and Nagerl, U. V. (2014). Stimulated emission depletion (STED) microscopy reveals nanoscale defects in the developmental trajectory of dendritic spine morphogenesis in a mouse model of fragile X syndrome. J. Neurosci. 34, 6405–6412. doi: 10.1523/JNEUROSCI.5302-13.2014
Williams, M. E., Wilke, S. A., Daggett, A., Davis, E., Otto, S., Ravi, D., et al. (2011). Cadherin-9 regulates synapse-specific differentiation in the developing hippocampus. Neuron 71, 640–655. doi: 10.1016/j.neuron.2011.06.019
Willsey, A. J., Sanders, S. J., Li, M., Dong, S., Tebbenkamp, A. T., Muhle, R. A., et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Wilson, B. M., and Cox, C. L. (2007). Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc. Natl. Acad. Sci. U.S.A. 104, 2454–2459. doi: 10.1073/pnas.0610875104
Winther, M., Berezin, V., and Walmod, P. S. (2012). NCAM2/OCAM/RNCAM: cell adhesion molecule with a role in neuronal compartmentalization. Int. J. Biochem. Cell Biol. 44, 441–446. doi: 10.1016/j.biocel.2011.11.020
Wisniowiecka-Kowalnik, B., Nesteruk, M., Peters, S. U., Xia, Z., Cooper, M. L., Savage, S., et al. (2010). Intragenic rearrangements in NRXN1 in three families with autism spectrum disorder, developmental delay, and speech delay. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 983–993. doi: 10.1002/ajmg.b.31064
Wong, W. T., Faulkner-Jones, B. E., Sanes, J. R., and Wong, R. O. (2000). Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. J. Neurosci. 20, 5024–5036.
Wu, G. Y., and Cline, H. T. (1998). Stabilization of dendritic arbor structure in vivo by CaMKII. Science 279, 222–226. doi: 10.1126/science.279.5348.222
Wu, G. Y., Zou, D. J., Rajan, I., and Cline, H. (1999). Dendritic dynamics in vivo change during neuronal maturation. J. Neurosci. 19, 4472–4483.
Wu, K., Hanna, G. L., Easter, P., Kennedy, J. L., Rosenberg, D. R., and Arnold, P. D. (2013). Glutamate system genes and brain volume alterations in pediatric obsessive-compulsive disorder: a preliminary study. Psychiatry Res. 211, 214–220. doi: 10.1016/j.pscychresns.2012.07.003
Wylie, S. R., and Chantler, P. D. (2003). Myosin IIA drives neurite retraction. Mol. Biol. Cell 14, 4654–4666. doi: 10.1091/mbc.E03-03-0187
Wylie, S. R., Wu, P. J., Patel, H., and Chantler, P. D. (1998). A conventional myosin motor drives neurite outgrowth. Proc. Natl. Acad. Sci. U.S.A. 95, 12967–12972. doi: 10.1073/pnas.95.22.12967
Xu, X., Xiong, Z., Zhang, L., Liu, Y., Lu, L., Peng, Y., et al. (2014). Variations analysis of NLGN3 and NLGN4X gene in Chinese autism patients. Mol. Biol. Rep. 41, 4133–4140. doi: 10.1007/s11033-014-3284-5
Yamamoto, Y., Mochida, S., Kurooka, T., and Sakisaka, T. (2009). Reciprocal intramolecular interactions of tomosyn control its inhibitory activity on SNARE complex formation. J. Biol. Chem. 284, 12480–12490. doi: 10.1074/jbc.M807182200
Yamamoto, Y., Mochida, S., Miyazaki, N., Kawai, K., Fujikura, K., Kurooka, T., et al. (2010). Tomosyn inhibits synaptotagmin-1-mediated step of Ca2+-dependent neurotransmitter release through its N-terminal WD40 repeats. J. Biol. Chem. 285, 40943–40955. doi: 10.1074/jbc.M110.156893
Yan, J., Noltner, K., Feng, J., Li, W., Schroer, R., Skinner, C., et al. (2008). Neurexin 1alpha structural variants associated with autism. Neurosci. Lett. 438, 368–370. doi: 10.1016/j.neulet.2008.04.074
Yanagi, K., Kaname, T., Wakui, K., Hashimoto, O., Fukushima, Y., and Naritomi, K. (2012). Identification of four novel synonymous substitutions in the X-Linked genes neuroligin 3 and neuroligin 4X in Japanese patients with autistic spectrum disorder. Autism Res. Treat. 2012:724072. doi: 10.1155/2012/724072
Yang, K., Trepanier, C., Sidhu, B., Xie, Y. F., Li, H., Lei, G., et al. (2012a). Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J. 31, 805–816. doi: 10.1038/emboj.2011.453
Yang, M. H., Yang, Y. H., Lu, C. Y., Jong, S. B., Chen, L. J., Lin, Y. F., et al. (2012b). Activity-dependent neuroprotector homeobox protein: a candidate protein identified in serum as diagnostic biomarker for Alzheimer’s disease. J. Proteom. 75, 3617–3629. doi: 10.1016/j.jprot.2012.04.017
Yap, A. S., Niessen, C. M., and Gumbiner, B. M. (1998). The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 141, 779–789. doi: 10.1083/jcb.141.3.779
Yashiro, K., Riday, T. T., Condon, K. H., Roberts, A. C., Bernardo, D. R., Prakash, R., et al. (2009). Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783. doi: 10.1038/nn.2327
Ye, H., Tan, Y. L., Ponniah, S., Takeda, Y., Wang, S. Q., Schachner, M., et al. (2008). Neural recognition molecules CHL1 and NB-3 regulate apical dendrite orientation in the neocortex via PTP alpha. EMBO J. 27, 188–200. doi: 10.1038/sj.emboj.7601939
Yi, J. J., Berrios, J., Newbern, J. M., Snider, W. D., Philpot, B. D., Hahn, K. M., et al. (2015). An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162, 795–807. doi: 10.1016/j.cell.2015.06.045
Yizhar, O., Matti, U., Melamed, R., Hagalili, Y., Bruns, D., Rettig, J., et al. (2004). Tomosyn inhibits priming of large dense-core vesicles in a calcium-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 101, 2578–2583. doi: 10.1073/pnas.0308700100
Ylisaukko-oja, T., Rehnstrom, K., Auranen, M., Vanhala, R., Alen, R., Kempas, E., et al. (2005). Analysis of four neuroligin genes as candidates for autism. Eur. J. Hum. Genet. 13, 1285–1292. doi: 10.1038/sj.ejhg.5201474
Yokoyama, K., Tezuka, T., Kotani, M., Nakazawa, T., Hoshina, N., Shimoda, Y., et al. (2011). NYAP: a phosphoprotein family that links PI3K to WAVE1 signalling in neurons. EMBO J. 30, 4739–4754. doi: 10.1038/emboj.2011.348
Yoo, H. J., Cho, I. H., Park, M., Yang, S. Y., and Kim, S. A. (2012). Family based association of GRIN2A and GRIN2B with Korean autism spectrum disorders. Neurosci. Lett. 512, 89–93. doi: 10.1016/j.neulet.2012.01.061
Yoshii, A., Zhao, J. P., Pandian, S., van Zundert, B., and Constantine-Paton, M. (2013). A myosin va mutant mouse with disruptions in glutamate synaptic development and mature plasticity in visual cortex. J. Neurosci. 33, 8472–8482. doi: 10.1523/JNEUROSCI.4585-12.2013
Youn, D. H., and Randic, M. (2004). Modulation of excitatory synaptic transmission in the spinal substantia gelatinosa of mice deficient in the kainate receptor GluR5 and/or GluR6 subunit. J. Physiol. 555, 683–698. doi: 10.1113/jphysiol.2003.057570
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., et al. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558. doi: 10.1073/pnas.0507856102
Yu, J., He, X., Yao, D., Li, Z., Li, H., and Zhao, Z. (2011). A sex-specific association of common variants of neuroligin genes (NLGN3 and NLGN4X) with autism spectrum disorders in a Chinese Han cohort. Behav. Brain Funct. 7:13. doi: 10.1186/1744-9081-7-13
Yu, T. W., Chahrour, M. H., Coulter, M. E., Jiralerspong, S., Okamura-Ikeda, K., Ataman, B., et al. (2013). Using whole-exome sequencing to identify inherited causes of autism. Neuron 77, 259–273. doi: 10.1016/j.neuron.2012.11.002
Yuen, R. K., Thiruvahindrapuram, B., Merico, D., Walker, S., Tammimies, K., Hoang, N., et al. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 21, 185–191. doi: 10.1038/nm.3792
Zamostiano, R., Pinhasov, A., Gelber, E., Steingart, R. A., Seroussi, E., Giladi, E., et al. (2001). Cloning and characterization of the human activity-dependent neuroprotective protein. J. Biol. Chem. 276, 708–714. doi: 10.1074/jbc.M007416200
Zappella, M., Meloni, I., Longo, I., Canitano, R., Hayek, G., Rosaia, L., et al. (2003). Study of MECP2 gene in Rett syndrome variants and autistic girls. Am. J. Med. Genet. B Neuropsychiatr. Genet. 119B, 102–107. doi: 10.1002/ajmg.b.10070
Zhang, L., He, J., Jugloff, D. G., and Eubanks, J. H. (2008). The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus 18, 294–309. doi: 10.1002/hipo.20389
Zhang, X. C., Piccini, A., Myers, M. P., Van Aelst, L., and Tonks, N. K. (2012). Functional analysis of the protein phosphatase activity of PTEN. Biochem. J. 444, 457–464. doi: 10.1042/BJ20120098
Zhao, Y., Zhang, X., Bao, X., Zhang, Q., Zhang, J., Cao, G., et al. (2014). Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients. BMC Med. Genet. 15:24. doi: 10.1186/1471-2350-15-24
Zheng, L., Ding, H., Lu, Z., Li, Y., Pan, Y., Ning, T., et al. (2008). E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 13, 285–294. doi: 10.1111/j.1365-2443.2008.01162.x
Zhong, X., Li, H., and Chang, Q. (2012). MeCP2 phosphorylation is required for modulating synaptic scaling through mGluR5. J. Neurosci. 32, 12841–12847. doi: 10.1523/JNEUROSCI.2784-12.2012
Zhou, J., Blundell, J., Ogawa, S., Kwon, C. H., Zhang, W., Sinton, C., et al. (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 29, 1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009
Zhou, Z., Hong, E. J., Cohen, S., Zhao, W. N., Ho, H. Y., Schmidt, L., et al. (2006). Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269. doi: 10.1016/j.neuron.2006.09.037
Zhu, Y. C., Li, D., Wang, L., Lu, B., Zheng, J., Zhao, S. L., et al. (2013). Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc. Natl. Acad. Sci. U.S.A. 110, 9118–9123. doi: 10.1073/pnas.1300003110
Keywords: autism-risk genes, neurite outgrowth, dendrite, dendritic spine, synapse formation, actin, adhesion molecule
Citation: Lin Y -C, Frei JA, Kilander MBC, Shen W and Blatt GJ (2016) A Subset of Autism-Associated Genes Regulate the Structural Stability of Neurons. Front. Cell. Neurosci. 10:263. doi: 10.3389/fncel.2016.00263
Received: 19 July 2016; Accepted: 28 October 2016;
Published: 17 November 2016.
Edited by:
Hansen Wang, University of Toronto, CanadaReviewed by:
Elva Diaz, University of California, Davis, USACopyright © 2016 Lin, Frei, Kilander, Shen and Blatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Chih Lin, eWNsaW5AaHVzc21hbmF1dGlzbS5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.