 Chiara Villa
Chiara Villa Romina Combi
Romina Combi- School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
Potassium (K+) channels are expressed in almost every cells and are ubiquitous in neuronal and glial cell membranes. These channels have been implicated in different disorders, in particular in epilepsy. K+ channel diversity depends on the presence in the human genome of a large number of genes either encoding pore-forming or accessory subunits. More than 80 genes encoding the K+ channels were cloned and they represent the largest group of ion channels regulating the electrical activity of cells in different tissues, including the brain. It is therefore not surprising that mutations in these genes lead to K+ channels dysfunctions linked to inherited epilepsy in humans and non-human model animals. This article reviews genetic and molecular progresses in exploring the pathogenesis of different human epilepsies, with special emphasis on the role of K+ channels in monogenic forms.
Introduction
Epilepsy is one of the most common neurological disorders characterized by abnormal electrical activity in the central nervous system (CNS) and recurrent seizures represent a cardinal clinical manifestation. The phenotypic expression of each seizure is determined by the original point of the hyperexcitability and its degree of spread in the brain (Steinlein, 2004). Several brain defects due to membrane instability could cause epilepsy.
In the last two decades, gene defects underlying different forms of epilepsy have been identified and most of these genes code for ion channels, which thus appear as important players in the etiopathogenesis of idiopathic epilepsy. Indeed, several epileptic phenotypes have been associated to dysfunctions of potassium (K+) channels (Brenner and Wilcox, 2012). It has been recently proposed to name such epilepsies as “K+ channelepsies” (D’Adamo et al., 2013). These channels play a major role in neuronal excitability and their importance is related to the level of their expression in subcellular domain, individual cell, or circuit (Cooper, 2012). K+ channels are also involved in setting the inward-negative resting membrane potential. Based on their structures, biophysical characteristics, pharmacological sensitivities and physiology, these channels are classified as voltage-gated (Kv), inwardly rectifying (Kir), sodium (Na)-activated channels or Ca2+-activated channels (Table 1; González et al., 2012).
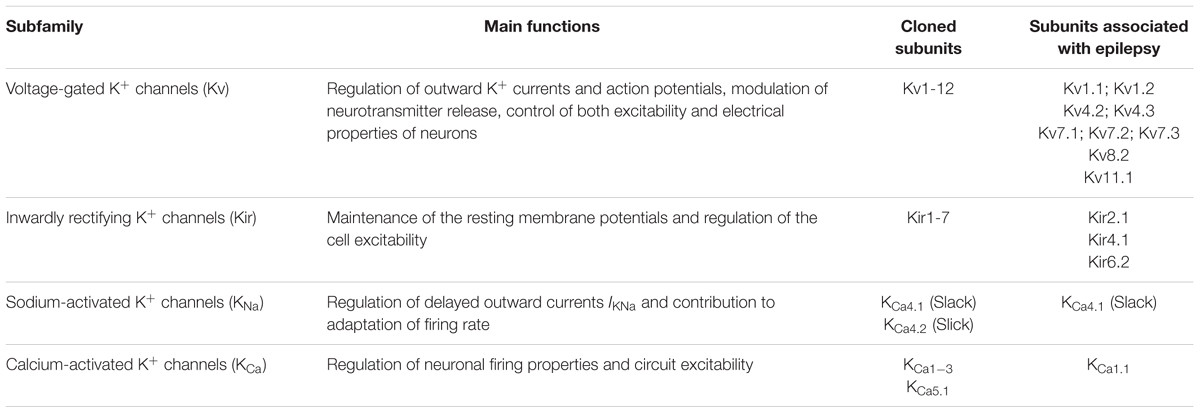
TABLE 1. Summary of human K+ channels subfamilies involved in epilepsies.
Herein we report an updated discussion on the role of mutations in K+ channels (Table 2) in the pathogenesis of human epilepsy.
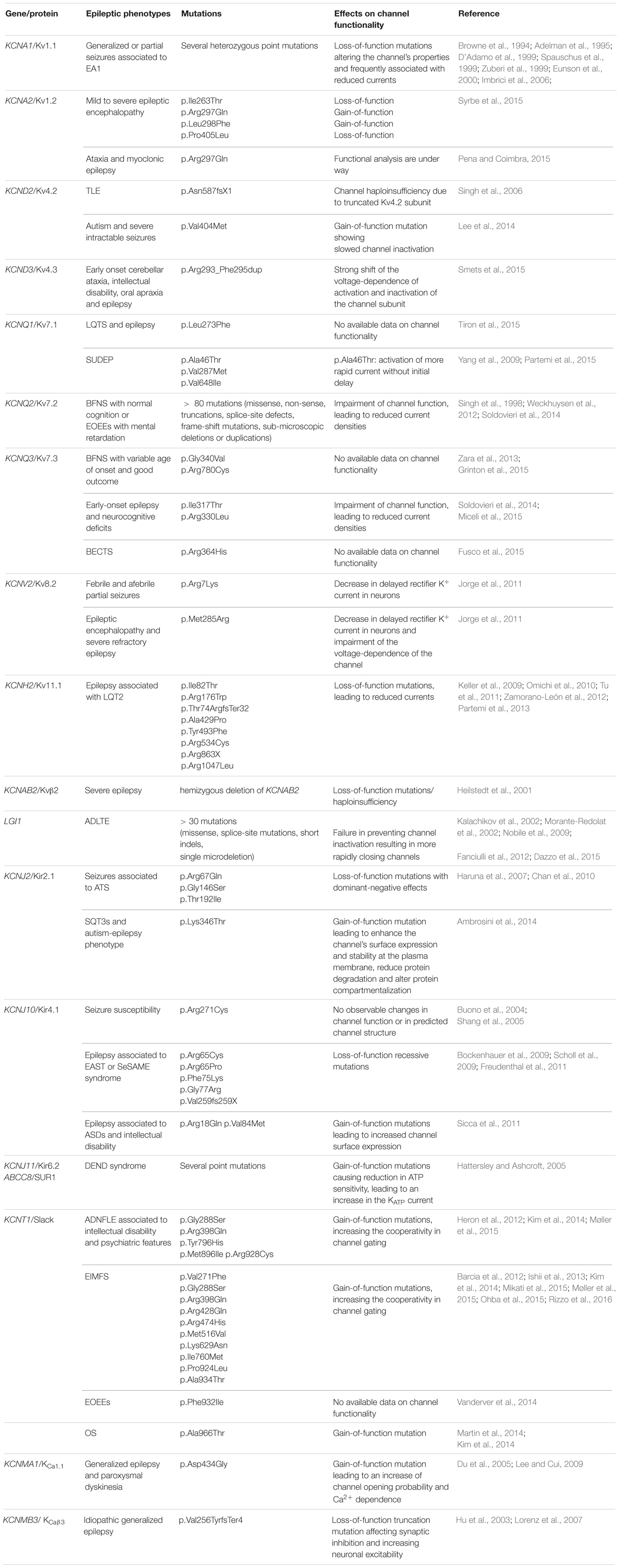
TABLE 2. Mutations in K+ channels associated with human epileptic phenotypes.
Voltage-Gated K+ Channels (Kv)
The Kv channels are widely expressed both in the central and in peripheral nervous system where they are involved in several processes (e.g., the regulation of the duration of action potentials, the modulation of the neurotransmitter release, the control of the electrical properties and the firing of neurons). Kv channels generally regulate outward K+ currents that contribute to membrane repolarization and hyperpolarization, thus limiting the neuronal excitability. Moreover, they actively participate in cellular and molecular signaling pathways that regulate the life and death of neurons, such as apoptosis, channel phosphorylation, or cell proliferation (Shah and Aizenman, 2014). In particular, neuronal cell apoptosis is correlated to an increased expression of Kv channels at the plasma membrane, thus facilitating more K+ efflux and a loss of cytosolic K+. This drop in the intracellular K+ concentration actives pro-apoptotic enzymes, such as nuclease or caspase that can trigger downstream apoptotic signals culminating in DNA fragmentation or degradation (Leung, 2010).
In human genome, forty different genes encoding for Kv channels were reported and subdivided into twelve sub-families (Kv1 through Kv12) (Gutman et al., 2005). Mammalian Kv channels are tetramers, composed of α-subunits that line an ion pore. Each α-subunit shows six α-helical transmembrane domains (S1–S6), a membrane-reentering P loop between S5 and S6, and cytosolic N-, and C-termini. The S5-P-S6 segments constitute the ion conduction pore, while the S1–S4 sequences are critical for the voltage-sensing and gating of the channel (Brenner and Wilcox, 2012).
Furthermore, α-subunits can bind to regulatory β subunits (Kvβ1, Kvβ2, and Kvβ3) as well as to other Kv channel-interacting proteins. This variability in the channel interactions results in strong modifications of the channel properties (McKeown et al., 2008).
The following Kv subfamilies have been associated with either epilepsy or other disorders showing seizures.
Kv1
The Kv1 subfamily plays an essential role in the initiation and shaping of action potentials. These channels are expressed at the soma, axons, synaptic terminals, and proximal dendrites. The most abundant Kv1 α-subunits are Kv1.1, Kv1.2, and Kv1.4. These subunits are differentially expressed and their composition is dependent upon the brain region, cell type and subcellular localization (Robbins and Tempel, 2012).
Heterozygous mutations in the KCNA1 gene, encoding the Kv1.1 α subunit, were associated with episodic ataxia type 1 (EA1), a dominantly inherited disorder characterized by generalized ataxia attacks and spontaneous muscle quivering (Browne et al., 1994). Interestingly, a subset of patients with familial EA1 shows epileptic seizures, suggesting that Kv1.1 dysfunctions may play a role in the pathophysiology of epilepsy (Spauschus et al., 1999; Zuberi et al., 1999; Eunson et al., 2000). Loss-of function mutations reported in the KCNA1 gene of EA1 patients cause reduced current amplitude thus contributing to seizures susceptibility (Browne et al., 1994; Adelman et al., 1995; D’Adamo et al., 1999; Imbrici et al., 2006).
In support of the hypothesis of an epileptogenic role of KCNA1 mutations, several knock-out mouse models for this gene developed an epileptic phenotype (Smart et al., 1998; Rho et al., 1999). Biochemical and biophysical studies demonstrated a colocalization of Kv1.1 and Kv1.2 subunits in several subcellular brain regions and that they could form heteromeric channels, which are reported as profoundly altered by EA1 mutations (D’Adamo et al., 1999).
Notably, a Kv1.2 knock-out mouse model displayed increased seizure susceptibility (Brew et al., 2007). In this regard, Syrbe et al. (2015) recently identified de novo loss or gain-of-function mutations in KCNA2 gene (Table 2), encoding the Kv1.2 channel, in patients showing mild to severe epileptic encephalopathy. A role of Kv1.2 was also suggested by another case report describing a de novo mutation, leading to the p.Arg297Gln amino acid substitution in a patient affected by ataxia and myoclonic epilepsy (Pena and Coimbra, 2015).
Kv4
The Kv4 channels are highly expressed in the brain and mediate the main dendritic A-currents which critically regulate action potential back-propagation and the induction of specific forms of synaptic plasticity. In particular, the Kv4.2 subunit is a key component of the A-type potassium current in the CNS (IA) (Birnbaum et al., 2004).
In 2006, Singh and collaborators described a truncation mutation (p.Asn587fsX1) in the Kv4.2 channel encoded by the KCND2 gene, in a patient affected by temporal lobe epilepsy (TLE). This mutation causes a frame-shift, leading to a premature termination codon and consequently to a Kv4.2 channel haploinsufficiency (Singh et al., 2006). Recently, a whole exome sequencing study identified a de novo gain-of-function mutation (p.Val404Met) in KCND2. The mutation was found in monozygotic twins affected by autism and severe intractable seizures and occurred at a highly conserved residue within the C-terminus of the S6 transmembrane region of the ion pore. A functional analysis of mutated channels revealed a significantly slowed channel inactivation (Lee et al., 2014).
Very recently, an involvement of Kv4.3 subunits in epilepsy was also suggested by the identification of a de novo mutation (p.Arg293_Phe295dup) in the relevant KCND3 gene causing a severe channel dysfunction in a patient with complex early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. This mutation results in the duplication of a RVF (Arginine–Valine–Phenylalanine) motif in the S4 segment and leads to a more positively charged voltage-sensor domain, altering the voltage-dependent gating properties of the channel. In details, the p.Arg293_Phe295dup mutation induced a strong depolarizing shift in the voltage dependence of both the activation (about +59.3 mV) and inactivation (+62 mV) of the channel (Smets et al., 2015).
Kv7
KCNQ (Kv7) channels are low-threshold activated voltage-gated potassium channels. Among the five known isoforms, KCNQ2–5 are expressed throughout the nervous system, whereas KCNQ1 is mostly expressed in cardiac tissue. The KCNQ2 gene is the most commonly reported as mutated in epilepsy. Its mutations cause neonatal epilepsies with wide phenotypic heterogeneity, ranging from benign familial neonatal seizures (BFNS) with normal cognition and unremarkable neuroimaging to early onset epileptic encephalopathies (EOEEs) with mental retardation, suppression-burst electroencephalography (EEG) and distinct neuroradiologic features (Singh et al., 1998; Weckhuysen et al., 2012; Soldovieri et al., 2014). More than 80 different mutations in KCNQ2, consisting of missense, non-sense, truncations, splice site defects and frame-shift mutations, as well as sub-microscopic deletions or duplications, were described and most of them are found in the pore region and the large intracellular C-terminal domain (Lee et al., 2009). Functional studies suggested a strict phenotype/genotype correlation between disease severity and functional properties of mutant channels (Miceli et al., 2013). KCNQ2 is a primary player that mediates neuronal muscarinic (M) currents: the opening of this channel or of heterogeneous KCNQ2/KCNQ3 complexes inhibits initiation of action potential and thus suppresses neuronal excitability (Brown and Passmore, 2009).
Mutations in KCNQ3 gene have been described in families affected with benign epilepsy with variable age of onset and good outcome (Zara et al., 2013; Grinton et al., 2015) or in a patient with benign childhood epilepsy with centrotemporal spikes (BECTS) (Fusco et al., 2015). However, two recent reports suggested that mutations in KCNQ3, similarly to KCNQ2, can be also found in patients with more severe phenotypes, including intellectual disability. In particular, they described KCNQ3 mutations in patients with early onset epilepsy and neurocognitive deficits (Soldovieri et al., 2014; Miceli et al., 2015; Table 2).
Mutations in the KCNQ1 gene were associated with a particular form of long QT syndrome, the LQT1 (Wang et al., 1996). Interestingly, some authors observed that epilepsy occurred in mouse lines bearing dominant human LQT1 mutations in this channel, which caused syncope and sudden death (Goldman et al., 2009). Moreover, genetic variants in the KCNQ1 gene were reported in three cases of sudden unexpected death in epilepsy (SUDEP), a catastrophic complication of human idiopathic epilepsy with unknown causes. However, the relationship of these variants to the disease remains to be elucidated (Yang et al., 2009; Partemi et al., 2015). The evidence that KCNQ1 genetic variations may confer susceptibility for recurrent seizure activity increasing the risk of sudden death is further supported by the description of a pathogenic KCNQ1 variant (p.Leu273Phe) in a family featuring LQTS and epilepsy (Tiron et al., 2015).
Kv8
The KCNV2 gene encodes the voltage-gated K+ channel Kv8.2. This subunit is electrophysiologically silent when assembled in homotetramer. Otherwise, it significantly reduces the surface expression of the resulting channels and influences their biophysical properties when involved in the formation of functional heterotetramers with Kv2 subunits (Czirják et al., 2007). Kv2.1 and Kv8.2 show significant regional overlap: within the hippocampus, transcripts for both KCNV2 and KCNB1, which encodes Kv2.1, are detected in excitatory neurons of the pyramidal cell layers and the dentate gyrus. Similarly, both of them are abundantly expressed in the cortex (Maletic-Savatic et al., 1995). Their regional colocalization is consistent with an effect of Kv8.2 variants on Kv2.1 channels within cells critically important for seizure generation and propagation.
A support of the involvement of KCNV2 in seizure pathogenesis was provided by the identification of non-synonymous variants in two unrelated children showing epilepsy: p.Arg7Lys and p.Met285Arg. In particular, the p.Arg7Lys was found in a patient affected by febrile and afebrile partial seizures, whereas the p.Met285Arg was reported in a case of epileptic encephalopathy and severe refractory epilepsy. The functional characterization of these variants demonstrated that they both enhanced Kv8.2-mediated suppression of Kv2.1 currents, suggesting a role in decreasing delayed rectifier K+ current in neurons, therefore increasing cells excitability. Moreover, the p.Met285Arg caused a shift in the voltage-dependence of activation as well as slower activation kinetics, in accordance with the more severe clinical phenotype of the patient (Jorge et al., 2011).
Kv11-HERG
The human ether-a-go-go-related gene (hERG, also known as KCNH2) encodes the pore-forming subunit of the rapid component of the delayed rectifier K+ channels, Kv11.1, which are expressed in several tissues, mostly in brain and heart. In the brain, Kv11.1 channels regulate neuronal firing and modulate the excitability of GABAergic and dopaminergic neurons. The same channel exerts a different function in the heart being involved in the regulation of membrane potentials in the ventricles (Vandenberg et al., 2012).
Mutations in the KCNH2 gene were reported to cause type 2 long QT syndrome (LQT2), a rare inherited ion channel disorder characterized by prolonged QT interval and predisposing patients to ventricular arrhythmias that can lead to syncope and sudden cardiac death (SCD). LQT2 syndrome is frequently misdiagnosed as epilepsy due to seizures that are triggered by cerebral hypoperfusion during a ventricular arrhythmia, therefore suggesting a possible link between epilepsy and cardiac arrhythmias, as described by several clinical reports (Johnson et al., 2009; Keller et al., 2009; Omichi et al., 2010; Tu et al., 2011; Zamorano-León et al., 2012; Partemi et al., 2013). In particular, a seizure phenotype was reported in about 30% of unrelated LQTS patients carrying pathogenic variants in the KCNH2 gene, suggesting that mutations in the Kv11.1 channel associated with LQTS may also predispose to seizure activity (Johnson et al., 2009). Moreover, a post-mortem study identified nearly 13% of LQTS pathogenic variants in the KCNH2 and SCN5A genes in epileptic samples. In particular, regarding KCNH2, two non-synonymous mutations have been identified: p.Arg176Trp and p.Arg1047Leu (Tu et al., 2011). Another study on three families showing a history of seizures and LQTS2 lead to the identification of three novel KCNH2 mutations: p.Tyr493Phe, Ala429Pro and Thr74ArgfsTer32 (also named p.del234-241). In vitro functional analyses of all these variants showed a loss of hERG potassium channel function with a reduction of the current, suggesting a dominant negative effect (Keller et al., 2009). Omichi et al. (2010) reported a case of a man with long history of epilepsy and referred for cardiologic evaluation, showing the p.Arg534Cys mutation. In addition, other authors identified a nonsense mutation (p.Arg863X) leading to a 296-amino acid deletion (Zamorano-León et al., 2012) while a loss-of-function mutation (p.Ile82Thr) was reported in a pedigree featuring LQTS, idiopathic epilepsy and increased risk of sudden death (Partemi et al., 2013).
Auxiliary Subunits of Kv Channels
Kv channel functional diversity is enhanced by coassembly with a wide array of auxiliary subunits, which cannot form functional channels alone but which can greatly impact channel function upon coassembly with α-subunits to form hetero-oligomeric complexes (Trimmer, 1998). Defects in these subunits may affect Kv channel function and network excitability, resulting thus in an increase of seizure susceptibility. Several subunits have been identified, including β-subunit (Kvβ), leucine-rich glioma-inactivated-1 (KvLGI1) and K+ channel-interacting protein (KvKChIP).
Kvβ
Kvβ subunits are cytoplasmatic proteins critical for the correct membrane localization and normal biophysical properties of voltage-gated K+ channels. Variations in the expression of different Kvβ genes and their isoforms could significantly impact K+ channel function, especially with respect to inactivation kinetics. In the mammalian genome three genes encode Kvβ subunits: Kvβ1, Kvβ2, and Kvβ3 (Pongs and Schwarz, 2010). Interestingly, Kvβ2 knockout mouse models were characterized by cold-swim induced tremors and occasional seizures, suggesting thus a role of this subunit in the regulation of neuronal excitability (McCormack et al., 2002). An association between the severity of seizures and the loss-of-function of the KCNAB2 gene that encodes the β2 subunit was reported (Heilstedt et al., 2001). In particular, the hemizygous deletion of KCNAB2 identified in this manuscript in epileptic patients suggested that haploinsufficiency of this gene may represent a significant risk factor for epilepsy: the lack of the β subunit would reduce K+ channel-mediate membrane repolarization and increase neuronal excitability (Heilstedt et al., 2001).
KvLGI1
The leucine-rich glioma-inactivated-1 (LGI1) is the best characterized LGI family protein, highly expressed in neurons, which encodes a secreted protein containing two domains [a leucine-rich repeat domain (LRR) and a β-propeller domain called EPTP] that mediate protein-protein interactions. LGI1 binds to the presynaptic voltage-gated potassium channel Kv1.1 and prevents Kv channel inactivation mediated by the β subunit of the channel (Schulte et al., 2006). The LG1 gene was find to be mutated in approximately 50% of ADLTE (autosomal dominant lateral TLE) families: more than 30 disease-causing mutations in LGI1 gene have been associated so far with this focal epilepsy that is characterized by good response to antiepileptic drugs and with a juvenile onset (Kalachikov et al., 2002; Morante-Redolat et al., 2002; Dazzo et al., 2015). In particular, almost all mutations are missense, splice-site, or short indels (Nobile et al., 2009; Ho et al., 2012) while only a single microdeletion has been reported (Fanciulli et al., 2012). Certain LGI1 mutants (typically non-secreted mutants) fail to prevent channel inactivation resulting in more rapidly closing channels, which extends presynaptic depolarization and leads to increased calcium (Ca2+) influx. Consequently, release of neurotransmitter is increased excessively and may induce focal seizures (Nobile et al., 2009). Moreover, it was demonstrated that the loss of LGI1 gene in mice induced lethal epilepsy, suggesting its essential role as an antiepileptogenic ligand. LGI1 may serve as a major determinant of brain excitation and the LGI1 gene-targeted mouse could provide a good model for human epilepsy (Fukata et al., 2010).
KvKChIP
The K+ channel-interacting proteins (KChIPs 1–4) compose a subfamily of neuronal Ca2+ sensor proteins that modulate trafficking, targeting to the plasma membrane, as well as turnover and endocytosis of Kv4 channels (An et al., 2000). Among KChIPs, KChIP2 is abundantly expressed in hippocampal pyramidal cells and represents the major target of Kv4 α subunits to form a complex essential for IA regulation in hippocampal neurons (Rhodes et al., 2004). This current has been found to be reduced in the presence of a deletion in the KChIP2 gene by Wang and collaborators. The authors thus suggested that it may increase susceptibility to seizures (Wang et al., 2013). Moreover, they also hypothesized a role of KChIP2 in SUDEP risk (Wang et al., 2013), since KChIP2 knockout mice were previously shown to be highly susceptible to induced arrhythmias (Kuo et al., 2001). In conclusion, these data suggested that loss-of-function mutations in modulatory subunits could increase the susceptibility to seizures and cardiac arrhythmias, thereby providing a unified mechanism for a neurocardiac syndrome such as SUDEP.
Inwardly Rectifying Potassium Channels
Inwardly rectifying K+ (Kir) channels are widely expressed in several excitable and non-excitable tissues playing a key role in the maintenance of the resting membrane potential and consequently in the regulation of cell excitability. Approximately 15 Kir clones forming either homotetramers or heterotetramers were identified and grouped in seven different families based on sequence similarity and functional properties: Kir1-Kir7 (Hibino et al., 2010). Generally, Kir channels showed the greater conductance at negative potentials in respect to the equilibrium potential for K+ (EK), while an inhibition of the outward flow of K+ ions caused by both Mg2+ and polyamines was reported at more positive values (Lopatin et al., 1994). Several Kir channels have been associated with epileptic phenotypes and, in particular, Kir2.1, Kir3, Kir4 and Kir6.
Kir2.1
The Kir2.1 channel is encoded by the KCNJ2 gene whose expression is reported in several brain areas (Karschin et al., 1996) as well as in astrocytes where they control astrocyte-mediated K+ buffering in combination with Kir4.1 (Jabs et al., 2008; Chever et al., 2010).
Several mutations impairing the channel functionality were reported in the KCNJ2 of Andersen–Tawil syndrome (ATS) patients (Haruna et al., 2007; Chan et al., 2010; Guglielmi et al., 2015; see Table 2 for mutation details). On the other hand, Kir2.1 gain-of-function mutations cause the type-3 variant of the short QT syndrome (SQT3s) which results in QT shortening and increased risk of sudden cardiac death (Priori et al., 2005). Recently, some authors detected a novel mutation (p.Lys346Thr) in the KCNJ2 in monozygotic twins displaying SQT3s and autism-epilepsy phenotype, suggesting the existence of a Kir2.1 role in neuropsychiatric disorders and epilepsy. Functional studies revealed that this mutation causes an increase of the channel’s surface expression and stability at the plasma membrane, a reduction in protein degradation and an altered protein compartmentalization (Ambrosini et al., 2014).
Kir3-GIRK
The G-protein-coupled Kir (GIRK) channels belong to the subfamily of Kir3 that are important regulators of electrical excitability in both cardiomyocytes and neurons (Slesinger et al., 1995). Different types of neurotransmitters, such as acetylcholine, dopamine, opioids, serotonin, somatostatin, adenosine, and GABA, activate these channels by stimulating their G-protein coupled receptors (GPCRs). This results in a final membrane hyperpolarization and inhibition of cell excitability due to the activation of an outward flux of K+ ions (Krapivinsky et al., 1995; Slesinger et al., 1995). Mammals express four GIRK channel subunits (GIRK1-4, also named Kir3.1-3.4), encoded by KCNJ3, KCNJ6, KCNJ9, and KCNJ5, respectively. These four subunits can form homo or heterotetramers with unique biophysical properties, regulation and distribution (Lüscher and Slesinger, 2010).
Alterations in GIRK channel function have been associated with pathophysiology of severe brain disorders, including epilepsy. In this regard, a GIRK2 knockout mouse model resulted to be more susceptible to develop both spontaneous or induced seizures in respect to wild type mice (Signorini et al., 1997). In particular, mice carrying a p.Gly156Ser mutation displayed an epileptic phenotype (Patil et al., 1995). Indeed, this mutation has been found to alter the putative ion-permeable, pore-forming domain of the channel, inducing Ca2+ overload in cells and reducing channel availability, leading thus to neurodegeneration and seizures susceptibility (Slesinger et al., 1996).
An increased expression of GIRK channels was observed in rat brain after an electroconvulsive shock, probably altering the excitability of granule cells and the functions of neurotransmitter receptors which are coupled to these channels (Pei et al., 1999). Another evidence in support of a role of GIRK channels in epilepsy was provided by the demonstration that ML297, a potent and selective activator of GIRK channels, showed epileptogenic properties in mice (Kaufmann et al., 2013). On the other hand, the inhibition of GIRK channel activity by drugs causes seizures (Mazarati et al., 2006). All these considerations imply that changes in Kir3 channel activity may alter the susceptibility to seizures.
Kir4
Among Kir4 channels, the Kir4.1, encoded by the KCNJ10 gene, is the only one that has been associated to epilepsy. This subunit can assemble itself in homomeric channels or it can constitute heterotetramers in combination with Kir5.1 (KCNJ16) (Pessia et al., 2001). Kir4.1 expression has been detected primarily in the thalamus, cortex, brainstem and hippocampus (Higashi et al., 2001). Kir4.1 channels play a key role in maintaining resting membrane potential by transporting K+ from the extracellular space into glial cells in the CNS (Nishida and MacKinnon, 2002).
Alterations of Kir4.1 channels have been linked to seizure susceptibility in both mice (Ferraro et al., 2004) and humans (Buono et al., 2004). Conditional Kir4.1 knockout mice in astrocytes have been found to display premature lethality and severe seizures prior to death (Djukic et al., 2007), supporting the idea of a pathophysiological relationship of the Kir4.1 impairment with epilepsy. Concerning human Kir4.1, a linkage study identified a missense variation (p.Arg271Cys) as associated with epileptic phenotypes (Buono et al., 2004). However, the variant did not result to have functional effects in vitro (Shang et al., 2005). Mutations in this gene were also reported in EAST syndrome (also named SeSAME) patients, a rare condition showing epileptic seizures among other signs (Bockenhauer et al., 2009; Scholl et al., 2009; Freudenthal et al., 2011; see Table 2 for mutation details).
Single nucleotide variations in Kir4.1 were detected in the DNA of TLE patients presenting with hippocampal sclerosis and antecedent febrile seizures, supporting the importance of KCNJ10 as a candidate gene for seizures susceptibility (Heuser et al., 2010).
Interestingly, several authors reported a strong association between epilepsy and autism spectrum disorders (ASDs) and an “autism-epilepsy phenotype” has been proposed (Tuchman et al., 2009; Lee et al., 2015). Indeed, a mutational screening of KCNJ10 in 52 children affected by cryptogenic epilepsy identified two heterozygous mutations (p.Arg18Gln and p.Val84Met) in three children of two unrelated families displaying seizures, ASDs, and intellectual disability. The functional consequences of these mutations appeared to be a gain-of-function mechanism. These findings suggest that an abnormal K+ homeostasis in the brain may increase the susceptibility to this “autism-epilepsy phenotype” (Sicca et al., 2011). A common mechanism between autism and epilepsy could be the impairment of astrocytic-dependent K+ buffering, altering neuronal excitability and synaptic function.
Kir6-KATP
The adenosine triphosphate (ATP)-sensitive K+ (KATP) channels are widely distributed in various tissues where they couple cell metabolism to cell excitability. These channels are assembled by an inward rectifier K+ channel pore (Kir6.1/Kir6.2) and an ATP-binding regulatory subunit, named sulfonylurea receptor (SUR1/SUR2A/SUR2B) (Olson and Terzic, 2010). Neuronal KATP channels are mainly constituted by a coassembly of Kir6.2/SUR1 subunits. (Inagaki et al., 1995).
Several gain-of-function mutations were detected in the Kir6.2 (KCNJ11) or the SUR1 subunit (ABCC8). These mutations are responsible for developmental delay, epilepsy and neonatal diabetes (DEND), accounting for approximately 40% of cases and caused a decrease in the ability of ATP to block the KATP channel. This results in more fully openings of the channel at physiologically relevant concentrations of ATP, thus increasing the KATP current (Hattersley and Ashcroft, 2005). Nevertheless, the pathophysiological mechanism leading to epilepsy remains to be elucidated. Probably, elevated levels of extracellular glucose and intracellular ATP attenuate KATP channels, producing a more excitable state (Huang et al., 2007). Moreover, mice lacking Kir6.2 are vulnerable to hypoxia, exhibiting a reduced threshold for generalized seizure (Yamada et al., 2001). Transgenic mice, overexpressing the SUR1 gene in the forebrain, show a significant increase in the threshold for kainate-induced seizures (Hernández-Sánchez et al., 2001).
Sodium-Activated Potassium Channels (KNa)
The Na+-activated K+ channels (KNa) are found in neurons throughout the brain and are responsible for delayed outward currents named IKNa. These currents regulate neuronal excitability and the rate of adaption in response to repeated stimulation at high frequencies. In many cases, IKNa is mediated by the phylogenetically related KNa channel subunits Slack and Slick (Bhattacharjee and Kaczmarek, 2005). Like the Kv channels, these subunits have six hydrophobic, transmembrane segments (S1–S6) with a pore P-domain between S5 and S6 and a large cytoplasmatic C-terminal domain containing two regulators of K+ conductance (RCK) domains that are likely to be sites for Na+-binding and channel gating. The Slack subunit binds with Slick to form heterotetrameric channel complexes (Kaczmarek, 2013). Slack has been associated with different epilepsy phenotypes.
Slack
The KCNT1 gene encodes the KNa channel subunit KCNT1, called Slack (sequence like a calcium-activated potassium channel, also known as KCa4.1 or Slo2.2). KCNT1 is highly expressed in the brain but also in the heart and the kidney at lower levels. Concerning brain, it is not widely expressed in the cortex but it is found in neurons of the frontal cortex (Bhattacharjee et al., 2002), consistent with its known role in the pathogenesis of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (Heron et al., 2012). While KCNT1 channels are thought to play important roles in modulating the firing patterns and general excitability of many types of neurons, their precise function is yet to be resolved.
Mutations in KCNT1 gene have been found in different epilepsy syndromes: ADNFLE (Heron et al., 2012; Kim et al., 2014; Møller et al., 2015), epilepsy of infancy with migrating focal seizures (EIMFS, previously known as malignant migrating partial seizures in infancy, MMPSI or also more recently as malignant migrating focal seizures of infancy, MMFSI) (Barcia et al., 2012; Ishii et al., 2013; Ohba et al., 2015; Rizzo et al., 2016) and other types of EOEEs, (Vanderver et al., 2014; Ohba et al., 2015), including Ohtahara syndrome (OS) (Martin et al., 2014). The involvement of KCNT1 in these distinct disorders suggests that KCNT1 mutations may cause a spectrum of focal epilepsies (Møller et al., 2015). Patients displaying KCNT1 mutations have a very high occurrence of severe mental and intellectual disability.
Four missense mutations (p.Arg398Gln, p.Tyr796His, p.Met896Ile, and p.Arg928Cys) in KCNT1 gene were reported to be associated with ADNFLE cases showing comorbidities of intellectual disability and psychiatric features (Heron et al., 2012). This is in contrast to ADNFLE patients without mutations in KCNT1 gene, where intelligence and other neurologic functions are largely unimpaired (Phillips et al., 1998). Mutations are clustered around the RCK and cytoplasmatic NAD+ binding domain (Heron et al., 2012), the site that regulates the channel sensitivity to Na+ intracellular concentrations (Tamsett et al., 2009). A complete penetrance is reported in ADNFLE families showing KCNT1 mutations (Heron et al., 2012) with the exception of a non-penetrant case (Møller et al., 2015).
Interestingly, Møller et al. (2015) reported that a KCNT1 mutation (p.Arg398Gln) can lead to either ADNFLE or EIMFS within the same family, indicating that genotype-phenotype correlations are not straightforward). Similarly, a more recent study showed that the p.Gly288Ser mutation could cause both phenotypes, probably due to genetic modifiers or environmental factors (Kim et al., 2014). Nevertheless, this association was unexpected since in vitro studies demonstrated that mutations associated with MMFSI caused a significantly larger increase in current amplitude than those associated with ADNFLE (Milligan et al., 2014).
Concerning EIMFS, in addition to the above mentioned p.Gly288Ser and p.Arg398Gln, several additional mutations have been identified, including p.Val271Phe, p.Arg428Gln, p.Arg474Gln, p.Met516Val, p.Lys629Asn, p.Ile760Met, p.Pro924Leu and p.Ala934Thr (Barcia et al., 2012; Ishii et al., 2013; Mikati et al., 2015; Ohba et al., 2015; Rizzo et al., 2016). These are clustered not only around the RCK and NAD+ binding domain of the protein, but also within its S5 transmembrane segment, indicating that the alteration of other regions of KCNT1 could also be pathogenic (Ishii et al., 2013; McTague et al., 2013; Kim et al., 2014)
Finally, two KCNT1 mutations were associated with other forms of EOEEs, strengthening once again the existence of a wide phenotypic spectrum of KCNT1 mutations. In particular, the p.Phe932Ile was detected in a patient affected by EOEEs whereas the p.Ala966Thr was found in one showing OS. Both of them are clustered around the RCK and NAD+ binding domains of the protein (Martin et al., 2014; Vanderver et al., 2014; Ohba et al., 2015).
The effect of nine different mutations in KCNT1 gene that give rise to these distinct forms of epilepsy was examined and it was demonstrated that they all result in channels displaying a strong gain-of-function phenotype: all of them produced many-fold increases in current amplitude as compared with the wild-type channel. This could greatly increase the cooperativity in channel gating that is detected in clusters of multiple channels (Kim et al., 2014).
Calcium-Activated Potassium Channels (KCa)
Ca2+-activated K+ channels are highly conserved complexes thought to play a critical role in neuronal firing properties and circuit excitability in the human brain. Three groups of Ca2+-activated K+ channels can be distinguished: large conductance (BKCa), intermediate conductance (IKCa), and small conductance (SKCa) channels (N’Gouemo, 2011). The opening of these channels is in response to an increase in Ca2+ concentration and a depolarization of the membrane potential, which in turn causes a secondary hyperpolarization reestablishing the membrane potential as well as Ca2+ levels. Otherwise it can produce an afterhyperpolarization to potentials more negative than the resting membrane potential (Latorre and Brauchi, 2006; Nardi and Olesen, 2008). To date, only the association between KCa1.1 channel and epilepsy has been demonstrated.
KCa1.1
KCNMA1 gene encoded the α-subunit of the large conductance KCa1.1 channels. They show the typical tetrameric structure of K+ channels, with four α-subunits each displaying seven transmembrane segments, with a unique S0 segment, and the charged S4 segment conferring the voltage-dependence. Ca2+ sensitivity comes instead from the bulky C-terminal tail that includes a negatively charged, high-affinity Ca2+ binding region (Jiang et al., 2001) and the double negative charged RCK-domain. These channels could associate with four different types of β subunits (β1-β4, each encoded by a specific gene KCNMB1-4) which modulated channel function uniquely (Orio et al., 2002).
KCa1.1 channels play a role in promoting high neuronal frequency firing which is consistent with their predominant expression in axon and presynaptic terminals of neurons located in brain regions (e.g., hippocampus and cortex) frequently involved in epilepsy (Gu et al., 2007; Martire et al., 2010). The involvement of these channels in epilepsy was suggested not only by their localization but also by studies on animal models. In this regard, it has been demonstrated in mice highly susceptible to convulsions that the inhibition of KCa1.1 channels is sufficient to block cortical bursting activity (Jin et al., 2000). Moreover, the loss of β4 subunits in KCaβ4 knockout mice promoted the excitatory synaptic transmission, resulting in temporal cortex seizures (Brenner et al., 2005). Finally, Ermolinsky et al. (2008) demonstrated a deficit of KCNMA1 expression in the dentate gyrus in animal models, hypothesizing therefore its critical role in the pathogenesis of mesial temporal lobe epilepsy (mTLE).
An association between KCa1.1 channels and epilepsy has also been observed in humans. A missense mutation in KCNMA1 (p.Asp434Gly) was detected in a large family with generalized epilepsy and paroxysmal dyskinesia. Functional studies revealed an increased Ca2+ sensitivity predicting a gain-of-function and neuronal hyperexcitability by a presumably faster action potential repolarization (Du et al., 2005). Additional studies suggested that depending on the distribution of the various β subunits in the brain, this mutation can differently modulate KCa1.1 channels contributing to the pathophysiology of epilepsy and dyskinesia (Lee and Cui, 2009). As far as genes different from KCNMA1, a polymorphism in KCNMB4, named rs398702, was also associated with mTLE in an Irish cohort population (Cavalleri et al., 2007) but the study failed to be replicated (Manna et al., 2013), while a truncation mutation in KCNMB3 (p.Val256TyrfsTer4) affecting synaptic inhibition and thereby increasing neuronal excitability and seizure susceptibility, was associated with idiopathic generalized epilepsy (Hu et al., 2003; Lorenz et al., 2007).
Concluding Remarks
Epilepsy is one of the most common chronic and heterogeneous neurological disorders, affecting 1–2% of the population, characterized by recurrent unprovoked seizures due to abnormal synchronized electrical discharges within the CNS (Steinlein, 2004). Since ion channels mediate the axonal conduction of action potentials and transduction through synaptic transmission, increasing evidence suggests that any mutation-induced channel malfunction directly alter brain excitability and can induce epileptic seizures. Therefore, the discovery of genetic defects and, in particular, the electrophysiological characterization of mutant ion channels in hereditary forms of epilepsy may elucidate pathophysiological concepts of hyperexcitability in the CNS. This knowledge could enable new therapeutic strategies by antagonizing the epilepsy-causing mechanisms using the defective proteins as pharmacological targets. Given these considerations, we present an overview of mutations in K+ channels and their related accessory subunits underlying different human epileptic phenotypes. Several families of K+ channels have been involved in the pathogenesis of epilepsy or other syndromes showing seizures as a clinical sign. For each channel family, the effect of reported mutations is different: loss-of-function as well as gain-of-function could be observed. The common effect of all mutations is to determine membrane hyperexcitability, thus increasing the susceptibility to seizures. Our review highlights the pleiotropic effects of some mutations in K+ channels and the lack of a direct genotype–phenotype correlation. Interestingly, K+ channels dysfunctions seem to be mainly observed in epileptic patients with neurological comorbidities, such as ASDs, intellectual disabilities or psychiatric features, in which they are associated with more clinical severity. This observation could suggest to perform a mutation screening of K+ channels in patients showing intellectual disabilities.
In conclusion, the discovery of K+ channels encoding genes that influence susceptibility and disease progression will provide insight into the molecular events of epileptogenesis, improve molecular diagnostic utility, and identify novel therapeutic targets for treatment of human epilepsy.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adelman, J. P., Bond, C. T., Pessia, M., and Maylie, J. (1995). Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 15, 1449–1454. doi: 10.1016/0896-6273(95)90022-5
Ambrosini, E., Sicca, F., Brignone, M. S., D’Adamo, M. C., Napolitano, C., Servettini, I., et al. (2014). Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum. Mol. Genet. 23, 4875–4886. doi: 10.1093/hmg/ddu201
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Barcia, G., Fleming, M. R., Deligniere, A., Gazula, V. R., Brown, M. R., Langouet, M., et al. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259. doi: 10.1038/ng.2441
Bhattacharjee, A., Gan, L., and Kaczmarek, L. K. J. (2002). Localization of the Slack potassium channel in the rat central nervous system. Comp. Neurol. 454, 241–254. doi: 10.1002/cne.10439
Bhattacharjee, A., and Kaczmarek, L. K. (2005). For K+ channels, Na+ is the new Ca2+. Trends Neurosci. 28, 422–428. doi: 10.1016/j.tins.2005.06.003
Birnbaum, S. G., Varga, A. W., Yuan, L. L., Anderson, A. E., Sweatt, J. D., and Schrader, L. A. (2004). Structure and function of Kv4-family transient potassium channels. Physiol. Rev. 84, 803–833. doi: 10.1152/physrev.00039.2003
Bockenhauer, D., Feather, S., Stanescu, H. C., Bandulik, S., Zdebik, A. A., Reichold, M., et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970. doi: 10.1056/NEJMoa0810276
Brenner, R., Chen, Q. H., Vilaythong, A., Toney, G. M., Noebels, J. L., and Aldrich, R. W. (2005). BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat. Neurosci. 8, 1752–1759. doi: 10.1038/nn1573
Brenner, R., and Wilcox, K. S. (2012). “Potassium channelopathies of epilepsy,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. Olsen, and A. Delgado-Escueta (Bethesda: Oxford University Press).
Brew, H. M., Gittelman, J. X., Silverstein, R. S., Hanks, T. D., Demas, V. P., Robinson, L. C., et al. (2007). Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J. Neurophysiol. 98, 1501–1525. doi: 10.1152/jn.00640.2006
Brown, D. A., and Passmore, G. M. (2009). Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 156, 1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x
Browne, D. L., Gancher, S. T., Nutt, J. G., Brunt, E. R., Smith, E. A., Kramer, P., et al. (1994). Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 8, 136–140. doi: 10.1038/ng1094-136
Buono, R. J., Lohoff, F. W., Sander, T., Sperling, M. R., O’Connor, M. J., Dlugos, D. J., et al. (2004). Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res. 58, 175–183. doi: 10.1016/j.eplepsyres.2004.02.003
Cavalleri, G. L., Weale, M. E., Shianna, K. V., Singh, R., Lynch, J. M., Grinton, B., et al. (2007). Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol. 6, 970–998. doi: 10.1016/S1474-4422(07)70247-8
Chan, H. F., Chen, M. L., Su, J. J., Ko, L. C., Lin, C. H., and Wu, R. M. (2010). A novel neuropsychiatric phenotype of KCNJ2 mutation in one Taiwanese family with Andersen-Tawil syndrome. J. Hum. Genet. 55, 186–188. doi: 10.1038/jhg.2010.2
Chever, O., Djukic, B., McCarthy, K. D., and Amzica, F. (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010
Cooper, E. C. (2012). “Potassium channels (including KCNQ) and epilepsy,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. Olsen, and A. Delgado-Escueta (Bethesda: Oxford University Press).
Czirják, G., Tóth, Z. E., and Enyedi, P. (2007). Characterization of the heteromeric potassium channel formed by kv2.1 and the retinal subunit kv8.2 in Xenopus oocytes. J. Neurophysiol. 98, 1213–1222. doi: 10.1152/jn.00493.2007
D’Adamo, M. C., Catacuzzeno, L., Di Giovanni, G., Franciolini, F., and Pessia, M. (2013). K(+) channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front. Cell. Neurosci. 7:134. doi: 10.3389/fncel.2013.00134
D’Adamo, M. C., Imbrici, P., Sponcichetti, F., and Pessia, M. (1999). Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J. 13, 1335–1345.
Dazzo, E., Santulli, L., Posar, A., Fattouch, J., Conti, S., Lodén-van Straaten, M., et al. (2015). Autosomal dominant lateral temporal epilepsy (ADLTE): novel structural and single-nucleotide LGI1 mutations in families with predominant visual auras. Epilepsy Res. 110, 132–138. doi: 10.1016/j.eplepsyres.2014.12.004
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007
Du, W., Bautista, J. F., Yang, H., Diez-Sampedro, A., You, S. A., Wang, L., et al. (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 37, 733–738. doi: 10.1038/ng1585
Ermolinsky, B., Arshadmansab, M. F., Pacheco Otalora, L. F., Zarei, M. M., and Garrido-Sanabria, E. R. (2008). Deficit of Kcnma1 mRNA expression in the dentate gyrus of epileptic rats. Neuroreport 19, 1291–1294. doi: 10.1097/WNR.0b013e3283094bb6
Eunson, L. H., Rea, R., Zuberi, S. M., Youroukos, S., Panayiotopoulos, C. P., Liguori, R., et al. (2000). Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 48, 647–656. doi: 10.1002/1531-8249(200010)48:4<647::AID-ANA12>3.0.CO;2-Q
Fanciulli, M., Santulli, L., Errichiello, L., Barozzi, C., Tomasi, L., Rigon, L., et al. (2012). LGI1 microdeletion in autosomal dominant lateral temporal epilepsy. Neurology 78, 1299–1303. doi: 10.1212/WNL.0b013e3182518328
Ferraro, T. N., Golden, G. T., Smith, G. G., Martin, J. F., Lohoff, F. W., Gieringer, T. A., et al. (2004). Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm. Genome 15, 239–251. doi: 10.1007/s00335-003-2270-3
Freudenthal, B., Kulaveerasingam, D., Lingappa, L., Shah, M. A., Brueton, L., Wassmer, E., et al. (2011). KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron Physiol. 119, 40–48. doi: 10.1159/000330250
Fukata, Y., Lovero, K. L., Iwanaga, T., Watanabe, A., Yokoi, N., Tabuchi, K., et al. (2010). Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc. Natl. Acad. Sci. U.S.A. 107, 3799–3804. doi: 10.1073/pnas.0914537107
Fusco, C., Frattini, D., and Bassi, M. T. (2015). A novel KCNQ3 gene mutation in a child with infantile convulsions and partial epilepsy with centrotemporalspikes. Eur. J. Paediatr. Neurol. 19, 102–103. doi: 10.1016/j.ejpn.2014.08.006
Goldman, A. M., Glasscock, E., Yoo, J., Chen, T. T., Klassen, T. L., and Noebels, J. L. (2009). Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci. Transl. Med. 1:2ra6. doi: 10.1126/scitranslmed.3000289
González, C., Baez-Nieto, D., Valencia, I., Oyarzún, I., Rojas, P., Naranjo, D., et al. (2012). K(+) channels: function-structural overview. Compr. Physiol. 2, 2087–2149. doi: 10.1002/cphy.c110047
Grinton, B. E., Heron, S. E., Pelekanos, J. T., Zuberi, S. M., Kivity, S., Afawi, Z., et al. (2015). Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia 56, 1071–1080. doi: 10.1111/epi.13020
Gu, N., Vervaeke, K., and Storm, J. F. (2007). BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J. Physiol. 580, 859–882. doi: 10.1113/jphysiol.2006.126367
Guglielmi, L., Servettini, I., Caramia, M., Catacuzzeno, L., Franciolini, F., D’Adamo, M. C., et al. (2015). Update on the implication of potassium channels in autism: K(+) channelautism spectrum disorder. Front. Cell. Neurosci. 9:34. doi: 10.3389/fncel.2015.00034
Gutman, G. A., Chandy, K. G., Grissmer, S., Lazdunski, M., McKinnon, D., Pardo, L. A., et al. (2005). International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508. doi: 10.1124/pr.57.4.10
Haruna, Y., Kobori, A., Makiyama, T., Yoshida, H., Akao, M., Doi, T., et al. (2007). Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum. Mutat. 28:208. doi: 10.1002/humu.9483
Hattersley, A. T., and Ashcroft, F. M. (2005). Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes 54, 2503–2513. doi: 10.2337/diabetes.54.9.2503
Heilstedt, H. A., Burgess, D. L., Anderson, A. E., Chedrawi, A., Tharp, B., Lee, O., et al. (2001). Loss of the potassium channel beta-subunit gene, KCNAB2, is associated with epilepsy in patients with 1p36 deletion syndrome. Epilepsia 42, 1103–1111. doi: 10.1046/j.1528-1157.2001.08801.x
Hernández-Sánchez, C., Basile, A. S., Fedorova, I., Arima, H., Stannard, B., Fernandez, A. M., et al. (2001). Mice transgenically overexpressing sulfonylurea receptor 1 in forebrain resist seizure induction and excitotoxic neuron death. Proc. Natl. Acad. Sci. U.S.A. 98, 3549–3554. doi: 10.1073/pnas.051012898
Heron, S. E., Smith, K. R., Bahlo, M., Nobili, L., Kahana, E., Licchetta, L., et al. (2012). Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44, 1188–1190. doi: 10.1038/ng.2440
Heuser, K., Nagelhus, E. A., Taubøll, E., Indahl, U., Berg, P. R., Lien, S., et al. (2010). Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 88, 55–64. doi: 10.1016/j.eplepsyres.2009.09.023
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., et al. (2001). An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931.
Ho, Y. Y., Ionita-Laza, I., and Ottman, R. (2012). Domain-dependent clustering and genotype-phenotype analysis of LGI1 mutations in ADPEAF. Neurology 78, 563–568. doi: 10.1212/WNL.0b013e318247ccbf
Hu, S., Labuda, M. Z., Pandolfo, M., Goss, G. G., McDermid, H. E., and Ali, D. W. (2003). Variants of the KCNMB3 regulatory subunit of maxi BK channels affect channel inactivation. Physiol. Genom. 15, 191–198. doi: 10.1152/physiolgenomics.00110.2003
Huang, C. W., Huang, C. C., Cheng, J. T., Tsai, J. J., and Wu, S. N. (2007). Glucose and hippocampal neuronal excitability: role of ATP-sensitive potassium channels. J. Neurosci. Res. 85, 1468–1477. doi: 10.1002/jnr.21284
Imbrici, P., D’Adamo, M. C., Kullmann, D. M., and Pessia, M. (2006). Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur. J. Neurosci. 24, 3073–3083. doi: 10.1111/j.1460-9568.2006.05186.x
Inagaki, N., Gonoi, T., Clement, J. P. 4th, Namba, N., Inazawa, J., Gonzalez, G., et al. (1995). Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270, 1166–1170. doi: 10.1126/science.270.5239.1166
Ishii, A., Shioda, M., Okumura, A., Kidokoro, H., Sakauchi, M., Shimada, S., et al. (2013). A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene 531, 467–471. doi: 10.1016/j.gene.2013.08.096
Jabs, R., Seifert, G., and Steinhäuser, C. (2008). Astrocytic function and its alteration in the epileptic brain. Epilepsia 49, 3–12. doi: 10.1111/j.1528-1167.2008.01488.x
Jiang, Y., Pico, A., Cadene, M., Chait, B. T., and MacKinnon, R. (2001). Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron 29, 593–601.
Jin, W., Sugaya, A., Tsuda, T., Ohguchi, H., and Sugaya, E. (2000). Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res. 860, 21–28. doi: 10.1016/S0006-8993(00)01943-0
Johnson, J. N., Hofman, N., Haglund, C. M., Cascino, G. D., Wilde, A. A., and Ackerman, M. J. (2009). Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology 72, 224–231. doi: 10.1212/01.wnl.0000335760.02995.ca
Jorge, B. S., Campbell, C. M., Miller, A. R., Rutter, E. D., Gurnett, C. A., Vanoye, C. G., et al. (2011). Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility. Proc. Natl. Acad. Sci. U.S.A. 108, 5443–5448. doi: 10.1073/pnas.1017539108
Kaczmarek, L. K. (2013). Slack, slick and sodium-activated potassium channels. ISRN Neurosci. 2013:354262. doi: 10.1155/2013/354262
Kalachikov, S., Evgrafov, O., Ross, B., Winawer, M., Barker-Cummings, C., Martinelli Boneschi, F., et al. (2002). Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat. Genet. 30, 335–341. doi: 10.1038/ng832
Karschin, C., Dissmann, E., Stühmer, W., and Karschin, A. (1996). IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J. Neurosci. 16, 3559–3570.
Kaufmann, K., Romaine, I., Days, E., Pascual, C., Malik, A., Yang, L., et al. (2013). ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 4, 1278–1286. doi: 10.1021/cn400062a
Keller, D. I., Grenier, J., Christé, G., Dubouloz, F., Osswald, S., Brink, M., et al. (2009). Characterization of novel KCNH2 mutations in type 2 long QT syndrome manifesting as seizures. Can. J. Cardiol. 25, 455–462. doi: 10.1016/S0828-282X(09)70117-5
Kim, G. E., Kronengold, J., Barcia, G., Quraishi, I. H., Martin, H. C., Blair, E., et al. (2014). Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep. 9, 1661–1672. doi: 10.1016/j.celrep.2014.11.015
Krapivinsky, G., Gordon, E. A., Wickman, K., Velimirović, B., Krapivinsky, L., and Clapham, D. E. (1995). The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature 374, 135–141. doi: 10.1038/374135a0
Kuo, H. C., Cheng, C. F., Clark, R. B., Lin, J. J., Lin, J. L., Hoshijima, M., et al. (2001). A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107, 801–813. doi: 10.1016/S0092-8674(01)00588-8
Latorre, R., and Brauchi, S. (2006). Large conductance Ca2+-activated K+ (BK) channel: activation by Ca2+ and voltage. Biol. Res. 39, 385–401. doi: 10.4067/S0716-97602006000300003
Lee, B. H., Smith, T., and Paciorkowski, A. R. (2015). Autism spectrum disorder and epilepsy: disorders with a shared biology. Epilepsy Behav. 47, 191–201. doi: 10.1016/j.yebeh.2015.03.017
Lee, H., Lin, M. C., Kornblum, H. I., Papazian, D. M., and Nelson, S. F. (2014). Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum. Mol. Genet. 23, 3481–3489. doi: 10.1093/hmg/ddu056
Lee, I. C., Chen, J. Y., Chen, Y. J., Yu, J. S., and Su, P. H. (2009). Benign familial neonatal convulsions: novel mutation in a newborn. Pediatr. Neurol. 40, 387–391. doi: 10.1016/j.pediatrneurol.2008.12.004
Lee, U. S., and Cui, J. (2009). {beta} subunit-specific modulations of BK channel function by a mutation associated with epilepsy and dyskinesia. J. Physiol. 587, 1481–1498. doi: 10.1113/jphysiol.2009.169243
Leung, Y. M. (2010). Voltage-gated K+ channel modulators as neuroprotective agents. Life Sci. 86, 775–780. doi: 10.1016/j.lfs.2010.04.004
Lopatin, A. N., Makhina, E. N., and Nichols, C. G. (1994). Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372, 366–369. doi: 10.1038/372366a0
Lorenz, S., Heils, A., Kasper, J. M., and Sander, T. (2007). Allelic association of a truncation mutation of the KCNMB3 gene with idiopathic generalized epilepsy. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 10–13. doi: 10.1002/ajmg.b.30369
Lüscher, C., and Slesinger, P. A. (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315. doi: 10.1038/nrn2834
Maletic-Savatic, M., Lenn, N. J., and Trimmer, J. S. (1995). Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J. Neurosci. 15, 3840–3851.
Manna, I., Labate, A., Mumoli, L., Ferlazzo, E., Aguglia, U., Quattrone, A., et al. (2013). Failure to confirm association of a polymorphism in KCNMB4 gene with mesial temporal lobe epilepsy. Epilepsy Res. 106, 284–287. doi: 10.1016/j.eplepsyres.2013.03.014
Martin, H. C., Kim, G. E., Pagnamenta, A. T., Murakami, Y., Carvill, G. L., Meyer, E., et al. (2014). Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 23, 3200–3211. doi: 10.1093/hmg/ddu030
Martire, M., Barrese, V., D’Amico, M., Iannotti, F. A., Pizzarelli, R., Samengo, I., et al. (2010). Pre-synaptic BK channels selectively control glutamate versus GABA release from cortical and hippocampal nerve terminals. J. Neurochem. 115, 411–422. doi: 10.1111/j.1471-4159.2010.06938.x
Mazarati, A., Lundström, L., Sollenberg, U., Shin, D., Langel, U., and Sankar, R. (2006). Regulation of kindling epileptogenesis by hippocampal galanin type 1 and type 2 receptors: the effects of subtype-selective agonists and the role of G-protein-mediated signaling. J. Pharmacol. Exp. Ther. 318, 700–708. doi: 10.1124/jpet.106.104703
McCormack, K., Connor, J. X., Zhou, L., Ho, L. L., Ganetzky, B., Chiu, S. Y., et al. (2002). Genetic analysis of the mammalian K+ channel beta subunit Kvbeta 2 (Kcnab2). J. Biol. Chem. 277, 13219–13228. doi: 10.1074/jbc.M111465200
McKeown, L., Swanton, L., Robinson, P., and Jones, O. T. (2008). Surface expression and distribution of voltage-gated potassium channels in neurons (Review). Mol. Membr. Biol. 25, 332–343. doi: 10.1080/09687680801992470
McTague, A., Appleton, R., Avula, S., Cross, J. H., King, M. D., Jacques, T. S., et al. (2013). Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain 136, 1578–1591. doi: 10.1093/brain/awt073
Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., Migliore, M., Cilio, M. R., et al. (2013). Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc. Natl. Acad. Sci. U.S.A. 110, 4386–4391. doi: 10.1073/pnas.1216867110
Miceli, F., Striano, P., Soldovieri, M. V., Fontana, A., Nardello, R., Robbiano, A., et al. (2015). A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 56, e15–e20. doi: 10.1111/epi.12887
Mikati, M. A., Jiang, Y. H., Carboni, M., Shashi, V., Petrovski, S., Spillmann, R., et al. (2015). Quinidine in the treatment of KCNT1-positive epilepsies. Ann. Neurol. 78, 995–999. doi: 10.1002/ana.24520
Milligan, C. J., Li, M., Gazina, E. V., Heron, S. E., Nair, U., Trager, C., et al. (2014). KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 75, 581–590. doi: 10.1002/ana.24128
Møller, R. S., Heron, S. E., Larsen, L. H., Lim, C. X., Ricos, M. G., Bayly, M. A., et al. (2015). Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56, e114–e120. doi: 10.1111/epi.13071
Morante-Redolat, J. M., Gorostidi-Pagola, A., Piquer-Sirerol, S., Sáenz, A., Poza, J. J., Galán, J., et al. (2002). Mutations in theLGI1/Epitempin gene on 10q24 cause autosomal dominant lat-eral temporal epilepsy. Hum. Mol. Genet. 11, 1119–1128. doi: 10.1093/hmg/11.9.1119
Nardi, A., and Olesen, S. P. (2008). BK channel modulators: a comprehensive overview. Curr. Med. Chem. 15, 1126–1146. doi: 10.2174/092986708784221412
N’Gouemo, P. (2011). Targeting BK (big potassium) channels in epilepsy. Expert Opin. Ther. Targets 15, 1283–1295. doi: 10.1517/14728222.2011.620607
Nishida, M., and MacKinnon, R. (2002). Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111, 957–965. doi: 10.1016/S0092-8674(02)01227-8
Nobile, C., Michelucci, R., Andreazza, S., Pasini, E., Tosatto, S. C., and Striano, P. (2009). LGI1 mutations in autosomal dominant and sporadic lateral temporal epilepsy. Hum. Mutat. 30, 530–536. doi: 10.1002/humu.20925
Ohba, C., Kato, M., Takahashi, N., Osaka, H., Shiihara, T., Tohyama, J., et al. (2015). De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia 56, e121–e128. doi: 10.1111/epi.13072
Olson, T. M., and Terzic, A. (2010). Human K(ATP) channelopathies: diseases of metabolic homeostasis. Pflugers. Arch. 460, 295–306. doi: 10.1007/s00424-009-0771-y
Omichi, C., Momose, Y., and Kitahara, S. (2010). Congenital long QT syndrome presenting with a history of epilepsy: misdiagnosis or relationship between channelopathies of the heart and brain? Epilepsia 51, 289–292. doi: 10.1111/j.1528-1167.2009.02267.x
Orio, P., Rojas, P., Ferreira, G., and Latorre, R. (2002). New disguises for an old channel: MaxiK channel beta-subunits. News Physiol. Sci. 17, 156–161.
Partemi, S., Cestèle, S., Pezzella, M., Campuzano, O., Paravidino, R., Pascali, V. L., et al. (2013). Loss-of-function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia 54, e112–e116. doi: 10.1111/epi.12259
Partemi, S., Vidal, M. C., Striano, P., Campuzano, O., Allegue, C., Pezzella, M., et al. (2015). Genetic and forensic implications in epilepsy and cardiac arrhythmias: a case series. Int. J. Legal Med. 129, 495–504. doi: 10.1007/s00414-014-1063-4
Patil, N., Cox, D. R., Bhat, D., Faham, M., Myers, R. M., and Peterson, A. S. (1995). A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat. Genet. 11, 126–129. doi: 10.1038/ng1095-126
Pei, Q., Lewis, L., Grahame-Smith, D. G., and Zetterström, T. S. (1999). Alteration in expression of G-protein-activated inward rectifier K+-channel subunits GIRK1 and GIRK2 in the rat brain following electroconvulsive shock. Neuroscience 90, 621–627. doi: 10.1016/S0306-4522(98)00453-9
Pena, S. D., and Coimbra, R. L. (2015). Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy. Clin. Genet. 87, e1–e3. doi: 10.1111/cge.12542
Pessia, M., Imbrici, P., D’Adamo, M. C., Salvatore, L., and Tucker, S. J. (2001). Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J. Physiol. 532, 359–367. doi: 10.1111/j.1469-7793.2001.0359f.x
Phillips, H. A., Scheffer, I. E., Crossland, K. M., Bhatia, K. P., Fish, D. R., Marsden, C. D., et al. (1998). Autosomal dominant nocturnal frontal-lobe epilepsy: genetic heterogeneity and evidence for a second locus at 15q24. Am. J. Hum. Genet. 63, 1108–1116. doi: 10.1086/302047
Pongs, O., and Schwarz, J. R. (2010). Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90, 755–796. doi: 10.1152/physrev.00020.2009
Priori, S. G., Pandit, S. V., Rivolta, I., Berenfeld, O., Ronchetti, E., Dhamoon, A., et al. (2005). A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 96, 800–807. doi: 10.1161/01.RES.0000162101.76263.8c
Rho, J. M., Szot, P., Tempel, B. L., and Schwartzkroin, P. A. (1999). Developmental seizure susceptibility of kv1.1 potassium channel knockout mice. Dev. Neurosci. 21, 320–327. doi: 10.1159/000017381
Rhodes, K. J., Carroll, K. I., Sung, M. A., Doliveira, L. C., Monaghan, M. M., Burke, S. L., et al. (2004). KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J. Neurosci. 24, 7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004
Rizzo, F., Ambrosino, P., Guacci, A., Chetta, M., Marchese, G., Rocco, T., et al. (2016). Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell. Neurosci. 16, 54–63. doi: 10.1016/j.mcn.2016.01.004
Robbins, C. A., and Tempel, B. L. (2012). Kv1.1 and Kv1.2: similar channels, different seizure models. Epilepsia 53(Suppl. 1), 134–141. doi: 10.1111/j.1528-1167.2012.03484.x
Scholl, U. I., Choi, M., Liu, T., Ramaekers, V. T., Häusler, M. G., Grimmer, J., et al. (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847. doi: 10.1073/pnas.0901749106
Schulte, U., Thumfart, J. O., Klöcker, N., Sailer, C. A., Bildl, W., Biniossek, M., et al. (2006). The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron 49, 697–706. doi: 10.1016/j.neuron.2006.01.033
Shah, N. H., and Aizenman, E. (2014). Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 5, 38–58. doi: 10.1007/s12975-013-0297-7
Shang, L., Lucchese, C. J., Haider, S., and Tucker, S. J. (2005). Functional characterisation of missense variations in the Kir4.1 potassium channel (KCNJ10) associated with seizure susceptibility. Brain Res. Mol. Brain Res. 139, 178–183. doi: 10.1016/j.molbrainres.2005.05.003
Sicca, F., Imbrici, P., D’Adamo, M. C., Moro, F., Bonatti, F., Brovedani, P., et al. (2011). Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol. Dis. 43, 239–247. doi: 10.1016/j.nbd.2011.03.016
Signorini, S., Liao, Y. J., Duncan, S. A., Jan, L. Y., and Stoffel, M. (1997). Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc. Natl. Acad. Sci. U.S.A. 94, 923–927. doi: 10.1073/pnas.94.3.923
Singh, B., Ogiwara, I., Kaneda, M., Tokonami, N., Mazaki, E., Baba, K., et al. (2006). A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol. Dis. 24, 245–253. doi: 10.1016/j.nbd.2006.07.001
Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., Melis, R., et al. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29.
Slesinger, P. A., Patil, N., Liao, Y. J., Jan, Y. N., Jan, L. Y., and Cox, D. R. (1996). Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+ channels. Neuron 16, 321–331. doi: 10.1016/S0896-6273(00)80050-1
Slesinger, P. A., Reuveny, E., Jan, Y. N., and Jan, L. Y. (1995). Identification of structural elements involved in G protein gating of the GIRK1 potassium channel. Neuron 15, 1145–1156. doi: 10.1016/0896-6273(95)90102-7
Smart, S. L., Lopantsev, V., Zhang, C. L., Robbins, C. A., Wang, H., Chiu, S. Y., et al. (1998). Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron 20, 809–819. doi: 10.1016/S0896-6273(00)81018-1
Smets, K., Duarri, A., Deconinck, T., Ceulemans, B., van de Warrenburg, B. P., Züchner, S., et al. (2015). First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med. Genet. 16:51. doi: 10.1186/s12881-015-0200-3
Soldovieri, M. V., Boutry-Kryza, N., Milh, M., Doummar, D., Heron, B., Bourel, E., et al. (2014). Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: first evidence for an altered channel regulation by syntaxin-1A. Hum. Mutat. 35, 356–367. doi: 10.1002/humu.22500
Spauschus, A., Eunson, L., Hanna, M. G., and Kullmann, D. M. (1999). Functional characterization of a novel mutation in KCNA1 in episodic ataxia type 1 associated with epilepsy. Ann. N. Y. Acad. Sci. 868, 442–446. doi: 10.1111/j.1749-6632.1999.tb11310.x
Steinlein, O. K. (2004). Genetic mechanisms that underlie epilepsy. Nat. Rev. Neurosci. 5, 400–408. doi: 10.1038/nrn1388
Syrbe, S., Hedrich, U. B., Riesch, E., Djémié, T., Müller, S., Møller, R. S., et al. (2015). De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 47, 393–399. doi: 10.1038/ng.3239
Tamsett, T. J., Picchione, K. E., and Bhattacharjee, A. (2009). NAD+ activates KNa channels in dorsal root ganglion neurons. J. Neurosci. 29, 5127–5134. doi: 10.1523/JNEUROSCI.0859-09.2009
Tiron, C., Campuzano, O., Pérez-Serra, A., Mademont, I., Coll, M., Allegue, C., et al. (2015). Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure. 25, 65–67. doi: 10.1016/j.seizure.2015.01.003
Trimmer, J. S. (1998). Regulation of ion channel expression by cytoplasmic subunits. Curr. Opin. Neurobiol. 8, 370–374. doi: 10.1016/S0959-4388(98)80063-9
Tu, E., Bagnall, R. D., Duflou, J., and Semsarian, C. (2011). Post-mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol. 21, 201–208. doi: 10.1111/j.1750-3639.2010.00438.x
Tuchman, R., Moshé, S. L., and Rapin, I. (2009). Convulsing toward the pathophysiology of autism. Brain Dev. 31, 95–103. doi: 10.1016/j.braindev.2008.09.009
Vandenberg, J. I., Perry, M. D., Perrin, M. J., Mann, S. A., Ke, Y., and Hill, A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478. doi: 10.1152/physrev.00036.2011
Vanderver, A., Simons, C., Schmidt, J. L., Pearl, P. L., Bloom, M., Lavenstein, B., et al. (2014). Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr. Neurol. 50, 112–114. doi: 10.1016/j.pediatrneurol.2013.06.024
Wang, H. G., He, X. P., Li, Q., Madison, R. D., Moore, S. D., McNamara, J. O., et al. (2013). The auxiliary subunit KChIP2 is an essential regulator of homeostatic excitability. J. Biol. Chem. 288, 13258–13268. doi: 10.1074/jbc.M112.434548
Wang, Q., Curran, M. E., Splawski, I., Burn, T. C., Millholland, J. M., VanRaay, T. J., et al. (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23. doi: 10.1038/ng0196-17
Weckhuysen, S., Mandelstam, S., Suls, A., Audenaert, D., Deconinck, T., Claes, L. R., et al. (2012). KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25. doi: 10.1002/ana.22644
Yamada, K., Ji, J. J., Yuan, H., Miki, T., Sato, S., Horimoto, N., et al. (2001). Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science 292, 1543–1546. doi: 10.1126/science.1059829
Yang, T., Chung, S. K., Zhang, W., Mullins, J. G., McCulley, C. H., Crawford, J., et al. (2009). Biophysical properties of 9 KCNQ1 mutations associated with long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2, 417–426. doi: 10.1161/CIRCEP.109.850149
Zamorano-León, J. J., Yañez, R., Jaime, G., Rodriguez-Sierra, P., Calatrava-Ledrado, L., Alvarez-Granada, R. R., et al. (2012). KCNH2 gene mutation: a potential link between epilepsy and long QT-2 syndrome. J. Neurogenet. 26, 382–386. doi: 10.3109/01677063.2012.674993
Zara, F., Specchio, N., Striano, P., Robbiano, A., Gennaro, E., Paravidino, R., et al. (2013). Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia 54, 425–436. doi: 10.1111/epi.12089
Keywords: K+ channels, epilepsy, mutation, KCNT1, Kir channels, Kv channels
Citation: Villa C and Combi R (2016) Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front. Cell. Neurosci. 10:81. doi: 10.3389/fncel.2016.00081
Received: 28 September 2015; Accepted: 15 March 2016;
Published: 30 March 2016.
Edited by:
Laura Cancedda, Istituto Italiano di Tecnologia, ItalyReviewed by:
Satpal Singh, State University of New York at Buffalo, USALuca Imeri, University of Milan, Italy
Copyright © 2016 Villa and Combi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Villa, Y2hpYXJhLnZpbGxhQHVuaW1pYi5pdA==