 Bruno Di Marco Vieira
Bruno Di Marco Vieira Rowan A. Radford
Rowan A. Radford Roger S. Chung
Roger S. Chung Gilles J. Guillemin
Gilles J. Guillemin Dean L. Pountney
Dean L. Pountney
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 12 November 2015
Sec. Non-Neuronal Cells
Volume 9 - 2015 | https://doi.org/10.3389/fncel.2015.00437
This article is part of the Research Topic Glial Cells: Managers of Neuro-immunity View all 18 articles
Multiple system atrophy (MSA) is a progressive neurodegenerative disease presenting with combinations of autonomic dysfunction, parkinsonism, cerebellar ataxia and/or pyramidal signs. Oligodendroglial cytoplasmic inclusions (GCIs) rich in α-synuclein (α-syn) constitute the disease hallmark, accompanied by neuronal loss and activation of glial cells which indicate neuroinflammation. Recent studies demonstrate that α-syn may be released from degenerating neurons to mediate formation of abnormal inclusion bodies and to induce neuroinflammation which, interestingly, might also favor the formation of intracellular α-syn aggregates as a consequence of cytokine release and the shift to a pro-inflammatory environment. Here, we critically review the relationships between α-syn and astrocytic and microglial activation in MSA to explore the potential of therapeutics which target neuroinflammation.
Multiple System Atrophy (MSA) is a complex progressive neurodegenerative disease which affects 3.4–4.9 cases/100,000 with 0.6–0.7/100,000 new cases each year. It has no significant gender bias with disease onset typically over the age of 60 and a mean survival of ~7.9 years following diagnosis (Watanabe et al., 2002; Wenning et al., 2013; Longo et al., 2015). Although there are no strong genetic determinants, studies have associated MSA cases with the H1 haplotype of MAPT, also linked to tauopathies such as Progressive Supranuclear Palsy (PSP; Vilariño-Güell et al., 2011), COQ2 loss of function linked mutations (Multiple-System Atrophy Research Collaboration, 2013) and hexanucleotide repeat expansions in C9orf72 (also associated with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia; Goldman et al., 2014). Clinically, a predominance of Parkinsonism (MSA-P) or Cerebellar Ataxia (MSA-C) plus a heterogenous combination of pyramidal signs, autonomic and urogenital dysfunctions may be detected (Longo et al., 2015). Due to this complex phenotype, definite MSA diagnosis requires autopsy to detect glial cytoplasmic inclusions (GCIs) immunopositive for α-synuclein (α-syn) and neurodegeneration in striatonigral or olivopontocerebellar structures (Lantos, 1998; Trojanowski and Revesz, 2007; Gilman et al., 2008).
Alpha-synuclein is a 14.4 kDa protein of predominant neuronal pre-synaptic location where it is believed to chaperone the assembly of synaptic vesicles for exocytosis via interaction with synaptotagmin (SNARE complex component) and has characteristic conformational plasticity. It normally exists as a soluble monomer/tetramer in equilibrium with a membrane-bound α-helical multimer (Narayanan and Scarlata, 2001; Tong et al., 2009; Burré et al., 2014). However, for reasons not yet fully elucidated, α-syn may misfold into abnormal dimers, oligomers, or fibrils/protofibrils that aggregate and constitute the pathological hallmark of several neurodegenerative conditions, including Parkinson’s disease (PD) and Dementia with Lewy Bodies (DLB), where they primarily occur in neurons (McKeith et al., 2005; Shulman et al., 2011). As mature human oligodendrocytes do not express α-syn normally (Miller et al., 2005), the origin of α-syn glial aggregates in MSA is unclear, whether as a consequence of primary oligodendrogliopathy followed by neuronal degeneration or a neuronal α-synucleinopathy leading to glial inclusions (Nishie et al., 2004; Wenning et al., 2008). Thus, it has been hypothesized that intercellular transmission of α-syn might be occurring via mechanisms such as endocytosis, direct penetration, micropinocytosis, pore formation, nanotube tunneling or diffusion (Ubhi et al., 2011; Konno et al., 2012). Nevertheless, the proposed mechanisms of release and subsequent cellular uptake suggest that at α-syn pathology transmission may occur in a putative prion-like manner (Prusiner et al., 2015).
In addition to α-syn rich GCIs as the central pathological feature, MSA also exhibits neuronal loss and strong neuroinflammation which both correlate with the density of inclusions and disease duration (Gai et al., 2003; Ozawa et al., 2004; Ahmed et al., 2012) as well as expression of inflammatory markers (Chen et al., 2015). Neuroinflammation is a dynamic response that involves changes in glial cell morphology, number, function and concomitant production of signaling molecules (O’Callaghan et al., 2008; Shastri et al., 2013). In the context of neurodegenerative diseases, persistent intra- and extracellular imbalances (such as those caused by misfolded proteins, oxidative stress, and neuronal death) are known to trigger and chronically perpetuate this response, which is dominated by microglia and astrocytes (Takeuchi, 2013). Gliosis is the term that indicates the phenotypic changes of glia and is exemplified in Figures 1A–C, where activated astrocytes and microglia co-localize with GCI pathology. This manuscript explores the role of α-syn and its relationship to neuroinflammation mediated by astrocytes and microglia in MSA.
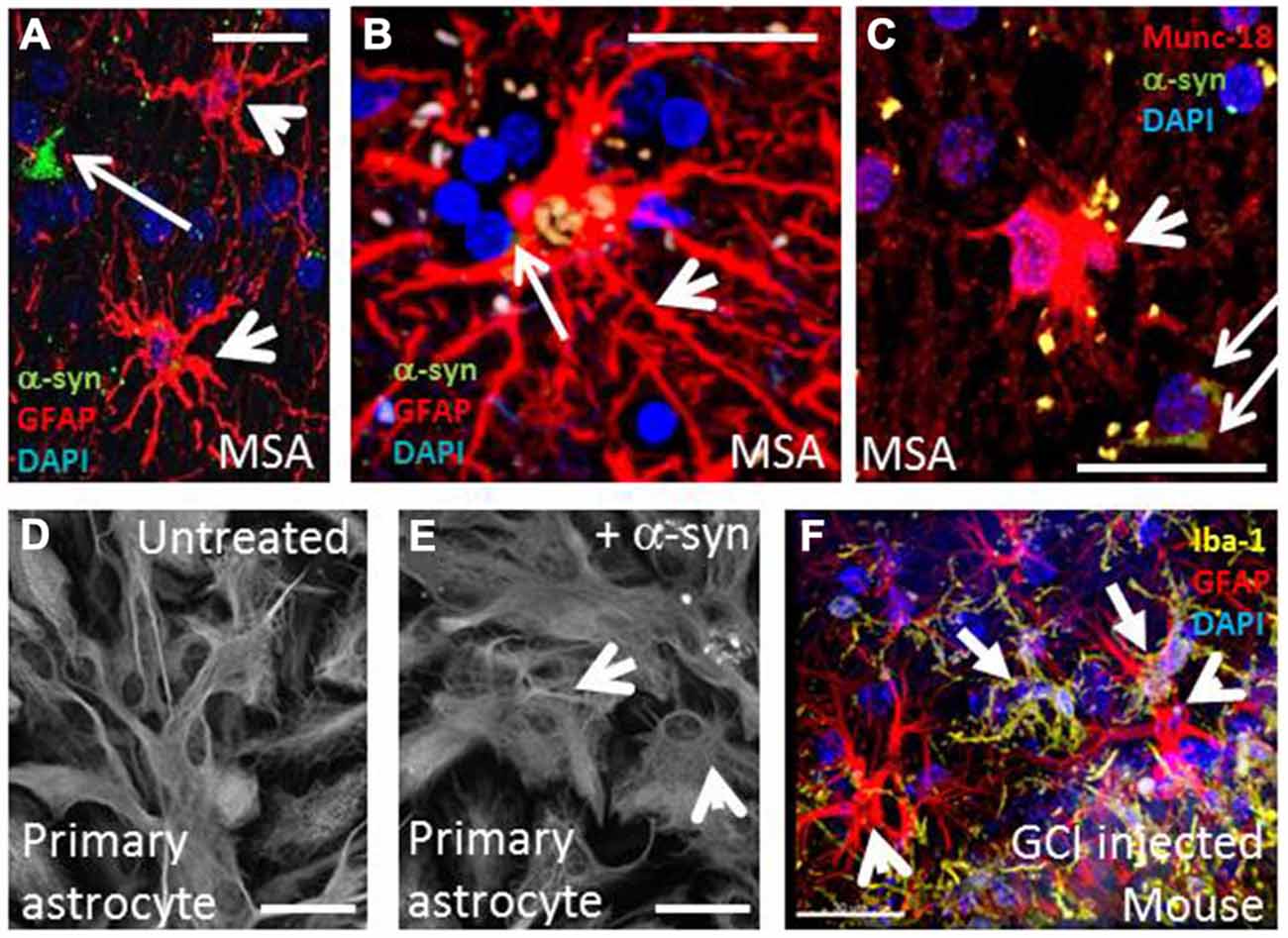
Figure 1. Multiple system atrophy is characterized by widespread oligodendroglial α-syn inclusion bodies, astrogliosis and microgliosis. (A,B) MSA putamen (A) and visual (B) showing activated astrocytes (arrowheads, GFAP, red) in close proximity to GCIs (arrows, α-syn, green). (C) A subset of activated astrocytes are also intensely immunopositive for the exocytic vesicle marker, munc18 (Radford et al., 2015). (D,E) Rat primary astrocyte cells adopt activated morphology when treated with α-syn. (E) compared to control cells (D) Scale bars, 20 μm. (F) Frequent activated microglia (solid arrow, yellow, Iba-1) and activated astrocytes (red, GFAP) occur near to the site of GCI injection in unilateral-lesioned mice (Radford et al., 2015). Scale bar, 30 μm.
That α-syn is the pathogenic root of neurodegeneration in MSA has been given further credence by the discovery of SNCA mutations in α-synucleinopathies (mostly PD), but where G51D and A53E α-syn mutations have been described in patients with atypical parkinsonism and may overlap with MSA (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004; Lesage et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014). In MSA, it remains unclear which α-syn form/s is the principal mediator of toxicity but their compact and insoluble structure allows them to resist intracellular cleavage, accumulate and disrupt otherwise normal downstream processes (e.g., the ubiquitin-proteasome system, synaptic exocytosis, mitochondrial metabolism and ER-Golgi transport; Burré et al., 2015). Multiple modes of α-syn toxicity are reviewed elsewhere, including membrane permeabilization by annular oligomers and disrupting protein degradation pathways by inhibition of the proteasome and autophagy (Cuervo et al., 2004; Winner et al., 2011; Daturpalli et al., 2013; Roberts and Brown, 2015), but it is accepted that this α-syn-induced dysfunction can lead to the death of central nervous system (CNS) cells that defines neurodegeneration (Radford et al., 2014).
In addition, the exchange of amorphous α-syn between neurons and glial cells via exo- and endocytosis characterizes the cell-to-cell transmission and uptake that may ultimately lead to pathology spread (Reyes et al., 2014), with evidence for unconventional exocytosis (independent from ER-Golgi) and exosomes as mechanisms of release (Emmanouilidou et al., 2010; Jang et al., 2010). Key studies have recently provided evidence for α-syn transmission to occur in a prion-like manner. Thus, α-syn isolated post-mortem from MSA cases was transferred to HEK cells (modified to express the A53T mutation and tagged with yellow fluorescent protein, namely α-syn 140*A53T-YFP), that were then found to increase aggregate formation and increase YFP aggregate expression from the infected single-cells group, when compared to untransfected group (indicating a de novo prion formation in the transfected group; Woerman et al., 2015). In line with these findings, when brain extracts from MSA cases were injected intracerebrally in transgenic mice (M83 carrying the A53T mutation, namely tgM83), phenotypic changes (e.g., dysmetria and circling behavior) manifested 100–150 days post-inoculation in the homozygous group and pathological aggregates of phosphorylated α-syn and astrogliosis were detected in regions including brainstem. Interestingly, tgM83 mice inoculated with PD homogenates did not exhibit specific α-syn deposition or manifest clinical alterations significantly different from those of controls (Prusiner et al., 2015). These findings provide experimental evidence for MSA as a possible prion disease, with different α-syn strains being able to spread and promote tissue pathology in contrast with those of PD.
Astrocytes are key players in CNS homeostasis and pathology and are involved in a wide array of functions including modulating CNS immunity and inflammation, synaptic pruning and degradation of neuronal organelles (De Keyser et al., 2008; Sofroniew and Vinters, 2010; Chung et al., 2013; Hostenbach et al., 2013; Davis et al., 2014; Ben Haim et al., 2015). In MSA, the influence of α-syn on astrogliosis has been investigated by several studies. Treatment of primary astrocytes with α-syn promoted astrogliotic changes, as shown in Figures 1D,E (Radford et al., 2015). In astrocytes transfected with an inhibitor of endocytic vesicle formation (dominant negative dynamin-1 K44A mutant) and co-cultured with neuron-derived cell lines expressing α-syn, endocytosis was shown as a mechanism for direct uptake, which strongly correlates with the production of cytokines (such as IL-1α, -1β, -6, -18), colony-stimulating factors (CSF-1, -2, -3), and chemokines [(CCL-3, -4, -5, -12, -20), (CXCL-1, -2, -5, -10, -11, -12, -16)] (Lee et al., 2010). Moreover, in transgenic mice overexpressing oligodendroglial α-syn, exposure to oxidative stress (using the mitochondrial inhibitor 3-nitropropionic acid) led to astrogliosis and degeneration in close proximity to GCIs (Stefanova et al., 2005). Accordingly, the morphometric analysis of human cases and mouse models of MSA reveals that the degree of astrogliosis increased with proximity to α-syn deposits, as seen in Figures 1A,B (Song et al., 2009; Radford et al., 2015).
Microglia account for approximately 10% of all brain cells and derive from a primitive myeloid lineage of macrophages that migrate to cerebral regions during intrauterine life, after which they are distributed unevenly throughout the brain hemispheres, mostly concentrating in the hippocampus, basal ganglia and substantia nigra (Prinz et al., 2011). Normally, in the healthy brain, resident microglia adopt a resting (surveillant) phenotype, which is maintained by feedback of signaling molecules such as neuronal fractalkine and astrocytic glial-derived neurotropic factor, with perturbations of homeostasis triggering microglial activation into effector phenotypes, namely M1 and M2 (Tang and Le, 2015). This requires interaction of the noxious stimulus with immune response receptors such as complement factors, pattern recognition receptors and scavenger receptors (Husemann et al., 2002; Scheffel et al., 2012). Once activated, the M1 phenotype produces pro-inflammatory and cytotoxic molecules, such as TNF-α, IL-6, IL-1β, superoxide, NO, reactive oxygen species (ROS) and excitatory amino acids, which can induce more neuronal damage and progression of cellular dysfunction (Kettenmann et al., 2011). The other activated state, M2, also manifests phagocytic activity, but performs anti-inflammatory responses through release of IL-10 and transforming growth factor beta (TGF-β). It may also be induced by anti-inflammatory cytokines (e.g., IL-13 and IL-14) and acts in tissue repair via release of growth factors such as major histocompatibility complex 5, monocyte chemoattractant protein 1 and insulin-like growth factor 1 (Colton and Wilcock, 2010; Welser-Alves and Milner, 2013).
Morphologically, cellular hypertrophy and branching are the most commonly described changes in microgliosis which, depending on nature and intensity of the damage, may be detected as early as minutes to hours after acute injuries, with rapid process extension occurring in an ATP-dependent manner through P2Y12 receptors (Jensen et al., 1999; Davalos et al., 2005; Nimmerjahn et al., 2005; Parkhurst and Gan, 2010). In primary mesencephalic neuron-glia culture systems, extracellular α-syn was shown to be directly phagocytosed by microglia producing microgliosis, upregulation of NADPH oxidase and secretion of ROS, enhancing neurodegeneration (Zhang et al., 2005). Microgliosis can be identified clustering around α-syn years to decades after α-syn accumulation (Ishizawa et al., 2004; Graeber and Streit, 2010), or colocalizing with α-syn-rich neurons after direct stereotactic injection of α-syn ribbons or fibrils (Peelaerts et al., 2015).
In the extracellular environment, the abnormal presence of α-syn can be sensed and internalized by glial cells, leading to a cascade of reactive gliosis, secretion of pro-inflammatory cytokines and subsequent cell recruitment; characterizing the amplification of a localized deposit of protein. Localized micro- and astrogliosis resulted from injection of purified GCI material into mouse medial forebrain bundle after 23 days, as shown in Figure 1F (Radford et al., 2015). In microglia, pathogen pattern recognition receptors in the membrane surface enable the initial identification of foreign structural motifs on multiple arrays of pathogens (in the case of infectious disease), but they are also capable of recognizing changes in homeostatic cellular conditions and endogenous molecules, such as misfolded proteins in neurodegenerative diseases (Stefanova et al., 2005; Block et al., 2007). In particular, the Toll-like receptors (TLRs) 2 and 4 are known to interact with α-syn. In a cellular model, purified microglial cultures from brains of wild type (TLR4+/+) and deficient (TLR4−/−) postnatal mice were treated with wild-type and abnormal α-syn forms (fibrillar, truncated). The results revealed prolific microgliosis in the TLR4+/+ groups, increased phagocytic activity, upregulation of nuclear factor-kappa B (NF-κB), and increased production of CXCL1, IL-6, and TNF-α. Furthermore, TLR4-deficient microglia showed a reduced production of ROS upon α-syn treatments. In line with these findings, both human cases and transgenic mouse models of MSA also exhibit upregulation of TLRs (Béraud et al., 2011; Brudek et al., 2013; Fellner et al., 2013).
The temporal relationship between astro- and microgliosis in MSA is still poorly understood, as preclinical studies cannot reproduce the long timescale of MSA pathogenesis. Clinical radiology could overcome this limitation but, to date, tagging neuroinflammation with specific markers (Gerhard et al., 2003) has not been performed (Schrag et al., 1998; Schocke et al., 2002; Ozawa et al., 2004; Watanabe et al., 2004; Brooks et al., 2009; Chandran and Stoessl, 2014). Despite limitations related to preclinical and clinical assessment of neuroinflammation, it may be that, because glial cells express no or very little α-syn, glial uptake of α-syn occurs and α-syn triggers the neuroinflammatory process, which may then operate in waves of incremental feed-forward damage. This process, combined with the prion-like behavior of α-syn, implicates the role of neuroinflammation in worsening/perpetuating MSA. Figure 2 represents schematically the interplay between α-syn aggregation and release, astrogliosis and microgliosis and the feedback of pro-inflammatory factors that may in turn result in additional neuronal stress.
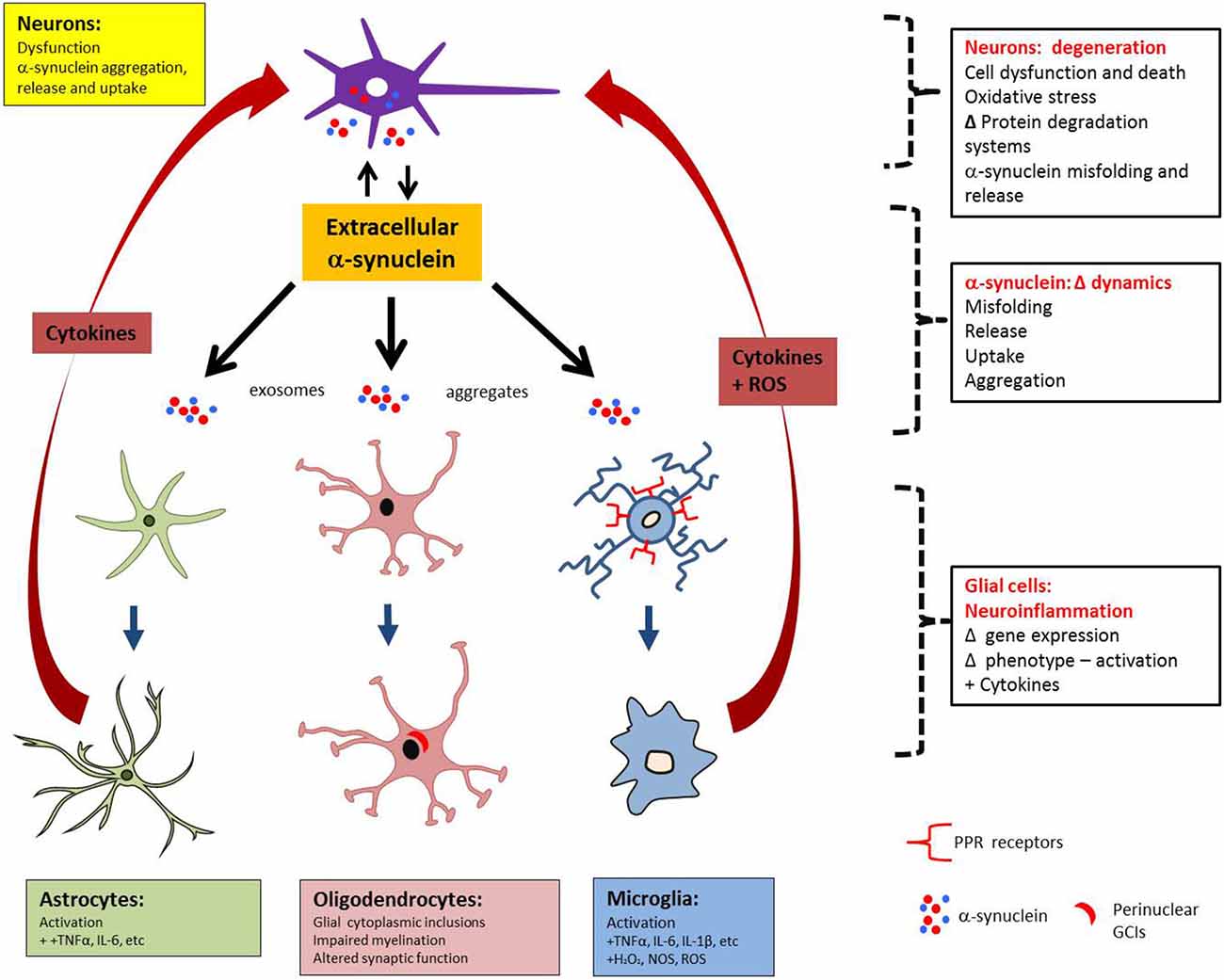
Figure 2. MSA pathology may spread between anatomically connected regions as a result of reciprocal rounds of α-syn release and neuroinflammation. Neuronal dysfunction can lead to α-syn aggregation and release of α-syn aggregates, which can then interact directly with astrocytes and microglia to mediate activation. In turn, the release of pro-inflammatory factors by activated glia can act back on neurons to cause stress, thereby stimulating the formation and release of additional α-syn aggregates.
It is known that extracellular α-syn may directly act upon astrocytes, microglia and oligodendrocytes. However, to date, there is a lack of studies specifically addressing gliosis as a trigger for α-syn misfolding and/or release (Croisier et al., 2005). Because of their role in surveillance and in reaction to pathogens, microglia may exert an indirect effect on α-syn by secreting a variety of toxic factors, which disrupt basic intracellular protein degradation systems and ultimately affect α-syn dynamics (Fellner and Stefanova, 2013). For example, it is known that α-syn oxidation and nitration inhibits chaperone-mediated autophagy (CMA; Kiffin et al., 2004; Martinez-Vicente et al., 2008; Xilouri et al., 2009, 2013) and that α-syn phosphorylation alters macroautophagy (Tenreiro et al., 2014) which, unlike CMA, can eliminate larger protein species, such as oligomers and aggregates (Engelender, 2012; Tanik et al., 2013). Moreover, neuron-glia cultures treated with lipopolysaccharide (LPS), a potent stimulator of microgliosis, have increased H2O2-mediated chemoattraction towards α-syn aggregates (Ejlerskov et al., 2015), which can be enhanced by pre-injection of LPS in an oxidative-stress (rotenone) mouse model (Tien et al., 2013). Although a unified body of data is required to define a single model of MSA, the overproduction of cytotoxic by-products by microgliosis may contribute to α-syn misfolding/aggregation.
MSA may also share common pathological features with the tauopathy PSP, although astrocytes display a degenerative phenotype in PSP tissue rather than a reactive one (Togo and Dickson, 2002; Radford et al., 2015). Studies with microglial fractalkine receptor deficient hTauCx3cr1−/− mice have shown that enhanced microglial activation led to accelerated tau pathology and could be transferred to non-transgenic recipient mice by adoptive microglia; which was blocked by the interleukin1 receptor antagonist, Kineret (Bhaskar et al., 2010; Maphis et al., 2015). Recently, microglia have been shown to be directly linked to hTau propagation between non-synaptically connected neuronal populations in vivo via exosome release following phagocytosis of hTau (Asai et al., 2015). As most experiments to date have used models of PD to address α-syn diseases, future experiments will need to focus on specific MSA animal and cell culture models, such as by direct injection of purified GCI material (Radford et al., 2015), that may better mimic the disease’s specific cellular features (Halliday and Stevens, 2011), especially to further elucidate the role of both astrocytes and microglia in α-syn misfolding/aggregation and spreading.
Due to the possible cyclic nature of α-syn aggregation/release and gliosis in MSA, interventions that target neuroinflammation have the potential to slow the progression of disease and increase quality of life. Recent studies have approached α-synucleinopathies, including MSA, by use of immunotherapy (Valera and Masliah, 2013). For example, short immunogenic peptides mimicking the C-terminus of α-syn were administered to MBP-α-syn trangenic mice, followed by measurements of inducible anti-α-syn antibodies, cellular and tissue outcomes over time. Interestingly, as the peptides used did not carry the native epitope but instead a variation of it, this approach did not produce autoimmunity reactions via T-cells, leading to a beneficial prolonged response. Moreover, the induced antibodies were able to cross the blood-brain barrier where they could detect intracellular α-syn (monomer, oligomers and aggregates) after being internalized by microglia, oligodendrocytes and astrocytes. This reduced α-syn colocalization in oligodendrocytes and astrocytes but not in microglia, suggesting increased microglial uptake whilst preserving the oligodendrocyte population and decreasing demyelination, neuronal death and motor deficits (Mandler et al., 2015). Indeed, as in animal models of α-synucleinopathy the microglial response occurs prior to neuronal loss (Sanchez-Guajardo et al., 2015), strategies such as pre-immunization with α-syn peptides or approaches aimed at priming the CNS against α-syn immune insult may provide a glial memory and lessen subsequent neuroinflammatory responses to therapeutic benefit. Moreover, as activated microglia could participate actively in the spread of α-syn pathology, therapies that promote microgliosis to facilitate the clearance of extracellular pathological protein aggregates may have unwanted side effects. Furthermore, a recent study has shown that peripheral vector administration of the protease neurosin could degrade extracellular α-syn and thereby may reduce microglial and astrocyte activation (Spencer et al., 2015).
Some studies also aimed at microgliosis as a therapeutic target. For example, the inhibition of pro-inflammatory (iNOS or NAPH oxidase) enzymes from activated microglia is followed by decreased degeneration of neurons upon treatments with 7-nitroindazole and apocynin (Gao et al., 2003). Also, the treatment of the PLP-α-syn mouse model with minocycline decreased the density of activated microglia in the SNpc, reduced iNOS and TLR4-immunoreactivity and reduced dopaminergic degeneration (Stefanova et al., 2007).
As their differentiation capacity and immunomodulatory properties can address both neurodegeneration and neuroinflammation, mesenchymal stem-cells (MSCs) have been investigated as therapeutic options. In a study using the transgenic PLP mouse model of MSA (Stemberger et al., 2011), intravenous transplantation of MSCs promoted neuroprotection in the SNpc (as determined by TH+ neurons in the treated group) and down regulation of cytokines (IL-1α, IL-2, Il-10, TGF-β1 and TNF-α) in midbrain-brainstem lysates 4 weeks post-injection. Similarly, in the double-toxin-induced MSA-P mouse model, treatment with human MSC (hMSC) improved motor behavior (pole-descending test), increased neuronal survival (TH- and NeuN-positive markers), and decreased astro- and microgliosis (anti-Iba1 and anti-GFAP immunostaining, respectively) in the SNpc and striatum (Park et al., 2011). Lastly, a clinical trial using autologous MSC transplantation in patients with MSA-C improved symptoms severity [as observed on the baseline unified MSA rating scale (UMSARS)], and attenuated the declines of cerebral glucose metabolism and gray matter density (assessed by neuroimaging) along a 360-day follow-up (Lee et al., 2012). The immunosuppressive actions of MSC are believed to operate in a non-MHC-restricted manner, for example via secretion of soluble factors such as TGF-β1, PGE2 and indoleamine 2,3-dioxygenase (IDO; Krampera et al., 2006b). As glial cells are known to use IDO in the conversion of tryptophan to kynurenine, the finding that interferon-γ can upregulate the enzyme points to the kynurenine pathway as another therapeutic target in neuroinflammation related and non-related to MSCs (Krampera et al., 2006a). Recently, mice lacking anti-inflammatory interferon-β were shown to develop motor and cognitive deficits and α-syn pathology similar to a DLB/PD phenotype (Ejlerskov et al., 2015). This strengthens the link between dysfunctional inflammation and α-synucleinopathies and indicates dysfunction in inflammation can induce α-syn toxicity and vice versa.
Therapies targeting neuroinflammation in other α-syn disease models may warrant investigation in MSA. One example is the induction of heat shock proteins (HSPs) which are known to act via multiple pathways such as protein misfolding, neuroinflammation and mitochondrial oxidative phosphorylation. Recently, a HSP inducer, carbenoxolone, was shown to decrease astrogliosis, pro-inflammatory cytokines and oxidative stress in a rotenone model of PD (Thakur and Nehru, 2015). Additionally, HSPs are intimately involved in the degradation of α-syn by CMA (Wong et al., 2013; Vijayakumaran et al., 2015) and their induction may provide dual benefits by reducing neuroinflammation and toxic α-syn aggregates. The renin-angiotensin system may also be targeted to reduce the microglial inflammatory response via angiotensin II antagonists (Labandeira-Garcia et al., 2011). Microglial β-nicotinamide adenine dinucleotide phosphate oxidase 2 inhibitors may be useful by decreasing microglial O2- generation (Zhang et al., 2014). Other alternatives may be to combat oxidative stress, such as by the naturally occurring Acetyl-L-Carnitine (Singh et al., 2015). Counterbalancing neuroinflammation in glia and immune cells with naturally occurring lipids or hormonal modulation (Herrera et al., 2015; Skaper et al., 2015) may also be promising therapeutic strategies for MSA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by Griffith University and Menzies Health Institute Queensland.
α-syn, α-synuclein; CMA, Chaperone-mediated autophagy; CNP, Cyclic nucleotide phosphodiesterase; CNS, Central nervous system; CSFs, Colony-stimulating factors; DLB, Dementia with Lewy bodies; ER, Endoplasmic reticulum; GCI, Glial cytoplasmic inclusion; GFAP, Glial fibrillary acidic protein; HSP, Heat shock protein; IDO, Indoleamine 2,3-dioxygenase; IL, Interleukin; iNOS, Inducible nitric oxide synthase; LPS, Lipopolysaccharide; MBP, Myelin basic protein; MSA, Multiple system atrophy; MSC, Mesenchymal stem-cells; NADPH, Nicotinamide adenine dinucleotide phosphate hydroxylase; NF-κB, Nuclear factor kappa B; NO, Nitric oxide; PD, Parkinson’s disease; PET, Positron Emission Tomography; PGE2, Prostaglandin E2; PLP, Proteolipid protein; PSP, Progressive Supranuclear Palsy; ROS, Reactive oxygen species; SNARE, Soluble NSF Attachment Protein receptor; SNpc, Substantia nigra pars compacta; TGF, Transforming growth factor; TH, Tyrosine hydroxylase; TLR, Toll-like receptors; TNF, Tumour necrosis factor; YFP, Yellow fluorescent protein.
Ahmed, Z., Asi, Y., Sailer, A., Lees, A. J., Houlden, H., Revesz, T., et al. (2012). The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol. App. Neurobiol. 38, 4–24. doi: 10.1111/j.1365-2990.2011.01234.x
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. doi: 10.1038/nn.4132 [Epub ahead of print].
Ben Haim, L., Carrillo-de Sauvage, M. A., Ceyzériat, K., and Escartin, C. (2015). Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 9:278. doi: 10.3389/fncel.2015.00278
Béraud, D., Twomey, M., Bloom, B., Mittereder, A., Ton, V., Neitzke, K., et al. (2011). α-Synuclein alters toll-like receptor expression. Front. Neurosci. 5:80. doi: 10.3389/fnins.2011.00080
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Block, M. L., Zecca, L., and Hong, J.-S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Brooks, D. J., Seppi, K., and Neuroimaging Working Group on, M. S. A. (2009). Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov. Disord. 24, 949–964. doi: 10.1002/mds.22413
Brudek, T., Winge, K., Agander, T. K., and Pakkenberg, B. (2013). Screening of Toll-like receptors expression in multiple system atrophy brains. Neurochem. Res. 38, 1252–1259. doi: 10.1007/s11064-013-1020-5
Burré, J., Sharma, M., and Südhof, T. C. (2014). α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U. S. A. 111, E4274–E4283. doi: 10.1073/pnas.1416598111
Burré, J., Sharma, M., and Südhof, T. C. (2015). Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 35, 5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015
Chandran, V., and Stoessl, A. J. (2014). Imaging in multiple system atrophy. Neurol. Clin. Neurosci. 2, 178–187. doi: 10.1111/ncn3.125
Chen, D., Wei, X., Zou, J., Wang, R., Liu, X., Xu, X., et al. (2015). Contra-directional expression of serum homocysteine and uric acid as important biomarkers of multiple system atrophy severity: a cross-sectional study. Front. Cell. Neurosci. 9:247. doi: 10.3389/fncel.2015.00247
Chung, W.-S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Colton, C. A., and Wilcock, D. M. (2010). Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191. doi: 10.2174/187152710791012053
Croisier, E., Moran, L., Dexter, D., Pearce, R., and Graeber, M. (2005). Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J. Neuroinflammation 2:14. doi: 10.1186/1742-2094-2-14
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Daturpalli, S., Waudby, C. A., Meehan, S., and Jackson, S. E. (2013). Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 425, 4614–4628. doi: 10.1016/j.jmb.2013.08.006
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Davis, C. H., Kim, K.-Y., Bushong, E. A., Mills, E. A., Boassa, D., Shih, T., et al. (2014). Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. U S A 111, 9633–9638. doi: 10.1073/pnas.1404651111
De Keyser, J., Mostert, J. P., and Koch, M. W. (2008). Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J. Neurol. Sci. 267, 3–16. doi: 10.1016/j.jns.2007.08.044
Ejlerskov, P., Hultberg, J. G., Wang, J., Carlsson, R., Ambjørn, M., Kuss, M., et al. (2015). Lack of neuronal IFN-β-IFNAR causes lewy body- and parkinson’s disease-like dementia. Cell 163, 324–339. doi: 10.1016/j.cell.2015.08.069
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Engelender, S. (2012). α-synuclein fate: proteasome or autophagy? Autophagy 8, 418–420. doi: 10.4161/auto.19085
Fellner, L., and Stefanova, N. (2013). The role of glia in alpha-synucleinopathies. Mol. Neurobiol. 47, 575–586. doi: 10.1007/s12035-012-8340-3
Fellner, L., Irschick, R., Schanda, K., Reindl, M., Klimaschewski, L., Poewe, W., et al. (2013). Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360. doi: 10.1002/glia.22437
Gai, W., Pountney, D. L., Power, J., Li, Q., Culvenor, J., McLean, C., et al. (2003). α-Synuclein fibrils constitute the central core of oligodendroglial inclusion filaments in multiple system atrophy. Exp. Neurol. 181, 68–78. doi: 10.1016/s0014-4886(03)00004-9
Gao, H.-M., Liu, B., and Hong, J.-S. (2003). Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 23, 6181–6187.
Gerhard, A., Banati, R., Goerres, G., Cagnin, A., Myers, R., Gunn, R., et al. (2003). [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 61, 686–689. doi: 10.1212/01.wnl.0000078192.95645.e6
Gilman, S., Wenning, G., Low, P. A., Brooks, D., Mathias, C., Trojanowski, J., et al. (2008). Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71, 670–676. doi: 10.1212/01.wnl.0000324625.00404.15
Goldman, J. S., Quinzii, C., Dunning-Broadbent, J., Waters, C., Mitsumoto, H., Brannagan, T. H., et al. (2014). Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol. 71, 771–774. doi: 10.1001/jamaneurol.2013.5762
Graeber, M. B., and Streit, W. J. (2010). Microglia: biology and pathology. Acta Neuropathol. 119, 89–105. doi: 10.1007/s00401-009-0622-0
Halliday, G. M., and Stevens, C. H. (2011). Glia: initiators and progressors of pathology in parkinson’s disease. Mov. Disord. 26, 6–17. doi: 10.1002/mds.23455
Herrera, A. J., Espinosa-Oliva, A. M., Carrillo-JiméNez, A., Oliva-Martín, M. J., García-Revilla, J., GarcíA-Quintanilla, A., et al. (2015). Relevance of chronic stress and the two faces of microglia in parkinson’s disease. Front. Cell. Neurosci. 9:312. doi: 10.3389/fncel.2015.00312
Hostenbach, S., Cambron, M., D’haeseleer, M., Kooijman, R., and De Keyser, J. (2013). Astrocyte loss and astrogliosis in neuroinflammatory disorders. Neurosci. Lett. 565, 39–41. doi: 10.1016/j.neulet.2013.10.012
Husemann, J., Loike, J. D., Anankov, R., Febbraio, M., and Silverstein, S. C. (2002). Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40, 195–205. doi: 10.1002/glia.10148
Ishizawa, K., Komori, T., Sasaki, S., Arai, N., Mizutani, T., and Hirose, T. (2004). Microglial activation parallels system degeneration in multiple system atrophy. J. Neuropathol. Exp. Neurol. 63, 43–52.
Jang, A., Lee, H. J., Suk, J. E., Jung, J. W., Kim, K. P., and Lee, S. J. (2010). Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x
Jensen, M., Hegelund, I., Poulsen, F., Owens, T., Zimmer, J., and Finsen, B. (1999). Microglial reactivity correlates to the density and the myelination of the anterogradely degenerating axons and terminals following perforant path denervation of the mouse fascia dentata. Neuroscience 93, 507–518. doi: 10.1016/s0306-4522(99)00139-6
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kiffin, R., Christian, C., Knecht, E., and Cuervo, A. M. (2004). Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 15, 4829–4840. doi: 10.1091/mbc.e04-06-0477
Konno, M., Hasegawa, T., Baba, T., Miura, E., Sugeno, N., Kikuchi, A., et al. (2012). Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol. Neurodegener. 7:38. doi: 10.1186/1750-1326-7-38
Krampera, M., Cosmi, L., Angeli, R., Pasini, A., Liotta, F., Andreini, A., et al. (2006a). Role for interferon-γ in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem cells 24, 386–398. doi: 10.1634/stemcells.2005-0008
Krampera, M., Pasini, A., Pizzolo, G., Cosmi, L., Romagnani, S., and Annunziato, F. (2006b). Regenerative and immunomodulatory potential of mesenchymal stem cells. Curr. Opin. Pharmacol. 6, 435–441. doi: 10.1016/j.coph.2006.02.008
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). AlaSOPro mutation in the gene encoding α-synuclein in parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Labandeira-Garcia, J. L., Rodriguez-Pallares, J., Villar-Cheda, B., Rodríguez-Perez, A. I., Garrido-Gil, P., and Guerra, M. J. (2011). Aging, angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis. 2, 257–274.
Lantos, P. L. (1998). The definition of multiple system atrophy: a review of recent developments. J. Neuropathol. Exp. Neurol. 57, 1099–1111. doi: 10.1097/00005072-199812000-00001
Lee, H.-J., Kim, C., and Lee, S.-J. (2010). Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxid. Med. Cell. Longev. 3, 283–287. doi: 10.4161/oxim.3.4.12809
Lee, P. H., Lee, J. E., Kim, H.-S., Song, S. K., Lee, H. S., Nam, H. S., et al. (2012). A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann. Neurol. 72, 32–40. doi: 10.1002/ana.23612
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honoré, A., Rozas, N., et al. (2013). G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Longo, D. L., Fanciulli, A., and Wenning, G. K. (2015). Multiple-system atrophy. New Engl. J. Med. 372, 249–263. doi: 10.1056/NEJMra1311488
Mandler, M., Valera, E., Rockenstein, E., Mante, M., Weninger, H., Patrick, C., et al. (2015). Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol. Neurodegen. 10, 1–15. doi: 10.1186/s13024-015-0008-9
Maphis, N., Xu, G., Kokiko-Cochran, O. N., Jiang, S., Cardona, A., Ransohoff, R. M., et al. (2015). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755. doi: 10.1093/brain/awv081
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118:777. doi: 10.1172/JCI32806
McKeith, I., Dickson, D., Lowe, J., Emre, M., O’Brien, J., Feldman, H., et al. (2005). Diagnosis and management of dementia with lewy bodies third report of the DLB consortium. Neurology 65, 1863–1872. doi: 10.1212/wnl.65.12.1992-a
Miller, D., Johnson, J., Solano, S., Hollingsworth, Z., Standaert, D., and Young, A. (2005). Absence of α-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural Transm. 112, 1613–1624. doi: 10.1007/s00702-005-0378-1
Multiple-System Atrophy Research Collaboration. (2013). Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 369:233. doi: 10.1056/NEJMoa1212115
Narayanan, V., and Scarlata, S. (2001). Membrane binding and self-association of α-synucleins. Biochemistry 40, 9927–9934. doi: 10.1021/bi002952n
Nimmerjahn, A. K., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dinamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nishie, M., Mori, F., Yoshimoto, M., Takahashi, H., and Wakabayashi, K. (2004). A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol. App. Neurobiol. 30, 546–554. doi: 10.1111/j.1365-2990.2004.00564.x
O’Callaghan, J. P., Sriram, K., and Miller, D. B. (2008). Defining "neuroinflammation". Ann. N. Y. Acad. Sci. 1139, 318–1130. doi: 10.1196/annals.1432.032
Ozawa, T., Paviour, D., Quinn, N. P., Josephs, K. A., Sangha, H., Kilford, L., et al. (2004). The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 127, 2657–2671. doi: 10.1093/brain/awh303
Park, H.-J., Bang, G., Lee, B. R., Kim, H. O., and Lee, P. H. (2011). Neuroprotective effect of human mesenchymal stem cells in an animal model of double toxin-induced multiple system atrophy parkinsonism. Cell Transplant. 20, 827–835. doi: 10.3727/096368910x540630
Parkhurst, C. N., and Gan, W.-B. (2010). Microglia dynamics and function in the CNS. Curr. Opin. Neurobiol. 20, 595–600. doi: 10.1016/j.conb.2010.07.002
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180.e1–2180.e5. doi: 10.1016/j.neurobiolaging.2014.03.024
Peelaerts, W., Bousset, L., Van der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Prinz, M., Priller, J., Sisodia, S. S., and Ransohoff, R. M. (2011). Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 14, 1227–1235. doi: 10.1038/nn.2923
Proukakis, C., Dudzik, C. G., Brier, T., MacKay, D. S., Cooper, J. M., Millhauser, G. L., et al. (2013). A novel α-synuclein missense mutation in parkinson disease. Neurology 80, 1062–1064. doi: 10.1212/WNL.0b013e31828727ba
Prusiner, S. B., Woerman, A. L., Mordes, D. A., Watts, J. C., Rampersaud, R., Berry, D. B., et al. (2015). Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U S A 112, E5308–E5317. doi: 10.1073/pnas.1514475112
Radford, R., Rcom-H’Cheo-Gauthier, A., Wong, M. B., Eaton, E. D., Quilty, M., Blizzard, C., et al. (2015). The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α-synuclein inclusions. Mol. Cell. Neurosci. 65, 68–81. doi: 10.1016/j.mcn.2015.02.015
Radford, R., Wong, M., and Pountney, D. L. (2014). Neurodegenerative aspects of multiple system atrophy. Handb. Neurotox. 2157–2180. doi: 10.1007/978-1-4614-5836-4_110
Reyes, J. F., Rey, N. L., Bousset, L., Melki, R., Brundin, P., and Angot, E. (2014). Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398. doi: 10.1002/glia.22611
Roberts, H. L., and Brown, D. R. (2015). Seeking a mechanism for the toxicity of oligomeric α-synuclein. Biomolecules 5, 282–305. doi: 10.3390/biom5020282
Sanchez-Guajardo, V., Tentillier, N., and Romero-Ramos, M. (2015). The relation between α-synuclein and microglia in parkinson’s disease: recent developments. Neuroscience 302, 47–58. doi: 10.1016/j.neuroscience.2015.02.008
Scheffel, J., Regen, T., Van Rossum, D., Seifert, S., Ribes, S., Nau, R., et al. (2012). Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia 60, 1930–1943. doi: 10.1002/glia.22409
Schocke, M., Seppi, K., Esterhammer, R., Kremser, C., Jaschke, W., Poewe, W., et al. (2002). Diffusion-weighted MRI differentiates the parkinson variant of multiple system atrophy from PD. Neurology 58, 575–580. doi: 10.1212/wnl.58.4.575
Schrag, A., Kingsley, D., Phatouros, C., Mathias, C., Lees, A., Daniel, S., et al. (1998). Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 65, 65–71. doi: 10.1136/jnnp.65.1.65
Shastri, A., Bonifati, D. M., and Kishore, U. (2013). Innate immunity and neuroinflammation. Mediators Inflamm. 2013:342931. doi: 10.1155/2013/342931
Shulman, J. M., De Jager, P. L., and Feany, M. B. (2011). Parkinson’s disease: genetics and pathogenesis. Annu. Rev. Pathol. 6, 193–222. doi: 10.1146/annurev-pathol-011110-130242
Singh, S., Mishra, A., and Shukla, S. (2015). Alcar exerts neuroprotective and pro-neurogenic effects by inhibition of glial activation and oxidative stress via activation of the wnt/β-catenin signaling in parkinsonian rats. Mol. Neurobiol. doi: 10.1007/s12035-015-9361-5 [Epub ahead of print].
Skaper, S., Facci, L., Barbierato, M., Zusso, M., Bruschetta, G., Impellizzeri, D., et al. (2015). N-Palmitoylethanolamine and neuroinflammation: a novel therapeutic strategy of resolution. Mol. Neurobiol. 52, 1034–1042. doi: 10.1007/s12035-015-9253-8
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Song, Y. J. C. P., Halliday, G. M. P., Holton, J. L. P. F., Lashley, T. P., O’Sullivan, S. S. M., McCann, H. B., et al. (2009). Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 68, 1073–1083. doi: 10.1097/nen.0b013e3181b66f1b
Spencer, B., Valera, E., Rockenstein, E., Trejo-Morales, M., Adame, A., and Masliah, E. (2015). A brain-targeted, modified neurosin (kallikrein-6) reduces α-synuclein accumulation in a mouse model of multiple system atrophy. Mol. Neurodegen. 10:48. doi: 10.1186/s13024-015-0043-6
Stefanova, N., Reindl, M., Neumann, M., Haass, C., Poewe, W., Kahle, P. J., et al. (2005). Oxidative stress in transgenic mice with oligodendroglial α-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am. J. Pathol. 166, 869–876. doi: 10.1016/s0002-9440(10)62307-3
Stefanova, N., Reindl, M., Neumann, M., Kahle, P. J., Poewe, W., and Wenning, G. K. (2007). Microglial activation mediates neurodegeneration related to oligodendroglial α-synucleinopathy: implications for multiple system atrophy. Mov. Disord. 22, 2196–2203. doi: 10.1002/mds.21671
Stemberger, S., Jamnig, A., Stefanova, N., Lepperdinger, G., Reindl, M., and Wenning, G. K. (2011). Mesenchymal stem cells in a transgenic mouse model of multiple system atrophy: immunomodulation and neuroprotection. PLoS One 6:e19808. doi: 10.1371/journal.pone.0019808
Takeuchi, H. (2013). Roles of glial cells in neuroinflammation and neurodegeneration. Clin. Exp. Neuroimmunol. 4, 2–16. doi: 10.1111/cen3.12059
Tang, Y., and Le, W. (2015). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. doi: 10.1007/s12035-014-9070-5 [Epub ahead of print].
Tanik, S. A., Schultheiss, C. E., Volpicelli-Daley, L. A., Brunden, K. R., and Lee, V. M. (2013). Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 288, 15194–15210. doi: 10.1074/jbc.M113.457408
Tenreiro, S., ReimÃo-Pinto, M. M., Antas, P., Rino, J., Wawrzycka, D., Macedo, D., et al. (2014). Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of parkinson’s disease. PLoS Genet. 10:e1004302. doi: 10.1371/journal.pgen.1004302
Thakur, P., and Nehru, B. (2015). Inhibition of neuroinflammation and mitochondrial dysfunctions by carbenoxolone in the rotenone model of parkinson’s disease. Mol. Neurobiol. 51, 209–219. doi: 10.1007/s12035-014-8769-7
Tien, L.-T., Kaizaki, A., Pang, Y., Cai, Z., Bhatt, A. J., and Fan, L.-W. (2013). Neonatal exposure to lipopolysaccharide enhances accumulation of α-synuclein aggregation and dopamine transporter protein expression in the substantia nigra in responses to rotenone challenge in later life. Toxicology 308, 96–103. doi: 10.1016/j.tox.2013.03.014
Togo, T., and Dickson, D. (2002). Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol. 104, 398–402.
Tong, J., Wong, H., Guttman, M., Ang, L. C., Forno, L. S., Shimadzu, M., et al. (2009). Brain α-synuclein accumulation in multiple system atrophy, parkinson’s disease and progressive supranuclear palsy: a comparative investigation. Brain 133, 172–188. doi: 10.1093/brain/awp282
Trojanowski, J., and Revesz, T. (2007). Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol. App. Neurobiol. 33, 615–620. doi: 10.1111/j.1365-2990.2007.00907.x
Ubhi, K., Low, P., and Masliah, E. (2011). Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 34, 581–590. doi: 10.1016/j.tins.2011.08.003
Valera, E., and Masliah, E. (2013). Immunotherapy for neurodegenerative diseases: focus on α-synucleinopathies. Pharmacol. Ther. 138, 311–322. doi: 10.1016/j.pharmthera.2013.01.013
Vijayakumaran, S., Wong, M., Antony, H., and Pountney, D. (2015). Direct and/or indirect roles for SUMO in modulating alpha-synuclein toxicity. Biomolecules 5, 1697–1716. doi: 10.3390/biom5031697
Vilariño-Güell, C., Soto-Ortolaza, A., Rajput, A., Mash, D., Papapetropoulos, S., Pahwa, R., et al. (2011). MAPT H1 haplotype is a risk factor for essential tremor and multiple system atrophy. Neurology 76, 670–672. doi: 10.1212/wnl.0b013e31820c30c1
Watanabe, H., Fukatsu, H., Katsuno, M., Sugiura, M., Hamada, K., Okada, Y., et al. (2004). Multiple regional (1)H-MR spectroscopy in multiple system atrophy: NAA/Cr reduction in pontine base as a valuable diagnostic marker. J. Neurol. Neurosurg. Psychiatry 75, 103–109.
Watanabe, H., Saito, Y., Terao, S., Ando, T., Kachi, T., Mukai, E., et al. (2002). Progression and prognosis in multiple system atrophy. Brain 125, 1070–1083. doi: 10.1093/brain/awf117
Welser-Alves, J. V., and Milner, R. (2013). Microglia are the major source of TNF-α and TGF-β1 in postnatal glial cultures; regulation by cytokines, lipopolysaccharide and vitronectin. Neurochem. Int. 63, 47–53. doi: 10.1016/j.neuint.2013.04.007
Wenning, G. K., Geser, F., Krismer, F., Seppi, K., Duerr, S., Boesch, S., et al. (2013). The natural history of multiple system atrophy: a prospective european cohort study. Lancet Neurol. 12, 264–274. doi: 10.1016/S1474-4422(12)70327-7
Wenning, G. K., Stefanova, N., Jellinger, K. A., Poewe, W., and Schlossmacher, M. G. (2008). Multiple system atrophy: a primary oligodendrogliopathy. Ann. Neurol. 64, 239–246. doi: 10.1002/ana.21465
Winner, B., Jappelli, R., Maji, S. K., Desplats, P. A., Boyer, L., Aigner, S., et al. (2011). In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U S A 108, 4194–4199. doi: 10.1073/pnas.1100976108
Woerman, A. L., Stöhr, J., Aoyagi, A., Rampersaud, R., Krejciova, Z., Watts, J. C., et al. (2015). Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. U S A 112, E4949–E4958. doi: 10.1073/pnas.1513426112
Wong, M., Goodwin, J., Norazit, A., Meedeniya, A. B., Richter-Landsberg, C., Gai, W., et al. (2013). SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox. Res. 23, 1–21. doi: 10.1007/s12640-012-9358-z
Xilouri, M., Brekk, O. R., Landeck, N., Pitychoutis, P. M., Papasilekas, T., Papadopoulou-Daifoti, Z., et al. (2013). Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 136, 2130–2146. doi: 10.1093/brain/awt131
Xilouri, M., Vogiatzi, T., Vekrellis, K., Park, D., and Stefanis, L. (2009). Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 4:e5515. doi: 10.1371/journal.pone.0005515
Zarranz, J. J., Alegre, J., Gómez-esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of α-synuclein causes parkinson and lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zhang, W., Yan, Z. F., Gao, J. H., Sun, L., Huang, X. Y., Liu, Z., et al. (2014). Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 49, 1153–1165. doi: 10.1007/s12035-013-8586-4
Keywords: multiple system atrophy, α-synuclein, neuroinflammation, astrocytes, microglia
Citation: Vieira BDM, Radford RA, Chung RS, Guillemin GJ and Pountney DL (2015) Neuroinflammation in Multiple System Atrophy: Response to and Cause of α-Synuclein Aggregation. Front. Cell. Neurosci. 9:437. doi: 10.3389/fncel.2015.00437
Received: 11 August 2015; Accepted: 19 October 2015;
Published: 12 November 2015.
Edited by:
Alessandro Tozzi, University of Perugia, ItalyReviewed by:
Björn Spittau, Albert Ludwigs University of Freiburg, GermanyCopyright © 2015 Vieira, Radford, Chung, Guillemin and Pountney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dean L. Pountney, ZC5wb3VudG5leUBncmlmZml0aC5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.