 Agnese De Mario1†
Agnese De Mario1† Angela Castellani1†
Angela Castellani1† Caterina Peggion1
Caterina Peggion1 Maria Lina Massimino2
Maria Lina Massimino2 Dmitry Lim3
Dmitry Lim3 Andrew F. Hill4
Andrew F. Hill4 M. Catia Sorgato1,2*
M. Catia Sorgato1,2* Alessandro Bertoli1*
Alessandro Bertoli1*- 1Department of Biomedical Science, University of Padova, Padova, Italy
- 2CNR Neuroscience Institute, University of Padova, Padova, Italy
- 3Department of Pharmaceutical Science, University of Piemonte Orientale, Novara, Italy
- 4Department of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC, Australia
The prion protein (PrPC) is a cell surface glycoprotein mainly expressed in neurons, whose misfolded isoforms generate the prion responsible for incurable neurodegenerative disorders. Whereas PrPC involvement in prion propagation is well established, PrPC physiological function is still enigmatic despite suggestions that it could act in cell signal transduction by modulating phosphorylation cascades and Ca2+ homeostasis. Because PrPC binds neurotoxic protein aggregates with high-affinity, it has also been proposed that PrPC acts as receptor for amyloid-β (Aβ) oligomers associated with Alzheimer’s disease (AD), and that PrPC-Aβ binding mediates AD-related synaptic dysfunctions following activation of the tyrosine kinase Fyn. Here, use of gene-encoded Ca2+ probes targeting different cell domains in primary cerebellar granule neurons (CGN) expressing, or not, PrPC, allowed us to investigate whether PrPC regulates store-operated Ca2+ entry (SOCE) and the implication of Fyn in this control. Our findings show that PrPC attenuates SOCE, and Ca2+ accumulation in the cytosol and mitochondria, by constitutively restraining Fyn activation and tyrosine phosphorylation of STIM1, a key molecular component of SOCE. This data establishes the existence of a PrPC-Fyn-SOCE triad in neurons. We also demonstrate that treating cerebellar granule and cortical neurons with soluble Aβ(1–42) oligomers abrogates the control of PrPC over Fyn and SOCE, suggesting a PrPC-dependent mechanizm for Aβ-induced neuronal Ca2+ dyshomeostasis.
Introduction
The prion protein (PrPC) is an ubiquitous cell surface protein mostly expressed in the central nervous system, particularly at synapses (Um et al., 2012). Although it is now established that a conformational conversion of PrPC generates the prion, the infectious particle causing fatal prion diseases in humans and animals (Prusiner, 1998), the physiological role played by PrPC is still undefined. However, most of the proposed functions emphasize an involvement of PrPC in surface receptor complexes transducing signals beneficial to the cell (Aguzzi et al., 2008; Linden et al., 2008). Accordingly, electrophysiological approaches and dye-based Ca2+ measurements in PrP-knockout (PrP-KO) or prion-infected cells have implicated PrPC in the regulation of Ca2+ signaling (reviewed in Sorgato and Bertoli, 2009), which controls a wide range of physiological processes but also triggers cell death events if the fine Ca2+ tuning is compromised. Convincing evidence of the PrPC-Ca2+ liaison comes from the physical interaction of PrPC with a subunit of the N-methyl-D-aspartate (NMDA)-sensitive glutamatergic receptors (NMDA-R), which reduces Ca2+ entry into hippocampal neurons and thus protects from glutamate excitotoxicity (Khosravani et al., 2008). PrPC was also found to modulate Ca2+ influx in primary cerebellar granule neurons (CGN) after inducing store-operated Ca2+ entry (SOCE; Lazzari et al., 2011). SOCE occurs in response to depletion of Ca2+ from endoplasmic reticulum (ER), which causes the redistribution of ER luminal Ca2+ sensors (stromal interaction molecules, STIM1 and 2) into punctate clusters that recruit and activate the plasma membrane (PM) pore-forming proteins (Orai1-3) of store-operated channels (SOC; Soboloff et al., 2012). SOC mediate exclusively Ca2+ currents, which primarily serve to refill intracellular Ca2+ reservoirs but also to regulate different cell functions, including programmed cell death (Dubois et al., 2014), and gene expression (Lalonde et al., 2014) and excitation in neurons (Hartmann et al., 2014). It is thus reasonable to speculate that Ca2+ serves to mediate the different cell functions ascribed to PrPC (Peggion et al., 2011).
It has also been claimed that the alleged receptor function of PrPC relates to the modulation of phosphorylation cascades, in particular that governed by Fyn (Linden et al., 2008; Sorgato et al., 2009), a member of the Src Tyr-kinase family (SFK) highly expressed in neurons (Um and Strittmatter, 2013). Interestingly, because Tyr-phosphorylation regulates fundamental components of SOCE (Lopez et al., 2012), and the pharmacological inhibition, or deletion, of Tyr-kinases reduces SOCE-induced Ca2+ transients (Lee et al., 2006; Chung et al., 2007; Zuo et al., 2011) in various cell lines, one can hypothesize a tripartite connection between PrPC, Fyn and SOCE.
Recently, PrPC was found to act as a cell surface binding partner for β-enriched protein aggregates, including amyloid-β (Aβ) oligomers (Laurén et al., 2009) related to Alzheimer’s disease (AD; Shankar et al., 2008), prions and other toxic oligomeric protein species (Resenberger et al., 2011). In this context, the PrPC-Fyn link has been proposed to mediate the effects of such neurotoxic entities. In particular, the PrPC-dependent activation of Fyn was identified as central to couple PrPC-Aβ complexes to NMDA-R redistribution, Ca2+ signaling alterations, tau hyper-phosphorylation, spine loss and death in neurons, and AD pathology in mice (Larson et al., 2012; Um et al., 2012).
To clarify PrPC patho-physiology, in this study we sought to determine the capacity of PrPC to govern local Ca2+ homeostasis in primary neuronal cultures after SOCE. We found that PrPC regulates SOCE and SOCE-related Ca2+ movements in several cell domains by controling Fyn activation and the Tyr-phosphorylation of STIM1. We also observed that the control of PrPC over Fyn and SOCE is subverted by soluble Aβ oligomers, suggesting that disruption of Ca2+ signaling mediates the PrPC-dependent effects of Aβ oligomers.
Materials and Methods
Animals
We used PrP-KO (line F10) mice and, as control, transgenic Tg46 (PrP-Tg) mice in which the expression of PrPC at normal levels was rescued over the PrP-KO genotype (Mallucci et al., 2002; both strains kindly provided by the M.R.C. Prion Unit, London, UK). All aspects of animal care and experimentation were performed in compliance with European and Italian (D.L. 116/92) laws concerning the care and use of laboratory animals. The authors’ Institution has been accredited for the use of experimental mice by the Italian Ministry of Health, and by the Ethical Committee of the University of Padova.
Primary Neuronal Cultures
CGN primary cultures were prepared (from 7 day-old mice killed by decapitation after anesthesia with methoxyflurane), cultured and transduced with lentiviral particles as previously described in Lazzari et al. (2011). Primary cultures of cortical neurons were obtained from cortices dissected from E18 mouse embryos. After carefully removing meningeal layers and blood vessels, the cortical tissue was gently minced and dissociated mechanically in B27 (2%)-added Hibernate E medium (Gibco). After centrifugation (180×g, 5 min), cells were suspended in Neurobasal medium (Gibco) supplemented with B27 (2%), horse serum (2%), glutamate (25 μM), L-glutamine (0.5 mM), and gentamycin [0.1 mg/ml (Gibco)], seeded on poly-L-lysine (100 μg/ml)-coated 13 mm-diameter glass coverslips at a density of 450,000 cell/cm2, and maintained at 37°C in a humidified incubator with 5% CO2. After 48 h, cells were infected with lentiviral particles and grown as previously described in Lazzari et al. (2011), except that cytosine-d-arabinofuranoside (10 μM) was added 72 h, and cells were used for experiments 144 h, after plating.
Cell Cultures
HeLa and HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium/High-Glucose (Euroclone), supplemented with 10% fetal bovine serum (Euroclone), 100 U/ml penicillin and 100 μg/ml streptomycin (Euroclone), and maintained at 37°C in a humidified incubator with 5% CO2. Twenty four hours before infection, HeLa cells were seeded onto 13 mm-diameter glass coverslips and allowed to grow to 70–80% confluence. For the production of lentiviral particles, HEK-293T cells were seeded onto 100 mm-diameter Petri dishes at ~40% confluence and transfected 24 h after plating, as described below.
Construction of Lentiviral Vectors for Aequorins and Cell Transduction
To follow [Ca2+] fluctuations in specific neuronal compartments, we exploited a lentiviral expression system to transduce cells with chimeric constructs encoding the Ca2+-probe aequorin (AEQ) tagged with the influenza virus hemagglutinin (HA) epitope, and linked to sequences addressing the protein to the cytosolic domains proximal to the PM (AEQpm; Marsault et al., 1997; Lazzari et al., 2011), the bulk cytosol (AEQcyt; Brini et al., 1995), the ER lumen (AEQer; Montero et al., 1995; Lazzari et al., 2011) and the mitochondrial matrix (AEQmit; Rizzuto et al., 1992). Lentiviral vectors for AEQpm, AEQer and AEQmit were generated as described in Lim et al. (2008) and Lazzari et al. (2011), using an AEQ mutant with reduced Ca2+ affinity allowing [Ca2+] measurements up to hundreds of μM (Kendall et al., 1992). Conversely, a chimeric construct of wild-type (WT) AEQ fused to the monomeric red fluorescent protein (mRFP) was used to detect [Ca2+]cyt. To generate the AEQcyt lentiviral vector, two PCR reactions were performed. In the first one, the mRFP sequence was amplified without the stop codon using the pCDNA3-mRFP plasmid (Clontech) as template, and the following primers: XbaI-mRFP (forward: CGTCTAGAATGGCCTCCTCCGAGGAC) and mRFP-BglII (reverse: GAGGCGCCGGTGGAGTG-GAGATCTCG). In the second PCR, the HA-AEQ cassette was amplified using the pCDNA1-cytAEQ plasmid (Brini et al., 1995) as template, and the following primers: BglII-AEQ (forward: CGAGATCTCGAGCTCAAGCTTTATGA) and AEQ-SalI (reverse: GGTATCGATAAGCTTGATGTCGACGC). PCR products were digested with XbaI and BglII (for mRFP), or with BglII and SalI (for HA-AEQ), and the resulting fragments were assembled into the XbaI- and SalI-digested backbone of the lentiviral vector pRRLsin.PPTs.hCMV.GFP.pre in a three-step ligation reaction, yielding pLV-AEQcyt. Lentiviral particles were obtained as described previously (Lazzari et al., 2011). Briefly, HEK-293T cells were co-transfected with one of the transgene plasmids (pLV-AEQcyt, pLV-AEQmit, pLV-AEQpm or pLV-AEQer) together with the three packaging plasmids pMDLg/pRRE, pMD2.VSVG, pRSV-Rev (Naldini et al., 1996; Lazzari et al., 2011), using the calcium-phosphate method. After 10 h, the transfection medium was replaced with fresh culture medium, and after 72 h the culture medium was collected, and lentiviral particles were harvested by ultracentrifugation (50,000×g, 2 h), resuspended in phosphate-buffered saline (PBS; 140 mM NaCl, 2 mM KCl, 1.5 mM KH2PO4, 8 mM Na2HPO4, pH 7.4) and stored at −80°C. Viral titer was assessed by infecting HeLa cells by serial viral dilutions. Minimal dilutions adequate for obtaining 100% of infected cells were used to infect primary neurons and HeLa cells. All procedures for the production and use of lentiviral particles were performed in a biosafety level-2 environment. The expression and correct cellular localization of the Ca2+ probes were ascertained by immunocytochemical procedures, as described below and reported in Supplementary Figure 2.
Measurements of Local Ca2+ Fluxes
The [Ca2+] range that can be reliably measured with the AEQ probes is defined by the type of AEQ (WT or mutated) and of the prosthetic group coelenterazine (Coe; normal (Coe-wt) or modified (Coe-n), Ottolini et al., 2014). The AEQ and Coe variants used in this work are listed in Supplementary Table 1.
For measuring [Ca2+]pm, [Ca2+]cyt and [Ca2+]mit transients, cells were firstly incubated (1 h, 37°C, 5% CO2) with Coe-wt (5 μM, Santa Cruz Biotechnology, cat. n. sc-205904) in a modified (Ca2+-free) Krebs-Ringer buffer (KRB, 125 mM NaCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, 5 mM KCl, 20 mM HEPES, pH 7.4) containing EGTA (100 μM) for simultaneously reconstituting functional AEQ and depleting intracellular Ca2+ stores. For the reconstitution of AEQer, cells were firstly incubated (10 min, 37°C) in the above Ca2+-free KRB containing EGTA (1 mM), and then incubated (50 min, 4°C) in Ca2+-free KRB supplemented with EGTA (500 μM), ionomycin (5 μM, Sigma) and Coe-n (5 μM, AnaSpec, cat. n. 82260).
Coverslips were then placed onto a recording chamber—equipped with a perfusion system—located into the luminometer. For the measurements of [Ca2+]pm, [Ca2+]cyt and [Ca2+]mit, after a 1 min-perfusion step with EGTA (100 μM)-containing KRB, SOCE was elicited by perfusing cells with Ca2+ (1 mM)-containing KRB, as previously described in Lazzari et al. (2011). For measuring [Ca2+]er, cells were successively perfused with Ca2+-free KRB containing: EGTA (500 μM, 2 min); EGTA (1 mM) and bovine serum albumin [BSA 2% (w/v), 3 min]; EGTA (500 μM, 2 min); EGTA (100 μM, 1 min). CGN were finally stimulated for SOCE by perfusion with Ca2+ (1 mM)-containing KRB.
If needed, the inhibitors of voltage-gated Ca2+ channels (VGCC) [nifedipine (10 μM) and NiCl2 (50 μM or 1 mM); both from Sigma] were added to the perfusing buffer before (1 min) and during VGCC activation. Instead, the SFK inhibitors [PP2 (10 μM, Tocris) and saracatinib (5 μM, Santa Cruz Biotechnology)], and the negative control of PP2, PP3 (10 μM, Santa Cruz Biotechnology), were added during the reconstitution step, and kept in the perfusion buffer throughout the entire experiment. With the exception of NiCl2, all the above molecules were dissolved in dimethylsulfoxide [DMSO 0.1% (v/v)].
AEQ light emission was collected by means of an in-house built luminometer, equipped with a low-noise photo-multiplier coupled by an A/D board to a computer-assisted acquisition system, with a 1 Hz sampling rate (Ottolini et al., 2014). At the end of the recording, cells were permeabilized and exposed to saturating Ca2+ concentration by perfusion with KRB containing digitonin (50 μM, Sigma) and CaCl2 (10 mM). This allowed the calibration of the recorded light signal with respect to the total AEQ content, and its conversion into Ca2+ concentration, using the algorithm and the custom-made software previously described in Brini et al. (1995) and Montero et al. (1995).
Immunocytochemistry
For immunocytochemical analyses, cells were washed in ice-cold PBS and fixed (20 min, RT) in 2% (w/v) paraformaldehyde (Sigma) in PBS. After extensive washing in PBS, cells were permeabilized with Triton X-100 [0.02% (w/v) in PBS, 1 h, RT], and then incubated (overnight, 4°C) with a mouse monoclonal antibody to the HA epitope [HA.11/clone16B12, Covance, cat. n. MMS-101P (1:250 in PBS)].
Cells were then washed in PBS, and incubated (1 h, 37°C) with AlexaFluor 488-conjugated anti-mouse antibody (1:500, Molecular Probes). Finally, coverslips were washed in PBS, mounted in Mowiol 40–88 [Sigma, 8% (w/v) in glycerol:PBS (1:3)] and observed with a confocal microscope system (Leica TCS SP5), which also allowed the acquisition and analysis of digital images.
Preparation and Characterization of Aβ Oligomers
Chemically synthesized human Aβ(1–42) peptides (Keck Laboratories) were dissolved (1 mg/ml), and incubated (1 h, RT), in 1,1,1,3,3,3-hexafluoro-2-propanol. The suspension was divided into solvent-free (by evaporation) aliquots (50 μg) and stored (−80°C). Just before use, peptides were dissolved in NaOH (20 mM, 50 μl), sonicated (15 min on ice), diluted with PBS (Vf, 250 μl), and centrifuged (14,000×g, 5 min) to remove insoluble aggregates. After determining their concentration spectrophotometrically (λ, 214 nm), Aβ(1–42) peptides were aged (1 h, 37°C) to form oligomers, and then administered to CGN or cortical neurons (1 h, 37°C) during the AEQ reconstitution step at a final concentration of 5 μM of monomer equivalents. Routinely, Aβ(1–42) oligomerization was tested by Western blot (WB). For SDS-PAGE, Aβ(1–42) samples (~300 ng), subjected, or not, to the aging protocol, were diluted in a buffer containing SDS [3% (w/v)], β-mercaptoethanol [1.5% (v/v)], glycerol [7.5% (w/v)], Coomassie blue G-250 [0.0125% (Serva)], Tris/HCl (37.5 mM, pH 7.0), and run in a urea (6M)-containing Tris-Tricine gel [16% (w/v) acrylamide-N,N′-methylenebisacrylamide (29:1)] (Schägger, 2006). Proteins were then electro-blotted onto polyvinylidene fluoride (PVDF) membranes [0.22 μm pore size (Biorad)], which were processed as described below, except for the blocking solution that contained non-fat dry milk [5% (w/v)] instead of BSA, and the primary mouse monoclonal antibody to Aβ(1–42) (Covance, clone 6E10, cat. n. SIG 39320-200).
Lysis of CGN and WB Analysis
To analyze the (activating auto-) Tyr-phosphorylation of SFK members (p-SFK) and of total neuronal proteins, CGN—treated, or not, with Aβ oligomers (see above)—were incubated (1 h, 37°C, 5% CO2) in KRB [supplemented with CaCl2 (1 mM), or EGTA (100 μM), to ensure protein analysis under the same conditions used to measure Ca2+ fluxes] in the absence, or in the presence, of the SFK inhibitors (PP2 and saracatinib), or PP3 (see above). CGN were lysed using an ice-cold buffer containing glycerol [10% (w/v)], SDS [2% (w/v)], Tris/HCl (62.5 mM, pH 6.8), urea (1.8 M), Na3VO4 (5 mM), and cocktails of protease and phosphatase inhibitors (Roche). Lysates, whose protein concentration was determined by a Lowry assay kit (Sigma, cat. n. TP0300), were then adjusted to an equal protein concentration using reducing (dithiothreitol, 50 mM) Laemmli sample buffer (Laemmli, 1970). After boiling (5 min), lysates were subjected to SDS–PAGE [10% (w/v) acrylamide-N,N′-methylenebisacrylamide (37.5:1)] and electro-blotted onto PVDF membranes [0.45 μm pore size (Millipore)], which were Coomassie-stained to verify equal loading and transfer (and for subsequent densitometric analyses, see below). PVDF membranes were then incubated (1 h, RT) with a blocking solution made of Tris-buffered saline added with Tween-20 (TBS-T) [0.1% (w/v)] and BSA [3% (w/v)], followed by addition of the appropriate primary antibody (4°C, over-night). In particular, to detect p-SFK, a rabbit polyclonal antibody (Cell Signaling Technology, cat. n. 2101) to p-Tyr416 was used. Instead, a mouse monoclonal antibody (clone P-Y20, Millipore, cat. n. 05-947) was used to detect the p-Tyr of total neuronal proteins. Simultaneously with these tests, the total amount of Fyn (in an equal CGN quantity) was determined using a specific rabbit polyclonal antibody (Cell Signaling Technology, cat. n. 4023). After three 10 min-washes (with TBS-T), membranes were incubated (1 h, RT) with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG secondary antibody (Santa Cruz Biotechnology, cat. n. sc-2004 and sc-2005, respectively). Immunoreactive bands were visualized and digitalized by means of a digital Kodak Image Station, using an enhanced chemiluminescence reagent kit (Millipore). For densitometric analyses, band intensities were normalized to the optical density of the corresponding lanes stained with Coomassie blue. To quantify the level of active Fyn, the normalized band intensity of p-SFK was divided by the normalized band intensity of Fyn. To electrophoretically separate different SFK members, equal amounts (10 μg) of CGN proteins were separated onto glycerol (35%)-supplemented SDS-PAGE gels [8% (w/v) acrylamide-N,N′-methylenebisacrylamide (37.5:1)]. After transferring proteins onto PVDF membranes [0.45 μm pore size (Millipore)], and the vertical cutting of membranes into strips, each strip was immunoprobed with a monoclonal antibody to either Src (Santa Cruz Biotechnology, cat. n. sc-5266), or Lck (Santa Cruz Biotechnology, cat. n. sc-433), while a polyclonal antibody was employed to label Lyn (Millipore, cat. n. 06-207), or Fyn, or p-SFK (indicated above). After incubation with the appropriate secondary antibody, PVDF membranes were accurately reconstructed before visualizing the immunoreactive signals. To detect PrPC, the mouse monoclonal antibody 8H4 was used (kindly provided by Prof. M.S. Sy, Case Western University, Cleveland, OH).
Immunoprecipitation of STIM1
CGN—incubated (1 h, 37°C, 5% CO2) in KRB [supplemented with EGTA (100 μM) in order to deplete Ca2+ stores]— were lysed (30 min on ice) at 1 × 107 cell/ml in a buffer containing Tris-HCl (20 mM, pH 7.4), NaCl (75 mM), EGTA (5 mM), Triton X-100 [1% (w/v)], sodium deoxycholate [1% (w/v)], Na3VO4, (5 mM) and cocktails of protease and phosphatase inhibitors. Cell lysates were sonicated and centrifuged (13,000×g, 15 min, 4°C) to remove insoluble materials, and the protein content of the collected supernatant was determined by a bicinchoninic acid-based assay kit (Thermo Scientific, cat. n. 23227). 300 μg of the supernatant proteins were then incubated (overnight, 4°C) with 1.5 μg of a monoclonal antibody to STIM1 (BD Bioscience, cat. n. 610954), followed by incubation with protein A-agarose (15 μl, SantaCruz Biotechnology, cat. n. sc-2001; 4 h, 4°C). The immunoadsorbant was centrifuged (4,000×g, 2 min, 4°C), washed twice with NaCl (100 mM), EDTA (2 mM), Na3VO4 (1 mM), and Tris-HCl (50 mM, pH 7.4), resuspended in reducing Laemmli buffer, boiled (3 min) and loaded onto Mini-protean TGX precast gels (4–15%, Biorad). Proteins were electro-blotted onto PVDF membranes [0.45 μm pore size (Millipore)], and immunodetected using, first, the antibody to p-Tyr (clone P-Y20, see above), and then, after stripping membranes with a commercial kit (Thermo Scientific, cat. n. 21059), a rabbit polyclonal antibody to STIM1 (Cell Signaling Technology, cat. n. 4916). Immunoreactive bands were visualized as described above, and for densitometric analyses the p-Tyr band intensity was normalized to that of STIM1.
Statistical Analysis
Values are reported as mean ± SEM. Data analysis was performed as described in Lazzari et al. (2011). Statistics were based on two-sample Student’s t-test, with a p-value < 0.05 being considered statistically significant.
Results
PrPC Attenuates SOCE and SOCE-Induced Mitochondrial Ca2+ Uptake
Previously, comparison of primary CGN cultures obtained from WT and PrP-KO mice has demonstrated that the presence of PrPC decreases SOCE-induced Ca2+ elevation in the cytosolic domains proximal to the PM (Lazzari et al., 2011). Here, we have extended the study to ascertain whether PrPC also impinges on Ca2+ fluxes in other domains of primary CGN derived from PrP-KO, and as control, isogenic PrP-Tg mice (Supplementary Figure 1). Preliminarily, the correct sub-cellular localization of the differently targeted AEQ probes was confirmed by immunocytochemical approaches (Supplementary Figure 2).
Figure 1 reports Ca2+ transients in PM microdomains (A), the bulk cytosol (B) and the mitochondrial matrix (C) following SOCE. While these data confirm that PrP-KO CGN are exposed to higher Ca2+ transients in PM microdomains than PrP-expressing neurons (Figure 1A), they also highlight that PrP-KO neurons display a significantly increased Ca2+ rise in the cytosol (Figure 1B) and in the mitochondrial matrix (Figure 1C). We argue that the higher Ca2+ levels observed in PrP-KO neurons are likely to be accounted for by both the more pronounced Ca2+ entry from the extracellular space (Figure 1A), and by the lower Ca2+-buffering capacity displayed by the ER (Figure 1D).
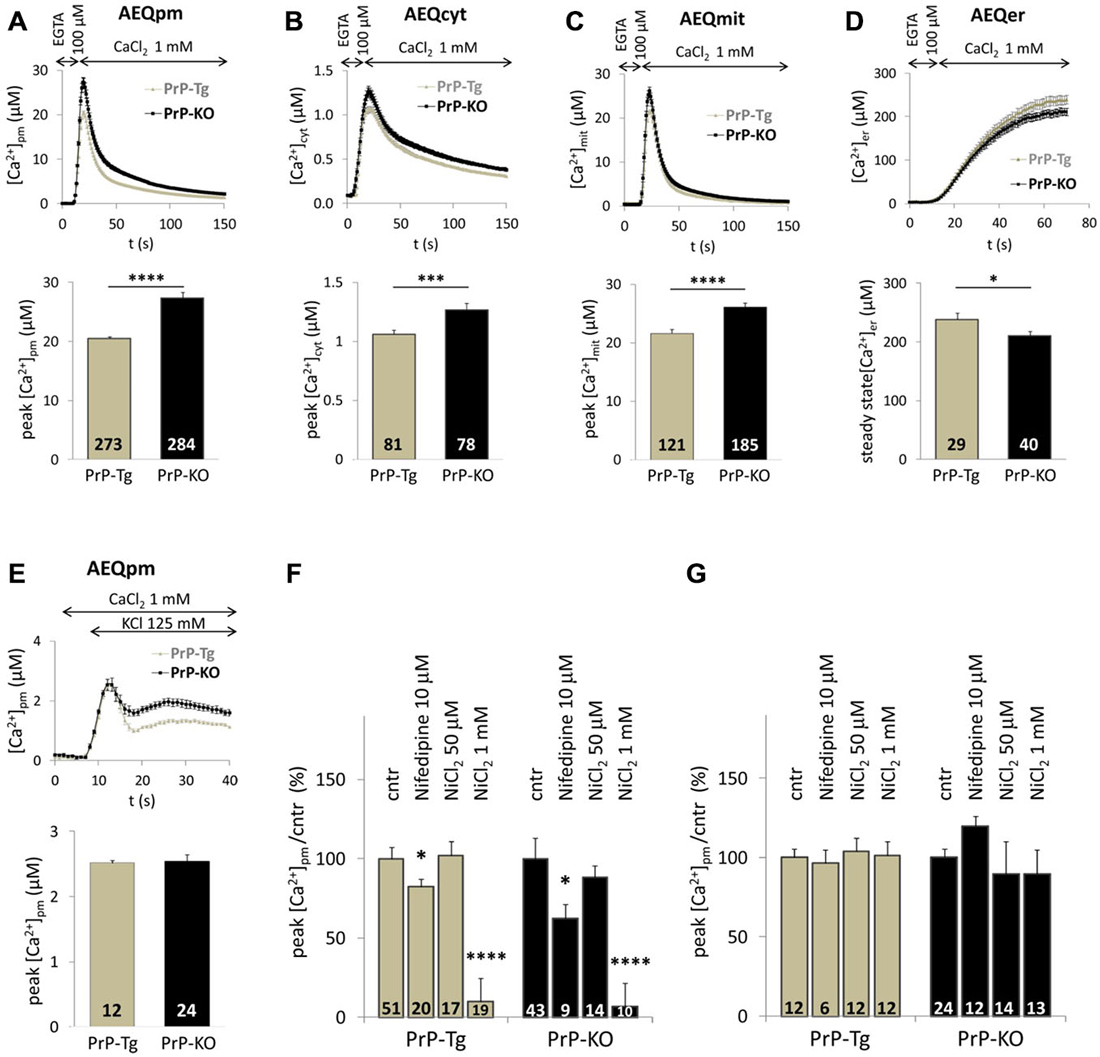
Figure 1. PrPC regulates Ca2+ movements in different cell domains of cerebellar granule neurons (CGN) after stimulation of SOCE. (A–C) PrP-KO CGN (black) have higher Ca2+ fluxes than control PrP-Tg CGN (gray) in the cytosolic domains proximal to the plasma membrane (PM) (A), the bulk cytosol (B) and the mitochondrial matrix (C), as measured by the appropriate AEQ probe. Upper panels report the kinetics of local [Ca2+] transients, while the bar diagrams of the corresponding [Ca2+] peaks are shown in the lower panels. Here and after, the number of replicates (n) is indicated inside each bar diagram. (D) The steady-state [Ca2+] level in the endoplasmic reticulum (ER) lumen of PrP-KO CGN is significantly lower than in PrP-Tg CGN. In (A–D), SOCE was activated by treating neurons with the Ca2+ chelator EGTA to deplete Ca2+ reservoirs. (E) Activation of VGCC by the 125 mM K+-depolarizing solution provokes similar [Ca2+]pm transients and peaks in both CGN genotypes. (F,G) VGCC inhibitors (nifedipine and NiCl2) lower VGCC-induced [Ca2+]pm peaks (F), but leave unchanged those following SOCE (G). *p < 0.05, ***p < 0.001, ****p < 10−5, Student’s t-test.
VGCC are highly abundant in neurons. To exclude their activation by local membrane depolarization following SOCE, we stimulated VGCC alone by perfusing neurons with either 25 mM or 125 mM K+ to mimic mild and strong depolarization, respectively. We found no detectable PM Ca2+ transients in CGN—expressing or not PrPC—under mild depolarizing conditions (data not shown). Conversely, the 125 mM K+ solution induced similar (<3 μM) PM Ca2+ peaks in both CGN types (Figure 1E) that, however, are much smaller than those observed upon SOCE. A second control envisaged treating neurons with inhibitors specific to L-type VGCC [nifedipine (10 μM)], or targeting all VGCC [NiCl2 (50 μM or 1 mM)]. We found that both inhibitors reduce Ca2+ entry stimulated by 125 mM K+ irrespective of the CGN genotype (~20% inhibition by nifedipine; ~95% inhibition by 1 mM NiCl2, Figure 1F). Instead, no significant diminution of PM Ca2+ peaks was observed in PrP-Tg and PrP-KO CGN when each inhibitor was added under the conditions employed to stimulate SOCE (Figure 1G). These results demonstrate that the contribution of VGCC to the observed Ca2+ transients is, if any, minimal.
PrPC Controls SOCE through Fyn
Because of multiple reports linking PrPC to Fyn (Mouillet-Richard et al., 2000; Santuccione et al., 2005; Pantera et al., 2009), we investigated whether Fyn signaling pathways critically acted in the observed regulation of PrPC over SOCE. To inspect its activating auto-phosphorylation, we monitored the Tyr phosphorylation of SFK (p-SFK), demonstrating that p-SFK was at higher levels in PrP-KO CGN than in PrP-Tg neurons both with Ca2+-depleted stores (to trigger SOCE; Figure 2A), and under basal (i.e., with Ca2+-filled stores) conditions (Supplementary Figure 3). Albeit Fyn is particularly abundant in neurons (Um and Strittmatter, 2013), other SFK members are present (Lck, Lyn, Src, and Yes; Salter and Kalia, 2004). However, because among all tested SFK members Fyn is the only one co-migrating with the immunolabelled p-SFK band (Supplementary Figure 4), we can conclude that the observed p-SFK (Figure 2A and Supplementary Figure 3) corresponds to p-Fyn, and consequently, that PrPC downregulates Fyn in CGN.
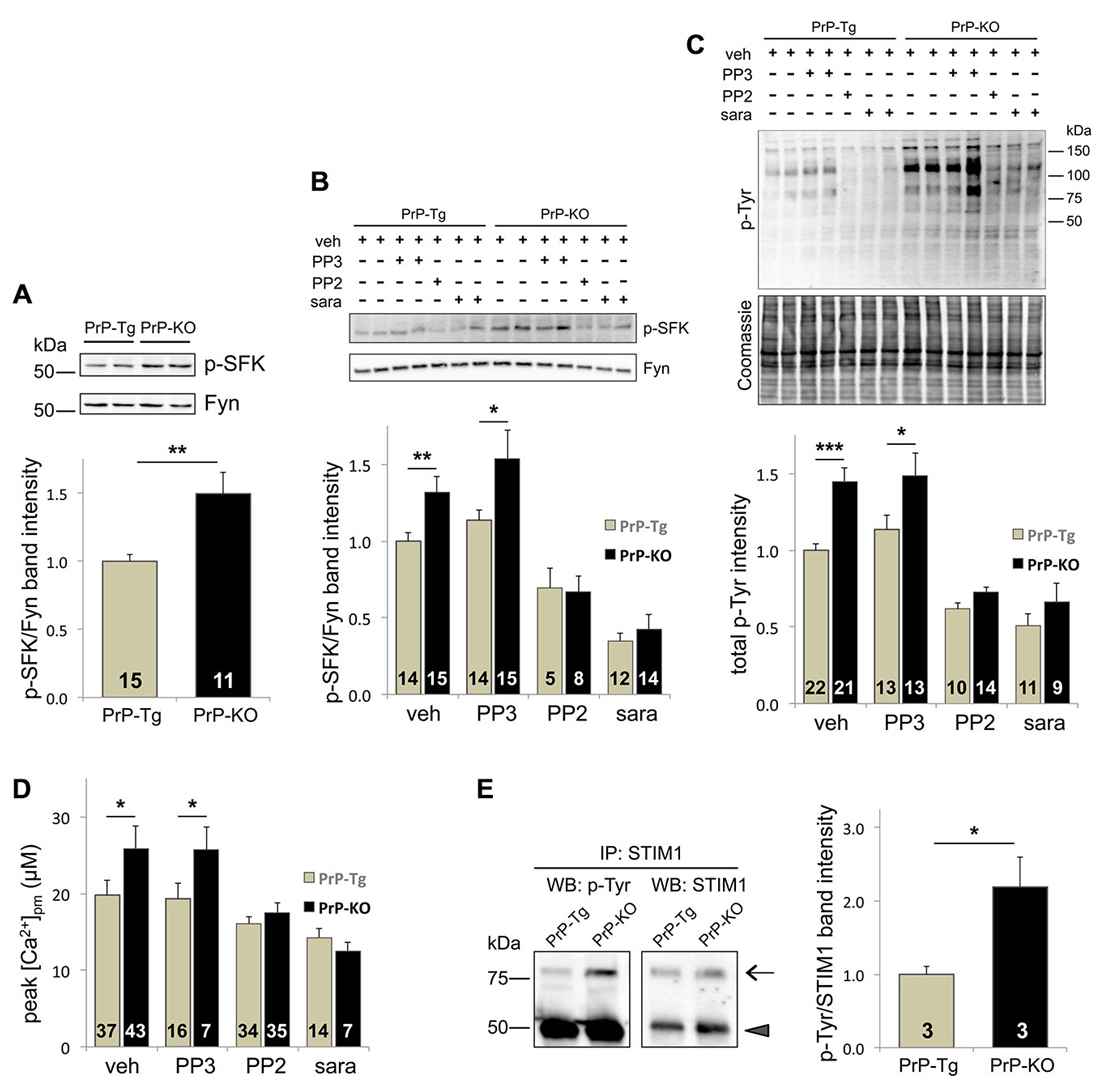
Figure 2. The Tyr-kinase Fyn is the link between PrPC and SOCE. (A) PrPC reduces the level of active Fyn, as evident from both the representative WB (upper panel) of PrP-Tg and PrP-KO CGN probed with an antibody to p-SFK or total Fyn (both run in duplicate), under SOCE-activating conditions (Ca2+-depleted stores), and the corresponding densitometric analysis of p-SFK immunosignals normalized to that of total Fyn (lower panel). Contrary to other neuronal SFK members (Supplementary Figure 4), the identical apparent mass of p-SFK and Fyn indicates that the Fyn band corresponds to the p-SFK band. Similar results were obtained under basal conditions, i.e., with Ca2+-filled stores (see Supplementary Figure 3). (B,C) Addition (+) of the SFK inhibitors saracatinib (sara, 5 μM) and PP2 (10 μM), but not of PP3 (10 μM), reduces the level of both p-SFK (B) and total Tyr-phosphorylated (p-Tyr) proteins (C) of CGN compared to the untreated (−) samples. (B) The upper panel reports a representative WB of the two neuronal genotypes treated with, or without, the SFK inhibitors, and immunostained as in (A), while the lower panel shows the corresponding densitometric analysis of p-SFK normalized to the band intensity of total Fyn. Veh (vehicle) indicates the control experiment run in the presence of DMSO (0.1%). (C) The upper panel reports the WB of total p-Tyr proteins present in the two CGN genotypes treated as in (B). In the corresponding densitometric analysis (lowest panel), the p-Tyr band intensity was normalized to that of the Coomassie blue-stained bands (middle panel). (D) Only saracatinib and PP2 decrease SOCE-induced [Ca2+]pm peaks, and abrogate the difference observed in control (veh-treated) PrP-Tg and PrP-KO CGN. (E) STIM1 is more abundantly Tyr-phosphorylated in PrP-KO CGN than in PrP-Tg CGN under SOCE-stimulating conditions. This is evident from the representative WB (left panel) of immunoprecipitated (IP) STIM1 probed with an antibody to either p-Tyr or total STIM1 (arrow), and from the corresponding densitometric analysis (right panel) reported as the ratio between the p-Tyr band intensity and the STIM1 band intensity. The arrowhead in the left panels indicates the immunosignal of the mouse IgG used in the immunoprecipitation assay. On the left of the WB, MW standards are indicated. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t-test. The analysis of the statistical significance (p-value, Student’s t-test) of data for the comparison between different treatments within each PrP genotype (B–D) is reported in Supplementary Table 2. Other details are as in the legend to Figure 1.
As expected, PP2 and saracatinib, two different selective inhibitors of SFK (but not PP3, the negative control of PP2), significantly diminish the level of p-Fyn (Figure 2B), and of all p-Tyr proteins (Figure 2C), in both CGN types. Importantly, these inhibitors also diminish SOCE-induced PM Ca2+ peaks, abrogating in this way the difference observed in untreated PrP-Tg and PrP-KO neurons (Figure 2D). The parallelism between SOCE and Fyn activations thus demonstrates that PrPC regulates SOCE by controling the Fyn signaling pathways.
A key issue to unravel was identification of the molecular mechanizm linking SOCE to the PrPC-dependent control of Fyn. We focused on STIM1, one of the two ER Ca2+-sensor isoforms responsible for SOCE activation (Muik et al., 2012), not only because its Tyr-phosphorylation upregulates SOCE (Lopez et al., 2012), but also because the expression STIM1, which reaches its higher level in the cerebellum (Klejman et al., 2009), is predominant over STIM2 in mouse CGN (Lalonde et al., 2014). To mention also that the protocol of severe Ca2+ store depletion used here to fully activate SOCE is ideal to engage STIM1, whose Ca2+ affinity is much higher compared to STIM2 (Hoth and Niemeyer, 2013). Immunoprecipitation assays showed that, under the above-mentioned conditions, PrP-KO CGN display significantly more Tyr-phosphorylated STIM1 than PrP-Tg neurons (Figure 2E).
Aβ(1–42) Oligomers Impair PrPC-Dependent Control of SOCE through Fyn
Following the hypothesis that PrPC-Aβ interaction could be crucial for AD-related neuronal impairment (Laurén et al., 2009; Um and Strittmatter, 2013), we investigated whether soluble Aβ(1–42) oligomers (Supplementary Figure 5) perturb the control of PrPC over SOCE by monitoring PM Ca2+ transients. Compared to the untreated counterpart, we found that addition of soluble Aβ(1–42) oligomers augments PM Ca2+ peaks of PrP-Tg CGN to the same value detected in untreated PrP-KO CGN (Figure 3A). Because no statistically significant effect was evident on Aβ-treated PrP-KO CGN, this result indicates that dysregulation of SOCE by oligomeric Aβ(1–42) is strictly dependent on the presence of PrPC. Importantly, an identical PrPC-dependent alteration of SOCE was also observed in primary cortical neurons (Figure 3B), which are a primary target of Aβ oligomers in AD (Haass and Selkoe, 2007).
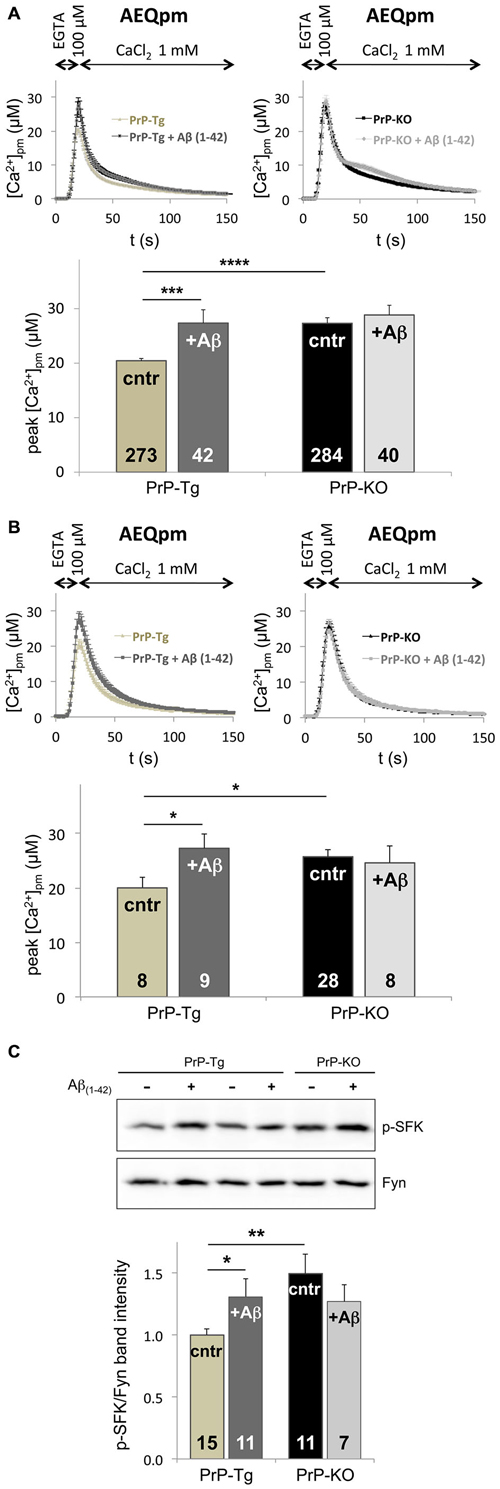
Figure 3. Soluble Aβ(1–42) oligomers increase SOCE and Fyn activation in a PrPC-dependent manner. (A,B) Exposure to Aβ(1–42) oligomers (at a concentration of 5 μM of monomer equivalents) increases the [Ca2+]pm peaks only in PrP-expressing CGN (A) and cortical neurons (B) upon SOCE stimulation, thus abrogating the difference between the two genotypes observed in the absence of Aβ(1–42) oligomers (cntr). (C) Under SOCE-triggering conditions, Aβ(1–42) oligomers significantly increase the auto-phosphorylation of Fyn in PrP-Tg CGN, but not in PrP-KO CGN, nullifying the difference between the two genotypes observed in the absence of Aβ(1–42) oligomers (cntr). This is apparent from both the representative WB of untreated (−), or Aβ(1–42)-treated (+), neurons (upper panel; probed with an antibody to p-SFK or total Fyn), and the densitometric analysis of the anti-p-SFK immunosignal normalized to that of total Fyn (lower panel). Similar results were obtained under basal conditions, i.e., with Ca2+-filled stores (see Supplementary Figure 6). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 10−5, Student’s t-test. Other details are as in the legend to Figures 1, 2.
The capacity of Aβ(1–42) oligomers to disturb SOCE only in PrP-Tg CGN was paralleled by the action on Fyn, because once again oligomeric Aβ(1–42) increases the level of active Fyn in these neurons, both under SOCE-activating (Figure 3C) and basal (Supplementary Figure 6) conditions. This data indicates that soluble oligomeric Aβ(1–42) increases SOCE by impairing the PrPC-dependent downregulation of Fyn.
Discussion
By the novel comparison of local Ca2+ oscillations in isogenic primary CGN expressing, or not, PrPC, we report here that PrPC restricts the accumulation of Ca2+ in the cytosol and mitochondria of neurons following SOCE. PrPC accomplishes this task by limiting SOC-mediated Ca2+ entry and by increasing Ca2+ uptake by the ER, which likely depends on the capacity of PrPC to upregulate the expression of the sarco/endoplasmic reticulum Ca2+-ATPase pump (Lazzari et al., 2011). We inferred that, within the used experimental paradigms, SOC are the only Ca2+ channels influenced by PrPC because our control experiments show that VGCC do not significantly contribute to the observed Ca2+ fluxes (see also Park et al., 2010), nor that PrPC changes the activity of VGCC in contrast to previous indications (Herms et al., 2000; Korte et al., 2003). Irrespective of this aspect, our results also highlight the considerable capacity of CGN mitochondria to buffer SOC-mediated Ca2+ influx, undisclosed in neurons thus far, in accord to the connection between the mitochondrial Ca2+ uniporter and SOCE observed in mast cells (Samanta et al., 2014).
Our data emphasize the protective function of PrPC towards perilous local Ca2+ overloads (Khosravani et al., 2008) that may undermine neuronal functions and plasticity, especially in neurodegenerative disorders (Berridge, 2014). Consistent with this view, while normal SOCE was found to maintain the structure of mushroom spines (pivotal to learning and memory; Sun et al., 2014), excessive SOCE was implicated in hypoxia-induced neuronal death (Berna-Erro et al., 2009). Likewise, the capacity of PrPC to shape local Ca2+ signals may shed light into neurodegenerative pathways such as those occurring during prion infection, where changes in the expression level and processing of PrPC (Mays et al., 2014) may contribute to Ca2+-induced neuronal damages.
The PrPC-dependent downregulation of SOCE was also observed in primary cortical neurons, suggesting that PrPC controls SOCE and intracellular Ca2+ transients in different neuronal cell types. In this context, it is to be mentioned that, although SOCE is a major pathway for Ca2+ entry in non-excitable cells (Parekh and Putney, 2005), the importance of SOCE is increasingly recognized also in excitable cells, including neurons (Moccia et al., 2015), in which its role is just beginning to be fully deciphered (Majewski and Kuznicki, 2015). In particular, a recent report suggests that SOCE could also regulate gene expression through the transcription factor Sp4 (Lalonde et al., 2014), which is known to contribute to complex neuronal processes including learning and memory (Zhou et al., 2010). Our results thus suggest that the modulation of SOCE could be one of the means connecting PrPC to the different neuronal functions attributed to the protein.
Mechanistically, we identified Fyn as a molecular intermediate enabling PrPC to control SOCE in light of the observed close correlation between Fyn and SOCE: both are upregulated in the absence of PrPC, while both are downregulated by selective SFK inhibitors (PP2 and saracatinib). In addition to the mandatory variation in ER Ca2+ levels, SOCE is regulated by different mechanizms that include the glutathionylation and phosphorylation of STIM proteins (Hawkins et al., 2010; Pozo-Guisado and Martin-Romero, 2013). In particular, the Tyr-phosphorylation of STIM1 by SFK members increases SOCE (Lopez et al., 2012). We report here that—under SOCE-triggering conditions—STIM1 is more Tyr-phosphorylated in PrP-KO than in PrP-expressing neurons, a result fully consistent with the higher activation of SOCE and Fyn observed in PrP-KO neurons. Although the precise site(s) of Tyr-phosphorylation on STIM1 is(are) unknown, Tyr361 in the cytosolic domain of the protein appears the most likely target of SFK members. This site was found to be phosphorylated by mass spectrometry studies (http://www.phosphosite.org/siteAction.do?id=25755096), is highly conserved among different mammalian species, and is also present in STIM2. Further studies will clarify if Tyr361 is the actual phosphorylation site by SFK and if this is a mechanizm to functionally regulate both STIM isoforms.
Multiple indications have implicated Fyn as a downstream effector of PrPC in the regulation of key cell processes, ranging from embryogenesis and neuritogenesis to, at large, neuroprotective signaling (Aguzzi et al., 2008; Linden et al., 2008). However, while most observations were obtained upon stimulation of cells [e.g., by antibody-mediated clustering of PrPC (Mouillet-Richard et al., 2000; Pantera et al., 2009), or upon binding of PrPC to NCAM (Santuccione et al., 2005)], for the first time to our knowledge we have shown that PrPC depresses Fyn activity under basal conditions. This result opens the possibility that PrPC is constitutively implicated in such a crucial aspect of cell physiology, and that dysregulation of this function may be particularly relevant for those disease-related species, like Aβ oligomers and prions, which have been proposed to exploit PrPC as surface binding partner for the downstream transduction of their toxicity (Barton and Caughey, 2011; Wang et al., 2013; Hirsch et al., 2014).
In AD, both Ca2+ dyshomeostasis (Green and LaFerla, 2008; Demuro et al., 2010) and aberrant Fyn signaling (Lambert et al., 1998; Roberson et al., 2011) were indicated to mediate the deleterious effects of oligomeric Aβ. Accordingly, it was shown that Fyn, which is part of a super-molecular complex with PrPC and the metabotropic glutamate receptor 5 (mGluR5, serving to connect Fyn and PrPC on the opposite sides of the PM), is activated after Aβ docking to PrPC (Larson et al., 2012; Um et al., 2012, 2013). We found that exposure to soluble Aβ(1–42) oligomers disrupts the PrPC-Fyn-SOCE triangle because such a treatment increases activation of both Fyn and SOCE in PrP-expressing, but not in PrP-KO, CGN. Importantly, the PrPC-dependent effect of Aβ oligomers on SOCE was also observed in cortical neurons that, together with hippocampal neurons, are the preferential target of Aβ toxicity (Haass and Selkoe, 2007).
Considering the influence of PrPC on Fyn, our findings are reminiscent of Fyn activation by PrPC cross-linking (Mouillet-Richard et al., 2000) or PrPC-NCAM clustering (Santuccione et al., 2005). However, they disclose a different underlying mechanism, whereby the interaction of PrPC with extracellular ligands releases the basal block of PrPC on Fyn rather than the ligand-PrPC complex directly promoting Fyn activation. Hence, one initial step of oligomeric Aβ(1–42) toxicity could involve PrPC displacement from the role of sentinel against neuronal Ca2+ overload. Additional studies are needed to clarify whether this effect is consequent to a structural modification of PrPC, or a dislodgment from natural interacting partners, or a modification of the membrane lipid architecture surrounding the protein. Likewise, also the link between Fyn and PrPC in CGN needs to be elucidated. We exclude the involvement of mGluR5 (Um et al., 2013), given that the neurons utilized here do not harbor detectable amounts of mGluR5 nor respond to archetypal mGluR5 agonists (unpublished observations).
In conclusion, we report here that exposure of neurons to oligomeric Aβ(1–42) results in increased activation of Fyn and SOCE. However, given that PrP-KO mice show no gross phenotype, nor overt signs of neurodegeneration, the alterations observed in this work cannot be sufficient to account for AD pathology. Nonetheless, they could act as necessary events that, combined with other (PrPC-dependent and/or PrPC-independent) insults by Aβ oligomers, eventually contribute to AD-related neuronal damage. Further studies will be necessary to demonstrate whether Aβ oligomers perturb local Ca2+ fluxes different from SOCE, in particular those arising from NMDA-R that have already been functionally associated to PrPC (Khosravani et al., 2008).
Author Contributions
MCS, AFH and AB designed research; ADM, AC, CP, MLM and DL performed research (ADM and AC contributed equally); ADM, CP and AB analyzed data; AB, AFH and MCS wrote the article.
Funding
This work was supported by the Italian Ministry of University and Research (PRIN/2010 to DL), the University of Padova (PRAT CPDA121988/12 to MCS), and the Australian Academy of Sciences (to AFH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Aβ, amyloid-β; AD, Alzheimer’s disease; AEQ, aequorin; CGN, cerebellar granule neurons; DMSO, dimethylsulfoxide; ER, endoplasmic reticulum; HA, hemagglutinin; KRB, Krebs-Ringer buffer; mRFP, monomeric red fluorescent protein; NMDA-R, N-methyl-D-aspartate-sensitive glutamatergic receptors; PBS, phosphate-buffered saline; PM, plasma membrane; PrPC, prion protein; PrP-KO, PrP-knockout; PrP-Tg, transgenic Tg46; PVDF, polyvinylidene fluoride; SFK, Src Tyr-kinase family; SOC, store-operated channels; SOCE, store-operated Ca2+ entry; STIM, stromal interaction molecules; VGCC, voltage-gated Ca2+ channels; WB, Western blot.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fncel.2015.00416/abstract
References
Aguzzi, A., Baumann, F., and Bremer, J. (2008). The prion’s elusive reason for being. Annu. Rev. Neurosci. 31, 439–477. doi: 10.1146/annurev.neuro.31.060407.125620
Barton, K. A., and Caughey, B. (2011). Is PrP the road to ruin? EMBO J. 30, 1882–1884. doi: 10.1038/emboj.2011.129
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitative Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2:ra67. doi: 10.1126/scisignal.2000522
Berridge, M. J. (2014). Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 592, 281–293. doi: 10.1113/jphysiol.2013.257527
Brini, M., Marsault, R., Bastianutto, C., Alvarez, J., Pozzan, T., and Rizzuto, R. (1995). Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). J. Biol. Chem. 270, 9896–9903. doi: 10.1074/jbc.270.17.9896
Chung, S. C., Limnander, A., Kurosaki, T., Weiss, A., and Korenbrot, J. I. (2007). Coupling Ca2+ store release to Icrac channel activation in B lymphocytes requires the activity of Lyn and Syk kinases. J. Cell Biol. 177, 317–328. doi: 10.1083/jcb.200702050
Demuro, A., Parker, I., and Stutzmann, G. E. (2010). Calcium signaling and amyloid toxicity in alzheimer disease. J. Biol. Chem. 285, 12463–12468. doi: 10.1074/jbc.r109.080895
Dubois, C., Vanden Abeele, F., Lehen’kyi, V., Gkika, D., Guarmit, B., Lepage, G., et al. (2014). Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 6, 19–32. doi: 10.1016/j.ccr.2014.04.025
Green, K. N., and LaFerla, F. M. (2008). Linking calcium to Abeta and Alzheimer’s disease. Neuron 59, 190–194. doi: 10.1016/j.neuron.2008.07.013
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hartmann, J., Karl, R. M., Alexander, R. P., Adelsberger, H., Brill, M. S., Rühlmann, C., et al. (2014). STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission and cerebellar motor behavior. Neuron 82, 635–644. doi: 10.1016/j.neuron.2014.03.027
Hawkins, B. J., Irrinki, K. M., Mallilankaraman, K., Lien, Y. C., Wang, Y., Bhanumathy, C. D., et al. (2010). S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 90, 391–405. doi: 10.1083/jcb.201004152
Herms, J. W., Korte, S., Gall, S., Schneider, I., Dunker, S., and Kretzschmar, H. A. (2000). Altered intracellular calcium homeostasis in cerebellar granule cells of prion protein-deficient mice. J. Neurochem. 75, 1487–1492. doi: 10.1046/j.1471-4159.2000.0751487.x
Hirsch, T. Z., Hernandez-Rapp, J., Martin-Lannerée, S., Launay, J. M., and Mouillet-Richard, S. (2014). PrP(C) signalling in neurons: from basics to clinical challenges. Biochimie 104, 2–11. doi: 10.1016/j.biochi.2014.06.009
Hoth, M., and Niemeyer, B. A. (2013). The neglected CRAC proteins: Orai2, Orai3 and STIM2. Curr. Top. Membr. 71, 237–271. doi: 10.1016/b978-0-12-407870-3.00010-x
Kendall, J. M., Dormer, R. L., and Campbell, A. K. (1992). Targeting aequorin to the endoplasmic reticulum of living cells. Biochem. Biophys. Res. Commun. 189, 1008–1016. doi: 10.1016/0006-291x(92)92304-g
Khosravani, H., Zhang, Y., Tsutsui, S., Hameed, S., Altier, C., Hamid, J., et al. (2008). Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 181, 551–565. doi: 10.1083/jcb.200711002
Klejman, M. E., Gruszczynska-Biegala, J., Skibinska-Kijek, A., Wisniewska, M. B., Misztal, K., Blazejczyk, M., et al. (2009). Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem. Int. 54, 49–55. doi: 10.1016/j.neuint.2008.10.005
Korte, S., Vassallo, N., Kramer, M. L., Kretzschmar, H. A., and Herms, J. (2003). Modulation of L-type voltage-gated calcium channels by recombinant prion protein. J. Neurochem. 87, 1037–1042. doi: 10.1046/j.1471-4159.2003.02080.x
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Lalonde, J., Saia, G., and Gill, G. (2014). Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci. Signal. 7:ra51. doi: 10.1126/scisignal.2005242
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, non fibrillar ligands derived from Abeta 1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U S A 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrPC -Fyn couples human oligomeric aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871. doi: 10.1523/JNEUROSCI.1858-12.2012
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Lazzari, C., Peggion, C., Stella, R., Massimino, M. L., Lim, D., Bertoli, A., et al. (2011). Cellular prion protein is implicated in the regulation of local Ca2+ movements in cerebellar granule neurons. J. Neurochem. 116, 881–890. doi: 10.1111/j.1471-4159.2010.07015.x
Lee, K. M., Son, S. W., Babnigg, G., and Villereal, M. L. (2006). Tyrosine phosphatase and cytochrome P450 activity are critical in regulating store-operated calcium channels in human fibroblasts. Exp. Mol. Med. 38, 703–717. doi: 10.1038/emm.2006.83
Lim, D., Fedrizzi, L., Tartari, M., Zuccato, C., Cattaneo, E., Brini, M., et al. (2008). Calcium homeostasis and mitochondrial dysfunction in striatal neurons of huntington disease. J. Biol. Chem. 283, 5780–5789. doi: 10.1074/jbc.m704704200
Linden, R., Martins, V. R., Prado, M. A. M., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Lopez, E., Jardin, I., Berna-Erro, A., Bermejo, N., Salido, G. M., Sage, S. O., et al. (2012). STIM1 tyrosine-phosphorylation is required for STIM1-Orai1 association in human platelets. Cell. Signal. 24, 1315–1322. doi: 10.1016/j.cellsig.2012.02.012
Majewski, L., and Kuznicki, J. (2015). SOCE in neurons: signaling or just refilling? Biochim. Biophys. Acta 1853, 1940–1952. doi: 10.1016/j.bbamcr.2015.01.019
Mallucci, G. R., Ratte, S., Asante, E. A., Linehan, J., Gowland, I., Jefferys, J. G. R., et al. (2002). Post-natal knock out of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 21, 202–210. doi: 10.1093/emboj/21.3.202
Marsault, R., Murgia, M., Pozzan, T., and Rizzuto, R. (1997). Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 16, 1575–1581. doi: 10.1093/emboj/16.7.1575
Mays, C. E., Kim, C., Haldiman, T., van der Merwe, J., Lau, A., Yang, J., et al. (2014). Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Invest. 124, 847–858. doi: 10.1172/jci72241
Moccia, F., Zuccolo, E., Soda, T., Tanzi, F., Guerra, G., Mapelli, L., et al. (2015). Stim and orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 9:153. doi: 10.3389/fncel.2015.00153
Montero, M., Brini, M., Marsault, R., Alvarez, J., Sitia, R., Pozzan, T., et al. (1995). Monitoring dynamic changes in free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO J. 14, 5467–5475.
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M., et al. (2000). Signal transduction through prion protein. Science 289, 1925–1928. doi: 10.1126/science.289.5486.1925
Muik, M., Schindl, R., Fahrner, M., and Romanin, C. (2012). Ca2+ release-activated Ca2+ (CRAC) current, structure and function. Cell. Mol. Life Sci. 69, 4163–4176. doi: 10.1007/s00018-012-1072-8
Naldini, L., Blömer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F. H., et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267. doi: 10.1126/science.272.5259.263
Ottolini, D., Calì, T., and Brini, M. (2014). Methods to measure intracellular Ca(2+) fluxes with organelle-targeted aequorin-based probes. Methods Enzymol. 543, 21–45. doi: 10.1016/b978-0-12-801329-8.00002-7
Pantera, B., Bini, C., Cirri, P., Paoli, P., Camici, G., Manao, G., et al. (2009). PrPC activation induces neurite outgrowth and differentiation in PC12 cells: role for caveolin-1 in the signal transduction pathway. J. Neurochem. 110, 194–207. doi: 10.1111/j.1471-4159.2009.06123.x
Parekh, A. B., and Putney, J. W. Jr. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810. doi: 10.1152/physrev.00057.2003
Park, C. Y., Shcheglovitov, A., and Dolmetsch, R. (2010). The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105. doi: 10.1126/science.1191027
Peggion, C., Bertoli, A., and Sorgato, M. C. (2011). Possible role for Ca2+ in the pathophysiology of the prion protein? Biofactors 37, 241–249. doi: 10.1002/biof.161
Pozo-Guisado, E., and Martin-Romero, F. J. (2013). The regulation of STIM1 by phosphorylation. Commun. Integr. Biol. 6:e26283. doi: 10.4161/cib.26283
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U S A 95, 13363–13393. doi: 10.1073/pnas.95.23.13363
Resenberger, U. K., Harmeier, A., Woerner, A. C., Goodman, J. L., Müller, V., Krishnan, R., et al. (2011). The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070. doi: 10.1038/emboj.2011.86
Rizzuto, R., Simpson, A. W. M., Brini, M., and Pozzan, T. (1992). Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327. doi: 10.1038/358325a0
Roberson, E. D., Halabisky, B., Yoo, J. W., Yao, J., Chin, J., Yan, F., et al. (2011). Amyloid-β/Fyn-induced synaptic, network and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31, 700–711. doi: 10.1523/JNEUROSCI.4152-10.2011
Salter, M. W., and Kalia, L. V. (2004). Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328. doi: 10.1038/nrn1368
Samanta, K., Douglas, S., and Parekh, A. B. (2014). Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS One 9:e101188. doi: 10.1371/journal.pone.0101188
Santuccione, A., Sytnyk, V., Leshchyns’ka, I., and Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169, 341–354. doi: 10.1083/jcb.200409127
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Soboloff, J., Rothberg, B. S., Mades, M., and Gill, D. L. (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565. doi: 10.1038/nrm3414
Sorgato, M. C., and Bertoli, A. (2009). From cell protection to death: may Ca2+ signals explain the chameleonic attributes of the mammalian prion protein? Biochem. Biophys. Res. Commun. 379, 171–174. doi: 10.1016/j.bbrc.2008.12.026
Sorgato, M. C., Peggion, C., and Bertoli, A. (2009). Is, indeed, the prion protein a Harlequin servant of "many" masters? Prion 3, 202–205. doi: 10.4161/pri.3.4.10012
Sun, S., Zhang, H., Liu, J., Popugaeva, E., Xu, N. J., Feske, S., et al. (2014). Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. doi: 10.1016/j.neuron.2014.02.019
Um, J. W., and Strittmatter, S. M. (2013). Amyloid-β induced signaling by cellular prion protein and Fyn kinase in alzheimer disease. Prion 7, 37–41. doi: 10.4161/pri.22212
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for alzheimer Aβ oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Wang, H., Ren, C. H., Gunawardana, C. G., and Schmitt-Ulms, G. (2013). Overcoming barriers and thresholds-signaling of oligomeric Aβ through the prion protein to Fyn. Mol. Neurodegener. 8:24. doi: 10.1186/1750-1326-8-24
Zhou, X., Nie, Z., Roberts, A., Zhang, D., Sebat, J., Malhotra, D., et al. (2010). Reduced NMDAR1 expression in the Sp4 hypomorphic mouse may contribute to endophenotypes of human psychiatric disorders. Hum. Mol. Genet. 19, 3797–3805. doi: 10.1093/hmg/ddq298
Keywords: prion protein, Ca2+, store-operated Ca2+ entry, Fyn kinase, cerebellar granule neurons, Alzheimer’s disease, Abeta oligomers, STIM1
Citation: De Mario A, Castellani A, Peggion C, Massimino ML, Lim D, Hill AF, Sorgato MC and Bertoli A (2015) The prion protein constitutively controls neuronal store-operated Ca2+ entry through Fyn kinase. Front. Cell. Neurosci. 9:416. doi: 10.3389/fncel.2015.00416
Received: 07 August 2015; Accepted: 02 October 2015;
Published: 28 October 2015.
Edited by:
Francesco Moccia, University of Pavia, ItalyReviewed by:
Euan Robert Brown, Heriot Watt University, UKEun-Kyoung Choi, Hallym University, South Korea
Copyright © 2015 De Mario, Castellani, Peggion, Massimino, Lim, Hill, Sorgato and Bertoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Catia Sorgato, Y2F0aWEuc29yZ2F0b0B1bmlwZC5pdA==; Alessandro Bertoli, YWxlc3NhbmRyby5iZXJ0b2xpQHVuaXBkLml0
† These authors have contributed equally to this work.