 Jorge Azpurua
Jorge Azpurua Benjamin A. Eaton
Benjamin A. Eaton- Department of Physiology, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA
Two of the most salient phenotypes of aging are cognitive decline and loss of motor function, both of which are controlled by the nervous system. Cognition and muscle contraction require that neuronal synapses develop and maintain proper structure and function. We review the literature on how normal physiological aging disrupts central and peripheral synapse function including the degradation of structure and/or control of neurotransmission. Here we also attempt to connect the work done on the epigenetics of aging to the growing literature of how epigenetic mechanisms control synapse structure and function. Lastly, we address possible roles of epigenetic mechanisms to explain why the basal rates of age-related dysfunction vary so widely across individuals.
Introduction
Synapses are the functional unit that processes signals between neurons, and between an innervating motor neuron and its target muscle. Neurotransmitter release at the synapse is required for planning and executive function, memory formation and recall, motor behavior, hormone release, and virtually every organismal function that feeds back into the central nervous system. Modulation of synaptic strength is heavily implicated in the persistence of memory and general cognitive functions. The synapse is a particularly interesting target for the effects of aging on nervous system function. The structure of the synapse is complex and many synapses must be maintained throughout the life of the animal. Additionally, the composition of ion channels at both pre- and post synaptic terminals is highly heterogeneous, crucial for function, and may cause synapse malfunction if altered. Lastly, synaptic dysfunction could affect neuroendocrine signaling. Coordination of synaptic vesicle release requires the neuron to maintain proper synaptic structure, membrane excitability and neurotransmission, as well as integrate retrograde signals from the post-synaptic terminal. Defects in these complex processes have been implicated in many diseases including Alzheimer’s disease (AD), and Parkinson’s disease (PD), which both feature a strong aging-associated risk component (Hebert et al., 2001; Levy et al., 2002).
Here we review the growing literature on the contribution of one hallmark of cellular aging—epigenetic alterations- on neuronal aging, with a strict focus on synaptic structure and function during non-pathological aging. The effects of adult onset neurodegenerative disease on synapse function have been extensively reviewed elsewhere (Wishart et al., 2006; Gillingwater and Wishart, 2013). We use “epigenetics” broadly referring to stable and heritable changes in gene expression that can be transmitted across cell divisions without DNA mutations. Major mechanisms of epigenetic control include DNA CpG methylation, histone modifications (acetylation and methylation), deposition of alternative histones, and RNA silencing. Aging organisms undergo genome-wide DNA CpG demethylation across their tissues, leading to wide transcriptional induction. Histone methylation and acetylation have also been reported to change with age (Calvanese et al., 2009). These changes are associated with various disease states, such as a cancer (Esteller et al., 2001). Proper synaptic function requires tight co-regulation of many genes, making epigenetic dysfunction an attractive candidate to explain age-associated declines in the nervous system. The growing recognition of neuroepigenetics has been previously reviewed (Sweatt, 2013). Here we will attempt to link what is known about epigenetic regulation in the nucleus, to non-pathological aging-related dysfunction of the synaptic terminal.
Synaptic Structure and Aging : Central and Sensory Synapses
Central Synapse Structure is Altered by Aging
A substantial body of evidence supports the idea that aging disrupts synaptic connections in the central nervous system. Synapses may be completely lost in some contexts and the evidence strongly points at synaptic loss being a key feature of general brain aging as well as a hallmark of AD pathology (DeKosky and Scheff, 1990; Cabalka et al., 1992; Masliah et al., 1993; Scheff and Price, 2001; Coleman and Yao, 2003). In humans, there is support for an association between age related mild-cognitive impairment and loss of synapses and there is evidence in rodents as well (Coggan et al., 2004; Scheff et al., 2006; Canas et al., 2009; Richard et al., 2010). In some tissues, loss of postsynaptic dendritic spines has been implicated as the primary mechanism of synapse loss (Feldman and Dowd, 1975; Geinisman et al., 1992; de Brabander et al., 1998). Changes in synaptic structure and synapse loss with age by neuroanatomical region have been thoroughly reviewed (Petralia et al., 2014) and the evidence is robust. A recent study found that age-dependent declines in cognition and memory may best be explained as a decline in synaptic stability (Grillo et al., 2013). Grillo and colleagues used two-photon microscopy and semi-automated image processing to analyze synapse structure across age. They found that as age increases, there is a loss of “synaptic tenacity” driven by increases in the rate of bouton loss and remodeling.
Age-dependent changes to synapse structure have been studied in other model organisms as well. In C. elegans, animals show an age-dependent increase in a number of dendritic defects of sensory neurons, including ectopic branch points, which is predicted to alter synaptic connectivity (Toth et al., 2012). Intriguingly, in Drosophila sensory neurons the change is opposite of that seen in C. elegans, with fewer branch points as age increases (Corfas and Dudai, 1991). Thus the synapse may be an Achilles’ heel that is vulnerable to aging across all metazoan taxa.
Histone Acetylation and Central Synapse Structure
New evidence suggests that epigenetic alterations during aging may be driving central synapse loss and alterations in structure. Modulating the expression level of histone deacetylase 2 (HDAC2) has been shown to cause changes in synaptic structure of the CA1 pyramidal neurons of the mouse hippocampus (Guan et al., 2009). Overexpression of HDAC2 led to a reduction in the number of spines as seen through Golgi staining and decreased synaptophysin staining, whereas HDAC2 knockout showed an increase in both. HDAC2 is a class 1 (NAD-independent) histone deacetylase, which generally acts as a transcriptional silencer in concert with YY1 (Yang et al., 1996) and DNA methyl transferase 1 (DMNT1; Rountree et al., 2000). Chromatin immunoprecipitation from whole mouse brains of HDAC2 and HDAC1 both showed enrichment for cell cycle genes, but HDAC2 also showed higher enrichment of genes involved in synaptic formation and plasticity (e.g., Nrxn3 and Synapsin2). This report strongly supports the involvement of HDAC2 in structural synaptic maintenance.
Synaptic Structure and Aging: Neuromuscular Junction
Neuromuscular Junction Synapse Structure is Altered by Aging
Motor decline is one the most predictive biomarkers of mortality risk in humans (Buchman et al., 2007). Age-related pathologies in the neuromuscular junction (NMJ) have been studied in rodents since the early 1970’s (Gutmann et al., 1971; Banker et al., 1983; Cardasis and LaFontaine, 1987). More recently, live imaging of SOD1 knock-out mice sternomastoid muscles revealed a pronounced retraction of the NMJ with over two-thirds of NMJs denervated, recapitulating the aging phenotype in an accelerated fashion (Jang et al., 2010). Conversely, caloric restriction (a conserved pathway known to slow aging rates) was found to also abrogate the denervation of mouse NMJs (Valdez et al., 2010). Nonetheless, aging does not seem to affect all motor neurons equally, as brainstem motor neurons are at least partially resistant to age-dependent denervation (Valdez et al., 2012). Synapse morphology may also be altered in response to muscle fiber loss and regeneration (Li et al., 2011). It is possible that fiber loss is followed by improper synapse innervations due to compromised synaptogenesis in aged neurons.
The NMJ synapses in both larval and adult Drosophila have been characterized with development and aging as well. Initial imaging studies in the longitudinal abdominal muscle of adult flies showed several age-dependent structural changes, including enlarged bouton area, decreased branch length and “naked boutons” directly facing the extracellular basal lamina (Beramendi et al., 2007). In the same study, the authors also showed a significant increase in synaptic vesicle size, as well as changes in mitochondrial structure and area. Together with mammalian studies, this supports that alterations to the NMJ structure are a conserved aging phenotype.
As with central synapses, there is evidence to support the involvement of epigenetic factors in age-dependent structural dysfunction of the NMJ. A Drosophila screen for altered synaptic structure in larvae found that the chromatin insulator bpd [also known as mod(mdg4)] was necessary for proper NMJ formation (Gorczyca et al., 1999). Non-lethal mutants of the gene were found to harbor additional axonal branch points, and boutons failed to form tight clusters. Bpd is a chromatin factor that regulates transcriptional silencing by binding DNA sequences known as gypsy insulators (Gerasimova et al., 1995, 2000; Chen and Corces, 2001). A screen for memory formation mutants also identified the gene as well as a later microarray screen for mushroom body-specific genes (Dubnau et al., 2003; Kobayashi et al., 2006). Taken together, these results imply this epigenetic regulator as necessary for proper synaptic structure formation. Notably, chromatin insulation is thought to become disrupted with age (Fu et al., 2008). Despite this, the exact genes regulated by Bpd in a neuronal context remain unknown.
Epigenetic Factors can Directly Modify Synaptic Proteins
A series of recent studies on the larval NMJ revealed that deacetylases may also play non-canonical roles in alterations of presynaptic active zone morphology. The protein ELP3 is a histone acetyl-transferase (HAT) that was shown to be able to acetylate the synaptic t-bar protein Bruchpilot (Brp) and thereby promote its degradation (Han et al., 2008; Miśkiewicz et al., 2011). Intriguingly, another histone-interacting protein, histone deacetylase 6 (HDAC6) was also found to regulate Brp acetylation in opposite fashion to ELP3 (Miskiewicz et al., 2014). These results imply that proteins involved in epigenetic regulation may have additional functions at the presynaptic terminal. Epigenetic changes in the nucleus may promote the shuttling of these proteins between the nucleus and cytoplasm. A different epigenetic regulator, HDAC4, changes its cellular localization in response to neuronal activity (Chawla et al., 2003). It is currently not known how the intracellular localization of these chromatin-modifying proteins changes with age.
Epigenetics of Synaptic Function and Aging
Plasticity is Altered by Aging
While the structure of the synapse clearly contributes to its proper function, failure of the neuron to properly maintain and adjust neurotransmitter release is possibly even more deleterious and wide-spread. The formation of memories requires synaptic plasticity in the form of Hebbian (“fire together, wire together”) synaptic facilitation, long-term potentiation (LTP), and long-term depression (LTD). Defects in LTP induction and maintenance with age have been correlated to declines in memory and cognitive ability (Penner et al., 2010). The extent of the effect of aging on the induction of LTP seems to depend on anatomical region and stimulation paradigm (Rosenzweig and Barnes, 2003). Maintenance of LTP, on the other hand, has been robustly shown to decrease with age (Landfield et al., 1978; Barnes, 2003). Studies have found that the slowing of aging by caloric restriction also preserved LTP maintenance in aged rats (Hori et al., 1992; McGahon et al., 1999; Eckles-Smith et al., 2000). Induction of LDP by low frequency stimulation in CA1 neurons of aged rats was able to reliably and rapidly reverse LTP, in contrast to what is observed in the neurons of younger rats (Norris et al., 1996).
Other forms of synaptic plasticity may also become disrupted with age. Non-Hebbian homeostatic mechanisms are believed to be required in neural networks to prevent a cascade of positive-feedback induced hyperactivity (Turrigiano, 2011). Homeostatic synaptic scaling has not been studied in the context of normal aging, however there is evidence it is impaired with age from studies of neurodegenerative disease. For example, in a mouse model of AD (with a double knock-in of presenilin 1 and amyloid precursor protein) AMPA receptor particles decreased in old age (Chang et al., 2006). There was also a downscaling of AMPA receptor mediated currents after middle age. This in turn led to defects in LTD and LTP. Despite a lack of change in control mice, this suggests that there is an age-component to the defects of homeostatic scaling seen in AD model mice. It is possible that these changes may mimic homeostatic dysfunctions that happen at very advanced ages.
Although many laboratories have studied the effects of pathological and non-pathological aging on the functions of the Drosophila nervous system using behavioral paradigms, the effects of non-pathological aging on synapse function has not been thoroughly studied (Jones and Grotewiel, 2011). It is clear that Drosophila also undergo age-dependent declines in memory formation and maintenance (Tamura et al., 2003; Simon et al., 2006). This has been found to occur in a PKA-dependent manner (Yamazaki et al., 2007). Given that PKA signaling is an important for changing the post-synaptic membrane channel composition, it implies that aging is causing central synapse dysfunction in Drosophila. (Silva et al., 1998) In addition, studies investigating the propagation of signals through the giant fiber circuit have demonstrated age-dependent degradation of circuit function that could be due to changes in neurotransmission (Zhao et al., 2010).
A new study in the adult Drosophila NMJ found that there is an age-dependent change in the homeostatic set point of the innervating neuron (Mahoney et al., 2014). Although the release properties of the cell are stable across 7 and 35 days of age, in 42-day-old animals the excitatory post-synaptic potential and quantal content (measures of synaptic vesicle release) consistently increases by approximately 70%. This shift was found to be distinct from canonical homeostatic signaling itself, as it was also seen in ephexin mutants (in which homeostasis is inhibited). This implies a change in cell-intrinsic properties with age-possibly an increase in active voltage-gated calcium channels or synaptic vesicle fusion proteins. The observed set-point change also has some potentially negative physiological consequences, as demonstrated by the enhanced synaptic depression observed at aged synapses. For an aged fly, this may cause motor behavior deficits in demanding environmental circumstances. This study is particularly intriguing because of the defined window during which there seems to be a shift in the properties of the innervating cell, potentially indicating wide changes in gene expression and neuronal identity.
Histone Acetylation and Plasticity
Epigenetics has emerged as a key regulator of long-term memory formation. A seminal study in the Aplysia snail by the Kandel laboratory found that by applying a long-term facilitation (a presynaptic form of synaptic strengthening) inducer and an LTD inducer at different terminals of a bidirectionally branched neuron the response to these inputs was integrated at the nucleus by the factors CREB1 and CREB2. CREB1 activates the HAT CPB, whereas CREB2 recruits HDAC5. These two chromatin remodeling complexes compete at the promoter of the C/EBP transcription factor which regulates synaptic structure and strength. In the case studied, LTD was found to epigenetically dominate facilitation (Guan et al., 2002).
Vertebrate studies have also found that histone modification is necessary for plasticity. Mice deficient in methyl CpG binding protein (MBD1) were found to harbor defects in neurogenesis and LTP (Zhao et al., 2003). A landmark study by Levenson and colleagues showed that memory formation transiently increases the acetylation of histone H3, and that LTP induction can be increased by administering HDAC inhibitors to mice (Levenson et al., 2004). Other studies have demonstrated the importance of HATs on LTP and memory (Alarcón et al., 2004; Korzus et al., 2004). Behavioral assays also implicate a synaptic role for histone acetylation. In mice, significant age-dependent declines in fear conditioning and memory were observed and histone H4K5 acetylation levels were altered more slowly in old mice. Peleg et al. (2010) and other studies have found evidence supporting epigenetic dysregulation in cognitive aging. Knocking out HDAC6 in a mouse model of AD ameliorated cognitive defects in 8-month old mice (Govindarajan et al., 2013).
CpG Methylation and Plasticity
Histone acetylation is not the only epigenetic mechanism that can affect synapse function. CpG methylation has been heavily implied in LTP maintenance. In VP16-CREB mice, which consolidate persistent LTP after weak stimulus, microarray comparisons to wild types revealed that brain derived neurotrophic growth factor (BDNF) was a key component (Barco et al., 2005). A study in rats found a similar result and also demonstrated that LTP maintenance could be abrogated by administration of DNMT and HDAC inhibitors (Sui et al., 2012). BDNF expression has previously been shown to be regulated by DNA CpG methylation (Martinowich et al., 2003). Recent studies have found that BDNF methylation is be required for fear memory consolidation, and can be altered in early life in mice by events such as early weaning or isolation (Lubin et al., 2008; Roth et al., 2009). Additionally, a recent study found that age-dependent changes to BDNF expression could be reversed by epigenetic manipulation (Zeng et al., 2011).
In aged rats with differential cognitive decline outcomes (that is, a population of the same age but with individuals of different cognitive capacities), individuals with less decline in cognition were shown to use an alternative LTD mechanism that was NMDA receptor–independent (Lee et al., 2005). Within-population variability suggests that epigenetic differences among individuals could play a role in aging outcomes. One potential source of variation among aged individuals is CpG methylation, which has been shown to diverge with age, even in isogenic backgrounds (Fraga et al., 2005; Kaminsky et al., 2009).
Intrinsic homeostatic plasticity has also been shown to be epigenetically regulated. Recent work has shown that phosphorylation of methyl-CpG binding protein 2 (MeCP2) was required for synaptic scaling in mouse hippocampal neurons (Zhong et al., 2012). Mutations in MeCP2 had previously been identified as causing the autism-spectrum disorder Rhett’s syndrome (Amir et al., 1999). The authors mutated the serine residues in MeCP2 that were phosphorylation targets to alanine residues and found that synaptic downscaling induced by bicuculline was disrupted, but not up scaling by tetrodotoxin (TTX). Together, these findings support CpG methylation deregulation as contributing to age-related decline in synaptic plasticity and homeostasis.
Concluding Remarks
If defects at the synapse are proximally caused by epigenetic dysregulation of key synaptic genes, then what are the distal causes that drive neuronal epigenetic dysfunction with age? One possibility is that changes in epigenetic regulation are the unavoidable consequence of species-specific noise in global transcription. Gene expression is not perfectly regulated and has large intrinsic stochasticity (Kaern et al., 2005). This would help explain why genetically identical conspecifics of the same chronological age can have dramatically different mortality risk and health. Aging-accumulated changes in expression of epigenetic regulators may cascade into larger changes culminating with alterations in cell function and identity. Synapses would be particularly sensitive to these disruptions.
Another possibility is that an insult or damage that normally accumulates with age (e.g., oxidation, advanced age glycation products, DNA damage, amyloid aggregation, etc.) gradually interferes with normal epigenetic control. DNA lesions are a particularly interesting candidate, as their repair requires histone remodeling and the epigenetic marks may not be fully reverted to their original state following repair (Polo et al., 2006; Groth et al., 2007). DNA damage can also create opportunities for retrotransposon activation (Van Meter et al., 2014) and insertion, which can potentially disrupt the epigenetic state of surrounding chromatin (Farkash and Luning Prak, 2006). Evidence for transposable element activity increasing with age has been reported in Drosophila neurons (Li et al., 2013) and more generally in mammalian neurons (Erwin et al., 2014). A sketch of these possibilities is shown in Figure 1.
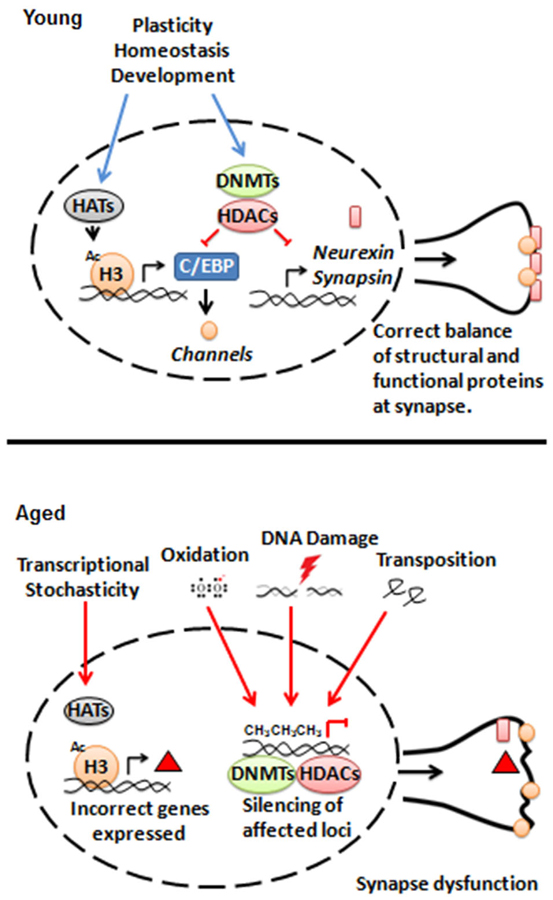
Figure 1. Model for interactions between epigenetic regulators and synaptic structure and function during aging.
Most regulatory hierarchies remain unknown, and the epigenetic markup of many tissues is still understudied. The nervous system has received special attention in this regard given the prerequisite for memory formation on dynamic adjustment of gene expression. Epigenetic marks and proteins have also received attention from those working in the aging field, ever since the discovery that HDACs and sirtuins could modulate lifespan. We suggest that a more thorough synthesis of the two fields is warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Rebekah E. Mahoney and Tabita Kreko-Pierce for useful discussion and input.
References
Alarcón, J. M., Malleret, G., Touzani, K., Vronskaya, S., Ishii, S., Kandel, E. R., et al. (2004). Chromatin acetylation, memory and LTP are impaired in CBP+/–- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42, 947–959. doi: 10.1016/j.neuron.2004.05.021
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Banker, B. Q., Kelly, S. S., and Robbins, N. (1983). Neuromuscular transmission and correlative morphology in young and old mice. J. Physiol. 339, 355–377. doi: 10.1113/jphysiol.1983.sp014721
Barco, A., Patterson, S. L., Alarcon, J. M., Gromova, P., Mata-Roig, M., Morozov, A., et al. (2005). Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron 48, 123–137. doi: 10.1016/j.neuron.2005.09.005
Barnes, C. A. (2003). Long-term potentiation and the ageing brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 765–772. doi: 10.1098/rstb.2002.1244
Beramendi, A., Peron, S., Casanova, G., Reggiani, C., and Cantera, R. (2007). Neuromuscular junction in abdominal muscles of Drosophila melanogaster during adulthood and aging. J. Comp. Neurol. 501, 498–508. doi: 10.1002/cne.21253
Buchman, A. S., Wilson, R. S., Boyle, P. A., Bienias, J. L., and Bennett, D. A. (2007). Change in motor function and risk of mortality in older persons. J. Am. Geriatr. Soc. 55, 11–19. doi: 10.1111/j.1532-5415.2006.01032.x
Cabalka, L. M., Hyman, B. T., Goodlett, C. R., Ritchie, T. C., and Van Hoesen, G. W. (1992). Alteration in the pattern of nerve terminal protein immunoreactivity in the perforant pathway in Alzheimer’s disease and in rats after entorhinal lesions. Neurobiol. Aging 13, 283–291. doi: 10.1016/0197-4580(92)90041-u
Calvanese, V., Lara, E., Kahn, A., and Fraga, M. F. (2009). The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 8, 268–276. doi: 10.1016/j.arr.2009.03.004
Canas, P. M., Duarte, J. M., Rodrigues, R. J., Köfalvi, A., and Cunha, R. A. (2009). Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiol. Aging 30, 1877–1884. doi: 10.1016/j.neurobiolaging.2008.01.003
Cardasis, C. A., and LaFontaine, D. M. (1987). Aging rat neuromuscular junctions: a morphometric study of cholinesterase-stained whole mounts and ultrastructure. Muscle Nerve 10, 200–213. doi: 10.1002/mus.880100303
Chang, E. H., Savage, M. J., Flood, D. G., Thomas, J. M., Levy, R. B., Mahadomrongkul, V., et al. (2006). AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc. Natl. Acad. Sci. U S A 103, 3410–3415. doi: 10.1073/pnas.0507313103
Chawla, S., Vanhoutte, P., Arnold, F. J., Huang, C. L., and Bading, H. (2003). Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 85, 151–159. doi: 10.1046/j.1471-4159.2003.01648.x
Chen, S., and Corces, V. G. (2001). The gypsy insulator of Drosophila affects chromatin structure in a directional manner. Genetics 159, 1649–1658.
Coggan, J. S., Grutzendler, J., Bishop, D. L., Cook, M. R., Gan, W., Heym, J., et al. (2004). Age-associated synapse elimination in mouse parasympathetic ganglia. J. Neurobiol. 60, 214–226. doi: 10.1002/neu.20022
Coleman, P. D., and Yao, P. J. (2003). Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 24, 1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001
Corfas, G., and Dudai, Y. (1991). Morphology of a sensory neuron in Drosophila is abnormal in memory mutants and changes during aging. Proc. Natl. Acad. Sci. U S A 88, 7252–7256. doi: 10.1073/pnas.88.16.7252
de Brabander, J. M., Kramers, R. J., and Uylings, H. B. (1998). Layer-specific dendritic regression of pyramidal cells with ageing in the human prefrontal cortex. Eur. J. Neurosci. 10, 1261–1269. doi: 10.1046/j.1460-9568.1998.00137.x
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Dubnau, J., Chiang, A. S., Grady, L., Barditch, J., Gossweiler, S., McNeil, J., et al. (2003). The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr. Biol. 13, 286–296. doi: 10.1016/s0960-9822(03)00064-2
Eckles-Smith, K., Clayton, D., Bickford, P., and Browning, M. D. (2000). Caloric restriction prevents age-related deficits in LTP and in NMDA receptor expression. Brain Res. Mol. Brain Res. 78, 154–162. doi: 10.1016/s0169-328x(00)00088-7
Erwin, J. A., Marchetto, M. C., and Gage, F. H. (2014). Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 15, 497–506. doi: 10.1038/nrn3730
Esteller, M., Corn, P. G., Baylin, S. B., and Herman, J. G. (2001). A gene hypermethylation profile of human cancer. Cancer Res. 61, 3225–3229.
Farkash, E. A., and Luning Prak, E. T. (2006). DNA damage and L1 retrotransposition. J. Biomed. Biotechnol. 2006:37285. doi: 10.1155/JBB/2006/37285
Feldman, M. L., and Dowd, C. (1975). Loss of dendritic spines in aging cerebral cortex. Anat. Embryol. (Berl) 148, 279–301. doi: 10.1007/bf00319848
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U S A 102, 10604–10609. doi: 10.1073/pnas.0500398102
Fu, Y., Sinha, M., Peterson, C. L., and Weng, Z. (2008). The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 4:e1000138. doi: 10.1371/journal.pgen.1000138
Geinisman, Y., deToledo-Morrell, L., Morrell, F., Persina, I. S., and Rossi, M. (1992). Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus 2, 437–444. doi: 10.1002/hipo.450020411
Gerasimova, T. I., Byrd, K., and Corces, V. G. (2000). A chromatin insulator determines the nuclear localization of DNA. Mol. Cell 6, 1025–1035. doi: 10.1016/s1097-2765(00)00101-5
Gerasimova, T. I., Gdula, D. A., Gerasimov, D. V., Simonova, O., and Corces, V. G. (1995). A Drosophila protein that imparts directionality on a chromatin insulator is an enhancer of position-effect variegation. Cell 82, 587–597. doi: 10.1016/0092-8674(95)90031-4
Gillingwater, T. H., and Wishart, T. M. (2013). Mechanisms underlying synaptic vulnerability and degeneration in neurodegenerative disease. Neuropathol. Appl. Neurobiol. 39, 320–334. doi: 10.1111/nan.12014
Gorczyca, M., Popova, E., Jia, X. X., and Budnik, V. (1999). The gene mod(mdg4) affects synapse specificity and structure in Drosophila. J. Neurobiol. 39, 447–460. doi: 10.1002/(sici)1097-4695(19990605)39:3<447::aid-neu10>3.0.co;2-q
Govindarajan, N., Rao, P., Burkhardt, S., Sananbenesi, F., Schlüter, O. M., Bradke, F., et al. (2013). Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 5, 52–63. doi: 10.1002/emmm.201201923
Grillo, F. W., Song, S., Teles-Grilo Ruivo, L. M., Huang, L., Gao, G., Knott, G. W., et al. (2013). Increased axonal bouton dynamics in the aging mouse cortex. Proc. Natl. Acad. Sci. U S A 110, E1514–E1523. doi: 10.1073/pnas.1218731110
Groth, A., Rocha, W., Verreault, A., and Almouzni, G. (2007). Chromatin challenges during DNA replication and repair. Cell 128, 721–733. doi: 10.1016/j.cell.2007.01.030
Guan, Z., Giustetto, M., Lomvardas, S., Kim, J. H., Miniaci, M. C., Schwartz, J. H., et al. (2002). Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111, 483–493. doi: 10.1016/s0092-8674(02)01074-7
Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H., Joseph, N., Gao, J., et al. (2009). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. doi: 10.1038/nature07925
Gutmann, E., Hanzlíková, V., and Vysokocil, F. (1971). Age changes in cross striated muscle of the rat. J. Physiol. 216, 331–343. doi: 10.1113/jphysiol.1971.sp009528
Han, Q., Lu, J., Duan, J., Su, D., Hou, X., Li, F., et al. (2008). Gcn5- and Elp3-induced histone H3 acetylation regulates hsp70 gene transcription in yeast. Biochem J. 409, 779–788. doi: 10.1042/bj20070578
Hebert, L. E., Beckett, L. A., Scherr, P. A., and Evans, D. A. (2001). Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord. 15, 169–173. doi: 10.1097/00002093-200110000-00002
Hori, N., Hirotsu, I., Davis, P. J., and Carpenter, D. O. (1992). Long-term potentiation is lost in aged rats but preserved by calorie restriction. Neuroreport 3, 1085–1088. doi: 10.1097/00001756-199212000-00013
Jang, Y. C., Lustgarten, M. S., Liu, Y., Muller, F. L., Bhattacharya, A., Liang, H., et al. (2010). Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 24, 1376–1390. doi: 10.1096/fj.09-146308
Jones, M. A., and Grotewiel, M. (2011). Drosophila as a model for age-related impairment in locomotor and other behaviors. Exp. Gerontol. 46, 320–325. doi: 10.1016/j.exger.2010.08.012
Kaern, M., Elston, T. C., Blake, W. J., and Collins, J. J. (2005). Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464. doi: 10.1038/nrg1615
Kaminsky, Z. A., Tang, T., Wang, S. C., Ptak, C., Oh, G. H., Wong, A. H., et al. (2009). DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 41, 240–245. doi: 10.1038/ng.286
Kobayashi, M., Michaut, L., Ino, A., Honjo, K., Nakajima, T., Maruyama, Y., et al. (2006). Differential microarray analysis of Drosophila mushroom body transcripts using chemical ablation. Proc. Natl. Acad. Sci. U S A 103, 14417–14422. doi: 10.1073/pnas.0606571103
Korzus, E., Rosenfeld, M. G., and Mayford, M. (2004). CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972. doi: 10.1016/j.neuron.2004.06.002
Landfield, P. W., McGaugh, J. L., and Lynch, G. (1978). Impaired synaptic potentiation processes in the hippocampus of aged, memory-deficient rats. Brain Res. 150, 85–101. doi: 10.1016/0006-8993(78)90655-8
Lee, H. K., Min, S. S., Gallagher, M., and Kirkwood, A. (2005). NMDA receptor-independent long-term depression correlates with successful aging in rats. Nat. Neurosci. 8, 1657–1659. doi: 10.1038/nn1586
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559. doi: 10.1074/jbc.m402229200
Levy, G., Schupf, N., Tang, M. X., Cote, L. J., Louis, E. D., Mejia, H., et al. (2002). Combined effect of age and severity on the risk of dementia in Parkinson’s disease. Ann. Neurol. 51, 722–729. doi: 10.1002/ana.10219
Li, Y., Lee, Y., and Thompson, W. J. (2011). Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J. Neurosci. 31, 14910–14919. doi: 10.1523/JNEUROSCI.3590-11.2011
Li, W., Prazak, L., Chatterjee, N., Grüninger, S., Krug, L., Theodorou, D., et al. (2013). Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci. 16, 529–531. doi: 10.1038/nn.3368
Lubin, F. D., Roth, T. L., and Sweatt, J. D. (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 28, 10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008
Mahoney, R. E., Rawson, J. M., and Eaton, B. A. (2014). An age-dependent change in the set point of synaptic homeostasis. J. Neurosci. 34, 2111–2119. doi: 10.1523/JNEUROSCI.3556-13.2014
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Masliah, E., Mallory, M., Hansen, L., DeTeresa, R., and Terry, R. D. (1993). Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 43, 192–197. doi: 10.1212/wnl.43.1_part_1.192
McGahon, B. M., Murray, C. A., Horrobin, D. F., and Lynch, M. A. (1999). Age-related changes in oxidative mechanisms and LTP are reversed by dietary manipulation. Neurobiol. Aging 20, 643–653. doi: 10.1016/s0197-4580(99)00027-5
Miśkiewicz, K., Jose, L. E., Bento-Abreu, A., Fislage, M., Taes, I., Kasprowicz, J., et al. (2011). ELP3 controls active zone morphology by acetylating the ELKS family member Bruchpilot. Neuron 72, 776–788. doi: 10.1016/j.neuron.2011.10.010
Miskiewicz, K., Jose, L. E., Yeshaw, W. M., Valadas, J. S., Swerts, J., Munck, S., et al. (2014). HDAC6 is a Bruchpilot deacetylase that facilitates neurotransmitter release. Cell Rep. 8, 94–102. doi: 10.1016/j.celrep.2014.05.051
Norris, C. M., Korol, D. L., and Foster, T. C. (1996). Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J. Neurosci. 16, 5382–5392.
Peleg, S., Sananbenesi, F., Zovoilis, A., Burkhardt, S., Bahari-Javan, S., Agis-Balboa, R. C., et al. (2010). Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756. doi: 10.1126/science.1186088
Penner, M. R., Roth, T. L., Barnes, C. A., and Sweatt, J. D. (2010). An epigenetic hypothesis of aging-related cognitive dysfunction. Front. Aging Neurosci. 2:9. doi: 10.3389/fnagi.2010.00009
Petralia, R. S., Mattson, M. P., and Yao, P. J. (2014). Communication breakdown: the impact of ageing on synapse structure. Ageing Res. Rev. 14, 31–42. doi: 10.1016/j.arr.2014.01.003
Polo, S. E., Roche, D., and Almouzni, G. (2006). New histone incorporation marks sites of UV repair in human cells. Cell 127, 481–493. doi: 10.1016/j.cell.2006.08.049
Richard, M. B., Taylor, S. R., and Greer, C. A. (2010). Age-induced disruption of selective olfactory bulb synaptic circuits. Proc. Natl. Acad. Sci. U S A 107, 15613–15618. doi: 10.1073/pnas.1007931107
Rosenzweig, E. S., and Barnes, C. A. (2003). Impact of aging on hippocampal function: plasticity, network dynamics and cognition. Prog. Neurobiol. 69, 143–179. doi: 10.1016/s0301-0082(02)00126-0
Roth, T. L., Lubin, F. D., Funk, A. J., and Sweatt, J. D. (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 65, 760–769. doi: 10.1016/j.biopsych.2008.11.028
Rountree, M. R., Bachman, K. E., and Baylin, S. B. (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277. doi: 10.1038/77023
Scheff, S. W., and Price, D. A. (2001). Alzheimer’s disease-related synapse loss in the cingulate cortex. J. Alzheimers Dis. 3, 495–505.
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Silva, A. J., Kogan, J. H., Frankland, P. W., and Kida, S. (1998). CREB and memory. Annu. Rev. Neurosci. 21, 127–148. doi: 10.1146/annurev.neuro.21.1.127
Simon, A. F., Liang, D. T., and Krantz, D. E. (2006). Differential decline in behavioral performance of Drosophila melanogaster with age. Mech. Ageing Dev. 127, 647–651. doi: 10.1016/j.mad.2006.02.006
Sui, L., Wang, Y., Ju, L. H., and Chen, M. (2012). Epigenetic regulation of reelin and brain-derived neurotrophic factor genes in long-term potentiation in rat medial prefrontal cortex. Neurobiol. Learn. Mem. 97, 425–440. doi: 10.1016/j.nlm.2012.03.007
Sweatt, J. D. (2013). The emerging field of neuroepigenetics. Neuron 80, 624–632. doi: 10.1016/j.neuron.2013.10.023
Tamura, T., Chiang, A. S., Ito, N., Liu, H. P., Horiuchi, J., Tully, T., et al. (2003). Aging specifically impairs amnesiac-dependent memory in Drosophila. Neuron 40, 1003–1011. doi: 10.1016/s0896-6273(03)00732-3
Toth, M. L., Melentijevic, I., Shah, L., Bhatia, A., Lu, K., Talwar, A., et al. (2012). Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 32, 8778–8790. doi: 10.1523/JNEUROSCI.1494-11.2012
Turrigiano, G. (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89–103. doi: 10.1146/annurev-neuro-060909-153238
Valdez, G., Tapia, J. C., Kang, H., Clemenson, G. D. Jr., Gage, F. H., Lichtman, J. W., et al. (2010). Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. U S A 107, 14863–14868. doi: 10.1073/pnas.1002220107
Valdez, G., Tapia, J. C., Lichtman, J. W., Fox, M. A., and Sanes, J. R. (2012). Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. doi: 10.1371/journal.pone.0034640
Van Meter, M., Kashyap, M., Rezazadeh, S., Geneva, A. J., Morello, T. D., Seluanov, A., et al. (2014). SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 5:5011. doi: 10.1038/ncomms6011
Wishart, T. M., Parson, S. H., and Gillingwater, T. H. (2006). Synaptic vulnerability in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 65, 733–739. doi: 10.1097/01.jnen.0000228202.35163.c4
Yamazaki, D., Horiuchi, J., Nakagami, Y., Nagano, S., Tamura, T., and Saitoe, M. (2007). The Drosophila DCO mutation suppresses age-related memory impairment without affecting lifespan. Nat. Neurosci. 10, 478–484. doi: 10.1038/nn1863
Yang, W. M., Inouye, C., Zeng, Y., Bearss, D., and Seto, E. (1996). Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. U S A 93, 12845–12850. doi: 10.1073/pnas.93.23.12845
Zeng, Y., Tan, M., Kohyama, J., Sneddon, M., Watson, J. B., Sun, Y. E., et al. (2011). Epigenetic enhancement of BDNF signaling rescues synaptic plasticity in aging. J. Neurosci. 31, 17800–17810. doi: 10.1523/JNEUROSCI.3878-11.2011
Zhao, X., Ueba, T., Christie, B. R., Barkho, B., McConnell, M. J., Nakashima, K., et al. (2003). Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. U S A 100, 6777–6782. doi: 10.1073/pnas.1131928100
Zhao, X. L., Wang, W. A., Tan, J. X., Huang, J. K., Zhang, X., Zhang, B. Z., et al. (2010). Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J. Neurosci. 30, 1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010
Keywords: aging, epigenetics, synapses, acetylation, neurotransmission
Citation: Azpurua J and Eaton BA (2015) Neuronal epigenetics and the aging synapse. Front. Cell. Neurosci. 9:208. doi: 10.3389/fncel.2015.00208
Received: 31 March 2015; Accepted: 13 May 2015;
Published online: 27 May 2015.
Edited by:
Yogesh P. Wairkar, University of Texas Medical Branch, USAReviewed by:
Daniel Marenda, Drexel University, USAChunlai Wu, Louisiana State University Health Sciences Center-Neuroscience, USA
Copyright © 2015 Azpurua and Eaton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin A. Eaton, Department of Physiology, University of Texas Health Science Center at San Antonio, 7703 Floyd Curl, MC 7756, San Antonio, TX 78229, USA,ZWF0b25iQHV0aHNjc2EuZWR1