 Takashi Shichita
Takashi Shichita Minako Ito1
Minako Ito1 Akihiko Yoshimura
Akihiko Yoshimura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 14 October 2014
Sec. Cellular Neuropathology
Volume 8 - 2014 | https://doi.org/10.3389/fncel.2014.00319
This article is part of the Research TopicMechanisms of neuroinflammation and inflammatory neurodegeneration in acute brain injuryView all 25 articles
Post-ischemic inflammation is important in ischemic stroke pathology. However, details of the inflammation process, its resolution after stroke and its effect on pathology and neural damage have not been clarified. Brain swelling, which is often fatal in ischemic stroke patients, occurs at an early stage of stroke due to endothelial cell injury and severe inflammation by infiltrated mononuclear cells including macrophages, neutrophils, and lymphocytes. At early stage of inflammation, macrophages are activated by molecules released from necrotic cells [danger-associated molecular patterns (DAMPs)], and inflammatory cytokines and mediators that increase ischemic brain damage by disruption of the blood–brain barrier are released. After post-ischemic inflammation, macrophages function as scavengers of necrotic cell and brain tissue debris. Such macrophages are also involved in tissue repair and neural cell regeneration by producing tropic factors. The mechanisms of inflammation resolution and conversion of inflammation to neuroprotection are largely unknown. In this review, we summarize information accumulated recently about DAMP-induced inflammation and the neuroprotective effects of inflammatory cells, and discuss next generation strategies to treat ischemic stroke.
Inflammation is implicated in almost all of central nervous system (CNS) diseases (Lo, 2010; Moskowitz et al., 2010; Iadecola and Anrather, 2011). Neurodegeneration, infection, trauma, and ischemia stimulate immune responses in the brain, although to varying degrees. The process in neuronal injury involves various intracellular mechanisms (abnormal metabolism and degeneration of protein, dysfunction of organelles, etc.), which cause the activation of microglia and the infiltration of circulating immune cells (Lo, 2010). Inflammation may not be always main process in the pathology of CNS diseases; nonetheless, the distinct characteristics of ischemic stroke are large amount of necrotic neuronal death and extreme infiltration of immune cells (Moskowitz et al., 2010; Iadecola and Anrather, 2011).
Severe inflammation causes cerebral swelling, which is often fatal in ischemic stroke patients. Broad necrotic lesion generates abundant inflammatory mediators and damage-associated molecular patterns (DAMPs), which enhance the chemotaxis of circulating immune cells and make them more efficient participants to promote inflammation (Moskowitz et al., 2010; Iadecola and Anrather, 2011). Cerebral inflammation exaggerates vascular dysfunction and induces further neuronal cell death (Dirnagl et al., 1999). Thus, post-ischemic inflammation is an essential process in the pathophysiology of ischemic stroke and is closely related to the prognosis after stroke (Dirnagl et al., 1999; Lo, 2010; Moskowitz et al., 2010; Iadecola and Anrather, 2011). In addition, inflammation is generally considered useful for the clearance of the large amount of debris caused by brain cell necrotic death (Moskowitz et al., 2010; Iadecola and Anrather, 2011). Inflammation, resolution of inflammation, and repair of neural damage are sequential pivotal events after stroke. To clarify the detailed mechanisms of each step of cerebral inflammation is indispensable to develop next generation therapies for ischemic stroke. The molecular basis of these steps is now being clarified by the recent accumulating evidences. We summarize these findings and discuss the principles of post-ischemic inflammation from beginning to end.
Brain ischemia induces various large metabolic changes in brain cells. Hypoxic stress, nutrients stress, and ER stress will cause cell death and trigger post-ischemic inflammation. Although receptors for pathogens such as Toll-like receptors (TLRs) are thought to be involved in early step of inflammation, brain is a sterile organ. Thus, endogenous molecules, i.e., DAMPs derived from injured brain cells, must trigger the inflammatory response in immune cells (Table 1). These DAMPs induce the activation of TLRs and other pattern recognition receptors [receptor for advanced glycation end products (RAGE) and c-type lectin receptors], which promote inflammatory mediator expression and tissue injury (Figure 1; Tang et al., 2007; Yanai et al., 2009; Suzuki et al., 2013). Recent scientific advances have suggested the existence of various types of DAMPs in ischemic brain.
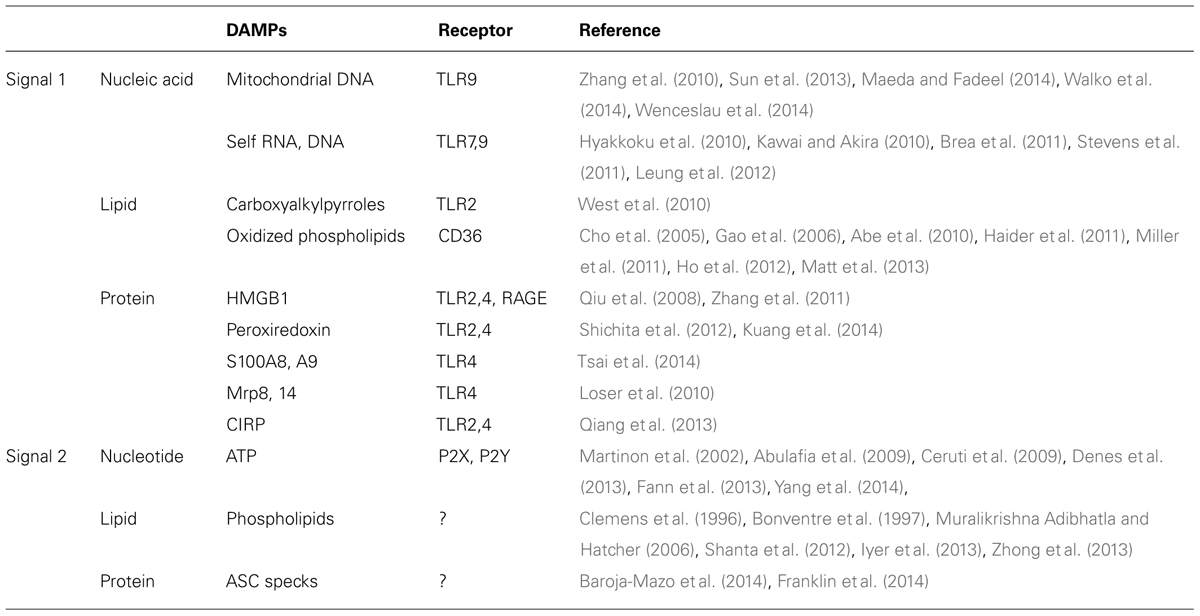
TABLE 1. List of inflammatory DAMPs.
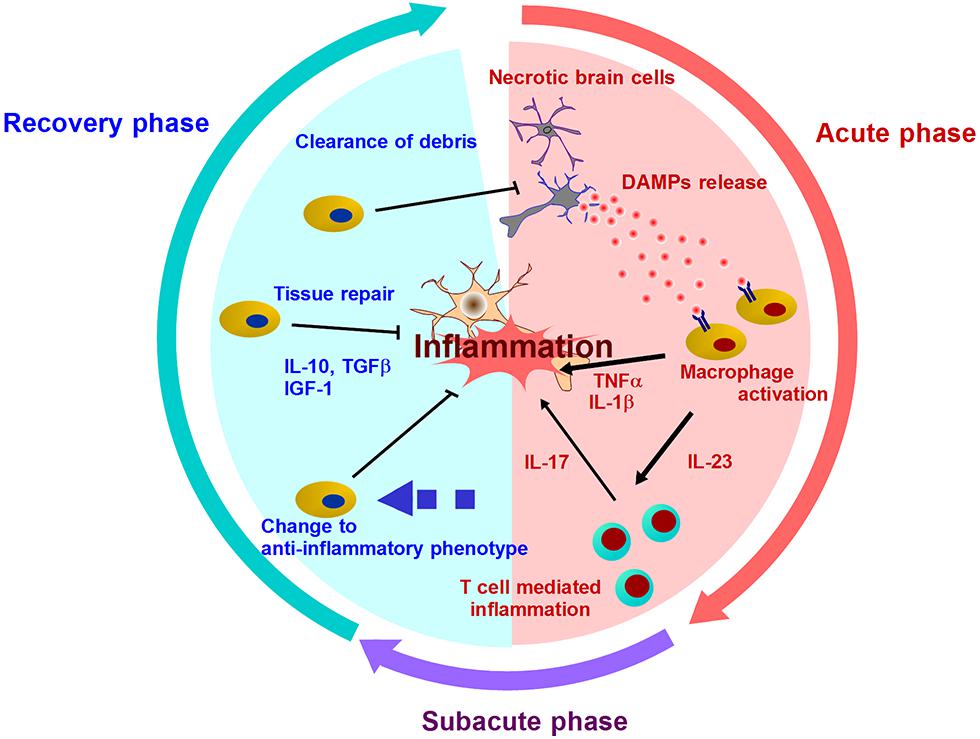
FIGURE 1. Mechanisms of post-ischemic inflammation. DAMPs are released into extracellular compartment and activate infiltrating immune cells by two ways: Signal 1 (via the activation of pattern recognition receptor) and Signal 2 (via the activation of inflammasome). Various inflammatory cytokines promote neuronal injury, and induce further inflammation mediated by T cells in subacute phase. After days and week after stroke onset, the resolution of post-ischemic inflammation is brought by the clearance of debris including DAMPs or inflammatory mediators, and the production of anti-inflammatory molecules or neurotrophic factors. In this recover phase, inflammatory immune cells turn into neuroprotective cells.
Various intracellular components are released into the extracellular space by necrotic brain cell death. Among these, nucleic acids and nucleotides are major DAMPs that have recently received much attention. Mitochondrial DNA released by cellular injury can be detected as DAMPs by immune cells, because mitochondria are considered to have a symbiotic origin that carries numerous characteristics resembling bacteria. Mitochondrial DNA is a sensor molecule of innate immunity by activating TLR9 and can be detected in cerebrospinal fluid after traumatic brain injury (Zhang et al., 2010; Walko et al., 2014). Vascular permeability is also increased by circulating mitochondrial DNA after injury (Sun et al., 2013; Wenceslau et al., 2014). Recently accumulated data indicates that mitochondrial DAMPs could be an important candidate for the trigger of post-ischemic inflammation, even if there is not yet any direct evidence (Maeda and Fadeel, 2014).
Self RNA and DNA (in complex with LL37 peptide) activate immune cells via TLR7 or TLR9 (Kawai and Akira, 2010). TLR7 is associated with the deterioration in ischemic stroke patients; in contrast, ischemic brain damage was not reduced in TLR9-deficient mice (Hyakkoku et al., 2010; Brea et al., 2011). Several reports demonstrate the implications of TLR7 and TLR9 in ischemic preconditioning. In these articles, the pretreatment using a TLR7 or TLR9 agonist reveals significant neuroprotection after cerebral ischemia by activating interferon regulatory factor 3/7- (IRF3/7)-induced type I interferon (IFN) signaling pathway (Stevens et al., 2011; Leung et al., 2012). Although the interaction between self nucleic acids and TLRs in the ischemic brain remains controversial, ischemic preconditioning via the TLR7 or TLR9 signaling pathway may represent a therapeutic strategy.
Purines (ATP and UTP) released from injured brain cells and their receptors, P2X and P2Y, function as alerting signals in CNS (Ceruti et al., 2009). Importantly, ATP also activates inflammasomes, which are large multimolecular complexes that control the activity of the proteolytic enzyme caspase-1 that cleaves pro-IL-1β to an active 17 kDa form (Martinon et al., 2002). The activation of the NLRP1 or NALP3 inflammasome has been recently reported to promote post-ischemic inflammation and neuronal death (Abulafia et al., 2009; Fann et al., 2013; Yang et al., 2014). Because IL-1β produced from both infiltrating immune cells and brain cells is important (Denes et al., 2013), it should be clarified how the inflammasome is activated in ischemic brain or hematopoietic cells. Inhibition of the inflammasome activation pathway may be a possible therapeutic strategy for ischemic stroke.
Various types of lipids are also important regulators of innate immunity. For example, oxidized low density lipoprotein (oxLDL) is a popular inflammatory mediator, which activates TLRs through binding with its receptor, CD36 (Stewart et al., 2010). Although the function of oxLDL in ischemic brain remains unclear, recent research has indicated that end products of lipid oxidation may be implicated in cerebral post-ischemic inflammation (Uchida, 2013). Carboxyalkylpyrroles, which are generated in inflammatory tissue, activate TLR2 and promote angiogenesis in ischemic organs (West et al., 2010). Oxidized phospholipids are also generated during cerebral inflammation and are considered to be DAMPs (Gao et al., 2006; Haider et al., 2011; Ho et al., 2012). Oxidized phospholipids are CD36 ligands that promote inflammation via TLR2 activation in ischemic brain (Cho et al., 2005; Abe et al., 2010). The recognition and endocytosis of oxidized lipids by pattern recognition receptors could regulate post-ischemic inflammation (Miller et al., 2011; Matt et al., 2013).
Phospholipids could also be inflammasome activators. Phospholipid metabolism is drastically altered by cerebral ischemia (Shanta et al., 2012). There are several reports showing the activation of phospholipase A2 (PLA2) in ischemic brain, which results in hydrolysis of membrane phospholipids (Clemens et al., 1996; Bonventre et al., 1997; Muralikrishna Adibhatla and Hatcher, 2006). Phospholipid hydrolysis and mitochondrial dysfunction induced by cerebral ischemia generate reactive oxygen species (ROS). Two recent studies have identified both ROS-dependent and ROS-independent pathways for inflammasome activation. The former is demonstrated by a charged phospholipid liposome that consecutively induces ROS-dependent calcium influx and NLRP3 inflammasome activation (Zhong et al., 2013). In the latter case, mitochondrial cardiolipin has been reported to directly bind to and activate the NLRP3 inflammasome (Iyer et al., 2013). Thus, the metabolism and modification of lipids during cerebral ischemia may be closely associated with the post-ischemic inflammation start signal.
High mobility group box 1 (HMGB1) and peroxiredoxin (Prx) family proteins are two major DAMPs in ischemic brain. There is a difference in the functional phase of these two proteins (Shichita et al., 2012). HMGB1, which is included in the nucleus of brain cells, is released extracellularly at the hyperacute phase (several hours after the stroke onset; Qiu et al., 2008). On the other hand, Prx family proteins function at the acute and subacute phases (12–72 h after the onset), especially in the penumbral area (Shichita et al., 2012). This is because Prx family protein expression is induced by an intracellular increase in ROS, which results from ischemic change. HMGB1 directly breaks down the blood–brain barrier and increases vascular permeability (Zhang et al., 2011). However, Prx directly induces the activation of infiltrating immune cells via TLR signaling. Ligustilide has been reported as a therapeutic candidate that suppresses cerebral post-ischemic inflammation by inhibiting the Prx/TLR4 signaling pathway (Kuang et al., 2014).
S100A8, S100A9, Mrp8, Mrp14, and cold-inducible RNA binding protein (CIRP) have also been reported to be protein DAMPs, although their relevance in post-ischemic inflammation has not yet been clarified (Loser et al., 2010; Qiang et al., 2013; Tsai et al., 2014). Inflammatory responses by these protein DAMPs occur through the activation of TLR2, TLR4, and RAGE. TLR2 and TLR4 signaling pathways are essential for sterile inflammation, including ischemic stroke (Chen et al., 2007). TLR2-blocking antibody is neuroprotective against ischemic brain injury (Ziegler et al., 2011). Similarly, resatorvid, which inhibits the TLR4 signaling pathway, attenuates ischemic brain injury and also suppresses Nox4-induced oxidative stress and neuronal apoptosis (Suzuki et al., 2012). It is also possible that DAMP-mediated TLR activation requires other adaptor molecules (Chun and Seong, 2010). CD14, a TLR4 co-receptor, may be implicated in post-ischemic inflammation (Reed-Geaghan et al., 2009). Heat shock protein gp96 is another candidate molecule that functions as an adaptor for both TLR2 and TLR4 (Yang et al., 2007).
It is not known whether protein DAMPs can activate inflammasomes. Recently, aggregated ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) has been reported to be released into the extracellular space after cell death and it activates inflammasomes in the surrounding immune cells (Baroja-Mazo et al., 2014; Franklin et al., 2014). Inflammasome activation, which occurs through ASC polymerization, results in caspase-1 activation and pyroptotic cell death. Extracellularly released ASC is internalized by surrounding macrophages and induces lysosomal damage and inflammasome activation. These mechanisms of inflammasome activation remain to be elucidated in ischemic brain injury.
Basic research may neglect the influence of aging and life habits by using healthy young rodents. These are important factors for the generation of DAMPs. Aging and continuous high serum glucose levels increase lipid peroxidation and AGEs in body systems (Basta et al., 2004; Cai et al., 2014). AGEs are proteins that are modified by sugar, through the Amadori and Maillard reaction. AGEs are found in chronic lesions; for example, the amyloid deposits that are surrounded by macrophages in patients with dialysis-related amyloidosis (Miyata et al., 1993). Thus, AGEs usually take a long time (more than a month) to generate; however, AGEs can be generated in a short period of time during inflammation (Weil, 2012). Glyoxal and glyceraldehyde induce AGE formation within 1 week (Takeuchi et al., 2001). In addition, the pivotal role of the RAGE in post-ischemic inflammation has been demonstrated (Muhammad et al., 2008). AGEs can be a potential DAMP, especially in aged human ischemic stroke patients.
Activated immune cells and brain cells are the major players after various DAMPs trigger post-ischemic inflammation. These cells produce inflammatory cytokines, chemokines, and other cytotoxic mediators, and this leads to prolonged inflammation and progressive brain edema during several days after the stroke onset (Figure 1). However, post-ischemic inflammation rarely lasts for a long period of time, and the most intense inflammatory phase takes place within 7 days after stroke onset (Dirnagl et al., 1999; Iadecola and Anrather, 2011). In this phase, the number of infiltrating immune cells decreases remarkably, and remaining immune cells in ischemic brain produce anti-inflammatory or neurotrophic factors (Shichita et al., 2009; Smirkin et al., 2010). For example, the detailed mechanisms about the infiltration and the change to anti-inflammatory phenotype of neutrophils have been recently clarified (Cuartero et al., 2013; Gorina et al., 2014). The period of cerebral post-ischemic inflammation always ends, and thus, the mechanisms of its resolution must exist in ischemic brain.
Three major points on the resolution of inflammation have been discussed in a recent publication (Buckley et al., 2012). These points are the production of anti-inflammatory mediators, the depletion of inflammatory mediators, and the induction of anti-inflammatory immune cells. After post-ischemic inflammation, infiltrating macrophages turn into anti-inflammatory macrophages, which produce neurotropic factors and clear necrotic debris. Inflammatory DAMPs will also be implicated in the induction of anti-inflammatory macrophages, although its mechanism still remains to be clarified. We introduce recent advantages of the relationship between post-ischemic inflammation and its resolution.
Many molecules have been reported to be neuroprotective factors. However, most of these molecules failed to improve neurological deficits in ischemic stroke patients, even if they are effective in animal stroke models. It has been suggested that neuroprotection alone is not sufficient to improve the prognosis of human ischemic stroke. Anti-inflammatory mechanisms in the entire brain and how these mechanisms are triggered needs to be determined. Because most brain cells are dead in the ischemic region several days after the stroke onset, infiltrating immune cells and reactive glial cells could be major players in the tissue repair. In practice, accelerating their effect is a potential next generation therapeutic strategy, and direct in vivo reprogramming of reactive glial cells into functional neurons after cerebral injury by retroviral transduction of the NeuroD1 gene was recently reported (Guo et al., 2014).
IL-10 and TGF-β are major anti-inflammatory molecules in various organ injuries. Both are produced by infiltrating immune cells and reactive glial cells after ischemic brain injury. Viral overexpression of IL-10 in ischemic brain is neuroprotective (Ooboshi et al., 2006). One recent report demonstrated the anti-inflammatory effect of TGF-β by inhibiting excessive neuroinflammation during the subacute phase of brain ischemia (Cekanaviciute et al., 2014). Although the anti-inflammatory effects of IL-10 and TGF-β have been pivotal, it remains to be clarified whether these effects last up to 1 week after the stroke onset (Pál et al., 2012). If the mechanisms for stimulating TGF-β and IL-10 expression can be controlled, this may become a strong therapeutic method.
Infiltrating immune cells and reactive glial cells produce various inflammatory mediators. TNF-α and IL-1β directly induce neuronal cell death. IL-23 and IL-1β activate T cell-mediated innate immunity and promote secondary ischemic damage during the subacute phase of ischemic brain injury (Shichita et al., 2009; Konoeda et al., 2010). The existence of these inflammatory mediators, including DAMPs, prolongs post-ischemic inflammation and will be a threat to neuronal survival and repair. However, inflammatory mediator degradation mechanisms remain mostly unknown. Inflammatory molecules may be degraded by some enzymes or consumed by receptor-mediated endocytosis. It is expected that nucleotides and lipids are rapidly metabolized in the ischemic brain, and transfer by the blood stream or cerebrospinal fluid (CSF) will help to scavenge inflammatory mediator. Further research should clarify the detailed mechanisms to scavenge inflammatory molecules produced in the ischemic brain, and antibody therapy will be a pivotal therapeutic method targeting this potential mechanism. TNF-α and IL-23 neutralizing antibody have been used clinically for rheumatoid arthritis and psoriasis patients, respectively. Natalizumab is the neutralizing antibody for integrin-α4, which is necessary for T cell infiltration into the inflammatory tissue, and has already been used for multiple sclerosis (Yednock et al., 1992). T cell depletion from ischemic brain has received attention as a potential next generation therapy for ischemic stroke (Meisel and Meisel, 2011; Wei et al., 2011). Thus, antibody therapies may be used to help treat ischemic stroke patients in the near future.
Activation of inflammasomes in immune cells induces the production of IL-1β in its mature form, and finally results in the rapid cell death of the same cells, which is called pyroptosis. Pyroptosis may be a possible mechanism for the clearance of inflammatory immune cells. This is supported by the fact that dying cells detected using the TdT-mediated dUTP nick end labeling (TUNEL) staining method include macrophages and glial cells in the ischemic brain (Mabuchi et al., 2000).
The repair process for damaged brain tissues and regeneration of neural cells takes place during resolution of inflammation. It is difficult to separate this process from the anti-inflammatory mechanism, because they may overlap each other. We will further discuss neuroprotective factors and repairing the damage to immune cells.
Various growth factors are also produced by immune cells and glial cells (Gudi et al., 2011). Among these, IGF-1 and FGF-2 are produced by infiltrating macrophages and microglia during the recovery phase of ischemic brain injury (which occurs 1 week after the stroke onset). IGF-1 and FGF-2 improve the neurological outcome by saving neuron and glial cells from cell death (Ness et al., 2004; Ikeda et al., 2005; Zhu et al., 2008; Hill et al., 2012; Lalancette-Hébert et al., 2012). IGF-1 also enhances repair after ischemic stroke by promoting neural regeneration, remyelination, and synaptogenesis (Lecker et al., 2007; Zhu et al., 2008, 2009; Kooijman et al., 2009). Further investigation should clarify the mechanisms of IGF-1 and FGF-2 induction in the ischemic brain. Recently, transfer of mesenchymal stem cells (MSCs) has been explored as a next generation therapy for ischemic stroke (Kalladka and Muir, 2014). MSCs produce various growth factors and promote neuronal survival and neurogenesis (Calió et al., 2014). By improving the transfer method, cell therapy may become a pivotal therapeutic strategy (Guo et al., 2013).
The neuroprotective effect of prostaglandin E2 (PGE2) and its receptor signaling pathway has received recent attention. PGE2 has an effect via four distinct G protein-coupled EP receptors (E-prostanoid: EP1, EP2, EP3, and EP4). The activation of EP2 signaling has a neuroprotective effect in ischemic brain injury, which was shown in the significant increase in infarct volume in mice lacking the EP2 receptor (McCullough et al., 2004). Similarly, signaling via the EP4 receptor, which is expressed in both neurons and ischemic endothelial cells, has neuroprotective effects against ischemic brain injury (Liang et al., 2011). The administration of an EP4 agonist reduces infarct volume and neurological deficits. In a neonatal hypoxic-ischemic encephalopathy model, the inhibition of EP1 receptor signaling or the activation of EP2, EP3, and EP4 receptor signaling reveals attenuation of the ischemic injury (Taniguchi et al., 2011). PGE2 and EP receptor signaling pathways have various functions, which are dependent on distinct pathology of cerebral diseases (Furuyashiki and Narumiya, 2011). Targeting specific EP receptors in ischemic brain may become a novel therapeutic method.
Similar to PGE2, some lipids have been reported to have anti-inflammatory effects and promote neuroregeneration (Serhan, 2014). Cerebral ischemia increases PLA2 activity, which results in the hydrolysis of phospholipids in the cellular membrane (Shanta et al., 2012). Although the PLA2 effect itself is cytotoxic because it disrupts the cellular membrane, PLA2 also generates docosahexaenoic acid (DHA) derivatives and lysophospholipids through phospholipid hydrolysis (Bonventre et al., 1997). Resolvin and Neuroprotectin have been investigated for their anti-inflammatory function in ischemic stroke (Marcheselli et al., 2003; Bazan, 2009). Lysophospholipids also increase in ischemic brain and promote neurite outgrowth (Ikeno et al., 2005; Spohr et al., 2011; Shanta et al., 2012). Regulating the effect of these lipids is expected for the resolution of post-ischemic inflammation.
Inflammatory DAMPs activate glial cells and infiltrating immune cells to promote post-ischemic inflammation. Paradoxically, this mechanism results in the infiltrating macrophage cell death and also induces anti-inflammatory and tissue-repairing immune cells.
Immune cell activation also induces anti-inflammatory cells. These cells have been called M2 macrophages, in contrast to the inflammatory M1 macrophages. Many researches have described the M2 macrophage markers; these markers include: arginase-1 (Arg1), chitinase3l3 (Ym), and Relmα (Fizz1). These markers are intracellular enzymes that are implicated in collagen synthesis and cell division; therefore, M2 enzymes are considered to promote tissue repair. Arg1 is the only marker that was reported to function as a neuroprotective enzyme (Estévez et al., 2006). However, these M2 markers may not be a good indicator for recovery after ischemic stroke. M2 markers are rapidly expressed in macrophages by TLR activation or other pattern recognition receptors, which also induce inflammatory cytokine expression (Hu et al., 2012). M2 markers appear in ischemic brain mostly during the same phase as the inflammatory mediators, the M1 markers, are expressed. In addition, the transfer of M2 marker positive-macrophages has not been reported to be sufficiently neuroprotective (Desestret et al., 2013).
During post-ischemic inflammation, some populations of macrophages and microglia become neuroprotective (Lalancette-Hébert et al., 2007). Galectin-1 has been suggested to be an inducer of anti-inflammatory macrophage/microglial cells (Starossom et al., 2012; Quintá et al., 2014). Galectin-1 is produced by astrocytes and has a neuroprotective effect against ischemic brain damage (Qu et al., 2011). Thus, the resolution of post-ischemic inflammation can be enhanced by the induction of a specific macrophage/microglial cell population. However, it is not clear whether the M2 markers truly reflect the neuroprotective function of macrophages and microglial cells. Suppressing inflammation alone is not enough to protect the brain from ischemic injury. IGF-1 and FGF-2 production seems to be a good index of repairing function (Lalancette-Hébert et al., 2012).
Further study is required to clarify whether sufficient clearance of inflammatory mediators (including DAMPs) begins neuronal regeneration after ischemic stroke. A recent study has suggested that there is a relationship between TLR activation and neuronal repair (Bohacek et al., 2012). It is possible that DAMPs triggers the secondary signals, which lead to resolution of post-ischemic inflammation, even if the primary signals via pattern recognition receptors promote ischemic damage. What is this mechanism? The role of immune cells, other than macrophages and microglia in part of the repair process, is not fully understood. This understanding may be critical for the establishment of next generation therapies for ischemic stroke.
Immunity and various physiological mechanisms are implicated in the triggering, persistence, and resolution of post-ischemic inflammation. Recent accumulating evidences clarify the complexity of these mechanisms to understand the entire mechanisms. They will show promising potential targets to develop therapies for ischemic stroke.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abe, T., Shimamura, M., Jackman, K., Kurinami, H., Anrather, J., Zhou, P.,et al. (2010). Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke 41, 898–904. doi: 10.1161/STROKEAHA.109.572552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abulafia, D. P., de Rivero Vaccari, J. P., Lozano, J. D., Lotocki, G., Keane, R. W., and Dietrich, W. D. (2009). Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 29, 534–544. doi: 10.1038/jcbfm.2008.143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baroja-Mazo, A., Martín-Sánchez, F., Gomez, A., Martínez, C. M., Amores-Iniesta, J., Compan, V.,et al. (2014). The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 15, 738–748. doi: 10.1038/ni.2919
Basta, G., Schmidt, A. M., and De Caterina, R. (2004). Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 63, 582–592. doi: 10.1016/j.cardiores.2004.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bazan, N. G. (2009). Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J. Lipid Res. 50, S400–S405. doi: 10.1194/jlr.R800068-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohacek, I., Cordeau, P., Lalancette-Hébert, M., Gorup, D., Weng, Y. C., Gajovic, S.,et al. (2012). Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J. Neuroinflammation 9, 191. doi: 10.1186/1742-2094-9-191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonventre, J. V., Huang, Z., Taheri, M. R., O’Leary, E., Li, E., Moskowitz, M. A.,et al. (1997). Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 390, 622–625. doi: 10.1038/37635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brea, D., Sobrino, T., Rodríguez-Yáñez, M., Ramos-Cabrer, P., Agulla, J., Rodríguez-González, R.,et al. (2011). Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin. Imunol. 139, 193–198. doi: 10.1016/j.clim.2011.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buckley, C. D., Gilroy, D. W., Serhan, C. N., Stockinger, B., and Tak, P. P. (2012). The resolution of inflammation. Nat. Rev. Immunol. 13, 59–66. doi: 10.1038/nri3362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cai, W., Uribarri, J., Zhu, L., Chen, X., Swamy, S., Zhao, Z.,et al. (2014). Oral glycotoxins are a modifiable cause of dementia and the metabolic syndrome in mice and humans. Proc. Natl. Acad. Sci. U.S.A. 111, 4940–4945. doi: 10.1073/pnas.1316013111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calió, M. L., Marinho, D. S., Ko, G. M., Ribeiro, R. R., Carbonel, A. F., Oyama, L. M.,et al. (2014). Transplantation of bone marrow mesenchymal stem cells decreases oxidative stress, apoptosis, and hippocampal damage in brain of a spontaneous stroke model. Free Radic. Biol. Med. 70, 141–154. doi: 10.1016/j.freeradbiomed.2014.01.024
Cekanaviciute, E., Fathali, N., Doyle, K. P., Williams, A. M., Han, J., and Buckwalter, M. S. (2014). Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 62, 1227–1240. doi: 10.1002/glia.22675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ceruti, S., Villa, G., Genovese, T., Mazzon, E., Longhi, R., Rosa, P.,et al. (2009). The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain 132(Pt 8), 2206–2218. doi: 10.1093/brain/awp147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, C. J., Kono, H., Golenbock, D., Reed, G., Akira, S., and Rock, K. L. (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856. doi: 10.1038/nm1603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, S., Park, E. M., Febbraio, M., Anrather, J., Park, L., Racchumi, G.,et al. (2005). The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J. Neurosci. 25, 2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chun, K. H., and Seong, S. Y. (2010). CD14 but not MD2 transmit signals from DAMP. Int. Immunopharmaol. 10, 98–106. doi: 10.1016/j.intimp.2009.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clemens, J. A., Stephenson, D. T., Smalstig, E. B., Roberts, E. F., Johnstone, E. M., Sharp, J. D.,et al. (1996). Reactive glia express cytosolic phospholipase A2 after transient global forebrain ischemia in the rat. Stroke 27, 527–535. doi: 10.1161/01.STR.27.3.527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cuartero, M. I., Ballesteros, I., Moraga, A., Nombela, F., Vivancos, J., Hamilton, J. A.,et al. (2013). N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARg agonist rosiglitazone. Stroke 44, 3498–3508. doi: 10.1161/STROKEAHA.113.002470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denes, A., Wilkinson, F., Bigger, B., Chu, M., Rothwell, N. J., and Allan, S. M. (2013). Central and haematopoietic interleukin-1 both contribute to ischaemic brain injury. Dis. Model. Mech. 6, 1043–1048. doi: 10.1242/dmm.011601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Desestret, V., Riou, A., Chauveau, F., Cho, T. H., Devillard, E., Marinescu, M.,et al. (2013). In vitro and in vivo models of cerebral ischemia show discrepancy in therapeutics effects of M2 macrophages. PLoS ONE. 8:e67063. doi: 10.1371/journal.pone.0067063
Dirnagl, U., Iadecola, C., and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397. doi: 10.1016/S0166-2236(99)01401-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Estévez, A. G., Sahawneh, M. A., Lange, P. S., Bae, N., Egea, M., and Ratan, R. R. (2006). Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neurons. J. Neurosci. 26, 8512–8516. doi: 10.1523/JNEUROSCI.0728-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fann, D. Y., Lee, S. Y., Manzanero, S., Tang, S. C., Gelderblom, M., Chunduri, P.,et al. (2013). Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 4, e790. doi: 10.1038/cddis.2013.326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Franklin, B. S., Bossaller, L., De Nardo, D., Ratter, J. M., Stutz, A., Engels, G.,et al. (2014). The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 15, 727–737. doi: 10.1038/ni.2913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Furuyashiki, T., and Narumiya, S. (2011). Stress responses: the contribution of prostaglandin E2 and its receptors. Nat. Rev. Endocrinol. 7, 163–175. doi: 10.1038/nrendo.2010.194
Gao, S., Zhang, R., Greenberg, M. E., Sun, M., Chen, X., Levison, B. S.,et al. (2006). Phospholipid hydroxyalkenals, a subset of recently discovered endogenous CD36 ligands, spontaneously generate novel furan-containing phospholipids lacking CD36 binding activity in vivo. J. Biol. Chem. 281, 31298–31308. doi: 10.1074/jbc.M604039200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorina, R., Lyck, R., Vestweber, D., and Engelhardt, B. (2014). β2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood–brain barrier. J. Immunol. 192, 324–337. doi: 10.4049/jimmunol.1300858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gudi, V., Škuljec, J., Yildiz, Ö., Frichert, K., Skripuletz, T., Moharregh-Khiabani, D.,et al. (2011). Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS ONE 6:e22623. doi: 10.1371/journal.pone.0022623
Guo, L., Ge, J., Wang, S., Zhou, Y., Wang, X., and Wu, Y. (2013). A novel method for efficient delivery of stem cells to the ischemic brain. Stem Cell Res. Ther. 4, 116. doi: 10.1186/scrt327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, Z., Zhang, L., Wu, Z., Chen, Y., Wang, F., and Chen, G. (2014). In Vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 14, 188–202. doi: 10.1016/j.stem.2013.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haider, L., Fischer, M. T., Frishcer, J. M., Bauer, J., Höftberger, R., Botond, G.,et al. (2011). Oxidative damage in multiple sclerosis lesions. Brain 134(Pt 7), 1914–1924. doi: 10.1093/brain/awr128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hill, J. J., Jin, K., Mao, X. O., Xie, L., and Greenberg, D. A. (2012). Intracerebral chondroitinase ABC and heparan sulfate proteoglycan glypican improve outcome from chronic stroke in rats. Proc. Natl. Acad. Sci. U.S.A. 109, 9155–9160. doi: 10.1073/pnas.1205697109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ho, P. P., Kanter, J. L., Johnson, A. M., Srinagesh, H. K., Chang, E. J., Purdy, T. M.,et al. (2012). Identification of naturally occurring fatty acids of the myelin sheath that resolve neuroinflammation. Sci. Transl. Med. 4, 137ra73. doi: 10.1126/scitranslmed.3003831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, X., Li, P., Guo, Y., Wang, H., Leak, R. K., Chen, S.,et al. (2012). Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. doi: 10.1161/STROKEAHA.112.659656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hyakkoku, K., Hamanaka, J., Tsuruma, K., Shimazawa, M., Tanaka, H., Uematsu, S.,et al. (2010). Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 171, 258–267. doi: 10.1016/j.neuroscience.2010.08.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., and Anrather, J. (2011). The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808. doi: 10.1038/nm.2399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ikeda, N., Nonoguchi, N., Zhao, M. Z., Watanabe, T., Kajimoto, Y., Furutama, D.,et al. (2005). Bone marrow stromal cells that enhanced fibroblast growth factor-2 secretion by herpes simplex virus vector improve neurological outcome after transient focal cerebral ischemia in rats. Stroke 36, 2725–2730. doi: 10.1161/01.STR.0000190006.88896.d3
Ikeno, Y., Konno, N., Cheon, S. H., Bolchi, A., Ottonello, S., Kitamoto, K.,et al. (2005). Secretory phospholipases A2 induce neurite outgrowth in PC12 cells through lysophosphatidylcholine generation and activation of G2A receptor. J. Biol. Chem. 280, 28044–28052. doi: 10.1074/jbc.M503343200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K.,et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalladka, D., and Muir, K. W. (2014). Brain repair: cell therapy in stroke. Stem Cells Cloning 7, 31–44.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konoeda, F., Shichita, T., Yoshida, H., Sugiyama, Y., Muto, G., Hasegawa, E.,et al. (2010). Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochem. Biophys. Res. Commun. 402, 500–506. doi: 10.1016/j.bbrc.2010.10.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kooijman, R., Sarre, S., Michotte, Y., and De Keyser, J. (2009). Insulin-like growth factor I: a potential neuroprotective compound for the treatment of acute ischemic stroke? Stroke 40, e83–e88. doi: 10.1161/STROKEAHA.108.528356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuang, X., Wang, L. F., Yu, L., Li, Y. J., Wang, Y. N., He, Q.,et al. (2014). Ligustilide ameliorates neuroinflammation and brain injury in focal cerebral ischemia/reperfusion rats: involvement of inhibition of TLR4/peroxiredoxin 6 signaling. Free Radic. Biol. Med. 71, 165–175. doi: 10.1016/j.freeradbiomed.2014.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lalancette-Hébert, M., Gowing, G., Simard, A., Weng, Y. C., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lalancette-Hébert, M., Swarup, V., Beaulieu, J. M., Bohacek, I., Abdelhamid, E., Weng, Y. C.,et al. (2012). Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 32, 10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012
Lecker, R. R., Soldner, F., Velasco, I., Gavin, D. K., Androutsellis-Theotokis, A., and McKay, R. D. (2007). Long-lasting regeneration after ischemia in the cerebral cortex. Stroke 38, 153–161. doi: 10.1161/01.STR.0000252156.65953.a9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leung, P. Y., Stevens, S. L., Packard, A. E., Lessov, N. S., Yang, T., Conrad, V. K.,et al. (2012). Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke 43, 1383–1389. doi: 10.1161/STROKEAHA.111.641522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, X., Lin, L., Woodling, N. S., Wang, Q., Anacker, C., Pan, T.,et al. (2011). Signaling via the prostaglandin E2 receptor EP4 exerts neuronal and vascular protection in a mouse model of cerebral ischemia. J. Clin. Invest. 121, 4362–4371. doi: 10.1172/JCI46279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lo, E. H. (2010). Degeneration and repair in central nervous system disease. Nat. Med. 16, 1205–1209. doi: 10.1038/nm.2226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loser, K., Vogl, T., Voskort, M., Leuken, A., Kupas, V., Nacken, W.,et al. (2010). The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat. Med. 16, 713–717. doi: 10.1038/nm.2150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mabuchi, T., Kitagawa, K., Ohtsuki, T., Kuwabara, K., Yagita, Y., Yanagihara, T.,et al. (2000). Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 31, 1735–1743. doi: 10.1161/01.STR.31.7.1735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maeda, A., and Fadeel, B. (2014). Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 5, e1312. doi: 10.1038/cddis.2014.277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcheselli, V. L., Hong, S., Lukiw, W. J., Tian, X. H., Gronert, K., Musto, A.,et al. (2003). Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278, 43807–43817. doi: 10.1074/jbc.M305841200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
Matt, U., Sharif, O., Martins, R., Furtner, T., Langeberg, L., Gawish, R.,et al. (2013). WAVE1 mediates suppression of phagocytosis by phospholipid-derived DAMPs. J. Clin. Invest. 123, 3014–3024. doi: 10.1172/JCI60681
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCullough, L., Wu, L., Haughey, N., Liang, X., Hand, T., Wang, Q.,et al. (2004). Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J. Neurosci. 24, 257–268. doi: 10.1523/JNEUROSCI.4485-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meisel, C., and Meisel, A. (2011). Suppressing immunosuppression after stroke. N. Engl. J. Med. 365, 2134–2136. doi: 10.1056/NEJMcibr1112454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, Y. I., Choi, S. H., Wiesner, P., Fang, L., Harkewicz, R., Hartvigsen, K.,et al. (2011). Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 108, 235–248. doi: 10.1161/CIRCRESAHA.110.223875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyata, T., Oda, O., Inagi, R., Iida, Y., Araki, N., Yamada, N.,et al. (1993). beta 2-Microglobulin modified with advanced glycation end products is a major component of hemodialysis-associated amyloidosis. J. Clin. Invest. 92, 1243–1252. doi: 10.1172/JCI116696
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muhammad, S., Barakat, W., Stoyanov, S., Murikinati, S., Yang, H., Tracey, K. J.,et al. (2008). The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 28, 12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muralikrishna Adibhatla, R., and Hatcher, J. F. (2006). Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 40, 376–387. doi: 10.1016/j.freeradbiomed.2005.08.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ness, J. K., Scaduto, R. C. Jr., and Wood, T. L. (2004). IGF-I prevents glutamate-mediated bax translocation and cytochrome C release in O4+ oligodendrocyte progenitors. Glia 46, 183–194. doi: 10.1002/glia.10360
Ooboshi, H., Ibayashi, S., Shichita, T., Kumai, Y., Takada, J., Ago, T.,et al. (2006). Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 111, 913–919. doi: 10.1161/01.CIR.0000155622.68580.DC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pál, G., Vincze, C., Renner, É., Wappler, E. A., Nagy, Z., Lovas, G., et al. (2012). Time course, distribution and cell types of induction of transforming growth factor betas following middle cerebral artery occlusion in the rat brain. PLoS ONE 7:e46731. doi: 10.1371/journal.pone.0046731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiang, X., Yang, W. L., Wu, R., Zhou, M., Jacob, A., Dong, W.,et al. (2013). Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 19, 1489–1495. doi: 10.1038/nm.3368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, J., Nishimura, M., Wang, Y., Sims, J. R., Qiu, S., Savitz, S. I.,et al. (2008). Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 28, 927–938. doi: 10.1038/sj.jcbfm.9600582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qu, W. S., Wang, Y. H., Ma, J. F., Tian, D. S., Zhang, Q., Pan, D. J.,et al. (2011). Galectin-1 attenuates astrogliosis-associated injuries and improves recovery of rats following focal cerebral ischemia. J. Neurochem. 116, 217–226. doi: 10.1111/j.1471-4159.2010.07095.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quintá, H. R., Pasquini, J. M., Rabinovich, G. A., and Pasquini, L. A. (2014). Glycan-dependent binding of galectin-1 to neuropilin-1 promotes axonal regeneration after spinal cord injury. Cell Death. Differ. 21, 941–955. doi: 10.1038/cdd.2014.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G., and Landreth, G. E. (2009). CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 29, 11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serhan, C. N. (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. doi: 10.1038/nature13479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shanta, S. R., Choi, C. S., Lee, J. H., Shin, C. Y., Kim, Y. J., Kim, K. H.,et al. (2012). Global changes in phospholipids identified by MALDI MS in rats with focal cerebral ischemia. J. Lipid Res. 53, 1823–1831. doi: 10.1194/jlr.M022558
Shichita, T., Hasegawa, E., Kimura, A., Morita, R., Sakaguchi, R., Takada, I.,et al. (2012). Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 18, 911–917. doi: 10.1038/nm.2749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shichita, T., Sugiyama, Y., Ooboshi, H., Sugimori, H., Nakagawa, R., Takada, I.,et al. (2009). Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat. Med. 215, 946–950. doi: 10.1038/nm.1999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smirkin, A., Matsumoto, H., Takahashi, H., Inoue, A., Tagawa, M., Ohue, S.,et al. (2010). Iba1+/NG2+ macrophage-like cells expressing a variety of neuroprotective factors ameliorate ischemic damage of the brain. J. Cereb. Blood Flow Metab. 30, 603–615. doi: 10.1038/jcbfm.2009.233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spohr, T. C., Dezonne, R. S., Rehen, S. K., and Gomes, F. C. (2011). Astrocytes treated by lysophosphatidic acid induce axonal outgrowth of cortical progenitors through extracellular matrix protein and epidermal growth factor signaling pathway. J. Neurochem. 119, 113–123. doi: 10.1111/j.1471-4159.2011.07421.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Starossom, S. C., Mascanfroni, I. D., Imitola, J., Cao, L., Raddassi, K., Hernandez, S. F.,et al. (2012). Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 37, 249–263. doi: 10.1016/j.immuni.2012.05.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevens, S. L., Leung, P. Y., Vartanian, K. B., Gopalan, B., Yang, T., Simon, R. P.,et al. (2011). Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J. Neurosci. 31, 8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A.,et al. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161. doi: 10.1038/ni.1836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, S., Sursal, T., Adibnia, Y., Zhao, C., Zheng, Y., Li, H.,et al. (2013). Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS ONE 8:e59989. doi: 10.1371/journal.pone.0059989
Suzuki, Y., Hattori, K., Hamanaka, J., Murase, T., Egashira, Y., Mishiro, K.,et al. (2012). Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci. Rep. 2, 896. doi: 10.1038/srep00896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, Y., Nakano, Y., Mishiro, K., Takagi, T., Tsuruma, K., Nakamura, M.,et al. (2013). Involvement of Mincle and Syk in the changes to innate immunity after ischemic stroke. Sci. Rep. 3, 3177. doi: 10.1038/srep03177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takeuchi, M., Yanase, Y., Matsuura, N., Yamagishi Si, S., Kameda, Y., Bucala, R.,et al. (2001). Immunological detection of a novel advanced glycation end-product. Mol. Med. 7, 783–791.
Tang, S. C., Arumugam, T. V., Xu, X., Cheng, A., Mughal, M. R., Jo, D. G.,et al. (2007). Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. U.S.A. 104, 13798–13803. doi: 10.1073/pnas.0702553104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taniguchi, H., Anacker, C., Suarez-Mier, G. B., Wang, Q., and Andreasson, K. (2011). Function of prostaglandin E2 EP receptors in the acute outcome of rodent hypoxic ischemic encephalopathy. Neurosci. Lett. 504, 185–190. doi: 10.1016/j.neulet.2011.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, S. Y., Segovia, J. A., Chang, T. H., Morris, I. R., Berton, M. T., Tessier, P. A.,et al. (2014). DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 10:e1003848. doi: 10.1371/journal.ppat.1003848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uchida, K. (2013). Redox-derived damage-associated molecular patterns: ligand function of lipid peroxidation adducts. Redox Biol. 1, 94–96. doi: 10.1016/j.redox.2012.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walko, T. D. 3rd, Bola, R. A., Hong, J. D., Au, A. K., Bell, M. J., Kochanek, P. M.,et al. (2014). Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock 41, 499–503. doi: 10.1097/SHK.0000000000000160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, Y., Yemisci, M., Kim, H. H., Yung, L. M., Shin, H. K., Hwang, S. K.,et al. (2011). Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 69, 119–129. doi: 10.1002/ana.22186
Weil, Z. M. (2012). Ischemia-induced hyperglycemia: consequences, neuroendocrine regulation, and a role for RAGE. Horm. Behav. 62, 280–285. doi: 10.1016/j.yhbeh.2012.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wenceslau, C. F., McCarthy, C. G., Szasz, T., Spitler, K., Goulopoulou, S., Webb, R. C.,et al. (2014). Mitochondrial damage-associated molecular patterns and vascular function. Eur. Heart J. 35, 1172–1177. doi: 10.1093/eurheartj/ehu047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
West, X. Z., Malinin, N. L., Merkulova, A. A., Tischenko, M., Kerr, B. A., Borden, E. C.,et al. (2010). Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976. doi: 10.1038/nature09421
Yanai, H., Ban, T., Wang, Z., Choi, M. K., Kawamura, T., Negishi, H.,et al. (2009). HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103. doi: 10.1038/nature08512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, F., Wang, Z., Wei, X., Han, H., Meng, X., Zhang, Y.,et al. (2014). NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 34, 660–667. doi: 10.1038/jcbfm.2013.242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Liu, B., Dai, J., Srivastava, P. K., Zammit, D. J., Lefrançois, L.,et al. (2007). Heat shock protein gp96 is a master chaperone for Toll-like receptors and is important in the innate function of macrophages. Immunity 26, 215–226. doi: 10.1016/j.immuni.2006.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yednock, T. A., Cannon, C., Fritz, L. C., Sanchez-Madrid, F., Steinman, L., and Karin, N. (1992). Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 356, 63–66. doi: 10.1038/356063a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Takahashi, H. K., Liu, K., Wake, H., Liu, R., Maruo, T.,et al. (2011). Anti-high mobility group box-1 monoclonal antibody protects the blood–brain barrier from ischemia-induced disruption in rats. Stroke 42, 1420–1428. doi: 10.1161/STROKEAHA.110.598334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W.,et al. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. doi: 10.1038/nature08780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhong, Z., Zhai, Y., Liang, S., Mori, Y., Han, R., Sutterwala, F. S.,et al. (2013). TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 4, 1611. doi: 10.1038/ncomms2608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, W., Fan, Y., Frenzel, T., Gasmi, M., Bartus, R. T., Young, W. L.,et al. (2008). Insulin growth factor-1 gene transfer enhances neurovascular remodeling and improves long-term stroke outcome in mice. Stroke 39, 1254–1261. doi: 10.1161/STROKEAHA.107.500801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, W., Fan, Y., Hao, Q., Shen, F., Hashimoto, T., Yang, G. Y.,et al. (2009). Postischemic IGF-1 gene transfer promotes neurovascular regeneration after experimental stroke. J. Cereb. Blood Flow Metab. 29, 1528–1537. doi: 10.1038/jcbfm.2009.75
Ziegler, G., Freyer, D., Harhausen, D., Khojasteh, U., Nietfeld, W., and Trendelenburg, G. (2011). Blocking TLR2 in vivo protects against accumulation of inflammatory cells and neuronal injury in experimental stroke. J. Cereb. Blood Flow Metab. 31, 757–766. doi: 10.1038/jcbfm.2010.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: damage-associated molecular patterns (DAMPs), inflammation, cytokines, inflammasome, resolution of inflammation
Citation: Shichita T, Ito M and Yoshimura A (2014) Post-ischemic inflammation regulates neural damage and protection. Front. Cell. Neurosci. 8:319. doi: 10.3389/fncel.2014.00319
Received: 28 July 2014; Accepted: 23 September 2014;
Published online: 14 October 2014.
Edited by:
Arthur Liesz, University Hospital Munich, GermanyReviewed by:
Mathias Gelderblom, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2014 Shichita, Ito and Yoshimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akihiko Yoshimura, Department of Microbiology and Immunology, School of Medicine, Keio University, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan e-mail:eW9zaGltdXJhQGE2LmtlaW8uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.