 Ujval Anilkumar
Ujval Anilkumar Jochen H. M. Prehn
Jochen H. M. Prehn
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Neurosci. , 30 September 2014
Sec. Cellular Neurophysiology
Volume 8 - 2014 | https://doi.org/10.3389/fncel.2014.00281
This article is part of the Research Topic Neuronal Self-Defense: Compensatory Mechanisms in Neurodegenerative Disorders View all 19 articles
Cells under stress activate cell survival and cell death signaling pathways. Cell death signaling frequently converges on mitochondria, a process that is controlled by the activities of pro- and anti-apoptotic B-cell lymphoma 2 (BCL-2) proteins. In this review, we summarize current knowledge on the control of neuronal survival, development and injury by anti-apoptotic BCL-2 family proteins. We discuss overlapping and differential effects of the individual family members BCL-2, BCL-extra long (BCL-XL), myeloid cell leukemia 1 (MCL-1), and BCL2-like 2 (BCL-W) in the control of survival during development and pathophysiological processes such as trophic factor withdrawal, ischemic injury, excitotoxicity, oxidative stress and energy stress. Finally we discuss recent evidence that several anti-apoptotic BCL-2 proteins influence mitochondrial bioenergetics and control neuronal Ca2+ homeostasis independent of their classical role in cell death signaling.
The BCL-2 gene was first identified in B-cell follicular lymphomas (Tsujimoto et al., 1985). The BCL-2 protein family act as key regulators in the intrinsic or “mitochondrial” apoptosis pathway. The different BCL-2 protein family either trigger or constrain apoptosis (Youle and Strasser, 2008). They are classified into three different classes depending on their structural and functional properties: (a) anti-apoptotic BCL-2 proteins including BCL-2 itself, BCL-XL (BCL-extra long), MCL-1 and BCL-W which contain four BH(1-4) domains (Czabotar et al., 2014); (b) pro pro-apoptotic proteins BAX (BCL-2-associated × protein), BAK (BCL-2-antagonist/killer-1) (Wei et al., 2001) and potentially BOK (BCL-2 related ovarian killer) that contain three conserved BH domains and interacts strongly with some anti-apoptotic proteins (Hsu et al., 1997); and (c) BH3-only proteins including BIM (BCL-2 interacting mediator), PUMA (p53 upregulated modulator of apoptosis), BID (BH3 interacting domain death agonist), BIK (BCL-2 interacting killer), BAD (BCL-2 associated death promoter), BMF (BCL-2 modifying factor, Hrk (Hara-kiri) and NOXA (Latin for “damage”) that have homology to the BCL-2 family proteins in only a single domain, the BH3 domain (Giam et al., 2008; Happo et al., 2012). While anti-apoptotic BCL-2 family proteins as well as BAX and BAK are often constitutively expressed in cells, BH3-Only proteins are typically transcriptionally or post-translationally activated in response to apoptotic stress signaling (Engel et al., 2011). Activation of the mitochondrial apoptosis pathway through pro-apoptotic BCL-2 proteins is able to activate different cell death pathways including apoptosis (Kilbride and Prehn, 2013). The key upstream event that leads to the activation of these different pathways is mitochondrial outer membrane permeabilization (MOMP). This process is triggered by the membrane insertion and oligomerization of the pro-apoptotic members BAX and BAK, with subsequent release of apoptosis-activating factors such as cytochrome c (cyt c) from the mitochondrial intermembrane space to the cytosol. Two models have been proposed for the activation of BAX and BAK during apoptosis: (a) the direct activation model where BAX and BAK activation occurs directly through conformational changes induced by BH3-only proteins (Letai et al., 2002) and (b) indirect activation model where cell death signals induce the binding of BH3-only pro-apoptotic initiators to anti-apoptotic BCL-2 proteins, facilitating the release and activation of BAX and BAK (Uren et al., 2007; Willis et al., 2007). Anti-apoptotic BCL-2 proteins are integral membrane proteins, possessing a C-terminal transmembrane domain that localizes these proteins to intracellular membranes, notably the mitochondrial outer membrane (MOM), but also the endoplasmic reticulum and the nuclear envelope (Cory and Adams, 2002; Chipuk et al., 2006; Brunelle and Letai, 2009; Tait and Green, 2010) and where they are also able to inhibit the process of MOMP by binding to pro-apoptotic BCL-2 proteins (Gonzalez-Garcia et al., 1994; Yang et al., 1995, 1997).
Neurons are highly specialized, excitable cells that communicate to other target neurons through the process of synaptic transmission. Glutamate is the principal excitatory neurotransmitter in the CNS. Glutamate release from presynaptic nerve terminals activates post-synaptic glutamate receptors including NMDA, AMPA, and Kainate receptors (Fykse and Fonnum, 1996). However, overactivation of glutamate receptors can be neurotoxic, a process termed excitotoxicity (Olney et al., 1972). Excitotoxic neuronal cell death is primarily mediated by excessive Ca2+ influx via NMDA receptors (Choi, 1985, 1987, 1988) and has been implicated in neurological disorders including stroke, traumatic brain injury, ischemia, Huntington's disease and amyotrophic lateral sclerosis (Dirnagl et al., 1999; Waggie et al., 1999; Mehta et al., 2013). Glutamate neurotoxicity but also other processes such as ischemia/reperfusion injury also invoke oxidative stress (Barnham et al., 2004).
Recent studies have shown that excitotoxic and oxidative stress-induced neuronal injury involves BCL-2 family proteins. Specifically, injury conditions that produce a more delayed neuronal injury are often triggered by the transcriptional and post-translational activation of BH-3 only proteins such as BIM, PUMA, and BID (Konig et al., 2007; Steckley et al., 2007; Concannon et al., 2010). Similarly, the pro-apoptotic BAX protein has been shown to be implicated in excitotoxicity, oxidative stress and trophic factor deprivation-induced neuronal apoptosis (Deckwerth et al., 1996; D'Orsi et al., 2012). In this review we focus on recent advancements in describing the role of individual anti-apoptotic BCL-2 family proteins during neuronal development, injury and neurodegeneration.
BCL-2 is widely expressed in the developing brain including, neuroepithelial cells of the ventricular zones as well as the post-mitotic cells of the cortical plate, cerebellum, hippocampus and spinal cord (Merry et al., 1994). BCL-2 knockout mice show normal embryonic development but present with lymphoid apoptosis, neuronal and intestinal lesions and terminal kidney disease (Veis et al., 1993). High expression of BCL-2 mRNA was observed in the developing nervous system and reduced significantly in the post-natal brain (Abe-Dohmae et al., 1993; Merry et al., 1994). Interestingly, high level of BCL-2 expression was maintained in sensory and sympathetic adult neurons (Merry et al., 1994). BCL-2 acts as an important regulator of cell death in developing sympathetic neurons after neuronal growth factor deprivation, whereas BCL-2 is not involved in the survival of mature sympathetic neurons (Greenlund et al., 1995). Overexpression of BCL-2 inhibited BAX-mediated cytochrome-c release, caspase activation and cell death in nerve growth factor-deprived sympathetic neurons (Putcha et al., 1999). These results taken together indicate that BCL-2 plays an important role specifically during development of the nervous system.
Functional studies in primary neuron cultures and animal models indicated that BCL-2 overexpression protected hippocampal neurons against glutamate-mediated excitotoxicity, and significantly reduced lesion size in the hippocampus resulting from NMDA induced excitotoxic damage (Wong et al., 2005). Overexpression of BCL-2 blocked translocation of apoptosis inducing factor (AIF) from mitochondria to the nucleus, resulting in improved cortical neuron survival following focal cerebral ischemia (Zhao et al., 2004). BCL-2 deficient mice show enhanced oxidative stress and alterations in antioxidants in the brain (Hochman et al., 1998) and up-regulation of BCL-2 may aid DNA repair following oxidative stress (Deng et al., 1999). Interestingly, BCL-2 expression also inhibited apoptosis of newborn neurons following MCAO in adult rat brains (Zhang et al., 2006). Transgenic mice overexpressing BCL-2 in neurons resulted in hypertrophy of the nervous system caused by reduced naturally occurring cell death but also showed a 50% reduction in brain infarct volume compared to wild type mice after permanent ischemia induced by MCAO(Martinou et al., 1994). Furthermore, transplantation of embryonic stem cells overexpressing BCL-2 into the post-infarct brain cavity of adult rats after MCAO resulted in neuronal differentiation and improvements in functional recovery and behavioral testing (Wei et al., 2005). Of note, alterations in endoplasmic reticulum Ca2+ homeostasis have been shown to induce apoptosis in neurons (Mattson et al., 2000). BCL-2 also modulates ER Ca2+ content by decreasing ER Ca2+ uptake (Ferrari et al., 2002; Rudner et al., 2002) which supports axon regeneration and neurite outgrowth during energy stress and mobilizes intracellular calcium signaling (Jiao et al., 2005). These results suggest that BCL-2 may represent an interesting target in stroke recovery therapy.
The BCL-X gene can be alternatively spliced to produce two protein isoforms, BCL-XL and BCL-XS (Gonzalez-Garcia et al., 1994). BCL-XL acts as an anti-apoptotic protein whereas BCL-Xs exhibits pro-apoptotic properties. BCL-XL is found in post-mitotic cells in the adult brain whereas BCL-XS expression is predominantly expressed in developing cells with a high turnover rate such as lymphocytes (Boise et al., 1993). BCL-XL is highly expressed in developing neurons as they migrate away from ventricular zone, and remains up-regulated in post-mitotic neurons in the adult brain (Motoyama et al., 1995; Roth et al., 2000). BCL-XL shows close homology to BCL-2 (Gonzalez-Garcia et al., 1994). Deletion of BCL-X induces massive apoptotic cell death in developing neurons throughout the nervous system and results in lethality at embryonic day 13 (Motoyama et al., 1995; Akhtar et al., 2004). BCL-XL also protects cultured sympathetic neurons against nerve growth factor withdrawal (Gonzalez-Garcia et al., 1995). These data suggest that BCL-XL plays important roles during development of the nervous system; in addition BCL- XL is also expressed at high levels in the adult nervous system.
Overexpression of BCL-XL protected neurons in the hippocampus and cortex against hypoxic-ischemia (Parsadanian et al., 1998). Systemic delivery of BCL-XL fusion protein inhibited caspase-3 and -9 activities and also prevented translocation of AIF into the nucleus following hypoxic-ischemic brain injury (Yin et al., 2006). Interestingly, ischemic preconditioning blocked the assembly of BAD with BCL-XL, cleavage of BCL-XL to a pro-apoptotic form, and release of pro-apoptotic factors from mitochondria (Miyawaki et al., 2008). Overexpression of BCL-XL also protected primary rat septal neurons against oxygen glucose deprivation and hypoglycaemic stress (Panickar et al., 2005). Furthermore, decreased expression of BCL-XL has been implicated in spinal cord injury induced neuronal cell death. This was attenuated by exogenous administration of a BCL-XL fusion protein into the spinal cord (Nesic-Taylor et al., 2005). In addition, transplantation of neural stem cells overexpressing BCL-XL enhanced graft survival by supplying tropic factors essential for survival, and improved locomotor recovery in rats following spinal cord injury (Lee et al., 2009).
BCL-XL is also involved in non-apoptotic processes such as synapse formation. Overexpression of BCL-XL in hippocampal neurons increased synapse numbers and localization of mitochondria to synapses, a process that was modulated through the mitochondrial fission protein, dynamin related protein 1 (Li et al., 2008). Interestingly, BCL-XL demonstrated a dual role in synaptic transmission under hypoxia (Hickman et al., 2008). Inhibition of BCL-XL resulted in reduced recovery of synaptic responses under hypoxia, but exerted neuroprotective effect (Hickman et al., 2008). Recently, it has been demonstrated that BCL-XL is important in maintaining mitochondrial fission, fusion and biomass (Berman et al., 2009), and directly interacts with ATP synthase to stabilize the mitochondrial membrane potential (Chen et al., 2011). BCL-XL has also been demonstrated to influence Ca2+ signaling in astrocytes induced by the activation of inositol 1,4,5-triphosphate (IP3)-generating metabotropic type 5 glutamate receptors (mGluR5) during the process of motoneuron degeneration. Administration of the BH4 domain of BCL-X(L) fused to the protein transduction domain of the HIV-1 TAT protein was sufficient to restore Ca2+ homeostasis in astrocytes overexpressing the ALS-associated SOD1 (G93A) mutation, and chronic treatment of SOD1(G93A) transgenic mice with the TAT-BH4 peptide delayed the onset of the disease and improved motor function and lifespan (Martorana et al., 2012).
BCL-w also known as BCL2-like 2 (Bcl2l2) is a highly conserved gene located on the mouse chromosome 14 and the human chromosome 14 at band q11. BCL-w is highly expressed particularly in the brain, colon and testes and is also associated with intracellular membranes (O'Reilly et al., 2001). Although BCL-w is widely expressed, mice deficient in BCL-w failed to show large abnormalities with the exception of increased apoptosis in sperm cells during spermatogenesis, resulting in a sterile male phenotype (Print et al., 1998). The level of BCL-w has been shown to increase during neuronal development and BCL-w has been localized to specific regions of the mature brain where it may play a crucial role in maintaining their neuronal survival (Hamner et al., 1999). In this context, BCL-w overexpression increased neuronal survival in NGF-dependent trigerminal neurons and BDNF- dependent nodose neurons over the period of development in response to neurotrophin withdrawal (Middleton et al., 2001).
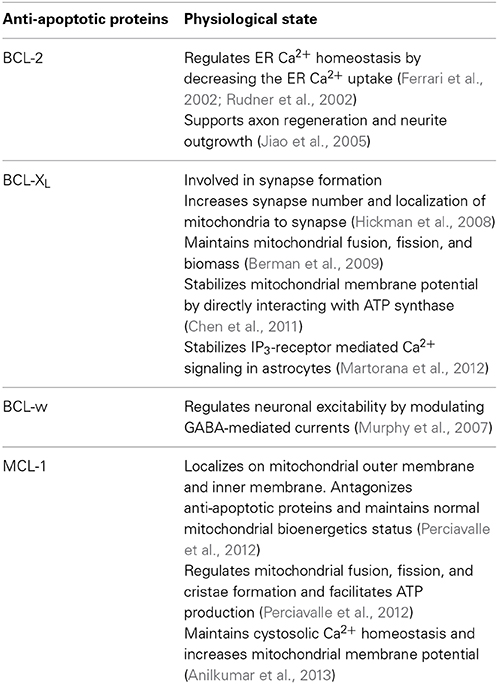
Table 1. Non-cell death related functions of anti-apoptotic proteins.
Functional studies revealed that BCL-w interacts with the pro-apoptotic protein BAD and blocks neuronal death induced by growth factor deprivation in sympathetic neurons (Hamner et al., 2001). Interestingly, BCL-w expression was found to be increased in mouse neurons up to 72 h after transient middle cerebral artery occlusion (MCAO). Moreover, BCL-w co-localized with mitochondria in non-fragmented neurons and protected neurons against Ca2+ mediated brain injury by inhibiting cytochrome-c release and maintaining mitochondrial membrane potential following MCAO (Yan et al., 2000). Mouse deficient in BCL-w showed an increased neuronal loss and nuclear fragmentation in the hippocampus after status epilepticus with a neurophysiological phenotype leading to earlier onset of seizure, and was suggested to influence neuronal excitability (Murphy et al., 2007). This effect may be regulated through an effect of BCL-w on GABA-mediated currents, a disruption of which may lead to seizure induction (Murphy et al., 2007). Furthermore, in an experimental model of Alzheimer's disease, BCL-w blocked the mitochondrial release of Smac and inhibited neuronal apoptosis induced by β-amyloid (Yao et al., 2005). Using a rat model of transient MCAO and oxygen glucose deprivation in neurons, it was demonstrated that BCL-w plays an important role in neuroprotection following ischemic injury and is directly regulated by microRNA-29b (Shi et al., 2012). There results taken together suggest that BCL-w plays an important role in various neurological conditions to protect neurons and therefore also presents an attractive target to development of therapeutic agents.
The human MCL-1 gene is located on chromosome 1q21, and MCL-1 proteins were originally isolated from myeloid leukemia cells (Kozopas et al., 1993). MCL-1 is prominently expressed in neuroendocrine cells, sympathetic neurons, cardiac and skeletal muscles (Krajewski et al., 1995). Importantly, diffuse expression and rapid induction of transcription of MCL-1 is found in neurons (Mori et al., 2004). MCL-1 germline deletion in mice resulted in peri-implantation lethality at embryonic day 3.5, showing the most severe phenotype amongst the anti-apoptotic BCL-2 family members (Rinkenberger et al., 2000). MCL-1 is associated with membranes through its C-terminal hydrophobic tail and has a predominant mitochondrial localization (Yang et al., 1995). Recently, it has been demonstrated that MCL-1 is spliced into two variants and resides in two distinct mitochondrial regions: outer membrane (MCL-1OM) and matrix (MCL-1MATRIX), and has been proposed to exert differential effects depending on its location: MCL-1OM antagonizes apoptosis, whereas MCL-1MATRIX facilitates ATP production, membrane potential, respiration, cristae ultrastructure and mitochondrial fusion (Perciavalle et al., 2012).
MCL-1 is essential for neuronal development. Conditional deletion of MCL-1 in mice induced apoptosis of neuronal progenitors and newly committed neurons as they commence their migration away from the ventricular zone (Arbour et al., 2008). In addition, conditional deletion of MCL-1 in vitro in neuronal precursor cells showed a two-fold increase in apoptosis (Malone et al., 2012). These results suggested that MCL-1 is crucial for survival of neuronal precursor cells. Recently, it has been demonstrated that MCL-1 can act as a switch between autophagy and apoptosis in a developmentally regulated manner under energetic stress conditions (Germain et al., 2011). Being an anti-apoptotic BCL-2 family member, MCL-1 inhibited apoptosis by binding and sequestering the pro-apoptotic BCL-2 family member BAK (Willis et al., 2005) and blocking the activation and translocation of BAX (Chen et al., 2007). Interestingly, MCL-1 protected cortical neurons against NMDA-mediated excitotoxicity, but also increased mitochondrial bioenergetics of cortical neurons and normalized neuronal Ca2+ homeostasis during NMDA excitation (Anilkumar et al., 2013). Such dual effects of MCL-1 may make it an attractive target for the treatment of neurodegenerative disorders such as Alzheimer's and Parkinson's disease, in which altered bioenergetics and increased neuronal loss are so prominent. MCL-1 heterozygous mice also demonstrated increased neuronal sensitivitiy against pilocarpine-induced status epilepticus (Mori et al., 2004). Furthermore, sustained expression of MCL-1 protected and loss of MCL-1 increased DNA damage- induced neuron death (Arbour et al., 2008). A recent study has also shown that apoptosis induced by serum and KCl deprivation in cerebellar granule neuron was mediated by proteasomal degradation of MCL-1, and that stabilization of MCL-1 by blocking its ubiquitination and degradation was protective (Magiera et al., 2013). In addition, neurons depleted of Parkin, mutated gene in juvenile onset and familial forms of Parkinson's disease, became acutely sensitive to oxidative stress, and this was attributed to decreased MCL-1 levels (Ekholm-Reed et al., 2013). Although the role of MCL-1 in neurons and neurodegenrative disorders warrants further investigation, emerging evidence suggests that it may represent the most promising therapeutic target of all BCL-2 family proteins studied so far.
BCL-2 family proteins have been firmly established to play a significant role in initiating or inhibiting apoptosis during neuronal development and injury. Emerging evidence suggests that some of the anti-apoptotic protein family members are involved in maintaining both mitochondrial bioenergetics and neuronal survival, in particular BCL-XL and MCL-1. BCL-2, BCL-XL and MCL-1 modulation of Ca2+ signaling during acute neuronal injury may also play a vital role in neuroprotection. Hence manipulating the pro-survival BCL-2 family members may be beneficial in developing future therapies for neurological and neurodegenerative disorders. In addition, exploiting neural stem cells overexpressing anti-apoptotic BCL-2 family proteins may provide an attractive and powerful tool for post-neuronal injury therapies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by Science Foundation Ireland (13/IA/1881 and 08/IN1/B1949) and the European Union FP7 Marie Curie IAPP program (OXY-SENSE; Contract 230641) to Jochen H. M. Prehn.
Abe-Dohmae, S., Harada, N., Yamada, K., and Tanaka, R. (1993). Bcl-2 gene is highly expressed during neurogenesis in the central nervous system. Biochem. Biophys. Res. Commun. 191, 915–921. doi: 10.1006/bbrc.1993.1304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Akhtar, R. S., Ness, J. M., and Roth, K. A. (2004). Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim. Biophys. Acta 1644, 189–203. doi: 10.1016/j.bbamcr.2003.10.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anilkumar, U., Weisova, P., Dussmann, H., Concannon, C. G., Konig, H., and Prehn, J. H. (2013). AMP-activated protein kinase (AMPK)-induced preconditioning in primary cortical neurons involves activation of MCL-1. J. Neurochem. 124, 721–734. doi: 10.1111/jnc.12108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arbour, N., Vanderluit, J. L., Le Grand, J. N., Jahani-Asl, A., Ruzhynsky, V. A., Cheung, E. C., et al. (2008). Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci. 28, 6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barnham, K. J., Masters, C. L., and Bush, A. I. (2004). Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214. doi: 10.1038/nrd1330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berman, S. B., Chen, Y. B., Qi, B., McCaffery, J. M., Rucker, E. B. 3rd. Goebbels, S., et al. (2009). Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 184, 707–719. doi: 10.1083/jcb.200809060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boise, L. H., Gonzalez-Garcia, M., Postema, C. E., Ding, L., Lindsten, T., Turka, L. A., et al. (1993). bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608. doi: 10.1016/0092-8674(93)90508-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brunelle, J. K., and Letai, A. (2009). Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 122(Pt 4), 437–441. doi: 10.1242/jcs.031682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S., Dai, Y., Harada, H., Dent, P., and Grant, S. (2007). Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 67, 782–791. doi: 10.1158/0008-5472.CAN-06-3964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y. B., Aon, M. A., Hsu, Y. T., Soane, L., Teng, X., McCaffery, J. M., et al. (2011). Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 195, 263–276. doi: 10.1083/jcb.201108059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chipuk, J. E., Bouchier-Hayes, L., and Green, D. R. (2006). Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 13, 1396–1402. doi: 10.1038/sj.cdd.4401963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, D. W. (1985). Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 58, 293–297. doi: 10.1016/0304-3940(85)90069-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, D. W. (1988). Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 11, 465–469. doi: 10.1016/0166-2236(88)90200-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Concannon, C. G., Tuffy, L. P., Weisova, P., Bonner, H. P., Davila, D., Bonner, C., et al. (2010). AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J. Cell Biol. 189, 83–94. doi: 10.1083/jcb.200909166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cory, S., and Adams, J. M. (2002). The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2, 647–656. doi: 10.1038/nrc883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Czabotar, P. E., Lessene, G., Strasser, A., and Adams, J. M. (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63. doi: 10.1038/nrm3722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deckwerth, T. L., Elliott, J. L., Knudson, C. M., Johnson, E. M. Jr. Snider, W. D., and Korsmeyer, S. J. (1996). BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17, 401–411. doi: 10.1016/S0896-6273(00)80173-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, G., Su, J. H., Ivins, K. L., van Houten, B., and Cotman, C. W. (1999). Bcl-2 facilitates recovery from DNA damage after oxidative stress. Exp. Neurol. 159, 309–318. doi: 10.1006/exnr.1999.7145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dirnagl, U., Iadecola, C., and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397. doi: 10.1016/S0166-2236(99)01401-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Orsi, B., Bonner, H., Tuffy, L. P., Dussmann, H., Woods, I., Courtney, M. J., et al. (2012). Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J. Neurosci. 32, 1847–1858. doi: 10.1523/JNEUROSCI.2345-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ekholm-Reed, S., Goldberg, M. S., Schlossmacher, M. G., and Reed, S. I. (2013). Parkin-dependent degradation of the F-box protein Fbw7beta promotes neuronal survival in response to oxidative stress by stabilizing Mcl-1. Mol. Cell. Biol. 33, 3627–3643. doi: 10.1128/MCB.00535-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Engel, T., Plesnila, N., Prehn, J. H., and Henshall, D. C. (2011). In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J. Cereb. Blood Flow Metab. 31, 1196–1210. doi: 10.1038/jcbfm.2011.26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrari, D., Pinton, P., Szabadkai, G., Chami, M., Campanella, M., Pozzan, T., et al. (2002). Endoplasmic reticulum, Bcl-2 and Ca2+ handling in apoptosis. Cell Calcium 32, 413–420. doi: 10.1016/S0143416002002014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fykse, E. M., and Fonnum, F. (1996). Amino acid neurotransmission: dynamics of vesicular uptake. Neurochem. Res. 21, 1053–1060. doi: 10.1007/BF02532415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Germain, M., Nguyen, A. P., Le Grand, J. N., Arbour, N., Vanderluit, J. L., Park, D. S., et al. (2011). MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 30, 395–407. doi: 10.1038/emboj.2010.327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Giam, M., Huang, D. C., and Bouillet, P. (2008). BH3-only proteins and their roles in programmed cell death. Oncogene 27(Suppl. 1), S128–S136. doi: 10.1038/onc.2009.50
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Garcia, M., Garcia, I., Ding, L., O'Shea, S., Boise, L. H., Thompson, C. B., et al. (1995). bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 92, 4304–4308.
Gonzalez-Garcia, M., Perez-Ballestero, R., Ding, L., Duan, L., Boise, L. H., Thompson, C. B., et al. (1994). bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development 120, 3033–3042.
Greenlund, L. J., Korsmeyer, S. J., and Johnson, E. M. Jr. (1995). Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron 15, 649–661. doi: 10.1016/0896-6273(95)90153-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamner, S., Arumae, U., Li-Ying, Y., Sun, Y. F., Saarma, M., and Lindholm, D. (2001). Functional characterization of two splice variants of rat bad and their interaction with Bcl-w in sympathetic neurons. Mol. Cell. Neurosci. 17, 97–106. doi: 10.1006/mcne.2000.0905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamner, S., Skoglosa, Y., and Lindholm, D. (1999). Differential expression of bcl-w and bcl-x messenger RNA in the developing and adult rat nervous system. Neuroscience 91, 673–684. doi: 10.1016/S0306-4522(98)00642-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Happo, L., Strasser, A., and Cory, S. (2012). BH3-only proteins in apoptosis at a glance. J. Cell Sci. 125(Pt 5), 1081–1087. doi: 10.1242/jcs.090514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickman, J. A., Hardwick, J. M., Kaczmarek, L. K., and Jonas, E. A. (2008). Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J. Neurophysiol. 99, 1515–1522. doi: 10.1152/jn.00598.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hochman, A., Sterin, H., Gorodin, S., Korsmeyer, S., Ziv, I., Melamed, E., et al. (1998). Enhanced oxidative stress and altered antioxidants in brains of Bcl-2-deficient mice. J. Neurochem. 71, 741–748. doi: 10.1046/j.1471-4159.1998.71020741.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsu, S. Y., Kaipia, A., McGee, E., Lomeli, M., and Hsueh, A. J. (1997). Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc. Natl. Acad. Sci. U.S.A. 94, 12401–12406. doi: 10.1073/pnas.94.23.12401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiao, J., Huang, X., Feit-Leithman, R. A., Neve, R. L., Snider, W., Dartt, D. A., et al. (2005). Bcl-2 enhances Ca(2+) signaling to support the intrinsic regenerative capacity of CNS axons. EMBO J. 24, 1068–1078. doi: 10.1038/sj.emboj.7600589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kilbride, S. M., and Prehn, J. H. (2013). Central roles of apoptotic proteins in mitochondrial function. Oncogene 32, 2703–2711. doi: 10.1038/onc.2012.348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konig, H. G., Rehm, M., Gudorf, D., Krajewski, S., Gross, A., Ward, M. W., et al. (2007). Full length Bid is sufficient to induce apoptosis of cultured rat hippocampal neurons. BMC Cell Biol. 8:7. doi: 10.1186/1471-2121-8-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kozopas, K. M., Yang, T., Buchan, H. L., Zhou, P., and Craig, R. W. (1993). MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. U.S.A. 90, 3516–3520. doi: 10.1073/pnas.90.8.3516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krajewski, S., Bodrug, S., Krajewska, M., Shabaik, A., Gascoyne, R., Berean, K., et al. (1995). Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am. J. Pathol. 146, 1309–1319.
Lee, S. I., Kim, B. G., Hwang, D. H., Kim, H. M., and Kim, S. U. (2009). Overexpression of Bcl-XL in human neural stem cells promotes graft survival and functional recovery following transplantation in spinal cord injury. J. Neurosci. Res. 87, 3186–3197. doi: 10.1002/jnr.22149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Letai, A., Bassick, M. C., Walensky, L. D., Sorcinelli, M. D., Weiler, S., and Korsmeyer, S. J. (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. doi: 10.1016/S1535-6108(02)00127-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., Chen, Y., Jones, A. F., Sanger, R. H., Collins, L. P., Flannery, R., et al. (2008). Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 2169–2174. doi: 10.1073/pnas.0711647105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Magiera, M. M., Mora, S., Mojsa, B., Robbins, I., Lassot, I., and Desagher, S. (2013). Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ. 20, 281–292. doi: 10.1038/cdd.2012.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malone, C. D., Hasan, S. M., Roome, R. B., Xiong, J., Furlong, M., Opferman, J. T., et al. (2012). Mcl-1 regulates the survival of adult neural precursor cells. Mol. Cell. Neurosci. 49, 439–447. doi: 10.1016/j.mcn.2012.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinou, J. C., Dubois-Dauphin, M., Staple, J. K., Rodriguez, I., Frankowski, H., Missotten, M., et al. (1994). Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 13, 1017–1030. doi: 10.1016/0896-6273(94)90266-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martorana, F., Brambilla, L., Valori, C. F., Bergamaschi, C., Aronica, E., Volterra, A., et al. (2012). The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum. Mol. Genet. 21, 826–840. doi: 10.1093/hmg/ddr513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattson, M. P., LaFerla, F. M., Chan, S. L., Leissring, M. A., Shepel, P. N., and Geiger, J. D. (2000). Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 23, 222–229. doi: 10.1016/S0166-2236(00)01548-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehta, A., Prabhakar, M., Kumar, P., Deshmukh, R., and Sharma, P. L. (2013). Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 698, 6–18. doi: 10.1016/j.ejphar.2012.10.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merry, D. E., Veis, D. J., Hickey, W. F., and Korsmeyer, S. J. (1994). bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development 120, 301–311.
Middleton, G., Wyatt, S., Ninkina, N., and Davies, A. M. (2001). Reciprocal developmental changes in the roles of Bcl-w and Bcl-x(L) in regulating sensory neuron survival. Development 128, 447–457.
Miyawaki, T., Mashiko, T., Ofengeim, D., Flannery, R. J., Noh, K. M., Fujisawa, S., et al. (2008). Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4892–4897. doi: 10.1073/pnas.0800628105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mori, M., Burgess, D. L., Gefrides, L. A., Foreman, P. J., Opferman, J. T., Korsmeyer, S. J., et al. (2004). Expression of apoptosis inhibitor protein Mcl1 linked to neuroprotection in CNS neurons. Cell Death Differ. 11, 1223–1233. doi: 10.1038/sj.cdd.4401483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Motoyama, N., Wang, F., Roth, K. A., Sawa, H., Nakayama, K., Negishi, I., et al. (1995). Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267, 1506–1510. doi: 10.1126/science.7878471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, B., Dunleavy, M., Shinoda, S., Schindler, C., Meller, R., Bellver-Estelles, C., et al. (2007). Bcl-w protects hippocampus during experimental status epilepticus. Am. J. Pathol. 171, 1258–1268. doi: 10.2353/ajpath.2007.070269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nesic-Taylor, O., Cittelly, D., Ye, Z., Xu, G. Y., Unabia, G., Lee, J. C., et al. (2005). Exogenous Bcl-xL fusion protein spares neurons after spinal cord injury. J. Neurosci. Res. 79, 628–637. doi: 10.1002/jnr.20400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olney, J. W., Sharpe, L. G., and Feigin, R. D. (1972). Glutamate-induced brain damage in infant primates. J. Neuropathol. Exp. Neurol. 31, 464–488. doi: 10.1097/00005072-197207000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O'Reilly, L. A., Print, C., Hausmann, G., Moriishi, K., Cory, S., Huang, D. C., et al. (2001). Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ. 8, 486–494. doi: 10.1038/sj.cdd.4400835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Panickar, K. S., Nonner, D., and Barrett, J. N. (2005). Overexpression of Bcl-xl protects septal neurons from prolonged hypoglycemia and from acute ischemia-like stress. Neuroscience 135, 73–80. doi: 10.1016/j.neuroscience.2005.02.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parsadanian, A. S., Cheng, Y., Keller-Peck, C. R., Holtzman, D. M., and Snider, W. D. (1998). Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J. Neurosci. 18, 1009–1019.
Perciavalle, R. M., Stewart, D. P., Koss, B., Lynch, J., Milasata, S., Bathina, M., et al. (2012). Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 14, 575–583. doi: 10.1038/ncb2488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Print, C. G., Loveland, K. L., Gibson, L., Meehan, T., Stylianou, A., Wreford, N., et al. (1998). Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. U.S.A. 95, 12424–12431. doi: 10.1073/pnas.95.21.12424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Putcha, G. V., Deshmukh, M., and Johnson, E. M. Jr. (1999). BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 19, 7476–7485.
Rinkenberger, J. L., Horning, S., Klocke, B., Roth, K., and Korsmeyer, S. J. (2000). Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 14, 23–27. doi: 10.1101/gad.14.1.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roth, K. A., Kuan, C., Haydar, T. F., D'Sa-Eipper, C., Shindler, K. S., Zheng, T. S., et al. (2000). Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc. Natl. Acad. Sci. U.SA. 97, 466–471. doi: 10.1073/pnas.97.1.466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rudner, J., Jendrossek, V., and Belka, C. (2002). New insights in the role of Bcl-2 Bcl-2 and the endoplasmic reticulum. Apoptosis 7, 441–447. doi: 10.1023/A:1020087108926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, G., Liu, Y., Liu, T., Yan, W., Liu, X., Wang, Y., et al. (2012). Upregulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Exp. Brain Res. 216, 225–230. doi: 10.1007/s00221-011-2925-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steckley, D., Karajgikar, M., Dale, L. B., Fuerth, B., Swan, P., Drummond-Main, C., et al. (2007). Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 27, 12989–12999. doi: 10.1523/JNEUROSCI.3400-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tait, S. W., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsujimoto, Y., Cossman, J., Jaffe, E., and Croce, C. M. (1985). Involvement of the bcl-2 gene in human follicular lymphoma. Science 228, 1440–1443. doi: 10.1126/science.3874430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uren, R. T., Dewson, G., Chen, L., Coyne, S. C., Huang, D. C., Adams, J. M., et al. (2007). Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J. Cell Biol. 177, 277–287. doi: 10.1083/jcb.200606065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veis, D. J., Sorenson, C. M., Shutter, J. R., and Korsmeyer, S. J. (1993). Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75, 229–240. doi: 10.1016/0092-8674(93)80065-M
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waggie, K. S., Kahle, P. J., and Tolwani, R. J. (1999). Neurons and mechanisms of neuronal death in neurodegenerative diseases: a brief review. Lab. Anim. Sci. 49, 358–362.
Wei, L., Cui, L., Snider, B. J., Rivkin, M., Yu, S. S., Lee, C. S., et al. (2005). Transplantation of embryonic stem cells overexpressing Bcl-2 promotes functional recovery after transient cerebral ischemia. Neurobiol. Dis. 19, 183–193. doi: 10.1016/j.nbd.2004.12.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., et al. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730. doi: 10.1126/science.1059108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Willis, S. N., Chen, L., Dewson, G., Wei, A., Naik, E., Fletcher, J. I., et al. (2005). Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305. doi: 10.1101/gad.1304105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Willis, S. N., Fletcher, J. I., Kaufmann, T., van Delft, M. F., Chen, L., Czabotar, P., et al. (2007). Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859. doi: 10.1126/science.1133289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, L. F., Ralph, G. S., Walmsley, L. E., Bienemann, A. S., Parham, S., Kingsman, S. M., et al. (2005). Lentiviral-mediated delivery of Bcl-2 or GDNF protects against excitotoxicity in the rat hippocampus. Mol. Ther. 11, 89–95. doi: 10.1016/j.ymthe.2004.08.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, C., Chen, J., Chen, D., Minami, M., Pei, W., Yin, X. M., et al. (2000). Overexpression of the cell death suppressor Bcl-w in ischemic brain: implications for a neuroprotective role via the mitochondrial pathway. J. Cereb. Blood Flow Metab. 20, 620–630. doi: 10.1097/00004647-200003000-00020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., et al. (1997). Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132. doi: 10.1126/science.275.5303.1129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, T., Kozopas, K. M., and Craig, R. W. (1995). The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J. Cell Biol. 128, 1173–1184. doi: 10.1083/jcb.128.6.1173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yao, M., Nguyen, T. V., and Pike, C. J. (2005). Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 25, 1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, W., Cao, G., Johnnides, M. J., Signore, A. P., Luo, Y., Hickey, R. W., et al. (2006). TAT-mediated delivery of Bcl-xL protein is neuroprotective against neonatal hypoxic-ischemic brain injury via inhibition of caspases and AIF. Neurobiol. Dis. 21, 358–371. doi: 10.1016/j.nbd.2005.07.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. doi: 10.1038/nrm2308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, R., Xue, Y. Y., Lu, S. D., Wang, Y., Zhang, L. M., Huang, Y. L., et al. (2006). Bcl-2 enhances neurogenesis and inhibits apoptosis of newborn neurons in adult rat brain following a transient middle cerebral artery occlusion. Neurobiol. Dis. 24, 345–356. doi: 10.1016/j.nbd.2006.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, H., Yenari, M. A., Cheng, D., Barreto-Chang, O. L., Sapolsky, R. M., and Steinberg, G. K. (2004). Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J. Cereb. Blood Flow Metab. 24, 681–692. doi: 10.1097/01.WCB.0000127161.89708.A5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: BCL-2, apoptosis, mitochondria, neuronal injury, neuronal development, neurodegeneration, ischemia, excitotoxcity
Citation: Anilkumar U and Prehn JHM (2014) Anti-apoptotic BCL-2 family proteins in acute neural injury. Front. Cell. Neurosci. 8:281. doi: 10.3389/fncel.2014.00281
Received: 24 June 2014; Accepted: 25 August 2014;
Published online: 30 September 2014.
Edited by:
Rosanna Parlato, Ulm University, GermanyReviewed by:
Barbara Wroblewska, Georgetown University, USACopyright © 2014 Anilkumar and Prehn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jochen H. M. Prehn, Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, 123 St. Stephen's Green, Dublin, Ireland e-mail:anByZWhuQHJjc2kuaWU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.