 Jianling Chen
Jianling Chen Shunying Yu
Shunying Yu Yingmei Fu
Yingmei Fu Xiaohong Li
Xiaohong Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Neurosci., 11 September 2014
Sec. Cellular Neuropathology
Volume 8 - 2014 | https://doi.org/10.3389/fncel.2014.00276
This article is part of the Research TopicNeural and Synaptic Defects in Autism Spectrum DisordersView all 24 articles
Recent studies have found that hundreds of genetic variants, including common and rare variants, rare and de novo mutations, and common polymorphisms contribute to the occurrence of autism spectrum disorders (ASDs). The mutations in a number of genes such as neurexin, neuroligin, postsynaptic density protein 95, SH3, and multiple ankyrin repeat domains 3 (SHANK3), synapsin, gephyrin, cadherin, and protocadherin, thousand-and-one-amino acid 2 kinase, and contactin, have been shown to play important roles in the development and function of synapses. In addition, synaptic receptors, such as gamma-aminobutyric acid receptors and glutamate receptors, have also been associated with ASDs. This review will primarily focus on the defects of synaptic proteins and receptors associated with ASDs and their roles in the pathogenesis of ASDs via synaptic pathways.
Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental disorders characterized by social communication deficits and stereotyped behaviors with restricted interests (American Psychiatry Association [APA], 2013). Autism was first reported by Kanner (1943), who described seven boys and four girls who exhibited “extreme aloneness from the very beginning of life, not responding to anything that comes to them from the outside world.” Asperger (1944) described four boys with social communication difficulties. During the past 70 years, the definition of autism has developed as understanding of the disorder increased. It was first introduced as infantile autism in the official diagnostic nomenclature in the third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III), was referred to as Pervasive Developmental Disorders (PDD) in DSM-IV and was defined as ASDs in the latest revision of the DSM, DSM-V (American Psychiatry Association [APA], 2013), which was published in May 2013. In DSM-V, ASDs includes disorders that were previously diagnosed separately, such as autistic disorder, Asperger’s disorder, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified. The decision to merge the three disorders was taken because they could not be easily distinguished from each other. Studies have shown that more than half of adults with autism have poor or very poor outcomes (Howlin et al., 2004; Billstedt et al., 2005) in terms of independent living, educational attainment, employment, and peer relationships.
Autism was considered as a rare childhood disorder, and in the first epidemiological study conducted in the UK in 1966, Lotter (1966) reported a prevalence rate of autism of 4.5 in 10,000 children. However, the prevalence of ASDs has steadily increased in the past two decades; for example, in the USA the estimated prevalence was reported to be 19 in 10,000 children in 1992 increasing to 1 in 150 in 2002, 1 in 110 in 2006, and 1 in 88 in 2008 (Rice, 2012). ASDs are recognized as a common disorder today, with a median worldwide prevalence of 0.62% (Elsabbagh et al., 2012), and boys are affected by ASDs four times more frequently than girls. The increased prevalence of ASDs is most likely because of broadened diagnostic criteria and heightened awareness, but may also partially reflect a true increase due to environmental factors acting upon a genetically vulnerable background (King and Bearman, 2009; Lintas and Persico, 2009; Li et al., 2012).
In addition to a variable severity of the core deficits, ASDs patients also present other psychiatric and medical conditions, such as intellectual disability, epilepsy, motor control problems, attention-deficit/hyperactivity disorder, tics, anxiety, sleep disorders, and gastrointestinal problems (Simonoff et al., 2008; Lai et al., 2014).
For the past several decades, ASDs have been recognized as a complex brain disorder with high heritability, except with rare pedigrees, usually accompanied with other neurodevelopmental conditions shown to have Mendelian inheritance (Morrow et al., 2008; Novarino et al., 2012). Recent genomic and genetic studies have found that hundreds of genetic variants, including common and rare variants, contribute to the occurrence of ASDs. Rare and de novo mutations may pose a substantial risk for ASDs and play a substantial role in population risk, and common polymorphisms also contribute to ASDs. The role of individual alleles remains elusive and underestimated due to their small effect sizes (Murdoch and State, 2013). Many genes associated with ASDs play roles in the development and function of synapses, such as neuroligin 3 (NLGN3), NLGN4X, neurexin 1 (NRXN1), and SH3, and multiple ankyrin repeat domains 3 (SHANK3).
Post-mortem studies of ASDs patients have shown a reduction in the number of neurons in the amygdala, fusiform gyrus, and cerebellum and signs of persistent neuroinflammation (Lai et al., 2014). In addition, reduction in the density of serotonin transporters (5-HTT) was also found in the deep layers of the fusiform gyrus in autistic subjects (Oblak et al., 2013). Transcriptome analyses showed that genes involved in synaptic function were downregulated in the ASDs post-mortem brain. Moreover, the emergence of various types of genetically modified mouse models targeting ASDs-associated genes or loci in recent years have provided insights into particular aspects of ASDs. Therefore, it may be proposed that ASDs are a synaptic defect disease. In this review, we will focus on the role of synaptic-related genes in ASDs.
Neurexins (NRXN) are a family of synaptic adhesion proteins that are located on the presynaptic membrane and bind to their postsynaptic counterpart, NLGNs. The NRXN family consists of three genes (NRXN1, NRXN2, and NRXN3), each of them generating a long mRNA encoding α-NRXN and a short mRNA encoding β-NRXN from two independent promoters. The intracellular domains of α-NRXNs and β-NRXNs are identical, whereas the extracellular domains are different. Specifically, the extracellular domains of α-NRXNs contain six laminin, nectin, and sex-hormone binding globulin (LNS) domains and three epidermal growth factor (EGF) domains, which form three repeated LNS (A)-EGF-LNS (B) structure. However, β-NRXNs have no EGF domain and only one LNS domain (Tabuchi and Sudhof, 2002). NRXN1, NRXN2, and NRXN3 are located on chromosomes 2p16.3, 11q13, and 14q31, respectively. α-NRXN triple knockout mice had reduced synaptic Ca2+ channel function, which causes impaired spontaneous and evoked neurotransmitter release (Missler et al., 2003).
Rare copy number variations and/or point mutations in NRXN genes have been repeatedly reported to be associated with ASDs (Table 1). Friedman and Luiselli (2008) first reported a 320 kb de novo heterozygous deletion of the NRXN-1α promoter and exons 1–5 in a boy with cognitive impairment, autistic features and physical dysmorphism. Later, the Autism Genome Project Consortium et al. (2007) identified a de novo heterozygous deletion that eliminated several NRXN1 exons, including 1α and 1β, in two affected female siblings in one ASDs family. Several other studies have also reported deletions in NRXN1 in ASDs patients (Kim et al., 2008; Marshall et al., 2008; Morrow et al., 2008; Glessner et al., 2009; Pinto et al., 2010; Bremer et al., 2011; Levy et al., 2011). To date, no homozygous deletions in NRXN1 have been found, which may suggest that the dosage of NRXN1 is very important for neurological development. P300P, an NRXN1 common variant, was associated with ASDs in a Chinese ASDs patient. In addition to being associated with ASDs, NRXN1 deletions have also been reported in other psychiatric conditions, such as schizophrenia, bipolar disorder, attention deficit hyperactivity disorder, and Tourette syndrome (International Schizophrenia Consortium, 2008; Walsh et al., 2008; Guilmatre et al., 2009; Zhang et al., 2009; Sundaram et al., 2010; Lionel et al., 2011).
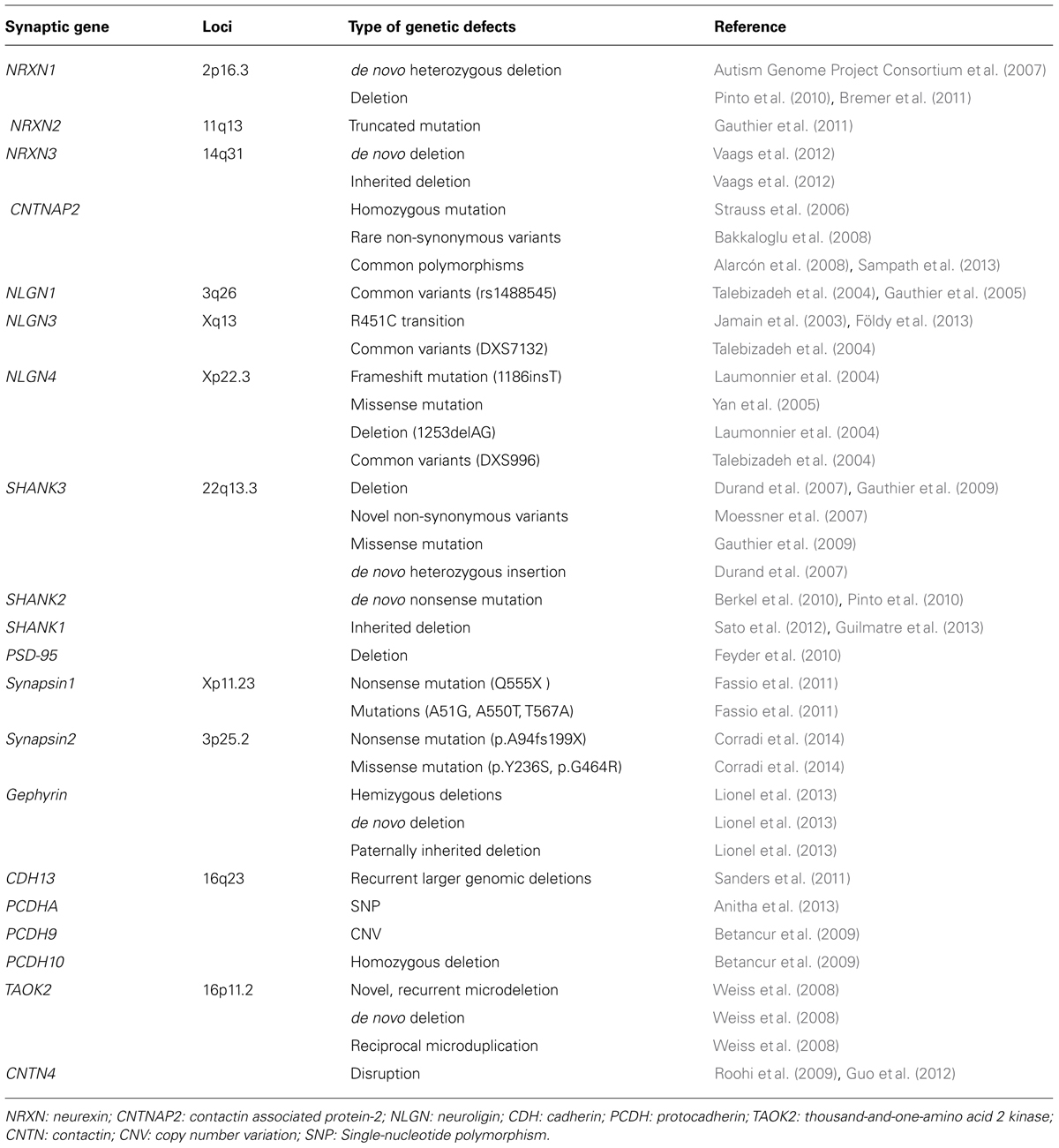
TABLE 1. Summary of different defects in gene encoding for synaptic proteins in autism spectrum disorders.
A truncated mutation of NRXN2 inherited from a father with severe language delay and a family history of schizophrenia was identified by Gauthier et al in an ASDs patient (Gauthier et al., 2011). NRXN3 deletions have also been found in four ASD individuals: one was a de novo mutation, two were inherited from a non-affected mother or father, and one was inherited from a father with subclinical autism (Vaags et al., 2012).
NRXN animal models have provided evidence supporting the role of NRXN in ASDs pathology. NRXN1α KO mice showed a defect in excitatory synaptic strength, with a decrease in miniature excitatory postsynaptic current frequency and in the input–output relation of evoked postsynaptic potentials (Etherton et al., 2009). Behavioral studies have shown that NRXN-1 deficient mice display decreased prepulse inhibition and increased grooming behaviors but no obvious changes in social behaviors or spatial learning (Etherton et al., 2009). Studies in an α-NRXN triple KO mice with all three α-NRXNs (Nrxn1α/2α/3α) deleted have shown that α-NRXNs were not required for synapse formation but were essential for Ca2+-triggered neurotransmitter release (Missler et al., 2003).
Contactin associated protein-2 (CNTNAP2, also known as Caspr2) is a member of the NRXN superfamily and is involved in neuron–glia interactions and clustering K+ channels in myelinated axons. Strauss et al. (2006) identified a homozygous mutation of CNTNAP2 in Amish children with PDD, seizures, and language regression (Strauss et al., 2006; Table 1). Bakkaloglu et al. (2008) found 13 rare non-synonymous variants unique to ASDs patients, which suggests that ASDs patients carry more CNTNAP2 rare variants. Several other studies have also found other common polymorphisms of CNTNAP2 that are associated with ASDs (Alarcón et al., 2008; Arking et al., 2008; Sampath et al., 2013). Interestingly, Alarcón et al. (2008) found that CNTNAP2 provided a strong male affection bias in ASDs.
CNTNAP2 KO mice exhibited deficits in the core ASDs behavioral domains, such as stereotypic motor movements, behavioral inflexibility, communication, and social behavior abnormalities (Peñagarikano et al., 2011).
Neuroligins (NLGN) are a different type of synaptic cell adhesion proteins that are located in the postsynaptic membrane. NLGNs bind to their adhesive counterpart NRXNs and play an important role in synapse formation and function (Figure 1). The human NLGN family includes five NLGN genes (NLGN1, 2, 3, 4, 4Y), which are localized at 3q26 (NLGN1), 17p13 (NLGN2), Xq13 (NLGN3), Xp22.3 (NLGN4), and Yq11.2 (NLGN4Y). NLGNs contain a large extracellular domain that shares sequence homology with acetylcholinesterase and that is necessary for β-NRXN binding and synaptogenic activity, two EF-hand motifs that bind to Ca2+, an O-glycosylation region, a transmembrane domain, and a cytoplasmic C-terminal tail that contains a PSD-95/Dlg/ZO-1 (PDZ) interaction site (Dean and Dresbach, 2006; Figure 1). NLGNs-1, -3, and -4 localize mainly to glutamate synaptic sites, whereas NLGN-2 localizes primarily to gamma-aminobutyric acid (GABA) synapses (Missler et al., 2003; Figure 1). In vitro and in vivo, NLGN-1 overexpression increases excitatory synaptic responses and potentiates synaptic NMDA receptor (NMDAR)/AMPAR ratios. In contrast, NLGN-2 overexpression increases inhibitory synaptic responses. Accordingly, the inhibition of NLGN-1 expression selectively decreases the NMDAR/AMPAR ratio, whereas the deletion of NLGN-2 selectively decreases inhibitory synaptic responses. Furthermore, NLGN-1 expression selectively increases the maturation but not initiation of excitatory synapse formation in adult-born neurons (Chubykin et al., 2007; Schnell et al., 2012).
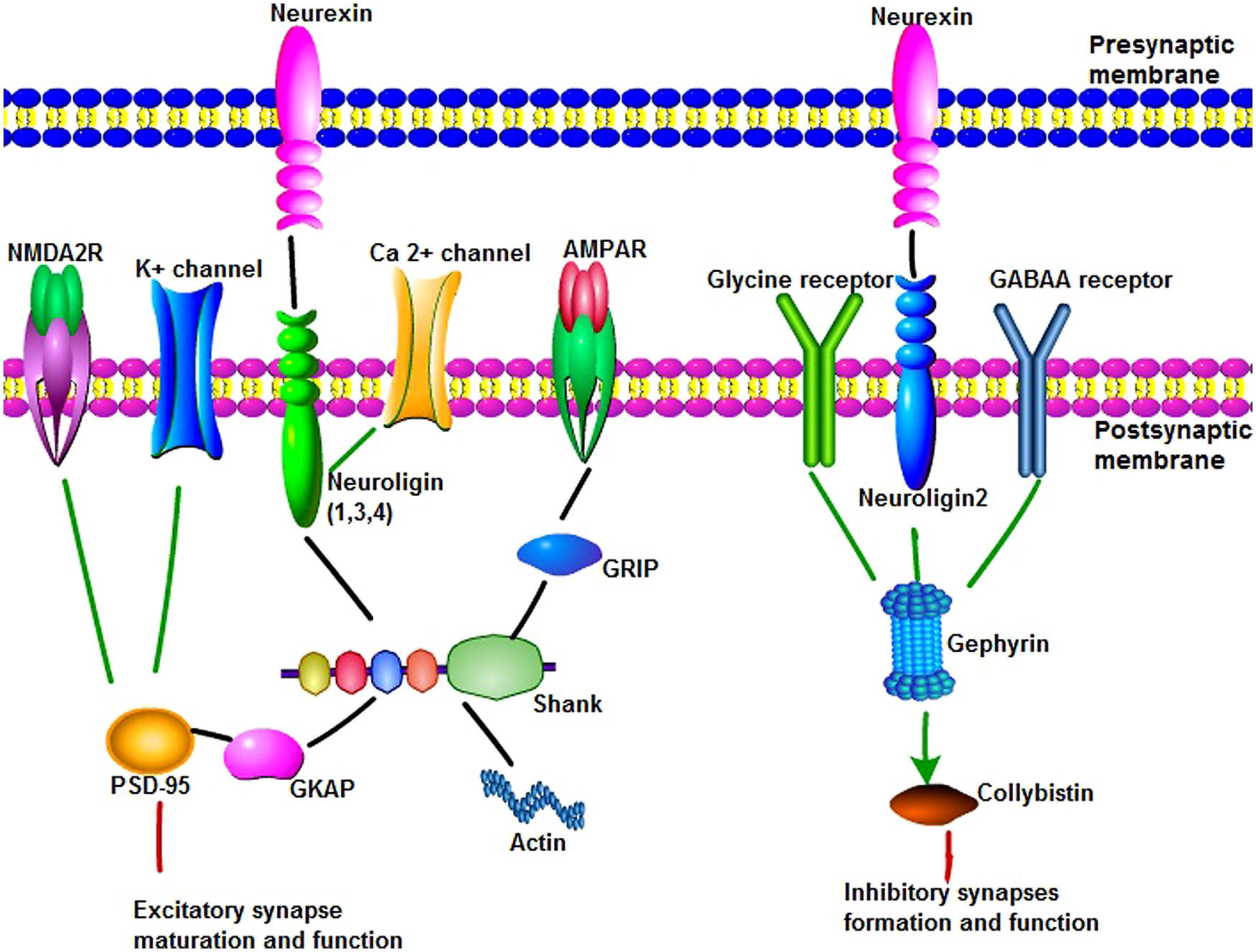
FIGURE 1. The main synaptic proteins and receptors involved in this review in synaptogenesis and function. (1) Neuroligins(NLGNs) can bind to neurexins (NRXNs), Ca2+ channel and shank through their different domains to play an important role in synapse maturation and function. NLGNs 1, 3, and 4 localize to excitatory synaptic sites, whereas NLGN-2 localizes primarily to inhibitory synapses. Shank are synaptic scaffold proteins that bind NLGN–NRXN and NMDA receptor complexes at the postsynaptic density of excitatory glutamatergic synapses. Multiple domains of shank are useful for protein–protein interaction, such as linking to actin cytoskeleton, binding to glutamate receptor-interacting protein to link with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Postsynaptic density protein 95 (PSD-95) serves as a major functional bridge interconnecting the NRXN-NLGN-SHANK pathway. (2) Gephyrin is a key scaffold molecule of the postsynaptic membrane at inhibitory synapses. Gephyrin can interact with glycine receptor and alpha and beta subunits of the gamma-aminobutyric acid A (GABAA) receptor to mediate inhibition of synapse formation and function. NLGN 2 can bind to protein gephyrin and activate collybistin. NLGN 2, gephyrin, and collybistin complexes are sufficient to inhibitory neurotransmitter receptors clustering.
The earliest report regarding the potential association of NLGN genes and ASDs is Jamain et al. (2003; Table 1). In one ASDs multiplex family, the group found three ASDs siblings carrying one frameshift mutation (1186insT) of NLGN4 that was inherited from the non-affected mother, creating a stop codon that led to a premature termination of the protein. In another family, a R451C transition in NLGN3 that changed a highly conserved arginine residue into cysteine within the esterase domain was identified in two affected siblings. This point mutation was inherited from the non-affected mother. A study in a large French family found a 2 bp deletion (1253delAG) that resulted in a premature stop codon in the middle of the sequence of the normal NLGN4 gene (Laumonnier et al., 2004). In this family, 10 males had non-specific X-linked mental retardation, two had autism, and one had pervasive developmental disorder. All affected patients had the same frameshift mutation. Missense changes in NLGN4 were also found in Portuguese ASDs families (Yan et al., 2005). A study in a small Finnish autism sample did not find any functional mutation of NLGN1, NLGN3, NLGN4, or NLNG4Y, although three common variants (rs1488545 in NLGN1, DXS7132 in NLGN3, and DXS996 in NLGN4) that showed minor association with ASDs were found. Despite the evidence that NLGN is associated with ASDs, several studies have failed to find associations between rare mutations and common variants and ASDs (Talebizadeh et al., 2004; Vincent et al., 2004; Gauthier et al., 2005; Blasi et al., 2006; Wermter et al., 2008; Liu et al., 2013).
Several NLGN mutant mouse models have been developed to investigate the role of NLGN mutations in ASDs. Nlgn3 R451C knock-in mice, which corresponded to the human non-synonymous SNP (R451C) in NLGN3 found in ASDs patients (Jamain et al., 2003), showed social interaction deficits and increased spatial memory and an electrophysiological phenotype consisting of increased inhibitory synaptic transmission in the somatosensory cortex (Tabuchi et al., 2007; Etherton et al., 2011a). Furthermore, researchers found that both NLGN-3 R451C-knockin and NLGN-3 knockout mutations in mice showed impairment in tonic endocannabinoid signaling (Földy et al., 2013). Another NLGN3 mutation, R704C, was introduced into mouse NLGN-3 by homologous recombination, and electrophysiological and morphological studies have shown that although the NLGN-3 R704C mutation did not significantly alter synapse formation, it dramatically impaired synapse function. Moreover, the R704C mutation caused a major and selective decrease in AMPA receptor-mediated synaptic transmission in pyramidal neurons of the hippocampus, without similarly changing NMDA or GABA receptor-mediated synaptic transmission and without detectably altering presynaptic neurotransmitter release (Etherton et al., 2011b). Mice lacking the human NLGN4 (NLGN4-KOs) ortholog exhibited highly selective deficits in reciprocal social interactions and communication reminiscent of ASDs (Jamain et al., 2008).
Shank family proteins, also known as ProSAP, are synaptic scaffold proteins that bind NLGN-NRXN and NMDAR complexes at the postsynaptic density (PSD) of excitatory glutamatergic synapses (Figure 1). There are three genes that encode Shank proteins (SHANK1, SHANK2, and SHANK3). All Shank proteins are expressed in the brain but exhibit different patterns. Shank1 is expressed in most parts of the brain, except for the striatum, and it is highly expressed in the cortex and hippocampus. Shank2 and Shank3 are also present in the cortex and hippocampus. Shank2 is mostly absent from the thalamus and striatum, whereas Shank3 appears to be predominantly expressed in those regions. In the cerebellum, Shank2 is restricted to Purkinje cells, whereas Shank3 is restricted to granule cells (Sheng and Kim, 2000). SHANK directly or indirectly binds to NLGNs in the PSD. In vitro and in vivo studies highlight the important role of Shank3 for synaptic function. SHANK3 functions as a scaffolding protein in spine morphogenesis and synaptic plasticity. Knockdown of Shank3 in cultured hippocampal neurons leads to a reduced number and increased length of dendritic spines. When overexpressed in cultured hippocampal neurons, Shank3 promotes the maturation and enlargement of dendritic spines (Betancur et al., 2009). Knock-down of Shank3 in hippocampal neurons decreases spiny density, whereas transfection of Shank3 in aspiny neurons induces the formation of dendritic spines with functional synapses (Betancur et al., 2009).
Shank contains multiple domains for protein–protein interaction, including ankyrin repeats (binding to α-fodrin to link to the actin cytoskeleton and calpain/calmodulin-mediated Ca2+ signaling), an SH3 domain (binding to glutamate receptor-interacting protein to link with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors to the postsynaptic scaffold), a PDZ domain (binding different molecules within the PSD, including GKAP, to allow Shank to attach to PSD-95), a proline-rich region (which contains sites for Homer and cortactin), and a sterile alpha motif domain (which is involved in the polymerization of Shank molecules; Lim et al., 1999; Yoo et al., 2014; Figure 1).
SHANK3 was the first gene in the SHANK family reported to be associated with ASDs (Table 1). The SHANK3 gene is located on chromosome 22q13.3 within the critical region of 22q13.3 deletion syndrome (also known as Phelan-McDermid syndrome, PMS). 22q13.3 deletion syndrome is characterized by neonatal hypotonia, global developmental delay, absent or severely delayed speech, autistic behaviors, and intellectual disability (Phelan, 2008). The size of the deleted segments varied widely in individuals with this syndrome, but deletions of SHANK3 were present in nearly all cases (Wilson et al., 2003, 2008; Dhar et al., 2010). Three ASDs families were observed to carry alterations of 22q and/or the SHANK3 gene (Durand et al., 2007). In one family, an individual carried a de novo deletion of 22q13, in which the deletion breakpoint was located in intron 8 of SHANK3 and a 142 kb of the terminal 22q13 was removed. In the second family, two affected siblings were heterozygous for an insertion of a guanine nucleotide in exon 21, and the mutation was a de novo mutation. In the third family, a terminal 22q deletion was identified in a girl with autism who exhibited severe language delay. A 22qter partial trisomy in her brother who had Asperger syndrome was also identified, although the boy demonstrated precocious language development and fluent speech. These unbalanced cytogenetic abnormalities were inherited from a paternal translocation, t(14;22) (p11.2;q13.33). This finding suggests a gene dosage effect of SHANK3. A study of SHANK3 variants by screening SHANK’s exon sequence in 400 ASDs families found 10 novel non-synonymous variants in ASDs (Moessner et al., 2007). Among these mutations, one was a de novo mutation, and the other nine were all inherited from one unaffected parent. Rare functional mutations of SHANK3 have been identified in two ASDs families. One was a de novo deletion at an intronic donor splice site, and one was a missense mutation inherited from an epileptic father (Gauthier et al., 2009). Moreover, three deleterious variants (one 6-amino acid deletion upstream of the SH3 domain, one missense variant in the PDZ domain, and one insertion/deletion of a repeated 10 bp GC sequence located 9-bp downstream from the 3′ end of exon 11) were found in Japanese ASDs families (Waga et al., 2011). It has been reported that 2.3% of ASDs patients carry deleterious mutations in SHANK3 (Boccuto et al., 2013).
Several studies have identified de novo deleterious mutations of SHANK2 in ASDs (Berkel et al., 2010; Pinto et al., 2010; Leblond et al., 2012; Table 1). In one study, a de novo nonsense mutation, seven rare inherited changes and additional variants specific to ASDs were identified by sequencing SHANK2 in 396 ASDs patients. In a separate study, SHANK2 was sequenced in 455 patients. When combining the results of these two studies, a significant enrichment of variants affecting conserved amino acids in 29 of 851 (3.4%) patients and 16 of 1090 (1.5%) controls was observed. In neuronal cell cultures, the variants identified in patients were associated with reduced synaptic density at dendrites compared with the variants only detected in controls (Leblond et al., 2012).
The SHANK1 gene rare mutation has also been associated with ASDs (Sato et al., 2012; Table 1). An inherited deletion of 63.8 kb encompassing SHANK1 and the CLEC11A gene was found in a multigenerational family. In this family, four males carrying this deletion showed high-functioning autism or a broader autism phenotype, whereas the two females carrying the same deletion were not affected by ASDs (Sato et al., 2012). SHANK1 deletions may be associated with high-functioning autism in males (Guilmatre et al., 2013).
Genetic mouse models resembling different SHANK mutations have been created to investigate the role of Shank in synapse conformation and function and its contribution to autistic pathology. Shank1 mutant mice (with a deletion of exons 14 and 15, which includes most of the PDZ region, resulted in the knockout of all detectable Shank1 protein in these animals) showed decreased movement in the open field and deficits in motor learning and contextual fear conditioning. Although these animals did not show apparent repetitive behaviors and seemed to have normal levels of social interaction, they showed a general deficit in social communicative behaviors by both ultrasonic vocalizations and urine-based communicative behaviors (Hung et al., 2008; Silverman et al., 2011; Wöhr et al., 2011). Shank2 mutant mice, mimicking the human microdeletion of exons 6 and 7, which targeted the PDZ domain and knocked out of all Shank2 isoforms, also showed alterations in behavior and synaptic plasticity (Schmeisser et al., 2012; Won et al., 2012). Shank3 exons 4–9 KO mice (resulting in the loss of the longest two isoforms of Shank3) were used to mimic human SHANK3 mutation by several research groups. Although the deficits in social interaction in these models were not consistent (Bozdagi et al., 2010; Peça et al., 2011; Yang et al., 2012), all of the models showed a repetitive self-grooming phenotype (Wang et al., 2011; Yang et al., 2012).
Postsynaptic density protein 95 (PSD-95; also known as DLG4, SAP90), is a member of the membrane-associated guanylate kinase family of synaptic molecules and serves as a major functional bridge interconnecting the NRXN-NLGN-SHANK pathway (Figure 1). PSD-95 contains three PDZ domains, a single interior SH3 domain, and a COOH-terminal guanylate kinase domain. The cytoplasmic domains of all three NLGNs bind to the third PDZ domain of PSD-95, whereas NMDA2 receptors and K+channels bind to the first and second PDZ domains. PSD-95 is localized at excitatory synapses and has been implicated in promoting synapse stability. PSD-95 makes synaptic contacts more stable in older neurons than in younger neurons.
PSD-95 knockout mice exhibit reduced AMPAR function and a decreased frequency of AMPAR-mediated miniature EPSCs, suggesting that PSD-95 may regulate synaptic maturation through postsynaptic AMPA-type glutamate receptors (GluARs).
Thus far, no rare point mutation or CNV or common variants have been reported to be associated with ASDs, but PSD-95 deletion (Dlg4-/-) mice have been shown to exhibit a complex range of behavioral and molecular abnormalities relevant to ASDs (Table 1). Dlg4-/- mice showed increased repetitive behaviors, abnormal communication and social behaviors, impaired motor coordination, increased stress reactivity, and anxiety-related responses. Dlg4-/- mice also had subtle dysmorphology of amygdala dendritic spines and altered forebrain expression of various synaptic genes (Feyder et al., 2010).
The synapsins are a family of presynaptic phosphoproteins that account for 9% of the vesicle protein and can regulateneurotransmitter release and neurite outgrowth (Rosahl et al., 1995). Synapsins contain a mosaic of conserved (A–C, E) and individual domains (D, F–J). They have three family members in mammals (synapsin 1, synapsin 2, and synapsin 3), which locate on chromosomes Xp11.23, 3p25.2, and 22q12.3, respectively. Cultured neurons from synapsin 1,2,3(–/–) triple knock-out mice exhibit severely dispersed synaptic vesicles and considerably reduced synaptic vesicles number (Fornasiero et al., 2012).
Mutations in synapsin 1 (Q555X, A51G, A550T, and T567A) were found in a large French-Canadian family with epilepsy and ASDs (Fassio et al., 2011; Table 1). Furthermore, the nonsense Q555X mutation can reduce the phosphorylation caused by CaMKII and Mapk/Erk, which regulate synaptic vesicles trafficking and neurite outgrowth. The missense mutation of A550T and T567A can impair the targeting to nerve terminals (Fassio et al., 2011).
Synapsin 2 has also been identified as an autism predisposing gene. In a study involving 190 individuals with ASDs, researchers found one nonsense mutation (p.A94fs199X) and two missense mutations of synapsin 2 (p.Y236S and p.G464R; Corradi et al., 2014; Table 1).
Synapsin knockout mice may be identified as a useful experimental model of ASDs and epilepsy. Researchers found that synapsin knockout mice exhibit social novelty abnormality and avoidance behavior in social approach which are reminiscent of ASDs. Specifically, synapsin 2 deletion mice display deficits in short-term social recognition and increased repetitive self-grooming behavior. Synapsin 1and synapsin 3 deletion mice display an impaired social transmission of food preference. Synapsin 1and synapsin 2 deletion mice display a decreased environmental interest (Greco et al., 2013). The results demonstrate an involvement of synapsins in the development of the behavioral traits of ASDs.
Gephyrin is a key scaffold molecule of the postsynaptic membrane at inhibitory synapses (Figure 1). It contains three domains, G domain in N-terminal, E-domain in C-terminal, and a large linker domain of the two. Gephyrin can interact with glycine receptor and alpha and beta subunits of the GABAA receptor to mediate inhibition. NLGN 2 can bind to protein gephyrin through a conserved cytoplasmic motif and activate collybistin. NLGN 2, gephyrin, and collybistin complexes are sufficient to inhibitory neurotransmitter receptors clustering. Deletion of NLGN 2 in mice leads to a loss of recruitment of gephyrin at perisomatic but not dendritic sites (Poulopoulos et al., 2009; Table 1). Gephyrin-deficient mice die early postnatally and display loss of postsynaptic GABA(A) receptor and glycine receptors clustering, whereas glutamate receptor subunits were normally localized (Kneussel et al., 1999; Grosskreutz et al., 2003).
Exonic microdeletions in gephyrin gene have been reported a correlation with neurodevelopmental disorders including ASDs (Lionel et al., 2013). In one family, the proband with ASDs has a 357 kb de novo deletion in gephyrin and exhibits limited movement, slow motor development, and language delay. The second family has a 319 kb paternally inherited deletion in gephyrin and exhibits mild global developmental delay in early life, social difficulties, and repetitive behaviors. The third family has a 273 kb de novo deletion in gephyrin gene and exhibits developmental delay, cyclical seizures, and behavioral issues including anxiety, obsessive compulsive disorders, tics, and impulsive behaviors (Lionel et al., 2013).
Cadherins (CDHs) are a family of glycosylated transmembrane proteins that mediate cell–cell adhesion, neuronal migration, spine morphology, synapse formation, and synaptic remodeling (Redies et al., 2012). Because the function of CDHs is dependent on the presence of Ca2+, they are named for the Ca2+-dependent cell adhesion molecule family. The CDH family is classified into classical CDHs, desmosomal cadherins, and protocadherins (PCDHs). Genome-wide association studies on a cohort of 4305 autistic subjects have shown that common variants between the CDH9 and CDH10 genes on chromosome 5p14.1 are associated with autism (Wang et al., 2009). Similarly, recurrent larger genomic deletions in 16q23 in CDH13 was also observed in ASDs patients in 1124 ASDs families participating in genome-wide analyses (Sanders et al., 2011; Table 1). Furthermore, after the detection of 14 SNPs of protocadherin α in DNA samples of 3211 individuals with autism, 5 SNPs were showed significantly associated with autism (Anitha et al., 2013). In addition, CNVs in PCDH9 and homozygous deletions in PCDH10 have also been reported in ASDs (Betancur et al., 2009; Table 1).
In situ hybridization analysis in the embryonic and postnatal mouse demonstrated that CDH8 expression is restricted to specific developing gray matter structures. Later, a study using the PPL statistical framework identified that CDH8 is expressed in the developing human cortex of ASDs family, which implicates CDH8 in susceptibility to autism (Redies et al., 2012).
Thousand-and-one-amino acid 2 kinase (TAOK2), also known as TAO2, is a serine/threonine-protein kinase that is encoded by the TAOK2 gene in humans. It can activate mitogen-activated protein kinase (MAPK) pathways to regulate gene transcription. TAOK2 interact with semaphorin 3A receptor neuropilin 1, which regulates basal dendrite arborization. In addition, TAOK2 can be phosphorylated and activated by Sema3A. In cultured cortical neurons, TAOK2 downregulation can decrease JNK phosphorylation and cause its inactivation. Furthermore, basal dendrite formation in cortical neurons caused by TAOK2 downregulation can be rescued by active JNK1 overexpression. TAOK2 is involved in membrane blebbing, the DNA damage response, and the MAPK14/p38 MAPK stress-activated MAPK cascade. Recently, TAOK2 has been shown to play a role in basal dendrite formation (de Anda et al., 2012).
The TAOK2 gene is located in the 16p11.2 chromosomal region. Approximately 1% of autistic subjects have been shown to have a novel, recurrent microdeletion, a de novo deletion of 593 kb on chromosome 16p11.2, and a reciprocal microduplication on chromosome 16p11.2 (Weiss et al., 2008), suggesting that 16p11.2 or the TAOK2 gene is involved in susceptibility to ASDs (Table 1).
Contactins (CNTNs) are members of the immunoglobulin superfamily. They are glycosylphosphatidylinositol-anchored neuronal membrane proteins and play important roles in axon growth and guidance and synapse formation and plasticity.
Array-based comparative genomic hybridization identified a paternally inherited chromosome 3 copy number variation in three autistic subjects. Specifically, a deletion in two siblings and a duplication in an unrelated individual were detected. Furthermore, these variations were mediated by disruptions of CNTN4 (Roohi et al., 2009), suggesting that CNTN4 may be involved in ASDs (Table 1). Recently, a study conducted in a Chinese population also came to the same conclusion (Guo et al., 2012). Although the sample sizes of these two studies were small, a CNV analysis involving 2195 autistic subjects indicated that CNTN4 deletions and duplications are associated with ASDs (Betancur et al., 2009). In a cohort of ASDs subjects, a CNV in the CNTN5 gene was identified in one individual. In addition, a CNTN6 deletion has also been found in an autistic family (Zuko et al., 2013).
Cntn6 knockout mice exhibited slower learning in terms of equilibrium and vestibular senses (Zuko et al., 2013), indicating that Cntn6-deficiency leads to defects in motor coordination. Other characteristics of ASDs, such as social interaction and social communication, remain to be determined in these mice. Cntn5 knockout mice exhibited decreased susceptibility to audiogenic seizures and impaired hearing, which may be related to the impairment of sensory information integration reminiscent of ASDs (Zuko et al., 2013).
Gamma-aminobutyric acid is the major inhibitory neurotransmitter in the human brain and is synthesized from excitatory neurotransmitter glutamate via the action of glutamate decarboxylase (GAD) enzymes, which have two main isoforms, GAD65 and GAD67. There are two main types of GABA receptors, ionotropic GABAA receptors and metabotropic GABAB receptors. GABAB receptors are localized at pre-, post-, or extrasynaptic sites as functional heterodimers, whereas GABAA receptors are the major mediators of fast inhibitory neurotransmission in the mammalian brain.
There are three GABAA receptor genes (GABRB3, GABRA5, and GABRG3) localized on the human chromosome 15q11–q13, a part of the genome which is involved with genome instability, gene expression, imprinting and recombination and is one of the most complex regions in the genome (Martin et al., 2000). Duplications of the 15q11–13 locus have been observed in ASDs in several studies (Bolton et al., 2001; Kwasnicka-Crawford et al., 2007; Depienne et al., 2009). Duplication of the region containing GABAA receptor subunits may lead to excessive inhibitory neurotransmission due to gene dosage; however, an in vitro study using a human neuronal cell line carrying a maternal 15q duplication showed that this variant leads to reduced GABRB3 expression via impaired homologous pairing (Meguro-Horike et al., 2011), suggesting that 15q11–q13 genes are regulated epigenetically at the level of both inter- and intra-chromosomal associations and that chromosome imbalance disrupts the epigenetic regulation of genes in 15q11–q13.
Moreover, mouse models mimicking human 15q11–q13 duplication have exhibited features of autism, such as poor social interaction, behavioral inflexibility, and abnormal ultrasonic vocalizations (Nakatani et al., 2009).
GABAB receptors play an important role in maintaining excitatory-inhibitory balance in brain. In autistic brain subjects, researchers have found that the expression of GABAB receptor subunits GABAB receptor 1 (GABBR1) and GABAB receptor 2 (GABBR2) were significantly reduced (Fatemi et al., 2009). Furthermore, clinical trials show that the selective GABAB receptor agonist STX209 (arbaclofen) has a potential to improve social function and behavior in patients with fragile X syndrome and was generally well-tolerated in ASDs individuals (Berry-Kravis et al., 2012; Erickson et al., 2014).
In vitro, STX209 (arbaclofen, R-baclofen) can correct the elevated basal protein synthesis in the hippocampus of Fmr1-knockout mice, an animal model of Fragile X syndrome. In vivo, acute administration of STX209 can decrease mRNA translation in the cortex of Fmr1-knockout mice. Furthermore, the chronic administration of STX209 in juvenile mice can improve the increased spine density in Fmr1-knockout mice (Henderson et al., 2012). Since ASDs individuals have something in common with Fragile X syndrome, this implies that GABAB receptor agonist STX209 may also improve synaptic abnormalities in ASDs.
Consistent with the genetic evidence for the involvement of GABAergic genes in ASDs, the expression of GABAergic genes and related proteins have been reported to be reduced in the post-mortem ASDs brain. GAD65 and GAD67 proteins were reduced in the cerebellum and parietal cortex (Fatemi et al., 2002), GAD67 mRNA was reduced in cerebellar Purkinje cells (Yip et al., 2007), and GABAA receptor binding was reduced in the hippocampus (Blatt et al., 2001) and anterior and posterior cingulate cortices (Oblak et al., 2009, 2011).
Although technical difficulties still exist, researchers have attempted to measure GABA function in vivo, and these results support the presence of GABAergic defects in ASDs patients. Using proton magnetic resonance spectroscopy ([1H]MRS; Harada et al., 2011) reported that GABA concentrations were reduced in the frontal cortex of ASDs children, whereas no differences were observed in the basal ganglia (Harada et al., 2011). Two studies using SPECT (Single Photon Emission Computed Tomography) found reductions in GABAA receptors in both ASDs adults and children (Mori et al., 2012; Mendez et al., 2013).
In addition to the genes/proteins involved directly in GABA synthesis and transmission, many other factors exert indirect effects on GABA functioning through the regulation of gene expression, receptor trafficking, and downstream signaling pathways; therefore, GABAergic dysfunction could also be a downstream consequence of mutations in the genes involved in the increase or decrease of GABA transmission. CNTNAP2, which is a part of the NRXN family, has been associated with autism (Gregor et al., 2011; Stein et al., 2011). CNTNAP2 knockout mice showed specific deficits in inhibitory signaling, with reduced GAD1 expression and a reduced number of GABAergic interneurons (Peñagarikano et al., 2011). Similar findings have been observed in another ASDs candidate gene model; CADPS2 knockout mice showed reduced cortical parvalbumin GABA interneurons and a reduced number of cerebellar Purkinje cells (Sadakata et al., 2007).
Glutamate is the major excitatory neurotransmitter in the human brain. Glutamate receptors (GluARs) are composed of ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs). Findings from genetic studies, post-mortem brain studies, animal models, and clinical drug trials have implicated a dysfunctional glutamatergic system in ASDs; however, hypo- and hyperfunction coexists in different forms of ASDs.
Ionotropic glutamate receptors are classified into NMDA (N-methyl-D-aspartate), AMPA (2-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid), and kainate receptors based on structural, pharmacological, and physiological properties. iGluRs are tetramers encoded by 18 genes. NMDARs are obligate heteromers formed as tetramers from the co-assembly of GluN1, GluN2A-GluN2D, GluN3A, and GluN3B subunits. Each NMDAR channel contains a combination of two GluN1 and two GluN2A–GluN2D subunits or two GluN1 with one GluN2 and one GluN3 subunit. AMPARs are homo- or hetero-tetramers formed from the GluA1–GluA4 subunits and are Mg2+-insensitive. Kainate receptors are tetramers formed from combinations of the GluK1–GluK5 subunits.
Several genetic studies have reported that NMDARs genes are associated with ASDs. Two studies that sequenced ASDs patients identified rare disruptive mutations in the GluN2B (GRIN2B) gene (Tarabeux et al., 2011; O’Roak et al., 2012). Common polymorphisms in GRIN2B and GRIN2A have also been associated with ASDs (Barnby et al., 2005; Yoo et al., 2012). Interestingly, the NMDAR subunits have differential expression during development, with GluN2B expressed early in development, followed by GluN2A during later development and synapse maturation (Sanz-Clemente et al., 2013).
Ramanathan et al. (2004) identified a 19 mb deletion of chromosome 4q in an ASDs child, which included the AMPA 2 gene that encodes the glutamate receptor GluR2 sub-unit (Ramanathan et al., 2004). One study identified chromosome 6q21 as a candidate region for autism and found a functional SNP in glutamate receptor 6 (GluR6 or GRIK2) gene associated with ASDs (Jamain et al., 2002).
A post-mortem brain study also found that ASDs patients have specific abnormalities in AMPA receptors and glutamate transporters in the cerebellum (Purcell et al., 2001). The mRNA levels of excitatory amino acid transporter 1 and glutamate receptor AMPA1 (GluA1) were significantly increased in autism subjects, and AMPAR density was decreased in the ASDs cerebellum (Purcell et al., 2001).
Parvalbumin-selective NMDAR 1 knockout (NR1 KO) mice exhibited autism-like phenotypes compared with wild-type mice; the N1 ERP latency was delayed, sociability was reduced, and mating USVs were impaired (Saunders et al., 2013).
The administration of acute PCP and ketamine, NMDAR antagonists, has been shown to mimic the symptoms of autism in humans (Carlsson, 1998). Based on this phenomenon and neuroimaging and neuroanatomical studies, Carlsson (1998) proposed that infantile autism is a hypoglutamatergic disorder. Recently, both the use of an NMDAR agonist and antagonist has been reported in ASDs patients. Daily doses of D-cycloserine, an NMDAR glycine site partial agonist, significantly improved social withdrawal (Posey et al., 2004), and daily doses of amantadine (memantine), an NMDAR non-competitive antagonist, reduced some negative symptoms of autism, such as hyperactivity (King et al., 2001; Chez et al., 2007).
mGluRs are members of the group C family of G-protein-coupled receptors. mGluRs have seven transmembrane domains that span the cell membrane. Differently to iGluRs, they are not ion channels. There are eight different types of mGluRs, namely mGluR1 to mGluR8, which are divided into three groups, group 1, group 2, and group 3. mGluR1 and mGluR5 belong to group 1 family, mGluR2, mGluR3, and mGluR4 belong to group 2 family, and mGluR6, mGluR7, and mGluR8 belong to group 3 family. They can regulate neuronal excitability, learning, and memory.
A study using high-throughput multiplex sequencing revealed significant enrichment of rare functional variants in the mGluR pathway in non-syndromic autism cases. (Kelleher et al., 2012). Most recently, in a valproate-induced rat model of autism, the expressions of mGluR2/3 protein and mGluR2 mRNA were found significantly reduced. N-acetylcysteine (NAC) recued social interaction and anxiety-like behaviors of the VPA-exposed rats. In addition, these effects can be blocked by intra-amygdala infusion of mGluR2/3 antagonist LY341495 (Chen et al., 2014). These results indicate that the disruption of social interaction in VPA induced rats could be restored by NAC, which may depend on the activation of mGluR2/3.
A decrease in mGluR has been found in PTEN knockout mice showing autism-like behavioral deficits (Lugo et al., 2014). By reducing 50% of mGluR5 expression, several abnormalities of Fmr1 knockout mice can be rescued. For example, density of dendritic spines on cortical pyramidal neurons and basal protein synthesis in hippocampus are increased, inhibitory avoidance extinction and audiogenic seizures are improved (Dolen et al., 2007).
In this review, we have summarized findings about some synapse proteins and receptors linked to ASDs. Due to different sample sizes and research methods, some results need further replication in additional and larger samples. For some of the synapse protein defects described in this review, animal model studies are lacking. Furthermore, genetic mutations only have been found in some ASDs subjects. Many patients do not exhibit these types of changes. Other signaling pathways, such as MAPK/JNK, have been correlated with synapse pathways in the pathogenesis of ASDs. Therefore, an intriguing question for future work is whether other signaling pathways have crosstalk with synapse pathways during the occurrence of ASDs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author gratefully acknowledges the support of Shanghai Jiao Tong University K.C. Wong Medical Fellowship Fund and the National Basic Research Program 973 of China (Grant No. 2012CB 517900).
Alarcón, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M.,et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
American Psychiatry Association [APA]. (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Arlington, VA: American Psychiatric Publishing.
Anitha, A., Thanseem, I., Nakamura, K., Yamada, K., Iwayama, Y., Toyota, T.,et al. (2013). Protocadherin alpha (PCDHA) as a novel susceptibility gene for autism. J. Psychiatry Neurosci. 38, 192–198. doi: 10.1503/jpn.120058
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M.,et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164. doi: 10.1016/j.ajhg.2007.09.015
Asperger, H. (1944). Die ‘autistischen psychopathen’ im kindesalter. Arch. Psychiatr. Nervenkr. 117, 76–136. doi: 10.1007/BF01837709
Autism Genome Project Consortium, Szatmari, P., Paterson, A. D., Zwaigenbaum, L., Roberts, W., Brian, J.,et al. (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328. doi: 10.1038/ng1985
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M.,et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Barnby, G., Abbott, A., Sykes, N., Morris, A., Weeks, D. E., Mott, R.,et al. (2005). Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 76, 950–966. doi: 10.1086/430454
Berkel, S., Marshall, R. C., Weiss, B., Howe, J., Roeth, R., Moog, U.,et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Berry-Kravis, E. M., Hessl, D., Rathmell, B., Zarevics, P., Cherubini, M., Walton-Bowen, K.,et al. (2012). Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci. Transl. Med. 4:152ra127. doi: 10.1126/scitranslmed.3004214
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412. doi: 10.1016/j.tins.2009.04.003
Billstedt, E., Gillberg, I. C., and Gillberg, C. (2005). Autism after adolescence: population-based 13- to 22-year follow-up study of 120 individuals with autism diagnosed in childhood. J. Autism Dev. Disord. 35, 351–360. doi: 10.1007/s10803-005-3302-5
Blasi, F., Bacchelli, E., Pesaresi, G., Carone, S., Bailey, A. J., Maestrini, E.,et al. (2006). Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 220–221. doi: 10.1002/ajmg.b.30287
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., and Bauman, M. L. (2001). Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J. Autism Dev. Disord. 31, 537–543. doi: 10.1023/A:1013238809666
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A.,et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Bolton, P. F., Dennis, N. R., Browne, C. E., Thomas, N. S., Veltman, M. W., Thompson, R. J.,et al. (2001). The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am. J. Med. Genet. 105, 675–685. doi: 10.1002/ajmg.1551
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N.,et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1:15. doi: 10.1186/2040-2392-1-15
Bremer, A., Giacobini, M., Eriksson, M., Gustavsson, P., Nordin, V., Fernell, E.,et al. (2011). Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 115–124. doi: 10.1002/ajmg.b.31142
Carlsson, M. L. (1998). Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate–serotonin interactions for pharmacotherapy. J. Neural. Transm. 105, 525–535. doi: 10.1007/s007020050076
Chen, Y. W., Lin, H. C., Ng, M. C., Hsiao, Y. H., Wang, C. C., Gean, P. W.,et al. (2014). Activation of mGluR2/3 underlies the effects of N-acetylcystein on amygdala-associated autism-like phenotypes in a valproate-induced rat model of autism. Front. Behav. Neurosci. 8:219. doi: 10.3389/fnbeh.2014.00219
Chez, M. G., Burton, Q., Dowling, T., Chang, M., Khanna, P., and Kramer, C. (2007). Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J. Child Neurol. 22, 574–579. doi: 10.1177/0883073807302611
Chubykin, A. A., Atasoy, D., Etherton, M. R., Brose, N., Kavalali, E. T., Gibson, J. R.,et al. (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. doi: 10.1016/j.neuron.2007.05.029
Corradi, A., Fadda, M., Piton, A., Patry, L., Marte, A., Rossi, P.,et al. (2014). SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 23, 90–103. doi: 10.1093/hmg/ddt401
de Anda, F. C., Rosario, A. L., Durak, O., Tran, T., Gräff, J., Meletis, K.,et al. (2012). Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 15, 1022–1031. doi: 10.1038/nn.3141
Dean, C., and Dresbach, T. (2006). Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 29, 21–29. doi: 10.1016/j.tins.2005.11.003
Depienne, C., Moreno-De-Luca, D., Heron, D., Bouteiller, D., Gennetier, A., Delorme, R.,et al. (2009). Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol. Psychiatry 66, 349–359. doi: 10.1016/j.biopsych.2009.01.025
Dhar, S. U., del Gaudio, D., German, J. R., Peters, S. U., Ou, Z., Bader, P. I.,et al. (2010). 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. Am. J. Med. Genet. A 152A, 573–581. doi: 10.1002/ajmg.a.33253
Dolen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S.,et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F.,et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Elsabbagh, M., Divan, G., Koh, Y. J., Kim, Y. S., Kauchali, S., Marcín, C.,et al. (2012). Global prevalence of autism and other pervasive developmental disorders. Autism Res. 5, 160–179. doi: 10.1002/aur.239
Erickson, C. A., Veenstra-Vanderweele, J. M., Melmed, R. D., McCracken, J. T., Ginsberg, L. D., Sikich, L.,et al. (2014). STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J. Autism Dev. Disord. 44, 958–964. doi: 10.1007/s10803-013-1963-z
Etherton, M. R., Blaiss, C. A., Powell, C. M., and Südhof, T. C. (2009). Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. U.S.A. 106, 17998–18003. doi: 10.1073/pnas.0910297106
Etherton, M., Földy, C., Sharma, M., Tabuchi, K., Liu, X., Shamloo, M.,et al. (2011a). Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. U.S.A. 108, 13764–13769. doi: 10.1073/pnas.1111093108
Etherton, M. R., Tabuchi, K., Sharma, M., Ko, J., and Südhof, T. C. (2011b). An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 30, 2908–2919. doi: 10.1038/emboj.2011.182
Fassio, A., Patry, L., Congia, S., Onofri, F., Piton, A., Gauthier, J.,et al. (2011). SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 20, 2297–2307. doi: 10.1093/hmg/ddr122
Fatemi, S. H., Folsom, T. D., Reutiman, T. J., and Thuras, P. D. (2009). Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum 8, 64–69. doi: 10.1007/s12311-008-0075-3
Fatemi, S. H., Halt, A. R., Stary, J. M., Kanodia, R., Schulz, S. C., and Realmuto, G. R. (2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810. doi: 10.1016/S0006-3223(02)01430-0
Feyder, M., Karlsson, R. M., Mathur, P., Lyman, M., Bock, R., Momenan, R.,et al. (2010). Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am. J. Psychiatry 167, 1508–1517. doi: 10.1176/appi.ajp.2010.10040484
Földy, C., Malenka, R. C., and Südhof, T. C. (2013). Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 78, 498–509. doi: 10.1016/j.neuron.2013.02.036
Fornasiero, E. F., Raimondi, A., Guarnieri, F. C., Orlando, M., Fesce, R., Benfenati, F.,et al. (2012). Synapsins contribute to the dynamic spatial organization of synaptic vesicles in an activity-dependent manner. J. Neurosci. 32, 12214–12227. doi: 10.1523/JNEUROSCI.1554-12.2012
Friedman, A., and Luiselli, J. K. (2008). Excessive daytime sleep: behavioral assessment and intervention in a child with autism. Behav. Modif. 32, 548–555. doi: 10.1177/0145445507312187
Gauthier, J., Bonnel, A., St-Onge, J., Karemera, L., Laurent, S., Mottron, L.,et al. (2005). NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 132B, 74–75. doi: 10.1002/ajmg.b.30066
Gauthier, J., Siddiqui, T. J., Huashan, P., Yokomaku, D., Hamdan, F. F., Champagne, N.,et al. (2011). Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 130, 563–573. doi: 10.1007/s00439-011-0975-z
Gauthier, J., Spiegelman, D., Piton, A., Lafrenière, R. G., Laurent, S., St-Onge, J.,et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 421–424. doi: 10.1002/ajmg.b.30822
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S.,et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Greco, B., Managò, F., Tucci, V., Kao, H. T., Valtorta, F., and Benfenati, F. (2013). Autism-related behavioral abnormalities in synapsin knockout mice. Behav. Brain Res. 251, 65–74. doi: 10.1016/j.bbr.2012.12.015
Gregor, A., Albrecht, B., Bader, I., Bijlsma, E. K., Ekici, A. B., Engels, H.,et al. (2011). Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med. Genet. 12:106. doi: 10.1186/1471-2350-12-106
Grosskreutz, Y., Betz, H., and Kneussel, M. (2003). Rescue of molybdenum cofactor biosynthesis in gephyrin-deficient mice by a Cnx1 transgene. Biochem. Biophys. Res. Commun. 301, 450–455. doi: 10.1016/S0006-291X(02)03062-0
Guilmatre, A., Dubourg, C., Mosca, A. L., Legallic, S., Goldenberg, A., Drouin-Garraud, V.,et al. (2009). Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry 66, 947–956. doi: 10.1001/archgenpsychiatry.2009.80
Guilmatre, A., Huguet, G., Delorme, R., and Bourgeron, T. (2013). The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 74, 113–122. doi: 10.1002/dneu.22128
Guo, H., Xun, G., Peng, Y., Xiang, X., Xiong, Z., Zhang, L.,et al. (2012). Disruption of Contactin 4 in two subjects with autism in Chinese population. Gene 505, 201–205. doi: 10.1016/j.gene.2012.06.051
Harada, M., Taki, M. M., Nose, A., Kubo, H., Mori, K., Nishitani, H.,et al. (2011). Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 tesla instrument. J. Autism Dev. Disord. 41, 447–454. doi: 10.1007/s10803-010-1065-0
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R.,et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4:152ra128. doi: 10.1126/scitranslmed.3004218
Howlin, P., Goode, S., Hutton, J., and Rutter, M. (2004). Adult outcome for children with autism. J. Child Psychol. Psychiatry 45, 212–229. doi: 10.1111/j.1469-7610.2004.00215.x
Hung, A. Y., Futai, K., Sala, C., Valtschanoff, J. G., Ryu, J., Woodworth, M. A.,et al. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708. doi: 10.1523/JNEUROSCI.3032-07.2008
International Schizophrenia Consortium. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. doi: 10.1038/nature07239
Jamain, S., Betancur, C., Quach, H., Philippe, A., Fellous, M., Giros, B.,et al. (2002). Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 7, 302–310. doi: 10.1038/sj.mp.4000979
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I. C.,et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jamain, S., Radyushkin, K., Hammerschmidt, K., Granon, S., Boretius, S., Varoqueaux, F.,et al. (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. U.S.A. 105, 1710–1715. doi: 10.1073/pnas.0711555105
Kelleher, R. J. III, Geigenmüller, U., Hovhannisyan, H., Trautman, E., Pinard, R., Rathmell, B.,et al. (2012). High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS ONE 7:e35003. doi: 10.1371/journal.pone.0035003
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y.,et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
King, B. H., Wright, D. M., Handen, B. L., Sikich, L., Zimmerman, A. W., McMahon, W.,et al. (2001). Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 40, 658–665. doi: 10.1097/00004583-200106000-00010
King, M., and Bearman, P. (2009). Diagnostic change and the increased prevalence of autism. Int. J. Epidemiol. 38, 1224–1234. doi: 10.1093/ije/dyp261
Kneussel, M., Brandstätter, J. H., Laube, B., Stahl, S., Müller, U., and Betz, H. (1999). Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J. Neurosci. 19, 9289–9297.
Kwasnicka-Crawford, D. A., Roberts, W., and Scherer, S. W. (2007). Characterization of an autism-associated segmental maternal heterodisomy of the chromosome 15q11-13 region. J. Autism Dev. Disord. 37, 694–702. doi: 10.1007/s10803-006-0225-8
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi: 10.1016/S0140-6736(13)61539-1
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P.,et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi: 10.1086/382137
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G.,et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Levy, D., Ronemus, M., Yamrom, B., Lee, Y. H., Leotta, A., Kendall, J.,et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897. doi: 10.1016/j.neuron.2011.05.015
Li, X., Zou, H., and Brown, W. T. (2012). Genes associated with autism spectrum disorder. Brain Res. Bull. 88, 543–552. doi: 10.1016/j.brainresbull.2012.05.017
Lim, S., Naisbitt, S., Yoon, J., Hwang, J. I., Suh, P. G., Sheng, M.,et al. (1999). Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 274, 29510–29518. doi: 10.1074/jbc.274.41.29510
Lintas, C., and Persico, A. M. (2009). Autistic phenotypes and genetic testing: state-of-the-art for the clinical geneticist. J. Med. Genet. 46, 1–8. doi: 10.1136/jmg.2008.060871
Lionel, A. C., Crosbie, J., Barbosa, N., Goodale, T., Thiruvahindrapuram, B., Rickaby, J.,et al. (2011). Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 3:95ra75. doi: 10.1126/scitranslmed.3002464
Lionel, A. C., Vaags, A. K., Sato, D., Gazzellone, M. J., Mitchell, E. B., Chen, H. Y.,et al. (2013). Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 22, 2055–2066. doi: 10.1093/hmg/ddt056
Liu, Y., Du, Y., Liu, W., Yang, C., Liu, Y., Wang, H.,et al. (2013). Lack of association between NLGN3, NLGN4, SHANK2 and SHANK3 gene variants and autism spectrum disorder in a Chinese population. PLoS ONE 8:e56639. doi: 10.1371/journal.pone.0056639
Lotter, V. (1966). Epidemiology of autistic conditions in young children. Soc. Psychiatry Psychiatr. Epidemiol. 1, 124–135.
Lugo, J. N., Smith, G. D., Arbuckle, E. P., White, J., Holley, A. J., Floruta, C. M.,et al. (2014). Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front. Mol. Neurosci. 7:27. doi: 10.3389/fnmol.2014.00027
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J.,et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
Martin, E. R., Menold, M. M., Wolpert, C. M., Bass, M. P., Donnelly, S. L., Ravan, S. A.,et al. (2000). Analysis of linkage disequilibrium in gamma-aminobutyric acid receptor subunit genes in autistic disorder. Am. J. Med. Genet. 96, 43–48. doi: 10.1002/(SICI)1096-8628(20000207)96:1<43::AID-AJMG9>3.0.CO;2-3
Meguro-Horike, M., Yasui, D. H., Powell, W., Schroeder, D. I., Oshimura, M., Lasalle, J. M.,et al. (2011). Neuron-specific impairment of inter-chromosomal pairing and transcription in a novel model of human 15q-duplication syndrome. Hum. Mol. Genet. 20, 3798–3810. doi: 10.1093/hmg/ddr298
Mendez, M. A., Horder, J., Myers, J., Coghlan, S., Stokes, P., Erritzoe, D.,et al. (2013). The brain GABA-benzodiazepine receptor alpha-5 subtype in autism spectrum disorder: a pilot [(11)C]Ro15-4513 positron emission tomography study. Neuropharmacology 68, 195–201. doi: 10.1016/j.neuropharm.2012.04.008
Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R. E., Gottmann, K.,et al. (2003). Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948. doi: 10.1038/nature01755
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J.,et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297. doi: 10.1086/522590
Mori, T., Mori, K., Fujii, E., Toda, Y., Miyazaki, M., Harada, M.,et al. (2012). Evaluation of the GABAergic nervous system in autistic brain: (123)I-iomazenil SPECT study. Brain Dev. 34, 648–654. doi: 10.1016/j.braindev.2011.10.007
Morrow, E. M., Yoo, S. Y., Flavell, S. W., Kim, T. K., Lin, Y., Hill, R. S.,et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. doi: 10.1126/science.1157657
Murdoch, J. D., and State, M. W. (2013). Recent developments in the genetics of autism spectrum disorders. Curr. Opin. Genet. Dev. 23, 310–315. doi: 10.1016/j.gde.2013.02.003
Nakatani, J., Tamada, K., Hatanaka, F., Ise, S., Ohta, H., Inoue, K.,et al. (2009). Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246. doi: 10.1016/j.cell.2009.04.024
Novarino, G., El-Fishawy, P., Kayserili, H., Meguid, N. A., Scott, E. M., Schroth, J.,et al. (2012). Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338, 394–397. doi: 10.1126/science.1224631
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P.,et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Oblak, A., Gibbs, T. T., and Blatt, G. J. (2009). Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2, 205–219. doi: 10.1002/aur.88
Oblak, A., Gibbs, T. T., and Blatt, G. J. (2013). Reduced serotonin receptor subtypes in a limbic and a neocortical region in autism. Autism Res. 6, 571–583. doi: 10.1002/aur.1317
Oblak, A. L., Gibbs, T. T., and Blatt, G. J. (2011). Reduced GABAA receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Res. 1380, 218–228. doi: 10.1016/j.brainres.2010.09.021
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N.,et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peñagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H.,et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Phelan, M. C. (2008). Deletion 22q13.3 syndrome. Orphanet J. Rare Dis. 3:14. doi: 10.1186/1750-1172-3-14
Pinto, D. A., Pagnamenta, T., Klei, L., Anney, R., Merico, D., Regan, R.,et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Posey, D. J., Kem, D. L., Swiezy, N. B., Sweeten, T. L., Wiegand, R. E., and McDougle, C. J. (2004). A pilot study of D-cycloserine in subjects with autistic disorder. Am. J. Psychiatry 161, 2115–2117. doi: 10.1176/appi.ajp.161.11.2115
Poulopoulos, A., Aramuni, G., Meyer, G., Soykan, T., Hoon, M., Papadopoulos, T.,et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. doi: 10.1016/j.neuron.2009.08.023
Purcell, A. E., Jeon, O. H., Zimmerman, A. W., Blue, M. E., and Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628. doi: 10.1212/WNL.57.9.1618
Ramanathan, S., Woodroffem, A., Flodman, P. L., Mays, L. Z., Hanouni, M., Modahl, C. B.,et al. (2004). A case of autism with an interstitial deletion on 4q leading to hemizygosity for genes encoding for glutamine and glycine neurotransmitter receptor sub-units (AMPA 2, GLRA3, GLRB) and neuropeptide receptors NPY1R, NPY5R. BMC Med. Genet. 5:10. doi: 10.1186/1471-2350-5-10
Redies, C., Hertel, N., and Hübner, C. A. (2012). Cadherins and neuropsychiatric disorders. Brain Res. 1470, 130–144. doi: 10.1016/j.brainres.2012.06.020
Rice, C. (2012). Prevalence of autism spectrum disorders – Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill. Summ. 61, 1–19.
Roohi, J., Montagna, C., Tegay, D. H., Palmer, L. E., DeVincent, C., Pomeroy, J. C.,et al. (2009). Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 46, 176–182. doi: 10.1136/jmg.2008.057505
Rosahl, T. W., Spillane, D., Missler, M., Herz, J., Selig, D. K., Wolff, J. R.,et al. (1995). Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493. doi: 10.1038/375488a0
Sadakata, T., Washida, M., Iwayama, Y., Shoji, S., Sato, Y., Ohkura, T.,et al. (2007). Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Invest. 117, 931–943. doi: 10.1172/JCI29031
Sampath, S., Bhat, S., Gupta, S., O’Connor, A., West, A. B., Arking, D. E.,et al. (2013). Defining the contribution of CNTNAP2 to autism susceptibility. PLoS ONE 8:e77906. doi: 10.1371/journal.pone.0077906
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D.,et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Sanz-Clemente, A., Nicoll, R. A., and Roche, K. W. (2013). Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75. doi: 10.1177/1073858411435129
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S.,et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Saunders, J. A., Tatard-Leitman, V. M., Suh, J., Billingslea, E. N., Roberts, T. P., and Siegel, S. J. (2013). Knockout of NMDA receptors in parvalbumin interneurons recreates autism-like phenotypes. Autism Res. 6, 69–77. doi: 10.1002/aur.1264
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A.,et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Schnell, E., Bensen, A. L., Washburn, E. K., and Westbrook, G. L. (2012). Neuroligin-1 overexpression in newborn granule cells in vivo. PLoS ONE 7:e48045. doi: 10.1371/journal.pone.0048045
Sheng, M., and Kim, E. (2000). The Shank family of scaffold proteins. J. Cell Sci. 113(Pt 11), 1851–1856.
Silverman, J. L., Turner, S. M., Barkan, C. L., Tolu, S. S., Saxena, R., Hung, A. Y.,et al. (2011). Sociability and motor functions in Shank1 mutant mice. Brain Res. 1380, 120–137. doi: 10.1016/j.brainres.2010.09.026
Simonoff, E., Pickles, A., Charman, T., Chandler, S., Loucas, T., and Baird, G. (2008). Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry 47, 921–929. doi: 10.1097/CHI.0b013e318179964f
Stein, M. B., Yang, B. Z., Chavira, D. A., Hitchcock, C. A., Sung, S. C., Shipon-Blum, E.,et al. (2011). A common genetic variant in the neurexin superfamily member CNTNAP2 is associated with increased risk for selective mutism and social anxiety-related traits. Biol. Psychiatry 69, 825–831. doi: 10.1016/j.biopsych.2010.11.008
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M.,et al. (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 354, 1370–1377. doi: 10.1056/NEJMoa052773
Sundaram, S. K., Huq, A. M., Wilson, B. J., and Chugani, H. T. (2010). Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 74, 1583–1590. doi: 10.1212/WNL.0b013e3181e0f147
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M.,et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Tabuchi, K., and Sudhof, T. C. (2002). Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics 79, 849–859. doi: 10.1006/geno.2002.6780
Talebizadeh, Z., Bittel, D. C., Veatch, O. J., Butler, M. G., Takahashi, T. N., and Miles, J. H. (2004). Do known mutations in neuroligin genes (NLGN3 and NLGN4) cause autism? J. Autism Dev. Disord. 34, 735–736. doi: 10.1007/s10803-004-5295-x
Tarabeux, J., Kebir, O., Gauthier, J., Hamdan, F. F., Xiong, L., Piton, A.,et al. (2011). Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 1:e55. doi: 10.1038/tp.2011.52
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S.,et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141. doi: 10.1016/j.ajhg.2011.11.025
Vincent, J. B., Kolozsvari, D., Roberts, W. S., Bolton, P. F., Gurling, H. M., and Scherer, S. W. (2004). Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am. J. Med. Genet. B Neuropsychiatr. Genet. 129B, 82–84. doi: 10.1002/ajmg.b.30069
Waga, C., Okamoto, N., Ondo, Y., Fukumura-Kato, R., Goto, Y., Kohsaka, S.,et al. (2011). Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr. Genet. 21, 208–211. doi: 10.1097/YPG.0b013e328341e069
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M.,et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S.,et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C.,et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R.,et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675. doi: 10.1056/NEJMoa075974
Wermter, A. K., Kamp-Becker, I., Strauch, K., Schulte-Körne, G., and Remschmidt, H. (2008). No evidence for involvement of genetic variants in the X-linked neuroligin genes NLGN3 and NLGN4X in probands with autism spectrum disorder on high functioning level. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 535–537. doi: 10.1002/ajmg.b.30618
Wilson, H. L., Crolla, J. A., Walker, D., Artifoni, L., Dallapiccola, B., Takano, T.,et al. (2008). Interstitial 22q13 deletions: genes other than SHANK3 have major effects on cognitive and language development. Eur. J. Hum. Genet. 16, 1301–1310. doi: 10.1038/ejhg.2008.107
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C.,et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40, 575–584. doi: 10.1136/jmg.40.8.575
Wöhr, M., Roullet, F. I., Hung, A. Y., Sheng, M., and Crawley, J. N. (2011). Communication impairments in mice lacking Shank1: reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS ONE 6:e20631. doi: 10.1371/journal.pone.0020631
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J.,et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Yan, J., Oliveira, G., Coutinho, A., Yang, C., Feng, J., Katz, C.,et al. (2005). Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 10, 329–332. doi: 10.1038/sj.mp.4001629
Yang, M., Bozdagi, O., Scattoni, M. L., Wöhr, M., Roullet, F. I., Katz, A. M.,et al. (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 32, 6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012
Yip, J., Soghomonian, J. J., and Blatt, G. J. (2007). Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol. 113, 559–568. doi: 10.1007/s00401-006-0176-3
Yoo, H. J., Cho, I. H., Park, M., Yang, S. Y., and Kim, S. A. (2012). Family based association of GRIN2A and GRIN2B with Korean autism spectrum disorders. Neurosci. Lett. 512, 89–93. doi: 10.1016/j.neulet.2012.01.061
Yoo, J., Bakes, J., Bradley, C., Collingridge, G. L., and Kaang, B. K. (2014). Shank mutant mice as an animal model of autism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130143. doi: 10.1098/rstb.2013.0143
Zhang, D., Cheng, L., Qian, Y., Alliey-Rodriguez, N., Kelsoe, J. R., Greenwood, T.,et al. (2009). Singleton deletions throughout the genome increase risk of bipolar disorder. Mol. Psychiatry 14, 376–380. doi: 10.1038/mp.2008.144
Keywords: autism spectrum disorders, synaptic protein, GABA, PSD-95, SHANK3, TAOK2
Citation: Chen J, Yu S, Fu Y and Li X (2014) Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 8:276. doi: 10.3389/fncel.2014.00276
Received: 03 June 2014; Accepted: 21 August 2014;
Published online: 11 September 2014.
Edited by:
Laurie Doering, McMaster University, CanadaReviewed by:
Enrico Cherubini, International School for Advanced Studies, ItalyCopyright © 2014 Chen, Yu, Fu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shunying Yu, Shanghai Key Laboratory of Psychotic Disorders, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, 600 Wanping Nan Road, Shanghai 200030, China e-mail:eXVzaHVueUBnbWFpbC5jb20=
†Jianling Chen and Shunying Yu have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.