 Mauricio O. Nava-Mesa
Mauricio O. Nava-Mesa Lydia Jiménez-Díaz
Lydia Jiménez-Díaz Javier Yajeya
Javier Yajeya Juan D. Navarro-Lopez
Juan D. Navarro-Lopez- 1Neuroscience Research Group, University of Rosario, Bogotá, Colombia
- 2Neurophysiology and Behavior Lab, Centro Regional de Investigaciones Biomédicas, School of Medicine of Ciudad Real, University of Castilla-La Mancha, Ciudad Real, Spain
- 3Department of Physiology and Pharmacology, University of Salamanca, Salamanca, Spain
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by cognitive decline, brain atrophy due to neuronal and synapse loss, and formation of two pathological lesions: extracellular amyloid plaques, composed largely of amyloid-beta peptide (Aβ), and neurofibrillary tangles formed by intracellular aggregates of hyperphosphorylated tau protein. Lesions mainly accumulate in brain regions that modulate cognitive functions such as the hippocampus, septum or amygdala. These brain structures have dense reciprocal glutamatergic, cholinergic, and GABAergic connections and their relationships directly affect learning and memory processes, so they have been proposed as highly susceptible regions to suffer damage by Aβ during AD course. Last findings support the emerging concept that soluble Aβ peptides, inducing an initial stage of synaptic dysfunction which probably starts 20–30 years before the clinical onset of AD, can perturb the excitatory–inhibitory balance of neural circuitries. In turn, neurotransmission imbalance will result in altered network activity that might be responsible of cognitive deficits in AD. Therefore, Aβ interactions on neurotransmission systems in memory-related brain regions such as amygdaloid complex, medial septum or hippocampus are critical in cognitive functions and appear as a pivotal target for drug design to improve learning and dysfunctions that manifest with age. Since treatments based on glutamatergic and cholinergic pharmacology in AD have shown limited success, therapies combining modulators of different neurotransmission systems including recent findings regarding the GABAergic system, emerge as a more useful tool for the treatment, and overall prevention, of this dementia. In this review, focused on inhibitory systems, we will analyze pharmacological strategies to compensate neurotransmission imbalance that might be considered as potential therapeutic interventions in AD.
Introduction
Along last three decades, dementias are becoming a worldwide epidemiological problem. The importance of understanding the molecular basis of dementias and designing rational therapies for its treatment is of growing interest for populations where life expectancy along with concerns for a better quality of life are increasing. In December 2005, it was estimated that there were 24.3 million people living with dementia, there would be 31 million in 2010, and people affected by dementia will double every 20 years, rising to 81.1 million in 2040. But the reality is even worse than those approximations. People with dementia need a great amount of support and care that imply a high cost in terms of emotional, social, and financial resources that are mainly provided by their families with the help of local governments or insurance companies. Being ailments that run over a significant time period, the direct and indirect cost of medical care, employment of domestic caregivers, lost productivity in the immediate family, etc., is enormous. A joint effort from researches and health authorities needs to be made to deepen understanding of the etiology and physiopathology of these diseases and therefore develop therapies that improve health and welfare of people with dementia.
Alzheimer’s disease (AD) is the most prevalent cause of dementia among more than a hundred dementia types, and is the major cause of dementia in the elderly (around 50% for age range of 80–89 years old). According to the World Alzheimer Report 2010–2012 (Alzheimer’s Disease International), in 2010 there were about 36 million cases of AD and other dementias in the world, which will increase to 115.4 million in 2050. AD is a devastating progressive neurodegenerative disease characterized by cognitive decline, brain atrophy due to neuronal and synapse loss, and two neuropathological lesions firstly described by Alois Alzheimer in 1907: extracellular amyloid plaques and neurofibrillary tangles formation, composed of amyloid-beta peptide (Aβ) and intracellular aggregates of hyperphosphorylated tau protein, respectively (Goedert and Spillantini, 2006). Together with Parkinson’s disease, Huntington’s disease, transmissible spongiform encephalopathies and amyotrophic lateral sclerosis, AD is one of the neurodegenerative diseases that presents a pathological common mechanism (Soto, 2003) consistent on conformational disorders of a particular protein which can fold into a stable alternative conformation. In most cases, this alteration results in its aggregation and accumulation in tissues as fibrillar deposits that finally induce neuronal death (Bucciantini et al., 2002).
The events that trigger the main pathological changes in AD take place in regions of the temporal lobe, including the medial septum, hippocampus, amygdala, and entorhinal cortex. The early onset of AD is manifested as an inability to form new memories. However, the multiple structural and biochemical changes which are already documented in the mid-to-late stages of AD (such as synapse loss, plaque accumulation, tangle formation, and neurodegeneration) do not explain the memory deficits observed in the early stages of the illness (Selkoe, 2002). For example, the loss of synapses appears to be the best morphological correlate for functional deficits observed in the middle and late stages of AD, but many patients in early stages do not show a significant decline in number of synapses (Terry, 2004; Kelly et al., 2005; Spires-Jones and Knafo, 2012).
Based on these findings, attempts have been made to find an explanation for cognitive deficits observed at early stages of the disease when no significant decline in the synapse and cell number has been detected. It has been proposed that misfolded oligomeric forms or small Aβ aggregates that are not deposited in the tissue might induce an initial state of synaptic dysfunction in early AD patients. Numerous genetic, biochemical, and animal model studies have implicated the gradual contribution of Aβ, as a medium for AD. In this sense, it has also been suggested that insoluble amyloid plaques would also have a pathogenic role serving as relatively inert reservoirs of soluble toxic Aβ aggregates that could readily be activated and disassembled by exposure to biological lipids (Martins et al., 2008). This synaptic dysfunction scenario could explain the cognitive deficits observed in the early stages of AD and, thus, precede synapse loss, plaque accumulation, tangle formation, and neurodegeneration (Klein, 2002; Selkoe, 2002; Soto, 2003). However, the mechanisms underlying functional deficits are not known yet.
During the last decade it has been suggested that an imbalance between excitatory and inhibitory neurotransmission systems might underlie the synaptic dysfunction caused by Aβ (Palop et al., 2007; Sun et al., 2009; Palop and Mucke, 2010a; Verret et al., 2012). Pharmacological treatments based on modulating excitatory and/or inhibitory neurotransmission have shown to improve AD symptoms (Farlow, 2009; McKeage, 2009), so that strategies aimed to reestablish the balance between both systems, particularly in early stages of the disease, seem to be the most appropriate to act on the functional deficits caused by Aβ (Huang and Mucke, 2012; Mucke and Selkoe, 2012; Verret et al., 2012).
In this regard, the present paper will review the state of the art of Aβ interactions on excitatory and mainly inhibitory neurotransmission in memory-related brain systems such as amygdaloid complex and septohippocampal system. These regions have shown to be critical in cognitive functions and their neurotransmission systems, particularly the inhibitory one, emerge as pivotal targets for drug design studies to improve learning processes and cognitive dysfunctions that manifest with age.
Aβ and Excitatory Neurotransmission
Several hypotheses have been postulated to explain the neurotoxicity of soluble Aβ aggregates on excitatory neurotransmission systems. Some of these proposals include a cascade of reactions that could involve the blockade of the glutamate recruitment by microglia (Hickman et al., 2008), alteration of the glutamatergic neurotransmission (Ashenafi et al., 2005; Santos-Torres et al., 2007), or modification of both glutamate N-methyl-D-aspartate (NMDA) and/or α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/Kainate (AMPA/Kainate) receptors endocytosis process (Hsieh et al., 2006; Uemura et al., 2007). Other authors consider intracellular calcium increase as the neurotoxic mechanism (Rovira et al., 2002; Resende et al., 2007). Data from Gu et al. (2003) support the cholinergic AD theory and therefore affectation of muscarinic receptors (Kar et al., 1996), suggesting alterations of potassium channels as Aβ action mechanism (Zhang and Yang, 2006). None of these possible mechanisms have completely been discarded at the moment.
The hypothesis explaining Aβ neurotoxic effects through actions on glutamatergic receptors have received important supports. Neuroprotection against Aβ toxic effects has been described by NMDA receptor blockade with MK801. This result supports the idea that a persistent hyperpolarization can reduce the Aβ neurotoxicity due to inactivation of NMDA receptors (Harkany et al., 1999). In accordance, reduction of clinical deterioration in the initial AD phases has also been described using the NMDA glutamatergic non-competitive antagonist memantine (O’Mahony et al., 1998). Depression of glutamatergic response by Aβ perfusion has been reported using electrophysiological recordings in amygdala and septum (Ashenafi et al., 2005; Santos-Torres et al., 2007). Other authors support that Aβ alters glutamatergic transmission affecting both metabotropic (Shankar et al., 2008; Um et al., 2013) or AMPA/Kainate receptors endocytosis (Hsieh et al., 2006). It has also been proposed that Aβ effect would be mediated by increased NMDA receptors endocytosis (Uemura et al., 2007) and other authors even postulate that Aβ acting on metabotropic receptors (mGluRI) interferes with the regulation of GABAergic transmission (Tyszkiewicz and Yan, 2005).
On the other hand, results supporting the cholinergic theory to explain the mechanisms that underlie AD have also been shown by many authors (Langmead et al., 2008). Muscarinic receptors, specifically M1 subtype, have been broadly related to AD. M1 subtype receptor is widely distributed in the brain and is expressed postsynaptically in cortex and hippocampus (Levey et al., 1991, 1995), important areas for learning and memory. In vitro studies have demonstrated that activation of muscarinic receptors induces an alternative pathway for amyloid precursor protein (APP) processing which increases secretion of APP soluble fraction and then reduces Aβ toxicity (Fisher et al., 2002). In particular, selective M1 agonist, AF267B, attenuates the major hallmarks of AD and reverses deficits in cognition (Caccamo et al., 2006, 2009). However, recent results suggest that a decrease in IM (the potassium current activated by muscarinic receptor stimulation) may be an integral part of AD pathophysiology (Leao et al., 2012; Duran-Gonzalez et al., 2013), explaining why IM blockers fail to improve cognition in AD clinical trials (Rockwood et al., 1997). Evidence also points out that the initial injurious effects of the fragment of Aβ, Aβ1-42, on M1 muscarinic receptor-mediated transmission is due to compromised coupling of the receptor with Gq/11 G-protein (Janickova et al., 2013). Nicotinergic neurotransmission has also been involved in AD early stages, not only through an activation of presynaptic α7-nicotinic acetylcholine receptors (α7-nAChR; Dougherty et al., 2003) but also by interaction with GABAergic (Spencer et al., 2006) and glutamatergic (Wang et al., 2009) systems.
Then in synaptic dysfunction processes, Aβ has been found to present differential effects on AMPA and NMDA receptors. NMDA has been related to Aβ neurotoxicity phenomena. However, cholinergic disruption induced by Aβ can be established at different levels, including cholinergic neurodegeneration, alterations in acetylcholine release, direct modulation of muscarinic receptors and associated effectors, or nicotinergic system.
Glial Cells in the Physiopathology of AD
Recent reports support the new concept that cognitive function arises from a cooperative activity between both neurons and glia (Perea et al., 2009; Fields et al., 2014). This neuron–glia network integrates information and controls synaptic transmission and plasticity in an active way. The term “tripartite synapse” was proposed in order to describe this cellular configuration which involves presynaptic neuron, postsynaptic neuron, and astrocytes (Araque et al., 1999; Perea et al., 2009). Astrocyte-induced neuromodulation has been described in particular brain structures such as hippocampus (Araque et al., 1998; Jourdain et al., 2007), cortex (Ding et al., 2007), and hypothalamus (Gordon et al., 2005). Several gliotransmitters released from astrocytes modulate synaptic plasticity in those brain structures (Yang et al., 2003; Pascual et al., 2005; Panatier et al., 2006) and participate in learning and memory processes.
Disruption of astrocytic functions and therefore in gliotransmission may underline several brain disorders (i.e., depression, schizophrenia, and epilepsy; Rajkowska et al., 1999; Cohen-Gadol et al., 2004; Fellin et al., 2004; Webster et al., 2005), as well as specific neurodegenerative diseases (i.e., parkinsonism; Forman et al., 2005; Halassa et al., 2007). In fact, enhanced astrocytic tau expression in aged transgenic animals results in glutamate-transporter activity reduction and consequent neurodegeneration (Komori, 1999; Dabir et al., 2004). In the context of AD, Aβ can disrupt both astrocytic calcium signaling and glutamate uptake capacity (Vincent et al., 2010; Matos et al., 2012). It has been recently shown in an ex vivo astrocyte preparation, that Aβ1-42 reduces the expression of the two major glutamate transporters in astroglia, GLT-1 and GLAST, through Adenosine A2A receptors (de Vivo et al., 2010; Matos et al., 2012). Taking into account that glutamate transporters are necessary for the clearance of excitatory neurotransmitters, the resulting excitotoxic neuronal damage induced by higher levels of Aβ through this mechanism is reasonable in AD. On the other hand, the neuromodulatory function of astrocytes in particular brain structures may explain specific vulnerability to excitotoxicity and neurodegeneration.
Aβ pathological increases induce multiple glial morphological changes. In fact, astrocytes and microglia become activated close to senile plaques in order to internalize and degrade Aβ (Mohamed and Posse de, 2011). Oxidative stress and inflammatory response induced by astrocytes and microglia activation may have a dual role in pathophysiology of AD with neuroprotective and detrimental consequences (Maccioni et al., 2001; Schipper et al., 2006). This is the basis of the therapeutic use of non-steroidal anti-inflammatory drugs (NSAID) in order to delay AD onset, as well as to reduce the rate of disease progression (Cudaback et al., 2014). Unfortunately, no clinical trials are available to support and recommend its use to prevent AD. However, a novel compound (CHF5074) with both anti-inflammatory and gamma-secretase (an enzyme involved in APP processing) modulatory activities in animal models may have a possible therapeutic role to prevent AD (Calza et al., 2013).
As mentioned previously, AMPA and NMDA receptors have been widely implicated in the physiopathology of AD (for review Parameshwaran et al., 2008). Several studies reported that astrocytes express functional NMDA receptors (Kommers et al., 2002; Lalo et al., 2006; Verkhratsky and Kirchhoff, 2007) which are involved in neuronal–glial signaling, synaptic transmission and cerebral vasodilation (Lalo et al., 2006; Palygin et al., 2010; Parfenova et al., 2012). Therefore, Aβ-induced dysfunction of glutamate receptors might affect NMDA receptors expressed in glial cells and, as a consequence, disrupt neuron–glial signal transmission (Mota et al., 2014). NMDA receptor antagonists, MK801 and memantine, might attenuate glutamate mediated cell excitotoxicity by excessive stimulation of NMDA receptors in astrocytes and neurons (Lee et al., 2010). In addition, regarding differences between glial and neuronal NMDA receptors, a new selective antagonist (UBP141) of astroglial NMDA receptors with potential therapeutic role in neurodegenerative diseases has been developed (Palygin et al., 2011). Finally, Talantova et al. (2013) showed that Aβ was able to induce astrocytic glutamate release which led to extrasynaptic NMDA receptor activation. In this case, nitromemantine, improved NMDA receptor antagonist which selectively inhibits extrasynaptic over physiological synaptic NMDA receptors activity, may protect against Aβ-induced synaptic dysfunction in hippocampus through selective extrasynaptic NMDA receptors blocking. In addition, Aβ has been shown to disrupt gliotransmission by enhancing calcium signaling through astrocytic α7-nAChRs which could as well underlie glial-based AD pathology (Lee et al., 2014).
Thus, devolvement of novel drugs targeting glial signaling may have a possible therapeutic role in AD. In fact, antiepileptic drugs such as levetiracetam reversed synaptic dysfunction and learning and memory deficits in human APP (hAPP) transgenic mice (Sanchez et al., 2012). One of the action mechanisms of levetiracetam is glutamate and GABA transporters increase in neurons and astrocytes (Ueda et al., 2007). Reduction in glutamate excitotoxicity and enhancement of inhibitory neurotransmission after chronic levetiracetam administration demonstrates a molecular mechanism which involves glial cells, to attenuate cognitive abnormalities in AD.
Finally, astrocytes and neurons work together through several metabolic pathways in order to perform new synthesis of glutamate and GABA (Bak et al., 2006). At inhibitory synapses this pathway is called the GABA–glutamine cycle and it depends on GABA transporters and a multi-enzyme machinery that coordinates this process (i.e., GABA transaminase, glutamate decarboxylase, and glutamine synthetase; Bak et al., 2006; Hertz, 2013). Several studies indicate that the activity of glutamine synthetase is decreased in AD. Dysfunction of astrocyte metabolism and therefore glutamate and GABA–glutamine cycles may underlie cognitive impairment in AD (Le Prince et al., 1995; Robinson, 2000; Nilsen et al., 2014). Drugs targeting GABA-metabolizing enzyme and neurotransmitter transporters are of therapeutical interest in GABA-related neurological disorders (Sarup et al., 2003). However, taking into account the different functional roles of glial and neuronal neurotransmitter transporters and the overlapping in GABA/glutamate metabolic pathways, developing of high selective cell-specific drugs is necessary in order to avoid pharmacological interactions and unpredictable side effects.
In summary, glial cells are dramatically affected in AD. Aβ-induced dysfunction of glutamate receptors (NMDA) in astrocytes disrupts neuron–glial signal transmission. On the other hand, Aβ interaction with cholinergic receptors (i.e., α7-nAChR) and glutamate transporters in glial cells may explain neurotoxicity and selective neurodegeneration. Reducing the activation of astrocytes and microglia is the basis of anti-inflammatory drugs in AD. Finally, metabolism and new synthesis of GABA in glial cells might be an interesting target to selective pharmacological modulation.
Aβ and Excitatory Neurotransmission: Amygdaloid Complex
Amyloid depositions are found not only in the hippocampus but in other subcortical brain structures. In fact, brain amyloidosis in subjects with higher vulnerability to AD pathology (i.e., individuals with mild cognitive impairment, MCI) includes structures such as parietal association cortices, posterior cingulate, precuneus, amygdale, and caudate (Tosun et al., 2013). Among these brain regions, the number of senile plaques has been reported to be the highest in amygdala (Arriagada et al., 1992). Accordingly, a recent diffusion-tensor imaging study has revealed significant decrease in the relative volume of amygdale in early stage AD subjects (Li et al., 2013). The amygdala has long been known to be vulnerable to Alzheimer-type pathology (Hopper and Vogel, 1976), and it has been described as one of earliest locations to develop Alzheimer pathology in Down syndrome (Mann et al., 1986). AD subjects develop brain pathology similar to that of Down syndrome, including Aβ depositions (Nardone et al., 2006). Asymmetrical neuronal loss in the amygdala ranges from 35% to 70% in AD (Scott et al., 1992; Vereecken et al., 1994). In addition, it has been reported that neuronal loss was more severe in the corticomedial regions than in the basolateral region of the amygdala (Tsuchiya and Kosaka, 1990). The distribution pattern of neuronal loss was similar to that of neurofibrillary tangles instead of the distribution of senile plaques (Tsuchiya and Kosaka, 1990). Severity of amygdala pathology correlates with disease duration in AD (Arriagada et al., 1992), and amygdala pathology has been associated with emotional and memory disturbances (Zald, 2003). Despite the importance that amygdaloid complex seems to have, few studies have investigated how Aβ induces injury and may contribute to underlie the emotional and cognitive symptoms typically observed in AD patients.
Based on the amygdala’s cytoarchitecture its subnuclei can be classified in superficial amygdaloid nuclei, centromedial group, and the basolateral complex (Heimer et al., 1999; Amunts et al., 2005). The basolateral amygdaloid complex is formed by the lateral, basolateral, and basomedial nuclei (Swanson and Petrovich, 1998), and innervated by cortical projections across the external capsule. In the amygdala, the excitatory synaptic activity evoked by stimulation of the external capsule is fundamentally mediated through the action of glutamic acid on AMPA/Kainate and NMDA receptors (Rainnie et al., 1991; Smith and Dudek, 1996). The amplitude of these responses is significantly depressed by Aβ without changes in membrane resistance values, which confirms that Aβ effect is localized at synaptic level (Ashenafi et al., 2005). Specifically, the Aβ effect seems to be located at presynaptic level since it could be prevented by calcicludine or nifedipine, both selective antagonists of presynaptic L-type calcium channels (Ashenafi et al., 2005).
Regarding cholinergic neurotransmission, the magnocellular division of the basal nucleus presents a high density of acetylcholinesterase positive fibers (Amaral and Bassett, 1989). The activation of these fibers generates depolarization of long duration in pyramidal cells, which is blocked by atropine (a competitive muscarinic receptor antagonist; Washburn and Moises, 1992; Moises et al., 1995). Muscarinic agonists, such as carbachol, mimic this type of response in the amygdaloid pyramidal neurons (Washburn and Moises, 1992; Yajeya et al., 2000). This effect is mediated by the closure of potassium channels and/or opening of non-specific cationic channels (Yajeya et al., 1997, 1999, 2000). On the other hand, data from Wang et al. (2000) showed that Aβ1-42 may block presynaptic α7-nAChR in neurons derived from human brain tissues and neuroblastoma cells. It can be assumed that as a consequence of such blocking, the concentration of calcium in the synaptic terminal diminishes, producing a decrease in the amount of neurotransmitter released when the terminal is activated. Although the existence of these receptors has been verified in presynaptic terminals of the amygdala (Girod et al., 2000), a previous study in our group has shown that α7-nAChR was not involved in the Aβ25-35 short-term neuromodulatory effects in basolateral amygdaloid complex (Ashenafi et al., 2005). In addition, nicotine stimulates mRNA expression of APP in the amygdala (Gutala et al., 2006). However, its functional implications in amygdaloid complex have not been deeply studied.
Aβ and Excitatory Neurotransmission: Septohippocampal System
Septum and hippocampus are structures dense and reciprocally interconnected through fimbria–fornix complex, and are functionally coupled to form the septohippocampal system, which shows a critical involvement in generating certain oscillatory activity, such as theta rhythm, necessary for fundamental processes in learning and memory (Stewart and Fox, 1990; Bland and Oddie, 2001; Buzsaki, 2002; Sotty et al., 2003; Colom, 2006; Colom et al., 2010; Rubio et al., 2012). Theta oscillation coordinates septohippocampal network and depends on interconnections, which include well known cholinergic and GABAergic components (Lynch et al., 1977; Kohler et al., 1984; Bland and Colom, 1993) and recently described glutamatergic projections (Sotty et al., 2003; Huh et al., 2010).
The initial symptoms of AD involve memory impairment and disorientation (McKhann et al., 1984; Swanberg et al., 2004). Damages found in septum and hippocampus could explain those cognitive deficits (Moreno et al., 2007; Palop et al., 2007; Villette et al., 2010; Rubio et al., 2012). Functional images applied in AD subjects have detected defects in the hippocampal formation, a brain structure where the disease begins (Bland and Colom, 1993; Gonzalez et al., 1995; Harris et al., 1998; Wu and Small, 2006). It has been shown that cholinergic cells of the medial septum/diagonal band of Broca (MS–DBB) and the enzymes necessary for the synthesis of acetylcholine, are particularly susceptible to disturbance, with consequent dysfunction in cognitive processes (Yamaguchi and Kawashima, 2001). Intracerebroventricular (i.c.v.) injection of Aβ25-35: (i) reduces the activity of acetylcholinesterase in the medial septum, cortex, and hippocampus of rats (Yamaguchi and Kawashima, 2001); (ii) decreases performance in passive avoidance and water maze tests (well established learning and memory tests); (iii) and also reduces neuronal loss and appearance of Aβ deposits in cortex, hippocampus, and caudate nucleus (Maurice et al., 1996). Moreover, the septal injection of different fragments of Aβ (Aβ18-28, Aβ25-35, or Aβ1-40) produces a marked reduction in basal or induced release of acetylcholine (Kar et al., 1996). Inhibition of acetylcholine release is also observed in the hippocampus and cortex in in vitro rat brain slices using different Aβ fragments (Kar et al., 1996). It has also been shown that Aβ inhibits some of the effects mediated by acetylcholine through septohippocampal muscarinic receptors (Kar et al., 1996; Santos-Torres et al., 2007). This capability could contribute to the particular Aβ vulnerability of cholinergic neuronal populations. However, the low concentrations (nanomolar) with which such effects are obtained, along with the fact that nervous system cells actively secrete Aβ product, suggest the possibility that this peptide may have physiological activity, acting as a neuromodulator not only on the cholinergic, but also on other neurotransmission systems.
In vitro studies in septum slices have found that Aβ induces a deficit in glutamatergic synaptic transmission (Santos-Torres et al., 2007). It has recently been observed in vivo that septal glutamatergic neurons are vulnerable to Aβ through excitotoxic mechanisms (Colom et al., 2010). The two characteristics of the theta rhythm, frequency and amplitude, are affected by modulation of septal NMDA receptors (Puma and Bizot, 1999; Bland et al., 2007). Furthermore, injection of NMDA antagonists in MS–DBB decreases the amplitude of hippocampal theta rhythm (Leung and Shen, 2004). This indicates that glutamatergic MS–DBB circuits are also affected by Aβ and are important for the generation and maintenance of septohippocampal rhythmic activity amplitude at theta frequencies (Colom et al., 2010).
Aβ has also shown to induce dysfunction of septal glutamatergic neurons involving muscarinic receptor effectors, the potassium voltage-gate channels, KCNQ (Leao et al., 2012). In this case, Aβ diminishes septal rhythmicity by decreasing KCNQ conductance, which negatively affects hippocampal rhythmogenesis and could underlie the memory loss observed in AD (Leao et al., 2012). In this sense, it has been reported that in the septohippocampal system Aβ reduces not only KCNQ2 subunit expression and then, KCNQ conductance, altering the neuronal excitability but also the expression of the oxidative stress-related genes superoxide dismutase 1 (SOD1), 8-oxoguanine DNA glycosylase (OGG1), and monamine oxidase A (MAOA). This situation leads to a neuronal dysfunction and damage that could not be fixed because of the decreased expression of repairing genes (Duran-Gonzalez et al., 2013).
Recent studies have shown that Aβ binds to α7-nAChR in several brain structures including the hippocampus (Spencer et al., 2006; Soderman et al., 2008). On the other hand, it has been proposed that α7-nAChR is functionally blocked in hippocampal CA1 neurons due to an interaction between the receptor and Aβ (Soderman et al., 2011). The functional consequences of such interaction may lead to impairments in both cognitive function (Soderman et al., 2008) and synaptic plasticity (Soderman et al., 2011). α 7-nAChR inhibition has also been explained by sustained increase in presynaptic Ca2+ evoked by Aβ which may underlie disruption of neuronal signaling via nAChRs in the early stages of AD (Dougherty et al., 2003). Since nicotine is able to induce LTP in CA1 hippocampal region probably due to reducing GABAergic inhibition and therefore, increasing the excitability of pyramidal neurons (Fujii et al., 2000), is plausible a functional interaction between α7-nAChR and GABAergic system. However, hippocampal GABAergic interneurons exposed to high levels of amyloid still presented α7-nAChR-mediated activity (Spencer et al., 2006). On the other hand, α7-nAChR and NMDA glutamatergic receptor activities are impaired in synaptosomes derived from AD postmortem tissue and in presence of high Aβ1-42 levels (Wang et al., 2000). Hence it will be essential to advance in the knowledge of α7-nAChR as therapeutic target for the treatment of Aβ-induced pathology and AD (Kem, 2000; Chen et al., 2006; Dziewczapolski et al., 2009).
Aβ and GABAergic Neurotransmission
The regulation of many physiological and cognitive processes is depends on a fine tuning between excitatory and inhibitory systems. In order to maintain neural network stability, GABA, the main inhibitory neurotransmitter in the mammalian central nervous system (Cardinali and Golombek, 1998), is known to regulate excitatory activity preventing neuronal hyperexcitation as well as oscillatory activity and firing rate impairments in neural networks (Oren et al., 2006; Zemankovics et al., 2013).
GABAergic neurons are the principal inhibitory neurons and one of the major local circuit neurons (Moore, 1993) which has been implicated in the regulation of a variety of behavioral functions such as learning and memory (Chapouthier, 1989; Vinogradova et al., 1998). In fact, GABAergic influence is the key to generate rhythmic synchronization of neurons during theta and gamma activity in different brain regions, contributing to neuronal communication and memory processing (Somogyi and Klausberger, 2005; Gong et al., 2009). Cortical and hippocampal function depends on optimum levels of inhibition (Borhegyi et al., 2004; Kaifosh et al., 2013; Xu et al., 2013; Bissonette et al., 2014) to maintain an adequate synaptic plasticity activity. For a long time, GABAergic neurotransmission has been considered well preserved in AD (Rissman et al., 2007). However, cumulative evidence indicates that changes in GABAergic neurotransmission are involved in the physiopathology of AD and may be very important as a possible target to pharmacological intervention previous to cognitive dysfunction in early AD. Below, we have reviewed the effect of Aβ on inhibitory GABA system in the different brain structures which relate to cognitive deficits in AD.
It has been already discussed in this review that excitatory neurotransmission contributes to the pathogenesis and progression of AD and as a result could serve to disrupt the excitatory/inhibitory balance in brain structures, participating in memory processing and therefore taking part of mechanisms that could explain cognitive dysfunction. It is important to consider that alterations on inhibitory neurotransmission or GABA receptors may also induce a significant impact on brain structures and functions, and might also participate in the dysregulation of the balance between excitatory and inhibitory neurotransmission seen in AD patients (Palop et al., 2007; Palop and Mucke, 2010b; Mucke and Selkoe, 2012). In fact, increased epileptiform activity and non-convulsive seizure induced by Aβ in both, animals models of AD (Palop et al., 2007) and AD in elderly people (Palop and Mucke, 2009, 2010a; De Simone et al., 2010) suggests that disruption of excitoratory/inhibitory balance by Aβ involves different neurotransmitter systems including the GABAergic.
Cognitive deficits in AD are explained by selective vulnerability, neurodegeneration, and loss of function of neuronal populations and neurotransmitter systems in particular brain regions such as hippocampus, septohippocampal system, and amygdala. Acetylcholine-releasing neurons and glutamatergic neurons in basal forebrain and hippocampus, respectively, are particularly vulnerable to Aβ neurotoxic effects (Coyle et al., 1983; Emre et al., 1992; Danysz et al., 2000; Giannakopoulos et al., 2009) while GABAergic neurons are relatively resistant to neurodegeneration in AD (Rissman et al., 2007). Recently, in a mouse model of AD it has been shown that glutamatergic hippocampal terminals decrease after Aβ1-42 perfusion while no significant changes in GABAergic terminals were observed (Canas et al., 2014). GABAergic synapses are preserved in human AD and APP/PS1 transgenic mice (Mitew et al., 2013). However, Aβ might have indirect effects on the inhibitory GABAergic transmission as a result of the dynamic GABAergic balance modulation of the other two excitatory systems (cholinergic and glutamatergic neurotransmission). It has recently been suggested that the imbalance between excitatory and inhibitory systems underlies the synaptic dysfunction caused by Aβ (Sun et al., 2009; Palop and Mucke, 2010a). As a consequence of relative sparing of GABAA receptors in AD, GABAergic sprouting in cortical and hippocampal networks enhance synaptic inhibition in order to compensate aberrant increases in network excitability (Palop et al., 2007). Hypersynchronous neuronal activity on those networks and Aβ-induced neurological deficits before neurodegeneration can be explained by this mechanism. Increase on glutamic acid decarboxylase (GAD, the rate-limiting enzyme synthesizing GABA) activity and consequently on the tone of the GABAergic system in AD brains has also been reported (Reinikainen et al., 1988). According to previous GABAergic hypothesis, this increase on extracellular GABA levels leads to alterations in neuronal membrane functions, abnormally enhanced synthesis of APP and facilitates neurodegeneration in basal forebrain system (Marczynski, 1998).
Controversially, selective somatostatin/NPY inhibitory interneurons neurodegeneration has been described in the hippocampus of a transgenic presenilin 1 PS1/APP AD model, with preservation of GABAergic mRNA synaptic markers (Ramos et al., 2006). In the same animal model, hyperactive neurons in cortical circuits are linked with a relative decrease in synaptic inhibition rather than increase in excitatory glutamatergic neurotransmission, suggesting impairments in GABAergic function (Busche et al., 2008; Palop and Mucke, 2010a). A recent report also shows that in mice expressing hAPP, the network dysfunction (hypersynchrony and reduced gamma oscillatory activity) and memory deficits in AD might arise from inhibitory interneuron deficit (Verret et al., 2012).
In line with the above scenario, therapies aimed at increasing GABAergic activity may reduce network/synaptic dysfunction on brain structures which participate in memory processing in AD subjects. Besides potential benefits of drugs which attenuate the Aβ-induced synaptic dysfunction, several previous studies have shown that selective GABAA receptor agonists (i.e., muscimol) are able to protect against Aβ-induced neurotoxicity in retinal, hippocampal, and cortical neurons in rodents (Gu et al., 2003; Paula-Lima et al., 2003; Louzada et al., 2004; Lee et al., 2005). Some neuroprotective effects of GABA modulators (i.e., Etazolate, selective GABAA receptor modulator) could be blocked by GABAA receptor antagonists (Marcade et al., 2008), indicating that these neuroprotective effects were due to GABAA receptor signaling and opening new therapeutic possibilities for AD treatment (Vandevrede et al., 2013). In fact, GABAA receptor agonists to treat age-related cognitive deficits were proposed as a new therapeutical approach in the 11th Alzheimer’s Disease Drug Discovery International Conference (Wolfe, 2010a,b). However, none precise action mechanism has been well described. Stimulation of GABA receptors by pentobarbital apparently restores neuronal maturation and neurogenesis in apolipoprotein E (APOE)4 knocking mice (Li et al., 2009). Apolipoprotein E4 is considered as a major genetic risk factor for early onset AD perhaps by accelerating Aβ plaque formation, or by impairing neuron repair (Baum et al., 2000; Lopez, 2011; Seppala et al., 2012). Despite the plausible neuroprotective effects of GABA agonists, the widely described side effects limit its long-term use (Lanctot et al., 2004, 2007). Furthermore, there is evidence of long-term effects of benzodiazepines (GABAA agonists) and their relationship with increased risk of dementia (Gallacher et al., 2012).
Cerebrospinal GABA studies as well as neuroimaging and postmortem studies have been useful to show the relationship between GABA system and AD (Jimenez-Jimenez et al., 1998; Yew et al., 1999; Lanctot et al., 2004). Postmortem autoradiographics and benzodiazepine binding studies of GABA receptors in cortex and hippocampus have been controversial to demonstrate changes in GABAA receptors levels; however, most of them have shown a relative decrease in GABAA receptors expression in frontal, temporal, and parietal cortical regions and limbic structures. GABAA subunits specific susceptibility could explain those dissimilar results. However, alteration in GABAA receptor subunits has shown paradoxical results probably due to compensatory mechanisms which are not well described (Mizukami et al., 1997, 1998; Howell et al., 2000; Rissman et al., 2003, 2007; Iwakiri et al., 2009). It is plausible that some effects on GABA dysfunction in AD induced by Aβ are not necessarily associated with a significant damage on GABA neurons or reduced expression of GABAA receptors, and could be explained by functional GABAA receptor activity changes. In fact, a conventional voltage-clamp study showed that Aβ may increase neuronal excitability by inhibiting GABA-induced Cl- current in the neurons of central nervous system (Sawada and Ichinose, 1996). This result suggests that GABA modulators and agonists can normalize Cl- flux and possibly restore the functional properties and excitability of theses neurons. In a series of elegant studies, the microtransplantation of functional receptors and channels from the human AD brain to frog oocytes showed an amplitude reduction of the currents elicited by GABA application, indicating that receptor-channel function was impaired (Miledi et al., 2004) or resulted from a diminished number of GABAA receptors in the membranes of AD brains (Bernareggi et al., 2007). Finally, GABA currents from AD brains have a faster and less sensitive rate of desensitization than those from control brains (Limon et al., 2011), which was explained by down regulation of α1 and γ2 receptor subunits while a compensatory up-regulation of α2, β1, and γ1 receptor subunits took place. Selective pharmacological modulators of GABAA subunits (i.e., α5-selective inverse agonist) may be effective to increase cognitive performance in memory disorders (Atack, 2010). Age-dependent reduction of GABA currents in AD brain from human postmortem tissue indicates a reduction of principal GABA receptors subunits (Limon et al., 2012). However, one of the major problems when GABAergic drugs are chronically used is the desensitization. It has been suggested that such phenomenon could be removed with phosphatase inhibitors or neurotrophic factors which positively modulate GABA currents (Palma et al., 2005; Limon et al., 2011) and both could be potential therapeutic targets for new AD drugs.
Hence, although some AD models have shown that the GABAergic system is relatively well preserved, Aβ might have indirect effects on GABAergic neurotransmission and induce inhibitory interneuron deficits, which could underlie neuronal hyperexcitability observed in AD. On the other hand, long-term Aβ exposition generates increased GABAergic activity and up/down regulation of specific GABAA subunits, as a compensatory mechanism. The understanding of those acute and chronic differential effects of Aβ on inhibitory systems is a pivotal point to develop novel therapeutical strategies to reduce cognitive impairment in early AD.
Aβ and GABAergic Neurotransmission: Amygdaloid Complex
As previously discussed in this review, there is general agreement to state that amygdala participates in emotional behavior processing. Afferent and efferent specific connections of the amygdala with a large variety of cortical and subcortical structures are the basis of cognitive functions and affective behaviors such as stress, defense, escape, pain, motivation, emotional discrimination, learning, and memory (Swanson and Petrovich, 1998; LeDoux, 2000; Gill and Grace, 2013). GABAergic afferents originating from amygdaloid interneurons of the basolateral complex which synapse on pyramidal cells probably modulate its function (McDonald, 1985; Carlsen, 1988; Carlsen and Heimer, 1988; McDonald and Augustine, 1993). In fact, altered signaling in GABAergic systems in amygdala produces impairments in emotional learning and memory tasks (Bolton et al., 2012). Regarding the above scenario, it has been proposed that inhibitory GABAergic activity in the basolateral amygdala cooperates to promote amygdala–hippocampal synchrony involved in emotional memory formation (Bienvenu et al., 2012). Amygdala dysfunction has been related to both AD (Bienvenu et al., 2012) and Aβ pathology in rodents (Devi and Ohno, 2010; Huang et al., 2010). Previously, our group demonstrated that GABAA inhibitory evoked responses decreased in amplitude after Aβ perfusion in basolateral amygdaloid complex through a presynaptic mechanism (Ashenafi et al., 2005). A transgenic mice study has shown that accumulation of Aβ in GABA neurons of the basolateral amygdaloid complex was related to enhanced innate and conditioned fear symptoms and spatial memory deficits (España et al., 2010). These results suggest that Aβ-induced dysfunction of GABAergic activity in key brain structures as amygdala might explain the emotional symptoms in AD such as anxiety and fear, as well as faster cognitive decline in memory processing.
A single neurotransmitter imbalance or Aβ-induced neurotoxic effects in a specific brain nucleus would not explain the pathology which involves whole brain regions and circuits. However, some psychological symptoms in AD subjects have been associated with GABAergic changes (Lanctot et al., 2007) and GABAA agonists have been widely used in the treatment of some behavioral and psychological symptoms of AD, such as aggression and agitation (Lanctot et al., 2004). Aβ-induced changes in GABAergic neurotransmitter system in amygdala would help us to understand the pathophysiology mechanism of emotional symptoms in AD subjects.
Aβ and GABAergic Neurotransmission: Septohippocampal System
The function of septal neurons of the basal forebrain is to modulate hippocampus and neocortex circuits’ activity in order to maintain sensory information and memory processes (Colom, 2006; Colom and Garrido-Sanabria, 2007). GABA neurons have been well described in that region in close proximity to cholinergic neurons in the MS–DBB complex (Kimura et al., 1980; Castañeda et al., 2005). In the same complex, burst-firing GABAergic neurons contribute to hippocampal theta rhythm in vivo (Sotty et al., 2003). Similarly, inhibitory neurons of the medial septum provide rhythmic drive to the hippocampus independently of intrahippocampal theta genesis (Hangya et al., 2009). GABA-containing afferents originating in the septum innervate most of the inhibitory interneurons in the hippocampus, which, in turn, control the activity of large populations of excitatory pyramidal cells (Freund and Antal, 1988). As a result, these septohippocampal GABAergic projections have a main role in modulating electrical rhythmic activity in the hippocampal formation.
In vivo experiments in rats have shown that cholinergic and GABAergic neurons from medial septum are involved in generating theta rhythmicity in the hippocampus (Simon et al., 2006). Predictive modeling of hippocampal microcircuits has shown that inhibitory interneurons from septum have a main role in spatial memories as well as in the maintenance of theta phase precession phenomenon of principal cells (Cutsuridis et al., 2010; Cutsuridis and Hasselmo, 2012). Because theta and gamma activity play a functional role in memory formation and retrieval (Bastiaansen and Hagoort, 2003; Hasselmo, 2005; Lisman, 2005), disruption of septohippocampal projection might explain cognitive deterioration in neurodegenerative diseases. Actually, several studies have demonstrated the relationship between Aβ-induced pathology and septohippocampal dysfunction as follows. In an interesting study, after single injection of Aβ1-40 into the medial septum, a significant reduced hippocampal theta rhythm was reported, associated with damage on cholinergic and glutamatergic neurons activity controlled by the GABAergic system (Colom et al., 2010). Similarly, Aβ1-42 injection into the MS–DBB complex preferentially injures septal cholinergic neurons but not inhibitory cells (Harkany et al., 1995). Despite these studies have proven that septal GABAergic neurons are spared after acute Aβ injection in septum, this consideration would not mean that Aβ induced septohippocampal dysfunction is not associated with inhibitory functional changes. By using in vivo preparations, it has been proposed that reduction of septal cholinergic and glutamatergic inputs onto GABAergic septal neurons may reduce the population of rhythmically bursting GABAergic neurons and suggest that GABAergic neurons are dysfunctional in Aβ-treated rats (Colom et al., 2010). Similarly, hippocampal Aβ1-40/31-35 injections induce a significant impairment of spatial memory in rats and concomitant reduction in the hippocampal theta rhythm (Liu et al., 2013). Aβ effects are associated with GABAergic neurons dysfunction and greatly weakened septal theta transmission to the hippocampus rather than interfere with its generation. Therefore, hippocampal Aβ-induced pathology reduces the bursting activity of septohippocampal GABAergic neurons (Villette et al., 2010). As previously stated, these neurons contact with hippocampal GABAergic interneurons, which in turn, control the pyramidal neurons rhythm. Since GABAergic septohippocampal neurons and hippocampal interneurons are relatively spared, these effects are probably due to functional regulation changes rather than neurodegeneration or reduction in GABA receptors expression.
Conversely, a study with a triple-transgenic mouse model of AD (TauPS2APP) revealed a significant neurodegeneration of GABAergic septohippocampal projection neurons as well as GABAergic hippocampal neurons (Loreth et al., 2012). Accordingly, loss of GABAergic septohippocampal axon terminals in the mouse model hAPP has been described (Rubio et al., 2012). However, some of these changes are not caused by neuronal loss. Recently, it has been reported in vivo that Aβ injection in dorsal hippocampus induces a selective death of GABAergic neuronal subpopulations projecting to the medial septum (Villette et al., 2012). In this sense, even with differences in the currently available models of AD, GABAergic decline in the septohippocampal projections may explain loss of hippocampus oscillatory activity necessary for learning and memory processes.
Hippocampal theta and gamma rhythms changes occur during the early stages of AD as a product of excitation–inhibition imbalance (Goutagny and Krantic, 2013). As described above, these changes may be related with Aβ effects on MS–DBB and afferents which in turn modulate hippocampus activity. However, hippocampal formation itself is a particularly vulnerable region to Aβ peptide accumulation (Moreno et al., 2007; Double et al., 2010). As a consequence, hippocampal atrophy and neurodegeneration are common features found in AD (Hof and Morrison, 1991; Cummings and Cole, 2002; Casas et al., 2004). Acute Aβ rise induces neurotoxic damage and synaptic dysfunction affecting GABAergic neurotransmitter system in the hippocampus and associative cortex by numerous mechanisms, affecting their function before neurodegeneration and accumulation of Aβ in senile plaques occurs (Selkoe, 2002; Klingner et al., 2003). In fact, cumulative evidence shows that soluble forms of Aβ can interfere with hippocampal synaptic plasticity responsible for learning and memory processing, inhibit long-term potentiation (LTP), and enhance long-term depression (LTD; Chen et al., 2000; Freir et al., 2001; Shankar et al., 2008; Ondrejcak et al., 2010). Hippocampal LTP in an AD animal model, the transgenic APP/PS1 mice, is larger after perfusion with GABAA receptor antagonist as a product of changes in synaptic protein levels. These data suggest that reduced LTP is associated to an enhanced GABAA receptor-mediated inhibition (Fernandez et al., 2007; Yoshiike et al., 2008). Several studies previously reported cognitive improvement in the presence of chronic systemic treatment with GABAA antagonists (i.e., flumazenil or picrotoxin; Marczynski, 1995, 1998; Fernandez et al., 2007; Yoshiike et al., 2008). According to these authors, those blockers are plausible useful therapeutic agents for age-related loss of cognitive functions in AD animal models. Nevertheless, other studies affirmed that Aβ-impairment effects on LTP appear to be independent of GABAA receptor-mediated synaptic inhibition because perfusion with picrotoxin had no effect on the inhibition of LTP (Raymond et al., 2003). Additionally, GABAergic synaptic transmission onto the hippocampus was affected by i.c.v. injection of Aβ1-40 and Aβ1-42 (Cullen et al., 1997) or Aβ25-35 (Sun and Alkon, 2002). Finally, several studies have reported specific GABAergic interneurons decrease (parvalbumin, calretinin, and somatostatin/NPY immunoreactive neurons) in the hippocampus of the APP/PS1 mice (Ramos et al., 2006; Baglietto-Vargas et al., 2010; Takahashi et al., 2010). Therefore, changes in the hippocampal inhibitory systems could explain the cognitive dysfunction in early stages of AD, but it is not clear whether is a consequence of functional decline in GABAergic neurotransmission, specific interneuron degeneration, or an imbalance in the excitatory/inhibitory activity, or a compound of these factors. Differences in the methodological approach (i.e., neuronal recording, genetically modified animal models, data analysis or in vitro vs. in vivo studies) may explain these paradoxical results. On the other hand, no clinical trial on GABAA receptor antagonists in AD or MCI has been successful or even performed, and therefore new studies are necessary in order to provide a link between basic science and clinical applications.
In summary, since septohippocampal GABAergic projections as well as intrahippocampal inhibitory interneurons have a main role in hippocampal oscillatory activity, inhibitory neurotransmission is an excellent candidate to be affected by Aβ in AD.
Aβ and GABAB Receptors
Despite the existence and widespread distribution of the GABA metabotropic type receptors (GABAB) in the central nervous system (Emson, 2007), their association with Aβ-induced pathology in AD have not been examined deeply yet. GABAB are coupled to intracellular signal transduction mechanisms via G proteins (Mott and Lewis, 1994; Kaupmann et al., 1998). These channels mediate slow and prolonged synaptic inhibition mainly by postsynaptic G protein-coupled activated inwardly rectifying potassium (GirK) channels (Luscher et al., 1997; Kaupmann et al., 1998). At presynaptic level, GABAB receptors also modulate (rather than generate) rhythmic activity in the MS–DBB (Henderson and Jones, 2005) and therefore have a possible role on memory function. GABAB receptor-mediated inhibition regulates the slow oscillation during gamma and theta oscillations in the control of cortical network activity (Kohl and Paulsen, 2010). Gamma oscillations have been associated with sensory processing (Singer, 1993), memory (Fell et al., 2001; Sederberg et al., 2007), attention (Fries et al., 2001), and finally, with consciousness (Llinas et al., 1998). GABAA and GABAB receptors are essential to learning and memory processes (Lasarge et al., 2009; Heinrichs et al., 2010; Mizoguchi and Yamada, 2011) and their pharmacological modulation affect the cognitive function. Since redistribution of hippocampus-dependent memories to neocortical sites is depending on slow oscillations (Diekelmann and Born, 2010), is plausible a role of GABAB receptors on this cognitive functions (Kohl and Paulsen, 2010). Autoradiography studies of hippocampus and cortex of postmortem AD brains (Chu et al., 1987; Young, 1987) have shown a significant reduction of GABAB receptors. In the same way, up-regulation of a novel non-coding RNA named 17A, RNA polymerase III-dependent embedded in the human G-protein-coupled receptor 51 gene (GPR51, GABAB2 receptor), affect GABAB2 intracellular signaling and enhances the secretion of Aβ (Massone et al., 2011). Therefore, the association between GABAB receptor dysfunction and Aβ-pathological metabolism is credible in AD.
On the other hand, the expression of GABAB receptors subunits may differ in accordance with the progression of AD. An immunohistochemical study in AD hippocampus demonstrated that in early stages of neurofibrillary tangle pathology the GABAB receptor subunit R1 expression could be stable or increased, and then decrease as the disease progresses (Iwakiri et al., 2005). These changes indicate that compensatory mechanisms are limited and the dysfunction of hippocampal inhibitory circuitry could involve GABAB receptors.
From a therapeutic point of view, some studies suggest that GABAB antagonists are more likely to have neuroprotective effects than agonists (Lafon-Cazal et al., 1999; Bowery, 2006). In fact, GABAB receptor antagonist may increase the expression of both neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) in rat hippocampus (Heese et al., 2000). Similarly, GABAB receptor agonist failed to inhibit Aβ-induced neuronal death (Lee et al., 2005; Marcade et al., 2008), while GABAB antagonist may improve cognitive performance in several animal models (Farr et al., 2000; Genkova-Papazova et al., 2000). New GABAB receptor antagonists have been developed for numerous in vitro and in vivo studies (Froestl, 2010). However, there is no clear evidence for its application in AD or any other cognitive dysfunction. Finally, high doses of GABAB receptor antagonists as well as GABAB receptor knock-out interrupt hippocampal and cortical oscillations leading to epileptiform activity and spontaneous seizures, respectively (Vergnes et al., 1997; Prosser et al., 2001; Schuler et al., 2001; Leung et al., 2005). In this sense, GABAB receptors modulation must have an important function in order to reach both, an effective neuroprotection and avoid epileptiform activity.
Aβ and GirK Channels
GABAB receptors and GirK channels are coupled and co-expressed in the postsynaptic membrane of pyramidal neurons in the hippocampus (Luscher et al., 1997; Kulik et al., 2003; Lujan et al., 2009) conforming an oligomeric stable molecular complex (Lujan et al., 2009; Ciruela et al., 2010). GirK channels act as key players in the control of cellular and network excitability by modulating synaptic activity in brain structures which participate in cognitive functions (Ciruela et al., 2010). GirK channels exhibit a tonic basal activity, even without receptor signaling, due to their direct binding to the Gα subunit of G proteins (Lujan et al., 2009). Therefore, it is plausible an Aβ-induced intracellular signaling impairment which compromises this effector system. In fact, Aβ has been shown to disrupt G protein-coupled receptors function (Thathiah and De, 2011), and compromise coupling of the receptor with G protein (Janickova et al., 2013) as well as several different secondary messenger systems (Thathiah and De, 2009; Yang et al., 2011; Fu et al., 2012; Zhang et al., 2014). In addition, we have recently showed that Aβ decrease GABAB currents in CA3 pyramidal neurons, a putative mechanism of Aβ-induced synaptic dysfunction observed in the septohippocampal system, which likely occur directly on GirK channels (Nava-Mesa et al., 2013).
GirK channels activity alteration may have multiple implications for synaptic activity, neuronal network function and cognitive processes. Numerous studies have emphasized its role in several pathological processes in the central nervous system such as epilepsy, pain, addiction, Parkinson or Down syndrome (Luscher and Slesinger, 2010). Deletion studies have revealed GirK channels role in learning and memory processes. GIRK4 knock-out mice exhibited impaired performance in a spatial learning and memory task (Wickman et al., 2000). Moreover, mutations in GIRK2 subunit reduced LTP and increased LTD in hippocampus (Sago et al., 1998; Siarey et al., 1999; Wickman et al., 2000) and it is especially relevant in Down syndrome, where cerebral Aβ accumulation is greatly accelerated and leads to invariant early onset AD neuropathology as well as learning and memory impairment (Lott and Head, 2005; Moncaster et al., 2010; Cooper et al., 2012).
Since loss-of-function of GirK channels might take part in the mechanisms that lead to excessive neural excitability and epilepsy that can be observed in hAPP transgenic mice model (Palop et al., 2007; Palop and Mucke, 2009, 2010a,b; Mucke and Selkoe, 2012), this type of potassium channels emerge as an interesting potential target to be studied, particularly now that its crystal structure has just been resolved (Whorton and MacKinnon, 2011).
GirK channels may be activated in a G protein-independent manner by different compounds (Kobayashi et al., 1999; Lewohl et al., 1999; Yamakura et al., 2001; Yow et al., 2011) and are blocked by different types of antidepressants (Kobayashi et al., 1999). However, there is little evidence for GirK subtype-specificity or pharmacokinetic advantages of most of those compounds, as they have other primary molecular targets (Lujan et al., 2013). However, a new class of subtype-selective agonists and antagonists has been identified (Kaufmann et al., 2013; Ramos-Hunter et al., 2013; Wen et al., 2013). An example is ML297, which has been found to be a potent, effective, and selective activator of GirK channels via a Gi/o-coupled receptor with preference for GIRK1/GIRK2 subunit combination (Days et al., 2010). This drug displays antiepileptic properties in animals models (Kaufmann et al., 2013) and might have a possible therapeutic potential for MCI or AD. As already discussed, seizures and epileptiform activity in AD subjects support the hypothesis that aberrant network activity contributes causally to synaptic and cognitive deficits (Palop and Mucke, 2009). Thus antiepileptic drugs (i.e., levetiracetam) might ameliorate those deficits (Sanchez et al., 2012; Vossel et al., 2013).
In summary, tonic GirK channel activity is necessary to control neuronal excitability and synaptic function. Therefore, GirK channels emerge as an interesting potential target to understand the physiopathology of the early stages of AD. A new class of selective GirK agonists with antiepileptic properties appears as a novel therapeutic tool to be tested in further studies.
Future Clinical Directions
The currently available therapy for AD (memantine, acetylcholinesterase inhibitors) may slow the progression of symptoms, but there are no existing treatments that reverse or stop disease progression even though the multiple advances in clinical and basic research.
As mentioned previously, GABAA receptor agonists have shown neuroprotective effects against Aβ-induced neurotoxicity in animal and in vitro models. Tramiprosate is an anti-amyloid compound for the treatment of AD. This drug has shown to reverse Aβ-induced synaptic plasticity dysfunction through activation of GABAA receptors (Aisen et al., 2006; Gervais et al., 2007). A phase III clinical trial showed a significant reduction in the hippocampus volume loss, but non-significant reduction in cognitive impairment (Aisen et al., 2011). Apparent divergence between neuroimaging lesions and cognitive deficits make difficult to determine reliable markers of AD. Other type of GABAA agonist (i.e., benzodiazepines) may have a significant role to manage psychological symptoms associated with AD. However, the undesirable side-effects associated and plausible receptor desensitization limit their chronic use (Lanctot et al., 2007). Furthermore, cognitive decline with lorazepam in individuals with higher risk for AD (carriers of APOE-epsilon4 allele, a common AD susceptibility gene) has been reported in a double-blind crossover study (Stonnington et al., 2009).
On the other hand, GABAB receptor is involved in the physiopathology of AD, and then is a pharmacological potential target. In a phase II trial, the GABAB receptor antagonist SGS742 improved cognition and memory performance in MCI possibly by up-regulation of GABAB receptors (Froestl, 2010) or through specific hippocampal protein expression (Sunyer et al., 2008; John et al., 2009). Nevertheless, in a more extensive phase IIb clinical trial in subjects with mild to moderate AD (http://www.clinicaltrials.gov/ct2/show/NCT00093951), the same drug was unsuccessful (Sabbagh, 2009).
Cumulative evidence indicates that the imbalance between excitatory and inhibitory neurotransmitter systems may underline the early cognitive deficits in AD. In this sense, antiepileptic drugs (i.e., levetiracetam and topiramate) may reverse the synaptic dysfunction associated with learning and memory impairment in animal models (Sanchez et al., 2012; Shi et al., 2013). A retrospective observational study, patients with amnestic MCI or early AD, treatment with lamotrigine and levetiracetam has shown clinical benefits and good tolerability (Vossel et al., 2013). In contrast, chronic treatment with valproate was associated with significant toxic effects including morphological brain changes in patients with moderate AD (Tariot et al., 2011).
Several clinical studies, including meta-analysis and clinical trials (Wei et al., 2007; Alvarez et al., 2011; Allegri and Guekht, 2012) reported the cognitive benefits of a mixture of neurotrophic factors (i.e., the nootropic agent cerebrolysin) as a therapeutical alternative for AD. In fact, those neurotrophic peptides can act as a neuroprotective agent or by synergically enhancing the effects of cholinesterase inhibitors (Alvarez et al., 2011). Finally, cerebrolysin may also regulate synaptic activity via presynaptic GABAB receptors on hippocampus (Xiong et al., 1996) and it also could improve cognitive performance in patients with mild to moderate AD according to several clinical trials (Wei et al., 2007).
In conclusion, since cholinergic or glutamatergic treatments in AD have shown limited success, therapies combining modulators of different neurotransmission systems seem to be a more useful tool for the treatment, and overall prevention, of this dementia. Pharmacological strategies to recover the unbalance between excitatory and inhibitory neurotransmitters have to take into account the GABAergic system. In this sense, recent data suggest that GABAB activity modulators which may control the neuronal excitability, as well as neurotrophic factors, are very interesting targets to be considered for further studies.
Concluding Remarks
It is reasonable to consider that Aβ-induced pathology on inhibitory synaptic activity might be explained by both their effects on specific inhibitory circuits or indirect effects on excitatory afferents to GABA neurons (Figure 1). Because of the availability of Aβ on synaptic cleft and the particular vulnerability of each neuronal circuit, some Aβ effects depend on the brain structure under study. On the other hand, there is increasing evidence to suggest that GABAergic neurons are relative spared in animal models of AD-like amyloid pathology. However, Aβ modulates inhibitory GABA activity through functional compensatory up-regulation mechanisms or neurodegeneration on cholinergic and glutamatergic neurons, which in turn innervate GABA interneurons. So GABAergic neurons are responsible for brain rhythmic activity necessary for learning and memory processing. It is necessary to note that many of the differences between studies are related to the experimental model used. The Aβ peptide fraction and also the exposure time or differences between acute, sub-acute and chronically models should be highlighted. In this manner, the effects of Aβ in the short-term tend to be more functional than structural while in the long or chronic –term effects induce activation of compensatory mechanisms that possibly involve increased expression of specific receptors subunits, which would be interesting molecular targets for drug development.
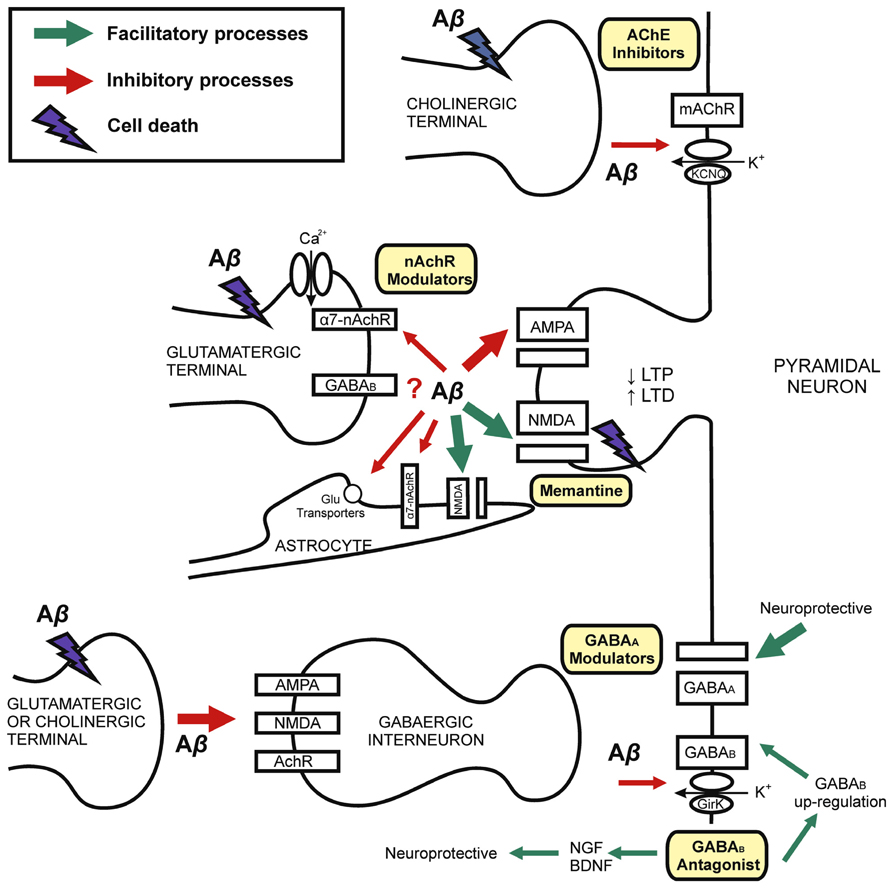
FIGURE 1. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Schematic drawing showing the role of Aβ in the physiopathology of AD and different targets for pharmacological modulation. AD is explained by Aβ-induced neurodegeneration, therefore with decline in synaptic terminals (left) on both principal cells and inhibitory interneurons. Chronic exposition to Aβ induces neuronal death. Synaptic loss is related with cognitive impairment in AD. Early memory decline is correlated with Aβ-synaptic dysfunction on excitatory and inhibitory receptors and neurotransmission. Current evidence for the treatment of AD involve both glutamatergic (NMDA-type glutamate receptors antagonist, i.e., memantine) and cholinergic (i.e., cholinesterase inhibitors, AChE inhibitors) transmission. The GABAergic system is a possible target for developing pharmacological interventions. GABAB receptors may be blocked by Aβ. However, GABAB antagonists induce GABAB receptors up-regulation and neurotrophic factors expression. GABAA modulation (i.e., by GABAA receptor agonist and selective inverse agonist) may have nootropic and neuroprotective effects. Although GABA interneurons are spared in several AD models, the imbalance between excitatory and inhibitory neurotransmission might underlie early AD cognitive dysfunction. Abbreviations: Aβ, Amyloid-β; AchR, acetylcholine receptor; AChE, acetyl-cholinesterase; α7-nAChR, α7-nicotinic acetylcholine receptor; BDNF, brain-derived neurotrophic factor; GirK, G protein-gated inwardly rectifying potassium channel; KCNQ, Kv7 family of voltage-gated potassium channels; LTP, long-term potentiation; LTD, long-term depression; NGF, nerve growth factor.
Hence, new therapeutical approaches must take into account the different drug’s action mechanisms, diverse neurotransmitters systems involved and finally, selective different cell targets in order to produce a better clinical results.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Spanish MINECO-FEDER (BFU2009-07341, SAF2010-14878 and BFU2011-22740) and Fundación Eugenio Rodríguez Pascual. JDNL held a Ramón y Cajal Reseach Fellow. We thank Dr. M. Gálvez for her enlighten comments and Eureka-science for its help in manuscript editing.
References
Aisen, P. S., Gauthier, S., Ferris, S. H., Saumier, D., Haine, D., Garceau, D., et al. (2011). Tramiprosate in mild-to-moderate Alzheimer’s disease–a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 7, 102–111. doi: 10.5114/aoms.2011.20612
Aisen, P. S., Saumier, D., Briand, R., Laurin, J., Gervais, F., Tremblay, P., et al. (2006). A phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology 67, 1757–1763. doi: 10.1212/01.wnl.0000244346.08950.64
Allegri, R. F., and Guekht, A. (2012). Cerebrolysin improves symptoms and delays progression in patients with Alzheimer’s disease and vascular dementia. Drugs Today (Barc.) 48(Suppl. A), 25–41. doi: 10.1358/dot.2012.48(Suppl.A).1739721
Alvarez, X. A., Cacabelos, R., Sampedro, C., Couceiro, V., Aleixandre, M., Vargas, M., et al. (2011). Combination treatment in Alzheimer’s disease: results of a randomized, controlled trial with cerebrolysin and donepezil. Curr. Alzheimer Res. 8, 583–591. doi: 10.2174/156720511796391863
Amaral, D. G., and Bassett, J. L. (1989). Cholinergic innervation of the monkey amygdala: an immunohistochemical analysis with antisera to choline acetyltransferase. J. Comp. Neurol. 281, 337–361. doi: 10.1002/cne.902810303
Amunts, K., Kedo, O., Kindler, M., Pieperhoff, P., Mohlberg, H., Shah, N. J., et al. (2005). Cytoarchitectonic mapping of the human amygdala, hippocampal region and entorhinal cortex: intersubject variability and probability maps. Anat. Embryol. (Berl.) 210, 343–352. doi: 10.1007/s00429-005-0025-5
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1998). Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur. J. Neurosci. 10, 2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215. doi: 10.1016/S0166-2236(98)01349-6
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. doi: 10.1212/WNL.42.3.631
Ashenafi, S., Fuente, A., Criado, J. M., Riolobos, A. S., Heredia, M., and Yajeya, J. (2005). Beta-amyloid peptide25-35 depresses excitatory synaptic transmission in the rat basolateral amygdala “in vitro.” Neurobiol. Aging 26, 419–428. doi: 10.1016/j.neurobiolaging.2004.05.008
Atack, J. R. (2010). Preclinical and clinical pharmacology of the GABAA receptor alpha5 subtype-selective inverse agonist alpha5IA. Pharmacol. Ther. 125, 11–26. doi: 10.1016/j.pharmthera.2009.09.001
Baglietto-Vargas, D., Moreno-Gonzalez, I., Sanchez-Varo, R., Jimenez, S., Trujillo-Estrada, L., Sanchez-Mejias, E., et al. (2010). Calretinin interneurons are early targets of extracellular amyloid-beta pathology in PS1/AbetaPP Alzheimer mice hippocampus. J. Alzheimers Dis. 21, 119–132. doi: 10.3233/JAD-2010-100066
Bak, L. K., Schousboe, A., and Waagepetersen, H. S. (2006). The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98, 641–653. doi: 10.1111/j.1471-4159.2006.03913.x
Bastiaansen, M., and Hagoort, P. (2003). Event-induced theta responses as a window on the dynamics of memory. Cortex 39, 967–992. doi: 10.1016/S0010-9452(08)70873-6
Baum, L., Chen, L., Ng, H. K., and Pang, C. P. (2000). Apolipoprotein E isoforms in Alzheimer’s disease pathology and etiology. Microsc. Res. Tech. 50, 278–281. doi: 10.1002/1097-0029(20000815)50:4<278::AID-JEMT5>3.0.CO;2-T
Bernareggi, A., Duenas, Z., Reyes-Ruiz, J. M., Ruzzier, F., and Miledi, R. (2007). Properties of glutamate receptors of Alzheimer’s disease brain transplanted to frog oocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 2956–2960. doi: 10.1073/pnas.0611513104
Bienvenu, T. C., Busti, D., Magill, P. J., Ferraguti, F., and Capogna, M. (2012). Cell-type-specific recruitment of amygdala interneurons to hippocampal theta rhythm and noxious stimuli in vivo. Neuron 74, 1059–1074. doi: 10.1016/j.neuron.2012.04.022
Bissonette, G. B., Bae, M. H., Suresh, T., Jaffe, D. E., and Powell, E. M. (2014). Prefrontal cognitive deficits in mice with altered cerebral cortical GABAergic interneurons. Behav. Brain Res. 259, 143–151. doi: 10.1016/j.bbr.2013.10.051
Bland, B. H., and Colom, L. V. (1993). Extrinsic and intrinsic properties underlying oscillation and synchrony in limbic cortex. Prog. Neurobiol. 41, 157–208. doi: 10.1016/0301-0082(93)90007-F
Bland, B. H., and Oddie, S. D. (2001). Theta band oscillation and synchrony in the hippocampal formation and associated structures: the case for its role in sensorimotor integration. Behav. Brain Res. 127, 119–136. doi: 10.1016/S0166-4328(01)00358-8
Bland, B. H., Derie-Gillespie, D., Mestek, P., Jackson, J., Crooks, R., and Cormican, A. (2007). To move or not: previous experience in a runway avoidance task determines the appearance of hippocampal Type 2 sensory processing theta. Behav. Brain Res. 179, 299–304. doi: 10.1016/j.bbr.2007.02.002
Bolton, M. M., Heaney, C. F., Sabbagh, J. J., Murtishaw, A. S., Magcalas, C. M., and Kinney, J. W. (2012). Deficits in emotional learning and memory in an animal model of schizophrenia. Behav. Brain Res. 233, 35–44. doi: 10.1016/j.bbr.2012.04.049
Borhegyi, Z., Varga, V., Szilagyi, N., Fabo, D., and Freund, T. F. (2004). Phase segregation of medial septal GABAergic neurons during hippocampal theta activity. J. Neurosci. 24, 8470–8479. doi: 10.1523/JNEUROSCI.1413-04.2004
Bowery, N. G. (2006). GABAB receptor: a site of therapeutic benefit. Curr. Opin. Pharmacol. 6, 37–43. doi: 10.1016/j.coph.2005.10.002
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., et al. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. doi: 10.1038/416507a
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Buzsaki, G. (2002). Theta oscillations in the hippocampus. Neuron 33, 325–340. doi: 10.1016/S0896-6273(02)00586-X
Caccamo, A., Fisher, A., and LaFerla, F. M. (2009). M1 agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr. Alzheimer Res. 6, 112–117. doi: 10.2174/156720509787602915
Caccamo, A., Oddo, S., Billings, L. M., Green, K. N., Martinez-Coria, H., Fisher, A., et al. (2006). M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 49, 671–682. doi: 10.1016/j.neuron.2006.01.020
Calza, L., Baldassarro, V. A., Giuliani, A., Lorenzini, L., Fernandez, M., Mangano, C., et al. (2013). From the multifactorial nature of Alzheimer’s disease to multitarget therapy: the contribution of the translational approach. Curr. Top. Med. Chem. 13, 1843–1852. doi: 10.2174/15680266113139990140
Canas, P. M., Simoes, A. P., Rodrigues, R. J., and Cunha, R. A. (2014). Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 76(Pt A), 51–56. doi: 10.1016/j.neuropharm.2013.08.026
Cardinali, D. P., and Golombek, D. A. (1998). The rhythmic GABAergic system. Neurochem. Res. 23, 607–614. doi: 10.1023/A:1022426519297
Carlsen, J. (1988). Immunocytochemical localization of glutamate decarboxylase in the rat basolateral amygdaloid nucleus, with special reference to GABAergic innervation of amygdalostriatal projection neurons. J. Comp. Neurol. 273, 513–526. doi: 10.1002/cne.902730407
Carlsen, J., and Heimer, L. (1988). The basolateral amygdaloid complex as a cortical-like structure. Brain Res. 441, 377–380. doi: 10.1016/0006-8993(88)91418-7
Casas, C., Sergeant, N., Itier, J. M., Blanchard, V., Wirths, O., van der Kolk, N., et al. (2004). Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 165, 1289–1300. doi: 10.1016/S0002-9440(10)63388-3
Castañeda, M. T., Sanabria, E. R., Hernandez, S., Ayala, A., Reyna, T. A., Wu, J. Y., et al. (2005). Glutamic acid decarboxylase isoforms are differentially distributed in the septal region of the rat. Neurosci. Res. 52, 107–119. doi: 10.1016/j.neures.2005.02.003
Chapouthier, G. (1989). The search for a biochemistry of memory. Arch. Gerontol. Geriatr. Suppl. 1, 7–19.
Chen, L., Yamada, K., Nabeshima, T., and Sokabe, M. (2006). alpha7 Nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in beta-amyloid infused rats. Neuropharmacology 50, 254–268. doi: 10.1016/j.neuropharm.2005.09.018
Chen, Q. S., Kagan, B. L., Hirakura, Y., and Xie, C. W. (2000). Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J. Neurosci. Res. 60, 65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q
Chu, D. C., Penney, J. B. Jr., and Young, A. B. (1987). Quantitative autoradiography of hippocampal GABAB and GABAA receptor changes in Alzheimer’s disease. Neurosci. Lett. 82, 246–252. doi: 10.1016/0304-3940(87)90264-3
Ciruela, F., Fernandez-Duenas, V., Sahlholm, K., Fernandez-Alacid, L., Nicolau, J. C., Watanabe, M., et al. (2010). Evidence for oligomerization between GABAB receptors and GIRK channels containing the GIRK1 and GIRK3 subunits. Eur. J. Neurosci. 32, 1265–1277. doi: 10.1111/j.1460-9568.2010.07356.x
Cohen-Gadol, A. A., Pan, J. W., Kim, J. H., Spencer, D. D., and Hetherington, H. H. (2004). Mesial temporal lobe epilepsy: a proton magnetic resonance spectroscopy study and a histopathological analysis. J. Neurosurg. 101, 613–620. doi: 10.3171/jns.2004.101.4.0613
Colom, L. V. (2006). Septal networks: relevance to theta rhythm, epilepsy and Alzheimer’s disease. J. Neurochem. 96, 609–623. doi: 10.1111/j.1471-4159.2005.03630.x
Colom, L. V., Castaneda, M. T., Banuelos, C., Puras, G., Garcia-Hernandez, A., Hernandez, S., et al. (2010). Medial septal beta-amyloid 1-40 injections alter septo-hippocampal anatomy and function. Neurobiol. Aging 31, 46–57. doi: 10.1016/j.neurobiolaging.2008.05.006
Colom, L. V., and Garrido-Sanabria, E. (2007). Modulation of normal and altered hippocampal excitability states by septal networks. J. Neurosci. Res. 85, 2839–2843. doi: 10.1002/jnr.21276
Cooper, A., Grigoryan, G., Guy-David, L., Tsoory, M. M., Chen, A., and Reuveny, E. (2012). Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 2642–2647. doi: 10.1073/pnas.1109099109
Coyle, J. T., Price, D. L., and DeLong, M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190. doi: 10.1126/science.6338589
Cudaback, E., Jorstad, N. L., Yang, Y., Montine, T. J., and Keene, C. D. (2014). Therapeutic implications of the prostaglandin pathway in Alzheimer’s disease. Biochem. Pharmacol. 88, 565–572. doi: 10.1016/j.bcp.2013.12.014
Cullen, W. K., Suh, Y. H., Anwyl, R., and Rowan, M. J. (1997). Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8, 3213–3217. doi: 10.1097/00001756-199710200-00006
Cummings, J. L., and Cole, G. (2002). Alzheimer disease. J. Am. Med. Assoc. 287, 2335–2338. doi: 10.1001/jama.287.18.2335
Cutsuridis, V., Cobb, S., and Graham, B. P. (2010). Encoding and retrieval in a model of the hippocampal CA1 microcircuit. Hippocampus 20, 423–446. doi: 10.1002/hipo.20661
Cutsuridis, V., and Hasselmo, M. (2012). GABAergic contributions to gating, timing, and phase precession of hippocampal neuronal activity during theta oscillations. Hippocampus 22, 1597–1621. doi: 10.1002/hipo.21002
Dabir, D. V., Trojanowski, J. Q., Richter-Landsberg, C., Lee, V. M., and Forman, M. S. (2004). Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am. J. Pathol. 164, 155–166. doi: 10.1016/S0002-9440(10)63106-9
Danysz, W., Parsons, C. G., Mobius, H. J., Stoffler, A., and Quack, G. (2000). Neuroprotective and symptomatological action of memantine relevant for Alzheimer’s disease–a unified glutamatergic hypothesis on the mechanism of action. Neurotox. Res. 2, 85–97. doi: 10.1007/BF03033787
Days, E., Kaufmann, K., Romaine, I., Niswender, C., Lewis, M., Utley, T., et al. (2010). Discovery and Characterization of a Selective Activator of the G-Protein Activated Inward-Rectifying Potassium (GIRK) Channel. Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23762945
De Simone, R., Puig, X. S., Gelisse, P., Crespel, A., and Genton, P. (2010). Senile myoclonic epilepsy: delineation of a common condition associated with Alzheimer’s disease in Down syndrome. Seizure 19, 383–389. doi: 10.1016/j.seizure.2010.04.008
de Vivo, L., Melone, M., Rothstein, J. D., and Conti, F. (2010). GLT-1 promoter activity in astrocytes and neurons of mouse hippocampus and somatic sensory cortex. Front. Neuroanat. 3:31. doi: 10.3389/neuro.05.031.2009
Devi, L., and Ohno, M. (2010). Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur. J. Neurosci. 31, 110–118. doi: 10.1111/j.1460-9568.2009.07031.x
Diekelmann, S., and Born, J. (2010). The memory function of sleep. Nat. Rev. Neurosci. 11, 114–126. doi: 10.1038/nrn2762
Ding, S., Fellin, T., Zhu, Y., Lee, S. Y., Auberson, Y. P., Meaney, D. F., et al. (2007). Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 27, 10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007
Double, K. L., Reyes, S., Werry, E. L., and Halliday, G. M. (2010). Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog. Neurobiol. 92, 316–329. doi: 10.1016/j.pneurobio.2010.06.001
Dougherty, J. J., Wu, J., and Nichols, R. A. (2003). Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740–6747.
Duran-Gonzalez, J., Michi, E. D., Elorza, B., Perez-Cordova, M. G., Pacheco-Otalora, L. F., Touhami, A., et al. (2013). Amyloid beta peptides modify the expression of antioxidant repair enzymes and a potassium channel in the septohippocampal system. Neurobiol. Aging 34, 2071–2076. doi: 10.1016/j.neurobiolaging.2013.02.005
Dziewczapolski, G., Glogowski, C. M., Masliah, E., and Heinemann, S. F. (2009). Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 29, 8805–8815. doi: 10.1523/JNEUROSCI.6159-08.2009
Emre, M., Geula, C., Ransil, B. J., and Mesulam, M. M. (1992). The acute neurotoxicity and effects upon cholinergic axons of intracerebrally injected beta-amyloid in the rat brain. Neurobiol. Aging 13, 553–559. doi: 10.1016/0197-4580(92)90055-3
Emson, P. C. (2007). GABA(B) receptors: structure and function. Prog. Brain Res. 160, 43–57. doi: 10.1016/S0079-6123(06)60004-6
España, J., Gimenez-Llort, L., Valero, J., Minano, A., Rabano, A., Rodriguez-Alvarez, J., et al. (2010). Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer’s disease transgenic mice. Biol. Psychiatry 67, 513–521. doi: 10.1016/j.biopsych.2009.06.015
Farlow, M. R. (2009). Treatment of mild cognitive impairment (MCI). Curr. Alzheimer Res. 6, 362–367. doi: 10.2174/156720509788929282
Farr, S. A., Uezu, K., Creonte, T. A., Flood, J. F., and Morley, J. E. (2000). Modulation of memory processing in the cingulate cortex of mice. Pharmacol. Biochem. Behav. 65, 363–368. doi: 10.1016/S0091-3057(99)00226-9
Fell, J., Klaver, P., Lehnertz, K., Grunwald, T., Schaller, C., Elger, C. E., et al. (2001). Human memory formation is accompanied by rhinal–hippocampal coupling and decoupling. Nat. Neurosci. 4, 1259–1264. doi: 10.1038/nn759
Fellin, T., Pascual, O., Gobbo, S., Pozzan, T., Haydon, P. G., and Carmignoto, G. (2004). Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43, 729–743. doi: 10.1016/j.neuron.2004.08.011
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10, 411–413. doi: 10.1038/nn1860
Fields, R. D., Araque, A., Johansen-Berg, H., Lim, S. S., Lynch, G., Nave, K. A., et al. (2014). Glial biology in learning and cognition. Neuroscientist doi: 10.1177/1073858413504465 [Epub ahead of print].
Fisher, A., Brandeis, R., Bar-Ner, R. H., Kliger-Spatz, M., Natan, N., Sonego, H., et al. (2002). AF150(S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J. Mol. Neurosci. 19, 145–153. doi: 10.1007/s12031-002-0025-3
Forman, M. S., Lal, D., Zhang, B., Dabir, D. V., Swanson, E., Lee, V. M., et al. (2005). Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J. Neurosci. 25, 3539–3550. doi: 10.1523/JNEUROSCI.0081-05.2005
Freir, D. B., Holscher, C., and Herron, C. E. (2001). Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J. Neurophysiol. 85, 708–713.
Freund, T. F., and Antal, M. (1988). GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature 336, 170–173. doi: 10.1038/336170a0
Fries, P., Reynolds, J. H., Rorie, A. E., and Desimone, R. (2001). Modulation of oscillatory neuronal synchronization by selective visual attention. Science 291, 1560–1563. doi: 10.1126/science.1055465
Froestl, W. (2010). Chemistry and pharmacology of GABAB receptor ligands. Adv. Pharmacol. 58, 19–62. doi: 10.1016/S1054-3589(10)58002-5
Fu, W., Ruangkittisakul, A., MacTavish, D., Shi, J. Y., Ballanyi, K., and Jhamandas, J. H. (2012). Amyloid beta (Abeta) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J. Biol. Chem. 287, 18820–18830. doi: 10.1074/jbc.M111.331181
Fujii, S., Ji, Z., and Sumikawa, K. (2000). Inactivation of alpha7 ACh receptors and activation of non-alpha7 ACh receptors both contribute to long term potentiation induction in the hippocampal CA1 region. Neurosci. Lett. 286, 134–138. doi: 10.1016/S0304-3940(00)01076-4
Gallacher, J., Elwood, P., Pickering, J., Bayer, A., Fish, M., and Ben-Shlomo, Y. (2012). Benzodiazepine use and risk of dementia: evidence from the Caerphilly Prospective Study (CaPS). J. Epidemiol. Community Health 66, 869–873. doi: 10.1136/jech-2011-200314
Genkova-Papazova, M. G., Petkova, B., Shishkova, N., and Lazarova-Bakarova, M. (2000). The GABA-B antagonist CGP 36742 prevent PTZ-kindling-provoked amnesia in rats. Eur. Neuropsychopharmacol. 10, 273–278. doi: 10.1016/S0924-977X(00)00082-1
Gervais, F., Paquette, J., Morissette, C., Krzywkowski, P., Yu, M., Azzi, M., et al. (2007). Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 28, 537–547. doi: 10.1016/j.neurobiolaging.2006.02.015
Giannakopoulos, P., Kovari, E., Gold, G., von, G. A., Hof, P. R., and Bouras, C. (2009). Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 24:20–29. doi: 10.1159/000197881
Gill, K. M., and Grace, A. A. (2013). Differential effects of acute and repeated stress on hippocampus and amygdala inputs to the nucleus accumbens shell. Int. J. Neuropsychopharmacol. 16, 2013–2025. doi: 10.1017/S1461145713000618
Girod, R., Barazangi, N., McGehee, D., and Role, L. W. (2000). Facilitation of glutamatergic neurotransmission by presynaptic nicotinic acetylcholine receptors. Neuropharmacology 39, 2715–2725. doi: 10.1016/S0028-3908(00)00145-3
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer’s disease. Science 314, 777–781. doi: 10.1126/science.1132814
Gong, N., Li, Y., Cai, G. Q., Niu, R. F., Fang, Q., Wu, K., et al. (2009). GABA transporter-1 activity modulates hippocampal theta oscillation and theta burst stimulation-induced long-term potentiation. J. Neurosci. 29, 15836–15845. doi: 10.1523/JNEUROSCI.4643-09.2009
Gonzalez, R. G., Fischman, A. J., Guimaraes, A. R., Carr, C. A., Stern, C. E., Halpern, E. F., et al. (1995). Functional MR in the evaluation of dementia: correlation of abnormal dynamic cerebral blood volume measurements with changes in cerebral metabolism on positron emission tomography with fludeoxyglucose F 18. Am. J. Neuroradiol. 16, 1763–1770.
Gordon, G. R., Baimoukhametova, D. V., Hewitt, S. A., Rajapaksha, W. R., Fisher, T. E., and Bains, J. S. (2005). Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci. 8, 1078–1086. doi: 10.1038/nn1498
Goutagny, R., and Krantic, S. (2013). Hippocampal oscillatory activity in Alzheimer’s disease: toward the identification of early biomarkers? Aging Dis. 4, 134–140.
Gu, Z., Zhong, P., and Yan, Z. (2003). Activation of muscarinic receptors inhibits beta-amyloid peptide-induced signaling in cortical slices. J. Biol. Chem. 278, 17546–17556. doi: 10.1074/jbc.M209892200
Gutala, R., Wang, J., Hwang, Y. Y., Haq, R., and Li, M. D. (2006). Nicotine modulates expression of amyloid precursor protein and amyloid precursor-like protein 2 in mouse brain and in SH-SY5Y neuroblastoma cells. Brain Res. 1093, 12–19. doi: 10.1016/j.brainres.2006.03.100
Halassa, M. M., Fellin, T., and Haydon, P. G. (2007). The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med. 13, 54–63. doi: 10.1016/j.molmed.2006.12.005
Hangya, B., Borhegyi, Z., Szilagyi, N., Freund, T. F., and Varga, V. (2009). GABAergic neurons of the medial septum lead the hippocampal network during theta activity. J. Neurosci. 29, 8094–8102. doi: 10.1523/JNEUROSCI.5665-08.2009
Harkany, T., De Jong, G. I., Soos, K., Penke, B., Luiten, P. G., and Gulya, K. (1995). Beta-amyloid (1–42) affects cholinergic but not parvalbumin-containing neurons in the septal complex of the rat. Brain Res. 698, 270–274. doi: 10.1016/0006-8993(95)01013-L
Harkany, T., Mulder, J., Sasvari, M., Abraham, I., Konya, C., Zarandi, M., et al. (1999). N-methyl-D-aspartate receptor antagonist MK-801 and radical scavengers protect cholinergic nucleus basalis neurons against beta-amyloid neurotoxicity. Neurobiol. Dis. 6, 109–121. doi: 10.1006/nbdi.1998.0230
Harris, G. J., Lewis, R. F., Satlin, A., English, C. D., Scott, T. M., Yurgelun-Todd, D. A., et al. (1998). Dynamic susceptibility contrast MR imaging of regional cerebral blood volume in Alzheimer disease: a promising alternative to nuclear medicine. Am. J. Neuroradiol. 19, 1727–1732.
Hasselmo, M. E. (2005). What is the function of hippocampal theta rhythm?–linking behavioral data to phasic properties of field potential and unit recording data. Hippocampus 15, 936–949. doi: 10.1002/hipo.20116
Heese, K., Otten, U., Mathivet, P., Raiteri, M., Marescaux, C., and Bernasconi, R. (2000). GABA(B) receptor antagonists elevate both mRNA and protein levels of the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) but not neurotrophin-3 (NT-3) in brain and spinal cord of rats. Neuropharmacology 39, 449–462. doi: 10.1016/S0028-3908(99)00166-5
Heimer, L., de Olmos, J. S., Alheid, G. F., Pearson, J., Sakamoto, N., Shinoda, K., et al. (1999). “The human basal forebrain Part II,” in Handbook of Chemical Neuroanatomy: The Primate Nervous System, eds T. Hokfelt, A. Bjorklund, and F. Bloom (Amsterdam: Elsevier Science), 57–226.
Heinrichs, S. C., Leite-Morris, K. A., Carey, R. J., and Kaplan, G. B. (2010). Baclofen enhances extinction of opiate conditioned place preference. Behav. Brain Res. 207, 353–359. doi: 10.1016/j.bbr.2009.10.013
Henderson, Z., and Jones, G. A. (2005). GABAB receptors in the medial septum/diagonal band slice from 16–25 day rat. Neuroscience 132, 789–800. doi: 10.1016/j.neuroscience.2005.01.027
Hertz, L. (2013). The glutamate–glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front. Endocrinol. (Lausanne) 4:59. doi: 10.3389/fendo.2013.00059
Hickman, S. E., Allison, E. K., and El, K. J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hof, P. R., and Morrison, J. H. (1991). Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer’s disease. Exp. Neurol. 111, 293–301. doi: 10.1016/0014-4886(91)90096-U
Hopper, M. W., and Vogel, F. S. (1976). The limbic system in Alzheimer’s disease. A neuropathologic investigation. Am. J. Pathol. 85, 1–20.
Howell, O., Atack, J. R., Dewar, D., McKernan, R. M., and Sur, C. (2000). Density and pharmacology of alpha5 subunit-containing GABA(A) receptors are preserved in hippocampus of Alzheimer’s disease patients. Neuroscience 98, 669–675. doi: 10.1016/S0306-4522(00)00163-9
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Huang, J., Chen, Y. J., Bian, W. H., Yu, J., Zhao, Y. W., and Liu, X. Y. (2010). Unilateral amyloid-beta25-35 injection into the rat amygdala increases the expressions of aberrant tau phosphorylation kinases. Chin. Med. J. (Engl.) 123, 1311–1314.
Huang, Y., and Mucke, L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. doi: 10.1016/j.cell.2012.02.040
Huh, C. Y., Goutagny, R., and Williams, S. (2010). Glutamatergic neurons of the mouse medial septum and diagonal band of Broca synaptically drive hippocampal pyramidal cells: relevance for hippocampal theta rhythm. J. Neurosci. 30, 15951–15961. doi: 10.1523/JNEUROSCI.3663-10.2010
Iwakiri, M., Mizukami, K., Ikonomovic, M. D., Ishikawa, M., Abrahamson, E. E., DeKosky, S. T., et al. (2009). An immunohistochemical study of GABA A receptor gamma subunits in Alzheimer’s disease hippocampus: relationship to neurofibrillary tangle progression. Neuropathology 29, 263–269. doi: 10.1111/j.1440-1789.2008.00978.x
Iwakiri, M., Mizukami, K., Ikonomovic, M. D., Ishikawa, M., Hidaka, S., Abrahamson, E. E., et al. (2005). Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: association with Braak staging. Acta Neuropathol. 109, 467–474. doi: 10.1007/s00401-005-0985-9
Janickova, H., Rudajev, V., Zimcik, P., Jakubik, J., Tanila, H., El-Fakahany, E. E., et al. (2013). Uncoupling of M1 muscarinic receptor/G-protein interaction by amyloid beta(1–42). Neuropharmacology 67, 272–283. doi: 10.1016/j.neuropharm.2012.11.014
Jimenez-Jimenez, F. J., Molina, J. A., Gomez, P., Vargas, C., de, B. F., Benito-Leon, J., et al. (1998). Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. J. Neural. Transm. 105, 269–277. doi: 10.1007/s007020050073
John, J. P., Sunyer, B., Hoger, H., Pollak, A., and Lubec, G. (2009). Hippocampal synapsin isoform levels are linked to spatial memory enhancement by SGS742. Hippocampus 19, 731–738. doi: 10.1002/hipo.20553
Jourdain, P., Bergersen, L. H., Bhaukaurally, K., Bezzi, P., Santello, M., Domercq, M., et al. (2007). Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 10, 331–339. doi: 10.1038/nn1849
Kaifosh, P., Lovett-Barron, M., Turi, G. F., Reardon, T. R., and Losonczy, A. (2013). Septo-hippocampal GABAergic signaling across multiple modalities in awake mice. Nat. Neurosci. 16, 1182–1184. doi: 10.1038/nn.3482
Kar, S., Seto, D., Gaudreau, P., and Quirion, R. (1996). Beta-amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal slices. J. Neurosci. 16, 1034–1040.
Kaufmann, K., Romaine, I., Days, E., Pascual, C., Malik, A., Yang, L., et al. (2013). ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 4, 1278–1286. doi: 10.1021/cn400062a
Kaupmann, K., Schuler, V., Mosbacher, J., Bischoff, S., Bittiger, H., Heid, J., et al. (1998). Human gamma-aminobutyric acid type B receptors are differentially expressed and regulate inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U.S.A. 95, 14991–14996. doi: 10.1073/pnas.95.25.14991
Kelly, B. L., Vassar, R., and Ferreira, A. (2005). Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer disease. J. Biol. Chem. 280, 31746–31753. doi: 10.1074/jbc.M503259200
Kem, W. R. (2000). The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer’s disease: studies with DMXBA (GTS-21). Behav. Brain Res. 113, 169–181. doi: 10.1016/S0166-4328(00)00211-4
Kimura, H., McGeer, P. L., Peng, F., and McGeer, E. G. (1980). Choline acetyltransferase-containing neurons in rodent brain demonstrated by immunohistochemistry. Science 208, 1057–1059. doi: 10.1126/science.6990490
Klein, W. L. (2002). ADDLs & protofibrils–the missing links? Neurobiol. Aging 23, 231–235. doi: 10.1016/S0197-4580(01)00312-8
Klingner, M., Apelt, J., Kumar, A., Sorger, D., Sabri, O., Steinbach, J., et al. (2003). Alterations in cholinergic and non-cholinergic neurotransmitter receptor densities in transgenic Tg2576 mouse brain with beta-amyloid plaque pathology. Int. J. Dev. Neurosci. 21, 357–369. doi: 10.1016/j.ijdevneu.2003.08.001
Kobayashi, T., Ikeda, K., Kojima, H., Niki, H., Yano, R., Yoshioka, T., et al. (1999). Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat. Neurosci. 2, 1091–1097. doi: 10.1038/16019
Kohl, M. M., and Paulsen, O. (2010). The roles of GABAB receptors in cortical network activity. Adv. Pharmacol. 58, 205–229. doi: 10.1016/S1054-3589(10)58009-8
Kohler, C., Chan-Palay, V., and Wu, J. Y. (1984). Septal neurons containing glutamic acid decarboxylase immunoreactivity project to the hippocampal region in the rat brain. Anat. Embryol. (Berl) 169, 41–44. doi: 10.1007/BF00300585
Kommers, T., Rodnight, R., Boeck, C., Vendite, D., Oliveira, D., Horn, J., et al. (2002). Phosphorylation of glial fibrillary acidic protein is stimulated by glutamate via NMDA receptors in cortical microslices and in mixed neuronal/glial cell cultures prepared from the cerebellum. Brain Res. Dev. Brain Res. 137, 139–148. doi: 10.1016/S0165-3806(02)00434-0
Komori, T. (1999). Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 9, 663–679. doi: 10.1111/j.1750-3639.1999.tb00549.x
Kulik, A., Vida, I., Lujan, R., Haas, C. A., Lopez-Bendito, G., Shigemoto, R., et al. (2003). Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J. Neurosci. 23, 11026–11035.
Lafon-Cazal, M., Viennois, G., Kuhn, R., Malitschek, B., Pin, J. P., Shigemoto, R., et al. (1999). mGluR7-like receptor and GABA(B) receptor activation enhance neurotoxic effects of N-methyl-D-aspartate in cultured mouse striatal GABAergic neurones. Neuropharmacology 38, 1631–1640. doi: 10.1016/S0028-3908(99)00124-0
Lalo, U., Pankratov, Y., Kirchhoff, F., North, R. A., and Verkhratsky, A. (2006). NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J. Neurosci. 26, 2673–2683. doi: 10.1523/JNEUROSCI.4689-05.2006
Lanctot, K. L., Herrmann, N., Mazzotta, P., Khan, L. R., and Ingber, N. (2004). GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can. J. Psychiatry 49, 439–453.
Lanctot, K. L., Herrmann, N., Rothenburg, L., and Eryavec, G. (2007). Behavioral correlates of GABAergic disruption in Alzheimer’s disease. Int. Psychogeriatr. 19, 151–158. doi: 10.1017/S1041610206003899
Langmead, C. J., Watson, J., and Reavill, C. (2008). Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 117, 232–243. doi: 10.1016/j.pharmthera.2007.09.009
Lasarge, C. L., Banuelos, C., Mayse, J. D., and Bizon, J. L. (2009). Blockade of GABA(B) receptors completely reverses age-related learning impairment. Neuroscience 164, 941–947. doi: 10.1016/j.neuroscience.2009.08.055
Le Prince, G., Delaere, P., Fages, C., Lefrancois, T., Touret, M., Salanon, M., et al. (1995). Glutamine synthetase (GS) expression is reduced in senile dementia of the Alzheimer type. Neurochem. Res. 20, 859–862. doi: 10.1007/BF00969698
Leao, R. N., Colom, L. V., Borgius, L., Kiehn, O., and Fisahn, A. (2012). Medial septal dysfunction by Abeta-induced KCNQ channel-block in glutamatergic neurons. Neurobiol. Aging 33, 2046–2061. doi: 10.1016/j.neurobiolaging.2011.07.013
LeDoux, J. E. (2000). “The amygdala and emotion: a view through fear,” in The Amygdala: A Functional Analysis, ed. J. P. Aggleton (New York: Oxford University Press), 289–310.
Lee, B. Y., Ban, J. Y., and Seong, Y. H. (2005). Chronic stimulation of GABAA receptor with muscimol reduces amyloid beta protein (25-35)-induced neurotoxicity in cultured rat cortical cells. Neurosci. Res. 52, 347–356. doi: 10.1016/j.neures.2005.04.008
Lee, L., Kosuri, P., and Arancio, O. (2014). Picomolar amyloid-beta peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis. 38, 49–62. doi: 10.3233/JAD-130740
Lee, M. C., Ting, K. K., Adams, S., Brew, B. J., Chung, R., and Guillemin, G. J. (2010). Characterisation of the expression of NMDA receptors in human astrocytes. PLoS ONE 5:e14123. doi: 10.1371/journal.pone.0014123
Leung, L. S., Canning, K. J., and Shen, B. (2005). Hippocampal afterdischarges after GABA(B)-receptor blockade in the freely moving rat. Epilepsia 46, 203–216. doi: 10.1111/j.0013-9580.2005.35804.x
Leung, L. S., and Shen, B. (2004). Glutamatergic synaptic transmission participates in generating the hippocampal EEG. Hippocampus 14, 510–525. doi: 10.1002/hipo.10199
Levey, A. I., Edmunds, S. M., Koliatsos, V., Wiley, R. G., and Heilman, C. J. (1995). Expression of m1–m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J. Neurosci. 15, 4077–4092.
Levey, A. I., Kitt, C. A., Simonds, W. F., Price, D. L., and Brann, M. R. (1991). Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 11, 3218–3226.
Lewohl, J. M., Wilson, W. R., Mayfield, R. D., Brozowski, S. J., Morrisett, R. A., and Harris, R. A. (1999). G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat. Neurosci. 2, 1084–1090. doi: 10.1038/16012
Li, G., Bien-Ly, N., Andrews-Zwilling, Y., Xu, Q., Bernardo, A., Ring, K., et al. (2009). GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5, 634–645. doi: 10.1016/j.stem.2009.10.015
Li, Y. D., He, H. J., Dong, H. B., Feng, X. Y., Xie, G. M., and Zhang, L. J. (2013). Discriminative analysis of early-stage Alzheimer’s disease and normal aging with automatic segmentation technique in subcortical gray matter structures: a multicenter in vivo MRI volumetric and DTI study. Acta Radiol. 54, 1191–1200. doi: 10.1177/0284185113492971
Limon, A., Reyes-Ruiz, J. M., and Miledi, R. (2011). GABAergic drugs and Alzheimer’s disease. Future Med. Chem. 3, 149–153. doi: 10.4155/fmc.10.291
Limon, A., Reyes-Ruiz, J. M., and Miledi, R. (2012). Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. U.S.A. 109, 10071–10076. doi: 10.1073/pnas.1204606109
Lisman, J. (2005). The theta/gamma discrete phase code occuring during the hippocampal phase precession may be a more general brain coding scheme. Hippocampus 15, 913–922. doi: 10.1002/hipo.20121
Liu, X. J., Yuan, L., Yang, D., Han, W. N., Li, Q. S., Yang, W., et al. (2013). Melatonin protects against amyloid-beta-induced impairments of hippocampal LTP and spatial learning in rats. Synapse 67, 626–636. doi: 10.1002/syn.21677
Llinas, R., Ribary, U., Contreras, D., and Pedroarena, C. (1998). The neuronal basis for consciousness. Philos. Trans. R. Soc. Lond. B Biol. Sci. 353, 1841–1849. doi: 10.1098/rstb.1998.0336
Lopez, O. L. (2011). The growing burden of Alzheimer’s disease. Am. J. Manag. Care 17(Suppl. 13), S339–S345.
Loreth, D., Ozmen, L., Revel, F. G., Knoflach, F., Wetzel, P., Frotscher, M., et al. (2012). Selective degeneration of septal and hippocampal GABAergic neurons in a mouse model of amyloidosis and tauopathy. Neurobiol. Dis. 47, 1–12. doi: 10.1016/j.nbd.2012.03.011
Lott, I. T., and Head, E. (2005). Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol. Aging 26, 383–389. doi: 10.1016/j.neurobiolaging.2004.08.005
Louzada, P. R., Paula Lima, A. C., Mendonca-Silva, D. L., Noel, F., De Mello, F. G., and Ferreira, S. T. (2004). Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 18, 511–518. doi: 10.1096/fj.03-0739com
Lujan, R., Marron, F. D. V., Aguado, C., and Wickman, K. (2013). New insights into the therapeutic potential of Girk channels. Trends Neurosci. 37, 20–29. doi: 10.1016/j.tins.2013.10.006
Lujan, R., Maylie, J., and Adelman, J. P. (2009). New sites of action for GIRK and SK channels. Nat. Rev. Neurosci. 10, 475–480. doi: 10.1038/nrn2668
Luscher, C., Jan, L. Y., Stoffel, M., Malenka, R. C., and Nicoll, R. A. (1997). G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695. doi: 10.1016/S0896-6273(00)80381-5
Luscher, C., and Slesinger, P. A. (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315. doi: 10.1038/nrn2834
Lynch, G., Rose, G., and Gall, C. (1977). Anatomical and functional aspects of the septo-hippocampal projections. Ciba Found. Symp. 5–24. doi: 10.1002/9780470720394.ch3
Maccioni, R. B., Munoz, J. P., and Barbeito, L. (2001). The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 32, 367–381. doi: 10.1016/S0188-4409(01)00316-2
Mann, D. M., Yates, P. O., Marcyniuk, B., and Ravindra, C. R. (1986). The topography of plaques and tangles in Down’s syndrome patients of different ages. Neuropathol. Appl. Neurobiol. 12, 447–457. doi: 10.1111/j.1365-2990.1986.tb00053.x
Marcade, M., Bourdin, J., Loiseau, N., Peillon, H., Rayer, A., Drouin, D., et al. (2008). Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J. Neurochem. 106, 392–404. doi: 10.1111/j.1471-4159.2008.05396.x
Marczynski, T. J. (1995). GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease; pharmacologic profile of the benzodiazepine antagonist, flumazenil. Rev. Neurosci. 6, 221–258. doi: 10.1515/REVNEURO.1995.6.3.221
Marczynski, T. J. (1998). GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease revisited. Brain Res. Bull. 45, 341–379. doi: 10.1016/S0361-9230(97)00347-X
Martins, I. C., Kuperstein, I., Wilkinson, H., Maes, E., Vanbrabant, M., Jonckheere, W., et al. (2008). Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 27, 224–233. doi: 10.1038/sj.emboj.7601953
Massone, S., Vassallo, I., Fiorino, G., Castelnuovo, M., Barbieri, F., Borghi, R., et al. (2011). 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 41, 308–317. doi: 10.1016/j.nbd.2010.09.019
Matos, M., Augusto, E., Machado, N. J., dos Santos-Rodrigues, A., Cunha, R. A., and Agostinho, P. (2012). Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J. Alzheimers Dis. 31, 555–567. doi: 10.3233/JAD-2012-120469
Maurice, T., Lockhart, B. P., and Privat, A. (1996). Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 706, 181–193. doi: 10.1016/0006-8993(95)01032-7
McDonald, A. J. (1985). Immunohistochemical identification of gamma-aminobutyric acid-containing neurons in the rat basolateral amygdala. Neurosci. Lett. 53, 203–207. doi: 10.1016/0304-3940(85)90186-7
McDonald, A. J., and Augustine, J. R. (1993). Localization of GABA-like immunoreactivity in the monkey amygdala. Neuroscience 52, 281–294. doi: 10.1016/0306-4522(93)90156-A
McKeage, K. (2009). Memantine: a review of its use in moderate to severe Alzheimer’s disease. CNS Drugs 23, 881–897. doi: 10.2165/11201020-000000000-00000
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. doi: 10.1212/WNL.34.7.939
Miledi, R., Duenas, Z., Martinez-Torres, A., Kawas, C. H., and Eusebi, F. (2004). Microtransplantation of functional receptors and channels from the Alzheimer’s brain to frog oocytes. Proc. Natl. Acad. Sci. U.S.A. 101, 1760–1763. doi: 10.1073/pnas.0308224100
Mitew, S., Kirkcaldie, M. T., Dickson, T. C., and Vickers, J. C. (2013). Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 34, 2341–2351. doi: 10.1016/j.neurobiolaging.2013.04.010
Mizoguchi, H., and Yamada, K. (2011). Pharmacologic treatment with GABA(B) receptor agonist of methamphetamine-induced cognitive impairment in mice. Curr. Neuropharmacol. 9, 109–112. doi: 10.2174/157015911795016976
Mizukami, K., Ikonomovic, M. D., Grayson, D. R., Rubin, R. T., Warde, D., Sheffield, R., et al. (1997). Immunohistochemical study of GABA(A) receptor beta2/3 subunits in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Exp. Neurol. 147, 333–345. doi: 10.1006/exnr.1997.6591
Mizukami, K., Ikonomovic, M. D., Grayson, D. R., Sheffield, R., and Armstrong, D. M. (1998). Immunohistochemical study of GABAA receptor alpha1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res. 799, 148–155. doi: 10.1016/S0006-8993(98)00437-5
Mohamed, A., and Posse de, C. E. (2011). Abeta internalization by neurons and glia. Int. J. Alzheimers Dis. 2011, 127984. doi: 10.4061/2011/127984
Moises, H. C., Womble, M. D., Washburn, M. S., and Williams, L. R. (1995). Nerve growth factor facilitates cholinergic neurotransmission between nucleus basalis and the amygdala in rat: an electrophysiological analysis. J. Neurosci. 15, 8131–8142.
Moncaster, J. A., Pineda, R., Moir, R. D., Lu, S., Burton, M. A., Ghosh, J. G., et al. (2010). Alzheimer’s disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS ONE 5:e10659. doi: 10.1371/journal.pone.0010659
Moore, R. Y. (1993). Principles of synaptic transmission. Ann. N. Y. Acad. Sci. 695, 1–9. doi: 10.1111/j.1749-6632.1993.tb23018.x
Moreno, H., Wu, W. E., Lee, T., Brickman, A., Mayeux, R., Brown, T. R., et al. (2007). Imaging the Abeta-related neurotoxicity of Alzheimer disease. Arch. Neurol. 64, 1467–1477. doi: 10.1001/archneur.64.10.1467
Mota, S. I., Ferreira, I. L., and Rego, A. C. (2014). Dysfunctional synapse in Alzheimer’s disease–a focus on NMDA receptors. Neuropharmacology 76(Pt A), 16–26. doi: 10.1016/j.neuropharm.2013.08.013
Mott, D. D., and Lewis, D. V. (1994). The pharmacology and function of central GABAB receptors. Int. Rev. Neurobiol. 36, 97–223. doi: 10.1016/S0074-7742(08)60304-9
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338. doi: 10.1101/cshperspect.a006338
Nardone, R., Marth, R., Ausserer, H., Bratti, A., and Tezzon, F. (2006). Reduced short latency afferent inhibition in patients with Down syndrome and Alzheimer-type dementia. Clin. Neurophysiol. 117, 2204–2210. doi: 10.1016/j.clinph.2006.07.134
Nava-Mesa, M. O., Jimenez-Diaz, L., Yajeya, J., and Navarro-Lopez, J. D. (2013). Amyloid-beta induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria–CA3 hippocampal synapse. Front. Cell Neurosci. 7:117. doi: 10.3389/fncel.2013.00117
Nilsen, L. H., Witter, M. P., and Sonnewald, U. (2014). Neuronal and astrocytic metabolism in a transgenic rat model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 34, 906–914. doi: 10.1038/jcbfm.2014.37
O’Mahony, S., Harkany, T., Rensink, A. A., Abraham, I., De Jong, G. I., Varga, J. L., et al. (1998). Beta-amyloid-induced cholinergic denervation correlates with enhanced nitric oxide synthase activity in rat cerebral cortex: reversal by NMDA receptor blockade. Brain Res. Bull. 45, 405–411. doi: 10.1016/S0361-9230(97)00405-X
Ondrejcak, T., Klyubin, I., Hu, N. W., Barry, A. E., Cullen, W. K., and Rowan, M. J. (2010). Alzheimer’s disease amyloid beta-protein and synaptic function. Neuromolecular Med. 12, 13–26. doi: 10.1007/s12017-009-8091-0
Oren, I., Mann, E. O., Paulsen, O., and Hajos, N. (2006). Synaptic currents in anatomically identified CA3 neurons during hippocampal gamma oscillations in vitro. J. Neurosci. 26, 9923–9934. doi: 10.1523/JNEUROSCI.1580-06.2006
Palma, E., Torchia, G., Limatola, C., Trettel, F., Arcella, A., Cantore, G., et al. (2005). BDNF modulates GABAA receptors microtransplanted from the human epileptic brain to Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 102, 1667–1672. doi: 10.1073/pnas.0409442102
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., and Mucke, L. (2009). Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 66, 435–440. doi: 10.1001/archneurol.2009.15
Palop, J. J., and Mucke, L. (2010a). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Palop, J. J., and Mucke, L. (2010b). Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: two faces of the same coin? Neuromolecular Med. 12, 48–55. doi: 10.1007/s12017-009-8097-7
Palygin, O., Lalo, U., and Pankratov, Y. (2011). Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br. J. Pharmacol. 163, 1755–1766. doi: 10.1111/j.1476-5381.2011.01374.x
Palygin, O., Lalo, U., Verkhratsky, A., and Pankratov, Y. (2010). Ionotropic NMDA and P2X1/5 receptors mediate synaptically induced Ca2+ signalling in cortical astrocytes. Cell Calcium 48, 225–231. doi: 10.1016/j.ceca.2010.09.004
Panatier, A., Theodosis, D. T., Mothet, J. P., Touquet, B., Pollegioni, L., Poulain, D. A., et al. (2006). Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784. doi: 10.1016/j.cell.2006.02.051
Parameshwaran, K., Dhanasekaran, M., and Suppiramaniam, V. (2008). Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp. Neurol. 210, 7–13. doi: 10.1016/j.expneurol.2007.10.008
Parfenova, H., Tcheranova, D., Basuroy, S., Fedinec, A. L., Liu, J., and Leffler, C. W. (2012). Functional role of astrocyte glutamate receptors and carbon monoxide in cerebral vasodilation response to glutamate. Am. J. Physiol. Heart Circ. Physiol. 302, H2257–H2266. doi: 10.1152/ajpheart.01011.2011
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J. Y., et al. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. doi: 10.1126/science.1116916
Paula-Lima, A. C., Louzada, P. R., De Mello, F. G., and Ferreira, S. T. (2003). Neuroprotection against Abeta and glutamate toxicity by melatonin: are GABA receptors involved? Neurotox. Res. 5, 323–327. doi: 10.1007/BF03033152
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. doi: 10.1016/j.tins.2009.05.001
Prosser, H. M., Gill, C. H., Hirst, W. D., Grau, E., Robbins, M., Calver, A., et al. (2001). Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol. Cell. Neurosci. 17, 1059–1070. doi: 10.1006/mcne.2001.0995
Puma, C., and Bizot, J. C. (1999). Hippocampal theta rhythm in anesthetized rats: role of AMPA glutamate receptors. Neuroreport 10, 2297–2300. doi: 10.1097/00001756-199908020-00014
Rainnie, D. G., Asprodini, E. K., and Shinnick-Gallagher, P. (1991). Excitatory transmission in the basolateral amygdala. J. Neurophysiol. 66, 986–998.
Rajkowska, G., Miguel-Hidalgo, J. J., Wei, J., Dilley, G., Pittman, S. D., Meltzer, H. Y., et al. (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 45, 1085–1098. doi: 10.1016/S0006-3223(99)00041-4
Ramos, B., Baglietto-Vargas, D., Del Rio, J. C., Moreno-Gonzalez, I., Santa-Maria, C., Jimenez, S., et al. (2006). Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging 27, 1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022
Ramos-Hunter, S. J., Engers, D. W., Kaufmann, K., Du, Y., Lindsley, C. W., Weaver, C. D., et al. (2013). Discovery and SAR of a novel series of GIRK1/2 and GIRK1/4 activators. Bioorg. Med. Chem. Lett. 23, 5195–5198. doi: 10.1016/j.bmcl.2013.07.002
Raymond, C. R., Ireland, D. R., and Abraham, W. C. (2003). NMDA receptor regulation by amyloid-beta does not account for its inhibition of LTP in rat hippocampus. Brain Res. 968, 263–272. doi: 10.1016/S0006-8993(03)02269-8
Reinikainen, K. J., Paljarvi, L., Huuskonen, M., Soininen, H., Laakso, M., and Riekkinen, P. J. (1988). A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer’s disease. J. Neurol. Sci. 84, 101–116. doi: 10.1016/0022-510X(88)90179-7
Resende, R., Pereira, C., Agostinho, P., Vieira, A. P., Malva, J. O., and Oliveira, C. R. (2007). Susceptibility of hippocampal neurons to Abeta peptide toxicity is associated with perturbation of Ca2+ homeostasis. Brain Res. 1143, 11–21. doi: 10.1016/j.brainres.2007.01.071
Rissman, R. A., De Blas, A. L., and Armstrong, D. M. (2007). GABA(A) receptors in aging and Alzheimer’s disease. J. Neurochem. 103, 1285–1292. doi: 10.1111/j.1471-4159.2007.04832.x
Rissman, R. A., Mishizen-Eberz, A. J., Carter, T. L., Wolfe, B. B., De Blas, A. L., Miralles, C. P., et al. (2003). Biochemical analysis of GABA(A) receptor subunits alpha 1, alpha 5, beta 1, beta 2 in the hippocampus of patients with Alzheimer’s disease neuropathology. Neuroscience 120, 695–704. doi: 10.1016/S0306-4522(03)00030-7
Robinson, S. R. (2000). Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 36, 471–482. doi: 10.1016/S0197-0186(99)00150-3
Rockwood, K., Beattie, B. L., Eastwood, M. R., Feldman, H., Mohr, E., Pryse-Phillips, W., et al. (1997). A randomized, controlled trial of linopirdine in the treatment of Alzheimer’s disease. Can. J. Neurol. Sci. 24, 140–145.
Rovira, C., Arbez, N., and Mariani, J. (2002). Abeta(25-35) and Abeta(1-40) act on different calcium channels in CA1 hippocampal neurons. Biochem. Biophys. Res. Commun. 296, 1317–1321. doi: 10.1016/S0006-291X(02)02072-7
Rubio, S. E., Vega-Flores, G., Martinez, A., Bosch, C., Perez-Mediavilla, A., Del, R. J., et al. (2012). Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer’s disease. FASEB J. 26, 4458–4467. doi: 10.1096/fj.12-208413
Sabbagh, M. N. (2009). Drug development for Alzheimer’s disease: where are we now and where are we headed? Am. J. Geriatr. Pharmacother. 7, 167–185. doi: 10.1016/j.amjopharm.2009.06.003
Sago, H., Carlson, E. J., Smith, D. J., Kilbridge, J., Rubin, E. M., Mobley, W. C., et al. (1998). Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. U.S.A. 95, 6256–6261. doi: 10.1073/pnas.95.11.6256
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. U.S.A. 109, E2895–E2903. doi: 10.1073/pnas.1121081109
Santos-Torres, J., Fuente, A., Criado, J. M., Riolobos, A. S., Heredia, M., and Yajeya, J. (2007). Glutamatergic synaptic depression by synthetic amyloid beta-peptide in the medial septum. J. Neurosci. Res. 85, 634–648. doi: 10.1002/jnr.21150
Sarup, A., Larsson, O. M., and Schousboe, A. (2003). GABA transporters and GABA-transaminase as drug targets. Curr. Drug Targets CNS Neurol. Disord. 2, 269–277. doi: 10.2174/1568007033482788
Sawada, M., and Ichinose, M. (1996). Amyloid beta proteins reduce the GABA-induced Cl-current in identified Aplysia neurons. Neurosci. Lett. 213, 213–215. doi: 10.1016/0304-3940(96)12847-0
Schipper, H. M., Bennett, D. A., Liberman, A., Bienias, J. L., Schneider, J. A., Kelly, J., et al. (2006). Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 27, 252–261. doi: 10.1016/j.neurobiolaging.2005.01.016
Schuler, V., Lüscher, C., Blanchet, C., Klix, N., Sansig, G., Klebs, K., et al. (2001). Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA[B(1)]. Neuron 31, 47–58. doi: 10.1016/S0896-6273(01)00345-2
Scott, S. A., DeKosky, S. T., Sparks, D. L., Knox, C. A., and Scheff, S. W. (1992). Amygdala cell loss and atrophy in Alzheimer’s disease. Ann. Neurol. 32, 555–563. doi: 10.1002/ana.410320412
Sederberg, P. B., Schulze-Bonhage, A., Madsen, J. R., Bromfield, E. B., McCarthy, D. C., Brandt, A., et al. (2007). Hippocampal and neocortical gamma oscillations predict memory formation in humans. Cereb. Cortex 17, 1190–1196. doi: 10.1093/cercor/bhl030
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Seppala, T. T., Nerg, O., Koivisto, A. M., Rummukainen, J., Puli, L., Zetterberg, H., et al. (2012). CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 78, 1568–1575. doi: 10.1212/WNL.0b013e3182563bd0
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shi, J. Q., Wang, B. R., Tian, Y. Y., Xu, J., Gao, L., Zhao, S. L., et al. (2013). Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci. Ther. 19, 871–881. doi: 10.1111/cns.12144
Siarey, R. J., Carlson, E. J., Epstein, C. J., Balbo, A., Rapoport, S. I., and Galdzicki, Z. (1999). Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38, 1917–1920. doi: 10.1016/S0028-3908(99)00083-0
Simon, A. P., Poindessous-Jazat, F., Dutar, P., Epelbaum, J., and Bassant, M. H. (2006). Firing properties of anatomically identified neurons in the medial septum of anesthetized and unanesthetized restrained rats. J. Neurosci. 26, 9038–9046. doi: 10.1523/JNEUROSCI.1401-06.2006
Singer, W. (1993). Synchronization of cortical activity and its putative role in information processing and learning. Annu. Rev. Physiol. 55, 349–374. doi: 10.1146/annurev.ph.55.030193.002025
Smith, B. N., and Dudek, F. E. (1996). Amino acid-mediated regulation of spontaneous synaptic activity patterns in the rat basolateral amygdala. J. Neurophysiol. 76, 1958–1967.
Soderman, A., Mikkelsen, J. D., West, M. J., Christensen, D. Z., and Jensen, M. S. (2011). Activation of nicotinic alpha(7) acetylcholine receptor enhances long term potentation in wild type mice but not in APP(swe)/PS1DeltaE9 mice. Neurosci. Lett. 487, 325–329. doi: 10.1016/j.neulet.2010.10.049
Soderman, A., Thomsen, M. S., Hansen, H. H., Nielsen, E. O., Jensen, M. S., West, M. J., et al. (2008). The nicotinic alpha7 acetylcholine receptor agonist ssr180711 is unable to activate limbic neurons in mice overexpressing human amyloid-beta1-42. Brain Res. 1227, 240–247. doi: 10.1016/j.brainres.2008.06.062
Somogyi, P., and Klausberger, T. (2005). Defined types of cortical interneurone structure space and spike timing in the hippocampus. J. Physiol. 562, 9–26. doi: 10.1113/jphysiol.2004.078915
Soto, C. (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. doi: 10.1038/nrn1007
Sotty, F., Danik, M., Manseau, F., Laplante, F., Quirion, R., and Williams, S. (2003). Distinct electrophysiological properties of glutamatergic, cholinergic and GABAergic rat septohippocampal neurons: novel implications for hippocampal rhythmicity. J. Physiol. 551, 927–943. doi: 10.1113/jphysiol.2003.046847
Spencer, J. P., Weil, A., Hill, K., Hussain, I., Richardson, J. C., Cusdin, F. S., et al. (2006). Transgenic mice over-expressing human beta-amyloid have functional nicotinic alpha 7 receptors. Neuroscience 137, 795–805. doi: 10.1016/j.neuroscience.2005.10.007
Spires-Jones, T., and Knafo, S. (2012). Spines, plasticity, and cognition in Alzheimer’s model mice. Neural Plast. 2012, 319836. doi: 10.1155/2012/319836
Stewart, M., and Fox, S. E. (1990). Do septal neurons pace the hippocampal theta rhythm? Trends Neurosci. 13, 163–168. doi: 10.1016/0166-2236(90)90040-H
Stonnington, C. M., Snyder, P. J., Hentz, J. G., Reiman, E. M., and Caselli, R. J. (2009). Double-blind crossover study of the cognitive effects of lorazepam in healthy apolipoprotein E (APOE)-epsilon4 carriers. J. Clin. Psychiatry 70, 1379–1384. doi: 10.4088/JCP.08m04593
Sun, B., Halabisky, B., Zhou, Y., Palop, J. J., Yu, G., Mucke, L., et al. (2009). Imbalance between GABAergic and Glutamatergic Transmission Impairs Adult Neurogenesis in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 5, 624–633. doi: 10.1016/j.stem.2009.10.003
Sun, M. K., and Alkon, D. L. (2002). Impairment of hippocampal CA1 heterosynaptic transformation and spatial memory by beta-amyloid(25–35). J. Neurophysiol. 87, 2441–2449. doi: 10.1152/jn.00230.2001
Sunyer, B., Diao, W. F., Kang, S. U., An, G., Boddul, S., and Lubec, G. (2008). Cognitive enhancement by SGS742 in OF1 mice is linked to specific hippocampal protein expression. J. Proteome Res. 7, 5237–5253. doi: 10.1021/pr800594b
Swanberg, M. M., Tractenberg, R. E., Mohs, R., Thal, L. J., and Cummings, J. L. (2004). Executive dysfunction in Alzheimer disease. Arch. Neurol. 61, 556–560. doi: 10.1001/archneur.61.4.556
Swanson, L. W., and Petrovich, G. D. (1998). What is the amygdala? Trends Neurosci. 21, 323–331. doi: 10.1016/S0166-2236(98)01265-X
Takahashi, H., Brasnjevic, I., Rutten, B. P., van der Kolk, N., Perl, D. P., Bouras, C., et al. (2010). Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer’s disease. Brain Struct. Funct. 214, 145–160. doi: 10.1007/s00429-010-0242-4
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S., et al. (2013). Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U.S.A. 110, E2518–E2527. doi: 10.1073/pnas.1306832110
Tariot, P. N., Schneider, L. S., Cummings, J., Thomas, R. G., Raman, R., Jakimovich, L. J., et al. (2011). Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch. Gen. Psychiatry 68, 853–861. doi: 10.1001/archgenpsychiatry.2011.72
Terry, R. D. (2004). Tangles precede plaques but don’t cause them. Neurobiol. Aging 25, 741–742. doi: 10.1016/j.neurobiolaging.2003.12.013
Thathiah, A., and De, S. B. (2009). G protein-coupled receptors, cholinergic dysfunction, and Abeta toxicity in Alzheimer’s disease. Sci. Signal. 2, re8. doi: 10.1126/scisignal.293re8
Thathiah, A., and De, S. B. (2011). The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat. Rev. Neurosci. 12, 73–87. doi: 10.1038/nrn2977
Tosun, D., Joshi, S., and Weiner, M. W. (2013). Neuroimaging predictors of brain amyloidosis in mild cognitive impairment. Ann. Neurol. doi: 10.1002/ana.23921 [Epub ahead of print].
Tsuchiya, K., and Kosaka, K. (1990). Neuropathological study of the amygdala in presenile Alzheimer’s disease. J. Neurol. Sci. 100, 165–173. doi: 10.1016/0022-510X(90)90029-M
Tyszkiewicz, J. P., and Yan, Z. (2005). beta-Amyloid peptides impair PKC-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J. Neurophysiol. 93, 3102–3111. doi: 10.1152/jn.00939.2004
Ueda, Y., Doi, T., Nagatomo, K., Tokumaru, J., Takaki, M., and Willmore, L. J. (2007). Effect of levetiracetam on molecular regulation of hippocampal glutamate and GABA transporters in rats with chronic seizures induced by amygdalar FeCl3 injection. Brain Res. 1151, 55–61. doi: 10.1016/j.brainres.2007.03.021
Uemura, K., Kuzuya, A., Aoyagi, N., Ando, K., Shimozono, Y., Ninomiya, H., et al. (2007). Amyloid beta inhibits ectodomain shedding of N-cadherin via down-regulation of cell-surface NMDA receptor. Neuroscience 145, 5–10. doi: 10.1016/j.neuroscience.2006.12.022
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Vandevrede, L., Tavassoli, E., Luo, J., Qin, Z., Yue, L., Pepperberg, D. R., et al. (2013). Novel analogues of chlormethiazole are neuroprotective in four cellular models of neurodegeneration by a mechanism with variable dependence on GABA receptor potentiation. Br. J. Pharmacol. 171, 389–402. doi: 10.1111/bph.12454
Vereecken, T. H., Vogels, O. J., and Nieuwenhuys, R. (1994). Neuron loss and shrinkage in the amygdala in Alzheimer’s disease. Neurobiol. Aging 15, 45–54. doi: 10.1016/0197-4580(94)90143-0
Vergnes, M., Boehrer, A., Simler, S., Bernasconi, R., and Marescaux, C. (1997). Opposite effects of GABAB receptor antagonists on absences and convulsive seizures. Eur. J. Pharmacol. 332, 245–255. doi: 10.1016/S0014-2999(97)01085-6
Verkhratsky, A., and Kirchhoff, F. (2007). NMDA Receptors in glia. Neuroscientist 13, 28–37. doi: 10.1177/1073858406294270
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Villette, V., Poindessous-Jazat, F., Bellessort, B., Roullot, E., Peterschmitt, Y., Epelbaum, J., et al. (2012). A new neuronal target for β-amyloid peptide in the rat hippocampus. Neurobiol. Aging 33, 1126.e1–1126.e14. doi: 10.1016/j.neurobiolaging.2011.11.024
Villette, V., Poindessous-Jazat, F., Simon, A., Lena, C., Roullot, E., Bellessort, B., et al. (2010). Decreased rhythmic GABAergic septal activity and memory-associated theta oscillations after hippocampal amyloid-beta pathology in the rat. J. Neurosci. 30, 10991–11003. doi: 10.1523/JNEUROSCI.6284-09.2010
Vincent, A. J., Gasperini, R., Foa, L., and Small, D. H. (2010). Astrocytes in Alzheimer’s disease: emerging roles in calcium dysregulation and synaptic plasticity. J. Alzheimers Dis. 22, 699–714. doi: 10.3233/JAD-2010-101089
Vinogradova, O. S., Kitchigina, V. F., and Zenchenko, C. I. (1998). Pacemaker neurons of the forebrain medical septal area and theta rhythm of the hippocampus. Membr. Cell Biol. 11, 715–725.
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Wang, H. Y., Lee, D. H., D’Andrea, M. R., Peterson, P. A., Shank, R. P., and Reitz, A. B. (2000). beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 275, 5626–5632. doi: 10.1074/jbc.275.8.5626
Wang, Y., Zhang, G., Zhou, H., Barakat, A., and Querfurth, H. (2009). Opposite effects of low and high doses of Abeta42 on electrical network and neuronal excitability in the rat prefrontal cortex. PLoS ONE 4:e8366. doi: 10.1371/journal.pone.0008366
Washburn, M. S., and Moises, H. C. (1992). Muscarinic responses of rat basolateral amygdaloid neurons recorded in vitro. J. Physiol. 449, 121–154.
Webster, M. J., O’Grady, J., Kleinman, J. E., and Weickert, C. S. (2005). Glial fibrillary acidic protein mRNA levels in the cingulate cortex of individuals with depression, bipolar disorder and schizophrenia. Neuroscience 133, 453–461. doi: 10.1016/j.neuroscience.2005.02.037
Wei, Z. H., He, Q. B., Wang, H., Su, B. H., and Chen, H. Z. (2007). Meta-analysis: the efficacy of nootropic agent cerebrolysin in the treatment of Alzheimer’s disease. J. Neural. Transm. 114, 629–634. doi: 10.1007/s00702-007-0630-y
Wen, W., Wu, W., Romaine, I. M., Kaufmann, K., Du, Y., Sulikowski, G. A., et al. (2013). Discovery of ‘molecular switches’ within a GIRK activator scaffold that afford selective GIRK inhibitors. Bioorg. Med. Chem. Lett. 23, 4562–4566. doi: 10.1016/j.bmcl.2013.06.023
Whorton, M. R., and MacKinnon, R. (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208. doi: 10.1016/j.cell.2011.07.046
Wickman, K., Karschin, C., Karschin, A., Picciotto, M. R., and Clapham, D. E. (2000). Brain localization and behavioral impact of the G-protein-gated K+ channel subunit GIRK4. J. Neurosci. 20, 5608–5615.
Wolfe, M. S. (2010a). Alzheimer’s disease drug discovery–11th international conference–promising new therapeutic approaches. 27–28 September 2010, Jersey City, NJ, USA. IDrugs 13, 825–827.
Wolfe, M. S. (2010b). Alzheimer’s disease drug discovery–11th international conference–targeting pathological tau. 27–28 September 2010, Jersey City, NJ, USA. IDrugs 13, 828–829.
Wu, W., and Small, S. A. (2006). Imaging the earliest stages of Alzheimer’s disease. Curr. Alzheimer Res. 3, 529–539. doi: 10.2174/156720506779025161
Xiong, H., Baskys, A., and Wojtowicz, J. M. (1996). Brain-derived peptides inhibit synaptic transmission via presynaptic GABAB receptors in CA1 area of rat hippocampal slices. Brain Res. 737, 188–194. doi: 10.1016/0006-8993(96)00731-7
Xu, X., An, L., Mi, X., and Zhang, T. (2013). Impairment of cognitive function and synaptic plasticity associated with alteration of information flow in theta and gamma oscillations in melamine-treated rats. PLoS ONE 8:e77796. doi: 10.1371/journal.pone.0077796
Yajeya, J., de la Fuente, J. A., Bajo, V. M., Riolobos, A. S., Heredia, M., and Criado, J. M. (1999). Muscarinic activation of a non-selective cationic conductance in pyramidal neurons in rat basolateral amygdala. Neuroscience 88, 159–167. doi: 10.1016/S0306-4522(98)00210-3
Yajeya, J., de la Fuente, J. A., Criado, J. M., Bajo, V., Sanchez-Riolobos, A., and Heredia, M. (2000). Muscarinic agonist carbachol depresses excitatory synaptic transmission in the rat basolateral amygdala in vitro. Synapse 38, 151–160. doi: 10.1002/1098-2396(200011)38:2<151::AID-SYN6>3.0.CO;2-K
Yajeya, J., de la Fuente, J. A., Merchan, M. A., Riolobos, A. S., Heredia, M., and Criado, J. M. (1997). Cholinergic responses of morphologically and electrophysiologically characterized neurons of the basolateral complex in rat amygdala slices. Neuroscience 78, 731–743. doi: 10.1016/S0306-4522(96)00614-8
Yamaguchi, Y., and Kawashima, S. (2001). Effects of amyloid-beta-(25–35) on passive avoidance, radial-arm maze learning and choline acetyltransferase activity in the rat. Eur. J. Pharmacol. 412, 265–272. doi: 10.1016/S0014-2999(01)00730-0
Yamakura, T., Lewohl, J. M., and Harris, R. A. (2001). Differential effects of general anesthetics on G protein-coupled inwardly rectifying and other potassium channels. Anesthesiology 95, 144–153. doi: 10.1097/00000542-200107000-00025
Yang, K., Belrose, J., Trepanier, C. H., Lei, G., Jackson, M. F., and MacDonald, J. F. (2011). Fyn, a potential target for Alzheimer’s disease. J. Alzheimers Dis. 27, 243–252. doi: 10.3233/JAD-2011-110353
Yang, Y., Ge, W., Chen, Y., Zhang, Z., Shen, W., Wu, C., et al. (2003). Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. U.S.A. 100, 15194–15199. doi: 10.1073/pnas.2431073100
Yew, D. T., Li, W. P., Webb, S. E., Lai, H. W., and Zhang, L. (1999). Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and Alzheimer patients: a comparison. Exp. Gerontol. 34, 117–133. doi: 10.1016/S0531-5565(98)00017-5
Yoshiike, Y., Kimura, T., Yamashita, S., Furudate, H., Mizoroki, T., Murayama, M., et al. (2008). GABA(A) receptor-mediated acceleration of aging-associated memory decline in APP/PS1 mice and its pharmacological treatment by picrotoxin. PLoS ONE 3:e3029. doi: 10.1371/journal.pone.0003029
Young, A. B. (1987). Cortical amino acidergic pathways in Alzheimer’s disease. J. Neural Transm. Suppl. 24, 147–152.
Yow, T. T., Pera, E., Absalom, N., Heblinski, M., Johnston, G. A., Hanrahan, J. R., et al. (2011). Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin-Q. Br. J. Pharmacol. 163, 1017–1033. doi: 10.1111/j.1476-5381.2011.01315.x
Zald, D. H. (2003). The human amygdala and the emotional evaluation of sensory stimuli. Brain Res. Brain Res. Rev. 41, 88–123. doi: 10.1016/S0165-0173(02)00248-5
Zemankovics, R., Veres, J. M., Oren, I., and Hajos, N. (2013). Feedforward inhibition underlies the propagation of cholinergically induced gamma oscillations from hippocampal CA3 to CA1. J. Neurosci. 33, 12337–12351. doi: 10.1523/JNEUROSCI.3680-12.2013
Zhang, C., Cheng, Y., Wang, H., Wang, C., Wilson, S. P., Xu, J., et al. (2014). RNA interference-mediated knockdown of long-form phosphodiesterase-4D (PDE4D) enzyme reverses amyloid-beta42-induced memory deficits in mice. J. Alzheimers Dis. 38, 269–280. doi: 10.3233/JAD-122236
Keywords: septohippocampal system, amyloid-β peptide, excitatory and inhibitory neurotransmission, learning and memory, Alzheimer’s disease
Citation: Nava-Mesa MO, Jiménez-Díaz L, Yajeya J and Navarro-Lopez JD (2014) GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer’s disease. Front. Cell. Neurosci. 8:167. doi: 10.3389/fncel.2014.00167
Received: 15 January 2014; Accepted: 02 June 2014;
Published online: 25 June 2014.
Edited by:
Ludovic Martin, Université de Nantes, FranceReviewed by:
Alexander Dityatev, German Center for Neurodegenerative Diseases, GermanyJacques Epelbaum, Institut National de la Santé et de la Recherche Médicale, France
Robert Nistico, University of Calabria, Italy
Copyright © 2014 Nava-Mesa, Jiménez-Díaz, Yajeya and Navarro-Lopez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan D. Navarro-López, Neurophysiology and Behavior Lab, Centro Regional de Investigaciones Biomédicas, School of Medicine of Ciudad Real, University of Castilla-La Mancha, Paseo Moledores s/n, 13071-Ciudad Real, Spain e-mail:anVhbi5uYXZhcnJvQHVjbG0uZXM=; Mauricio O. Nava-Mesa, Neuroscience Research Group, School of Medicine, University of Rosario, Kra 24 # 63C-69, Bogotá, Colombia e-mail:bW9uYXZhbUB1c2FsLmVz