 Miguel Medina
Miguel Medina Jesús Avila
Jesús Avila- 1Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Madrid, Spain
- 2Centro de Biología Molecular “Severo Ochoa” CSIC-UAM, Madrid, Spain
The microtubule-associated protein (MAP) tau plays a critical role in the pathogenesis of Alzheimer’s disease (AD) and several related disorders collectively known as tauopathies. Development of tau pathology is associated with progressive neuronal loss and cognitive decline. In the brains of AD patients, tau pathology spreads following an anatomically defined pattern. Mounting evidence strongly suggests that accumulation of abnormal tau is mediated through spreading of seeds of the protein from cell to cell and point at the involvement of extracellular tau species as the main agent in the interneuronal propagation of neurofibrillary lesions and spreading of tau toxicity throughout different brain regions in these disorders. That would support the concept that pathology initiates in a very small part of the brain many years before becoming symptomatic, spreading progressively to the whole brain within 10–20 years. Understanding the precise molecular mechanism underlying tau propagation is crucial for the development of therapeutics for this devastating disorder. In this work, we will discuss recent research on the role of extracellular tau in the spreading of tau pathology, through synaptic and non-synaptic mechanisms.
Introduction
A common pathological feature of many neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) or prion diseases, among others, is the abnormal deposition of proteins in the brain. Among these pathological proteins, the MAP tau forms intraneuronal filaments in a spectrum of neurological disorders collectively known as tauopathies.
Tau protein is a MAP that under physiological conditions regulates microtubules (MT) assembly, dynamic behavior, and spatial organization, and has also been shown to regulate the axonal transport of organelles, including mitochondria. The gene encoding tau protein MAPT is located on chromosome 17q21.3, spans approximately 150 kb and consists of 16 exons (Pittman et al., 2006) from which six major isoforms are expressed in adult brain through alternative splicing (reviewed in Andreadis, 2012). The interaction between tau and tubulin is mediated by four imperfect repeat domains (encompassing 31–32 residues) encoded by exons 9–12 (Lee et al., 1989). Alternative splicing of exon 10 results in the production of isoforms containing 3 or 4 binding domains (3R and 4R tau) (Himmler et al., 1989).
Adult human brain contains equal amounts of 3R and 4R isoforms whereas foetal brain, however, only expresses 3R tau, demonstrating developmental regulation of exon 10 splicing (Goedert et al., 1989). Different brain regions also differ in the relative levels of 3R and 4R isoforms with granule cells in the hippocampal formation reported to have only 3R tau. Disturbances, usually increases, in the 3R/4R ratio are a common feature in most neurodegenerative tauopathies. Furthermore, morphological differences exist among different diseases or disease types as different tau isoforms are accumulated in diseased brains, namely, six tau isoforms in AD, 3R tau isoforms in Pick’s disease, and 4R tau isoforms in profressive supranuclear palsy (PSP) and cortical basal degeneration (CBD; Goedert and Spillantini, 2011). Interestingly, a recent study has shown that the 4R/3R ration may have been underestimated in AD brains when compared with PSP or CBD, presumably due to extensive deamidation at Asn279 (Dan et al., 2013).
Within neurons, tau is predominantly found in axons (Hanger et al., 2009) as a highly soluble phosphoprotein (Iqbal et al., 2009). Phosphorylation is also developmentally regulated, with a high tau phosphorylation level during embryogenesis and early development, when only the shortest of the isoforms is being expressed. By contrast, adult brain expresses all six isoforms with relatively reduced phosphorylation levels compared with the foetal one (Liu et al., 2007).
The key discovery directly involving tau protein in neurodegeneration and dementia came from the finding that highly penetrant, dominant mutations in the MAPT gene encoding tau cause an inherited form of frontotemporal dementia and parkinsonism (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). A number of neurodegenerative disorders present prominent tau pathology in the CNS, predominantly within the neuronal compartment, but also within glial cells. Because of this shared histopathological feature, they are referred collectively as tauopathies, although they constitute a group of etiologically heterogeneous, clinically and neuropathologically overlapping disease entities (Ballatore et al., 2007; Spillantini and Goedert, 2013). In tauopathies, the intracellular soluble tau forms filamentous structures of aggregated, hyperphosphorylated tau, which are associated with synaptic loss and neuronal death. The occurrence of fibrillar tau inclusions in tauopathies strongly supports a key role in the observed clinical symptoms and pathology.
Further insights into the overlapping pathogenic and etiologic aspects of the discrete diseases will help to design (perhaps common) disease-modifying treatment strategies (Medina, 2011). To achieve that goal however, it is critical to understand the normal biological roles of tau, the specific molecular events that induce tau to become neurotoxic, the biochemical nature of pathogenic tau, the means by which pathogenic tau exerts neurotoxicity, and how tau pathology propagates.
As mentioned, the recognition of the MAP tau as a key player in the pathobiology of human neurodegenerative diseases has led to major efforts to understand its biological and pathological function(s). This has resulted in an improved understanding of tau cellular functions beyond its classical role in stabilizing MT (Morris et al., 2011) to unveil novel physiological tau functions that may also play a role in pathogenesis. Such functions include axonal transport (Terwel et al., 2002; Rodríguez-Martín et al., 2013), neuronal polarization (Caceres and Kosik, 1990; Dawson et al., 2010), axonogenesis (DiTella et al., 1994; Klein et al., 2002; Belkadi and LoPresti, 2008), interactions with the plasma membrane (Brandt et al., 1995; Lee et al., 1998; Maas et al., 2000), signal transduction (Lee et al., 2004; Ittner et al., 2010) and cell cycle (Andorfer et al., 2005). Furthermore, despite lacking an identified nuclear localization signal, tau has also been reported in nuclei in a number of cell lines (Loomis et al., 1990; Wang et al., 1993) and human brain (Brady et al., 1995) where it may play a role in DNA protection (Sultan et al., 2011).
Extracellular Tau
It has been over 20 years since the original report that intracellular tau levels are increased in the brains of AD patients when compared to non-demented controls (Barton et al., 1990; Khatoon et al., 1992). This increase in the amount of tau could be toxic to neurons since a reduction in the amount of intracellular tau has indeed a protective effect in mouse models of neurodegeneration (Rapoport et al., 2002; Roberson et al., 2007) and it has been suggested that reducing tau levels may be therapeutically beneficial (Götz et al., 2013). However, we must be cautious since other studies in similar tau-deficient mice point in the opposite direction, suggesting that loss of tau function can actually lead to neurodegeneration (Dawson et al., 2010).
Little is known about how tau synthesis is regulated although some factors such as fibroblast growth factor (Tatebayashi et al., 1999), Dyrk1A (Qian et al., 2013), or the haplotype H1 have been involved in increased synthesis whereas the miRNA-34 family (Dickson et al., 2013) seems to downregulate tau levels.
Conventional wisdom has suggested that the presence of tau in the brain parenchyma or in the cerebrospinal fluid (CSF) is a consequence of tau protein being released from dead cells. However, this has recently been challenged by a number of studies showing extracellular tau being released from cell lines and neurons via multiple pathways, strongly supporting the notion that secretion of tau protein may be an important biological function of tau protein, especially in disease. Despite the fact that tau lacks a signal sequence a number of reports have now shown that tau is released into culture medium from neuroblastoma cells, tau-expressing non-neuronal cells, induced pluripotent stem cell-derived human neurons, and mouse primary neurons (Kim et al., 2010; Shi et al., 2012). Thus, tau has been reported to be secreted unconventionally in naked form (Chai et al., 2012) or associated to exosomes (Saman et al., 2012) and/or other membrane vesicles (Simón et al., 2012a). Since increased tau cellular levels are detrimental, secretion has been proposed as a mechanism to eliminate the excess of tau protein thereby avoiding its toxicity (Simón et al., 2012b). Interestingly, while full length tau has been detected in the extracellular space, C-terminal cleavage of tau has been shown to enhance its secretion (Plouffe et al., 2012) which could have pathological relevance since some truncated tau species appears to be characteristic of particular tauopathies whereas other tau fragments may be common to several tauopathies (Hanger and Wray, 2010; Kovacech et al., 2010).
Extracellular tau has also been detected in the brain interstitial fluid of both wild-type and P301S tau-expressing mice in microdialysis studies (Yamada et al., 2011), as it has also been the case in patients following severe traumatic brain injury (Marklund et al., 2009; Magnoni et al., 2012). Actually, exosomal tau secretion has been suggested to account for the elevated CSF tau levels typically observed in early AD (Saman et al., 2012). Interestingly, tau mutations that are associated with the development of tauopathy appears to reduce tau release (Karch et al., 2012). Interestingly, physiological secretion of endogenous tau by cortical neurons appears to be regulated by neuronal activity, as tau release is enhanced by glutamate receptor stimulation induced by the agonist S-AMPA (Pooler et al., 2013). This process is calcium-dependent and modulated by phosphorylation and released tau is present in a relatively dephosphorylated state, compared to that of intracellular tau.
Thus, increasing evidence point out to extracellular tau as a physiological process independent of cell death (Figure 1), although the precise relationship between tau release under physiological conditions and the propagation of pathology in AD and other tauopathies remains to be determined.
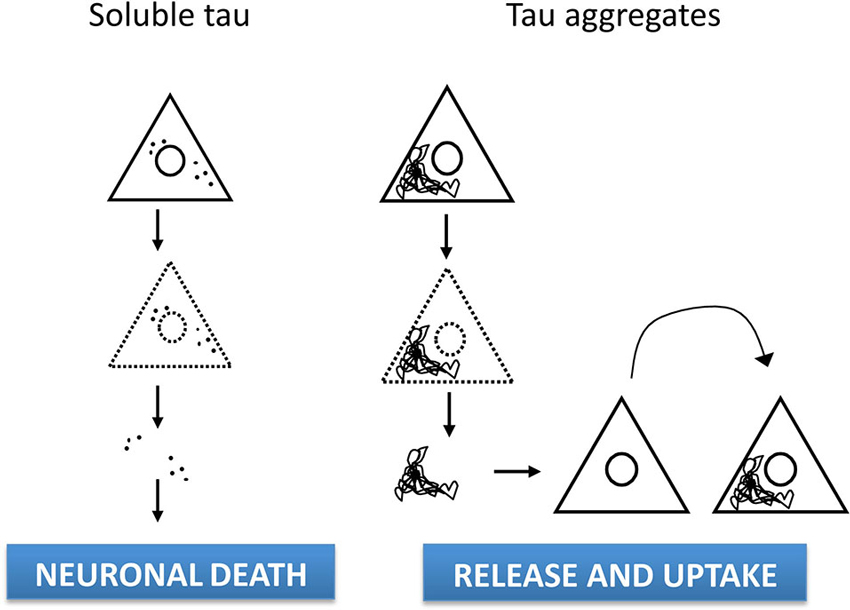
Figure 1. Cell-to-Sources of extracellular tau and -cell spreading. The established view has long considered the presence of tau in the brain parenchyma or in the CSF as a consequence of tau protein being released after cell lysis. However, extracellular tau appears to result from a physiological process independent of cell death, as tau being can be released from cell lines and neurons via multiple pathways, either in naked form or vesicle-associated. In addition, tau misfolding in diseased brain leads to abnormal conformations of tau that can be taken up by surrounding neurons. Thus, pathological progression could involve transmission of tau protein through a prion-like mechanism resulting in neurodegeneration in susceptible brain regions.
Tau can be toxic when applied extracellularly to cultured cells (Gómez-Ramos et al., 2006; Kopeikina et al., 2012). Several mechanisms for internalization of tau has been proposed, such as internalization of soluble, uncoated (“naked”) tau via receptor-mediated endocytosis (Gómez-Ramos et al., 2009), dynamin-driven endocytosis of non-fibrillar, soluble tau aggregates (Wu et al., 2013) or even actin-dependent, proteoglycan-mediated macropynocytosis (Holmes et al., 2013). Furthermore, it has been suggested that extracellular tau might provoke a receptor-activated increase in intracellular calcium through M1/M3 muscarinic receptor stimulation (Gómez-Ramos et al., 2008; Díaz-Hernández et al., 2010) and that such receptor activation could lead to endocytosis of extracellular tau. Remarkably, tau phosphorylation could inhibit its interaction with M1/M3 receptors and it has been proposed that such alterations might be involved in the transmission of tau pathology (Simón et al., 2013).
The discovery of extracellular tau as a physiological process that is independent of cell death (Pooler et al., 2013), indicates that tau release does not occur only as a result of reduced neuronal viability, and therefore that the increased tau observed in interstitial fluid and CSF in tauopathies may not be due solely to tau release from dying neurons (Yamada et al., 2011; Nedergaard, 2013). It is worth mentioning that tau phosphorylation at threonine 181 and total tau levels in CSF are considered useful biomarkers of neuronal degeneration or injury in the recent National Institute on Aging and Alzheimer Association (NIA-AA) revised criteria for the diagnosis of AD (Jack et al., 2012).
Propagation of Tau Pathology
Development of tau pathology is associated with progressive neuronal loss and cognitive decline. In the brains of AD patients, tau pathology propagates following an anatomically defined pattern described by the neuropathological Braak sequential staging (Braak et al., 2011). As a matter of fact, recent NIA-AA guidelines recommend the assessment of Braak and Braak staging of neurofibrillary degeneration as part of the so called “ABC score” for the neuropathological diagnosis of AD (Montine et al., 2012).
The originally staging system (Braak and Braak, 1991) defined six stages based on the presence and density of characteristic argyrophylic inclusions (neurofibrillary tangles (NFT), neuropil threads) in the medial temporal lobe and several brain isocortical regions. This system was subsequently adapted by the authors for routine use in paraffin-embedded tissue based on tau immunohistochemistry (Braak et al., 2006). Stages I–II (transentorhinal) correlate with the lengthy preclinical phase of the disease; whereas stages III–IV (limbic) do so with mild cognitive impairment (loss of episodic memory) or mild dementia; and advanced V–VI stages (isocortical) usually correspond to cases with moderate to severe dementia. Accurate staging of AD-related tau-positive pathology may be particularly important in the classification of preclinical disease and in the identification of atypical AD phenotypes. The above mentioned NIA-AA guidelines allow for standardization for diagnostic and research purposes.
Clinicopathological studies show that tau pathology progression from the entorhinal cortex through the hippocampus and into the limbic and association cortex is the main neuropathological variable that correlates with the clinical cognitive status of the patient (Arriagada et al., 1992; Nelson et al., 2012). Whether that pattern of accumulation reflects cell-to-cell spreading of disease, or simply successive involvement of differentially resistant neuronal populations, has been a matter of debate in recent years. Recent evidence from human studies suggests that tau pathology is actually linked to existing networks of neuronal connectivity. Thus, rather than diffuse, random, or confluent, tau pathology would target specific large-scale distributed networks that in the healthy brain feature convergent intrinsic functional and structural covariance (Seeley et al., 2009). However, the precise molecular and cellular mechanisms by which tau propagates and neuronal networks degenerate are still unknown.
Increasing evidence suggests that synaptic dysfunction is a key pathophysiological hallmark in neurodegenerative disorders, including AD which has been indeed considered a synaptopathy (Selkoe, 2002; Sheng et al., 2012), as synapse density best correlates with the cognitive decline observed in patients. Long regarded primarily as an axonal protein, when hyperphosphorylated tau also accumulates in the somatodendritic compartment during AD (Ballatore et al., 2007). Actually, tau mislocation in dendritic spines has been proposed to lead to synaptic dysfunction by various mechanisms, including regulating the amount of glutamate receptors in spines (Hoover et al., 2010), interacting with post-synaptic signaling complexes, targeting of synaptic mitochondria (Pooler et al., 2014) or destabilizing dendritic spines and dendritic arbor (Koleske, 2013). Presence of tau in the synapse in healthy brains suggests a role for tau in regulating normal synaptic function whereas during neurodegeneration, tau synaptotoxicity seems to be related to soluble forms rather than insoluble aggregates (Pooler et al., 2014).
On the other hand, emerging evidence strongly suggests that tau is essential for Aβ-induced synaptotoxicity (Ittner et al., 2010), a process that may involve EphB2, and NMDA receptors (Cissé et al., 2011; Sheng et al., 2012). Furthermore, studies in mouse organotypic hippocampal slice cultures from amyloid precurssor protein transgenics have demonstrated that extrasynaptic NR2B-containing NMDA receptors are required for tau-induced neurodegeneration (Tackenberg et al., 2013). Could then tau play a role in the transition between synaptic and extrasynaptic NMDA receptors? Although we do not have a definitive answer to that, the improved NMDA receptor antagonist nitromemantine protects against Aβ-induced synaptic dysfunction (Talantova et al., 2013). Nitromemantine selectively inhibits extrasynaptic over synaptic NMDA receptor activity (Kaufman et al., 2012), thus preventing the toxic effect of the activation of extrasynaptic NMDA receptors. Aβ binding to synaptic or extrasynaptic receptors may lead to different signaling and consequences (protection or death) (Li et al., 2011). Increase of extrasynaptic receptors with aging and AD may also explain the progression of the disease (Figure 2).
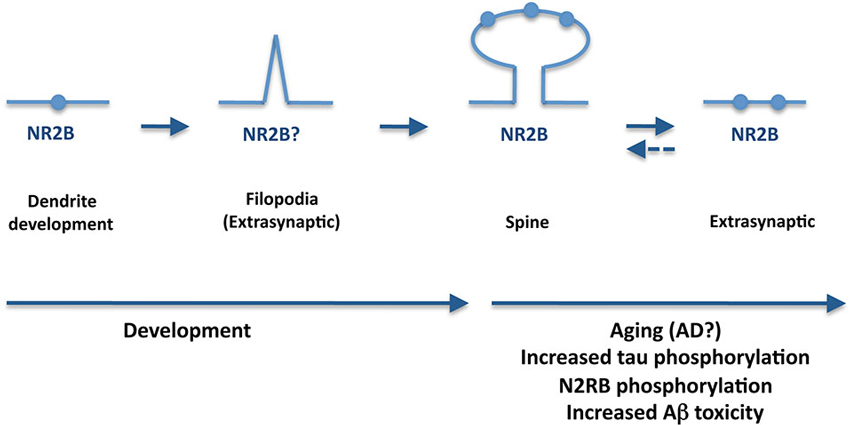
Figure 2. A potential role for tau in synapsis during aging and AD. During AD, tau is hyperphosphorylated and mislocates to the axonal compartment. Furthermore, tau appears essential for Aβ-induced synaptotoxicity whereas extrasynaptic NR2B-containing NMDA receptors are required for tau-induced neurodegeneration. An increase of extrasynaptic receptors with aging and AD may also explain the progression of the disease.
Interestingly, recent in vivo studies in tauopathy transgenic mouse models expressing human mutant tau specifically in the entorhinal cortex have shown relocation of tau from axons to the somatodendritic compartment as well as propagation of tau pathology to regions outside the entorhinal cortex, strongly suggesting a trans-synaptic mechanism of spreading of pathology through anatomically connected neuronal networks (de Calignon et al., 2012; Liu et al., 2012). These findings have been further supported by more recent neuropathological studies in post-mortem brains from argyrophylic grain disease (AGD), a sporadic tauopathy mainly involving the medial temporal lobe and the limbic region (Ferrer et al., 2008). This pathology exhibits a short number of closely related tau-positive inclusions and a highly homogeneous pattern of distribution and progression of pathology along several regions of the medial temporal lobe with known connectivity between them and with extra-temporal areas of involvement, leading to its proposition as a natural model for studying tau propagation in human brain (Rábano et al., 2014).
Recently, release and subsequent uptake of tau fibrils that directly contact native protein in recipient cells have been shown to mediate propagation of tau misfolding among cells, at least in vitro (Frost et al., 2009; Kfoury et al., 2012). Remarkably, intracerebral inoculation of synthetic preformed tau fibrils induced NFT-like inclusions that propagated from injected sites to connected brain regions in a time-dependent manner (Iba et al., 2013). Furthermore, conformation-specific trans-cellular propagation of tau fibrils after release into the extracellular space and subsequent triggering of aggregation in recipient cells by contacting native protein has been show in co-culture experiments (Kfoury et al., 2012). Thus, newly aggregated intracellular tau can transfer between co-cultured cells (Figure 1), thus providing a mechanism for tau-targeted immunotherapies as therapeutic strategy for AD and tauopathies (Gu and Sigurdsson, 2011; Medina, 2011; Golde et al., 2013). Actually, it has been suggested that the most likely mechanism of action for anti-tau antibodies is targeting tau released from cells (Yanamandra et al., 2013). The recent development of imaging-based biomarkers (Maruyama et al., 2013) will enable to track the progression of tau pathology in living patients and greatly facilitate the early phase testing of tau immunotherapy and other tau-based therapeutic strategies.
Conclusions
In summary, we have highlighted recent developments in tau biology relevant to AD and tauopathies. It has become increasingly clear that, apart from the well-established intracellular functions of tau in microtubule stabilization and axonal transport, intracellular and extracellular tau most likely have important signaling roles that could contribute to the neurodegenerative process in AD and related tauopathies. Furthermore, the presence of tau in synaptic regions of healthy brain suggest that tau may play a role in the regulation of normal synaptic function. In addition, recent studies have suggested that misfolding of tau in diseased brain leads to abnormal conformations of tau that can be taken up by surrounding neurons. Thus, pathological progression could involve transmission of tau protein through a potential prion-like, seeding mechanism resulting in neurodegeneration in susceptible brain regions. However, insufficient evidence exists yet to reliably determine whether there is a direct relationship between the recent identification of a physiological role for extracellular tau and the impairments in tau function associated with disease.
Key questions still remain open, such as the neuronal selectivity, the nature of the extracellular tau species involved, or the precise seeding/templating mechanisms, among many others. More research is needed to identify disease mechanisms driving release and propagation of tau pathology and to determine the impact of extracellular tau on cognitive decline during neurodegeneration. The observation that misfolded tau can be secreted and taken up by adjacent neurons calls for the development of novel strategies to block the propagation of tau pathology in the brain. Despite the fact that the presence of extensive tau pathology is central to the disease, tau-based therapeutic strategies have received little attention until recently (Medina and Avila, 2014). Next few years will certainly bring new insights into the cellular mechanisms underlying tau secretion and uptake, likely identifying novel therapeutic approaches intended to interfere early on in the process of propagation of tau pathology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454. doi: 10.1523/jneurosci.4637-04.2005
Andreadis, A. (2012). Tau splicing and the intricacies of dementia. J. Cell. Physiol. 227, 1220–1225. doi: 10.1002/jcp.22842
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. doi: 10.1212/wnl.42.3.631
Ballatore, C., Lee, V. M. Y., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672. doi: 10.1038/nrn2194
Barton, A. J., Harrison, P. J., Najlerahim, A., Heffernan, J., McDonald, B., Robinson, J. R., et al. (1990). Increased tau messenger RNA in Alzheimer’s disease hippocampus. Am. J. Pathol. 137, 497–502.
Belkadi, A., and LoPresti, P. (2008). Truncated Tau with the Fyn-binding domain and without the microtubule-binding domain hinders the myelinating capacity of an oligodendrocyte cell line. J. Neurochem. 107, 351–360. doi: 10.1111/j.1471-4159.2008.05600.x
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/bf00308809
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/nen.0b013e318232a379
Brady, R. M., Zinkowski, R. P., and Binder, L. I. (1995). Presence of tau in isolated nuclei from human brain. Neurobiol. Aging 16, 479–486. doi: 10.1016/0197-4580(95)00023-8
Brandt, R., Leger, J., and Lee, G. (1995). Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 131, 1327–1340. doi: 10.1083/jcb.131.5.1327
Caceres, A., and Kosik, K. S. (1990). Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 343, 461–463. doi: 10.1038/343461a0
Chai, X., Dage, J. L., and Citron, M. (2012). Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 48, 356–366. doi: 10.1016/j.nbd.2012.05.021
Cissé, M., Halabisky, B., Harris, J., Devidze, N., Dubal, D. B., Sun, B., et al. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. doi: 10.1038/nature09635
Dan, A., Takahashi, M., Masuda-Suzukake, M., Kametani, F., Nonaka, T., Kondo, H., et al. (2013). Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer’s disease brain. Acta Neuropathol. Commun. 1:54. doi: 10.1186/2051-5960-1-54
Dawson, H. N., Cantillana, V., Jansen, M., Wang, H., Vitek, M. P., Wilcock, D. M., et al. (2010). Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience 169, 516–531. doi: 10.1016/j.neuroscience.2010.04.037
de Calignon, A., Polydoro, M., Suárez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Díaz-Hernández, M., Gomez-Ramos, A., Rubio, A., Gomez-Villafuertes, R., Naranjo, J. R., Miras-Portugal, M. T., et al. (2010). Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 285, 32539–32548. doi: 10.1074/jbc.m110.145003
Dickson, J. R., Kruse, C., Montagna, D. R., Finsen, B., and Wolfe, M. S. (2013). Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 127, 739–749. doi: 10.1111/jnc.12437
DiTella, M., Feiguin, F., Morfini, G., and Caceres, A. (1994). Microfilament associated growth cone component depends upon Tau for its intracellular localization. Cell Motil. Cytoskeleton 29, 117–130. doi: 10.1002/cm.970290204
Ferrer, I., Santpere, G., and van Leeuwen, F. W. (2008). Argyrophilic grain disease. Brain 131, 1416–1432. doi: 10.1093/brain/awm305
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852. doi: 10.1074/jbc.m808759200
Goedert, M., and Spillantini, M. G. (2011). Pathogenesis of the tauopathies. J. Mol. Neurosci. 45, 425–431. doi: 10.1007/s12031-011-9593-4
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., and Crowther, R. A. (1989). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399.
Golde, T. E., Lewis, J., and McFarland, N. R. (2013). Anti-tauantibodies: hitting the target. Neuron 80, 254–256. doi: 10.1016/j.neuron.2013.10.009
Gómez-Ramos, A., Díaz-Hernández, M., Cuadros, R., Hernandez, F., and Avila, J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850. doi: 10.1016/j.febslet.2006.07.078
Gómez-Ramos, A., Díaz-Hernández, M., Rubio, A., Díaz-Hernández, J. I., Miras-Portugal, M. T., and Avila, J. (2009). Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 19, 708–717. doi: 10.1016/j.euroneuro.2009.04.006
Gómez-Ramos, A., Díaz-Hernández, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681. doi: 10.1016/j.mcn.2007.12.010
Götz, J., Xia, D., Leinenga, G., Chew, Y. L., and Nicholas, H. (2013). What renders TAU toxic. Front. Neurol. 4:72. doi: 10.3389/fneur.2013.00072
Gu, J., and Sigurdsson, E. M. (2011). Immunotherapy for tauopathies. J. Mol. Neurosci. 45, 690–695. doi: 10.1007/s12031-011-9576-5
Hanger, D. P., Anderton, B. H., and Noble, W. (2009). Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119. doi: 10.1016/j.molmed.2009.01.003
Hanger, D. P., and Wray, S. (2010). Tau cleavage and tau aggregation in neurodegenerative disease. Biochem. Soc. Trans. 38, 1016–10120. doi: 10.1042/bst0381016
Himmler, A., Drechsel, D., Kirschner, M. W., and Martin, D. W. Jr. (1989). Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 9, 1381–1388.
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U S A 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
Iba, M., Guo, J. L., McBride, J. D., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 33, 1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013
Iqbal, K., Liu, F., Gong, C. X., Alonso Adel, C., and Grundke-Iqbal, I. (2009). Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69. doi: 10.1007/s00401-009-0486-3
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jack, C. R. Jr., Knopman, D. S., Weigand, S. D., Wiste, H. J., Vemuri, P., Lowe, V., et al. (2012). An operational approach to National Institute on Aging-Alzheimer’s association criteria for preclinical Alzheimer disease. Ann. Neurol. 71, 765–775. doi: 10.1002/ana.22628
Karch, C. M., Jeng, A. T., and Goate, A. M. (2012). Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J. Biol. Chem. 287, 42751–42762. doi: 10.1074/jbc.m112.380642
Kaufman, A. M., Milnerwood, A. J., Sepers, M. D., Coquinco, A., She, K., Wang, L., et al. (2012). Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. J. Neurosci. 32, 3992–4003. doi: 10.1523/jneurosci.4129-11.2012
Kfoury, N., Holmes, B. B., Jiang, H., Holtzman, D. M., and Diamond, M. I. (2012). Trans-cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451. doi: 10.1074/jbc.m112.346072
Khatoon, S., Grundke-Iqbal, I., and Iqbal, K. (1992). Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 59, 750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x
Kim, W., Lee, S., Jung, C., Ahmed, A., Lee, G., and Hall, G. F. (2010). Interneuronal transfer of human tau between Lamprey central neurons in situ. J. Alzheimers Dis. 19, 647–664. doi: 10.3233/JAD-2010-1273
Klein, C., Kramer, E. M., Cardine, A. M., Schraven, B., Brandt, R., and Trotter, J. (2002). Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 22, 698–707.
Koleske, A. J. (2013). Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 14, 536–550. doi: 10.1038/nrn3486
Kopeikina, K. J., Hyman, B. T., and Spires-Jones, T. L. (2012). Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 3, 223–233. doi: 10.2478/s13380-012-0032-y
Kovacech, B., Skrabana, R., and Novak, M. (2010). Transition of tau protein from disordered to misordered in Alzheimer’s disease. Neurodegener. Dis. 7, 24–27. doi: 10.1159/000283478
Lee, G., Neve, R. L., and Kosik, K. S. (1989). The microtubule binding domain of tau protein. Neuron 2, 1615–1624. doi: 10.1016/0896-6273(89)90050-0
Lee, G., Newman, S. T., Gard, D. L., Band, H., and Panchamoorthy, G. (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111, 3167–3177.
Lee, G., Thangavel, R., Sharma, V. M., Litersky, J. M., Bhaskar, K., Fang, S. M., et al. (2004). Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J. Neurosci. 24, 2304–2312. doi: 10.1523/jneurosci.4162-03.2004
Li, S. F., Wu, M. N., Wang, X. H., Yuan, L., Yang, D., and Qi, J. S. (2011). Requirement of α7 nicotinic acetylcholine receptors for amyloid β protein-induced depression of hippocampal long-term potentiation in CA1 region of rats in vivo. Synapse 65, 1136–1143. doi: 10.1002/syn.20951
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. doi: 10.1371/journal.pone.0031302
Liu, F., Li, B., Tung, E. J., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2007). Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 26, 3429–3436. doi: 10.1111/j.1460-9568.2007.05955.x
Loomis, P. A., Howard, T. H., Castleberry, R. P., and Binder, L. I. (1990). Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. U S A 87, 8422–8426. doi: 10.1073/pnas.87.21.8422
Maas, T., Eidenmuller, J., and Brandt, R. (2000). Interaction of tau with the neural membrane cortex is regulated by phosphorylation sites that are modified in paired helical filaments. J. Biol. Chem. 275, 15733–15740. doi: 10.1074/jbc.m000389200
Magnoni, S., Esparza, T. J., Conte, V., Carbonara, M., Carrabba, G., Holtzman, D. M., et al. (2012). Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 135, 1268–1280. doi: 10.1093/brain/awr286
Marklund, N., Blennow, K., Zetterberg, H., Ronne-Engstrom, E., Enblad, P., and Hillered, L. (2009). Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J. Neurosurg. 110, 1227–1237. doi: 10.3171/2008.9.JNS08584
Maruyama, M., Shimada, H., Suhara, T., Shinotoh, H., Ji, B., Maeda, J., et al. (2013). Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79, 1094–1108. doi: 10.1016/j.neuron.2013.07.037
Medina, M. (2011). Recent developments in tau-based therapeutics for neurodegenerative diseases. Recent Pat. CNS Drug Discov. 6, 20–30. doi: 10.2174/157488911794079091
Medina, M., and Avila, J. (2014). New perspectives on the role of tau in Alzheimer’s disease. Implications for therapy. Biochem. Pharmacol. 88, 540–547. doi: 10.1016/j.bcp.2014.01.013
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Morris, M., Maeda, S., Vossel, K., and Mucke, L. (2011). The many faces of tau. Neuron 70, 410–426. doi: 10.1016/j.neuron.2011.04.009
Nedergaard, M. (2013). Garbage truck of the brain. Science 340, 1529–1530. doi: 10.1126/science.1240514
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Pittman, A. M., Fung, H. C., and de Silva, R. (2006). Untangling the tau gene association with neurodegenerative disorders. Hum. Mol. Genet. 15, R188–R195. doi: 10.1093/hmg/ddl190
Plouffe, V., Mohamed, N. V., Rivest-McGraw, J., Bertrand, J., Lauzon, M., and Leclerc, N. (2012). Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7:e36873. doi: 10.1371/journal.pone.0036873
Pooler, A. M., Noble, W., and Hanger, D. P. (2014). A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 76, 1–8. doi: 10.1016/j.neuropharm.2013.09.018
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Poorkaj, P., Bird, T. D., Wijsman, E., Nemens, E., Garruto, R. M., Anderson, L., et al. (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825. doi: 10.1002/ana.410430617
Qian, W., Jin, N., Shi, J., Yin, X., Jin, X., Wang, S., et al. (2013). Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) enhances tau expression. J. Alzheimers Dis. 37, 529–538. doi: 10.3233/JAD-130824
Rábano, A., Rodal, I., Cuadros, R., Calero, M., Hernández, F., and Ávila, J. (2014). Argyrophylic grain pathology as a natural model of tau propagation. J. Alzheimers Dis. doi: 10.4137/jen.s12202. [Epub ahead of print].
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tauis essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U S A 99, 6364–6369. doi: 10.1073/pnas.092136199
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rodríguez-Martín, T., Cuchillo-Ibáñez, I., Noble, W., Nyenya, F., Anderton, B. H., and Hanger, D. P. (2013). Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157. doi: 10.1016/j.neurobiolaging.2013.03.015
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Jackson, B., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Seeley, W. W., Crawford, R. K., Zhou, J., Miller, B. L., and Greicius, M. D. (2009). Neurodegenerative diseases target large-scale human brain networks. Neuron 62, 42–52. doi: 10.1016/j.neuron.2009.03.024
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sheng, M., Sabatini, B. L., and Südhof, T. C. (2012). Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 4:a005777. doi: 10.1101/cshperspect.a005777
Shi, Y., Kirwan, P., Smith, J., MacLean, G., Orkin, S. H., and Livesey, F. J. (2012). A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 4:124ra129. doi: 10.1126/scitranslmed.3003771
Simón, D., García-García, E., Gómez-Ramos, A., Falcón-Pérez, J. M., Díaz-Hernández, M., Hernández, F., et al. (2012b). Tau overexpression results in its secretion via membrane vesicles. Neurodegener. Dis. 10, 73–75. doi: 10.1159/000334915
Simón, D., Garcia-Garcia, E., Royo, F., Falcon-Perez, J. M., and Avila, J. (2012a). Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 586, 47–54. doi: 10.1016/j.febslet.2011.11.022
Simón, D., Hernández, F., and Avila, J. (2013). The involvement of cholinergic neurons in the spreading of tau pathology. Front. Neurol. 4:74. doi: 10.3389/fneur.2013.00074
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/S1474-4422(13)70090-5
Spillantini, M. G., Murrell, J. R., Goedert, M., Farlow, M. R., Klug, A., and Ghetti, B. (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U S A 95, 7737–7741. doi: 10.1073/pnas.95.13.7737
Sultan, A., Nesslany, F., Violet, M., Bégard, S., Loyens, A., Talahari, S., et al. (2011). Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 286, 4566–4575. doi: 10.1074/jbc.m110.199976
Tackenberg, C., Grinschgl, S., Trutzel, A., Santuccione, A. C., Frey, M. C., Konietzko, U., et al. (2013). NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 4:e608. doi: 10.1038/cddis.2013.129
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S., et al. (2013). Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation and synaptic loss. Proc. Natl. Acad. Sci. U S A 110, E2518–E2527. doi: 10.1073/pnas.1306832110
Tatebayashi, Y., Iqbal, K., and Grundke-Iqbal, I. (1999). Dynamic regulation of expression and phosphorylation of tau by fibroblast growth factor-2 in neural progenitor cells from adult rat hippocampus. J. Neurosci. 19, 5245–5254.
Terwel, D., Dewachter, I., and Van Leuven, F. (2002). Axonal transport, tau protein and neurodegeneration in Alzheimer’s disease. Neuromolecular Med. 2, 151–165. doi: 10.1385/nmm:2:2:151
Wang, Y., Loomis, P. A., Zinkowski, R. P., and Binder, L. I. (1993). A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J. Cell Biol. 121, 257–267. doi: 10.1083/jcb.121.2.257
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Yamada, K., Cirrito, J. R., Stewart, F. R., Jiang, H., Finn, M. B., Holmes, B. B., et al. (2011). In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 31, 13110–13117. doi: 10.1523/jneurosci.2569-11.2011
Keywords: Alzheimer, exosomes, vesicles, neurodegeneration, propagation, spreading, tau, tauopathies
Citation: Medina M and Avila J (2014) The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 8:113. doi: 10.3389/fncel.2014.00113
Received: 28 February 2014; Accepted: 05 April 2014
Published online: 23 April 2014
Edited by:
Ludovic Martin, Université de Nantes, FranceReviewed by:
Khalid Iqbal, New York State Institute for Basic Research in Developmental Disabilities, USAAmy Pooler, King’s College London, UK
Copyright © 2014 Medina and Avila. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Avila, Centro de Biología Molecular “Severo Ochoa” CSIC-UAM, Nicolás Cabrera 1, 28049 – Madrid, Spain e-mail:amF2aWxhQGNibS51YW0uZXM=