
 Enrica Maria Petrini1
Enrica Maria Petrini1
- 1 Department of Neuroscience and Brain Technologies, The Italian Institute of Technology, Genova, Italy
- 2 Laboratory of Neuroscience, Department of Biophysics, Wroclaw Medical University, Wroclaw, Poland
The time course of synaptic currents is a crucial determinant of rapid signaling between neurons. Traditionally, the mechanisms underlying the shape of synaptic signals are classified as pre- and post-synaptic. Over the last two decades, an extensive body of evidence indicated that synaptic signals are critically shaped by the neurotransmitter time course which encompasses several phenomena including pre- and post-synaptic ones. The agonist transient depends on neurotransmitter release mechanisms, diffusion within the synaptic cleft, spill-over to the extra-synaptic space, uptake, and binding to post-synaptic receptors. Most estimates indicate that the neurotransmitter transient is very brief, lasting between one hundred up to several hundreds of microseconds, implying that post-synaptic activation is characterized by a high degree of non-equilibrium. Moreover, pharmacological studies provide evidence that the kinetics of agonist transient plays a crucial role in setting the susceptibility of synaptic currents to modulation by a variety of compounds of physiological or clinical relevance. More recently, the role of the neurotransmitter time course has been emphasized by studies carried out on brain slice models that revealed a striking, cell-dependent variability of synaptic agonist waveforms ranging from rapid pulses to slow volume transmission. In the present paper we review the advances on studies addressing the impact of synaptic neurotransmitter transient on kinetics and pharmacological modulation of synaptic currents at inhibitory synapses.
Introduction
The timing, amplitude, and duration of synaptic currents are important determinants for specific neuronal network functions including signal integration, network oscillations, and selection of neuronal assemblies (Freund and Katona, 2007; Klausberger and Somogyi, 2008). Various GABAergic interneurons contact their target cells with synapses showing distinct kinetic features and precise sub-neuronal localization, suggesting that the concerted neuronal network functioning relies on the accurate spatial and temporal properties of synaptic inputs (Spruston, 2008; Klausberger, 2009; Karayannis et al., 2010). The kinetics of post-synaptic currents (PSCs) is commonly ascribed to the gating properties of post-synaptic receptors as exemplified by glutamatergic NMDA receptor- and AMPA receptor-mediated EPSCs showing slow and fast decay, respectively, due to different binding and gating properties of NMDA and AMPA glutamate receptors (Lester and Jahr, 1992). Likewise, GABAergic synapses are characterized by a marked kinetic diversity that is commonly ascribed to the variety of post-synaptic GABAA receptor (GABAAR) isoforms (Cherubini and Conti, 2001). For instance, receptors containing α1 or α3 subunits (along with β and γ2 subunits) have been demonstrated to underlie fast and slow IPSCs kinetics, respectively (Gingrich et al., 1995; Mozrzymas et al., 2003b; Barberis et al., 2007). In many cases, however, the kinetics of synaptic currents significantly differs from the prediction based solely on the gating properties of post-synaptic receptors. An increasing body of evidence suggests that both at excitatory and inhibitory synapses the profile of neurotransmitter concentration in the synaptic cleft plays a significant role in determining the amplitude and kinetics of synaptic currents. Typically, after release, neurotransmitter is present in the synaptic cleft for only a few hundreds of microseconds implying that activation of post-synaptic receptors occurs in conditions characterized by a high degree of non-equilibrium (Clements, 1996; Mozrzymas et al., 1999, 2003a). Moreover, such a particularly dynamic waveform of synaptic agonist concentration has been demonstrated to play a crucial role in determining the susceptibility of PSCs to various modulators. Importantly, recent studies revealed that for some types of GABAergic interneurons, neurotransmitter transient is particularly slow, lasting tens of milliseconds, raising novel concepts of volume transmission and paracrine modulation of local networks (Szabadics et al., 2007; Balakrishnan et al., 2009; Olah et al., 2009; Karayannis et al., 2010).
In the present review we discuss recent advancement in understanding the role of synaptic GABA concentration profile in shaping IPSCs kinetics and in pharmacological modulation of GABAergic currents.
Estimating the Neurotransmitter Time Course
Having focused this review on the role of agonist transient in synaptic signaling, it is important to bear in mind that the measurement of synaptic agonist time course remains a major challenge. Typically, at the central synapses, cleft width is ~20 nm, thus precluding placement of a measuring device within the synapse to directly monitor the agonist time course. Moreover, amperometry, a technique widely used for, e.g., monoamines detection, cannot be applied for major inhibitory and excitatory neurotransmitters in the brain (GABA and glutamate) as these aminoacids are not being oxidized on the carbon fibers. It needs to be taken into account that, even in the case of monoamines, amperometry senses these compounds after their spillover from the synapse, as the carbon fiber can only be placed in the release site vicinity. Taking into account these constrains, determination of synaptic neurotransmitter transient has been approached either with computer simulations or with tools enabling to indirectly infer its time course. The latter case exploits compounds that interfere with synaptic transmission in a way that depends on synaptic neurotransmitter waveform. Since the techniques used for estimation of synaptic agonist time course were extensively reviewed elsewhere (Clements, 1996; Mozrzymas, 2004; Scimemi and Beato, 2009), below we provide only a concise description of these methods.
Model Simulations
The spatiotemporal profile of neurotransmitter released from a vesicle can be described using the Fick’s equation:
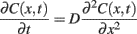
(1)Where C(x, t) – neurotransmitter concentration at time t and distance x from a reference point, D – diffusion coefficient. Application of this equation requires setting the boundary conditions that basically reflect the geometry of the synapse and diffusion constrains in its surroundings. Although electron microscopy may provide key ultrastructural information, it needs to be considered that this method requires tissue fixation, dehydration, and treatment with chemicals which may induce substantial cell swelling or shrinkage and thereby alter the structure of synapses. Moreover, the diffusion coefficient at the synapse may substantially differ (is typically much smaller) from that determined for bulk aqueous solutions. A number of other phenomena and system features may be considered in the Eq.1. The simplest model assumes instantaneous point release that is taken into account by setting appropriate initial condition, although more realistic release models (e.g., partial release due to transient releasing pore opening) can be considered. Moreover, neurotransmitter binding and/or buffering binding sites (e.g., by transporters involved in the uptake system and/or post-synaptic receptors) or enzymatic breakdown can be additionally included by adding the respective terms on the right hand side of Eq.1. This approach provides a macroscopic view of diffusion, as the neurotransmitter concentration is described by a continuous function C(x, t) which reflects a deterministic (zero fluctuation), local value of concentration. Formally, the lack of fluctuations corresponds to the limit behavior of a great number of diffusing particles. A complementary approach, which does account for local fluctuations, is to simulate the agonist diffusion using Monte Carlo method which is based on simulation of a stochastic “random walk” of released neurotransmitter molecules in the “environment” mimicking the 3D structure of the synaptic cleft and its close neighborhood.
Low Affinity Competitive Agonists
A clever and elegant method to estimate the agonist time course in glutamatergic synapses was to “perturbate” synaptic currents with competitive, quickly unbinding antagonists (Clements et al., 1992; Clements, 1996). If the synaptic agonist remained within the synapse for the time comparable to that needed for antagonist dissociation, a non-equilibrium displacement of competitive blocker would be unmasked by a current flowing through unblocked post-synaptic receptors. Such a displacement of the antagonist would be also manifested by a slowdown of synaptic current rising phase. Clearly, the longer the agonist presence in the synapse in comparison to the time needed for unbinding, the larger the displacement of antagonist, and the bigger the current. This approach has been initially used to probe the neurotransmitter concentration profile at glutamatergic synapses (Clements et al., 1992; Clements, 1996), at GABAergic synapses (Overstreet et al., 2003; Barberis et al., 2004, 2005; Karayannis et al., 2010) and, more recently, at glycinergic synapses (Beato et al., 2007; Beato, 2008; Scimemi and Beato, 2009). It is worth emphasizing that, as pointed out by Beato (2008) and Scimemi and Beato (2009), when a dose inhibition curve is fitted using the peak concentration and clearance time as free parameters, these two parameters cannot be unambiguously determined. This limitation can be overcome by either an experimental manipulation that prolongs the clearance time without affecting the peak (Beato, 2008) or by incorporating the time course of the PSCs in the fitting procedure (Overstreet et al., 2003).
Modifiers of Gating
A different option for the use of “perturbating” factors to get an insight into the synaptic agonist time course is represented by the use of compounds that affect post-synaptic receptor properties including binding and gating kinetics (Mozrzymas et al., 1999, 2003a, 2007b; Barberis et al., 2000). The agents influencing agonist binding were particularly useful in estimating the agonist transient. The rationale is that a relatively small up or down regulation of the binding rate exerts a particularly strong impact on the amplitude and time course of currents elicited by a very short exposure to synaptically released agonist. In practice, the pharmacological mechanism of a modulatory action was determined relying on the analysis of current responses to rapid agonist applications and, subsequently, the time course of the agonist transient was optimized to best reproduce the effect of the modulator on the synaptic currents. Thus, determination of agonist transient kinetics was not a unique goal of studies in which specific agents were investigated but rather it was a necessary step to reconcile the observed effects of modulators on synaptic and exogenously evoked currents.
A more detailed comparison of the techniques based on the use of rapidly dissociating antagonists and modifiers of gating is discussed below in the Section “Consideration of Synaptic Agonist Transient Sheds New Light on Pharmacological Modulation of Post-Synaptic Currents.”
Neurotransmitter Concentration Profile Shapes Synaptic Currents
Since the duration of the synaptic agonist pulse is much shorter than the mean open time of synaptic channels, the activation of post-synaptic receptors takes place in conditions of substantial non-equilibrium. An important consequence of this particular activation pattern is that, in contrast to what is assumed in classical equilibrium pharmacology, the strength of the synaptic agonist pulse depends on both the peak agonist concentration and pulse duration. This concept is illustrated in (Figure 1A), where the peak of simulated IPSCs evoked by the same GABA concentration (1 mM) strongly depends on the duration of GABA transient. During the last decade, in order to demonstrate that brief neurotransmitter synaptic exposures could limit the activation of post-synaptic receptors, different strategies have been adopted. One of them was the use of polymers such as dextran which slows down neurotransmitter diffusion thereby prolonging the neurotransmitter presence in the synaptic cleft. At glutamatergic and GABAergic synapses, synaptic currents recorded in the presence of dextran showed indeed higher amplitude (Min et al., 1998; Perrais and Ropert, 2000; Barberis et al., 2004) confirming that, generally, synaptic exposure may be a limiting factor for post-synaptic receptor activation. This observation has been corroborated by using an alternative approach based on pharmacological modulation of post-synaptic GABAAR (see also below). For instance, benzodiazepines (BDZ) that are believed to up regulate GABA binding rate (Rogers et al., 1994; Lavoie and Twyman, 1996), enhance mIPSCs amplitude as they effectively increase the occupancy of bound states following a short receptor exposure to synaptically released agonist (Frerking et al., 1995; Perrais and Ropert, 1999; Mozrzymas et al., 2007b). These two approaches, although useful to unveil non-equilibrium synaptic conditions, fail to provide quantitative details about agonist transient, especially when their use is limited to the analysis of synaptic currents. Moreover, it is not precisely known to what extent dextran penetrates the synaptic cleft and interferes with neurotransmitter diffusion within the synapse. In addition, as discussed in detail below, BDZs not only alter GABAAR affinity but also affect receptor gating thus complicating the assessment of their effect on agonist binding during synaptic transmission. While the influence of non-equilibrium conditions on the PSCs peak amplitude has been broadly acknowledged, the impact of neurotransmitter concentration profile in shaping the decay kinetics of synaptic currents is more elusive, and whether or not synaptic-like neurotransmitter exposures can unmask gating features not observed for longer agonist pulses is still a matter of debate. This is mainly due to the lack of knowledge on the gating mechanisms of GABAAR activated by synaptic-like GABA pulses (~1–3 mM, ~0.1 ms). The fastest ultra-rapid perfusion systems, indeed, cannot reliably reproduce such neurotransmitter pulse since, at best, they are able to deliver pulses in the range of ~0.4–1 ms (Jonas, 1995), although in most studies, including ours, minimal exposure is >1 ms. In order to overcome this limitation the following strategy has been adopted. If in non-equilibrium conditions the agonist pulse strength depends on both agonist concentration and application duration (C × t), it is possible to deliver pulses with a strength that is “equivalent” to the synaptic one simply by lowering the agonist concentration and keeping the value of C × t similar to that estimated for PSCs. Using this approach, Jones and Westbrook (1995) showed that ultra-weak GABA pulses (10 μM, 2 ms) evoked currents that deactivated faster than those elicited by saturating GABA pulses (10 mM, 2 ms). Similar results were reported by Banks and Pearce (2000). Which mechanisms underlie the current deactivation speed-up observed at ultra-weak GABA pulses? An increasing body of evidence points to the involvement of monoliganded states of GABAAR. Although GABAAR fully activates following the binding of two GABA molecules, it has been proposed that a single GABA molecule would be sufficient to induce a low probability open state (Macdonald et al., 1989; Jones and Westbrook, 1995; Baumann et al., 2003). This hypothesis was suggested by the observation that GABAAR mean open time critically depends on the agonist concentration being shifted toward short openings at low, submicromolar GABA doses. Based on this theory, Jones and Westbrook (1995) proposed that ultra-short applications of low GABA concentrations (10 μM, 2 ms) would largely limit the binding reaction to the monoliganded state(s), therefore promoting low probability openings leading to fast deactivation kinetics. More recently Petrini et al. (2011) applied a mutagenesis approach directed to impair a single GABA binding site and provided a more direct evidence that currents mediated by singly bound states of GABAAR show fast deactivation. In the same study, the possibility that monoliganded states of GABAAR could participate in shaping IPSCs kinetics during synaptic transmission was taken into consideration. In particular, model simulations of GABA release in the synaptic cleft suggested that, compared to disk center, receptors located at the disk periphery sense markedly lower GABA concentrations, thus activating preferentially in the singly bound state(s) (Figure 2). Thus, singly bound synaptic receptors characterized by a fast deactivation contributed to the acceleration of IPSCs decay kinetics. The largest contribution of singly bound states was found when the synapse works in conditions far from saturation, e.g., at low number of molecules released and/or large synaptic radius. In contrast, large amount of released neurotransmitter and/or small synapse radius generate saturation of post-synaptic receptors thus favoring receptor activation in the doubly bound state(s) (Figures 2B–E). These data, although strongly relying on model simulations, confirm that IPSCs kinetics could be finely tuned by the relative proportion of receptor activating in the mono- and doubly bound state, respectively, that in turn depends on both strength of pre-synaptic release and synapse geometry (Figure 2E). It has to be pointed out that the results of Petrini et al. (2011) predict, on average, ~11% IPSCs decay acceleration due to contribution of GABAAR activating in the monoliganded state (Figure 2D). Although this could appear as a moderate effect, it is expected to be relevant in network tasks strongly relying on the dynamic equilibrium between excitation and inhibition such as signal integration and fast oscillation. Worth of mention, in the case of non-equivalence between the two GABAAR binding sites (as reported for instance for glutamate kainate receptors, see below), during activation by short synaptic GABA exposure, the agonist would preferentially bind to the site with higher affinity thus determining larger probability of singly bound activation (Benke et al., 2004; Minier and Sigel, 2004). The aforementioned IPSCs speed-up due to GABAAR singly bound states could provide a partial explanation to the observation that GABAergic currents are systematically faster than currents evoked by brief exogenous GABA pulses. A potential role for monoliganded states in explaining such discrepancy has been previously studied by Banks and Pearce (2000) who compared the amplitude and the deactivation time constant of currents elicited by both exogenous brief saturating GABA pulses (2 ms, 10 mM) and “synaptic equivalent” pulses obtained by lowering the GABA dose (2 ms, 10 μM). In comparison to currents elicited by saturating pulses, those evoked by low GABA concentrations showed deactivation kinetics accelerated by 35% and peak amplitude reduced by 96%. Interestingly, currents evoked by low concentration GABA pulses still showed deactivation kinetics twofold slower than IPSCs. The strong sensitivity of the peak current to weak GABA pulses associated with a moderate dependence of the deactivation kinetics led Banks and Pearce (2000) to conclude that brief synaptic exposure could not account for the fast IPSCs kinetics. They suggested therefore that the reason for the slow response observed in excised patch (as compared to synaptic current) could be due to the presence of high affinity extra-synaptic receptors. Although this hypothesis is reasonable, several observations complicate this scenario. Currents elicited in excised patches containing recombinant GABAAR subtypes known to mediate synaptic currents (e.g., α1β2γ2) show slower deactivation than IPSCs decay (and similar to that obtained in patches pulled from neurons). It could be objected that receptors expressed in recombinant systems lack some important factors modulating the IPSCs kinetics. This hypothesis is certainly plausible, although manipulations of proteins known to compose or interact with scaffold proteins at GABAergic synapses show only a moderate effect on IPSCs decay time (Petrini et al., 2003; Marchionni et al., 2009). In keeping with this, Pugh and Raman (2005) proposed that the patch excision itself would stabilize GABA desensitization with consequent currents slow down, although the molecular determinants for such gating modification have not been identified yet. Other lines of evidence argue against the possibility that synaptic GABAAR would show different gating properties with respect to recombinant receptors or native receptors recorded in excised patches. For instance, currents elicited by UV laser photolysis of caged-GABA in diffraction limited spots (~400 nm) at individual intact GABA synapses still display deactivation kinetics markedly slower than IPSCs (Trigo et al., 2009; Matsuzaki et al., 2010). Similarly, focal micro-iontophoretic GABA applications restricted to ~600 nm diameter spots (Heine et al., 2008) fail to reproduce the IPSCs kinetics (Barberis et al., 2011). It has to be pointed out, however, that both uncaging and micro-iontophoresis deliver GABA pulses different from those occurring at the synapse. This is due to the fact that the “GABA spot” is bigger than the size of most of central synapses activating extra-synaptic receptors that typically do not participate in synaptic transmission. In addition, most of the commercially available GABA-caged compounds partially act as competitive blockers, potentially contributing to current slowdown (Trigo et al., 2009). In summary, although several lines of evidence show that neurotransmitter concentration profile plays a major role in shaping the amplitude and the kinetics of IPSCs, the precise assessment of its contribution will require the development of new devices capable to deliver, at intact synapses, neurotransmitter pulses in a temporal and spatial range relevant for synaptic transmission. It is important to mention that other factors are likely to affect the IPSC decay kinetics. These may include modulation of GABAAR gating by intracellular chloride concentration (Houston et al., 2009), phosphorylation state of various GABAAR subunits (β or γ), (Kittler and Moss, 2003; Houston et al., 2008) and interaction with yet unidentified accessory subunits or scaffolding proteins such as, e.g., gephyrin (Christie et al., 2002; Fritschy et al., 2003; Arancibia-Carcamo and Moss, 2006).
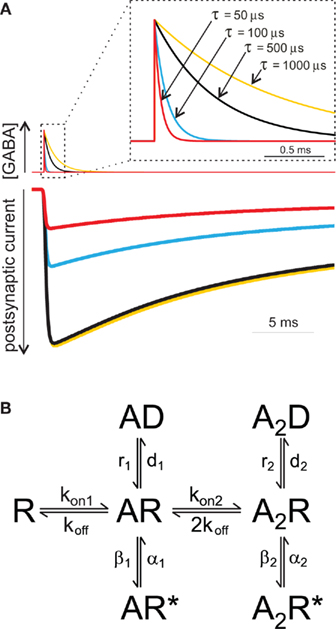
Figure 1. The amplitude of simulated IPSCs depends on the neurotransmitter time exposure. (A) Currents simulated using the Jones and Westbrook model (rate constants from Mozrzymas et al., 1999) elicited by four different GABA pulses. At GABA pulses with τ lower than ~500 μs, the current amplitude strongly depends on GABA pulse length. Please note that current evoked by fast clearance GABA pulses (τ = 50 μs) is markedly smaller that that obtained by longer GABA exposures. (B) Jones and Westbrook’s (1995) model. It assumes the sequential binding of two GABA molecules and includes open (AR*, A2R*) and desensitized (AD, A2D) states originating from both mono- and doubly liganded closed states (AR, A2R). This gating scheme has been extensively used in the investigation of the gating properties of GABAAR and, although oversimplified, this model reproduces the basic features of GABAAR (Jones and Westbrook, 1995; Mozrzymas et al., 2003b).
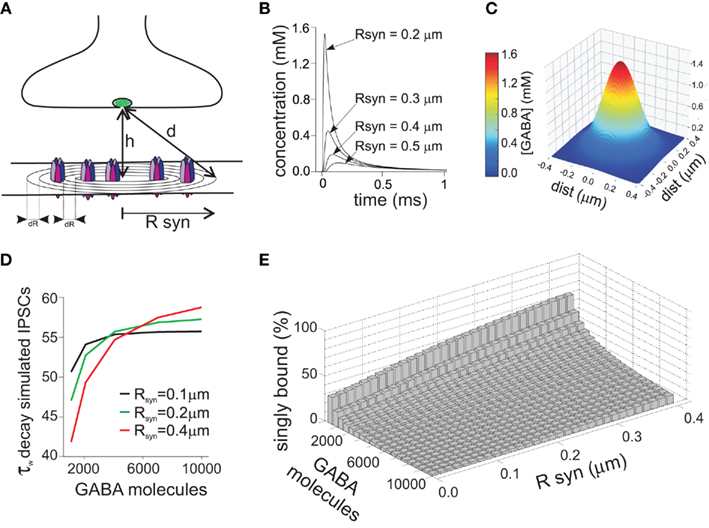
Figure 2. Receptors located at different distances from the disk center sense a different GABA concentration due to non-homogeneity of the neurotransmitter concentration profile. (A) Schematic representation of the GABAergic synapses. The distance from the releasing site to the synaptic site is indicated by d, while Rsyn represents the synaptic disk radius. (B) Simulated time course of the GABA concentration following release of 4000 GABA molecules, measured at different distances from the disk center. Please note that in the vicinity of the disk center the GABA concentration peak is markedly higher with respect to the disk periphery. (C) Simulated spatial profile of GABA diffusion in the cleft obtained 30 μs after the instantaneous release of 4000 GABA molecules. (D) Dependence of the decay time of simulated IPSCs on the number of GABA molecules released. At low number of GABA molecules released, receptors located at the disk periphery sense a low dose of GABA, activating preferentially in the monoliganded configuration, and contributing therefore to IPSCs speed-up. Please note that the IPSCs decay was also influenced by the synapse radius. (E) Summary of the relative weight of monoliganded GABAAR on the simulated IPSCs at different GABA molecules released and synaptic radiuses. Please note that conditions generating low saturation (low number of molecules released and/or large synaptic radiuses) favor the singly bound state activation, while high concentration pulses at small synapses determine almost complete activation in doubly bound state. Modified from Petrini et al. (2011) with permission.
Role of Desensitization in Synaptic Current
Desensitization process has been recognized to profoundly shape the deactivation process of GABAAR-mediated currents (Jones and Westbrook, 1995; Mozrzymas et al., 2003b). Indeed, due to slow unbinding rate, after GABA removal, the receptor may experience several transitions between bound open, closed, and desensitized state(s), implying that desensitization does prolong the deactivation kinetics. One of the most straightforward demonstrations of this phenomenon is that the deactivation time course depends on the degree of receptor desensitization induced by GABA pulses of different time durations. For instance, the kinetics of current relaxation after a 3 s pulse of saturating GABA is markedly slower than that observed at brief (2–3 ms) pulses (Jones and Westbrook, 1995; Mozrzymas et al., 2007b; Petrini et al., 2011). It could be hypothesized that shortening the GABA pulse length from 2 to 3 ms down to synaptic-like exposures (~0.1 ms) would induce even lower extent of desensitization with consequent faster deactivation kinetics. However, by considering only the doubly bound states, the deactivation time course predicted by the Jones and Westbrook’s (1995) model (Figure 1B) would not show major differences between currents evoked by ultra-short pulses (0.1 ms) and those elicited by longer (2–3 ms) pulses. The reason for this prediction is that the main mechanism of desensitization onset following brief exposure is receptor trapping into the desensitized state due to slow unbinding and resensitization rates (koff, r2, Figure 1B; Jones and Westbrook, 1995; Mozrzymas et al., 2003b, 2007b). Thus, although it might look at the first glance counterintuitive, most of rapid accumulation in the fast desensitized state takes place following application of brief GABA pulses depending weakly on the exposure time. On the other hand, pulses longer than 2–3 ms would favor accumulation of receptors in the desensitized states, including slower ones, contributing thus to observed slow down of deactivation kinetics. Glutamatergic synapses, in contrast, provide an excellent example of how synaptic exposure can efficiently shape the decay kinetics of synaptic currents by interfering with desensitization. The deactivation of AMPA and kainate receptors, contrary to GABAAR, is accelerated by desensitization that, in these receptors, is very fast and profound (Figure 3A). The GluK2/K5 subtype of kainate receptor (composing synaptic receptors) mediate fast deactivating currents (τ ~ 3 ms) when activated by long (1 mM, 100 ms) glutamate pulses, whereas short glutamate exposures (1 mM, ~1.5 ms) unmask markedly slower deactivation kinetics (τ ~ 46 ms, Figures 3A,B; Barberis et al., 2008). This behavior results from the fact that GluK2/K5 receptors possess two different types of binding sites showing distinct affinity and desensitization properties. In particular, glutamate binding to the high affinity sites induces poor desensitization while activation of low affinity binding sites determines fast and profound GluK2/K5 desensitization (Mott et al., 2010). During fast synaptic activation, therefore, the high affinity/poorly desensitizing binding site is expected to be preferentially activated mediating hence slow decaying responses.
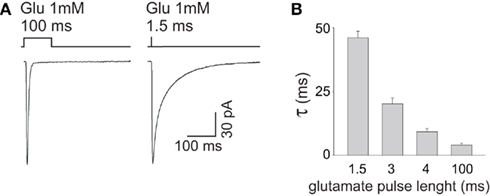
Figure 3. The decay kinetics of currents mediated by GluK2/K5 kainate receptors strictly depends on the length of glutamate pulse. (A) Representative traces of GluK2/K5 currents elicited by long (100 ms) and brief (1.5 ms) pulses of glutamate 1 mM. Please note that the current elicited by brief glutamate pulses (right panel) deactivates markedly slower that that obtained at long glutamate exposures (left panel). (B) Dependence of GluK2/K5 current deactivation kinetics upon glutamate pulse length. Modified from Barberis et al. (2008) with permission.
Consideration of Synaptic Agonist Transient Sheds New Light on Pharmacological Modulation of Post-Synaptic Currents
As discussed above, kinetic studies brought increased awareness that the time course of synaptic neurotransmitter transient plays a crucial role in determining the kinetics of synaptic currents. Moreover, pharmacological modulation of synaptic signals may critically depend on synaptic agonist waveform even in the cases in which drug action is purely post-synaptic. This is particularly evident for modulators affecting agonist binding to post-synaptic receptors. Indeed, for a brief and non-saturating agonist transient, even a slight modulation of the binding rate may result in a marked effect on bound receptor occupancy and therefore on the amplitude and time course of synaptic currents. Thus, the specificity of synaptic conditions may be responsible, at least partially, for kinetic and pharmacological differences observed between synaptic currents and responses evoked by exogenous agonist applications. As shown below, these differences may become a source of information on the time course of synaptic agonist. A critical limitation, when studying the pharmacology of PSCs, is that synaptic neurotransmitter transient is a priori unknown and may show substantial variability. Moreover, synaptic agonist cannot be easily manipulated thus precluding the use of classical pharmacological analyses based on dose-response relationships. For this reason, receptor kinetics and mechanisms of its modulation need to be determined relying on the analysis of rapid agonist applications. The weakness of this approach, however, is that the kinetics is determined not for synaptic receptors but for recombinant ones or for receptors in patches excised from neurons (containing a mixture of synaptic and extra-synaptic receptors). Moreover, while IPSCs are expected to be particularly sensitive to agents affecting binding, most modulators affect both binding and gating (opening/closing, desensitization, unbinding) of GABAAR. In general, even in a pharmacological study based on currents evoked by precisely defined agonist concentration “jumps,” it is not straightforward to “dissect” the effect on binding from modification of other transition rates. As shown in an elegant study by Jones et al. (1998) this task becomes a considerable challenge even when determining only two rate constants (binding and unbinding rates) that define the receptor affinity. In general, assessment of specific binding and gating rate constants from the analysis of evoked current time course is difficult because any current characteristics (e.g., rise time or decay kinetics) may potentially depend on all the rate constants and on occupancies of all the channel states (Colquhoun, 1998; Mozrzymas et al., 2003b). In the past decade, the idea to combine high resolution studies of receptor gating while considering the specificity of synaptic conditions dictated by rapid agonist transient has been implemented by us and others to explore the modulatory mechanisms of several pharmacological agents. Below we provide examples of such studies to emphasize important practical implications of agonist transient features in the context of PSC modulation. It is worth noting that determination of synaptic neurotransmitter waveform and description of pharmacological mechanisms can be regarded as synergistic components of these studies. Indeed, high resolution determination of receptor gating is crucial to describe the properties of post-synaptic receptors while assessment of synaptic neurotransmitter time course enables to extrapolate this knowledge to synaptic conditions. Conversely, optimization of agonist transient waveform offers the opportunity to verify to what extent the receptor gating determined from current responses (receptors in excised patches) is adequate for the analysis of synaptic currents. Importantly, besides relevant pharmacological information, most of these studies provided consistent indications regarding the time course of synaptic neurotransmitter transient.
Effect of Extracellular pH on mIPSCs and on Current Responses
In the context of the role of synaptic GABA time course, it has been particularly insightful to consider the effect of extracellular pH on mIPSCs and on current responses elicited by exogenous GABA. Strikingly, while currents evoked by saturating GABA showed a strong (several fold) and monotonic decrease in amplitude with increasing pH (Figures 4A,B), synaptic currents were characterized by a biphasic pH-dependence: an increase at pH = 6.5 (with respect to that at pH = 7.2), steep decrease at pH values below 6.5 and nearly no effect when alkalinizing the extracellular saline (Figures 4C,D). An intuitive explanation for this discrepancy could be that synaptic receptors and those in excised patches might show different kinetic and pharmacological properties (Wisden et al., 1996; Banks and Pearce, 2000; Cherubini and Conti, 2001; Fritschy and Brunig, 2003). However, the kinetic features such as rise time and decay kinetics of IPSCs and of current responses to saturating GABA showed a qualitatively similar pH-dependence (Mozrzymas et al., 2003a). This observation suggested that the major differences lay on receptor activation conditions rather than on receptor properties. Indeed, while rapid application was set to assure saturation (10–30 mM GABA applied for >1 ms), synaptic neurotransmitter was likely to show smaller peak concentration and faster clearance. Moreover, kinetic studies of current responses evoked by rapid GABA applications revealed that decrease in pH strongly down regulated the binding and desensitization rates. In conditions of saturating GABA application, it is thus expected that when decreasing pH, the current amplitude would increase as the open state occupancy would increase at the expense of desensitization and this prediction was consistent with experimental findings (Mozrzymas et al., 2003a; Figures 4A,B). In contrast, for synaptic currents which are evoked by non-saturating and very brief GABA transient, a decrease in the binding rate at low pH would reduce the occupancy of synaptic bound receptors. At pH = 6.5, mIPSC amplitude is enhanced because reduction of desensitization rate counter balanced the reduction in the occupancy of bound states. However, a further acidification and ensuing reduction of binding rate, largely suppressed the recruitment into bound states resulting eventually in a decrease of mIPSC amplitude. Thus, a particular pH-dependence of mIPSCs resulted from an interplay between proton-induced changes in the microscopic binding/gating and dynamic conditions of receptors activation by synaptically released neurotransmitter. Model simulations indicated that, as expected, the shorter the GABA transient the larger the discrepancies between pH-dependence of responses to saturating (GABA) and mIPSCs. Fitting the data to a modified Jones and Westbrook’s (1995) model indicated the time constant of agonist clearance of approximately 100 μs.
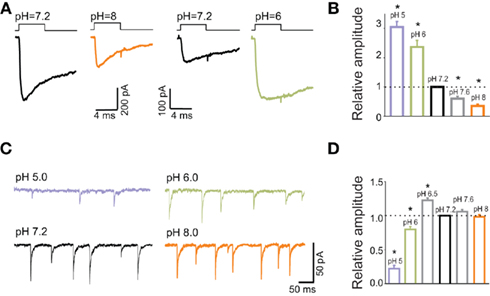
Figure 4. Current responses to applications of saturating GABA concentrations show different pH-sensitivity than mIPSCs. (A) Typical current responses to applications of saturating (GABA) at pH = 7.2 black) and at pH = 8 (orange) and pH = 6 (green). Traces are presented in pairs (pH = 7.2, 8.0 and pH = 7.2, 6.0) that were recorded from the same excised patches. (B) Statistics of relative amplitudes (normalized to that measured at pH = 7.2). Note a strong and monotonic pH-dependence. (C) Typical traces of mIPSCs recorded at various pH values as indicated. (D) pH-dependence of mIPSC amplitudes. Note a qualitatively different proton effect in comparison to that observed for current responses (B). Modified from Mozrzymas et al. (2003b) with permission.
Recently, Dietrich and Morad (2010) provided intriguing evidence that protons might be involved in modulation of synaptic currents in the scale of a single GABAergic synapse. Consistent with our previous reports, they have observed that mild acidification increased the mIPSC amplitude (Mozrzymas et al., 2003a, 2007a). However, when increasing the buffering power, mIPSC amplitude decreased and this effect was ascribed to alkalinization of synaptic milieu. These (and other) findings led Dietrich and Morad (2010) to the proposal that vesicular release is associated with a local acidification that boosts the synaptic currents. While the data obtained for current responses and mIPSCs in hippocampal culture turned out to be consistent, at least at a qualitative level, it needs to be considered that neurons might express several different types of receptors, characterized by potentially different pH-sensitivity. It seemed thus important to verify the observed pH-dependence on a homogeneous GABAAR population, representative for synaptic receptors. Analysis of protons effects on recombinant α1β2γ2 receptors (Mercik et al., 2006) qualitatively reproduced our major observations obtained on neuronal culture (Mozrzymas et al., 2003a) including a strong pH-dependence of amplitudes evoked by saturating (GABA) and a marked pH effect on the rising phase of current evoked by non-saturating GABA. An intriguing finding observed first in neuronal culture was that acidic pH (6.0) decreased and alkaline pH (8.0) increased current amplitude evoked by low (GABA; 1 μM; Figures 5A,B; Mozrzymas et al., 2003b) – i.e., the opposite to what observed for saturating (GABA; Figures 3A,B. Importantly, at qualitative level, these inverse effects of acidic and basic pH on currents evoked by low or high (GABA) was reproduced on recombinant α1β2γ2 receptors (Mercik et al., 2006) implying that such a peculiar proton sensitivity may be a feature of a homogeneous population of GABAARs (but see Pasternack et al., 1996). These observations are consistent with reduction of the binding and desensitization rates when lowering pH. Thus, at low (GABA) receptor occupancy is minimal and the impact of desensitization is low [contrary to the situation at saturating (GABA)]. Thus, in these conditions, acidification would lead to a decrease in current amplitude as it would further reduce the occupancy of bound receptors while down regulation of a weak desensitization would not be sufficient to counterbalance this reduction (Figures 5A,B; Mercik et al., 2006; Mozrzymas et al., 2007b). On the contrary, alkalinization would enhance both binding and desensitization, but in conditions of very low (GABA), gain in the binding rate would not be counterbalanced by increased desensitization giving rise eventually to enhancement of current amplitude (Figures 5A,B; Mercik et al., 2006; Mozrzymas et al., 2007a).
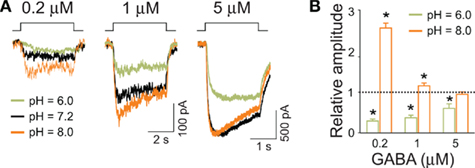
Figure 5. Amplitudes of currents elicited by low GABA concentrations are up regulated by basic and down regulated by acidic pH. (A) Typical current traces elicited by applications of GABA concentrations indicated above current traces at different pH values indicated by colors (green – 6.0, black – 7.2, orange – 8.0). Note that for (GABA) =1 μM, alkalinization of extracellular solution enhances the current amplitudes. (B) Statistics for relative changes in current amplitudes (with respect to that measured at pH = 7.2) for pH = 6.0 (green bars) and pH = 8.0 (orange bars). Note that especially at very low (GABA; 0.2 μM) basic pH strongly enhances the amplitude, the effect that is qualitatively opposite to that observed at saturating (GABA; Figure 4). Modified from Mozrzymas et al. (2003b) with permission.
Modulation of mIPSCs by Chlorpromazine
Chlorpromazine (CPZ) is a neuroleptic and its primary target are dopamine receptors (Snyder et al., 1974; Seeman, 1980) but several other receptors and channels are affected by this compound (Peroutka and Synder, 1980; Zorumski and Yang, 1988; Ogata et al., 1989; Muller et al., 1991; Bolotina et al., 1992; Benoit and Changeux, 1993; Lidsky et al., 1997). Since CPZ administration could be accompanied by seizures (Toone and Fenton, 1977; Itil and Soldatos, 1980) we were interested in describing the action of this drug on GABAergic transmission (Mozrzymas et al., 1999). CPZ strongly reduced mIPSC amplitude and accelerated its decay having no significant effect on the onset kinetics. Surprisingly, CPZ exerted a dramatically stronger inhibitory effect on mIPSCs than on the current responses elicited by exogenous GABA [especially when applying high (GABA)]. Moreover, at variance to mIPSCs, the rising phase of current responses to non-saturating (GABA) was clearly slowed down by CPZ. In attempt to clarify this discrepancy we have first determined the modulatory mechanism whereby CPZ affected GABAARs. Recordings of current responses to rapid GABA applications together with quantitative analysis based on model simulations indicated that the major CPZ effect was to reduce the binding rate and to increase the unbinding rate with only minor effects on other transition rates (opening/closing, desensitization). It may look surprising that such a simple mechanism (a “mere” weakening of binding) might produce such a variety of kinetic alterations of mIPSCs and of current responses (and that the CPZ effects were substantially different in these two current types). The key to reconcile these diverging findings was again to consider the differences between synaptic transient and exogenous agonist applications. Thus, similar to acidic pH, strong effect of CPZ on mIPSC amplitude was found to result mainly from reduction of binding rate and ensuing lower bound receptor occupancy. The lack of a significant CPZ effect on the mIPSC rising phase was a consequence of a very short receptor exposure to synaptic agonist (ca. 100 μs) for which reduction of binding rate resulted in a decrease in amplitude rather than in a change in the onset rate. On the contrary, longer exogenous applications of GABA (1 mM, 2–5 ms) revealed additionally a slowdown of the current rising phase. Taking altogether, this study revealed not only the mechanism whereby CPZ modulate GABAARs but also provided the first to our knowledge experimentally based estimation of GABA synaptic transient using, as a tool, the modifier of post-synaptic receptor gating. It is worth noting that inhibitory CPZ effect on GABAergic currents appears consistent with seizures induction by this drug although its causality remains to be proved.
Effect of Benzodiazepine Receptor Agonists on GABAergic Currents
Benzodiazepine receptor agonists (BRAs) are positive modulators of GABAergic currents that have been recognized for their sedative, anti-convulsant, anxiolytic, and hypnotic effects. At variance to another group of clinically relevant drugs, barbiturates, BRAs do not activate GABAARs by themselves (but see Campo-Soria et al., 2006), but they rather up regulate the activity of receptors activated by the agonist. Studies on recombinant receptors provided detailed information regarding sensitivity of different GABAAR subtypes to BRAs (Rudolph and Mohler, 2004, 2006; Wafford, 2005) and the localization of BDZ binding site has been indicated (Pritchett et al., 1989; Sigel et al., 1990; Duncalfe et al., 1996; Sigel and Buhr, 1997; Kloda and Czajkowski, 2007). The most widely accepted mechanism of BRA modulation of GABAAR is an increase in the receptor affinity while the effect on the kinetics of conformational transitions between bound states remains a matter of debate. Interestingly, the mechanism relying on BRA-induced increase in receptor affinity has been challenged by Rusch and Forman (2005) as well as by Downing et al. (2005) who studied spontaneously active GABAAR mutant and proposed that the major BRA effect is due to an up regulation of the receptor efficacy. In a series of experiments carried out in one of our laboratories (Mercik et al., 2007; Mozrzymas et al., 2007b; Wojtowicz et al., 2008) the modulatory mechanisms of GABAergic currents by BRAs with particular focus on the impact of specific synaptic conditions dictated by rapid agonist transient have been investigated. To our surprise, the potentiating effect of BRAs on current responses to exogenous agonist was limited to currents evoked by (GABA) several fold smaller than EC50. Moreover, at (GABA) higher than 30 μM, BRAs induced a reduction of current amplitude. BRA-induced down regulation of responses evoked by saturating (GABA) was confirmed on recombinant GABAARs (α1β2γ2) implying that this effect cannot be ascribed to a non-homogeneity of the receptor subtypes in the patches excised from cultured neurons (Mercik et al., 2007). This BRAs effect was attributed to an up modulation of receptor desensitization (Mozrzymas et al., 2007b). Moreover, these compounds prolonged the deactivation phase of the current responses but, again, this effect was limited to (GABA) much lower than EC50. At the same time, BRAs markedly enhanced mIPSC amplitudes and prolonged their decay kinetics. These results led to the conclusion that synaptically released GABA, in spite of reaching relatively high peak concentrations, is far from saturation mainly due to its very short duration. Moreover, as recently pointed out by Petrini et al. (2011), the spatial profile of synaptic agonist is likely to show marked non-homogeneities within the synaptic cleft raising the possibility that receptors located at the periphery of the synaptic disk could experience lower agonist concentration than the central ones. Thus, these peripheral receptors would be more susceptible to an up regulation by BRAs and thereby could boost mIPSC sensitivity to these compounds. Most importantly, experiments with BRAs (Mozrzymas et al., 2007b) confirmed a critical role of synaptic conditions dictated by a rapid agonist transient. Moreover, these studies indicated that in addition to the binding reaction (binding and unbinding rate) BRAs might up regulate the occupancy of the desensitized state (Mercik et al., 2007; Mozrzymas et al., 2007b). The latter finding is compatible with the observation of increased current fading in the presence of BDZ reported by Lavoie and Twyman (1996) and a similar observation by Mellor 1997. Moreover, in a recent report, Karayannis et al. (2010) showed that the decaying phase of slow IPSC recorded from the neurogliaform cells in the hippocampus is accelerated by zolpidem and diazepam providing an indirect evidence for BDA-induced up regulation of the desensitization process. In a recent report by Bianchi et al. (2009), it has been proposed that BDZs modulated GABAARs (α1β3γ2 subtype was examined) by reducing only the unbinding rate (koff). However, while data indicating BDZ-induced reduction of koff confirm previous reports, no evidence was presented for the lack of BDZ effect on the binding rate (kon). An up regulation of kon has been consistently reported in several studies addressing this problem (Macdonald and Olsen, 1994; Rogers et al., 1994; Lavoie and Twyman, 1996; Mozrzymas et al., 2007b).
Concomitant Modulation of GABAergic Currents by Protons and BDZ
As pointed out above, the major mechanism whereby BDZs modulate GABAARs is to increase binding and desensitization while protons exert an opposite action (Mozrzymas et al., 2003a, 2007a). It is thus interesting to check for the combined action of these two modulators on synaptic currents. While acidification strongly reduces desensitization, BDZ impact on this process is relatively weak. On the other hand, BDZ and protons exert the opposite action on binding and both effects are strong. It is thus expected that in conditions of a brief synaptic transient, combined effect of BDZ and acidic pH on agonist binding would be mutually compensatory but the current would be enhanced due to a net suppression of desensitization and ensuing higher occupancy of the open states. This prediction was confirmed in our recent study (Wojtowicz et al., 2008) in which BDZ impact on mIPSCs was tested in neurons in conditions of acidic and physiological pH (Figures 6A,B; Wojtowicz et al., 2008). In addition, analysis of the effect of BDZ and protons on current responses to rapid agonist application, provided evidence that the modulatory effects of these compounds are additive. It is worth noting that these results are likely to have some clinical implications as some brain disorders, such as hypoxia, ischemia, and hypoglycemia, are associated with acidosis of the extracellular fluid in the brain tissue (Kraig et al., 1986; Bengtsson et al., 1990; Katsura et al., 1993).
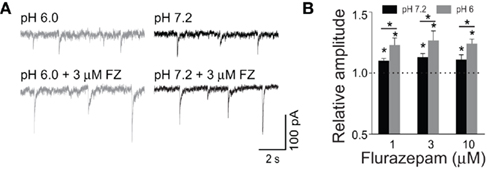
Figure 6. Low pH increases mIPSC susceptibility to modulation by flurazepam. (A) Examples of mIPSCs recorded at −70 mV at pH = 6.0 (gray) and pH = 7.2 (black) in the absence (upper traces) and presence of 3 μM flurazepam (lower traces). (B) Statistics of flurazepam effect on mIPSC amplitude at pH 7.2 (black bars), and at pH = 6.0 (gray bars). Note that the acidic pH increased the relative enhancement of mIPSC amplitude by flurazepam. Modified from Wojtowicz et al. (2008) with permission.
Zinc Effect on GABAergic Currents
Zinc ions are abundant in the CNS and have been found to be co-released with glutamate from mossy fiber boutons in the hippocampus (Slomianka, 1992). Several functions of zinc have been implicated in physiology and pathology and it has been proposed to perceive zinc as biological signal ion (Frederickson et al., 2005; Sensi et al., 2009). Zinc has been demonstrated to reduce the amplitude of GABAergic IPSCs (Defazio and Hablitz, 1998; Barberis et al., 2000; Mellor et al., 2000) and this effect was accompanied by substantial alterations in their time course (decrease in the onset rate and faster decay). In our study (Barberis et al., 2000), the mechanisms of zinc-mediated modulation were studied by analyzing the effect of this compound on synaptic currents and on current responses elicited by rapid GABA applications. The analysis of these currents revealed that the effect of zinc is complex and involves: decrease in binding (kon) and increase in unbinding rate (koff), slower entrance into the fully bound open state (β2) and modified desensitization (decrease in d2 and r2). Importantly, similar to what observed for other modulators reducing agonist binding, zinc ions caused a considerably larger inhibition of mIPSCs than of the current responses to exogenous GABA. Model simulations confirmed that a stronger inhibitory effect of these divalent cations on mIPSCs was a consequence of reduced binding rate in combination with short and non-saturating synaptic GABA transient.
Since it is known that the inhibitory effect of zinc is strongly attenuated by the presence of γ2 subunit, the impact of this ion on α1β2γ2 and on α1β2 receptors was considered (Barberis et al., 2002). Besides confirming that γ2 subunit-containing receptors are at least two order of magnitude more resistant to the blockade by zinc, profound differences were found in the effect of these ions on the onset and desensitization kinetics of these receptors. Importantly, the effect of zinc ions on the α1β2γ2 receptors closely resembled that observed for neuronal ones (Barberis et al., 2000) confirming thus that the pattern of current modulation of α1β2γ2 receptors may be representative, at least qualitatively, for neuronal population of GABAAR.
Voltage-Dependence of GABAergic Currents
Several studies provided evidence that the time course of GABAergic currents shows a substantial voltage-dependence (Barker and Harrison, 1988; Weiss et al., 1988; Jones and Harrison, 1993; Pearce, 1993; Lukatch and MacIver, 1997; Mellor and Randall, 1998; Krishek and Smart, 2001). Since the mechanisms underlying the modulation of GABAergic currents by voltage have not been fully elucidated, we have performed a kinetic analysis of current responses evoked by rapid applications of exogenous GABA (Pytel et al., 2006). I–V relationship for currents evoked by a saturating GABA concentration showed an inward rectification and this effect was accompanied by a faster onset as well as a larger rate and extent of desensitization at positive voltages. Moreover, at low GABA concentration (1 μM) currents were outwardly rectifying and at a non-saturating GABA concentrations (10–300 μM) current onset was significantly faster at positive voltages. Model simulations indicated that the major effect of membrane depolarization was to increase the rates of binding, desensitization, and of opening. The major factor implicated in the inward rectification was the increase in the occupancy of the desensitized state at positive voltages. Interestingly, this interpretation appears supported by the observation that at acidic pH (6.0), i.e., in conditions in which desensitization is markedly weakened, the inward rectification is nearly abolished (Pytel et al., 2005). However, when confronting the voltage-dependence of mIPSCs and of current responses to high (GABA), striking differences were found (Pytel and Mozrzymas, 2006). In contrast to the current responses, mIPSCs showed a strong outward rectification and decay kinetics of mIPSCs was substantially prolonged when changing the membrane voltage from negative to positive. A likely explanation for this observation is that positive voltage, by enhancing the binding rate, might increase the recruitment of synaptic receptors into the bound states after synaptic agonist release. Thus, the switch from a negative to positive polarization would exert a qualitatively similar effect as BRAs.
Oenanthotoxin
Oenanthotoxin (OETX) is a polyacetylenic alcohol found in plants of genus Oenanthe, known to be among the most poisonous in the European flora. It is known since pre-Roman times that it induced contraction of facial muscles giving rise to sinister smile (risus sardonicus). Since OETX action was found to induce also epileptic-like activity (Chauvel et al., 1978; Louvel and Heinemann, 1980, 1983) we have addressed the impact of this toxin on GABAergic currents (Appendino et al., 2009; Wyrembek et al., 2010). Interestingly, this compound exerted a complex action on GABAARs including strong reduction of amplitude, profound change in the current time course and use-dependent blocking action suggesting the open channel block. Most strikingly, however, the effect of this toxin was much more potent on mIPSCs than on current responses. While IC50 for mIPSCs was approximately 0.1 μM, for currents evoked by exogenous saturating or subsaturating GABA concentrations, IC50 was more than one order of magnitude higher (Wyrembek et al., 2010). Again, this discrepancy was attributed to a strong reduction of the binding rate by this toxin that resulted in a dramatic decrease in receptor occupancy during short-lasting synaptic application. Interestingly, OETX also down regulated receptor desensitization, an effect that would be expected to increase the occupancy of the open conformation. However, at submicromolar concentrations, OETX so strongly down regulated the agonist binding during synaptic exposure that the overall occupancy of bound receptors was greatly reduced thus obscuring the effect of a weakened desensitization, similarly to what observed for mIPSCs at strongly acidic pH (Mozrzymas et al., 2003a). Altogether, OETX affected GABAergic currents by a complex mechanism including allosteric modulation of binding and gating receptor properties and possibly by a direct block of the channel pore. Most importantly, a particularly large sensitivity of mIPSCs to OETX with respect to current responses to exogenous GABA was attributed to concomitant reduction of the receptor binding rate and short duration of GABA transient in the synapse.
Intermingled IPSC Modulation and Neurotransmitter Transient Dynamism
Although the nature of described above modulating factors was different (chemical compounds and electric field associated with membrane polarization) and the structures of pharmacological agents ranged from ions (protons or zinc) to complex molecules, it is worth noting some common features of their action and to propose some general observations emerging from these studies. The first and perhaps the most apparent is that the agonist transient time course needs to be perceived as a key regulator of synaptic current susceptibility to pharmacological modulation. It is noteworthy that for all modulators considered above, mIPSCs showed markedly larger sensitivity than the current responses, elicited by exogenous GABA applications, even if the latter ones were evoked using a technique designed to best reproduce synaptic conditions. Kinetic analysis carried out for all the modulators confirmed the general rule that the larger the impact on the agonist binding rate the larger the sensitivity of the synaptic currents to the modulator. Importantly, each time when confronting the effect of a given modulator on current responses evoked by rapid agonist applications and on the synaptic currents one gets a possibility to optimize the parameters describing the synaptic GABA transient time course. When performing such a procedure for data obtained on hippocampal neuronal culture for different modulators of binding and gating (Mozrzymas et al., 1999, 2003a; Barberis et al., 2000) the clearance time course was consistently estimated close to 100 μs. However, when applying the technique based on the use of rapid competitive blockers, predominant time constant for agonist clearance in similar models was considerably longer, ranging between a few hundreds of microseconds up to 1 ms (Clements et al., 1992; Scimemi and Beato, 2009). This discrepancy may result from several drawbacks of both approaches. In the case of available rapidly dissociating competitive antagonists, their unbinding time constant is at least several hundreds of microseconds, i.e., comparable with the agonist clearance time constants determined when using these compounds. Thus, one possibility is that determination of agonist transient waveform might be at the limit of the resolution of this method. A very fast clearance component of approximately 100 μs has been postulated by several Monte Carlo and model simulations (Holmes, 1995; Clements, 1996; Kleinle et al., 1996; Wahl et al., 1996; Glavinovic, 1999; Franks et al., 2002; Ventriglia and Maio, 2003; Petrini et al., 2011). It needs to be born in mind, however, that modeling of agonist transient as a pure diffusion might lead to overestimation of the clearance rate as in the synaptic and subsynaptic environment, the diffusion coefficient might be much lower than in the free solution due to, e.g., larger viscosity of extracellular fluid, interactions with extracellular matrix components or limited space (Nicholson and Sykova, 1998; Vorisek and Sykova, 2009). On the other hand, as already mentioned, the method of transient determination based on the use of binding and gating modifiers (Mozrzymas et al., 1999, 2003a; Barberis et al., 2000) might be biased by the fact that the modulators, besides modifying binding affect also gating and their effect might differ between synaptic and extra-synaptic receptors (and also between excised patch and whole-cell configurations). Synaptic currents can be modulated by cell adhesion molecules (Dalva et al., 2007; Huang and Scheiffele, 2008) and to some extent by a number of scaffolding proteins (Tyagarajan and Fritschy, 2010; Tyagarajan et al., 2011), the effects that are likely to be lost or severely impaired upon patch excision or intense cell dialysis. Thus, it seems reasonable to assume that basing on studies using both types of techniques, the predominant component of agonist clearance at the GABAergic synapse in the considered models (neuronal cell cultures) lies within the range between one hundred up to a few hundreds of microseconds.
Variety of Neuronal Types and a Variety of Synaptic Conditions
Pharmacokinetic studies outlined above were carried out either on cultured neurons or on cell lines expressing recombinant receptors. Several factors may alter the synaptic transmission in cultured neurons with respect to their counterparts in more physiological models (slices or in vivo). Cultured neurons, after a harmful enzymatic and mechanical treatment, are grown in artificial environment and, although some developmental mechanisms are maintained (Ortinski et al., 2004; Swanwick et al., 2006; Pytel et al., 2007), neuronal differentiation is altered, and synaptic connections are formed largely randomly due to dispersion of naturally occurring cues. Importantly, large volume of bulk aqueous solutions around neurons may greatly affect the diffusion conditions favoring faster neurotransmitter clearance from the synapses. Moreover, in neuronal cultures, glial cells are typically suppressed to avoid their overgrowth. Taking into consideration the extensive and rapidly growing body of evidence that astrocytes are strongly involved in the functioning of synapses, giving rise to the concept of a tri-partite synapse (Araque et al., 1999; Perea et al., 2009; Pereira and Furlan, 2010), it may be expected that the lack or deficit of astrocytes could profoundly affect the synaptic transmission. Astrocytes are known to set not only the physical barriers for free diffusion within and in the immediate surroundings of the synapse but also to play a pivotal role in the neurotransmitter uptake (Conti et al., 2004; Bergeles and Edwards, 2008). Especially in the case of glutamate, excitatory amino acid transporters (EAAT1 and EAAT2) are very efficient in removing free extracellular glutamate and are highly concentrated in astrocytes assuring an efficient control of the spatiotemporal profile of synaptically released glutamate (Diamond, 2005; Tzingounis and Wadiche, 2007; Bergeles and Edwards, 2008; Scimemi et al., 2009; Thomas et al., 2011). Electrogenic nature of glutamate transporters (2 Na+ ions, 1 H+ ion, 1 glutamate in, 1 K+ ion out) made it possible to track their kinetics by analyzing transporter-mediated currents evoked by exogenous applications of glutamate to excised patches (Bergles et al., 1997, 2002). These studies provided evidence that glutamate binding and its translocation to the inner side are rapid, although their full cycle (including glutamate release in the cytoplasm) requires approximately 10–20 ms (for the transporter EAAT2, Bergles et al., 2002). This implies that transporters would contribute to the fast clearance of synaptic neurotransmitter by rapidly binding the released molecules, acting therefore as neurotransmitter buffer (Scimemi et al., 2009).
Importantly, GABAergic interneurons are greatly diversified with respect to their morphology and function (Klausberger and Somogyi, 2008). The time duration (assessed as mean decay time constant) of GABAergic currents may range between little more than 1 ms for synapses formed by parvalbumin positive basket interneurons (Bartos et al., 2002) up to nearly 100 ms for currents in thalamic reticular nucleus (Huntsman and Huguenard, 2000, 2006; Mozrzymas et al., 2007a) or in neurogliaform cells in hippocampus and cortex (Szabadics et al., 2007; Olah et al., 2009; Zarnowska et al., 2009; Karayannis et al., 2010; Capogna and Pearce, 2011). This heterogeneity of synaptic currents resulting from interneuron type and target cell identity is often obscured when studying IPSCs in neuronal cultures and therefore the estimation of synaptic transient may be biased by data averaging for different types of neurons (both pre- and post-synaptic) while conditions of synaptic transmission are likely to be profoundly affected by artificial conditions in culture. A variety of synaptic conditions in a slice model has been observed by Hajos et al. (2000) who examined the impact of zolpidem on mIPSCs in different types of neurons in eight brain regions and provided evidence for different extent of occupancy of post-synaptic receptors (incomplete vs. complete) depending on cell type and localization in the brain.
Glutamatergic synapses are known for their particularly high pre- and post-synaptic morphological heterogeneity. Indeed, the shape of synaptic terminals may range between tiny “en passant” boutons and giant depolarizing terminals. Interestingly, these two extreme forms of pre-synaptic terminals may occur within the same axon (e.g., in hippocampal mossy fibers) which may additionally show a fascinating feature of target cell specific properties (Maccaferri et al., 1998; Pelkey and McBain, 2008). Similarly, the morphology of the post-synaptic structures may vary between small varicosities and filopodia to extensive thorny excrescences (Fiala et al., 2008). Taking additionally into account that synaptic structures can be tightly ensheathed by astrocytes that, as mentioned, are strongly involved in controlling the spatiotemporal profile of synaptic agonist, it is not surprising that the time course of synaptic currents may critically depend on the synaptic structure. Indeed, Barbour et al. (1994) have reported that AMPA/kainate receptor-mediated EPSCs recorded in parallel fibers are characterized by a slower decay than those measured from interneurons while synaptic receptor properties in these cell types were very similar. This difference was attributed to different spine and synapse geometry that favors prolonged presence of glutamate around dendritic spines in the case interneurons. More recently, Cathala et al. (2005) applied an elegant approach based on functional, pharmacological, morphological, and analytical techniques to address the mechanisms of developmental speeding of AMPA receptor-mediated mEPSCs in mouse cerebellar granule cells. Surprisingly, they found that developmental acceleration of mEPSCs is unrelated to any clear switch in the subunit stoichiometry of the post-synaptic receptors. However, 3D reconstruction of mossy fiber terminals and granule cell dendrites revealed that the post-synaptic density areas in young (P8) animals were more than four times larger than those in adult ones (P40). In contrast, P8 mossy fiber terminals were much smaller and less convoluted than those in adults and these differences in dendritic shape resulted in an increased number of diffusional sinks in P40. Model simulations confirmed that these morphological changes of synapses are sufficient to affect the synaptic glutamate concentration waveform and thereby to underlie the developmental acceleration of the glutamatergic mEPSCs.
Although most studies based on combined functional and morphological approaches concerned glutamatergic synaptic transmission, in the last years important reports appeared concerning also GABAergic synapses. Biro et al. (2006) have estimated quantal parameters for IPSCs measured in pairs of CCK-expressing interneurons and CA3 pyramidal cells and for the same pairs performed a post hoc EM determination of the number of boutons mediating IPSCs and the number of active zones per CCK-positive bouton. Surprisingly, quantal analysis indicated fivefold more functional releasing sites than those identified structurally. Biro et al. (2006) have proposed that this apparent discrepancy resulted from variation of synaptic GABA transient generated by multivesicular release in conditions of particularly low post-synaptic receptor occupancy. This mechanism may be regarded as a pre-synaptic, release probability-dependent scaling of PSCs at a single GABAergic synapse. Barberis et al. (2005) provided another example of pre-synaptic changes of GABA transient. By using the rapidly dissociating GABAAR antagonist, TPMPA they showed in an in vitro model, that in “young” neurons, displacement of TPMPA was more efficient than in “older” ones indicating that GABA uniquantal release is developmentally regulated.
As already mentioned, time duration of GABAergic synaptic currents measured in different projections may vary for nearly two orders of magnitudes. Initially, the phenomenon of IPSCs characterized by a particularly slow kinetics contrasting with predominant population of fast IPSCs has been observed in the hippocampus (Pearce, 1993; Banks et al., 1998). Later, IPSCs with slow kinetics were found also in other brain regions including the neocortex and thalamus as well as other brain regions (Kapur et al., 1997; Huntsman and Huguenard, 2000, 2006; Capogna and Pearce, 2011). Subsequent studies addressed the mechanisms underlying slow IPSCs and, not surprisingly, in different cell types distinct pre- or post-synaptic factors were implicated. Slow IPSCs in the thalamic RT interneurons are among the slowest GABAergic synaptic currents in the CNS (Huntsman and Huguenard, 2000, 2006; Browne et al., 2001). Extensive body of evidence based on slice recordings and recombinant receptors analysis consistently indicated that the slow IPSCs in these cells reflected a particular kinetics of post-synaptic α3 subunit-containing GABAAR (Verdoorn, 1994; Gingrich et al., 1995; Barberis et al., 2007; Mozrzymas et al., 2007a; Schofield and Huguenard, 2007). In contrast, in the cortical neurogliaform cells, slow IPSC kinetics was reported for synapses in which low affinity α1, β2/3, and γ2 subunit-containing receptors (typically implicated in fast transmission) were found to be predominant. In the case of these neurons, slow IPSC time course was attributed to a particularly slow GABA transient (Szabadics et al., 2007). This mechanism was recently further supported by an elegant functional and utrastructural study in which a remarkable observation was made that pre-synaptic boutons on axons of these cells were extremely dense but most of them did not form the classical synapse including a precisely juxtaposed post-synaptic density (Olah et al., 2009). Thus, wide synaptic clefts or GABA released from boutons distant from recipient GABAARs provide a consistent explanation for slow GABA transient described by Szabadics et al. (2007). Such a peculiar synaptic arrangement of neurogliaform cells was proposed to underlie the volume release of GABA, raising thus a novel concept that GABA released from these neurons might exert a paracrine effect on the local neuronal networks (Olah et al., 2009). In parallel, Karayannis et al. (2010) explored the mechanisms of slow IPSCs in the hippocampal neurogliaform cells. Importantly, using pharmacological tools, they have found that α1 and γ2 subunit-containing receptors were involved in mediating slow currents evoked by neurogliaform cell activation, similar to what reported by Szabadics et al. (2007) in the cortex. Elegant quantitative analysis and modeling led Karayannis et al. (2010) to the conclusion that slow kinetics IPSCs generated by neurogliaform cells in the hippocampus results from unusually slow GABA transient in the synapses formed by these cells. Moreover, their ultrastructural studies revealed that neurogliaform cells formed membrane-to-membrane appositions that were lacking a clear synaptic structure. This result corroborates the structural findings by Olah et al. (2009) in the cortical neurogliaform cells.
The above examples of studies carried out on the GABAergic transmission in models believed to closely reproduce physiological conditions, indicate an amazing variety of kinetic patterns that have not been previously observed in cultured neurons. It is worth to bear in mind that some GABAergic interneurons (e.g., parvalbumin positive basket cells) are responsible for the fastest point-to-point synaptic transmission (Bartos et al., 2002) while neurogliaform cells, which release the same neurotransmitter often in a close vicinity of basket neurons, give rise to the slowest GABAergic synaptic signals in the hippocampus, implicated in a paracrine action (Olah et al., 2009). Taken altogether, both post-synaptic receptor properties and conditions of synaptic activation, dictated by the spatiotemporal profile of synaptically released agonist, were found to play a crucial role in shaping the GABAergic synaptic currents including the fastest and the slowest signals.
Concluding remarks
Although tools allowing to directly monitoring the concentration of synaptic agonist in GABAergic and glutamatergic synapses are still lacking, our knowledge about its time course has been considerably enriched in the last two decades. Experimental approaches used to explore the dynamics of synaptic neurotransmitter are based on indirect information such as displacement of quickly unbinding competitive antagonists or modifiers of binding/gating receptor properties. While the former one was used intentionally to determine the agonist transient, the latter approach turned to be a necessary step to explore the mechanisms of pharmacological modulation. Indeed, for several agents, differences in pharmacological modulation of synaptic signals and of responses elicited by agonist applications could have been reconciled by considering the specific synaptic conditions set by the agonist transient. Investigations of the time course of synaptic neurotransmitters shed new light on our understanding of not only modulatory mechanisms but also extended our knowledge on the physiology of synaptic transmission in specific neurons. In particular, brief and spatially non-homogeneous GABA transient may favor openings of singly bound, peripherally localized synaptic receptors which would accelerate the IPSC decay kinetics. While early studies on cultured neurons implicated very rapid kinetics of synaptic agonist clearance, recent investigations carried out on brain slices revealed that in various types of interneurons, agonist transient kinetics may vary by nearly two orders of magnitude. It is worth emphasizing that synaptically released neurotransmitter, especially in the synapses showing slow transient, is spilling over, and affects the synapse neighborhood that may include other synapses, perisynaptic neuronal receptors mediating tonic transmission and astrocytes that emerged as powerful modulators of synaptic functions. Thus, synaptic agonist transient is not only setting the transmission conditions at a given synapse but is also largely determining the interaction of the synapse with its local environment.
Taking altogether, the precise description of the synaptic conditions resulting from the spatiotemporal neurotransmitter profile is a crucial step in exploring the synaptic transmission and its modulation. It is expected that development of new (e.g., optical) tools enabling high resolution tracking of agonist profiles at different synapses would mark an important step forward in deciphering the function of neuronal networks.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jerzy W. Mozrzymas was supported by the Foundation for Polish Science award “Mistrz” (contract No. 7/2008). Andrea Barberis and Enrica Maria Petrini were supported by EU/FP7 (FOCUS, to Fabio Benfenati and Andrea Barberis) and Telethon-Italy (GGP10135, to Laura Cancedda and Andrea Barberis).
References
Appendino, G., Pollastro, F., Verotta, L., Ballero, M., Romano, A., Wyrembek, P., Szczuraszek, K., Mozrzymas, J. W., and Taglialatela-Scafati, O. (2009). Polyacetylenes from sardinian Oenanthe fistulosa: a molecular clue to risus sardonicus. J. Nat. Prod. 72, 962–965.
Arancibia-Carcamo, I. L., and Moss, S. J. (2006). Molecular organization and assembly of the central inhibitory postsynapse. Results Probl. Cell Differ. 43, 25–47.
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215.
Balakrishnan, V., Kuo, S. P., Roberts, P. D., and Trussel, L. O. (2009). Slow glycinergic transmission mediated by transmitter pooling. Nat. Neurosci. 12, 286–294.
Banks, M. I., Li, T. B., and Pearce, R. A. (1998). The synaptic basis of GABAA, slow. J. Neurosci. 18, 1305–1317.
Banks, M. I., and Pearce, R. A. (2000). Kinetic differences between synaptic and extrasynaptic GABA(A) receptors in CA1 pyramidal cells. J. Neurosci. 20, 937–948.
Barberis, A., Cherubini, E., and Mozrzymas, J. W. (2000). Zinc inhibits miniature GABAergic currents by allosteric modulation of GABAA receptor gating. J. Neurosci. 20, 8618–8627.
Barberis, A., Lu, C., Vicini, S., and Mozrzymas, J. W. (2005). Developmental changes of GABA synaptic transient in cerebellar granule cells. Mol. Pharmacol. 67, 1221–1228.
Barberis, A., Mozrzymas, J. W., Ortinski, P. I., and Vicini, S. (2007). Desensitization and binding properties determine distinct alpha1beta2gamma2 and alpha3beta2gamma2 GABA(A) receptor-channel kinetic behavior. Eur. J. Neurosci. 25, 2726–2740.
Barberis, A., Petrini, E. M., and Cherubini, E. (2004). Presynaptic source of quantal size variability at GABAergic synapses in rat hippocampal neurons in culture. Eur. J. Neurosci. 20, 1803–1810.
Barberis, A., Petrini, E. M., Cherubini, E., and Mozrzymas, J. W. (2002). Allosteric interaction of zinc with recombinant alpha(1)beta(2)gamma(2) and alpha(1)beta(2) GABA(A) receptors. Neuropharmacology 43, 607–618.
Barberis, A., Sachidhanandam, S., and Mulle, C. (2008). GluR6/KA2 kainate receptors mediate slow-deactivating currents. J. Neurosci. 28, 6402–6406.
Barbour, B., Keller, B. U., Llano, I., and Marty, A. (1994). Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron 12, 1331–1343.
Barker, J. L., and Harrison, N. L. (1988). Outward rectification of inhibitory postsynaptic currents in cultured rat hippocampal neurones. J. Physiol. 403, 41–55.
Bartos, M., Vida, I., Frotscher, M., Meyer, A., Monyer, H., Geiger, J. R., and Jonas, P. (2002). Fast synaptic inhibition promotes synchronized gamma oscillations in hippocampal interneuron networks. Proc. Natl. Acad. Sci. U.S.A. 99, 13222–13227.
Baumann, S. W., Baur, R., and Sigel, E. (2003). Individual properties of the two functional agonist sites in GABA(A) receptors. J. Neurosci. 23, 11158–11166.
Beato, M. (2008). The time course of transmitter at glycinergic synapses onto motoneurons. J. Neurosci. 16, 7412–7425.
Beato, M., Burzomato, V., and Sivilotti, L. G. (2007). The kinetics of inhibition of rat recombinant heteromeric alpha1beta glycine receptors by the low-affinity antagonist SR-95531. J. Physiol. 580, 171–179.
Bengtsson, F., Boris-Moller, F., Hansen, A. J., and Siesjo, B. K. (1990). Extracellular pH in the rat brain during hypoglycemic coma and recovery. J. Cereb. Blood Flow Metab. 10, 262–269.
Benke, D., Fakitsas, P., Roggenmoser, C., Michel, C., Rudolph, U., and Mohler, H. (2004). Analysis of the presence and abundance of GABAA receptors containing two different types of alpha subunits in murine brain using point-mutated alpha subunits. J. Biol. Chem. 279, 43654–43660.
Benoit, P., and Changeux, J. P. (1993). Voltage dependencies of the effects of chlorpromazine on the nicotinic receptor channel from mouse muscle cell line So18. Neurosci. Lett. 160, 81–84.
Bergles, D. E., Dzubay, J. A., and Jahr, C. E. (1997). Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc. Natl. Acad. Sci. USA 94, 14821–14825.
Bergeles, D. E., and Edwards, R. H. (2008). “The role of glutamate transporters in synaptic transmission,” in Structural and Functional Organization of the Synapse, eds J. W. Hell, and M. D. Ehlers (New York, NY: Springer), 23–62.
Bergles, D. E., Tzigounis, A. V., and Jahr, C. E. (2002). Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J. Neurosci. 1, 10153–10162.
Bianchi, M. T., Botzolakis, E. J., Lagrange, A. H., and Macdonald, R. L. (2009). Benzodiazepine modulation of GABA(A) receptor opening frequency depends on activation context: a patch clamp and simulation study. Epilepsy Res. 85, 212–220.
Biro, A. A., Holderith, N. B., and Nusser, Z. (2006). Release probability-dependent scaling of the postsynaptic responses at single hippocampal GABAergic synapses. J. Neurosci. 26, 12487–12496.
Bolotina, V., Courtney, K. R., and Khodorov, B. (1992). Gate-dependent blockade of sodium channels by phenothiazine derivatives: structure-activity relationships. Mol. Pharmacol. 42, 423–431.
Browne, S. H., Kang, J., Akk, G., Chiang, L. W., Schulman, H., Huguenard, J. R., and Prince, D. A. (2001). Kinetic and pharmacological properties of GABA(A) receptors in single thalamic neurons and GABA(A) subunit expression. J. Neurophysiol. 86, 2312–2322.
Campo-Soria, C., Chang, Y., and Weiss, D. S. (2006). Mechanism of action of benzodiazepines on GABAA receptors. Br. J. Pharmacol. 148, 984–990.
Capogna, M., and Pearce, R. A. (2011). GABA A, slow: causes and consequences. Trends Neurosci. 34, 101–112.
Cathala, L., Holderith, N. B., Nusser, Z., DiGregorio, D. A., and Cull-Candy, S. G. (2005). Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat. Neurosci. 8, 1310–1318.
Chauvel, P., Louvel, J., Anger, J. P., and Chauvel, Y. (1978). Convulsions induced by enanthotoxin in the rat: correlation between electrophysiological modifications and clinical signs. C. R. Hebd. Seances Acad. Sci. Ser. D Sci. Nat. 286, 1833–1836.
Cherubini, E., and Conti, F. (2001). Generating diversity at GABAergic synapses. Trends Neurosci. 24, 155–162.
Christie, S. B., Li, R. W., Miralles, C. P., Riquelme, R., Yang, B. Y., Wendou-Yu, C. E., Daniels, S. B., Cantino, M. E., and De Blas, A. L. (2002). Synaptic and extrasynaptic GABAA receptors and gephyrin clusters. Prog. Brain Res. 136, 157–180.
Clements, J. D. (1996). Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 19, 163–171.
Clements, J. D., Lester, R. A., Tong, G., Jahr, C. E., and Westbrook, G. L. (1992). The time course of glutamate in the synaptic cleft. Science 258, 1498–1501.
Colquhoun, D. (1998). Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125, 924–947.
Conti, F., Minelli, A., and Melone, M. (2004). GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res. Brain Res. Rev. 45, 196–212.
Dalva, M. B., McClelland, A. C., and Kayser, M. S. (2007). Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220.
Defazio, T., and Hablitz, J. J. (1998). Zinc and zolpidem modulate mIPSCs in rat neocortical pyramidal neurons. J. Neurophysiol. 80, 1670–1677.
Diamond, J. S. (2005). Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J. Neurosci. 25, 2906–2916.
Dietrich, C. J., and Morad, M. (2010). Synaptic acidification enhances GABAA signaling. J. Neurosci. 30, 16044–16052.
Downing, S. S., Lee, Y. T., Farb, D. H., and Gibbs, T. T. (2005). Benzodiazepine modulation of partial agonist efficacy and spontaneously active GABA(A) receptors supports an allosteric model of modulation. Br. J. Pharmacol. 145, 894–906.
Duncalfe, L. L., Carpenter, M. R., Smillie, L. B., Martin, I. L., and Dunn, S. M. (1996). The major site of photoaffinity labeling of the gamma-aminobutyric acid type A receptor by [3H]flunitrazepam is histidine 102 of the alpha subunit. J. Biol. Chem. 271, 9209–9214.
Fiala, J. C., Harris, J., and Spacek, K. H. (2008). “Dendrite structure,” in Dendrites, eds. G. Stuart, N. Spruston, and M. Hausser (Oxford, UK: Oxford University Press), 1–41.
Franks, K. M., Bartol, T. M. Jr., and Sejnowski, T. J. (2002). A Monte Carlo model reveals independent signaling at central glutamatergic synapses. Biophys. J. 83, 2333–2348.
Frederickson, C. J., Koh, J. Y., and Bush, A. I. (2005). The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 6, 449–462.
Frerking, M., Borges, S., and Wilson, M. (1995). Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron 15, 885–895.
Fritschy, J. M., and Brunig, I. (2003). Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol. Ther. 98, 299–323.
Fritschy, J. M., Schweizer, C., Brunig, I., and Luscher, B. (2003). Pre- and post-synaptic mechanisms regulating the clustering of type-A gamma aminobutyric acid receptors (GABAA receptors). Biochem. Soc. Trans. 31, 889–892.
Gingrich, K. J., Roberts, W. A., and Kass, R. S. (1995). Dependence of the GABAA receptor gating kinetics on the alpha-subunit isoform: implications for structure-function relations and synaptic transmission. J. Physiol. 489(Pt 2), 529–543.
Glavinovic, M. I. (1999). Monte carlo simulation of vesicular release, spatiotemporal distribution of glutamate in synaptic cleft and generation of postsynaptic currents. Pflugers Arch. 437, 462–470.
Hajos, N., Nusser, Z., Rancz, E. A., Freund, T. F., and Mody, I. (2000). Cell type and synapse-specific variability in synaptic GABAA receptor occupancy. Eur. J. Neurosci. 12, 810–818.
Heine, M., Groc, L., Frischknecht, R., Beique, J. C., Lounis, B., Rumbaugh, G., Huganir, R. L., Cognet, L., and Choquet, D. (2008). Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science 320, 201–205.
Holmes, W. R. (1995). Modeling the effect of glutamate diffusion and uptake on NMDA and non-NMDA receptor saturation. Biophys. J. 69, 1734–1747.
Houston, C. M., Bright, D. P., Sivilotti, L. G., Beato, M., and Smart, T. G. (2009). Intracellular chloride ions regulate the time course of GABA-mediated inhibitory transmission. J. Neurosci. 19, 10416–10423.
Houston, C. M., Hosie, A. M., and Smart, T. G. (2008). Distinct regulation of beta2 and beta3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 23, 7574–7584.
Huang, Z. J., and Scheiffele, P. (2008). GABA and neuroligin signaling: linking synaptic activity and adhesion in inhibitory synapse development. Curr. Opin. Neurobiol. 18, 77–83.
Huntsman, M. M., and Huguenard, J. R. (2000). Nucleus-specific differences in GABA(A)-receptor-mediated inhibition are enhanced during thalamic development. J. Neurophysiol. 83, 350–358.
Huntsman, M. M., and Huguenard, J. R. (2006). Fast IPSCs in rat thalamic reticular nucleus require the GABAA receptor beta1 subunit. J. Physiol. 572, 459–475.
Itil, T. M., and Soldatos, C. (1980). Epileptogenic side effects of psychotropic drugs. Practical recommendations. J. Am. Med. Assoc. 244, 1460–1463.
Jonas, P. (1995). “Fast application of agonist to isolated membrane patches,” in Single Channel Recordings, eds B. Sakmann, and E. Neher (New York: Plenum Press), 132–243.
Jones, M. V., and Harrison, N. L. (1993). Effects of volatile anesthetics on the kinetics of inhibitory postsynaptic currents in cultured rat hippocampal neurons. J. Neurophysiol. 70, 1339–1349.
Jones, M. V., Sahara, Y., Dzubay, J. A., and Westbrook, G. L. (1998). Defining affinity with the GABAA receptor. J. Neurosci. 18, 8590–8604.
Jones, M. V., and Westbrook, G. L. (1995). Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15, 181–191.
Kapur, A., Pearce, R. A., Lytton, W. W., and Haberly, L. B. (1997). GABAA-mediated IPSCs in piriform cortex have fast and slow components with different properties and locations on pyramidal cells. J. Neurophysiol. 78, 2531–2545.
Karayannis, T., Elfant, D., Huerta-Ocampo, I., Teki, S., Scott, R. S., Rusakov, D. A., Jones, M. V., and Capogna, M. (2010). Slow GABA transient and receptor desensitization shape synaptic responses evoked by hippocampal neurogliaform cells. J. Neurosci. 30, 9898–9909.
Katsura, K., Folbergrova, J., Bengtsson, F., Kristian, T., Gido, G., and Siesjo, B. K. (1993). Recovery of mitochondrial and plasma membrane function following hypoglycemic coma: coupling of ATP synthesis, K+ transport, and changes in extra- and intracellular pH. J. Cereb. Blood Flow Metab. 13, 820–826.
Kittler, J. T., and Moss, S. J. (2003). Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol. 13, 341–347.
Klausberger, T. (2009). GABAergic interneurons targeting dendrites of pyramidal cells in the CA1 area of the hippocampus. Eur. J. Neurosci. 30, 947–957.
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57.
Kleinle, J., Vogt, K., Luscher, H. R., Muller, L., Senn, W., Wyler, K., and Streit, J. (1996). Transmitter concentration profiles in the synaptic cleft: an analytical model of release and diffusion. Biophys. J. 71, 2413–2426.
Kloda, J. H., and Czajkowski, C. (2007). Agonist-, antagonist-, and benzodiazepine-induced structural changes in the alpha1 Met113-Leu132 region of the GABAA receptor. Mol. Pharmacol. 71, 483–493.
Kraig, R. P., Pulsinelli, W. A., and Plum, F. (1986). Carbonic acid buffer changes during complete brain ischemia. Am. J. Physiol. 250, R348–R357.
Krishek, B. J., and Smart, T. G. (2001). Proton sensitivity of rat cerebellar granule cell GABAA receptors: dependence on neuronal development. J. Physiol. 530, 219–233.
Lavoie, A. M., and Twyman, R. E. (1996). Direct evidence for diazepam modulation of GABAA receptor microscopic affinity. Neuropharmacology 35, 1383–1392.
Lester, R. A., and Jahr, C. E. (1992). NMDA channel behavior depends on agonist affinity. J. Neurosci. 12, 635–643.
Lidsky, T. I., Yablonsky-Alter, E., Zuck, L. G., and Banerjee, S. P. (1997). Antipsychotic drug effects on glutamatergic activity. Brain Res. 764, 46–52.
Louvel, J., and Heinemann, U. (1980). Diminution of the extracellular concentration of calcium ions during focal epileptic crisis induced by enanthotoxin in the cat cortex. C. R. Acad. Sci. D Sci. Nat. 291, 997–1000.
Louvel, J., and Heinemann, U. (1983). Changes in [Ca2+]0, [K+]0 and neuronal activity during oenanthotoxin-induced epilepsy in cat sensorimotor cortex. Electroencephalogr. Clin. Neurophysiol. 56, 457–466.
Lukatch, H. S., and MacIver, M. B. (1997). Voltage-clamp analysis of halothane effects on GABA(A fast) and GABA(A slow) inhibitory currents. Brain Res. 765, 108–112.
Maccaferri, G., Toth, K., and McBain, C. J. (1998). Target-specific expression of presynaptic mossy fiber plasticity. Science 279, 1368–1370.
Macdonald, R. L., and Olsen, R. W. (1994). GABAA receptor channels. Annu. Rev. Neurosci. 17, 569–602.
Macdonald, R. L., Rogers, C. J., and Twyman, R. E. (1989). Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J. Physiol. 410, 479–499.
Marchionni, I., Kasap, Z., Mozrzymas, J. W., Sieghart, W., Cherubini, E., and Zacchi, P. (2009). New insights on the role of gephyrin in regulating both phasic and tonic GABAergic inhibition in rat hippocampal neurons in culture. Neuroscience 164, 552–562.
Matsuzaki, M., Hayama, T., Kasai, H., and Ellis-Davies, G. C. (2010). Two-photon uncaging of gamma-aminobutyric acid in intact brain tissue. Nat. Chem. Biol. 6, 255–257.
Mellor, J. R., and Randall, A. D. (1997). Frequency-dependent actions of benzodiazepines on GABAA receptors in cultured murine cerebellar granule cells. J. Physiol. 503, 353–369.
Mellor, J. R., and Randall, A. D. (1998). Voltage-dependent deactivation and desensitization of GABA responses in cultured murine cerebellar granule cells. J. Physiol. 506(Pt 2), 377–390.
Mellor, J. R., Wisden, W., and Randall, A. D. (2000). Somato-synaptic variation of GABA(A) receptors in cultured murine cerebellar granule cells: investigation of the role of the alpha6 subunit. Neuropharmacology 39, 1495–1513.
Mercik, K., Piast, M., and Mozrzymas, J. W. (2007). Benzodiazepine receptor agonists affect both binding and gating of recombinant alpha1beta2gamma2 gamma-aminobutyric acid-A receptors. Neuroreport 18, 781–785.
Mercik, K., Pytel, M., Cherubini, E., and Mozrzymas, J. W. (2006). Effect of extracellular pH on recombinant alpha1beta2gamma2 and alpha1beta2 GABAA receptors. Neuropharmacology 51, 305–314.
Min, M. Y., Rusakov, D. A., and Kullmann, D. M. (1998). Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron 21, 561–570.
Minier, F., and Sigel, E. (2004). Positioning of the alpha-subunit isoforms confers a functional signature to gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 101, 7769–7774.
Mott, D. D., Rojas, A., Fisher, J. L., Dingledine, R. J., and Benveniste, M. (2010). Subunit-specific desensitization of heteromeric kainate receptors. J. Physiol. 588, 683–700.
Mozrzymas, J. W. (2004). Dynamism of GABA(A) receptor activation shapes the “personality” of inhibitory synapses. Neuropharmacology 47, 945–960.
Mozrzymas, J. W., Barberis, A., Michalak, K., and Cherubini, E. (1999). Chlorpromazine inhibits miniature GABAergic currents by reducing the binding and by increasing the unbinding rate of GABAA receptors. J. Neurosci. 19, 2474–2488.
Mozrzymas, J. W., Barberis, A., and Vicini, S. (2007a). GABAergic currents in RT and VB thalamic nuclei follow kinetic pattern of alpha3- and alpha1-subunit-containing GABAA receptors. Eur. J. Neurosci. 26, 657–665.
Mozrzymas, J. W., Wojtowicz, T., Piast, M., Lebida, K., Wyrembek, P., and Mercik, K. (2007b). GABA transient sets the susceptibility of mIPSCs to modulation by benzodiazepine receptor agonists in rat hippocampal neurons. J. Physiol. 585, 29–46.
Mozrzymas, J. W., Zarnowska, E. D., Pytel, M., and Mercik, K. (2003a). Modulation of GABA(A) receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J. Neurosci. 23, 7981–7992.
Mozrzymas, J. W., Barberis, A., Mercik, K., and Zarnowska, E. D. (2003b). Binding sites, singly bound states, and conformation coupling shape GABA-evoked currents. J. Neurophysiol. 89, 871–883.
Muller, M., De Weille, J. R., and Lazdunski, M. (1991). Chlorpromazine and related phenothiazines inhibit the ATP-sensitive K+ channel. Eur. J. Pharmacol. 198, 101–104.
Nicholson, C., and Sykova, E. (1998). Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 21, 207–215.
Ogata, N., Nishimura, M., and Narahashi, T. (1989). Kinetics of chlorpromazine block of sodium channels in single guinea pig cardiac myocytes. J. Pharmacol. Exp. Ther. 248, 605–613.
Olah, S., Fule, M., Komlosi, G., Varga, C., Baldi, R., Barzo, P., and Tamas, G. (2009). Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature 461, 1278–1281.
Ortinski, P. I., Lu, C., Takagaki, K., Fu, Z., and Vicini, S. (2004). Expression of distinct alpha subunits of GABAA receptor regulates inhibitory synaptic strength. J. Neurophysiol. 92, 1718–1727.
Overstreet, L. S., Westbrook, G. L., and Jones, M. V. (2003). “Measuring and modeling the spatiotemporal profile of GABA at the synapse,” in Transmembrane Transporters, ed. M. W. Quick (New York: Wiley), 259–275.
Pasternack, M., Smirnov, S., and Kaila, K. (1996). Proton modulation of functionally distinct GABAA receptors in acutely isolated pyramidal neurons of rat hippocampus. Neuropharmacology 35, 1279–1288.
Pearce, R. A. (1993). Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron 10, 189–200.
Pelkey, K. A., and McBain, C. J. (2008). Target-cell-dependent plasticity within the mossy fibre-CA3 circuit reveals compartmentalized regulation of presynaptic function at divergent release sites. J. Physiol. 586, 1495–1502.
Perea, G., Navarrete, M., and Araque, A. (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431.
Pereira, A. Jr., and Furlan, F. A. (2010). Astrocytes and human cognition: modeling information integration and modulation of neuronal activity. Prog. Neurobiol. 92, 405–420.
Peroutka, S. J., and Synder, S. H. (1980). Relationship of neuroleptic drug effects at brain dopamine, serotonin, alpha-adrenergic, and histamine receptors to clinical potency. Am. J. Psychiatry 137, 1518–1522.
Perrais, D., and Ropert, N. (1999). Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J. Neurosci. 19, 578–588.
Perrais, D., and Ropert, N. (2000). Altering the concentration of GABA in the synaptic cleft potentiates miniature IPSCs in rat occipital cortex. Eur. J. Neurosci. 12, 400–404.
Petrini, E. M., Nieus, T., Ravasenga, T., Succol, F., Guazzi, S., Benfenati, F., and Barberis, A. (2011). Influence of GABAAR monoliganded states on GABAergic responses. J. Neurosci. 31, 1752–1761.
Petrini, E. M., Zacchi, P., Barberis, A., Mozrzymas, J. W., and Cherubini, E. (2003). Declusterization of GABAA receptors affects the kinetic properties of GABAergic currents in cultured hippocampal neurons. J. Biol. Chem. 278, 16271–16279.
Pritchett, D. B., Sontheimer, H., Shivers, B. D., Ymer, S., Kettenmann, H., Schofield, P. R., and Seeburg, P. H. (1989). Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature 338, 582–585.
Pugh, J. R., and Raman, I. M. (2005). GABAA receptor kinetics in the cerebellar nuclei: evidence for detection of transmitter from distant release sites. Biophys. J. 88, 1740–1754.
Pytel, M., Mercik, K., and Mozrzymas, J. W. (2005). The voltage dependence of GABAA receptor gating depends on extracellular pH. Neuroreport 16, 1951–1954.
Pytel, M., Mercik, K., and Mozrzymas, J. W. (2006). Membrane voltage modulates the GABA(A) receptor gating in cultured rat hippocampal neurons. Neuropharmacology 50, 143–153.
Pytel, M., and Mozrzymas, J. W. (2006). Membrane voltage differently affects mIPSCs and current responses recorded from somatic excised patches in rat hippocampal cultures. Neurosci. Lett. 393, 189–193.
Pytel, M., Wojtowicz, T., Mercik, K., Sarto-Jackson, I., Sieghart, W., Ikonomidou, C., and Mozrzymas, J. W. (2007). 17 beta-estradiol modulates GABAergic synaptic transmission and tonic currents during development in vitro. Neuropharmacology 52, 1342–1353.
Rogers, C. J., Twyman, R. E., and Macdonald, R. L. (1994). Benzodiazepine and beta-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J. Physiol. 475, 69–82.
Rudolph, U., and Mohler, H. (2004). Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu. Rev. Pharmacol. Toxicol. 44, 475–498.
Rudolph, U., and Mohler, H. (2006). GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr. Opin. Pharmacol. 6, 18–23.
Rusch, D., and Forman, S. A. (2005). Classic benzodiazepines modulate the open-close equilibrium in alpha1beta2gamma2L gamma-aminobutyric acid type A receptors. Anesthesiology 102, 783–792.
Schofield, C. M., and Huguenard, J. R. (2007). GABA affinity shapes IPSCs in thalamic nuclei. J. Neurosci. 27, 7954–7962.
Scimemi, A., and Beato, M. (2009). Determining the neurotransmitter concentration profile at active synapses. Mol. Neurobiol. 40, 289–306.
Scimemi, A., Tian, H., and Diamond, J. S. (2009). Neuronal transporters regulate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J. Neurosci. 18, 14581–14595.
Sensi, S. L., Paoletti, P., Bush, A. I., and Sekler, I. (2009). Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 10, 780–791.
Sigel, E., Baur, R., Trube, G., Mohler, H., and Malherbe, P. (1990). The effect of subunit composition of rat brain GABAA receptors on channel function. Neuron 5, 703–711.
Sigel, E., and Buhr, A. (1997). The benzodiazepine binding site of GABAA receptors. Trends Pharmacol. Sci. 18, 425–429.
Slomianka, L. (1992). Neurons of origin of zinc-containing pathways and the distribution of zinc-containing boutons in the hippocampal region of the rat. Neuroscience 48, 325–352.
Snyder, S. H., Banerjee, S. P., Yamamura, H. I., and Greenberg, D. (1974). Drugs, neurotransmitters, and schizophrenia. Science 184, 1243–1253.
Spruston, N. (2008). Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221.
Swanwick, C. C., Murthy, N. R., Mtchedlishvili, Z., Sieghart, W., and Kapur, J. (2006). Development of gamma-aminobutyric acidergic synapses in cultured hippocampal neurons. J. Comp. Neurol. 495, 497–510.
Szabadics, J., Tamas, G., and Soltesz, I. (2007). Different transmitter transients underlie presynaptic cell type specificity of GABAA, slow and GABAA, fast. Proc. Natl. Acad. Sci. U.S.A. 104, 14831–14836.
Thomas, C. G., Tian, H., and Diamond, J. S. (2011). The relative roles of diffusion and uptake in clearing synaptically released glutamate change during early postnatal development. J. Neurosci. 31, 4743–4754.
Toone, B. K., and Fenton, G. W. (1977). Epileptic seizures induced by psychotropic drugs. Psychol. Med. 7, 265–270.
Trigo, F. F., Papageorgiou, G., Corrie, J. E., and Ogden, D. (2009). Laser photolysis of DPNI-GABA, a tool for investigating the properties and distribution of GABA receptors and for silencing neurons in situ. J. Neurosci. Methods 181, 159–169.
Tyagarajan, S. K., and Fritschy, J. M. (2010). GABA(A) receptors, gephyrin and homeostatic synaptic plasticity. J. Physiol. 588, 101–106.
Tyagarajan, S. K., Ghosh, H., Yevenes, G. E., Nikonenko, I., Ebeling, C., Schwerdel, C., Sidler, C., Zeilhofer, H. U., Gerrits, B., Muller, D., and Fritschy, J. M. (2011). Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. U.S.A. 108, 379–384.
Tzingounis, A. V., and Wadiche, J. I. (2007). Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci. 8, 935–947.
Ventriglia, F., and Maio, V. D. (2003). Synaptic fusion pore structure and AMPA receptor activation according to Brownian simulation of glutamate diffusion. Biol. Cybern. 88, 201–209.
Verdoorn, T. A. (1994). Formation of heteromeric gamma-aminobutyric acid type A receptors containing two different alpha subunits. Mol. Pharmacol. 45, 475–480.
Vorisek, I., and Sykova, E. (2009). Measuring diffusion parameters in the brain: comparing the real-time iontophoretic method and diffusion-weighted magnetic resonance. Acta Physiol. (Oxf.) 195, 101–110.
Wafford, K. A. (2005). GABAA receptor subtypes: any clues to the mechanism of benzodiazepine dependence? Curr. Opin. Pharmacol. 5, 47–52.
Wahl, L. M., Pouzat, C., and Stratford, K. J. (1996). Monte Carlo simulation of fast excitatory synaptic transmission at a hippocampal synapse. J. Neurophysiol. 75, 597–608.
Weiss, D. S., Barnes, E. M. Jr., and Hablitz, J. J. (1988). Whole-cell and single-channel recordings of GABA-gated currents in cultured chick cerebral neurons. J. Neurophysiol. 59, 495–513.
Wisden, W., Korpi, E. R., and Bahn, S. (1996). The cerebellum: a model system for studying GABAA receptor diversity. Neuropharmacology 35, 1139–1160.
Wojtowicz, T., Wyrembek, P., Lebida, K., Piast, M., and Mozrzymas, J. W. (2008). Flurazepam effect on GABAergic currents depends on extracellular pH. Br. J. Pharmacol. 154, 234–245.
Wyrembek, P., Lebida, K., Mercik, K., Szczuraszek, K., Szczot, M., Pollastro, F., Appendino, G., and Mozrzymas, J. W. (2010). Block and allosteric modulation of GABAergic currents by oenanthotoxin in rat cultured hippocampal neurons. Br. J. Pharmacol. 160, 1302–1315.
Zarnowska, E. D., Keist, R., Rudolph, U., and Pearce, R. A. (2009). GABAA receptor alpha5 subunits contribute to GABAA, slow synaptic inhibition in mouse hippocampus. J. Neurophysiol. 101, 1179–1191.
Keywords: GABAergic synaptic transmission, GABA concentration time course at the synapse, GABAA receptors, non-equilibrium conditions, pharmacological modulation of GABAA-currents, modifiers of gating, fast-off competitive antagonists, GABAA receptor modeling
Citation: Barberis A, Petrini EM and Mozrzymas JW (2011) Impact of synaptic neurotransmitter concentration time course on the kinetics and pharmacological modulation of inhibitory synaptic currents. Front. Cell. Neurosci. 5:6. doi: 10.3389/fncel.2011.00006
Received: 18 April 2011;
Paper pending published: 28 April 2011;
Accepted: 05 June 2011;
Published online: 22 June 2011.
Edited by:
Enrico Chelubini, International School for Advanced Studies, ItalyReviewed by:
Marco Beato, University College London, UKAnnalisa Scimemi, National Institutes of Health, USA
Copyright: © 2011 Barberis, Petrini and Mozrzymas. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Andrea Barberis and Jerzy W. Mozrzymas, Laboratory of Neuroscience, Department of Biophysics, Wroclaw Medical University, ul. Chalubińskiego 3, 50-368 Wroclaw, Poland. e-mail:bW96cnp5QGJpb2Zpei5hbS53cm9jLnBs