 Riyuan Zhang
Riyuan Zhang Chong Chen
Chong Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 20 March 2025
Sec. Intestinal Microbiome
Volume 15 - 2025 | https://doi.org/10.3389/fcimb.2025.1532512
This article is part of the Research Topic Advances in Infectious Disease Research: Tackling Antimicrobial Resistance, Zoonoses, and More View all 4 articles
Background: Pigmented gallstone disease (PGS) is prevalent in China. Biliary microbiota is certified to be related to the PGS formation.
Methods: We performed 16S sequencing on both bile and gallstone samples in 16 patients with gallstone disease. We analyzed the microbial composition of the biliary tract and identified crucial bacteria related to the formation of PGS.
Results: Biliary tract bacterial composition analysis showed heterogeneity of dominated genus among individuals and correlation in bacterial composition between bile and gallstones. We screened 10 prevalent genera with significant abundance in the bile and gallstones. Actinomyces, Streptococcus, and Achromobacter had a significantly higher abundance in gallstones than in bile (P < 0.05). Furthermore, we identified 32 species that harbored uidA, pldA, and plc genes that encoded β-glucuronidase or phospholipase. Finally, we observed an enriched membrane transport for bile resistance through biliary microbiota.
Conclusion: β-glucuronidase-producing Streptococcus spp., including Streptococcus dysgalactiae and Streptococcus agalactiae, and Parabacteroides merdae, harbored both uidA and pldA genes and were found to be crucial bacterial species in PGS formation.
Gallstone disease is prevalent in China, especially pigmented gallstones (PGS). With age, the prevalence of gallstone disease reaches 16% and exceeds 20% in some regions (Su et al., 2020). Previous studies have linked biliary infection with gallstone development and indicated that bacteria may act as the nucleating factor initiating the formation of PGS (Stewart et al., 1987; Lee et al., 2010; Kose et al., 2018). Many bacteria, such as Escherichia coli, Klebsiella pneumoniae, Enterococcus faecium, Enterobacter cloacae, and Pseudomonas aeruginosa, have been identified in bile or gallstones through cultivation or polymerase chain reaction. Studies have shown a high prevalence of the phyla Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria in the primary choledocholithiasis group (Chen et al., 2019).The microbiome is involved in the pathogenesis of cholelithiasis through several pathways (Dan et al., 2023). The bacterial products, β-glucuronidase and phospholipase, and bacterial biofilm are the key points. Helicobacter species induce gallstone formation by precipitating calcium. Therefore, an improved understanding of the biliary microbiota would be helpful for studies on bacteria-related gallstone pathogenesis and the formation of PGS. In this study, based on 16S sequencing, we reconstructed the composition of biliary microbiota, identified bacteria related to PGS formation, and planned to further establish the relationship between biliary microbiota and PGS formation.
The study conformed to the ethical guidelines of the 1975 Declaration of Helsinki, and all individuals provided written informed consent upon enrollment. This study was approved by the Medical Ethics Committee of Pingyang County People’s Hospital. Sixteen individuals (average age: 69.00 ± 10.32 years; 6 women, 10 men) who were diagnosed with cholangitis and choledocholithiasis using B-mode ultrasonography and computed tomography were enrolled. Individuals 1, 4, 5, 7, and 14 had a history of surgery on the digestive system, and individuals 10, 13, 15, and 16 had a history of surgery on the biliary system for more than 4 years. The participants underwent choledochoscopy, choledocholithotomy, and lithotripsy at this time and 5 mL bile samples and brown GBS samples were collected from the common bile ducts simultaneously. The procedure for sample collection was performed under strict sterile conditions. Samples were immediately placed in sterile centrifuge tubes separately and stored at -80°C for subsequent study.
The surface of the gallstone was washed with phosphate-buffered saline. Each gallstone was cut from the center using a sterile blade and scraped to obtain the inner matrix. After grinding and drying to a constant weight, 200 mg of gallstone powder was incubated with 1 mL of 1% sodium lauryl sulfate at room temperature overnight. Lithium chloride (7 mol/L) was added to give a final concentration of 1.5 mol/L. The mixture was shaken for 1 minute, centrifuged at 12,000 g for 15 minutes, and the supernatant separated. The DNA was then extracted twice with an equal volume of saturated phenol and once with an equal volume of chloroform-isoamyl alcohol. A QIAamp DNA Mini kit (QIAGEN, Germany) was used for further purification.
A 500 µL volume of bile was centrifuged at 12,000 g for 10 minutes and the filtrate was collected. Added to the filtrate was an equal volume of lysis buffer [10 mM Tris-Hcl (pH = 8.5), 10 mM EDTA, 100 mM NaCl, 0.5% sodium lauryl sulfate) and 50 µL of proteinase K and this was incubated at 55°C for 8 hours. A QIAamp DNA Mini kit (QIAGEN, Germany) was used for further purification.
A two-step procedure for 16S rRNA library construction was used (Shen et al., 2015). The V3-V4 regions of the 16S rRNA gene cover a longer segment of the 16S rRNA, including two hypervariable regions (V3 and V4), which leads to improved bacterial classification resolution. The V3-V4 regions of the bacterial 16S rRNA gene were amplified using universal primer pairs 341F (5’- ACTCCTACGGGAGGCAGCA-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’). Sterile water was used as a negative control during the whole amplification and library preparation process. The 16S rRNA libraries were sequenced on the Illumina NovaSeq PE250 platform (Illumina, USA) to generate 2 × 250-bp paired-end reads.
The Qiime 2 analysis process was adopted and Divisive Amplicon Denoising Algorithm 2 (DADA2) was used to denoise the raw data, which was equivalent to clustering with 100% similarity. Furthermore, we used Qiime 2’s built-in DADA2 for low-quality filtering operations such as adapters, primer removal, and denoising by executing truncQ=2, F end maxEE=5, and R end maxEE=2. Finally, we used DADA2 to merge the double-ended sequences and de-chimerism to obtain clean reads.
Sequence data analysis mainly used Qiime 2 (Qiime 2-2020.2) and R (4.0.2). The denoised sequences were directly made de-redundant to obtain the feature information using DADA2. According to the silva-132-99 database and the primer pairs of the V3-V4 regions, Qiime2 was used to trim the reference genome’s, Silva132, 99% features to the V3-V4 region and train the classifier. The trained naive Bayes classifier was used to classify the features to obtain the corresponding species classification information. Each feature was defined as an amplicon sequence variant (ASV). Qiime 2 was used to calculate the alpha diversity index (Chao1, ACE, Shannon’s, and Simpson’s indices). R was used to perform the alpha diversity analysis between groups based on the Wilcoxon test (R phyloseq package). Principal coordinates analysis (PCoA) was conducted based on the Bray–Curtis distance (phyloseq/vegan package). ANOSIM analysis (R phyloseq/vegan package), Pearson’s correlation analysis, and analysis of differential bacteria based on the Wald test (R DESeq2 package) were conducted between groups. We used PICRUSt2 (https://github.com/picrust/picrust2) to predict the metabolic function of the flora by searching the KEGG GENOME Database and KEGG PATHWAY Database. P < 0.05 or adjusted P < 0.05 was considered to be statistically different.
Bile samples of individuals 9 and 14 failed to produce nucleic acid extract. We performed high throughput sequencing of the 16S rRNA V3-V4 regions of bacteria in 14 bile samples (group A) and 16 gallstone samples (group B), obtaining average raw reads of 177,548.50 ± 37,669.28 in group A and 147,736.69 ± 58,253.30 in group B. After multiple filtrations, the number of final clean reads was 144,080.70 ± 35,753.81 in group A and 131,720.90 ± 51,808.84 in group B. Furthermore, we performed species classification annotations using feature information generated by DADA2, with 43 to 1,142 ASVs obtained in group A and 43 to 1,392 ASVs obtained in group B. The number of ASVs among 16 samples in the group was quite different. After filtering out low-abundance ASVs (<10), a total of 813 ASVs were shared by the two groups, and the number of unique ASVs in group A and in group B was 1,615 and 1,142, respectively.
Diversity differences in bile microbiota and gallstone microbiota were observed. Analysis using the Chao1, ACE, Shannon’s, and Simpson’s indices indicated that the gallstone microbiota had a reduced bacterial diversity compared with the bile microbiota, without a statistical difference (P > 0.05) (Figure 1a). Wilcoxon rank sum test analysis based on the Bray–Curtis algorithm found that there was no statistical difference in the beta diversities index between the two groups (P = 0.321) (Figure 1b). PCoA indicated a similar microbial composition (Figure 1c). Rare faction curves showed the abundance and uniformity of the two groups (Figure 1d).
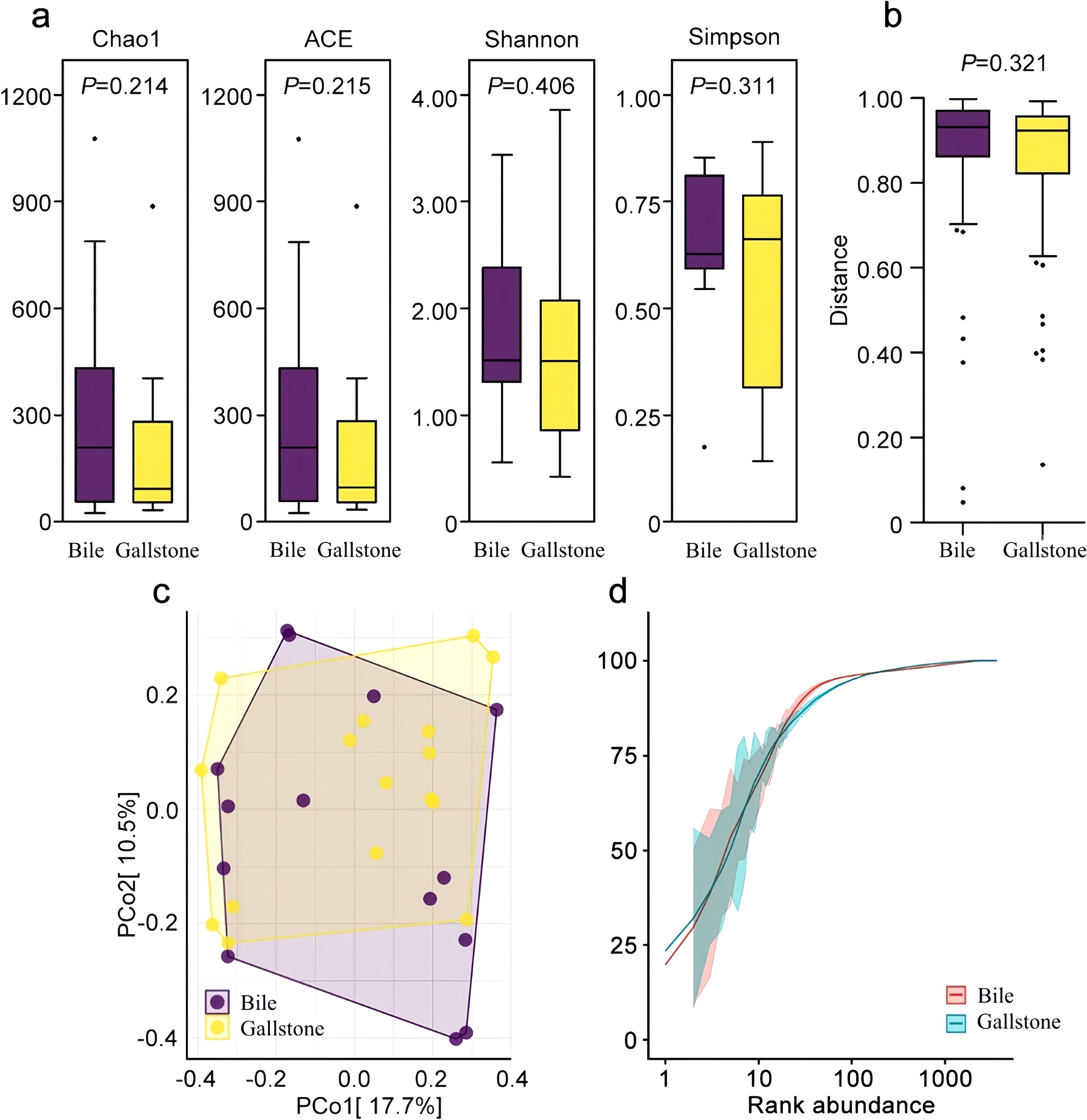
Figure 1. Microbial community characteristics of the bile and gallstone samples. (a) Four indexes of alpha diversity. Purple and yellow boxplots denote distributions of 16S sequencing of bile samples and gallstone samples, respectively. (b) Beta diversities based on the Wilcoxon rank sum test. (c) Beta diversities are presented as a PCoA matrix diagram. (d) Rare faction curves of the two groups.
Generally speaking, according to the abundance ranking of ASVs, Proteobacteria (14.78%), Firmicutes (9.66%), Bacteroidota (2.54%), Synergistota (1.97%), Actinobacteriota (0.51%), Fusobacteriota (0.17%), and Campilobacterota (0.09%) were the top seven phyla detected in the biliary tract samples. At the genus level, Escherichia Shigella (9.90%), Enterococcus (5.35%), Bacteroides (2.05%), Pseudomonas (1.97%), Clostridium sensu stricto 13 (1.79%), Pyramidobacter (1.72%) and a genus of Enterobacteriaceae (1.59%) were the top seven genera detected in the biliary tract samples. In this study, a total of 259 bacterial species were identified. Bacteroides fragilis (2.00%), Pyramidobacter piscolens (1.97%), Streptococcus dysgalactiae (0.36%), Streptococcus anginosus (0.15%), Campylobacter concisus (0.07%), Lactobacillus kefiranofaciens (0.07%), Actinomyces gerencseriae (0.05%), Vibrio fluvialis (0.05%), Bifidobacterium animalis (0.04%), Helicobacter typhlonius (0.01%), and Bacteroides sartorii (0.01%) had a relative abundance of more than 0.01%.
Biliary tract bacterial composition analysis showed heterogeneity among individuals. Individuals 4, 10, and 12 were dominated by Escherichia Shigella in both their bile and gallstone samples (over 90%), and followed by 3% to 5% of Pseudomonas. Enterococcus dominated in the bile and gallstone samples of individual 15, and only in the gallstone sample of individual 1, with a proportion of 90% approximately. Individual 13 had a large proportion of Pyramidobacter (bile sample, 29.19%; gallstone sample, 77.85%) and Hungatella (bile sample, 36.54%; gallstone sample, 5.39%). Pyramidobacter only dominated in the gallstone sample of individual 11 (88.01%) and not in their bile sample (0.31%). Individual 5 was dominated by Escherichia ShigellaI (bile sample, 25.70%; gallstone sample, 63.86%) and Clostridium sensu stricto 13 (bile sample, 67.59%; gallstone sample, 33.51%), and individual 2 had the same dominating bacteria in their bile sample (Escherichia ShigellaI, 23.65%; Clostridium sensu stricto 13, 75.51%) but not in their gallstone sample (Escherichia ShigellaI, 11.06%; Clostridium sensu stricto 13, 0.00%). Individuals 6, 8, and 16 had a relative abundance of Bacteroides (6, 39.24%; 8, 77.76%; 16, 76.13%) and Pseudomonas (6, 16.00%; 8, 15.03%; 16, 7.32%) in their bile samples, however, the bacteria were significantly different in their gallstone samples, dominated by Escherichia Shigella (6, 59.98%), Actinomyces (8, 30.00%), and Erysipelatoclostridium (16, 53.92%), respectively (Figure 2).
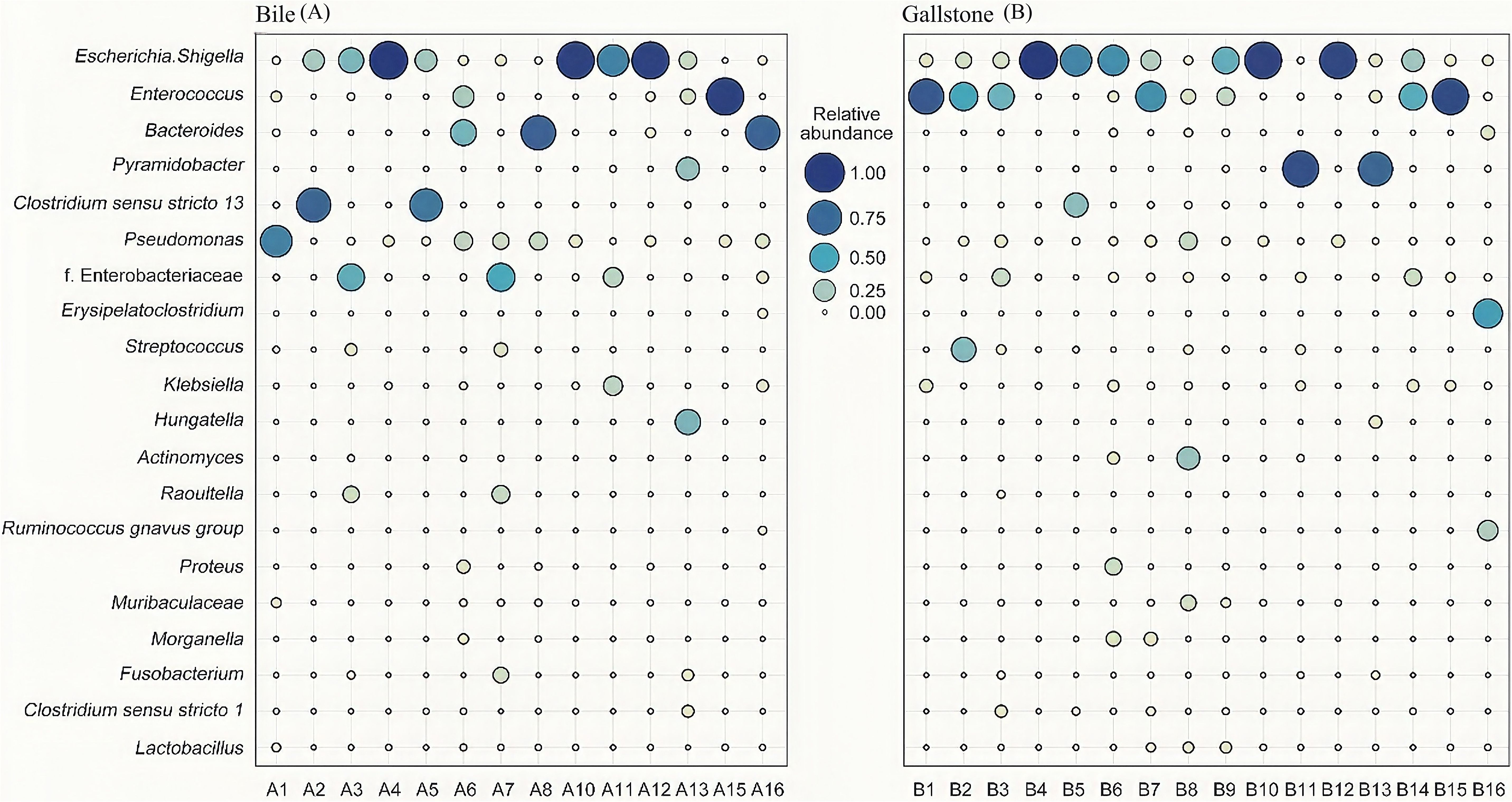
Figure 2. Microbial community characteristics generated by 16S sequencing. (A, B) Relative abundances of top 10 genera in the bile and gallstone samples. Lack of A9 and A14 due to failure of nucleic acid extraction. The genera names for (A, B) are shown on the y-axis of (A). f. Enterobacteriaceae represented an undefined genus of Enterobacteriaceae.
We did not observe a correlation between biliary microbiota and a history of previous surgery. The ANOSIM analysis showed that the difference in biliary microbial composition between the groups was slightly greater than the difference within the groups, without a statistical difference (R = 0.026, P = 0.227). Bacterial microbiota in the bile and gallstones within individuals exhibited a high correlation (P < 0.001 for all), except for individuals 1 (r = 0.050, P = 0.098), 2 (r = 0.051, P = 0.090), 7 (r = 0.055, P = 0.071), and 11 (r = 0.022, P = 0.475). In general, the microbial composition of bile and gallstones was the same. Thus, differential bacteria may be a key factor in PGS formation.
Although most genera that occurred in bile were also detected in gallstones, the abundance and prevalence of these bacteria differed greatly. Based on the Wald test, a total of 128 genera showed a statistical difference in abundance between bile and gallstones (P < 0.05). Twenty-nine genera were observed to be more abundant in gallstones than in bile.
Among them, 10 prevalent genera identified in at least 15/30 samples were selected, with a 4.23 to 29.19-fold difference between groups (Figure 3). Three genera (Actinomyces, Streptococcus, and Achromobacter) had a significantly higher abundance in gallstones than in bile. With respect to these 10 genera prevalent among individuals, ADurb.Bin063.1 was detected in 10 bile samples and in only 5 gallstone samples, with a statistical difference (χ2 = 4.821, P = 0.028). The prevalence of the other nine genera was not statistically different (P > 0.05).
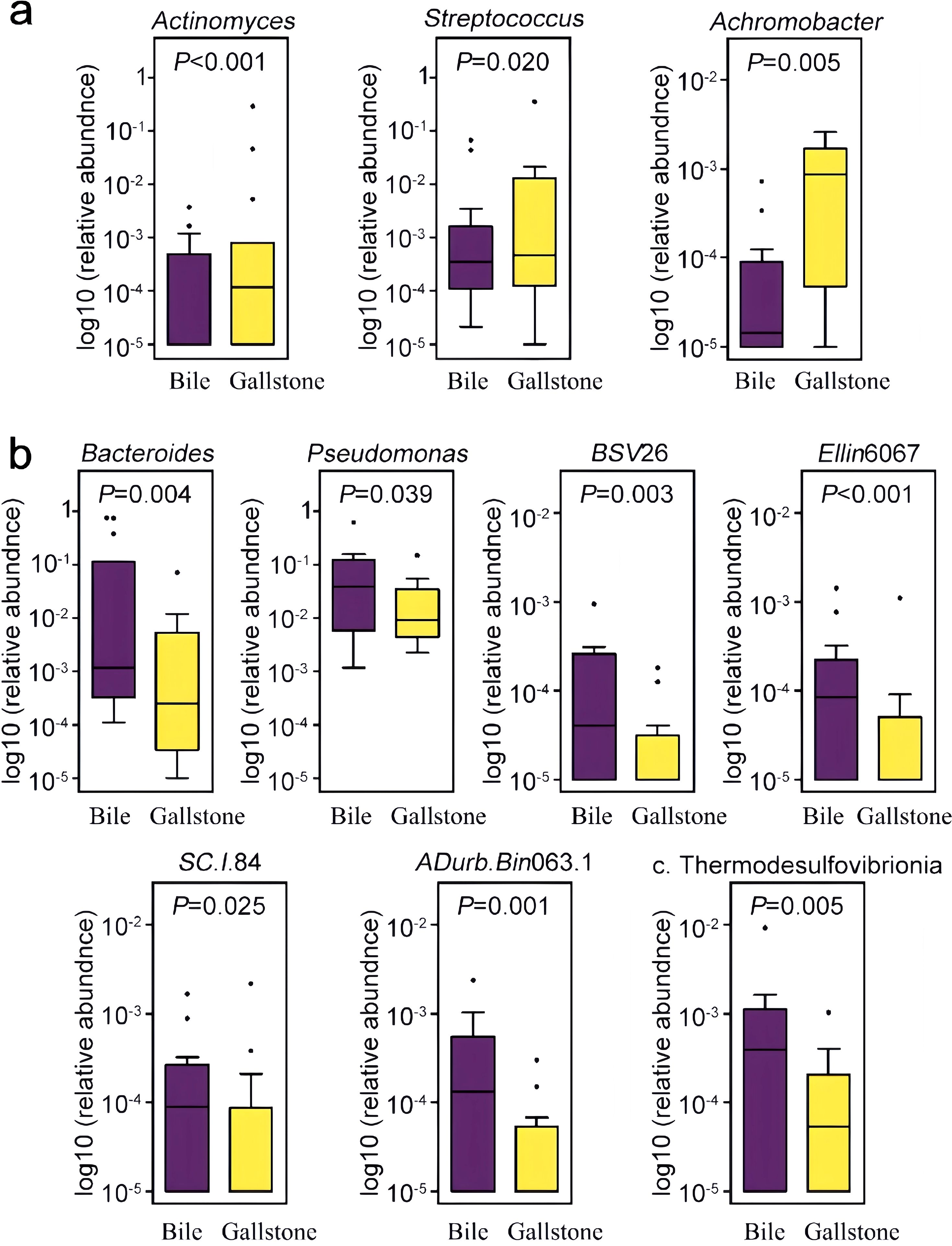
Figure 3. Distribution of differentially abundant genera in the bile and gallstone samples. Genera that were statistically increased (a) and statistically decreased (b) in relative abundance in the gallstone samples compared to bile samples are illustrated. Genera were filtered by the criterion of detection in at least 50% (15/30) of the samples. BSV26, a member of the Kryptoniales order; Ellin6067, a member of the Nitrosomonadaceae family; SC.I.84, a member of the Burkholderiales order; ADurb.Bin063.1, a member of the Pedosphaeraceae family; c. Thermodesulfovibrionia, an uncultured genera of the Thermodesulfovibrionia class.
We further investigated genes that might be related to PGS formation. Previous studies have demonstrated potential associations between PGS formation and bacterial products β-glucuronidase and phospholipase. Among the 259 identified biliary species, we extracted 32 species harboring genes encoding these enzymes by searching the KEGG GENOME database. Thus, 10 species were predicted to have the uidA gene encoding β-glucuronidase. All of the 10 species were detected in bile, and only 5 of the 10 species were detected in gallstones. The pldA gene was more abundant in bacteria in bile. In total, 20 species harbored the pldA gene encoding phospholipase A1/A2, with 12 of 20 species detected in bile and 15 of 20 species detected in gallstones. Parabacteroides merdae possessed both the uidA and pldA genes. Six species harbored the plc gene encoding phospholipase C (Figure 4).
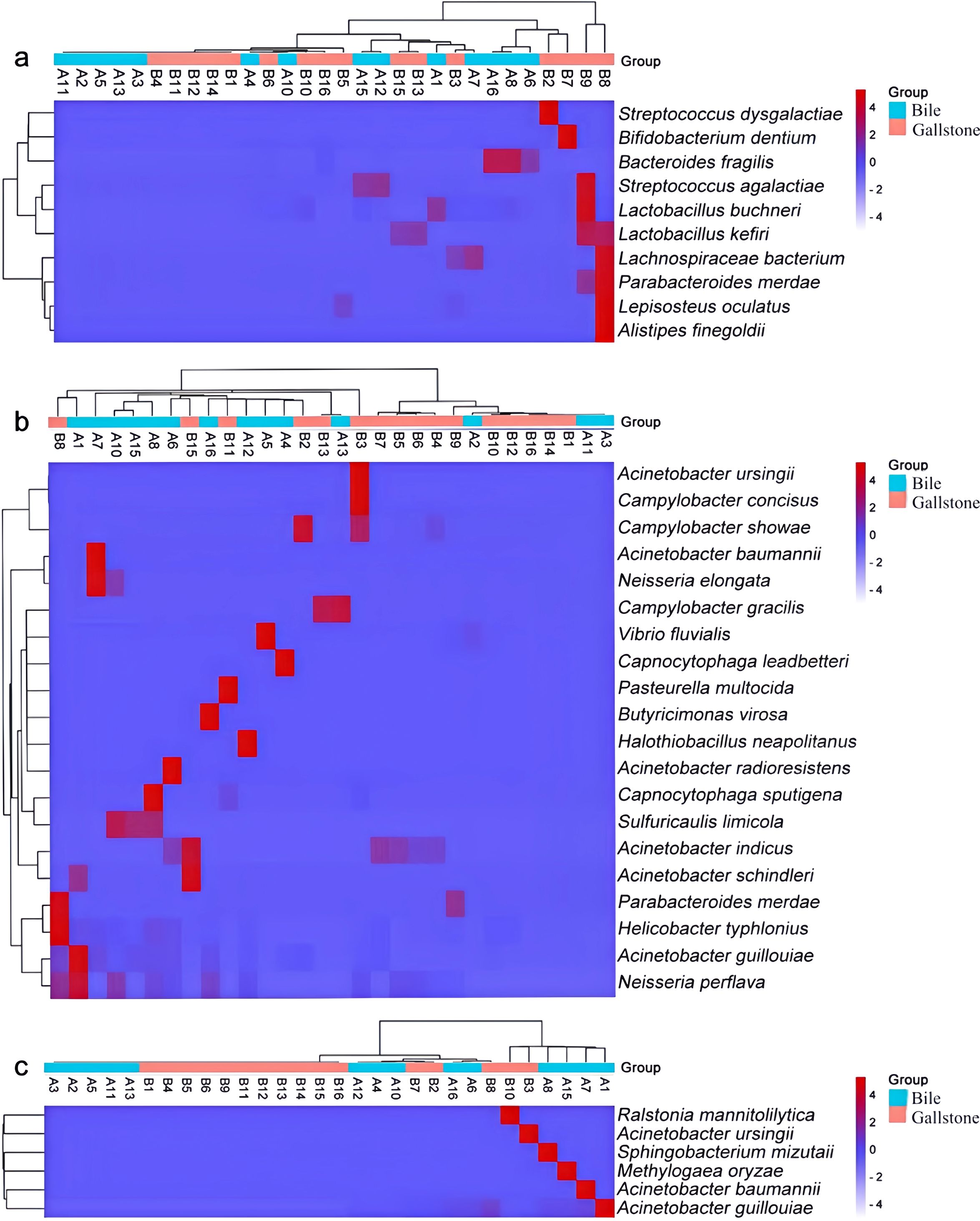
Figure 4. Heatmap with the crucial bacterial species harboring the uidA (a), plcA (b), and plc (c) genes. The heatmap color scale quantifies the log10 average relative abundance of species in the biliary tract, from purple (none or low abundance) to red (high abundance). The dendrograms illustrate the relationship between samples and groups, clustered by the Wald test.
Additionally, we used PICRUSt2 to predict the metabolic function of the biliary tract flora and clustered them based on the KEGG PATHWAY database. Metabolism was the basic life activity of biliary microbiota. There was an overall higher abundance of genes involved in carbohydrate metabolism than in amino acid metabolism, consistent with the report of Kose et al. (2018). Enriched membrane transport pathways maintain the exchange of substances between the bacterial organism and biliary environment, and were predicted to probably promote bile resistance (Figure 5).
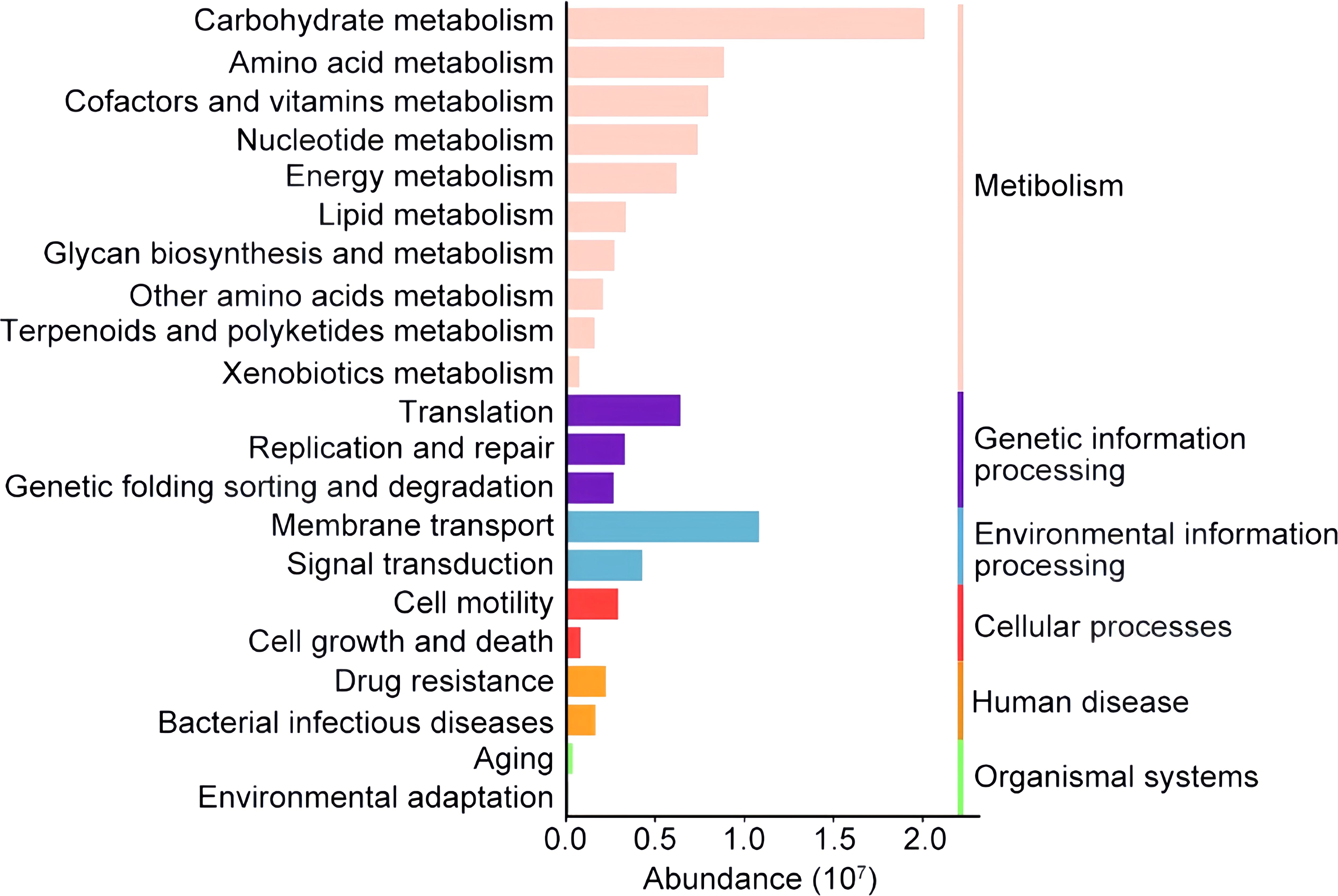
Figure 5. Histogram of the major functional pathways identified in the biliary tract of patients with PGS. The colored column showed the relative abundance of the metabolic pathway categories of microbiota in the biliary tract. The metabolic pathways are clustered according to the KEGG PATHWAY database on the right. Only the top 21 metabolic pathways are shown here.
Studies have identified bacteria in the majority of patients with PGS and conclude that bacteria play an important structural and functional role in the formation of PGS (Stewart et al., 1987; Wu et al., 2013; Shen et al., 2015). Bacteria detected in bile and gallstones frequently occur in the digestive tract. Ye et al. (2016) compared bacterial communities from the biliary tract and upper digestive tract of patients with gallstones and found that the biliary bacteria probably originated from a retrograde infection of gut bacteria. We found that all of the top seven genera in the biliary tract were intestinal microbiota, which explains the connection between biliary tract flora and intestinal flora (Kamada et al., 2013; Lin et al., 2018). Although bacteria come from the digestive tract, biliary microbiota have unique flora characteristics.
The dominant microbiota in the gut and intestines are abundant and uniform (Ruan et al., 2017; Lin et al., 2018). Different from them, the dominant biliary microbiota is relatively single and commonly dominated by several genera, generally one to three genera. Studies have reported that Escherichia Shigella and Enterococcus are most frequent whether detected by bile culture or high-throughput sequencing (Kaufman et al., 1989; Dyrhovden et al., 2020). The dominance was only found in the gallstone samples (12/16), but not in the bile samples (6/14) in this study. A consensus has not yet been reached on which type of microbiota is common to PGS. We found that Pyramidobacter, Hungatella, Clostridium sensu stricto 13, Bacteroides, and Pseudomonas comprise a relative proportion of genera in biliary tract in some individuals, which revealed that the microbial composition of the biliary tract exhibited heterogeneity (Wu et al., 2013; Ye et al., 2016). We did not observe a correlation between biliary tract flora and a history of previous surgery. Thus, the heterogeneity implies that the biliary microbiota may be shaped by more complicated factors, such as diet, lifestyle, or host immune responses. The possibility of accidental deviation in the reflux of biliary microbiota from the gut or duodenum cannot be ruled out. This difference may also be due to altered bile microbiota following stone nucleation. The microbiota in gallstones behaves like the bile microbiota ecology at a certain stage of stone formation. However, we found that the microbial composition of bile and gallstones was correlated at the time of detection, so the changes in bile during gallstone formation may not be as dramatic as previously thought in most individuals. It is possible that some bacteria were accidentally encapsulated in the stone during gallstone formation, resulting in the heterogeneity of the dominant genera.
There is no doubt that the microorganisms of gallstones are derived from bile. When microorganisms metabolize substances in bile, such as bilirubin, it promotes the deposition of bilirubin in bile. Nearby bacteria involved in bilirubin deposition may be adsorbed on the bilirubin crystals and then deposited together to form gallstones. Therefore, exploring the differential bacteria between gallstones and bile can clarify the microorganisms that are active during gallstone formation and those that are more associated with PGS formation. We confirmed a significant change in biliary microbiota between bile and gallstones. The prevalence of the genera Actinomyces, Streptococcus, and Achromobacter was observed to be more abundant in gallstones than in bile. Actinomyces infection can cause an exceedingly rare actinomycosis characterized by a tendency to form cholecystitis and abdominal abscesses (Vyas et al., 2007). Potti et al. (1999) reported a case of gallbladder carcinoma associated with biliary actinomycosis. Studies (Frey and Freter, 1968; Miquel et al., 1998) have shown that cholecystitis is not critical for the formation of gallstones, so it is speculated that the formation of gallstones and excessive overgrowth of Actinomyces may actually be the causes of cholecystitis or cholangitis. Achromobacter, a pigment-producing species, was detected in the bile of patients with benign and malignant pancreaticobiliary diseases (Poudel et al., 2023). Achromobacter catalyze the oxidation of L- tryptophan, yielding a red pigment, and probably play a key role in the color staining of brown gallstones (Krishnamurthi et al., 1969). Hancke and Marklein (1983) detected β-glucuronidase-producing group G Streptococcus in the bile, gallbladder wall, and livers of mongrel dogs with bile pigment gallstones. Previous studies also demonstrated that β-glucuronidase, a metabolite of Streptococcus, plays an important role in PGS formation (Wang et al., 2018). The deconjugation of β-glucuronidase can precipitate calcium bilirubinate which is conjugated with anionic glycoprotein, and cause the agglomeration of calcium bilirubinate crystals into stones (Maki, 1966). Thus, the overgrowth of Streptococcus, including Streptococcus dysgalactiae and Streptococcus agalactiae, may contribute to the gallstone formation in this study.
β-glucuronidase and phospholipase accelerate the precipitation of calcium bilirubinate. Some other β-glucuronidase and phospholipase-producing bacteria were detected in biliary tracts. β-glucuronidase induces the hydrolysis of bilirubin diglucuronides to produce unconjugated bilirubin, resulting in the precipitation of calcium bilirubinate (Osnes et al., 1997). Phospholipase-producing bacteria were abundant in the bile samples, contributing to the PGS formation. Studies prove that brown PGS contains calcium palmitate (Sharma et al., 2015). Bacterial phospholipase on biliary phosphatidylcholine can result in the release of palmitic acid, which then combines with ionized calcium to form calcium palmitate. The precipitation of calcium palmitate produces solids that may be incorporated into the stones. Nearly one-third of the cultured strains of cholesterol gallstones secrete β-glucuronidase and phospholipase (Hu et al., 2022). Parabacteroides merdae harbored both uidA and pldA genes, encoded these two enzymes, and played a crucial role in PGS formation. In addition, Parabacteroides merdae is involved in bile acid metabolism to produce isallo-lithocholic acid. This derivative exerts a potent antibacterial effect against gram-positive multidrug-resistant pathogens in bile (Stewart et al., 1987), which may cause the enrichment of gram-negative bacteria in PGS (Kose et al., 2018). Extracellular polymeric substances produced by these gram-negative bacteria exhibit anionic properties that enable calcium and magnesium ions to bind with polymer strands, providing a tightly bound biofilm architecture outside stones (Donlan, 2002).
Biliary microbiota are known to survive in a variety of extreme environments, including those containing sodium and potassium salts, bile acids, cholesterol, phospholipids, commonly used antibiotic drugs, and heavy metals. A variety of genes are involved in bile resistance to protect bacteria from the toxic agents in bile, such as multidrug export efflux pumps, DNA and cell wall repair proteins (Kose et al., 2018). In this study, we observed an enhanced membrane transportation that can transfer harmful substances outside bacteria, avoiding burden and damage. At the same time, pathways such as DNA repair were also highly abundant in biliary microbiota.
While our research has yielded several significant findings, it is essential to acknowledge certain limitations that warrant attention in future studies. Notably, the PCoA revealed that the gallstone and bile samples were not distinctly separated. This observation indicates that there may be factors not fully accounted for that could have influenced the differences between these samples. For example, the gallstone samples may have been contaminated by bile, or vice versa, leading to potential overlap that obscures the distinctions between the two sample types. Additionally, the similarity in microbial communities or chemical compositions between the gallstone and bile samples may have further contributed to their overlap in the PCoA plot. The next step will involve investigating whether this overlap arises from intra-patient variability or if it necessitates the development of visualizations for paired data. Moreover, the relatively small sample size of this investigation may not adequately represent the broader population of patients with gallstones. This limitation could result in biased outcomes and may overlook differences among various demographic groups. Furthermore, a small sample size may reduce statistical power, increasing the likelihood of undetected important biological effects. To validate and expand upon our findings, future research should pursue several key directions: conducting multi-center, large-sample studies to enhance the representativeness and reliability of the results.
In summary, we initially obtained the microbial community of the bile and gallstone samples and identified species that may play a role in the formation of PGS. β-glucuronidase-producing Streptococcus was enriched in gallstones and correlated with PGS formation. Furthermore, we identified 32 crucial species that harbored uidA, pldA, or plc genes encoding β-glucuronidase or phospholipase colonization in the biliary tract. The identification of PGS formation-related bacterial species provides a foundation for further research on the formation and treatment of PGS.
The data presented in our study have been deposited in the NCBI repository under the accession number PRJNA1236259.
The studies involving humans were approved by Medical Ethics Committee of Pingyang County People’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. An ethics statement is not applicable because this study is based exclusively on published literature.
RZ: Conceptualization, Writing – original draft, Writing – review & editing. CC: Conceptualization, Writing – original draft. SZ: Formal analysis, Investigation, Writing – original draft. JZ: Methodology, Writing – original draft. WC: Resources, Writing – original draft. ZC: Supervision, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
PGS, Pigmented gallstone disease; PCoA, Principal Co-ordinates Analysis.
Chen, B., Fu, S. W., Lu, L., Zhao, H. (2019). A preliminary study of biliary microbiota in patients with bile duct stones or distal cholangiocarcinoma. BioMed. Res. Int. 2019, 1092563. doi: 10.1155/2019/1092563
Dan, W. Y., Yang, Y. S., Peng, L. H., Sun, G., Wang, Z. K. (2023). Gastrointestinal microbiome and cholelithiasis: Current status and perspectives. World J. Gastroenterol. 29, 1589–1601. doi: 10.3748/wjg.v29.i10.1589
Donlan, R. M. (2002). Biofilms: microbial life on surfaces. Emerg. Infect. Dis. 8, 881–890. doi: 10.3201/eid0809.020063
Dyrhovden, R., Øvrebø, K. K., Nordahl, M. V., Nygaard, R. M., Ulvestad, E., Kommedal, Ø. (2020). Bacteria and fungi in acute cholecystitis. A prospective study comparing next generation sequencing to culture. J. Infect. 80, 16–23. doi: 10.1016/j.jinf.2019.09.015
Frey, C. F., Freter, R. (1968). Gallstone formation in the conventional mouse. The role of bacteria. Am. J. Surg. 116, 868–871. doi: 10.1016/0002-9610(68)90456-x
Hancke, E., Marklein, G. (1983). Experimental gallstone formation. Etiological significance of beta-glucuronidase producing bacteria and biliary obstruction. Langenbecks Arch. Chir. 359, 257–264. doi: 10.1007/bf01257312
Hu, H., Shao, W., Liu, Q., Liu, N., Wang, Q., Xu, J., et al. (2022). Gut microbiota promotes cholesterol gallstone formation by modulating bile acid composition and biliary cholesterol secretion. Nat. Commun. 13, 252. doi: 10.1038/s41467-021-27758-8
Kamada, N., Chen, G. Y., Inohara, N., Núñez, G. (2013). Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 14, 685–690. doi: 10.1038/ni.2608
Kaufman, H. S., Magnuson, T. H., Lillemoe, K. D., Frasca, P., Pitt, H. A. (1989). The role of bacteria in gallbladder and common duct stone formation. Ann. Surg. 209, 584–591. doi: 10.1097/00000658-198905000-00011
Kose, S. H., Grice, K., Orsi, W. D., Ballal, M., Coolen, M. J. L. (2018). Metagenomics of pigmented and cholesterol gallstones: the putative role of bacteria. Sci. Rep. 8, 11218. doi: 10.1038/s41598-018-29571-8
Krishnamurthi, V. S., Buckley, P. J., Duerre, J. A. (1969). Pigment formation from L-tryptophan by a particulate fraction from an Achromobacter species. Arch. Biochem. Biophys. 130, 636–645. doi: 10.1016/0003-9861(69)90081-2
Lee, J. W., Lee, D. H., Lee, J. I., Jeong, S., Kwon, K. S., Kim, H. G., et al. (2010). Identification of helicobacter pylori in gallstone, bile, and other hepatobiliary tissues of patients with cholecystitis. Gut Liver. 4, 60–67. doi: 10.5009/gnl.2010.4.1.60
Lin, L., Wen, Z. B., Lin, D. J., Dong, J. T., Jin, J., Meng, F. (2018). Correlations between microbial communities in stool and clinical indicators in patients with metabolic syndrome. World J. Clin. Cases. 6, 54–63. doi: 10.12998/wjcc.v6.i4.54
Maki, T. (1966). Pathogenesis of calcium bilirubinate gallstone: role of E. coli, beta-glucuronidase and coagulation by inorganic ions, polyelectrolytes and agitation. Ann. Surg. 164, 90–100. doi: 10.1097/00000658-196607000-00010
Miquel, J. F., Núñez, L., Amigo, L., González, S., Raddatz, A., Rigotti, A., et al. (1998). Cholesterol saturation, not proteins or cholecystitis, is critical for crystal formation in human gallbladder bile. Gastroenterology 114, 1016–1023. doi: 10.1016/s0016-5085(98)70322-1
Osnes, T., Sandstad, O., Skar, V., Osnes, M. (1997). Lipopolysaccharides and beta-glucuronidase activity in choledochal bile in relation to choledocholithiasis. Digestion 58, 437–443. doi: 10.1159/000201480
Potti, A., Chowfin, A., Tight, R. (1999). Actinomyces meyeri, gangrenous cholecystitis, and invasive carcinoma of the gallbladder. Infect. Dis. Clin. Pract. 8, 356–357. doi: 10.1097/00019048-199909000-00017
Poudel, S. K., Padmanabhan, R., Dave, H., Guinta, K., Stevens, T., Sanaka, M. R., et al. (2023). Microbiomic profiles of bile in patients with benign and Malignant pancreaticobiliary disease. PloS One 18, e0283021. doi: 10.1371/journal.pone.0283021
Ruan, L., Wu, D., Li, X., Huang, Q., Lin, L., Lin, J., et al. (2017). Analysis of microbial community composition and diversity in postoperative intracranial infection using high−throughput sequencing. Mol. Med. Rep. 16, 3938–3946. doi: 10.3892/mmr.2017.7082
Sharma, R., Soy, S., Kumar, C., Sachan, S. G., Sharma, S. R. (2015). Analysis of gallstone composition and structure in Jharkhand region. Indian J. Gastroenterol. 34, 29–37. doi: 10.1007/s12664-014-0523-6
Shen, H., Ye, F., Xie, L., Yang, J., Li, Z., Xu, P., et al. (2015). Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci. Rep. 5, 17450. doi: 10.1038/srep17450
Stewart, L., Smith, A. L., Pellegrini, C. A., Motson, R. W., Way, L. W. (1987). Pigment gallstones form as a composite of bacterial microcolonies and pigment solids. Ann. Surg. 206, 242–250. doi: 10.1097/00000658-198709000-00002
Su, Z., Gong, Y., Liang, Z. (2020). Prevalence of gallstone in Mainland China: A meta-analysis of cross-sectional studies. Clin. Res. Hepatol. Gastroenterol. 44, e69–e71. doi: 10.1016/j.clinre.2020.04.015
Vyas, J. M., Kasmar, A., Chang, H. R., Holden, J., Hohmann, E. (2007). Abdominal abscesses due to actinomycosis after laparoscopic cholecystectomy: case reports and review. Clin. Infect. Dis. 44, e1–e4. doi: 10.1086/510077
Wang, Y., Qi, M., Qin, C., Hong, J. (2018). Role of the biliary microbiome in gallstone disease. Expert Rev. Gastroenterol. Hepatol. 12, 1193–1205. doi: 10.1080/17474124.2018.1533812
Wu, T., Zhang, Z., Liu, B., Hou, D., Liang, Y., Zhang, J., et al. (2013). Gut microbiota dysbiosis and bacterial community assembly associated with cholesterol gallstones in large-scale study. BMC Genomics 14, 669. doi: 10.1186/1471-2164-14-669
Keywords: pigment gallstones, β-glucuronidase, Streptococcus, parabacteroides merdae, 16S rRNA sequencing
Citation: Zhang R, Chen C, Zheng S, Zhang J, Chen W and Chen Z (2025) Preliminary study of biliary microbiota and identification of bacterial species associated with pigmented gallstone formation. Front. Cell. Infect. Microbiol. 15:1532512. doi: 10.3389/fcimb.2025.1532512
Received: 22 November 2024; Accepted: 10 February 2025;
Published: 20 March 2025.
Edited by:
Priyanka Bhoj, Temple University, United StatesReviewed by:
Zhengrui Li, Shanghai Jiao Tong University, ChinaCopyright © 2025 Zhang, Chen, Zheng, Zhang, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Chen, cHl6cnkyMDEzQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.