 Benjamin Riocreux-Verney1
Benjamin Riocreux-Verney1 Marie Verneret1,2
Marie Verneret1,2 Rémi Diesler1Christine Dolmazon1Barbara Gineys1
Rémi Diesler1Christine Dolmazon1Barbara Gineys1 Jean-Luc Cadoré1,3
Jean-Luc Cadoré1,3 Jocelyn Turpin1†
Jocelyn Turpin1† Caroline Leroux1*†
Caroline Leroux1*†- 1Viral Infections and Comparative Pathology (IVPC) UMR754, Institut national de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), Universite Claude Bernard Lyon 1, École Pratique des Hautes Études (EPHE), Université Paris Sciences & Lettres (PSL), Lyon, France
- 2Universite Claude Bernard Lyon 1, Laboratory of Biometry and Evolutionary Biology (LBBE), UMR 5558, Centre national de la recherche scientifique (CNRS), VetAgro Sup, Villeurbanne, France
- 3VetAgro Sup, Veterinary Campus of Lyon, Marcy L’Etoile, France
Introduction: ENTV (Enzootic Nasal Tumor Virus) and JSRV (Jaagsiekte Sheep Retrovirus) are β-retroviruses responsible for respiratory cancers in sheep and goats. In this study, we analyzed the genetic features of the sheep and goat β-Retroviruses (29 JSRV and 24 ENTV strains) circulating in France to identify molecular signatures associated with disease severity in flocks.
Methods: We developed a highly specific PCR to amplify and sequence exogenous targeted regions or near full length proviruses based on limited discriminating motifs along their genomes.
Results: The phylogenetic reconstructions based on the Long Terminal Repeat (LTR) and env regions suggest that one major strain is circulating on the French territory for ENTV-1 and ENTV-2 while not clustering with already published Spanish, Canadian or Chinese strains. JSRV strains circulating in French sheep flocks were distributed in 2 distinct genetic clades clustering with sequences originating from North America, Africa and United-Kingdom. JSRV clade I was found to be associated with a higher incidence of cancer in French flocks. Specific motifs spanning the entire JSRV genome particularly in the LTRs and in the intracytoplasmic domain of the envelope were detected between the two genetic subtypes.
Discussion: This work represents the first nationwide study describing the circulation of the three closely related β-oncogenic retroviruses JSRV, ENTV-1 and ENTV-2 in French sheep and goat flocks. Better characterization of strain genetics is a critical step in monitoring circulating - retroviruses, especially those associated with higher cancer incidence in small ruminants.
1 Introduction
JSRV (jaagsiekte sheep retrovirus) and ENTV (enzootic nasal tumor virus) are two oncogenic β-retroviruses responsible for respiratory tumors of the lung and of the nasal mucosa, respectively, in domestic small ruminants. These diseases have been reported in many regions of the world, including Western Europe, Africa, Asia, and North America (York et al., 1992; Bai et al., 1999; DeMartini et al., 2001; Ortín et al., 2003; Caporale et al., 2005; Walsh et al., 2010; Lee et al., 2017; Sid et al., 2018; Nor El Houda et al., 2020; Mao et al., 2022). JSRV-induced cancer is associated with persistent cough, dyspnea, cachexia, and fluid production by the tumoral epithelial cells. Tumors arise from the transformed alveolar and bronchiolar epithelial cells. ENTV type 1 in sheep and type 2 in goats induce tumors by uncontrolled proliferation of nasal epithelial cells, resulting in respiratory distress and nasal discharge, which may be associated with facial deformities and osteolysis. These respiratory cancers lead to rapid death (within a few weeks) after the onset of the first clinical signs; thus, resulting in significant economic losses and a major impact on animal welfare. These viruses are transmitted by air, contact, via colostrum and milk (Grego et al., 2008), and in utero (Caporale et al., 2005).
The envelope (Env) of JSRV and ENTV has been shown to be the major oncogenic determinant. Env is cleaved into the surface (SU) and transmembrane (TM) glycoproteins, the former interacting with the cellular receptor Hyal2 (hyaluronidase 2) for viral entry and the latter responsible for cell transformation in vitro and in vivo (Palmarini et al., 2001; Alberti et al., 2002; Caporale et al., 2006; Wootton et al., 2006; Cousens et al., 2007; Maeda et al., 2021). Through its intracytoplasmic tail, JSRV and ENTV Env deregulates cell proliferation pathways (Protein kinase B (AkT) and Mitogen-activated protein kinase (MAPK)) by interacting with cytoplasmic cellular partners, thereby transforming infected cells (Maeda and Fan, 2008; Monot et al., 2015; Maeda et al., 2021). As shown for JSRV, the cellular protein RaLBP1 interacts with JSRV Env to promote AkT/mTOR activation and cell proliferation (Monot et al., 2015). Of particular importance, a YXXM motif located at the C-terminal end of the cytoplasmic tail is involved in cell transformation in numerous cell types and is critical for tumorigenesis in vivo (Palmarini et al., 2001; Alberti et al., 2002; Liu et al., 2003; Cousens et al., 2007).
Endogenous retroviruses (ERVs), which are closely related to JSRV and ENTVs, are present in high copy numbers in the small ruminant genomes. They result from iterative events of integration of retroviral copies into the germ cells and their vertical transmission to offspring. Endogenous β-retroviruses genetically close to exogenous JSRV and ENTV have been reported in small ruminants (Arnaud et al., 2007; Sistiaga-Poveda and Jugo, 2014; Cumer et al., 2019; Moawad et al., 2024; Verneret et al., 2024) and were initially named enJSRV and enENTV depending on the species from which they were characterized. Their integration into the genome occurred prior to the speciation of Ovis and Capra genera (Arnaud et al., 2007). The history of their endogenization including the role of JSRV and ENTVs in this process remains to be characterized. In contrast to their exogenous counterparts, the ERV envelope is not oncogenic in vitro (Palmarini et al., 2001), due to major differences in the oncogenic region at the end of the TM protein, in particular the absence of the YXXM motif. Because the exogenous viruses at the origin of ERVs are not known, enJSRV and enENTV nomenclature may be misleading. Sheep (sERV) or goat (gERV) ERVs related to oncogenic small ruminant β-retroviruses, focusing on the genome in which they have been described, may be more appropriate and will be used in this article. Recently, we and others have shown that these sERV and gERV are closely related and belong to the same genetic family II-5 (Verneret et al., 2024) or Cap ERV24 (Moawad et al., 2024).
Although known for decades and compared to other animal and human RNA viruses, the genetic diversity of JSRV and ENTVs is only partially known with only a handful of sequences reported, most of which are not associated with clinical status. Only nine complete sequences of JSRV have been reported originating from Africa, North America, Europe, and Asia (York et al., 1992; Palmarini et al., 1999; DeMartini et al., 2001). Fewer than 50 complete genomes of ENTV have been reported, originating from Canada and Europe for ENTV-1 (Cousens et al., 1999; Walsh et al., 2010) or from Europe and Asia for ENTV-2 (He et al., 2017; Ye et al., 2019). Nevertheless, genomic epidemiology is key to understanding the circulation of these oncogenic retroviruses in sheep and goat populations worldwide and, ultimately, to genetically tracing pathogenic strains. Specific amplification of the exogenous viruses at the origin of respiratory cancers is even more challenging due to the presence of the sERV and gERV, which are highly related to JSRV and ENTVs and should be carefully considered.
In France, lung and nasal cancers induced by oncogenic β-retroviruses are present in small ruminants but are often unrecognized, although some clinical signs such as clear and abundant secretions should alert (Leroux and Turpin, 2021; Devos et al., 2023). These virus-induced cancers are enzootic in France, but the frequency of clinical expression is variable, with flocks with low (sporadic) to high (outbreaks) incidence of cancers (Leroux and Turpin, 2021; Devos et al., 2023). In the latter case, as we have observed on several occasions, the high incidence of cancer can lead to the loss of the flock through uncontrollable mortality directly attributable to cancer or culling. This raises the issue of the reintroduction of retrovirus-free animals, which is difficult to achieve without routine detection and an established control program, and thus represents major threat to livestock production. Our ongoing work clearly demonstrates that ENTVs and JSRV are actively circulating in flocks but have never been genetically characterized.
In the present study, we addressed the diversity of the circulating strains of JSRV and ENTVs in France by developing highly specific PCRs targeting exogenous β-retroviruses in the LTR non-coding region that controls virus expression, in the env oncogene and by amplifying and sequencing the near full-length provirus. We have completed the description of 53 strains of JSRV and ENTVs and have observed JSRV genetic signatures that may be associated with disease severity.
2 Materials and methods
2.1 Biological samples
Twenty-four ENTV-induced nasal tumors and 29 JSRV-induced lung tumors collected between 2003 and 2023 originated from 29 different flocks or areas in France have been selected from our biobank (Table 1, Supplementary Figure S1). Based on information provided by Vets and breeders, the cancer frequency was graded as “low” (sporadic cases over a period of 1–2 years), “high” (multiple cases per year), or “unknown” (no information about the evolution of the number of cases). Cell lines IDO5 (dermal fibroblasts) obtained from Institut Mérieux, Lyon, France, and TIGEF (T-immortalized goat embryonic fibroblasts) (Da Silva Teixeira et al., 1997) were used as non-infected controls. Genomic DNAs were extracted from tissues and cells using the “Quick-DNA midiprep plus” kit (Zymo), after a mechanical grinding step (FastPrep device, MP Biomedicals) for tumor tissues.
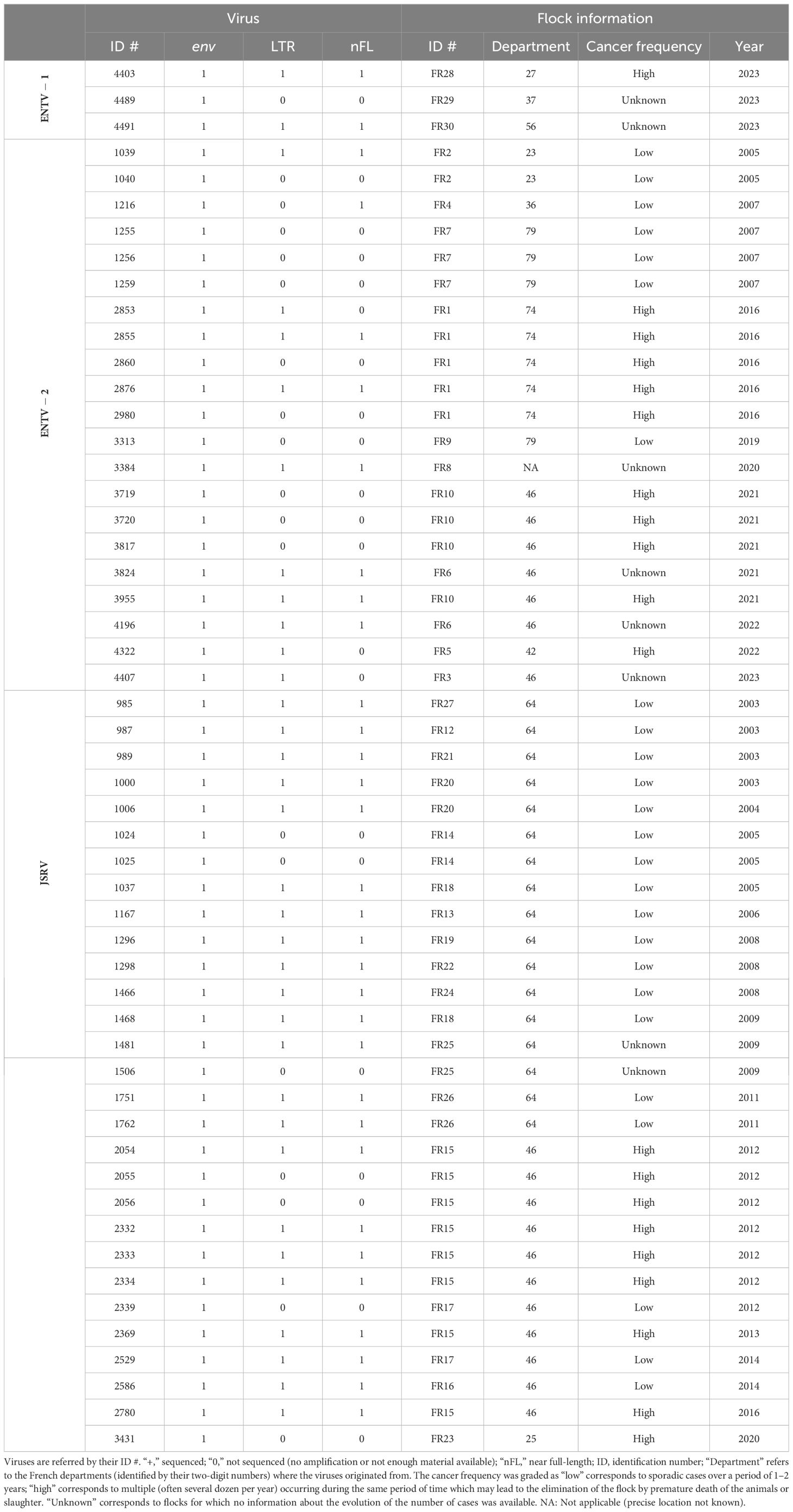
Table 1. Origin of the JSRVs and ENTV.
2.2 Amplification and Sanger sequencing of LTR and env
The LTR and env regions of JSRV, ENTV-1, and ENTV-2 were amplified by PCR (Table 2) using the high-fidelity DNA polymerase “PrimeStar GXL” (Takara), with primers targeting exogenous β-retroviruses. The specificity of the primers was tested first in silico by BLASTn analysis on sERV and gERV (see “Sequence used for the phylogenetic analysis” section of the Materials and methods) and on Ovis aries (Ramb2.0; GCA_016772045.1) and Capra hircus (ARS1.2; GCA_001704415.2) genomes. The primers used to amplify JSRV and ENTVs were iteratively improved throughout the study based on the sequenced strains (Table 2, Supplementary Figure S2). The specificity of the produced amplicons was confirmed by the absence of amplification from sheep (IDO5) and goat (TIGEF) DNA (Supplementary Figure S5).
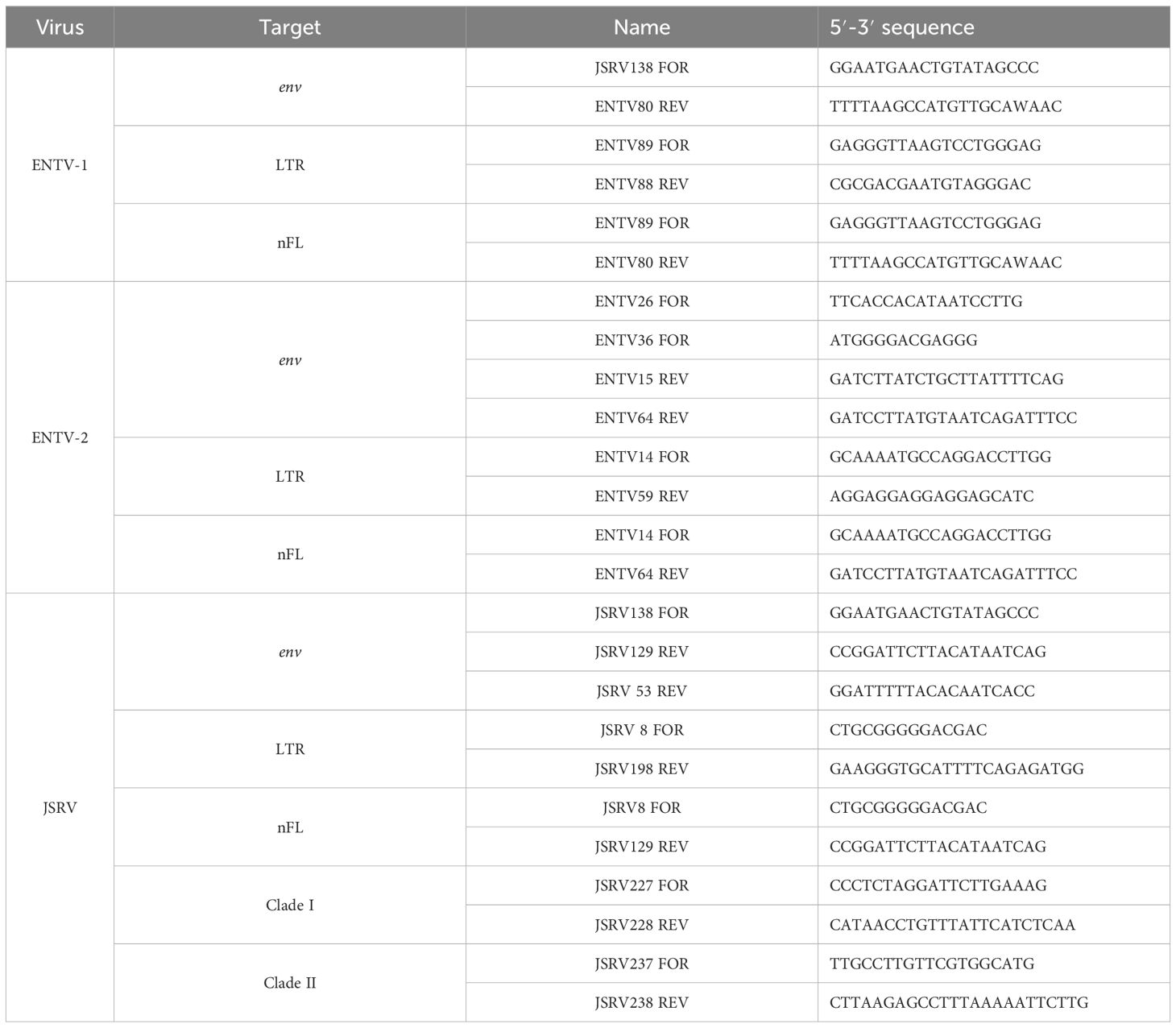
Table 2. Primers used to amplify LTR, env, or the near full-length (nFL) proviral genomes.
Sanger sequencing was performed on the env and LTR amplicons with a minimum coverage of 2X (Sequencing primers available in Supplementary Table S1). Contig sequences were generated and mapped to the reference sequences of JSRV (M80216.1 and AF357971.1), ENTV-1 (NC007015.1), and ENTV-2 (NC_004994.2) using the “Geneious Prime 2023.2.1” software. The complete U3-R-U5 LTRs were reconstructed by assembling sequences from the 5′ LTR amplicons and env amplicons carrying part of the 3′ LTR.
2.3 Whole provirus amplification and genetic analysis
Near full-length provirus amplicons were generated by PCR using “Prime STAR GXL” (Takara) with primers located in the U3 region of the 5′ and 3′ LTRs (Supplementary Figure S2) and purified using “1X AmpureXP beads” (Beckman Coulter). ONT (Oxford Nanopore Technologies) libraries were indexed and prepared using the “Native Barcoding kit” (SQK-NBD114.24) and sequenced on MinION Mk1c with R10.4.1 flow cells. All libraries were base-called using the High Accuracy Basecalling (HAC) model of Dorado v0.4.3 (Oxford Nanopore PLC, 2023). After demultiplexing, reads were mapped to JSRV (M80216.1 and AF357971.1), ENTV-1 (NC007015.1), and ENTV-2 (NC_004994.2) reference sequences using minimap2 v2.26 (Li, 2018). Consensus sequences were generated from mapped reads of at least 5,000 bp using “samtools consensus v1.18” (Danecek et al., 2021) with default settings for HAC basecall nanopore reads. Sequencing data generated in this study were deposited in the NCBI BioSample SRA database (project number PRJNA1103156).
Nucleotide and amino acid alignments were generated with the “MAFFT Alignment v7.490” tool (Katoh and Standley, 2013). JSRV and ENTVs’ Open Reading frames (ORFs) were determined by alignment with previously published sequences. As other retroviruses JSRV and ENTVs, Pro and Pol are suspected to be produced by a rimosomal frameshift. Thus, we determine the start of Pro and Pol as producing the longest ORF possible (York et al., 1992; Cousens et al., 1999; Walsh et al., 2010).
JSRV variability was determined from the nucleotide and predicted amino acid alignment matrix of each ENTVs and JSRV ORFs. The whole-genome identity variation along the JSRV and ENTV-2 genomes was calculated in “Geneious Prime 2023.2.1” on the reconstituted full-length nucleotide sequences alignment and then plotted in a window of 150 nt and steps of 10 nt. The JSRV amino acid pairwise identity variation was calculated on individual alignment for each ORF and plotted in a window of 10 aa and steps of 2 aa. Only one sequence per flock was conserved for the JSRV and ENTVs diversity assessment with the exception of flock FR26 where JSRV clade I and clade II were both detected (1751 and 1762 respectively).
2.4 Transcriptomic analysis of JSRV-infected lambs
Transcriptomic data generated by Karagianni et al (Karagianni et al., 2019) were re-analyzed to discriminate between mRNA originated from JSRV or from sERV in JSRV-infected lambs. Three sets of lung RNA-seq data were used, two from JSRV-infected lambs (ERR2683948 and ERR2683946) and one from a non-infected lamb (ERR2683952). The search for JSRV- or sERV-specific transcription signal was performed using BBduk v38.95 (BBMap, 2023), with specific JSRV and sERV k-mers (TCTGATTATGTAAGAATCCGG and GAGAGTTTTAATACATAAAAA, respectively), allowing two mismatches. The k-mers were designed from JSRV and sERV available in the public databases. Reads containing these k-mers were then mapped to the reference JSRV strain (AF105220.1) or sERV (DQ838493.1) as a validation step using BWA-mem v0.7.17 (Li and Durbin, 2009), and duplicate reads were removed using the “PICARD Markduplicates tool v4.0.11” (GATK) (Picard Tools - By Broad Institute).
2.5 Rapid genotyping of circulating clade I and II JSRVs
For JSRV clade–specific PCRs, primers were designed in the U3 region of the LTR for JSRV clade I and in the env-U3 region for clade II (Table 2). The mammalian thyroglobulin gene was used as a control to confirm the presence of amplifiable DNA.
2.6 Phylogenetic analysis and genetic diversity
LTR, env, and nFL (near full-length) proviral sequence alignments were performed using “MAFFT Alignment v7.490” (Katoh and Standley, 2013) in “Geneious Prime 2023.2.1.” Variability values were determined by the distance matrix of the alignment file, calculated as the percentage of bases differing between two sequences. Pairwise identity rate for multiple sequences were directly calculated from the alignment. Phylogenetic maximum likelihood trees in the env and LTR regions were constructed using the “IQ-TREE v1.5.3” tool (Nguyen et al., 2015) using an ultrafast bootstrap parameter of 10,000 iterations. Identical sequences from a same flock were removed for the env and LTR phylogenetic analysis. A minimal branch support of 80 was considered as a bootstrap threshold to define a phylogenetic clade.
2.7 Transcription factor binding site prediction
Consensus sequences were generated with “Geneious Prime 2023.2.1” on the French JSRV clade I or clade II LTR sequence alignment with a 75% threshold (Supplementary Material: JSRV_LTR_consensus.fasta). All the French LTR sequences were used for the consensus generation according to their classification in JSRV clade I or clade II (Table 1). Potential binding sites for transcription factors in JSRV and ENTV LTR were identified with the “EMBOSS 6.5.7 tool tfscan” on the eukaryotic transcription factor “Transfac” database (Matys et al., 2006) using both the “Vertebrate” and “Other” databases. The position of each transcription factor binding site was determined on the LTR clade I and clade II consensus alignment.
2.8 Nucleotide sequence accession numbers
The Env coding sequences obtained in the present study have been deposited in GenBank with the following accession numbers: PP575093 (#4403), PP719694 (#4489), PP575094 (#4491), PP575073 (#1039), PP575074 (#1040), PP575075 (#1216), PP575076 (#1255), PP575077 (#1256), PP575078 (#1259), PP575079 (#2853), PP575080 (#2855), PP575081 (#2860), PP575082 (#2876), PP575083 (#2980), PP575084 (#3313), PP575085 (#3384), PP575086 (#3719), PP719695 (#3720), PP575087 (#3817), PP575088 (#3824), PP575089 (#3955), PP575090 (#4196), PP575091(#4322), PP575092(#4407), PP575010 (#985), PP575009 (#987), PP575011 (#989), PP575012 (#1000), PP575013 (#1006), PP575014 (#1024), PP575015 (#1025), PP575016 (#1037), PP575017 (#1167), PP575018 (#1296), PP575019 (#1298), PP575020 (#1466), PP575021 (#1468), PP575022 (#1481), PP575023 (#1506), PP575024 (#1751), PP575025 (#1762), PP575026 (#2054), PP575027 (#2055), PP575028 (#2056), PP575029 (#2332), PP575030 (#2333), PP575031 (#2334), PP575032 (#2339), PP575033 (#2369), PP575034 (#2529), PP575035 (#2586), PP575036 (#2780), and PP575037 (#3431). Reconstructed LTRs are available on GenBank with the following accession numbers: PP575070 (#4403), PP575071 (#4489), PP575072 (#4491), PP575060 (#1039), PP575061 (#2853), PP575062 (#2855), PP575063 (#2876), PP575064 (#3384), PP575065 (#3824), PP575066 (#3955), PP575067 (#4196), PP575068 (#4322), PP575069 (#4407), PP575038 (#985), PP575039 (#987), PP575040 (#989), PP575041 (#1000), PP575042 (#1006), PP575043 (#1037), PP575044 (#1167), PP575045 (#1296), PP575046 (#1298), PP575047 (#1466), PP575048 (#1468), PP575049 (#1481), PP575050 (#1751), PP575051 (#1762), PP575052 (#2054), PP575053 (#2332), PP575054 (#2333), PP575055 (#2334), PP575056 (#2369), PP575057 (#2529), PP575058 (#2586), and PP575059 (#2780). Proviral consensus sequences are available on GenBank with the following accession numbers: PP669280 (#4403), PP707036 (#4491), PP707056 (#1039), PP707059 (#1216), PP707060 (#2855), PP707061 (#2876), PP669281 (#3824), PP707057 (#3955), PP707058 (#4196), PP707040 (#985), PP707043 (#987), PP707047 (#989), PP707039 (#1000), PP707041 (#1037), PP707044 (#1167), PP707037 (#1296), PP707046 (#1298), PP707038 (#1466), PP707042 (#1468), PP646154 (#1481), PP707055 (#1751), PP707045 (#1762), PP707050 (#2054), PP707051 (#2332), PP707054 (#2333), PP707053 (#2334), PP646155 (#2369), PP707052 (#2529), PP707049 (#2586), and PP707048 (#2780).
2.9 Sequence used for the phylogenetic analysis
Publicly available sequences used in the study were as follows: KU258881.1, KU258882.1, KU258883.1, KU258884.1, KU258885.1, KU258886.1, MN564749.1, MN564750.1, and MN564751.1 for gERV (Taxonomy ID: 2762664); FJ744146.1, FJ744147.1, FJ744148.1, FJ744149.1, FJ744150.1, GU292314.1, GU292315.1, GU292316.1, GU292317.1, GU292318.1, KC189895.1, and NC007015.1 for ENTV-1 (Taxonomy ID: 69576); KU179192.1, KU258870.1, KU258870.1, KU258871.1, KU258872.1, KU258873.1, KU258874.1, KU258875.1, KU258876.1, KU258877.1, KU258878.1, KU258879.1, KU258880.1, LC570918.1, LC762616.1, LC762617.1, MF033071.1, MK164396.1, MK164400.1, MK210250.1, MK559457.1, MT254061.1, MT254062.1, MT254063.1, MT254064.1, MT598195.1, NC_004994.2, ON843769.1, OQ989633.1, OR024676.1, OR965522.1, PP130116.1, PP130117.1, and PP130118.1 for ENTV-2 (Taxonomy IDs: 2913605, 2913605, and 2584748); AF136224.1, AF136225.1, AF153615.1, EF680296.1, EF680297.1, EF680298.1, EF680299.1, EF680300.1, EF680301.1, EF680302.1, EF680303.1, EF680304.1, EF680305.1, EF680306.1, EF680307.1, EF680308.1, EF680309.1, EF680310.1, EF680311.1, EF680312.1, EF680313.1, EF680314.1, EF680315.1, EF680316.1, EF680317.1, EF680318.1, and JQ995521.1 for sERV [Taxonomy ID: 9940 (ovis aries) defined as “enJSRV”]; and A27950.1, AF105220.1, AF357971.1, CQ964469.1, DQ838494.1, KP691837.1, KY041630.1, M80216.1, MN161849.1, MZ931278.1, ON204347.1, OR729406.1, Y18302.1, Y18303.1, Y18304.1, and Y18305.1 for JSRV (Taxonomy IDs: 11746 and 47898).
3 Results
3.1 Sequencing strategy to specifically amplify exogenous JSRV and ENTVs
Although sEVR or gERV are expressed in a variety of tissues in virus-free sheep, we questioned their expression in infected tissues based on transcriptomic dataset (Karagianni et al., 2019) from experimentally JSRV-infected lambs. We demonstrated the presence of both endogenous and exogenous RNAs in the lung (Table 3). This invalidates approaches that use RNA as a safe matrix to specifically detect exogenous ß-retroviruses. Phylogenetic trees were reconstructed from 87 complete sequences tagged as JSRV, ENTV-1, ENTV-2, sERV, and gERV found in public databases (Figure 1, Supplementary Figure S4). The tree based on complete genomes (Figure 1A) showed that several sequences identified as exogenous JSRV (DQ838494.1 CH and MZ931278 CH) or exogenous ENTVs (KU258870.1 CH and MK164396.1 CH) clearly clustered with ERVs, highlighting the challenge of specifically detecting exogenous oncogenic retroviruses.
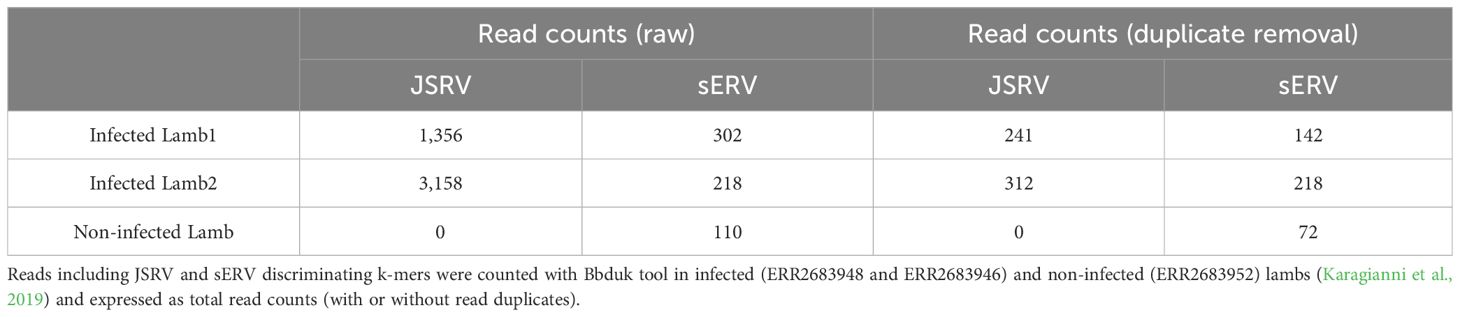
Table 3. Expression of JSRV and sERV in infected lambs.
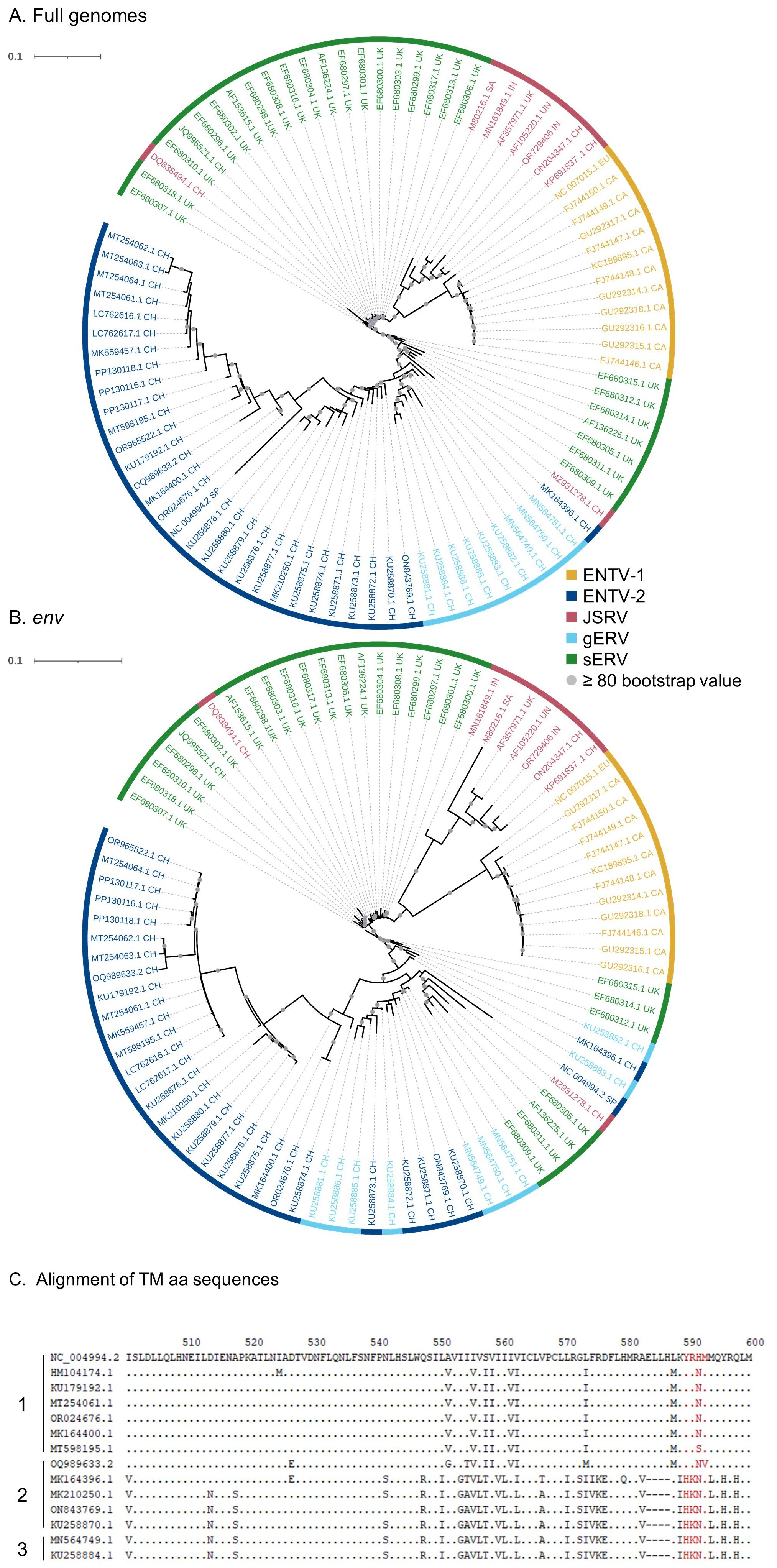
Figure 1. Complexity of distinguishing between the exogenous retroviruses and their endogenous counterparts in small ruminants. Phylogenetic analysis of ENTV-1, ENTV-2, and JSRV sequences and their related endogenous sequences from sheep and goats, available in public databases. Maximum likelihood tree with an ultrafast bootstrap parameter of 10,000 iterations using EF680307.1 (enJSRV10) as the root sequence for (A) the full-length proviral sequence and (B) the env region. The last two letters of each sequence define the geographic origin. CH, China; UK, United Kingdom; CA, Canada; IN, India; SA, South Africa; SP, Spain; EU, Europe; UN, unknown. Bootstrap values greater than 80% are represented by a gray dot and the branch length is indicative of the number of substitutions per site. The scale bar indicates the number of nucleotide substitutions per site. (C) Alignment of deduced amino acid sequences of the ENTV-2 and gERV Env TM region. The position of the YXXM motif, the molecular marker for the transforming exogenous strains of JSRV, ENTV-1, and ENTV-2, is indicated in red. “1” defines the sequences with ENTV-2 as a taxonomic assignment in GenBank (Taxonomy IDs: 239365, 2913605, and 2584748) and with a complete YXXM motif. “2” defines the sequences with ENTV-2 as a taxonomic assignment in GenBank but without the YXXM motif. “3” defines the sequences with gERV as a taxonomic assignment in GenBank (Taxonomy ID: 2762664). Dots represent identical amino acids to NC_004994.2 and dashes indicate gaps in the alignment.
Because the role of the YXXM motif in cell transformation has been established, we used the presence of the YXXM sequence as a specific signature of exogenous retroviruses. A consensus sERV sequence (Supplementary Material: Consensus_enJSRV-BDD.fasta) was generated from published sERV sequences to identify sERV and gERV copies present in 15 Caprinae genome assemblies using BLASTn (Supplementary Figure S3). Of the 667 endogenous sequences with an env part encoding the TM region, the majority (626) contained an Histidine Lysine Asparagine Methionine amino acid motif (HKNM) and none of them contained the canonical YXXM motif (Supplementary Figure S3). This confirms the exogenous signature of this short motif. Some sequences identified as exogenous ENTVs in the databases did not contain the YXXM motif (OQ989633.2, MK164396.1, MK210250.1, ON843769.1, and KU258870.1), indicating that they were likely amplified and sequenced from an ERV and were excluded from our subsequent JSRV and ENTV primer definition steps (Figure 1C). Specific amplification of exogenous retroviruses was confirmed using genomic DNA from nasal and lung tumors; the absence of amplification from uninfected IDO5 or TIGEF fibroblasts confirmed the exogenous nature of the amplicons with no cross-reaction with related endogenous sequences (Supplementary Figures S2, S5). Using this rigorous approach, we have developed molecular tools for the detection and amplification of strictly exogenous JSRVs and ENTVs based on the sERV and gERV genomes present in the genomic databases and on a specific signature that confirms their exogenous status.
3.2 ENTV-1 and ENTV-2 cluster in French phylogenetic groups
ENTV-1 and ENTV-2 were sequenced from 24 (3 from sheep and 21 from goats, respectively) nasal tumors collected from 13 flocks originating from different departments of France (Table 1). We demonstrated the circulation of a major genetic clade of ENTV-2, with sequences having a pairwise identity > 99.5% for both env and LTR regions (Figures 2A, B), except for 3384FR, which is the only French sequence found in an outgroup with between 98% and 99% pairwise identity in LTR and env with other French ENTV-2 sequences. All env sequences generated in this study had the YXXM motif confirming their exogenous nature. French ENTV-2 strains differed in the env region from strains reported in Spain (NC_004994.2, 5.5% nt divergence) or in China (KU258879.1, 16% nt divergence) (Figure 2A). Based on the LTR analysis, the French and Spanish ENTV-2 strains clustered together (Figure 2B).
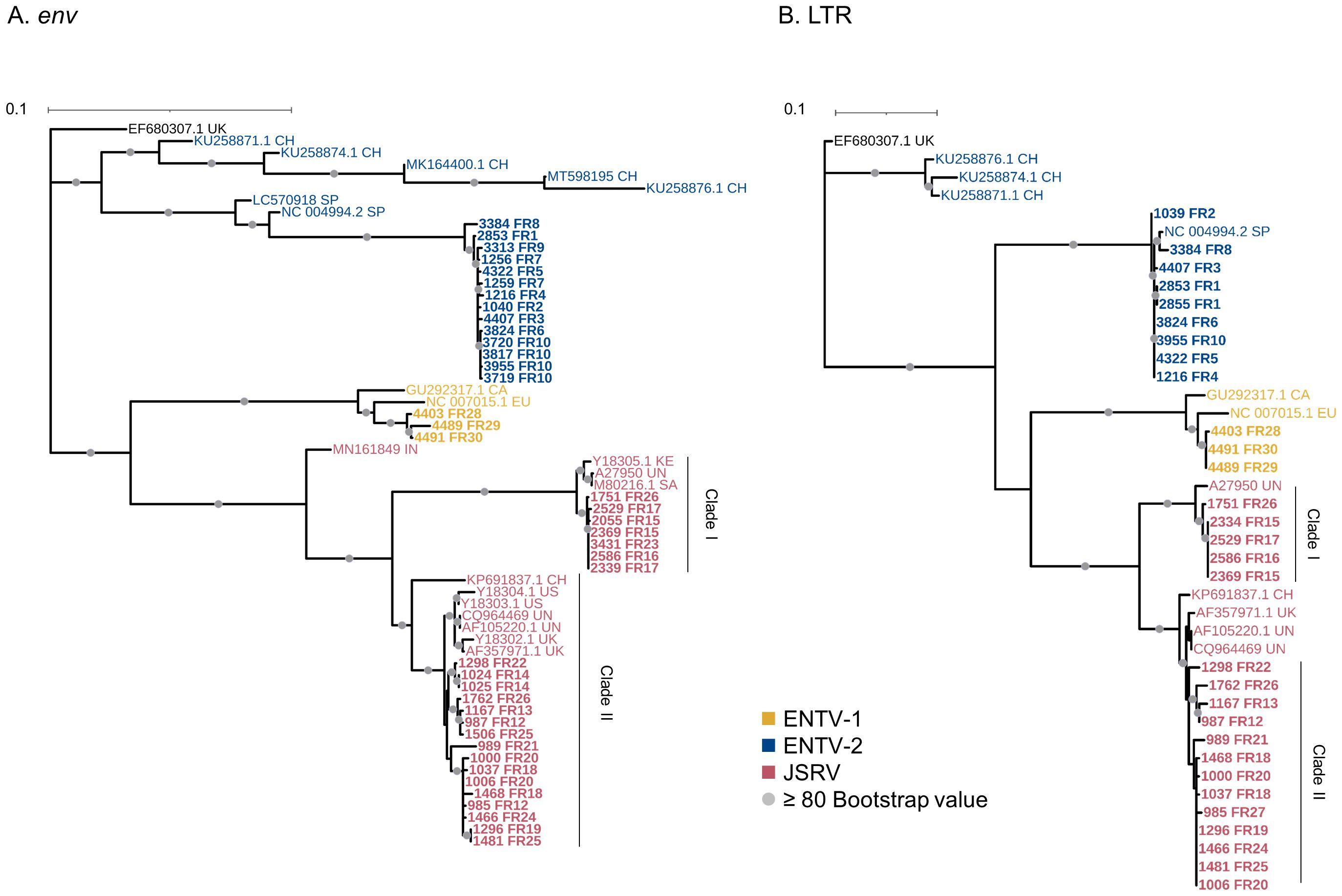
Figure 2. French JSRV and ENTV strains are distributed in distinct phylogenetic clades. (A, B) Maximum likelihood tree with an ultrafast bootstrap parameter of 10,000 iterations for (A) env (1851 bp) and (B) reconstituted LTR regions (458 bp). The French sequences described in this study are in bold. CH, China; UK, United Kingdom; CA, Canada; IN, India; SA, South Africa; SP, Spain; EU, Europe; KE, Kenya; US, United States; UN, unknown. Bootstrap values greater than 80% are represented by a gray dot. The scale bar indicates the number of nucleotide substitutions per site. Identical sequences from the same flock were removed from the phylogenetic analysis.
The three French ENTV-1 strains clustered together and were distinct from previously reported ENTV-1 in North America (GU292317.1) and Europe (NC007015.1). It should be noted that limited ENTV-1 sequences are available in the databases. Pairwise identity between French ENTV-1 strains was >98.9% for both LTR and env based on the alignment matrix used for the phylogenetic analysis (not shown).
3.3 Two distinct JSRV clades are circulating in France
Phylogenetic reconstruction based on env and LTR sequences obtained from JSRV present in 29 lung cancers revealed the circulation of at least two distinct clades I and II in the env and LTR phylogenetic analysis (Figures 2A, B). Clade I was genetically related to the South African JSRV reference (M80216.1), whereas clade II was more closely related to the UK JSRV reference (AF357971.1). A unique clade per flock was detected in all but one flock. Co-circulation of both clades was detected in flock FR26 for strains 1751 and 1762, sampled on the same day, and classified as clade I and II JSRVs respectively (Table 1, Figure 2). Within a clade, the nucleotide divergence was very low, with a mean pairwise identity of 98.9% (± 0.5%) for clade I and 99.9% (± 0.06%) for clade II in the env region. For two sequences found in the same flock (2334 FR15 and 2333 FR15), we observed a large deletion of 10 nucleotides in the U3 region of the LTR just before the TATA box.
3.4 JSRV and ENTV strains are stable between and within flocks
To explore the overall diversity across the entire viral genome, a near complete genome (nFL) sequencing strategy based on LTR and env targeted sequencing was developed. Amplicons of approximately 7.6 kb, from 2 ENTV-1, 7 ENTV-2, and 21 JSRV strains were obtained and sequenced using Oxford Nanopore technology (Supplementary Figures S2–S5, Table 1). The nFL proviral sequencing confirmed that the same major strains of JSRV and ENTV were circulating in the sheep and goat flocks. The ENTV strains have a low variability with 0.29% (± 0.08) for ENTV-2 and 0.46% for ENTV-1 for the nFL genome with less than 1% variability in the different genes (Figure 3A, Table 4). JSRV nFL provirus sequencing confirmed the circulation of JSRV clades I and II in France (Figure 3B), with 3.5% variability between the 16 fully sequenced strains (Figure 3B, Table 4). At the amino acid level, variability was observed in different regions of Gag (Figure 4A): in the phosphoprotein protein 24, p12, and nucleocapsid (NC) proteins. The N-terminal region encoding for the viral protease (PR) is also variable with a predicted potential smaller size of the Pro Coding Sequence (CDS) due to the presence of two premature stop codons for clade II in this region (Figure 4B). Variability is observed in the integrase protein (INT) with a premature stop codon at the C-terminus for five clade I strains, removing 4 aa (Figure 4C). A polymorphism hotspot was detected in the env region encoding for the cytoplasmic tail. In this 68–amino acid long region, the analysis showed 92.7% identity compared to over 98% identity when considering the full Env CDS (Figure 4D).
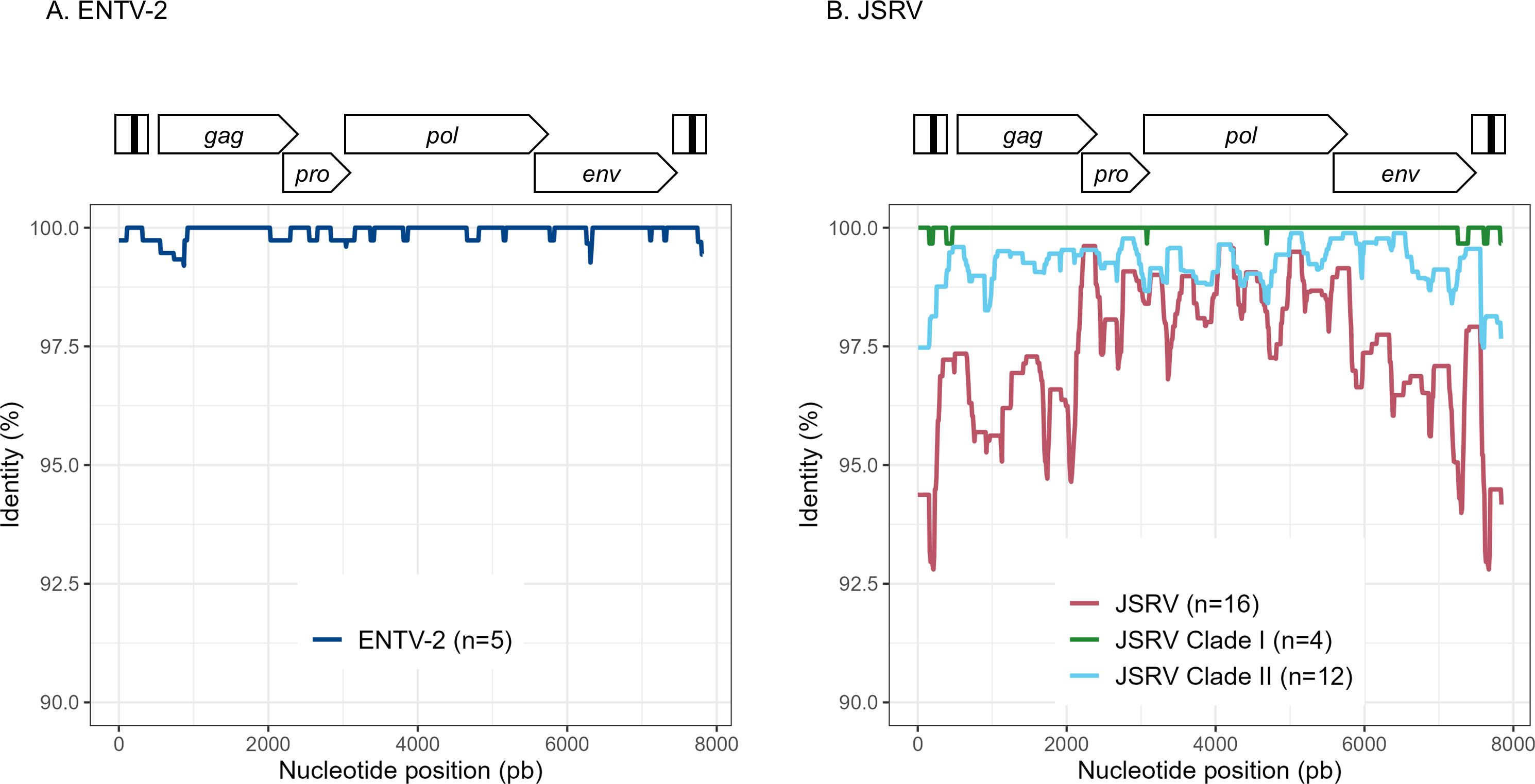
Figure 3. Variability ENTV-2 and JSRV. Nucleotide identity was determined along the reconstituted full-length proviruses of (A) ENTV-2 and (B) JSRV. Each sequence is from a different flock.
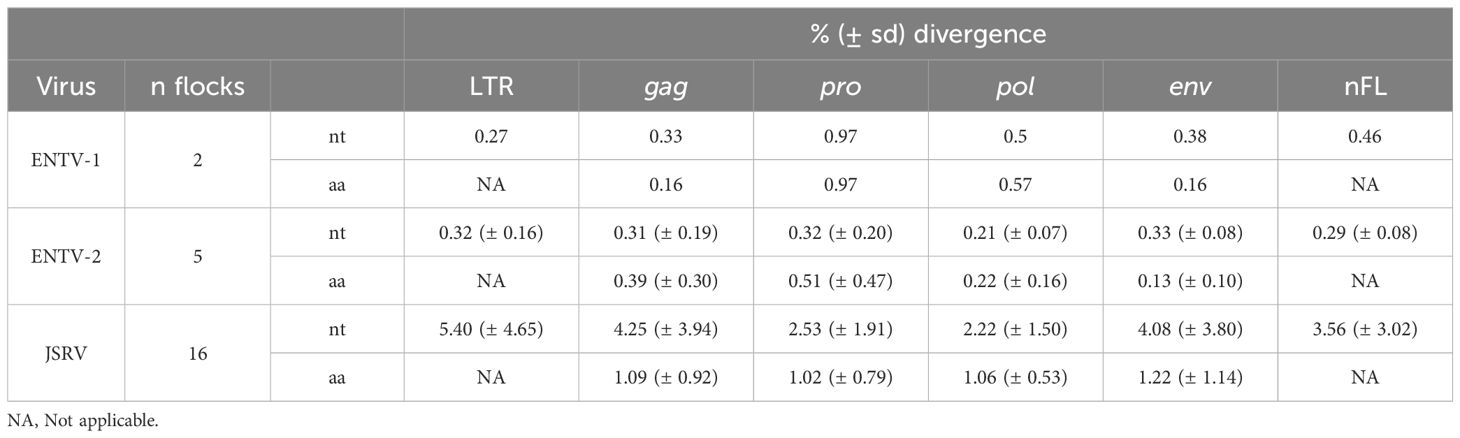
Table 4. JSRV and ENTV genetic diversity. NA: Not applicable.
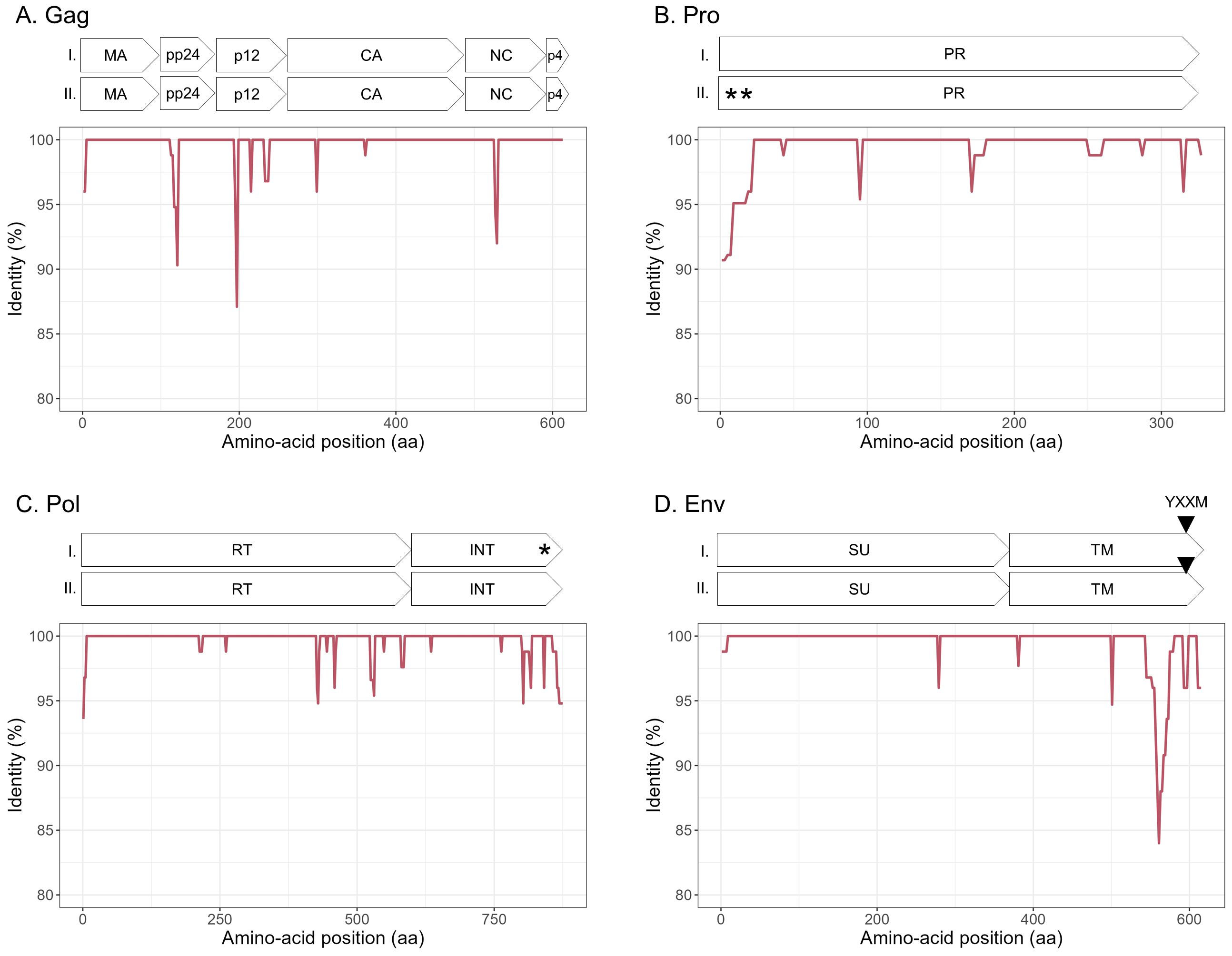
Figure 4. Amino acid sequence diversity of JSRV ORFs. (A–D) Calculated pairwise identity in deducted aa along the different ORFs of JSRV calculated with a sliding window of 10 aa and steps of 2 aa. The different protein/domains encoded by the ORF are shown above each graph. The YXXM motif is represented with a gray triangle. Stop codons are indicated by asterisks. (A) Gag, (B) Pro, (C) Pol, and (D) Env.
As for three flocks, we collected lung and nasal tumors over several months, we questioned the genetic stability of the viral strains at the origin of cancer over time. Over 1.5 months (flock FR1) or 11 months (flock FR6), ENTV-2 strains were highly stable with 0.03% and 0.1% nt variability, respectively, for the reconstituted full-length sequence with no variability in the env gene. We have collected JSRV tumor samples over 45 months (flock FR15), characterized by a high incidence of lung cancer associated with clade I viruses. Analysis confirmed the high stability of the JSRV genome, with only 0.2% nt variability across the six full-length proviral genomes. Because the time of infection is not known, the mutation rate cannot be estimated. These results highlight the persistence of the same viral strains over time for both JSRV and ENTV-2.
3.5 Clade I JSRV is associated with high prevalence of cancers
JSRV-induced lung cancers are generally reported as isolated cases, but flocks with a high incidence of cancer have been regularly observed in France, with a major impact on breeders through increased mortality rate of young adults. Among the 16 flocks from which we collected tumors, three (FR15, FR16, and FR23) showed a high incidence of cancer (Table 1) and were associated with the circulation of clade I JSRV (Figures 2A, B). The Env and LTR regions are the most divergent between the clade I and clade II and are involved in the transformation process and proviral expression respectively. The Env sequences of clades I and II showed differences, particularly in the region encoding for the intracytoplasmic tail of TM (Figure 5). The YXXM motif located in the CT region, known to be the major determinant of cell transformation, was present in both clades, but, whereas the clade I virus associated with cancer outbreaks carried a YRTM motif, clade II viruses carried a YRNM motif.

Figure 5. JSRVs clade I and clade II differ in the C-Terminal region of Env. Alignment of the deduced amino acid sequence of two representative JSRV clade I (2369 FR) and JSRV clade II (1481 FR) sequences. The localization of the YXXM motif is represented by a black square. The dots represent identical amino acids between the two sequences, and the asterisk represents the stop codon. YXXM motif starts at the 590th amino acid of JSRV Env.
In the LTR, the differences between JSRV clades I and II were mainly located in the U3 region with 31 mismatches. Prediction of transcription factor binding sites in the LTR using the EMBOSS tool tfscan clearly showed different profiles between the LTRs of the two clades (Figure 6, Supplementary Table S2). The consensus LTR sequences of JSRV clade I and clade II were aligned, and the position of each transcription factor was indicated from the beginning of U3. A total of 35 transcription factor binding sites were predicted to be common to JSRV clade I and clade II LTRs. Additionally, 11 and 17 transcription factors binding sites were, respectively, predicted solely in JSRV clade I and clade II LTR. Among them E2F1 (transcription factor E2F1) at position 105 and CREB (Cyclic Adenosine Monophosphate (cAMP) response element–binding protein) at position 156 were only predicted in JSRV clade I. Specific for JSRV clade II, two TMF (TATA element modulatory factor) at positions 104 and 174 were predicted. The presence of binding sites for the Hepatocyte Nuclear factor 3 (HNF-3), a transcription factor described as modulator of JSRV expression in the lung, at position 127 was confirmed for both clade I and clade II JSRV.
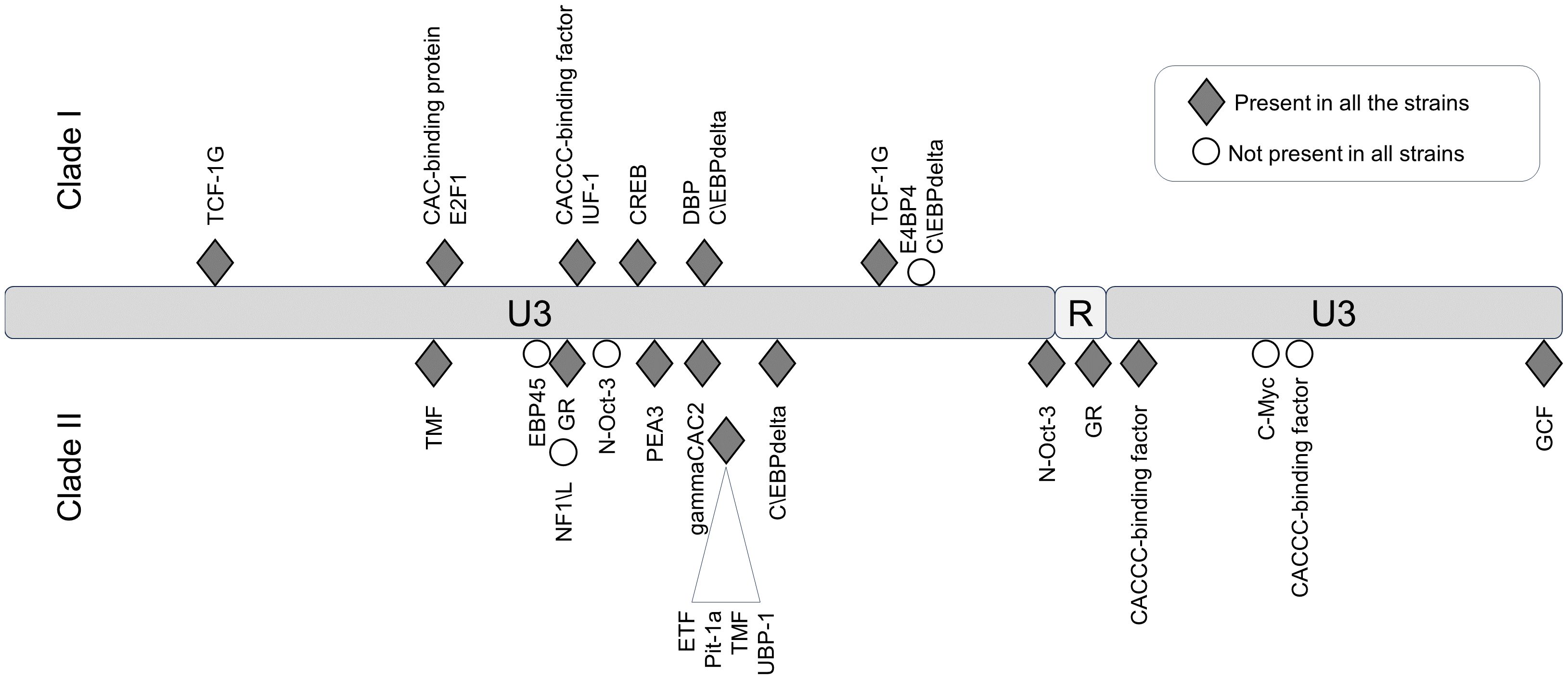
Figure 6. Differential transcription factor binding site prediction on JSRV LTR clade I and clade II. Predicted with the EMBOSS tool TF scan on LTR clade I and clade II sequences, transcription factor binding sites unique to each LTR were presented. Transcription factor binding site prediction was performed on consensus LTR sequences for clade I or clade II. The frequency of each site was tested for both clade using the EMBOSS tool TF scan on each individual LTR sequence (9 sequences for clade I and 13 sequences for clade II). U3, unique 3′; R, repeat; U5, unique 5′.
3.6 Rapid genotyping of JSRV clades
In the absence of routine diagnostics to identify infected sheep, we have developed a PCR-based genotyping tool using discriminating regions between clades I and II (Figure 7). This tool was tested on six JSRV strains of known genotype and the amplification of a fragment of 153 bp for clade I and 245 bp for clade II indicated the appropriate genotype.
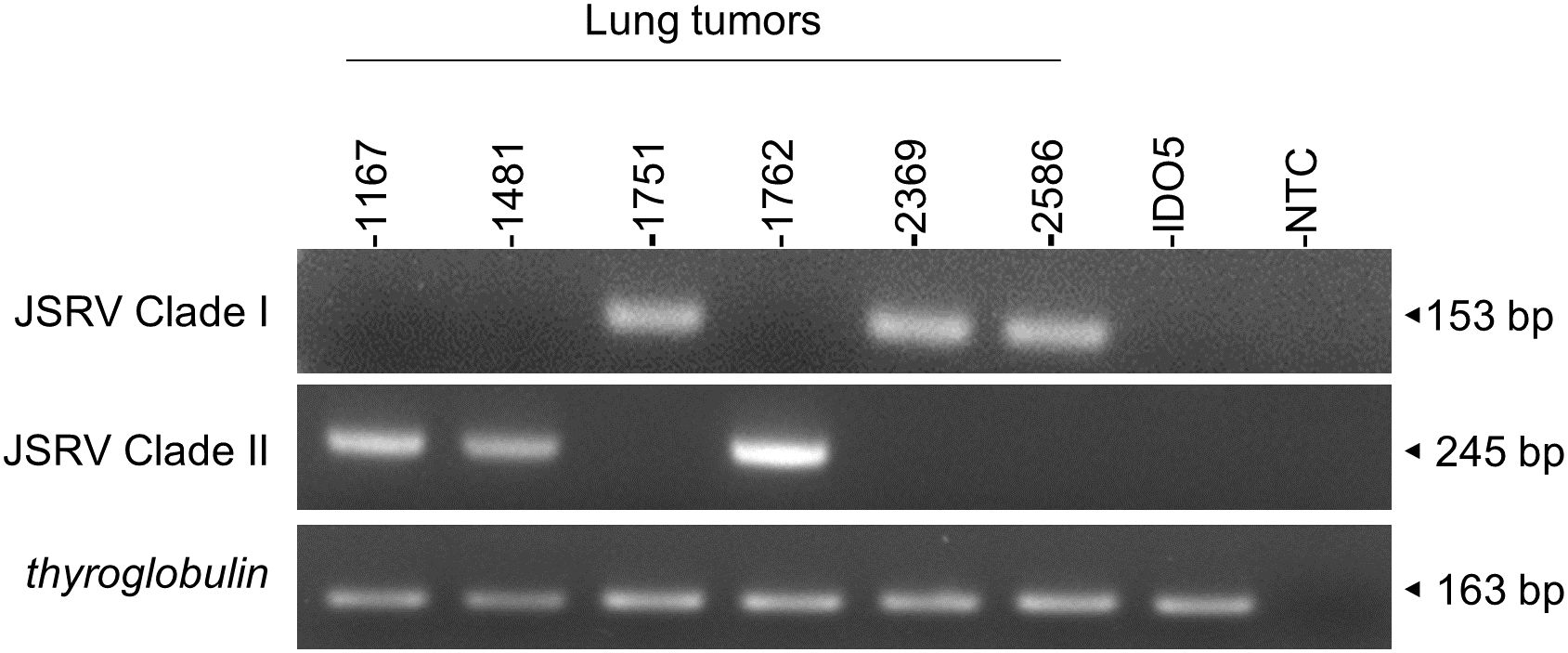
Figure 7. JSRV genotyping by PCR. Agarose gel migration of PCR amplicons corresponding to a region of the JSRV LTR measured at 153 bp for clade I and at 245 bp for clade II. DNA extracted from JSRV-induced tumors of known clade: clade I (1751, 2369, and 2586) and clade II (1169, 1481, and 1762) were used for this proof of concept. IDO5 is an ovine dermal fibroblast cell line used as a JSRV primer specificity control. Thyroglobulin: amplicon corresponding to a region of the thyroglobulin gene in the sheep genome, measured at 163 bp.
4 Discussion
To determine the diversity of JSRV and ENTV strains circulating in France, we developed both a targeted and near full-length sequencing approach. The detection of RNA from either endogenous or exogenous JSRV in the lung demonstrated the difficulty of performing short-read sequencing on total RNA to reconstruct the full-length genome of exogenous strains. With nearly as much sERV RNA as JSRV RNA, there is a high probability of artificially creating chimeric sequences of both endogenous and exogenous reads in non-discriminating regions. Our targeted sequencing and long read sequencing using primers located in discriminating regions allowed us to be confident in the nature of the strains being sequenced. Furthermore, the detection of endogenous RNA in the infected lung shed light on a possible interaction between endogenous and exogenous RNA/proteins in infected tissues that could modulate the pathogenicity of JSRV as it has already been described in vitro with the interaction between endogenous and exogenous Gag (Murcia et al., 2007) or on the possible recombination between endogenous and exogenous strains, as has been observed for feline leukemia virus to form a new pathogenic strain (Erbeck et al., 2021).
Our study has shown that at the national level, a limited number of major strains of JSRV and ENTVs circulate between flocks located in different regions of France, suggesting the introduction of strains that have established and persisted in the animal populations. The genetic stability observed is clearly different from that observed for lentiviruses in livestock, which replicate and mutate rapidly and eventually escape the immune system (Narayan et al., 1977; Leroux et al., 1997; Howe et al., 2002). The minimal diversity observed for ENTVs and JSRV is closer to that reported for HTLV-1 (human T-cell lymphotropic virus type 1) in humans, with 2%–8% nt diversity between geographic subtypes and low (0.5%) intra-strain variability (Desrames et al., 2014). Compared to other viruses, the number of sequences for JSRV and ENTVs in the public databases is still limited, hindering our understanding of their genetic epidemiology and global circulation. Our efforts to specifically characterize exogenous viruses using tools that exclude sERVs or gERVs, although we have not characterized all the viruses circulating in France, have helped us to define new clades or lineages (Figure 8). French ENTV-2 sequences structured a new lineage, together with the Spanish strain NC_004992.2 (Ortín et al., 2003), which was considered as an outgroup sequence before this work. French ENTV-1 clustered with another European ENTV-1 strain NC007015.1 (Cousens et al., 1999) in a new phylogenetic clade. Interestingly, we found that the French clade I viruses clustered with the JSRV strain first described in South Africa and to our knowledge only reported in Africa (Bai et al., 1999). This may illustrate the circulation of viruses between countries, probably through animal trade.
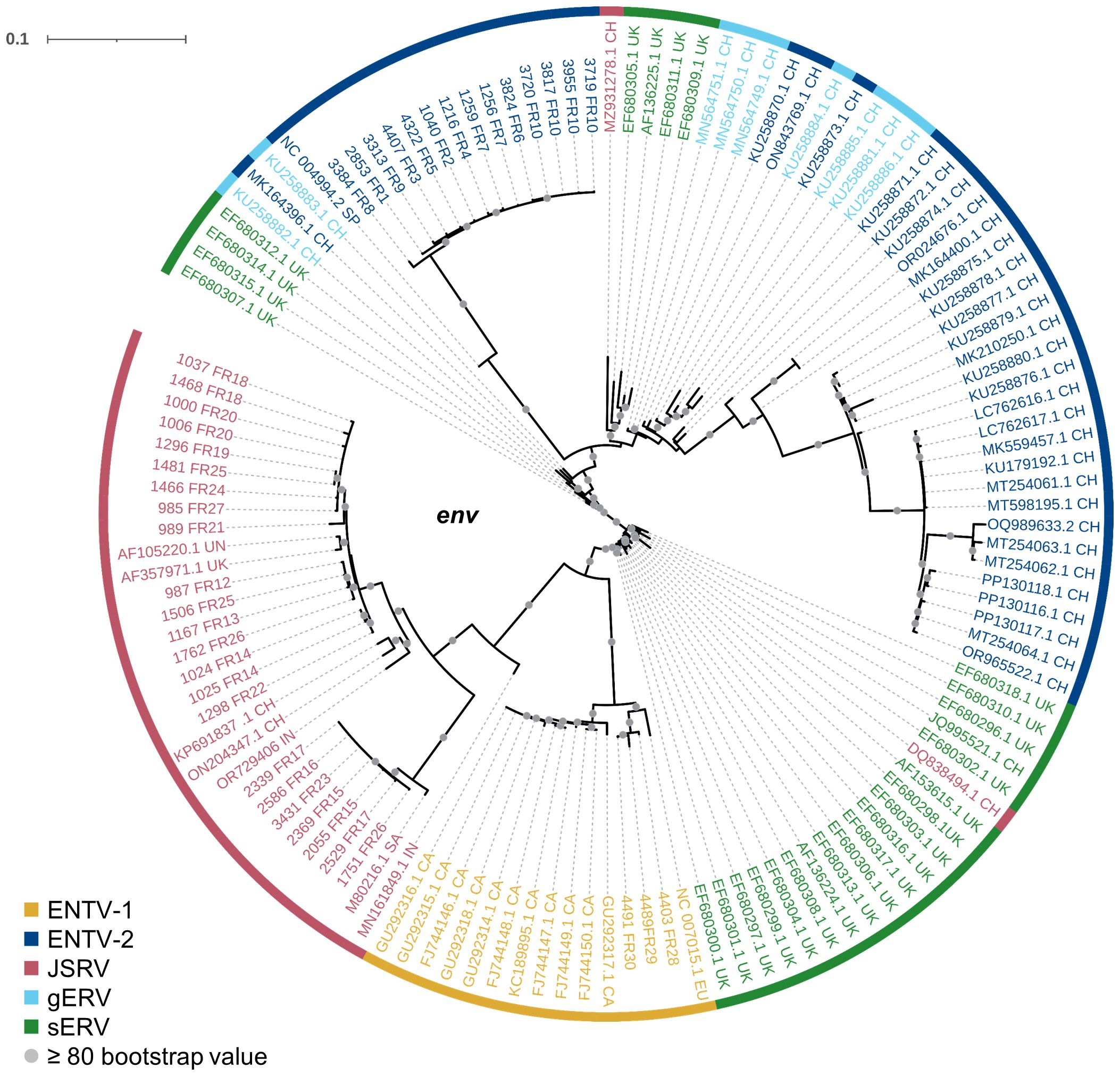
Figure 8. Phylogenetic relationships of the French JSRV and ENTV strains with other endogenous and exogenous small ruminant β-retrovirus. Maximum likelihood tree of the env region using 10,000 ultrafast bootstrap repeats with EF680307.1 (enJSRV10) as the rooted sequence. CH, China; UK, United Kingdom; CA, Canada; IN, India; SA, South Africa; SP, Spain; EU, Europe; UN, unknown. Bootstrap values greater than 80% are represented by a gray dot and the branch lengths indicate the number of substitutions per site.
We have shown that, at the country level and based on our virus collection, the genetic diversity is higher for JSRV than that for ENTVs with two distinct clades of JSRV circulating in France. As clade I was mostly associated with flocks with high incidence of cancers, we investigated whether a genetic signature could be detected between clades I and II. Polymorphism analysis along the different JSRV sequences showed a higher degree of polymorphism in Gag and Env. Most differences were observed in the p12 protein and in the nucleocapsid for Gag and in the TM domain for Env. In the p12 protein, an L domain involved in JSRV late restriction in vitro (Murcia et al., 2007) is found near the polymorphic region. Although no polymorphism is observed on this site, amino acid changes nearby could impact the restriction phenomenon and improve virus spreading in infected individuals. Polymorphism in the transmembrane protein of JSRV is also of interest as it is the major determinant in the transformation process (Palmarini et al., 2001; Hull and Fan, 2006). In the strains analyzed, most of the differences between JSRV clade I and clade II were in the transmembrane part of TM just before the CT domain. This region has been less investigated (Hull et al., 2012), and its impact on cell transformation remains to be determined. In the YXXM motif, a major determinant of cell transformation, amino acid 592 is found with a threonine/alanine or an asparagine for clades I and II, respectively. A glutamine is found at position 597 for clade I, whereas an arginine is present in clade II JSRV. Interestingly, these two amino acids at positions 592 and 597 of the CT domain have been reported to increase cell transformation in vitro (Hull and Fan, 2006). In addition for the amino acid at position 592, a change from an asparagine (clade II) to a threonine (clade I) increases the cell transformation in NIH 3T3 cells (Palmarini et al., 2001). The role of these mutations must be investigated to assess their impact on the transformation process and viral cycle. We observed differences in the predicted length of the integrase, four amino acids longer for clade I than that for clade II JSRVs. The LTRs of retroviruses are major determinants of viral expression, controlling cell specificity through the pattern of transcription factor binding to the non-coding sequence. We found transcription factors HNF-3, CCAAT-enhancer-binding proteins (C/EBP), and Nuclear factor I (NF-I) important for JSRV cell tropism and expression (McGee-Estrada et al., 2002; McGee-Estrada and Fan, 2006, 2007). Most nucleotide variations are in the viral promoter U3 region with transcription factor binding sites restricted to either the I or II LTR. In U5, the limited differentially predicted restriction factor binding sites could also be detrimental for JSRV expression as a previous study (Palmarini et al., 2000) showed that deletion of this region result in 50% to 80% decrease reporter expression. Altogether modification of LTR activity affects the viral cell cycle like oncogenic envelope protein expression or infectious particle production. Regarding the observations on the envelope or the effect of the LTR polymorphism on cell transformation or proviral expression, both need to be further confirmed by in vitro or in vivo studies
In conclusion, using highly specific sequencing methods, we demonstrated the circulation of major strains for ENTV-1 and ENTV-2 as opposed to two clades for JSRV. Genetic signature in the JSRV clade I and clade II could be associated with difference of pathogenicity manifested in flocks by differential cancer incidence. The JSRV polymorphism is mainly located in the TM of Env and the LTR regions involved in cell transformation and viral expression. We also developed a molecular tool to easily follow the circulation of JSRV clades in flocks and assess their potential link with disease prevalence. This study will facilitate the monitoring of JSRV and ENTV strains of concern in French flocks. Targeted genetic diagnostic through genomic epidemiology studies could help to reduce the circulation of more pathogenic strains and better understand the dynamics of infection between flocks.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the studies involving animals in accordance with the local legislation and institutional requirements because we used postmortem samples considered as waste. Written informed consent was not obtained from the owners for the participation of their animals in this study because there was oral consent and we collected the samples on the owner’s demand.
Author contributions
BR-V: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MV: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. RD: Formal analysis, Methodology, Validation, Writing – original draft, Writing – review & editing. CD: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. BG: Data curation, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. J-LC: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. JT: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. CL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by ANR, grant ANR-22-CE35-0002-01, and by INRAE Young Scientist Award. BR-V is the recipient of a VetAgro Sup PhD grant.
Acknowledgments
We thank our collaborators from the sheep and goat industry for the collection of biological samples and sharing the clinical information. This work was carried out using the computing facilities of the CC LBBE/PRABI. This study has benefited from the expertise and facilities of the DTAMB and PRABI-AMSB platforms of FR BioEEnViS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1466333/full#supplementary-material
Supplementary Figure 1 | Flock location on the French territory. The flocks from which JSRV and ENTV have been collected, are mapped according to their French department numbers.
Supplementary Figure 2 | Localization of PCR primers for the amplification of LTR, env and near full-length genome. For JSRV and ENTV-2 two different sets of primers were used for the amplification of env with the improvement throughout the study of the exo/endo amplification specificity.
Supplementary Figure 3 | Absence of the YXXM motif in the Env cytoplasmic tail of caprinae β-ERVs related to JSRV and ENTVs. sERV and gERV sequences within 15 caprinae assemblies (ARS1.2: GCA_001704415.1, ASM1117029: GCA_011170295.1, ASM1914517: GCA_019145175.1, ASM2665220: GCA_026652205.1, CapAeg_1.0: GCA_000978405.1, CAU_F1_maternal_1.0: GCA_023701675.1, CAU_O.aries_1.0: GCA_017524585.1, CVASU_BBG_1.0: GCA_004361675.1, Oar_v4.0: GCA_000298735.2, CAU_Oori_1.0: GCA_014523465.1, ARS-UI_Ramb_v2.0: GCA_016772045.1, Oar_ARS-UKY_Romanov_v1.0: GCA_022244705.1, Saanen_v1: GCA_015443085.1, Oar_ARS-UKY_WhiteDorper_v1.0: GCA_022244695.1, CHIR_2.0: GCA_000317765.2) were identified by BLASTn, using a consensus of sERV from sequences available in GenBank (see Figure 1 and Materials and methods). Sequences < 500 bp corresponding to solo LTRs were not included. MAFFT alignment of the different sequences allowed to select only sequences with a preserved TM region of Env.
Supplementary Figure 4 | Phylogenetic analysis of sequences of ENTV-1, ENTV-2 and JSRV and their related endogenous sequences from sheep and goats. Maximum likelihood tree with 10,000 ultrafast bootstrap replicated with EF680307.1 (enJSRV10) as root sequence for (A) gag (B) pol region. The scale bar indicates the nucleotide substitution per site. CH: China, UK: United Kingdom, CA: Canada, IN: India, SA: South Africa, SP: Spain, EU: Europe, PA: Pakistan, UN: Unknown.
Supplementary Figure 5 | Migration profile of ENTV and JSRV PCR amplification. For each PCR, the example of genomic DNA extracted from two tumors used as a template per virus is shown, in addition to DNA extracted from ovine (IDO5) and caprine (TIGEF) cell lines used as a control for endogenous amplification. nFL = near full-length.
Supplementary Table 1 | Primers used for the sequencing of JSRV, ENTV-1 and ENTV-2 env and LTR amplicons.
Supplementary Table 2 | Prediction of transcription factor binding sites in LTR of clades I and II JSRV. Sequences were scanned with the tfscan tool to identify transcription factors binding sites on clades I and II LTRs. (+): present, (-): absent. Positions start and end of the transcription factor binding sites are indicated, based on the alignment of the clade I and clade II LTR consensus sequences. The transcription factor frequency was calculated by scanning with the tfscan tool the 22 different LTR sequences of the study (9 LTR-I and 13 LTR-II sequences).
References
Alberti, A., Murgia, C., Liu, S.-L., Mura, M., Cousens, C., Sharp, M., et al. (2002). Envelope-induced cell transformation by ovine betaretroviruses. J. Virol. 76, 5387–5394. doi: 10.1128/jvi.76.11.5387-5394.2002
Arnaud, F., Caporale, M., Varela, M., Biek, R., Chessa, B., Alberti, A., et al. (2007). A paradigm for virus–host coevolution: sequential counter-adaptations between endogenous and exogenous retroviruses. PloS Pathog. 3, e170. doi: 10.1371/journal.ppat.0030170
Bai, J., Bishop, J. V., Carlson, J. O., DeMartini, J. C. (1999). Sequence comparison of JSRV with endogenous proviruses: envelope genotypes and a novel ORF with similarity to a G-protein-coupled receptor. Virology 258, 333–343. doi: 10.1006/viro.1999.9728
BBMap (2023). SourceForge. Available online at: https://sourceforge.net/projects/bbmap/ (Accessed March 26, 2024).
Caporale, M., Centorame, P., Giovannini, A., Sacchini, F., Di Ventura, M., de las Heras, M., et al. (2005). Infection of lung epithelial cells and induction of pulmonary adenocarcinoma is not the most common outcome of naturally occurring JSRV infection during the commercial lifespan of sheep. Virology 338, 144–153. doi: 10.1016/j.virol.2005.05.018
Caporale, M., Cousens, C., Centorame, P., Pinoni, C., De las Heras, M., Palmarini, M. (2006). Expression of the jaagsiekte sheep retrovirus envelope glycoprotein is sufficient to induce lung tumors in sheep. J. Virol. 80, 8030–8037. doi: 10.1128/jvi.00474-06
Cousens, C., Maeda, N., Murgia, C., Dagleish, M. P., Palmarini, M., Fan, H. (2007). In vivo tumorigenesis by Jaagsiekte sheep retrovirus (JSRV) requires Y590 in Env TM, but not full length orfX open reading frame. Virology 367, 413–421. doi: 10.1016/j.virol.2007.06.004
Cousens, C., Minguijon, E., Dalziel, R. G., Ortin, A., Garcia, M., Park, J., et al. (1999). Complete sequence of enzootic nasal tumor virus, a retrovirus associated with transmissible intranasal tumors of sheep. J. Virol. 73, 3986–3993. doi: 10.1128/JVI.73.5.3986-3993.1999
Cumer, T., Pompanon, F., Boyer, F. (2019). Old origin of a protective endogenous retrovirus (enJSRV) in the Ovis genus. Heredity 122, 187–194. doi: 10.1038/s41437-018-0112-z
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. doi: 10.1093/gigascience/giab008
Da Silva Teixeira, M. F., Lambert, V., Mselli-Lakahl, L., Chettab, A., Chebloune, Y., Mornex, J. F. (1997). Immortalization of caprine fibroblasts permissive for replication of small ruminant lentiviruses. Am. J. Vet. Res. 58, 579–584. doi: 10.2460/ajvr.1997.58.06.579
DeMartini, J. C., Bishop, J. V., Allen, T. E., Jassim, F. A., Sharp, J. M., de las Heras, M., et al. (2001). Jaagsiekte sheep retrovirus proviral clone JSRVJS7, derived from the JS7 lung tumor cell line, induces ovine pulmonary carcinoma and is integrated into the surfactant protein A gene. J. Virol. 75, 4239–4246. doi: 10.1128/JVI.75.9.4239-4246.2001
Desrames, A., Cassar, O., Gout, O., Hermine, O., Taylor, G. P., Afonso, P. V., et al. (2014). Northern african strains of human T-lymphotropic virus type 1 arose from a recombination event. J. Virol. 88, 9782–9788. doi: 10.1128/jvi.01591-14
Devos, J., Becker, C., Yanes, H., Turpin, J., Schelcher, F., Leroux, C. (2023). La tumeur nasale enzootique des caprins: une maladie méconnue. Bull. GTV Bull. Numéro 110, 75−79.
Erbeck, K., Gagne, R. B., Kraberger, S., Chiu, E. S., Roelke-Parker, M., VandeWoude, S. (2021). Feline leukemia virus (FeLV) endogenous and exogenous recombination events result in multiple feLV-B subtypes during natural infection. J. Virol. 95. doi: 10.1128/jvi.00353-21
Grego, E., De Meneghi, D., Álvarez, V., Benito, A. A., Minguijón, E., Ortín, A., et al. (2008). Colostrum and milk can transmit jaagsiekte retrovirus to lambs. Vet. Microbiol. 130, 247–257. doi: 10.1016/j.vetmic.2008.01.011
He, Y., Zhang, Q., Wang, J., Zhou, M., Fu, M., Xu, X. (2017). Full-length genome sequence analysis of enzootic nasal tumor virus isolated from goats in China. Virol. J. 14, 141. doi: 10.1186/s12985-017-0795-4
Howe, L., Leroux, C., Issel, C. J., Montelaro, R. C. (2002). Equine infectious anemia virus envelope evolution in vivo during persistent infection progressively increases resistance to in vitro serum antibody neutralization as a dominant phenotype. J. Virol. 76, 10588–10597. doi: 10.1128/jvi.76.21.10588-10597.2002
Hull, S., Fan, H. (2006). Mutational analysis of the cytoplasmic tail of jaagsiekte sheep retrovirus envelope protein. J. Virol. 80, 8069–8080. doi: 10.1128/JVI.00013-06
Hull, S., Lim, J., Hamil, A., Nitta, T., Fan, H. (2012). Analysis of jaagsiekte sheep retrovirus (Jsrv) envelope protein domains in transformation. Virus Genes 45, 508–517. doi: 10.1007/s11262-012-0793-y
Karagianni, A. E., Vasoya, D., Finlayson, J., Martineau, H. M., Wood, A. R., Cousens, C., et al. (2019). Transcriptional response of ovine lung to infection with jaagsiekte sheep retrovirus. J. Virol. 93, e00876–e00819. doi: 10.1128/JVI.00876-19
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Lee, A. M., Wolfe, A., Cassidy, J. P., McV. Messam, L. L., Moriarty, J. P., O’Neill, R., et al. (2017). First confirmation by PCR of Jaagsiekte sheep retrovirus in Ireland and prevalence of ovine pulmonary adenocarcinoma in adult sheep at slaughter. Ir. Vet. J. 70, 33. doi: 10.1186/s13620-017-0111-z
Leroux, C., Chastang, J., Greenland, T., Mornex, J. F. (1997). Genomic heterogeneity of small ruminant lentiviruses: existenceof heterogeneous populations in sheep and of the samelentiviral genotypes in sheep and goats. Arch. Virol. 142, 1125–1137. doi: 10.1007/s007050050147
Leroux, C., Turpin, J. (2021). Rétrovirus et maladies de l’appareil respiratoire chez les moutons et les chèvres. Bull. GTV Numéro Spéc. Pathol. Respir. L’air Temps, 23−34.
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinforma. Oxf. Engl. 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Liu, S.-L., Lerman, M. I., Miller, A. D. (2003). Putative phosphatidylinositol 3-kinase (PI3K) binding motifs in ovine betaretrovirus Env proteins are not essential for rodent fibroblast transformation and PI3K/Akt activation. J. Virol. 77, 7924–7935. doi: 10.1128/jvi.77.14.7924-7935.2003
Maeda, N., Fan, H. (2008). Signal transduction pathways utilized by enzootic nasal tumor virus (ENTV-1) envelope protein in transformation of rat epithelial cells resemble those used by jaagsiekte sheep retrovirus. Virus Genes 36, 147–155. doi: 10.1007/s11262-007-0193-x
Maeda, N., Inoshima, Y., De las Heras, M., Maenaka, K. (2021). Enzootic nasal tumor virus type 2 envelope of goats acts as a retroviral oncogene in cell transformation. Virus Genes 57, 50–59. doi: 10.1007/s11262-020-01808-7
Mao, L., Li, W., Hao, F., Yang, L., Li, J., Sun, M., et al. (2022). Research progress on emerging viral pathogens of small ruminants in China during the last decade. Viruses 14, 1288. doi: 10.3390/v14061288
Matys, V., Kel-Margoulis, O. V., Fricke, E., Liebich, I., Land, S., Barre-Dirrie, A., et al. (2006). TRANSFAC® and its module TRANSCompel®: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110. doi: 10.1093/nar/gkj143
McGee-Estrada, K., Fan, H. (2006). In vivo and in vitro analysis of factor binding sites in jaagsiekte sheep retrovirus long terminal repeat enhancer sequences: roles of HNF-3, NF-I, and C/EBP for activity in lung epithelial cells. J. Virol. 80, 332–341. doi: 10.1128/jvi.80.1.332-341.2006
McGee-Estrada, K., Fan, H. (2007). Comparison of LTR enhancer elements in sheep betaretroviruses: insights into the basis for tissue-specific expression. Virus Genes 35, 303–312. doi: 10.1007/s11262-007-0079-y
McGee-Estrada, K., Palmarini, M., Fan, H. (2002). HNF-3β Is a critical factor for the expression of the jaagsiekte sheep retrovirus long terminal repeat in type II pneumocytes but not in clara cells. Virology 292, 87–97. doi: 10.1006/viro.2001.1247
Moawad, A. S., Wang, F., Zheng, Y., Chen, C., Saleh, A. A., Hou, J., et al. (2024). Evolution of endogenous retroviruses in the subfamily of caprinae. Viruses 16, 398. doi: 10.3390/v16030398
Monot, M., Erny, A., Gineys, B., Desloire, S., Dolmazon, C., Aublin-Gex, A., et al. (2015). Early steps of jaagsiekte sheep retrovirus-mediated cell transformation involve the interaction between env and the RALBP1 cellular protein. J. Virol. 89, 8462–8473. doi: 10.1128/JVI.00590-15
Murcia, P. R., Arnaud, F., Palmarini, M. (2007). The transdominant endogenous retrovirus enJS56A1 associates with and blocks intracellular trafficking of jaagsiekte sheep retrovirus gag. J. Virol. 81, 1762–1772. doi: 10.1128/JVI.01859-06
Narayan, O., Griffin, D. E., Chase, J. (1977). Antigenic shift of visna virus in persistently infected sheep. Science 197, 376–378. doi: 10.1126/science.195339
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nor El Houda, B., Nassim, S., Bennoune, O., Soraya, O., De Las Heras, M., Leroux, C. (2020). Evidence of jaagsiekte sheep retrovirus-induced pulmonary adenocarcinoma in Ouled Djellal breed sheep in Algeria. Vet. Res. Forum 11, 93–95. doi: 10.30466/vrf.2019.107160.2549
Ortín, A., Cousens, C., Minguijón, E., Pascual, Z., Villarreal, M. P. D., Sharp, J. M., et al. (2003). Characterization of enzootic nasal tumour virus of goats: complete sequence and tissue distribution. J. Gen. Virol. 84, 2245–2252. doi: 10.1099/vir.0.19125-0
Palmarini, M., Datta, S., Omid, R., Murgia, C., Fan, H. (2000). The long terminal repeat of jaagsiekte sheep retrovirus is preferentially active in differentiated epithelial cells of the lungs. J. Virol. 74, 5776–5787. doi: 10.1128/JVI.74.13.5776-5787.2000
Palmarini, M., Maeda, N., Murgia, C., De-Fraja, C., Hofacre, A., Fan, H. (2001). A phosphatidylinositol 3-kinase docking site in the cytoplasmic tail of the jaagsiekte sheep retrovirus transmembrane protein is essential for envelope-induced transformation of NIH 3T3 cells. J. Virol. 75, 11002–11009. doi: 10.1128/JVI.75.22.11002-11009.2001
Palmarini, M., Sharp, J. M., de las Heras, M., Fan, H. (1999). Jaagsiekte sheep retrovirus is necessary and sufficient to induce a contagious lung cancer in sheep. J. Virol. 73, 6964–6972. doi: 10.1128/JVI.73.8.6964-6972.1999
Picard Tools - By Broad Institute (n.d). Available online at: https://broadinstitute.github.io/picard/ (Accessed March 25, 2024).
Sid, N., Belalmi, N. E. H., Benhamza, L., Ouhida, S., Zebiri, M. E., Aydoğan, A., et al. (2018). First case report of enzootic nasal adenocarcinoma in “Ouled Djellal“ ewe in Algeria. Open Vet. J. 8, 9. doi: 10.4314/ovj.v8i1.3
Sistiaga-Poveda, M., Jugo, B. M. (2014). Evolutionary dynamics of endogenous Jaagsiekte sheep retroviruses proliferation in the domestic sheep, mouflon and Pyrenean chamois. Heredity 112, 571–578. doi: 10.1038/hdy.2013.136
Verneret, M., Leroux, C., Faraut, T., Navratil, V., Lerat, E., Turpin, J. (2024). A genome-wide study of ruminants reveals two endogenous retrovirus families still active in goats. doi: 10.1101/2024.06.21.600049
Walsh, S. R., Linnerth-Petrik, N. M., Laporte, A. N., Menzies, P. I., Foster, R. A., Wootton, S. K. (2010). Full-length genome sequence analysis of enzootic nasal tumor virus reveals an unusually high degree of genetic stability. Virus Res. 151, 74–87. doi: 10.1016/j.virusres.2010.04.002
Wootton, S. K., Halbert, C. L., Miller, A. D. (2006). Envelope proteins of jaagsiekte sheep retrovirus and enzootic nasal tumor virus induce similar bronchioalveolar tumors in lungs of mice. J. Virol. 80, 9322–9325. doi: 10.1128/jvi.00865-06
Ye, C., Huang, Q., Chen, T., Jiang, J., Hou, F., Xu, D., et al. (2019). First detection and genotypic analysis of goat enzootic nasal tumor virus 2 in Chongqing, China. Arch. Virol. 164, 1647–1650. doi: 10.1007/s00705-019-04211-2
Keywords: JSRV, ENTV-1, ENTV-2, virulence factors, genomic epidemiology
Citation: Riocreux-Verney B, Verneret M, Diesler R, Dolmazon C, Gineys B, Cadoré J-L, Turpin J and Leroux C (2024) Association between genetic clades and cancer prevalence suggested by French-wide study of oncogenic small ruminant β-retrovirus diversity. Front. Cell. Infect. Microbiol. 14:1466333. doi: 10.3389/fcimb.2024.1466333
Received: 17 July 2024; Accepted: 01 October 2024;
Published: 08 November 2024.
Edited by:
Kazumi Nakano, The University of Tokyo, JapanReviewed by:
Philippe V. Afonso, Institut Pasteur, FranceNaoyoshi Maeda, Health Sciences University of Hokkaido, Japan
Copyright © 2024 Riocreux-Verney, Verneret, Diesler, Dolmazon, Gineys, Cadoré, Turpin and Leroux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Leroux, Y2Fyb2xpbmUubGVyb3V4QGlucmFlLmZy
†These authors have contributed equally to this work and share last authorship