 Yukun Lin
Yukun Lin Kunjing Liu
Kunjing Liu Fang Lu
Fang Lu Changming Zhai
Changming Zhai Fafeng Cheng
Fafeng Cheng- 1School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing, China
- 2School of Life Sciences, Beijing University of Chinese Medicine, Beijing, China
- 3Department of Rheumatism, Beijing University of Chinese Medicine Third Affiliated Hospital, Beijing, China
Programmed cell death (PCD) plays a crucial role in maintaining the normal structure and function of the digestive tract in the body. Infection with Helicobacter pylori (H. pylori) is an important factor leading to gastric damage, promoting the Correa cascade and accelerating the transition from gastritis to gastric cancer. Recent research has shown that several PCD signaling pathways are abnormally activated during H. pylori infection, and the dysfunction of PCD is thought to contribute to the development of gastric cancer and interfere with treatment. With the deepening of studies on H. pylori infection in terms of PCD, exploring the interaction mechanisms between H. pylori and the body in different PCD pathways may become an important research direction for the future treatment of H. pylori infection and H. pylori-related gastric cancer. In addition, biologically active compounds that can inhibit or induce PCD may serve as key elements for the treatment of this disease. In this review, we briefly describe the process of PCD, discuss the interaction between different PCD signaling pathways and the mechanisms of H. pylori infection or H. pylori-related gastric cancer, and summarize the active molecules that may play a therapeutic role in each PCD pathway during this process, with the expectation of providing a more comprehensive understanding of the role of PCD in H. pylori infection.
Introduction
Programmed cell death is an active, inherent phenomenon. It supports normal physiological activities during development and homeostasis after birth by eliminating damaged, infected or superannuated cells. In 1972, Kerr et al. made a groundbreaking discovery by describing a phenomenon known as apoptosis (Kerr et al., 1972). This phenomenon involves a programmed condensation of the nucleus and cytoplasm, leading to fragmentation of the cell into a series of membrane-bound, structurally well-preserved fragments. Subsequently, these fragments are either shed from the epithelial-lined surfaces or taken up by other cells and rapidly degraded by lysosomal enzymes from macrophages. Subsequently, different forms of PCD were gradually discovered (Table 1). Today, programmed cell death includes genetically programmed suicide mechanisms, such as apoptosis, necroptosis and pyroptosis (Newton et al., 2024). It can also result from highly conserved processes to degrade macromolecular structures and even entire organelles, as in autophagy (Cadwell, 2016), or from dysregulated metabolism, as observed in ferroptosis (Tang et al., 2021).

Table 1 Programmed cell death pathways with their morphological and biochemical features.
Although the PCD plays a crucial role in maintaining homeostasis in our body and keeping it in a stable state, its normal processes can be disrupted by the invasion of pathogens, especially H. pylori infection. This disruption can unbalance our internal environment and possibly even trigger gastric cancer (Meng et al., 2015). In the early stage of infection, H. pylori can induce the expression of programmed death ligand 1 (PD-L1) on the epithelial cells of the stomach, for example via the Shh signaling pathway (Holokai et al., 2019). PD-L1, which interacts with programmed death 1 (PD1) on the surface of cytotoxic T lymphocytes (CTLs), can inhibit CTLs to induce PCD (Sun et al., 2007). In this way, cells infected with H. pylori can be protected from the immune response. In addition, H. pylori infection causes visible damage to both the superficial epithelial cells and the cells that form pits and glands in the stomach, caused directly by the bacteria or by PCD-mediated mechanisms (Genta, 1997). Once the destroyed glands can no longer regenerate, fibroblasts and extracellular matrix fill the space they previously occupied in the lamina propria. This leads to an irreversible loss of functional structure known as atrophy, a condition thought to be a precursor to gastric cancer (Correa, 1995). In addition, H. pylori infection triggers several intracellular signaling pathways, such as mitogen-activated protein kinase (MAPK), nuclear factor kappa B (NF-κB) and phosphatidylinositol 3-kinase (PI3K). These signaling pathways in turn have a direct and indirect effect on the proliferation, differentiation and programmed cell death of the gastric epithelium and ultimately lead to the conversion of epithelial cells into oncogenic units (Kato et al., 2008; Yousefi et al., 2019).
Overall, it is clear that PCD plays an important role in H. pylori infection. However, the relationship between PCD and H. pylori infection is complicated and not simply linear. During the infection process, H. pylori can both promote and inhibit PCD. For example, H. pylori infection can trigger both ROS formation and DNA fragmentation, leading to the activation of caspase-3 and caspase-8. This suggests that oxidative stress can be exerted on gastric epithelial cells during H. pylori infection, leading to apoptosis (Ding et al., 2007). However, other studies have shown that the gamma-glutamyl transpeptidase (GGT) of H. pylori can inhibit apoptosis and induce proliferation of gastric epithelial cells through the induction of cyclooxygenase-2, epidermal growth factor-related peptides and interleukin-8 (Ricci et al., 2014).
In this review, we provide a brief overview of the different pathways of PCD, including apoptosis, necroptosis, pyroptosis, autophagy and ferroptosis. We discuss their known and proposed roles in H. pylori infection to elucidate the intricate relationships. In addition, we describe proposed therapeutic strategies for the treatment of H. pylori infection that target key regulators of various PCD signaling pathways.
Programmed cell death in H. pylori infection and related gastric cancer
Apoptosis, common in H. pylori infection
The term “apoptosis” comes from the Greek and means “falling away”, which reflects the characteristic property of apoptotic cells to disintegrate and fragment in a controlled manner. At the molecular level, apoptosis is triggered by either intrinsic or extrinsic signaling pathways, both of which converge in a common execution phase (Green, 2000). Intrinsic apoptosis is triggered by intracellular signals such as DNA damage, oxidative stress or growth factor deficiency and leads to the activation of pro-apoptotic proteins of the BCL-2 family, including BAX and BAK (Puthalakath and Strasser, 2002). These proteins promote permeabilization of the outer mitochondrial membrane, leading to the release of cytochrome c into the cytoplasm (Jost and Vucic, 2020). Cytochrome c then activates the caspase cascades, which ultimately leads to cell death. Extrinsic apoptosis, on the other hand, is triggered by extracellular signals, e.g. by the binding of death ligands to death receptors on the cell surface, such as tumor necrosis factor receptor 1 (TNFR1) or FAS. This interaction recruits and activates caspase-8, which can directly cleave and activate downstream effector caspases or trigger the mitochondrial signaling pathway through BID cleavage (Czabotar et al., 2014).
With the exponential increase in the number of publications on H. pylori as well as those written on apoptosis, it is clear that apoptosis is closely intertwined with H. pylori infection (Shirin and Moss, 1998). In the context of infection, there are changes in the extent of cell apoptosis, accompanied by changes in cell proliferation, migration and cytokine production (Alzahrani et al., 2014). In addition to directly triggering apoptosis, H. pylori induces sensitivity to various epithelial signaling pathways and events in the host. Certain mechanisms are triggered by H. pylori binding to cell surface receptors or by soluble virulence factors entering the epithelium, leading to the initiation of gastric pathologies, including inflammation, mucosal damage, and even the development of gastric cancer (Figure 1) (Tsai and Hsu, 2017).
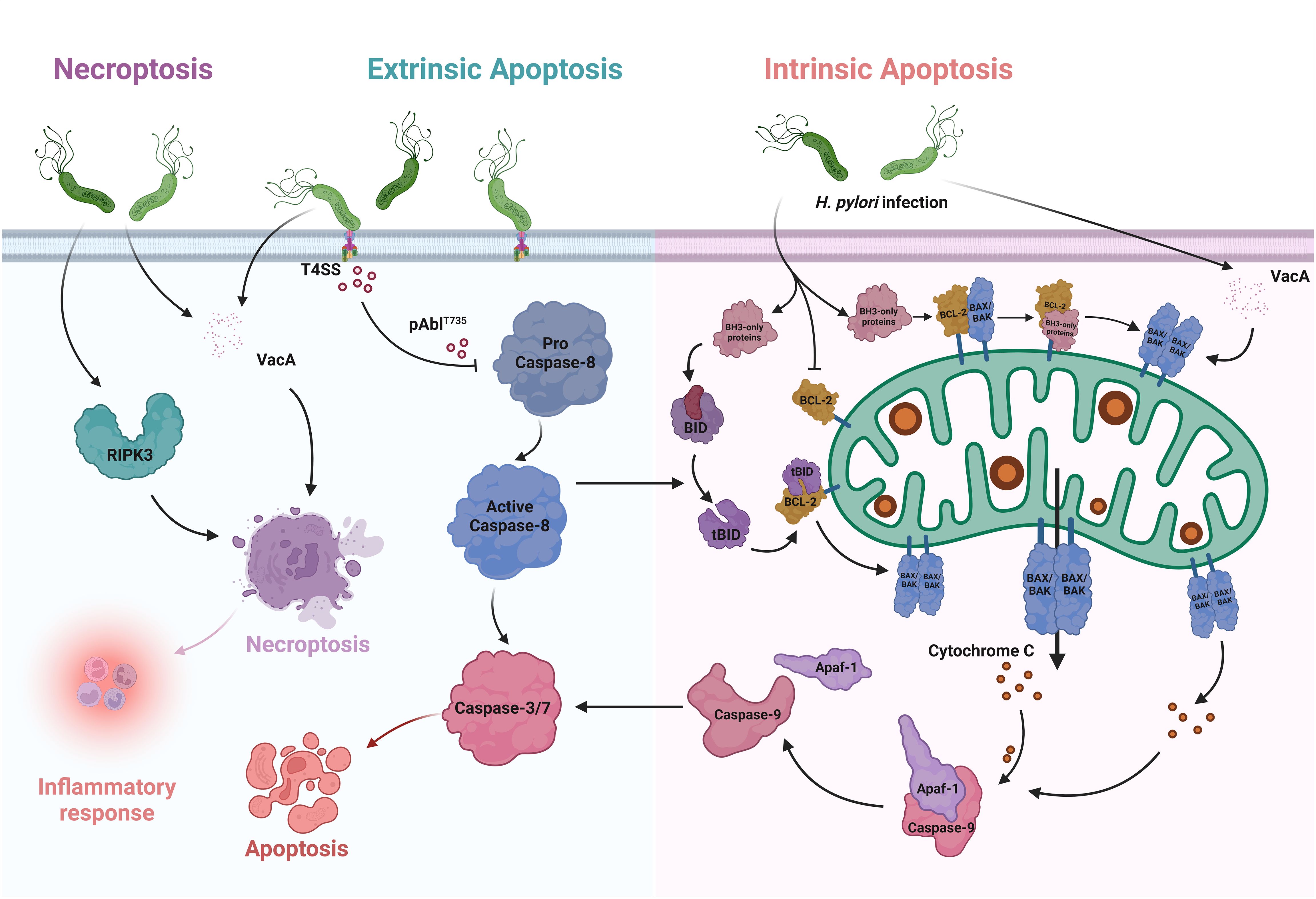
Figure 1 Apoptosis and Necroptosis in H. pylori infection. In the intrinsic pathway of apoptosis, only BH3 proteins are upregulated after H. pylori infection. They interact with the BCL-2 proteins, which are essential for survival, and thus override the inhibition of BAX and BAK. As a result, BAX and BAK oligomerize, leading to MOMP. After its release into the cytoplasm, cytochrome c binds to Apaf-1, which activates the initiator caspases and induces apoptosis. VacA, which causes the translocation of BAX and increases cytochrome c, can also enhance apoptosis. BID is one of the pure BH3 proteins. It can be cleaved by activated caspase-8 and releases BAX and BAK to activate intrinsic apoptosis via the MOMP pathway. In the extrinsic pathway, H. pylori promotes the formation of pAbIT735 via the type IV secretion system (T4SS), thereby attenuating caspase-8-dependent cell apoptosis. In necroptosis, infection with H. pylori leads to an increase in the key factor RIPK3, which promotes the occurrence of inflammatory reactions. VacA also triggers necroptosis and releases considerable amounts of inflammatory mediators.
Different apoptotic effects in H. pylori infection
Infection with H. pylori has different apoptotic effects on different types of host cells. In most cells, H. pylori infection increases apoptosis. In gastric epithelial cells, acute H. pylori infection significantly accelerated GES-1 apoptosis by increasing the expression of BAX and cleaved caspase-3, while decreasing the expression of BCL-2 (Liu et al., 2021). In lymphocytes, the researchers observed the transfer of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus. In addition, apoptosis of both T and B cells was significantly increased in H. pylori infected cells compared to uninfected controls (Singh et al., 2006). H. pylori induces apoptosis in T and B cell lines and translocates AIF. The destruction of T and B cells by apoptosis could explain the persistence of H. pylori infection. In the mononuclear phagocyte system, macrophages under infection have also shown the ability to induce apoptosis by the protein HP1286 secreted by H. pylori (Tavares and Pathak, 2017). Co-culturing H. pylori with peripheral blood monocytes, THP-1 or U937 cells leads to increased apoptosis (Zhang et al., 2017a). However, the researchers found that suppressing the expression of leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) on THP-1 cells can reduce this process (Zhang et al., 2017b). The results suggest that LAIR-1 modulates cell apoptosis and secretion of inflammatory cytokines in THP-1 cells, which may help to maintain inflammation and prevent the clearance of bacteria by the immune response. In gastric cancer cells, infection with H. pylori resulted in cell cycle arrest, decreased proliferation and increased apoptosis (Ding et al., 2008). H. pylori increased apoptosis in AGS cells with chromatin condensation and decreased BCL-2 levels, which was associated with NF-κB activation (Chu et al., 2003). In addition, H. pylori induced cullin 1-RING ubiquitin ligase (CRL1) and 26S proteasome-dependent degradation of STAMBPL1 in AGS (Chaithongyot and Naumann, 2022). STAMBPL1, which functions as a deubiquitinase of the anti-apoptotic protein survivin, balances the extent of survivin degradation with the E3 ligase CRL1 and thus regulates apoptotic cell death. Thus, the degradation of STAMBPL1 promotes apoptotic cell death.
However, there are always exceptions. Yang et al. found that H. pylori infection promotes apoptosis of MGC-803 cells, but high expression of FRA-1 induced by H. pylori suppresses this cell death (Yang et al., 2021). Liu et al. showed that acute infection with H. pylori significantly accelerated the apoptosis of GES-1, but the apoptosis rate of GES-1 cells with chronic H. pylori infection decreased significantly (Liu et al., 2021). Zhang et al. also demonstrated a novel mechanism by which H. pylori escapes from monocytes by upregulating early apoptosis and inhibiting late apoptosis (Zhang et al., 2017a). These studies have shown that inhibition of apoptosis of gastric epithelial cells due to chronic H. pylori infection is a contributing factor to severe gastric diseases, leading us to realize that the influence of H. pylori on cell apoptosis may not be unidirectional. An imbalance between apoptosis and proliferation may contribute to H. pylori-associated gastric carcinogenesis. Clinical research has also confirmed that dysregulation of apoptosis control in gastric intestinal metaplasia could further exacerbate gastric carcinogenesis, while elimination of H. pylori could potentially delay this process (Leung et al., 2001). It is worth noting that cell escape from apoptosis is an important link in the process of carcinogenesis.
Key virulence factors in apoptosis
H. pylori affects apoptosis through several virulence factors (Gonciarz et al., 2019). Among the numerous factors, the importance of Vacuolating cytotoxin A (VacA) and Cytotoxin-associated gene A (CagA) is obvious. Clinical studies have shown that gastric mucosal proliferation significantly correlates with the severity of acute gastritis in individuals infected with CagA+ VacA s1a strains of H. pylori. However, this increased proliferation was not accompanied by a parallel increase in apoptosis (Peek et al., 1997). The increased cell proliferation in the absence of a corresponding increase in apoptosis may explain the increased risk of gastric carcinoma associated with infection by CagA+ VacA s1a strains of H. pylori. VacA is observed in almost all clinical strains of H. pylori. While only certain strains produce the toxic and pathogenic VacA, this variant can induce vacuolization and apoptosis of cells (Ansari and Yamaoka, 2020). VacA can affect immune cells such as T cells and dendritic cells, leading to increased cell apoptosis (Winter et al., 2014; Kim et al., 2015). In addition, VacA can induce apoptosis in gastric cancer cells such as AGS and AZ-521 via low-density lipoprotein receptor-related protein-1 (LRP1) (Yahiro et al., 2012). But in some situations, the effect of VacA on cell apoptosis is also dual. In the response to H. pylori infection, infiltration of numerous inflammatory cells such as eosinophils occurs. VacA caused the translocation of cytoplasmic BAX to mitochondria and increased cytochrome c to facilitate apoptosis, while the expression of cellular apoptosis inhibitory protein (c-IAP)-2 was upregulated in the early phase of VacA stimulation (Kim et al., 2010). Another important virulence factor of H. pylori is the Cag secretion system. This system translocates CagA and peptidoglycan into the host cells, leading to the activation of PI3K signaling pathways. Activation of PI3K attenuated apoptosis in response to infection and was required for cell migration induced by H. pylori (Nagy et al., 2009). CagA can also inhibit cell apoptosis by reducing the expression of the tumor-suppressive E3 ubiquitin ligase proteins SIVA1 and ULF in SNU1 cells, possibly promoting the development of gastric cancer (Palrasu et al., 2022).
Having discussed the main virulence factors in H. pylori infection, we must not overlook other factors. H. pylori outer inflammatory protein A (OipA) is an outer membrane protein that contributes to gastric inflammation. It can trigger toxic events and initiate the apoptotic cascade in AGS through the intrinsic pathway (Teymournejad et al., 2017). In addition, Zhao et al. have shown that OipA affects apoptosis and cell cycle of AGS cells independent of its gene copy number (Zhao et al., 2020). However, Al-Maleki et al. reported that OipA “off” and ΔOipA cause a higher level of apoptosis in AGS cells than OipA “on” strains, and deletion of OipA increased bacterial VacA production (Al-Maleki et al., 2017). Virulence factors secreted by H. pylori, such as lipopolysaccharide (LPS) and the antigen complex glycic acid extract (GE), are also responsible for triggering apoptosis in gastric epithelial cells (Piotrowski et al., 1997; Gonciarz et al., 2020). In addition, the H. pylori T4SS effector D-glycero-β-D-manno-heptose-1,7-bisphosphate (βHBP) can trigger strong c-Abl threonine 735 phosphorylation and the process attenuates extrinsic apoptosis (Posselt et al., 2019). All these reports suggest that various virulence factors produced by H. pylori can regulate apoptosis through multiple signaling pathways. In addition to the bacterial virulence factors, the outer membrane vesicles (OMVs) secreted by H. pylori, which are involved in the transport of these factors, can also influence apoptosis through their various biologically active compounds (Chmiela et al., 2018). These metabolic by-products of H. pylori affect the migration, proliferation and apoptosis of normal gastric cells, while they do not affect the proliferation and migration of gastric cancer cells (He et al., 2020). This creates the conditions for the transition from gastritis to gastric cancer under H. pylori infection.
Dysregulation of apoptosis, from infection to gastric cancer
Apoptosis triggered by H. pylori may play a key role in the development of gastric cancer (Xia and Talley, 2001). Dysregulation of apoptosis within the gastric epithelium and the surrounding microenvironment is a characteristic feature of H. pylori infection and contributes to the development of gastric cancer (Lim et al., 2023). H. pylori leads to chronic inflammation because the host is unable to eradicate the infection. The chronic inflammation leads to oxidative stress originating from immune cells and within the epithelial cells of the stomach (Hardbower et al., 2013). H. pylori infection can synergistically interact with the tumor microenvironment (TME) and lead to DNA damage, abnormal gene expression and activation of signaling pathways. In addition, it affects the host immune system to promote tumor cell proliferation and metastasis, facilitate epithelial-mesenchymal transition (EMT), suppress apoptosis, and provide energy for tumor growth (Zhu et al., 2022). EMT is the most important biological event in epithelial cell invasion or metastasis. Yu et al. investigated that H. pylori CagA also triggers EMT in gastric cancer cells and promotes the mobility of gastric cancer cells by regulating PDCD4 (Yu et al., 2014).
The pathways by which H. pylori triggers carcinogenesis and progression of gastric cancer are complex and include nitration and oxidation of DNA by mutagenic factors, epigenetic alterations by H. pylori, disruption of the balance between cell proliferation and apoptosis, and promotion of cancer cell invasion and metastasis by H. pylori (Meng et al., 2015). A clinical report showed that dysregulation of the BAX/BCL-2 system was observed in gastric cancer with a significant down-regulation of the pro-apoptotic effect. This uncontrolled cell proliferation is coupled with various proto-oncogenes that contribute to this process, resulting in inhibition of apoptosis and increased cell survival, ultimately promoting tumor growth (Figure 2) (Konturek et al., 2001). Rosania et al. also reported that BCL-2 gene expression decreased in preneoplastic gastric lesions such as atrophic gastritis and intestinal metaplasia. However, they found that proliferation and apoptosis did not correlate with the status of H. pylori infection (Rosania et al., 2017).
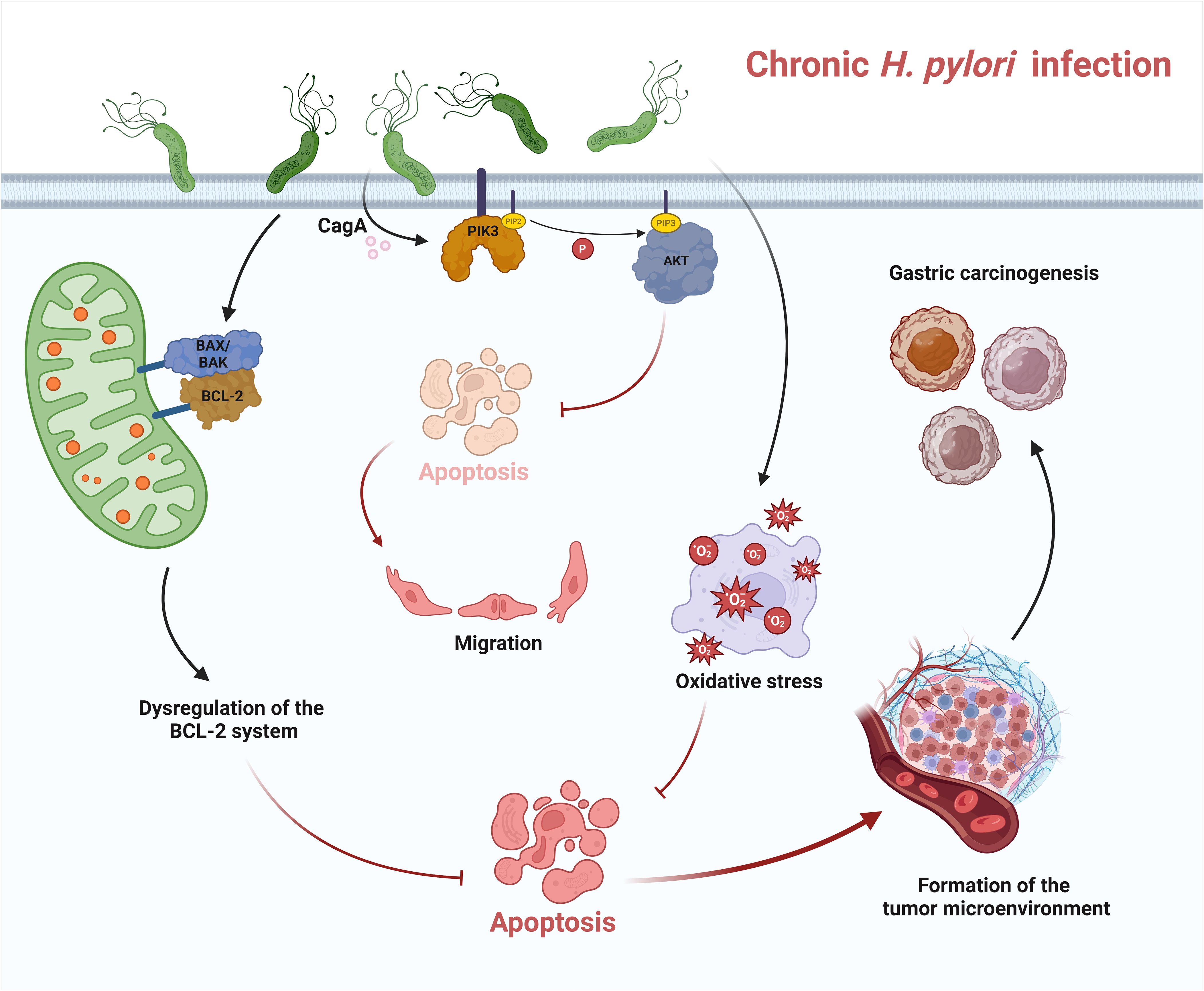
Figure 2 Apoptosis in chronic H. pylori infection. In chronic infection, H. pylori can trigger dysregulation of the BCL-2 system and downregulate apoptosis. H. pylori also leads to chronic inflammation, triggers oxidative stress and inhibits apoptosis. CagA, the virulence factor released by H. pylori, can regulate the activation of PI3K and AKT. AKT-dependent phosphorylation of caspase-9 attenuates apoptosis. All these dysregulations of apoptosis in chronic infection lead to the formation of a tumor microenvironment and ultimately contribute to the development of gastric cancer.
Necroptosis in H. pylori infection
Necroptosis is a form of PCD that differs from apoptosis in both its morphological and biochemical characteristics. While apoptosis is generally considered to be a process involving controlled shrinkage and fragmentation of the cell, necroptosis is a programmed form of necrosis leading to cell swelling, rupture of the plasma membrane and release of cell contents, which in turn results in inflammation and immune activation (Degterev et al., 2005). At the molecular level, necroptosis is initiated by the activation of RIPKs, in particular RIPK1 and RIPK3 (Sun et al., 2012). During necroptosis, these kinases form a complex known as the necrosome, which phosphorylates and activates the pseudokinase MLKL. The activated MLKL oligomerizes and translocates to the plasma membrane, where it disrupts membrane integrity and leads to cell lysis (Murphy et al., 2013). Necroptosis can be triggered in response to various stimuli, including death receptor activation, toll-like receptor signaling, or cellular stress such as DNA damage or viral infection (Mocarski et al., 2011). In contrast to apoptosis, which is often inhibited by the activation of caspases, necroptosis can occur when caspase-8 is blocked, making it a potential surrogate mechanism for cell death in conditions where apoptosis is impaired (Tummers and Green, 2017).
Signs of necroptosis were found in H. pylori infection. Virulence factors, such as VacA, induce necroptosis in immune cells and release high levels of inflammatory mediators. In the tissues of patients with clinical H. pylori infection, increased expression of the necroptosis key factor RIPK3-positive cells was found in both gastritis and atrophic lesions (Figure 1) (Cui et al., 2022). Phenotypic analysis showed that numerous RIPK3-positive cells in the gastric glands were identified as H + K+ ATPase-positive parietal cells, while in the lamina propria they were mainly CD3-positive T lymphocytes and CD68-positive macrophages.
Pyroptosis dysregulation in H. pylori infection
Pyroptosis is a form of PCD characterized by inflammatory reactions and cell lysis. It is distinct from apoptosis and necroptosis and plays a crucial role in the defense against microbial infections as well as in various inflammatory diseases (Pachathundikandi et al., 2016). Pyroptosis is triggered by the activation of specific intracellular signaling pathways that lead to the formation of large pores in the plasma membrane and subsequent cell swelling and lysis (Cookson and Brennan, 2001). At the molecular level, pyroptosis is mainly mediated by a group of proteins known as inflammasomes. These are intracellular multiprotein complexes that recognize microbial infections or cell damage (Martinon et al., 2002). In response to these stimuli, the inflammasomes assemble and activate caspase-1, also known as interleukin-1β (IL-1β)-converting enzyme, which cleaves GSDMD, a key executioner protein of pyroptosis (Shi et al., 2015). Cleaved GSDMD forms pores in the plasma membrane, leading to osmotic swelling and eventual cell lysis. The activation of inflammasomes can be triggered by various DAMPs (also called PAMPs in pathogen-infected cells) that originate from microbial infections or host cells (Rathinam et al., 2010). These include bacterial LPS, bacterial DNA and host-derived molecules such as ATP and uric acid crystals (Kayagaki et al., 2013). When these signals are sensed, oligomerization of inflammasomes occurs, particularly the NLRP3 inflammasome (Nucleotide-Binding Oligomerization Domain-like Receptor Family, Pyrin Domain-Containing 3), which recruits the adaptor protein ASC and leads to activation of caspase-1 and subsequent pyroptosis (Pachathundikandi et al., 2020). Pyroptosis is associated with the release of pro-inflammatory cytokines, including IL-1β and IL-18, which are synthesized as inactive precursors and require cleavage by activated caspase-1 for activation and secretion. The release of these cytokines amplifies the inflammatory response, recruits immune cells to the site of infection and helps eliminate pathogens (Galluzzi et al., 2018).
Pyroptosis dysregulation is associated with H. pylori infection. H. pylori can regulate the key steps of pyroptosis through various virulence factors such as UreB, CagA and VacA, thereby initiating the inflammatory cascade (Figure 3) (Kumar and Dhiman, 2018). Although various virulence factors can trigger pyroptosis, the flagellin protein of H. pylori, which can trigger NLRC4 phosphorylation, does not activate the inflammasome, suggesting that NLRC4 phosphorylation is not sufficient for inflammasome activation (Matusiak et al., 2015). These results were supported by the observation that S533 is phosphorylated in the inactive NLRC4 monomer (Hu et al., 2013).
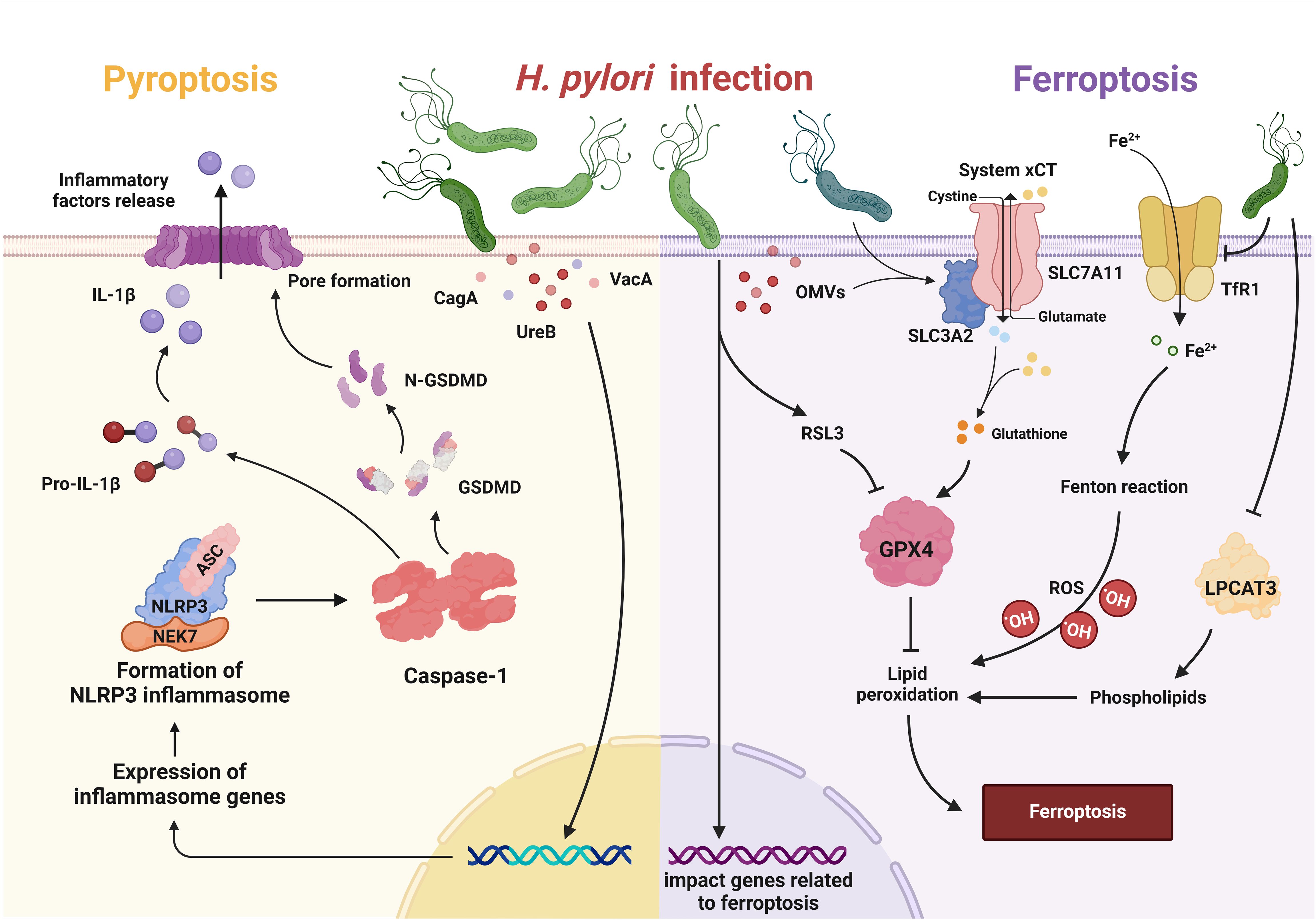
Figure 3 Pyroptosis and ferroptosis in H. pylori infection. In pyroptosis, H. pylori and its virulence factors (e.g. CagA, VacA, UreB) trigger the inflammatory cascade of NLRP3, which leads to the activation of caspase-1. Caspase-1 cleaves pro-IL-1β to generate its active form and also targets GSDMD, leading to recruitment of the N-terminal fragment of GSDMD to the plasma membrane. This leads to pore formation and the subsequent release of inflammatory factors. Upon ferroptosis, H. pylori and its OMVs upregulate Solute Carrier Family 3 Member 2 (SLC3A2) to attenuate ferroptosis, while repressing lysophosphatidylcholine acyltransferase 3 (LPCAT3) and downregulating transferrin receptor 1 (TfR1) to attenuate lipid peroxidation. H. pylori may also increase susceptibility to RAS-selective lethality 3 (RSL3)-induced ferroptosis by influencing associated genes.
The NLRP3 inflammasome
In H. pylori infection, IL-1β is highly expressed, leading to gastric acid inhibition, gastric cancer-associated gene methylation and angiogenesis. The NLRP3 inflammasome facilitates the maturation of IL-1β in various cell types such as macrophages, neutrophils and dendritic cells (Yuan et al., 2023). Another study showed that H. pylori infection indeed upregulates the expression of pro-IL-1β in human immune cells, but secretes only very low levels of mature IL-1β. However, administration of exogenous control activators such as nigericin or ATP to infected cells immediately triggered the formation of the NLRP3 inflammasome and the subsequent release of substantial amounts of mature IL-1β (Pachathundikandi et al., 2020). This suggests that chronic H. pylori infection manipulates inflammasome activation and pyroptosis to promote bacterial persistence.
Nevertheless, this deregulation of the inflammasome during H. pylori infection is susceptible to external stimulation by microbial, environmental, or host molecules that act as inflammasome activators, leading to the production of large amounts of mature IL-1β and signal-mediated gastric tumorigenesis in humans. Zhang et al. found that H. pylori infection leads to NLRP3 inflammasome activation, generation of intracellular ROS, and increased gastric cancer cell invasion and migration. In addition, ROS inhibition by N-acetyl-L-cysteine (NAC) effectively blocks NLRP3 activation and pyroptosis. Silencing of NLRP3 reduces the effects of H. pylori infection on gastric cancer cell migration and invasion (Zhang et al., 2022a). Taken together, this suggests that the role of the NLRP3 inflammasome in H. pylori infection deserves attention.
Programmed cell death associated with autophagy
Autophagy, derived from the Greek words “auto” meaning self and “phagy” meaning to eat, is a highly conserved cellular process that is essential for maintaining cellular health and homeostasis (Ohsumi, 2014). It plays a crucial role in various physiological processes such as development, immunity and energy metabolism, and its dysregulation has been found in numerous diseases such as cancer, neurodegenerative disorders and metabolic syndromes. The process of autophagy begins with the formation of a double-membrane structure, the autophagosome. This structure engulfs cellular components to be degraded, such as damaged organelles or protein aggregates. The autophagosome then fuses with lysosomes, organelles filled with digestive enzymes, to form an autolysosome. In the autolysosome, the engulfed cargo is broken down into its individual molecules such as amino acids, fatty acids and sugars, which can then be recycled by the cell to generate energy or build new cell components (Zhang et al., 2014). There are three different types of autophagy, each of which fulfills different functions within the cell. Macroautophagy is the best-studied form of autophagy and involves the massive degradation of cytoplasmic contents. In microautophagy, cytoplasmic components are taken up directly by the lysosomes by invagination of the lysosomal membrane. Chaperone-mediated autophagy (CMA) selectively targets specific proteins for degradation by directing them to the lysosomes with the help of chaperone proteins (Yang and Klionsky, 2020). Under normal circumstances, autophagy can be regarded as a PCD process in cells or organelles (Chesnokov et al., 2024). It is a renewal process in which cellular components are recycled and redistributed to enable a dynamic balance within the cell. It is like tearing down an old wall and using the bricks to build a new house. When autophagy is disrupted, impaired cellular components can aggregate, leading to cellular senescence or autophagic cell death (Nabavi-Rad et al., 2023). Normally, autophagy turns off apoptosis, while activation of pro-apoptotic caspases can interrupt the autophagy process. However, under certain circumstances, autophagy can consume too much cytoplasmic material and promote apoptosis or necrosis, ultimately leading to autophagic cell death (Mariño et al., 2014). Unfortunately, H. pylori infection can promote autophagic dysregulation.
Autophagy and H. pylori infection
Infection with H. pylori has different autophagic effects on various types of host cells. In macrophages, H. pylori can secrete cholesterol-α-glucosyltransferase (CGT), which inhibits the fusion of autophagosomes with lysosomes, leading to a significant increase in bacterial load within macrophages, thereby impairing the autophagic process of macrophage clearance (Lai et al., 2018). In gastric epithelial cells, H. pylori manipulates the NOX-ROS-Nrf2/HO-1-ROS loop to control intracellular oxidative stress and also affects ROS-mediated autophagy (Li et al., 2023a). In gastric cancer cells, autophagy is significantly altered after H. pylori infection and dysregulation of autophagy may be a causative factor for promoting the production of pro-inflammatory mediators in the human body (Halder et al., 2015; Sakatani et al., 2023). Initially, autophagy is a crucial pathway for controlling infection. However, prolonged exposure of the cells to the toxin VacA disrupts the induction of autophagy. This loss of autophagy leads to an accumulation of ROS, which can exacerbate inflammation and eventually lead to carcinogenesis (Raju et al., 2012). In H. pylori infection, the regulation of cellular autophagy can be mediated by microRNAs. Tang et al. showed that MIR30B was upregulated during H. pylori infection and favored bacterial replication by directly targeting ATG12 and Beclin1, important proteins involved in autophagy (Tang et al., 2012). Impairment of autophagy by MIR30B allows intracellular H. pylori to evade autophagic clearance and thus promotes the persistence of H. pylori infection. In another study, a significant correlation was found between MIR155 and immunohistochemical grade in H. pylori-positive patients. High expression of MIR155 could significantly reduce H. pylori survival by inducing autophagy (Wu et al., 2016). However, too much of a good thing is not always good. The increase of autophagy mediated by the Nrf2-HO-1 axis plays an important role in promoting H. pylori induced gastric carcinogenesis (Paik et al., 2019). VacA in H. pylori can also induce autophagy by promoting the formation of autophagosomes (Terebiznik et al., 2009). It can impair the activity of the lysosomal calcium channel MCOLN1/TRPML1, leading to the formation of enlarged, dysfunctional lysosomes and autophagosomes (Capurro et al., 2020). These autophagosomes are distinct from the induced large vacuoles and serve as an intracellular niche that allows the bacteria to evade eradication therapy. At this point, inhibition of autophagy stabilizes VacA and reduces vacuolization in the cells, limiting the damage caused by the toxin to the host cells (Raju and Jones, 2010). Thus, when autophagy is weakened, H. pylori can proliferate in the human body and its toxins cannot be effectively eliminated. Conversely, when autophagy is strengthened, H. pylori can also hide in abnormally increased autophagosomes and thus escape elimination by the host organism. It appears that H. pylori interferes with the normal autophagy process independently of the changes in autophagy and thus triggers negative reactions in the body. Furthermore, since there is evidence that autophagy associated with H. pylori depends on host cell types and bacterial strains, the ability of H. pylori to trigger autophagic responses should not be generalized (Deen et al., 2013).
Key virulence factors in autophagy
While the pathogenic mechanisms of H. pylori and its virulence factors are diverse, VacA and CagA play a crucial role in the interaction between H. pylori and the host autophagic machinery (Figure 4). VacA plays a key role in the pathogenesis of the disease by exerting pleiotropic effects on the host (Greenfield and Jones, 2013). One effect of acute VacA exposure is the induction of autophagy. However, prolonged exposure to the toxin impedes autophagy by inhibiting the maturation of autolysosomes. Kim et al. reported that the mitochondria-targeting bacterial toxin VacA inhibits a key sensor of host nutritional status, the mammalian target of rapamycin complex 1 (mTORC1), leading to a general cellular shift from biosynthetic to catabolic metabolism and further triggering the autophagic response (Kim et al., 2018). CagA can evade autophagic degradation in the host cells and thus exert its toxic effect (Tsugawa et al., 2019). Xie et al. found that autophagy initially increased and then gradually decreased during the duration of H. pylori infection in vitro, in a CagA-dependent manner. Moreover, dysregulation of autophagy promoted DNA damage in H. pylori-infected cells (Xie et al., 2020).
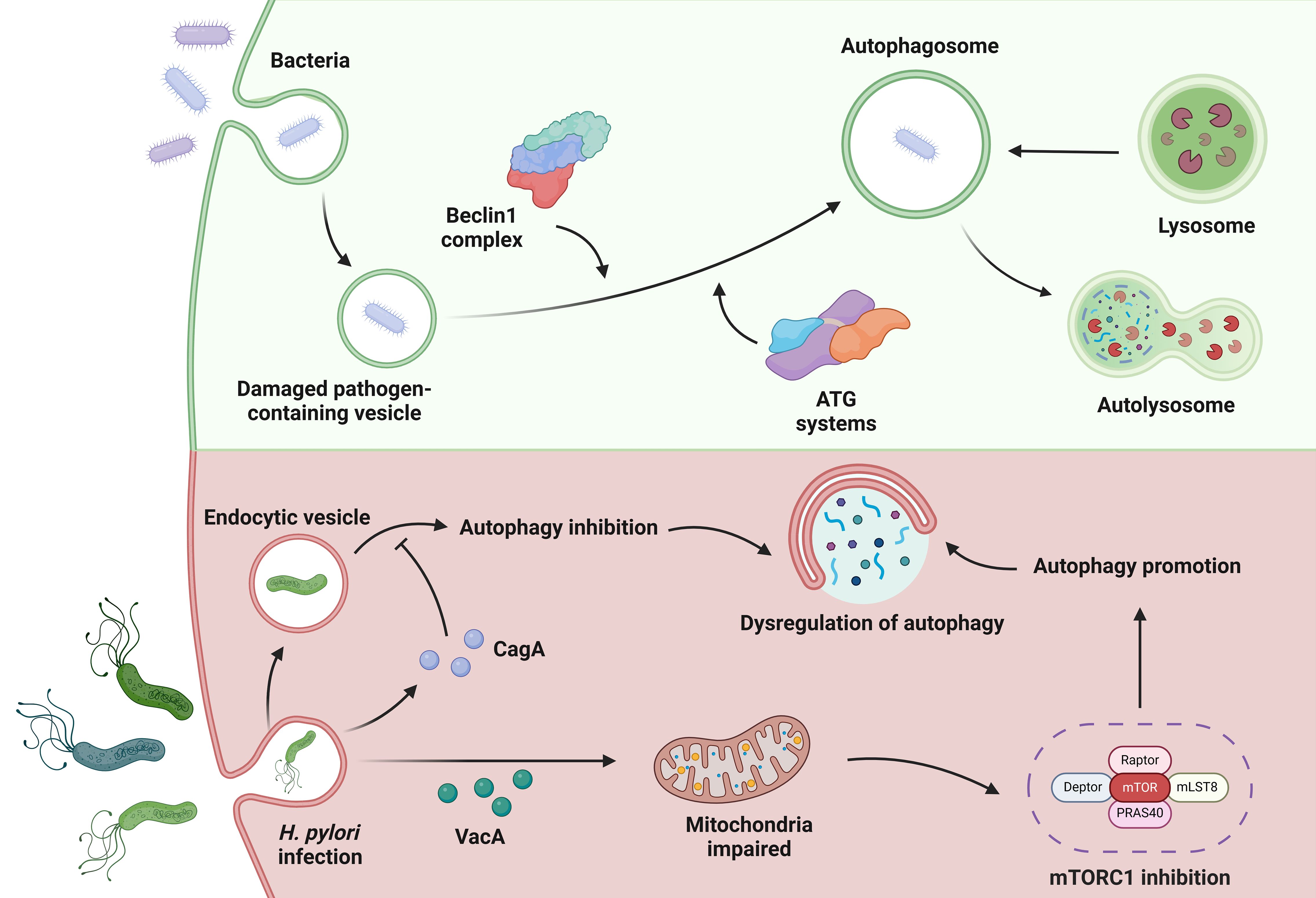
Figure 4 Autophagy in H. pylori infection. In autophagy, bacteria can be used to form damaged pathogen-containing vesicles. Under the action of the Beclin1 complex and the ATG systems, autophagosomes are formed that cooperate with the lysosomes in the cell to complete autophagy. When infected with H. pylori, the bacterial toxin VacA can target mitochondria to inhibit mTORC1 to promote cell autophagy. CagA, another toxin factor secreted by H. pylori, can evade autophagic degradation and gradually reduce cell autophagy. These complex circumstances during H. pylori infection lead to dysregulation of autophagy.
Dysregulation of autophagy leading to gastric cancer
Dysregulation of autophagy during H. pylori infection undoubtedly exacerbates the progression from gastritis to gastric cancer. For example, cagA and vacA can inhibit the activation of upstream signaling pathways of autophagy and thus inhibit autophagy of gastric mucosal cells in precancerous lesions of gastric cancer (Zhang et al., 2022b). The signaling pathways that link gastric cancer to H. pylori infection and are mediated by autophagy form a multi-layered protein network for regulation. Different signaling pathways have different functions and can interact with each other. Activation or abnormal induction of one of these pathways can lead to a cascade of cellular immune damage, ultimately culminating in the development of gastric cancer (Yang et al., 2022). For example, H. pylori can activate NF-κB and autophagy through nucleotide-binding oligomerization domain 1 (NOD1), allowing the bacterium to persist in the gastric niche and cause carcinogenic consequences (Suarez et al., 2015). In particular, H. pylori and its virulence factors can disrupt autophagy, leading to increased EMT (Shi et al., 2019). The occurrence of EMT and chronic inflammation leads to the emergence of cells with tumor stem cell (CSC) characteristics, such as the ability to migrate, invade and form tumor spheres (Courtois et al., 2021). In addition, the complex process of carcinogenesis triggered by H. pylori is closely linked to the genetic background of the host. Many of these genes influence cellular signaling pathways that contribute to inflammatory signals, inflammasome formation and autophagy (Mommersteeg et al., 2018). For example, clinical studies have shown that the autophagy gene ATG16L1 (rs2241880, G allele) was genotyped in subjects from different ethnic cohorts (Dutch and Australian) with premalignant gastric lesions of varying severity. The mechanism of the increased risk associated with ATG16L1 rs2441880 could be attributed to its modulation of H. pylori-induced ER stress signaling pathways and pro-inflammatory mediators (Mommersteeg et al., 2022).
Ferroptosis and H. pylori infection
Ferroptosis is a form of regulated cell death that is characterized by iron-dependent lipid peroxidation and differs from other forms of programmed cell death such as apoptosis and pyroptosis. This process was first described in 2012 and has since attracted considerable attention due to its role in various physiological and pathological conditions (Dixon et al., 2012). At the molecular level, ferroptosis involves the accumulation of lipid hydroperoxides, particularly phospholipids containing polyunsaturated fatty acids (PUFAs), resulting from the dysregulation of cellular antioxidant systems, including glutathione peroxidase 4 (GPX4) and the cystine/glutamate antiporter system xCT (Stockwell, 2022). GPX4 is a key enzyme responsible for the reduction of lipid hydroperoxides to non-toxic lipid alcohols, thus protecting cells from oxidative damage. Inhibition or depletion of GPX4 leads to an accumulation of lipid peroxides and ultimately triggers ferroptotic cell death (Yang et al., 2014). Several signaling pathways and molecules are involved in the regulation of ferroptosis. For example, the tumor suppressor p53 and the nuclear factor erythroid 2-related factor 2 (Nrf2) have been shown to modulate ferroptotic cell death through their effects on lipid peroxidation and antioxidant defense. In addition, proteins related to iron metabolism, such as ferritin, transferrin and TfR1, play a crucial role in regulating intracellular iron levels and thus influence susceptibility to ferroptosis (Stockwell, 2022).
Infection with H. pylori has a considerable influence on the process of ferroptosis (Figure 3). H. pylori and its components reduced the expression of LPCAT3, which plays a role in the generation of the lipid peroxide substrate. They also downregulated genes involved in iron uptake, such as TfR1. In addition, they upregulated the cystine/glutamate antiporter subunit SLC3A2 to counteract glutathione depletion, which attenuates ferroptosis (Melo et al., 2024). H. pylori infection can also influence the development of gastric cancer by affecting ferroptosis-related genes. Suppressor of cytokine signaling 1 (SOCS1), which is known to be a driver of ferroptosis, showed significant upregulation in both H. pylori-infected individuals and patients with gastric adenocarcinoma (STAD). Furthermore, increased SOCS1 expression correlated with an unfavorable prognosis in STAD patients. The increase in SOCS1 was associated with increased infiltration of immune cells and upregulation of immune checkpoints in STAD patients (Yan et al., 2023). Liu et al. demonstrated that the ferroptosis-related gene YWHAE is highly expressed in both H. pylori-associated gastritis and gastric cancer. The expression of YWHAE positively correlates with ferroptosis in gastric cancer and is associated with several cancer-related signaling pathways, including MAPK, NF-κB and PI3K (Liu et al., 2023). Zhu et al. also demonstrated that the molecular subtypes regulated by ferroptosis-associated genes correlate with TME cell infiltration and H. pylori infection increases the susceptibility of gastric cancer cells to RSL3-triggered ferroptosis (Zhu et al., 2023). To sum up, H. pylori appears to trigger pathways that can either enhance or block ferroptosis.
Therapy associated with PCD
Triple or quadruple therapies are the first choice in the treatment of H. pylori infections. Although conventional clinical therapies can effectively kill H. pylori, the continuous increase in antibiotic resistance has led to a decrease in the efficacy of standard triple and quadruple therapies. In addition, side effects related to H. pylori eradication, such as diarrhea, taste disturbance and nausea, have a certain incidence rate among patients (Liang et al., 2022). However, PCD such as autophagy, can play a role in eliminating intracellular H. pylori (Huang et al., 2018). With the increasing research on H. pylori infection related to PCD dysregulation, exploring the mechanisms of action of H. pylori virulence factors and their major targets in the different pathways of PCD may become an important research direction for future treatments of H. pylori infection and H. pylori-related gastric cancer. Establishing animal and cell models of H. pylori infection, exploring different cell death signaling pathways and gaining deeper insights into their importance in the pathogenesis of the disease, and searching for biologically active substances that can inhibit or induce PCD may be crucial for future treatments of H. pylori infection and associated gastric cancer (Table 2).
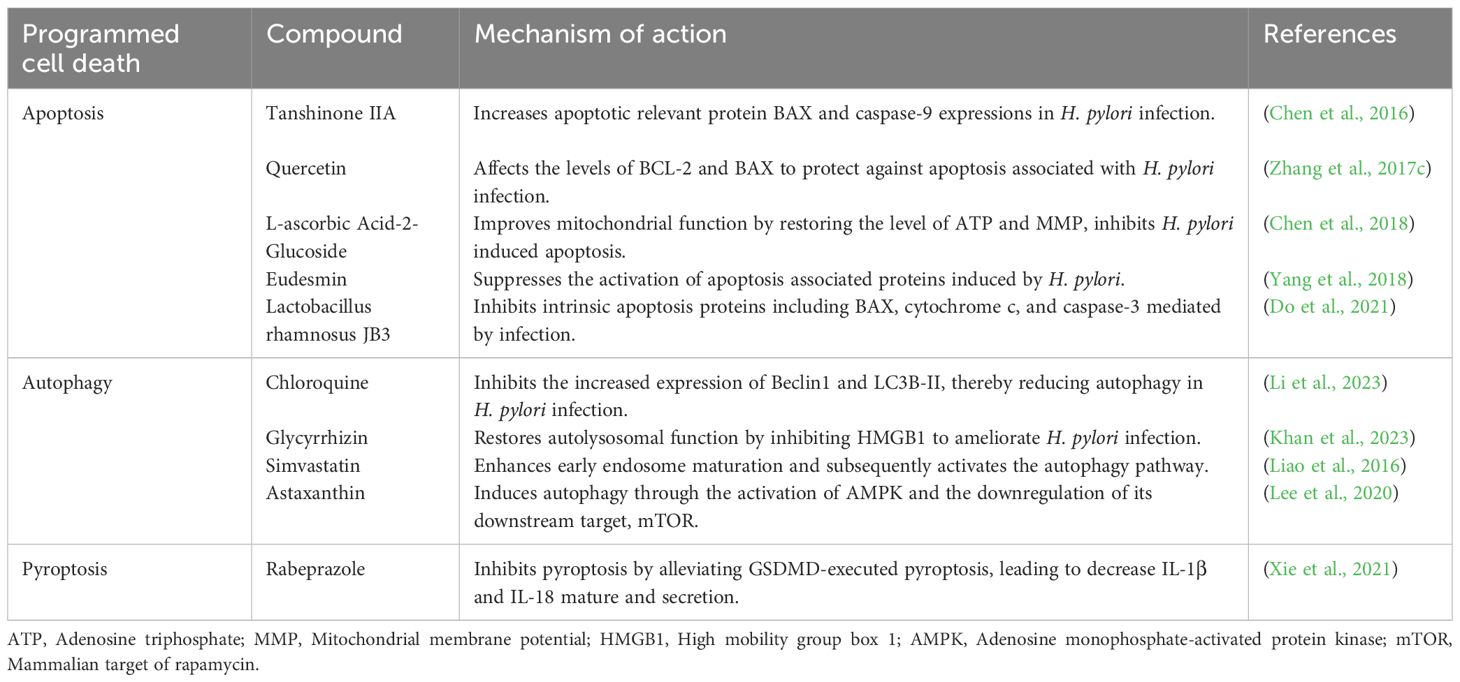
Table 2 Biologically active compounds targeting cell death pathways in H. pylori infection.
During apoptosis, several biologically active substances act in different phases, mainly influence intrinsic apoptosis. During the initiation phase of intrinsic apoptosis, upregulated BCL-2 homology region 3 proteins bind to survival-associated BCL-2 proteins and release BAX and BAK from their inhibition by survival-associated BCL-2 proteins. Quercetin modulates the balance between proliferation and apoptosis of gastric cells by affecting the levels of BCL-2 and BAX, thereby downregulating apoptosis induced by H. pylori infection and exerting a protective effect against gastritis-associated inflammation (Zhang et al., 2017c). In the subsequent steps of intrinsic apoptosis, BAX and BAK form oligomers that trigger mitochondrial events. L-ascorbic acid-2-glucoside (AA2G) reduces H. pylori-induced cell apoptosis by modulating the mitochondria-dependent apoptotic signaling pathway. The mechanism may be related to the restoration of mitochondrial ATP level and membrane potential, thereby improving mitochondrial function to protect gastric epithelial cells from H. pylori infection (Chen et al., 2018). After the mitochondrial phase, the assembly of the apoptosome scaffold is triggered and the caspase process is activated. Eudesmin inhibits the growth of H. pylori and suppresses the activation of caspase-associated proteins (caspase-3, -8, -9) induced by H. pylori (Yang et al., 2018). There are also preparations that can regulate intrinsic apoptosis in multiple steps, such as Lactobacillus rhamnosus JB3, which can dose-dependently inhibit proteins involved in the intrinsic apoptotic pathway, including BAX, cytochrome c and caspase-3, thus suppressing cell apoptosis induced by H. pylori (Do et al., 2021). The above preparations can all reduce the abnormal increase in apoptosis caused by H. pylori infection. However, in the chronic stage of H. pylori infection, apoptosis often decreases (Nagy et al., 2009). In chronic infection, treatment with tanshinone IIA significantly increases the expression of apoptosis-related proteins BAX and caspase-9, disrupts mitochondrial transmembrane potential, triggers the release of cytochrome c and activates caspase cascades. It markedly promotes intrinsic cellular apoptosis via the NF-κB and MAPK pathways and exerts a protective effect on host cells in severe inflammation and H. pylori-induced gastric cancer (Chen et al., 2016).
In autophagy, H. pylori infection can disrupt lysosomal function, leading to the development of enlarged and dysfunctional lysosomes and autophagosomes (Capurro et al., 2020). At the same time, infection leads to upregulation of autophagy-related proteins Beclin1 and LC3B-II, while chloroquine can inhibit this abnormal autophagic expression (Li et al., 2023b). In addition to suppressing abnormal autophagy during H. pylori infection, bioactive compounds can also promote the development of beneficial autophagy. The lysosomal autophagy pathway was impaired by an increase in lysosomal membrane permeabilization during H. pylori infection. However, glycyrrhizin preserves the integrity of the lysosomal membrane, which in turn facilitates the formation of autolysosomes. This restoration of lysosomal function leads to a reduction in intracellular H. pylori growth by eliminating the pathogenic niche (Khan et al., 2023). In addition, simvastatin can promote early endosome maturation and subsequently activate the autophagy pathway, which promotes lysosomal fusion and leads to the degradation of sequestered bacteria, thereby alleviating H. pylori-triggered inflammation (Liao et al., 2016).
Biologically active compounds can also comprehensively regulate several PCD processes to reduce the damage to normal PCD in the body caused by H. pylori infection. For example, astaxanthin induces autophagy by activating AMPK and downregulating its downstream target mTOR. When astaxanthin levels increase, it can also inhibit cell apoptosis induced by H. pylori (Lee et al., 2020). In addition, we should emphasize the use of the proton pump inhibitor rabeprazole in triple or quadruple drug regimens. In contrast to conventional acid-suppressive and antimicrobial drugs, rabeprazole acts by modulating PCD (Xie et al., 2021). NLRP3 and GSDMD are significantly increased in the gastric tissue of patients with H. pylori infection. Lansoprazole can attenuate GSDMD-induced cell pyroptosis, significantly inhibit the expression of ASC, NLRP3 and caspase-1, and thus lead to a reduction in the maturation and secretion of IL-1β.
Therapeutic interventions targeting regulators and effectors of various cell death pathways hold promise for improving treatment outcomes in patients with H. pylori infection. Given the complex nature of H. pylori infection, in which multiple cell death mechanisms interact with other cellular processes, effective therapies will likely include combinations of agents targeting different cell death programs, as well as molecules that affect additional cellular pathways. Therefore, the efficacy of the above agents at the animal and cellular level is promising, but their clinical utility needs to be further explored.
Discussion
A comprehensive understanding of programmed cell death has revealed ways to address aberrant situations in H. pylori infection. In apoptosis, H. pylori infection can cause dynamic changes in apoptosis levels in different cells. The increase in apoptosis in the acute phase may be an important mechanism by which the bacteria damage the stomach, while the decrease in apoptosis in the chronic phase leads to the formation of a tumor microenvironment that promotes the development of gastric cancer. In pyroptosis, the signaling pathway most closely associated with H. pylori infection is NLRP3. During infection, the pyroptotic inflammasome is susceptible to external stimuli such as microbes, environmental factors or inflammasome activators, leading to abundant production of inflammatory factors and promoting the occurrence of human gastric tumors. In autophagy, H. pylori and its virulence factors disrupt the normal level of autophagy, leading to an accumulation of damaged cellular components in the early stages and triggering autophagic cell death. In the later stages of infection, however, autophagic function is impaired, eventually leading to the development of gastric cancer. As for necroptosis and ferroptosis, there is relatively little research on their processes in H. pylori infection. However, animal studies, cell experiments and clinical observations have confirmed the importance of these two forms of cell death in H. pylori infection, and further investigation of their interaction is warranted.
The key question is whether blocking or promoting a specific PCD signaling pathway would be beneficial for the clinical treatment of H. pylori infection and associated gastric cancer. The prospects of such a therapy should be further investigated. We hope that research on PCD in the context of H. pylori infection will continue to progress and eventually lead to valuable life-saving treatments.
Author contributions
YL: Writing – original draft, Writing – review & editing. KL: Writing – original draft, Writing – review & editing. FL: Data curation, Visualization, Writing – review & editing. CZ: Conceptualization, Funding acquisition, Writing – review & editing. FC: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Our work is supported by National Natural Science Foundation of China (NSFC) #82205069 awarded to CZ.
Acknowledgments
Thanks to BioRender. All these figures are created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Maleki, A. R., Loke, M. F., Lui, S. Y., Ramli, N. S. K., Khosravi, Y., Ng, C. G., et al. (2017). Helicobacter pylori outer inflammatory protein A (OipA) suppresses apoptosis of AGS gastric cells in vitro. Cell. Microbiol. 19. doi: 10.1111/cmi.v19.12
Alzahrani, S., Lina, T. T., Gonzalez, J., Pinchuk, I. V., Beswick, E. J., Reyes, V. E. (2014). Effect of Helicobacter pylori on gastric epithelial cells. World J. Gastroenterol. 20, 12767–12780. doi: 10.3748/wjg.v20.i36.12767
Ansari, S., Yamaoka, Y. (2020). Role of vacuolating cytotoxin A in Helicobacter pylori infection and its impact on gastric pathogenesis. Expert Rev. Anti-Infective Ther. 18, 987–996. doi: 10.1080/14787210.2020.1782739
Bergmann, A. (2007). Autophagy and cell death: no longer at odds. Cell 131, 1032–1034. doi: 10.1016/j.cell.2007.11.027
Cadwell, K. (2016). Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat. Rev. Immunol. 16, 661–675. doi: 10.1038/nri.2016.100
Capurro, M. I., Prashar, A., Jones, N. L. (2020). MCOLN1/TRPML1 inhibition - a novel strategy used by Helicobacter pylori to escape autophagic killing and antibiotic eradication therapy in vivo. Autophagy 16, 169–170. doi: 10.1080/15548627.2019.1677322
Chaithongyot, S., Naumann, M. (2022). Helicobacter pylori-induced reactive oxygen species direct turnover of CSN-associated STAMBPL1 and augment apoptotic cell death. Cell. Mol. Life sciences: CMLS 79, 86. doi: 10.1007/s00018-022-04135-2
Chen, G.-Y., Shu, Y.-C., Chuang, D.-Y., Wang, Y.-C. (2016). Inflammatory and apoptotic regulatory activity of tanshinone IIA in helicobacter pylori -infected cells. Am. J. Chin. Med. 44, 1187–1206. doi: 10.1142/S0192415X1650066X
Chen, X., Liu, R., Liu, X., Xu, C., Wang, X. (2018). L-ascorbic Acid-2-Glucoside inhibits Helicobacter pylori-induced apoptosis through mitochondrial pathway in Gastric Epithelial cells. Biomedicine Pharmacotherapy = Biomedecine Pharmacotherapie 97, 75–81. doi: 10.1016/j.biopha.2017.10.030
Chesnokov, M. S., Mamedova, A. R., Zhivotovsky, B., Kopeina, G. S. (2024). A matter of new life and cell death: programmed cell death in the mammalian ovary. J. Biomed. Sci. 31, 31. doi: 10.1186/s12929-024-01017-6
Chmiela, M., Walczak, N., Rudnicka, K. (2018). Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. J. Biomed. Sci. 25, 78. doi: 10.1186/s12929-018-0480-y
Chu, S. H., Lim, J. W., Kim, K. H., Kim, H. (2003). NF-kappaB and Bcl-2 in Helicobacter pylori-induced apoptosis in gastric epithelial cells. Ann. New York Acad. Sci. 1010, 568–572. doi: 10.1196/annals.1299.106
Cookson, B. T., Brennan, M. A. (2001). Pro-inflammatory programmed cell death. Trends Microbiol. 9, 113–114. doi: 10.1016/S0966-842X(00)01936-3
Correa, P. (1995). Helicobacter pylori and gastric carcinogenesis. Am. J. Surg. Pathol. 19, S37–S43.
Courtois, S., Haykal, M., Bodineau, C., Sifré, E., Azzi-Martin, L., Ménard, A., et al. (2021). Autophagy induced by Helicobacter pylori infection is necessary for gastric cancer stem cell emergence. Gastric Cancer 24, 133–144. doi: 10.1007/s10120-020-01118-9
Cui, G., Yuan, A., Li, Z. (2022). Occurrences and phenotypes of RIPK3-positive gastric cells in Helicobacter pylori infected gastritis and atrophic lesions. Digestive Liver Dis. 54, 1342–1349. doi: 10.1016/j.dld.2022.04.013
Czabotar, P. E., Lessene, G., Strasser, A., Adams, J. M. (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature Reviews. Mol. Cell Biol. 15, 49–63. doi: 10.1038/nrm3722
Deen, N. S., Huang, S. J., Gong, L., Kwok, T., Devenish, R. J. (2013). The impact of autophagic processes on the intracellular fate of Helicobacter pylori: more tricks from an enigmatic pathogen? Autophagy 9, 639–652. doi: 10.4161/auto.23782
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi: 10.1038/nchembio711
Ding, S.-Z., Minohara, Y., Fan, X. J., Wang, J., Reyes, V. E., Patel, J., et al. (2007). Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infection Immun. 75, 4030–4039. doi: 10.1128/IAI.00172-07
Ding, S.-Z., Smith, M. F., Goldberg, J. B. (2008). Helicobacter pylori and mitogen-activated protein kinases regulate the cell cycle, proliferation and apoptosis in gastric epithelial cells. J. Gastroenterol. Hepatol. 23, e67–e78. doi: 10.1111/j.1440-1746.2007.04912.x
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Do, A. D., Chang, C.-C., Su, C.-H., Hsu, Y.-M. (2021). Lactobacillus rhamnosus JB3 inhibits Helicobacter pylori infection through multiple molecular actions. Helicobacter 26, e12806. doi: 10.1111/hel.12806
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differentiation 25, 486–541. doi: 10.1038/s41418-017-0012-4
Genta, R. M. (1997). Helicobacter pylori, inflammation, mucosal damage, and apoptosis: pathogenesis and definition of gastric atrophy. Gastroenterology 113, S51–S55. doi: 10.1016/S0016-5085(97)80012-1
Gonciarz, W., Krupa, A., Chmiela, M. (2020). Proregenerative activity of IL-33 in gastric tissue cells undergoing helicobacter pylori-induced apoptosis. Int. J. Mol. Sci. 21, 1801. doi: 10.3390/ijms21051801
Gonciarz, W., Krupa, A., Hinc, K., Obuchowski, M., Moran, A. P., Gajewski, A., et al. (2019). The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts. PloS One 14, e0220636. doi: 10.1371/journal.pone.0220636
Green, D. R. (2000). Apoptotic pathways: paper wraps stone blunts scissors. Cell 102, 1–4. doi: 10.1016/S0092-8674(00)00003-9
Greenfield, L. K., Jones, N. L. (2013). Modulation of autophagy by Helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol. 21, 602–612. doi: 10.1016/j.tim.2013.09.004
Halder, P., Datta, C., Kumar, R., Sharma, A. K., Basu, J., Kundu, M. (2015). The secreted antigen, HP0175, of Helicobacter pylori links the unfolded protein response (UPR) to autophagy in gastric epithelial cells. Cell. Microbiol. 17, 714–729. doi: 10.1111/cmi.12396
Hardbower, D. M., De Sablet, T., Chaturvedi, R., Wilson, K. T. (2013). Chronic inflammation and oxidative stress: the smoking gun for Helicobacter pylori-induced gastric cancer? Gut Microbes 4, 475–481. doi: 10.4161/gmic.25583
He, Y., Wang, C., Zhang, X., Lu, X., Xing, J., Lv, J., et al. (2020). Sustained exposure to helicobacter pylori lysate inhibits apoptosis and autophagy of gastric epithelial cells. Front. Oncol. 10. doi: 10.3389/fonc.2020.581364
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi: 10.1038/82732
Holokai, L., Chakrabarti, J., Broda, T., Chang, J., Hawkins, J. A., Sundaram, N., et al. (2019). Increased programmed death-ligand 1 is an early epithelial cell response to helicobacter pylori infection. PloS Pathog. 15, e1007468. doi: 10.1371/journal.ppat.1007468
Hu, Z., Yan, C., Liu, P., Huang, Z., Ma, R., Zhang, C., et al. (2013). Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175. doi: 10.1126/science.1236381
Huang, Y., Deng, X., Lang, J., Liang, X. (2018). Modulation of quantum dots and clearance of Helicobacter pylori with synergy of cell autophagy. Nanomedicine: Nanotechnology Biology Med. 14, 849–861. doi: 10.1016/j.nano.2017.12.016
Jost, P. J., Vucic, D. (2020). Regulation of cell death and immunity by XIAP. Cold Spring Harbor Perspect. Biol. 12, a036426. doi: 10.1101/cshperspect.a036426
Kato, K., Hasui, K., Wang, J., Kawano, Y., Aikou, T., Murata, F. (2008). Homeostatic mass control in gastric non-neoplastic epithelia under infection of Helicobacter pylori: an immunohistochemical analysis of cell growth, stem cells and programmed cell death. Acta Histochemica Et Cytochemica. 41, 23–38. doi: 10.1267/ahc.07021
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. doi: 10.1126/science.1240248
Kerr, J. F., Wyllie, A. H., Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. doi: 10.1038/bjc.1972.33
Khan, U., Karmakar, B. C., Basak, P., Paul, S., Gope, A., Sarkar, D., et al. (2023). Glycyrrhizin, an inhibitor of HMGB1 induces autolysosomal degradation function and inhibits Helicobacter pylori infection. Mol. Med. 29, 51. doi: 10.1186/s10020-023-00641-6
Kim, J. M., Kim, J. S., Kim, N., Ko, S. H., Jeon, J. I., Kim, Y.-J. (2015). Helicobacter pylori vacuolating cytotoxin induces apoptosis via activation of endoplasmic reticulum stress in dendritic cells. J. Gastroenterol. Hepatol. 30, 99–108. doi: 10.1111/jgh.12663
Kim, J. M., Kim, J. S., Lee, J. Y., Sim, Y.-S., Kim, Y.-J., Oh, Y.-K., et al. (2010). Dual effects of Helicobacter pylori vacuolating cytotoxin on human eosinophil apoptosis in early and late periods of stimulation. Eur. J. Immunol. 40, 1651–1662. doi: 10.1002/eji.200939882
Kim, I.-J., Lee, J., Oh, S. J., Yoon, M.-S., Jang, S.-S., Holland, R. L., et al. (2018). Helicobacter pylori Infection Modulates Host Cell Metabolism through VacA-Dependent Inhibition of mTORC1. Cell Host Microbe 23, 583–593.e588. doi: 10.1016/j.chom.2018.04.006
Konturek, P. C., Konturek, S. J., Pierzchalski, P., Bielański, W., Duda, A., Marlicz, K., et al. (2001). Cancerogenesis in Helicobacter pylori infected stomach–role of growth factors, apoptosis and cyclooxygenases. Med. Sci. Monitor: Int. Med. J. Exp. Clin. Res. 7, 1092–1107.
Kumar, S., Dhiman, M. (2018). Inflammasome activation and regulation during Helicobacter pylori pathogenesis. Microbial Pathogenesis 125, 468–474. doi: 10.1016/j.micpath.2018.10.012
Lai, C.-H., Huang, J.-C., Cheng, H.-H., Wu, M.-C., Huang, M.-Z., Hsu, H.-Y., et al. (2018). Helicobacter pylori cholesterol glucosylation modulates autophagy for increasing intracellular survival in macrophages. Cell. Microbiol. 20, e12947. doi: 10.1111/cmi.v20.12
Lee, H., Lim, J. W., Kim, H. (2020). Effect of astaxanthin on activation of autophagy and inhibition of apoptosis in helicobacter pylori-infected gastric epithelial cell line AGS. Nutrients 12, 1750. doi: 10.3390/nu12061750
Leung, W. K., Yu, J., To, K. F., Go, M. Y., Ma, P. K., Chan, F. K., et al. (2001). Apoptosis and proliferation in Helicobacter pylori-associated gastric intestinal metaplasia. Alimentary Pharmacol. Ther. 15, 1467–1472. doi: 10.1046/j.1365-2036.2001.01057.x
Li, M., Beg, A. A. (2000). Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J. Virol. 74, 7470–7477. doi: 10.1128/JVI.74.16.7470-7477.2000
Li, B., Du, Y., He, J., Lv, X., Liu, S., Zhang, X., et al. (2023a). Chloroquine inhibited Helicobacter pylori-related gastric carcinogenesis by YAP-β-catenin-autophagy axis. Microbial Pathogenesis 184, 106388. doi: 10.1016/j.micpath.2023.106388
Li, B., Lv, X., Xu, Z., He, J., Liu, S., Zhang, X., et al. (2023b). Helicobacter pylori infection induces autophagy via ILK regulation of NOXs-ROS-Nrf2/HO-1-ROS loop. World J. Microbiol. Biotechnol. 39, 284. doi: 10.1007/s11274-023-03710-4
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., et al. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. doi: 10.1016/S0092-8674(00)80434-1
Liang, B., Yuan, Y., Peng, X.-J., Liu, X.-L., Hu, X.-K., Xing, D.-M. (2022). Current and future perspectives for Helicobacter pylori treatment and management: From antibiotics to probiotics. Front. Cell. Infection Microbiol. 12. doi: 10.3389/fcimb.2022.1042070
Liao, W.-C., Huang, M.-Z., Wang, M. L., Lin, C.-J., Lu, T.-L., Lo, H.-R., et al. (2016). Statin decreases helicobacter pylori burden in macrophages by promoting autophagy. Front. Cell. Infection Microbiol. 6. doi: 10.3389/fcimb.2016.00203
Lim, M. C. C., Jantaree, P., Naumann, M. (2023). The conundrum of Helicobacter pylori-associated apoptosis in gastric cancer. Trends Cancer 9, 679–690. doi: 10.1016/j.trecan.2023.04.012
Liu, J.-F., Guo, D., Kang, E.-M., Wang, Y.-S., Gao, X.-Z., Cong, H.-Y., et al. (2021). Acute and chronic infection of H. pylori caused the difference in apoptosis of gastric epithelial cells. Microb. Pathogen. 150, 104717. doi: 10.1016/j.micpath.2020.104717
Liu, D., Peng, J., Xie, J., Xie, Y. (2024). Comprehensive analysis of the function of helicobacter-associated ferroptosis gene YWHAE in gastric cancer through multi-omics integration, molecular docking, and machine learning. Apoptosis: Int. J. Programmed Cell Death. 29 (3-4), 439–456. doi: 10.1007/s10495-023-01916-3
Mariño, G., Niso-Santano, M., Baehrecke, E. H., Kroemer, G. (2014). Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94. doi: 10.1038/nrm3735
Martinon, F., Burns, K., Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
Matusiak, M., Van Opdenbosch, N., Vande Walle, L., Sirard, J.-C., Kanneganti, T.-D., Lamkanfi, M. (2015). Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc. Natl. Acad. Sci. United States America 112, 1541–1546. doi: 10.1073/pnas.1417945112
Melo, J., Cavadas, B., Pereira, L., Figueiredo, C., Leite, M. (2024). Transcriptomic remodeling of gastric cells by Helicobacter pylori outer membrane vesicles. Helicobacter 29, e13031. doi: 10.1111/hel.13031
Meng, W., Bai, B., Sheng, L., Li, Y., Yue, P., Li, X., et al. (2015). Role of Helicobacter pylori in gastric cancer: advances and controversies. Discovery Med. 20, 285–293.
Mizushima, N., Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mocarski, E. S., Upton, J. W., Kaiser, W. J. (2011). Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 12, 79–88. doi: 10.1038/nri3131
Mommersteeg, M. C., Simovic, I., Yu, B., Van Nieuwenburg, S., Bruno, I. M. J., Doukas, M., et al. (2022). Autophagy mediates ER stress and inflammation in Helicobacter pylori-related gastric cancer. Gut Microbes 14, 2015238. doi: 10.1080/19490976.2021.2015238
Mommersteeg, M. C., Yu, J., Peppelenbosch, M. P., Fuhler, G. M. (2018). Genetic host factors in Helicobacter pylori-induced carcinogenesis: Emerging new paradigms. Biochim. Et Biophys. Acta Rev. Cancer 1869, 42–52. doi: 10.1016/j.bbcan.2017.11.003
Moujalled, D., Strasser, A., Liddell, J. R. (2021). Molecular mechanisms of cell death in neurological diseases. Cell Death Differentiation 28, 2029–2044. doi: 10.1038/s41418-021-00814-y
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J.-G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. doi: 10.1016/j.immuni.2013.06.018
Nabavi-Rad, A., Yadegar, A., Sadeghi, A., Aghdaei, H. A., Zali, M. R., Klionsky, D. J., et al. (2023). The interaction between autophagy, Helicobacter pylori, and gut microbiota in gastric carcinogenesis. Trends Microbiol. 31, 1024–1043. doi: 10.1016/j.tim.2023.04.001
Nagy, T. A., Frey, M. R., Yan, F., Israel, D. A., Polk, D. B., Peek, R. M. (2009). Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J. Infect. Dis. 199, 641–651. doi: 10.1086/596660
Newton, K., Strasser, A., Kayagaki, N., Dixit, V. M. (2024). Cell death. Cell 187, 235–256. doi: 10.1016/j.cell.2023.11.044
Ohsumi, Y. (2014). Historical landmarks of autophagy research. Cell Res. 24, 9–23. doi: 10.1038/cr.2013.169
Pachathundikandi, S. K., Blaser, N., Bruns, H., Backert, S. (2020). Helicobacter pylori avoids the critical activation of NLRP3 inflammasome-mediated production of oncogenic mature IL-1β in human immune cells. Cancers 12, 803. doi: 10.3390/cancers12040803
Pachathundikandi, S. K., Müller, A., Backert, S. (2016). Inflammasome activation by helicobacter pylori and its implications for persistence and immunity. Curr. Topics Microbiol. Immunol. 397, 117–131. doi: 10.1007/978-3-319-41171-2_6
Paik, J. Y., Lee, H. G., Piao, J.-Y., Kim, S.-J., Kim, D.-H., Na, H.-K., et al. (2019). Helicobacter pylori infection promotes autophagy through Nrf2-mediated heme oxygenase upregulation in human gastric cancer cells. Biochem. Pharmacol. 162, 89–97. doi: 10.1016/j.bcp.2019.02.003
Palrasu, M., Zaika, E., Paulrasu, K., Caspa Gokulan, R., Suarez, G., Que, J., et al. (2022). Helicobacter pylori pathogen inhibits cellular responses to oncogenic stress and apoptosis. PloS Pathog. 18, e1010628. doi: 10.1371/journal.ppat.1010628
Peek, R. M., Moss, S. F., Tham, K. T., Pérez-Pérez, G. I., Wang, S., Miller, G. G., et al. (1997). Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J. Natl. Cancer Institute 89, 863–868. doi: 10.1093/jnci/89.12.863
Piotrowski, J., Piotrowski, E., Skrodzka, D., Slomiany, A., Slomiany, B. L. (1997). Induction of acute gastritis and epithelial apoptosis by Helicobacter pylori lipopolysaccharide. Scandinavian J. Gastroenterol. 32, 203–211. doi: 10.3109/00365529709000195
Posselt, G., Wiesauer, M., Chichirau, B. E., Engler, D., Krisch, L. M., Gadermaier, G., et al. (2019). Helicobacter pylori-controlled c-Abl localization promotes cell migration and limits apoptosis. Cell communication signaling: CCS 17, 10. doi: 10.1186/s12964-019-0323-9
Puthalakath, H., Strasser, A. (2002). Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differentiation 9, 505–512. doi: 10.1038/sj.cdd.4400998
Raju, D., Hussey, S., Ang, M., Terebiznik, M. R., Sibony, M., Galindo-Mata, E., et al. (2012). Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 142, 1160–1171. doi: 10.1053/j.gastro.2012.01.043
Raju, D., Jones, N. L. (2010). Methods to monitor autophagy in H. pylori vacuolating cytotoxin A (VacA)-treated cells. Autophagy 6, 138–143. doi: 10.4161/auto.6.1.10222
Rathinam, V., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402. doi: 10.1038/ni.1864
Ricci, V., Giannouli, M., Romano, M., Zarrilli, R. (2014). Helicobacter pylori gamma-glutamyl transpeptidase and its pathogenic role. World J. Gastroenterol. 20, 630–638. doi: 10.3748/wjg.v20.i3.630
Rosania, R., Varbanova, M., Wex, T., Langner, C., Bornschein, J., Giorgio, F., et al. (2017). Regulation of apoptosis is impaired in atrophic gastritis associated with gastric cancer. BMC Gastroenterol. 17, 84. doi: 10.1186/s12876-017-0640-7
Sakatani, A., Hayashi, Y., Saiki, H., Kato, M., Uema, R., Inoue, T., et al. (2023). A novel role for Helicobacter pylori cytotoxin-associated gene A in negative regulation of autophagy in human gastric cells. BMC Gastroenterol. 23, 326. doi: 10.1186/s12876-023-02944-8
Shi, Y., Yang, Z., Zhang, T., Shen, L., Li, Y., Ding, S. (2019). SIRT1-targeted miR-543 autophagy inhibition and epithelial-mesenchymal transition promotion in Helicobacter pylori CagA-associated gastric cancer. Cell Death Dis. 10, 625. doi: 10.1038/s41419-019-1859-8
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shirin, H., Moss, S. F. (1998). Helicobacter pylori induced apoptosis. Gut 43, 592–594. doi: 10.1136/gut.43.5.592
Singh, M., Prasad, K. N., Saxena, A., Yachha, S. K. (2006). Helicobacter pylori induces apoptosis of T- and B-cell lines and translocates mitochondrial apoptosis-inducing factor to nucleus. Curr. Microbiol. 52, 254–260. doi: 10.1007/s00284-005-0103-1
Stennicke, H. R., Jürgensmeier, J. M., Shin, H., Deveraux, Q., Wolf, B. B., Yang, X., et al. (1998). Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 273, 27084–27090. doi: 10.1074/jbc.273.42.27084
Stockwell, B. R. (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421. doi: 10.1016/j.cell.2022.06.003
Suarez, G., Romero-Gallo, J., Piazuelo, M. B., Wang, G., Maier, R. J., Forsberg, L. S., et al. (2015). Modification of helicobacter pylori peptidoglycan enhances NOD1 activation and promotes cancer of the stomach. Cancer Res. 75, 1749–1759. doi: 10.1158/0008-5472.CAN-14-2291
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Sun, J., Xu, K., Wu, C., Wang, Y., Hu, Y., Zhu, Y., et al. (2007). PD-L1 expression analysis in gastric carcinoma tissue and blocking of tumor-associated PD-L1 signaling by two functional monoclonal antibodies. Tissue Antigens 69, 19–27. doi: 10.1111/j.1399-0039.2006.00701.x
Tang, D., Chen, X., Kang, R., Kroemer, G. (2021). Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125. doi: 10.1038/s41422-020-00441-1
Tang, B., Li, N., Gu, J., Zhuang, Y., Li, Q., Wang, H.-G., et al. (2012). Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori. Autophagy 8, 1045–1057. doi: 10.4161/auto.20159
Tavares, R., Pathak, S. K. (2017). Helicobacter pylori Secreted Protein HP1286 Triggers Apoptosis in Macrophages via TNF-Independent and ERK MAPK-Dependent Pathways. Front. Cell. Infection Microbiol. 7. doi: 10.3389/fcimb.2017.00058
Terebiznik, M. R., Raju, D., Vázquez, C. L., Torbricki, K., Kulkarni, R., Blanke, S. R., et al. (2009). Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5, 370–379. doi: 10.4161/auto.5.3.7663
Teymournejad, O., Mobarez, A. M., Hassan, Z. M., Talebi Bezmin Abadi, A. (2017). Binding of the Helicobacter pylori OipA causes apoptosis of host cells via modulation of Bax/Bcl-2 levels. Sci. Rep. 7, 8036. doi: 10.1038/s41598-017-08176-7
Tsai, H.-F., Hsu, P.-N. (2017). Modulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis by Helicobacter pylori in immune pathogenesis of gastric mucosal damage. J. Microbiology Immunology Infection = Wei Mian Yu Gan Ran Za Zhi 50, 4–9. doi: 10.1016/j.jmii.2016.01.002
Tsugawa, H., Mori, H., Matsuzaki, J., Sato, A., Saito, Y., Imoto, M., et al. (2019). CAPZA1 determines the risk of gastric carcinogenesis by inhibiting Helicobacter pylori CagA-degraded autophagy. Autophagy 15, 242–258. doi: 10.1080/15548627.2018.1515530
Tummers, B., Green, D. R. (2017). Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89. doi: 10.1111/imr.12541
Winter, J., Letley, D., Rhead, J., Atherton, J., Robinson, K. (2014). Helicobacter pylori membrane vesicles stimulate innate pro- and anti-inflammatory responses and induce apoptosis in Jurkat T cells. Infection Immun. 82, 1372–1381. doi: 10.1128/IAI.01443-13
Wu, K., Zhu, C., Yao, Y., Wang, X., Song, J., Zhai, J. (2016). MicroRNA-155-enhanced autophagy in human gastric epithelial cell in response to Helicobacter pylori. Saudi J. Gastroenterol. 22, 30–36. doi: 10.4103/1319-3767.173756
Xia, H. H., Talley, N. J. (2001). Apoptosis in gastric epithelium induced by Helicobacter pylori infection: implications in gastric carcinogenesis. Am. J. Gastroenterol. 96, 16–26. doi: 10.1111/j.1572-0241.2001.03447.x
Xie, J., Fan, L., Xiong, L., Chen, P., Wang, H., Chen, H., et al. (2021). Rabeprazole inhibits inflammatory reaction by inhibition of cell pyroptosis in gastric epithelial cells. BMC Pharmacol. Toxicol. 22, 44. doi: 10.1186/s40360-021-00509-7
Xie, C., Li, N., Wang, H., He, C., Hu, Y., Peng, C., et al. (2020). Inhibition of autophagy aggravates DNA damage response and gastric tumorigenesis via Rad51 ubiquitination in response to H. pylori infection. Gut Microbes 11, 1567–1589. doi: 10.1080/19490976.2020.1774311
Yahiro, K., Satoh, M., Nakano, M., Hisatsune, J., Isomoto, H., Sap, J., et al. (2012). Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J. Biol. Chem. 287, 31104–31115. doi: 10.1074/jbc.M112.387498
Yan, P., Cheng, M., Wang, L., Zhao, W. (2023). A ferroptosis-related gene in Helicobacter pylori infection, SOCS1, serves as a potential prognostic biomarker and corresponds with tumor immune infiltration in stomach adenocarcinoma: In silico approach. Int. Immunopharmacol. 119, 110263. doi: 10.1016/j.intimp.2023.110263
Yang, Y., Klionsky, D. J. (2020). Autophagy and disease: unanswered questions. Cell Death Differentiation 27, 858–871. doi: 10.1038/s41418-019-0480-9
Yang, Y., Shu, X., Xie, C. (2022). An overview of autophagy in helicobacter pylori infection and related gastric cancer. Front. Cell. Infection Microbiol. 12. doi: 10.3389/fcimb.2022.847716
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, J.-S., Wang, C.-M., Su, C.-H., Ho, H.-C., Chang, C.-H., Chou, C.-H., et al. (2018). Eudesmin attenuates Helicobacter pylori-induced epithelial autophagy and apoptosis and leads to eradication of H. pylori infection. Exp. Ther. Med. 15, 2388–2396. doi: 10.3892/etm.2018.5701
Yang, Y., You, B., Dong, S., Zhou, C. (2021). FRA-1 suppresses apoptosis of Helicobacter pylori infected MGC-803 cells. Mol. Biol. Rep. 48, 611–621. doi: 10.1007/s11033-020-06105-y
Yousefi, B., Mohammadlou, M., Abdollahi, M., Salek Farrokhi, A., Karbalaei, M., Keikha, M., et al. (2019). Epigenetic changes in gastric cancer induction by Helicobacter pylori. J. Cell. Physiol. 234, 21770–21784. doi: 10.1002/jcp.28925
Yu, H., Zeng, J., Liang, X., Wang, W., Zhou, Y., Sun, Y., et al. (2014). Helicobacter pylori promotes epithelial-mesenchymal transition in gastric cancer by downregulating programmed cell death protein 4 (PDCD4). PloS One 9, e105306. doi: 10.1371/journal.pone.0105306
Yuan, X.-Y., Zhang, Y., Zhao, X., Chen, A., Liu, P. (2023). IL-1β, an important cytokine affecting Helicobacter pylori-mediated gastric carcinogenesis. Microbial Pathogenesis. 174, 105933. doi: 10.1016/j.micpath.2022.105933
Zhang, S., Huang, J., Xie, X., He, Y., Mo, F., Luo, Z. (2017a). Quercetin from Polygonum capitatum Protects against Gastric Inflammation and Apoptosis Associated with Helicobacter pylori Infection by Affecting the Levels of p38MAPK, BCL-2 and BAX. Molecules 22, 744. doi: 10.3390/molecules22050744
Zhang, X., Li, C., Chen, D., He, X., Zhao, Y., Bao, L., et al. (2022b). H. pylori CagA activates the NLRP3 inflammasome to promote gastric cancer cell migration and invasion. Inflammation Res. 71, 141–155. doi: 10.1007/s00011-021-01522-6
Zhang, Y., Sun, H., Li, J., Rong, Q., Ji, X., Li, B. (2017b). The leukocyte-associated immunoglobulin (Ig)-like receptor-1 modulating cell apoptosis and inflammatory cytokines secretion in THP-1 cells after Helicobacter pylori infection. Microbial Pathogenesis 109, 292–299. doi: 10.1016/j.micpath.2017.06.012
Zhang, Y., Sun, H., Zhao, H., Chen, X., Li, J., Li, B. (2017c). Early apoptosis of monocytes induced by Helicobacter pylori infection through multiple pathways. Dev. Comp. Immunol. 73, 46–51. doi: 10.1016/j.dci.2017.03.010
Zhang, L., Sung, J. J. Y., Yu, J., Ng, S. C., Wong, S. H., Cho, C. H., et al. (2014). Xenophagy in Helicobacter pylori- and Epstein-Barr virus-induced gastric cancer. J. Pathol. 233, 103–112. doi: 10.1002/path.4351
Zhang, J., Wang, W., Yan, S., Li, J., Wei, H., Zhao, W. (2022a). CagA and VacA inhibit gastric mucosal epithelial cell autophagy and promote the progression of gastric precancerous lesions. Zhong Nan Da Xue Xue Bao. Yi Xue Ban 47, 942–951. doi: 10.11817/j.issn.1672-7347.2022.210779
Zhao, Q., Yin, W., Zhao, R., Wang, Y., Song, C., Wang, H., et al. (2020). Outer inflammatory protein of Helicobacter pylori impacts IL-8 expression, adherence, cell apoptosis and cell cycle of gastric cells independent of its copy number. Med. Microbiol. Immunol. 209, 621–630. doi: 10.1007/s00430-020-00688-w
Zhu, L., Huang, Y., Li, H., Shao, S. (2022). Helicobacter pylori promotes gastric cancer progression through the tumor microenvironment. Appl. Microbiol. Biotechnol. 106, 4375–4385. doi: 10.1007/s00253-022-12011-z
Keywords: Helicobacter pylori, programmed cell death, gastric cancer, infection, therapy
Citation: Lin Y, Liu K, Lu F, Zhai C and Cheng F (2024) Programmed cell death in Helicobacter pylori infection and related gastric cancer. Front. Cell. Infect. Microbiol. 14:1416819. doi: 10.3389/fcimb.2024.1416819
Received: 13 April 2024; Accepted: 08 July 2024;
Published: 31 July 2024.
Edited by:
Maurizio Sanguinetti, Catholic University of the Sacred Heart, ItalyReviewed by:
Diane Bimczok, Montana State University, United StatesGiuseppe Losurdo, University of Bari Medical School, Italy
Copyright © 2024 Lin, Liu, Lu, Zhai and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fafeng Cheng, NzAwNDc3QGJ1Y20uZWR1LmNu; Changming Zhai, ZHJ6Y21AYnVjbS5lZHUuY24=
†These authors have contributed equally to this work