 Chunhui Li1,2*†
Chunhui Li1,2*† Joshua Heuler2†
Joshua Heuler2† Duolong Zhu2†
Duolong Zhu2† Xiujuan Meng1
Xiujuan Meng1 Soumyadeep Chakraborty2Céline Harmanus3Shaohui Wang2Zhong Peng2
Soumyadeep Chakraborty2Céline Harmanus3Shaohui Wang2Zhong Peng2 Wiep Klaas Smits3
Wiep Klaas Smits3 Anhua Wu1
Anhua Wu1 Xingmin Sun2*
Xingmin Sun2*- 1Department of Infection Control Center of Xiangya Hospital, Central South University, Changsha, China
- 2Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 3Department of Medical Microbiology, Leiden University Medical Center, Leiden, Netherlands
Clostridioides difficile strains of sequence type (ST) 37, primarily including PCR ribotype (RT) 017, are prevalent in mainland China. Our study aimed to compare the major virulence factors of an epidemic C. difficile isolate of ST37 type (Xy06) from China with the well-characterized C. difficile reference strains R20291 (RT027) and CD630E (ST54), as well as a Chinese ST54 strain (Xy07) isolated from the same hospital. The Xy06 genome was predicted to harbor two complete prophages and several transposon-like elements. Comparative analysis of PaLoc revealed a truncated tcdA gene, a functional tcdB gene, a functional tcdC gene, and well-conserved tcdR and tcdE genes. Phenotypic comparisons showed that Xy06 was a robust producer of TcdB, readily sporulated and germinated, and strongly bound to human gut epithelial cells. In a mouse model of C. difficile infection, Xy06 was more virulent than strains CD630E and Xy07 and was comparable to strain R20291 in virulence. Our data suggest the potential threat of the epidemic ST37 strains in China.
1 Introduction
The symptoms of Clostridioides difficile infection (CDI) are mainly caused by toxin A (TcdA) and toxin B (TcdB), which are encoded by the tcdA and tcdB genes, respectively, in the pathogenicity locus (PaLoc). The PaLoc also contains three other genes (tcdC, tcdE, and tcdR) implicated in toxin expression or release (Martin-Verstraete et al., 2016). Besides TcdA and TcdB, certain strains, including the epidemic RT027 strains, also express the binary toxin (CDT), which is encoded on a different locus (CdtLoc). A small percentage of C. difficile strains produce a functional TcdB and a truncated, nonfunctional TcdA. These TcdA-negative, TcdB-positive (A-B+) strains are mostly typed as ST37/RT017 with few exceptions (Imwattana et al., 2019, Imwattana et al., 2022). C. difficile RT017 ranks among the most successful RTs of C. difficile in the world (Imwattana et al., 2019, Imwattana et al., 2022). Although C. difficile RT017 appears to originate from Asia, it has spread globally and caused multiple outbreaks worldwide (Cairns et al., 2017). Several publications reported that infections with strains of type ST37/RT017 are the leading epidemic healthcare-associated CDIs in China (Yan et al., 2013; Jin et al., 2017; Yang et al., 2017) and have attracted increasing attention (Hawkey et al., 2013; Du et al., 2014). Interestingly, ST81 (A-B+) strains have been reported to be dominant in some hospitals in Shanghai and Beijing in China. ST81 strains are genetically close to ST37 but produce less TcdB than ST37 strains (Senoh et al., 2015; Wang et al., 2018a; Yang et al., 2020; Su et al., 2021b; Wu et al., 2022). Several studies suggest that C. difficile RT017 can cause clinical disease symptoms that are indistinguishable from those caused by RTs of C. difficile strains producing both TcdA and TcdB (A+B+) (Goorhuis et al., 2011; Kim et al., 2012, Kim et al., 2016), and can cause disease as severe as that caused by “hypervirulent” C. difficile RT027 (Goorhuis et al., 2011). However, to date, no reports are available to evaluate the virulence characteristics of ST37 strains in vitro and in animal models of CDI in comparison with RT027 strains.
Here, we report a hypervirulent ST37/A-B+ C. difficile strain (Xy06) isolated from an ICU-hospitalized patient in China, where it caused severe infection symptoms. Xy07 is also an epidemic strain ST54 (A+B+) in our hospital and is included in this study as a clinical control strain from the same geographic region. We performed comparative genomic analyses of Xy06 with sequenced A-B+ C. difficile strains including two ST81 strains, and we further compared Xy06 with R20291, CD630E, and Xy07 in major virulence and pathogenicity in vitro and in vivo.
2 Materials and methods
2.1 Isolation and characterization of C. difficile strains
C. difficile Xy06 and Xy07 strains were isolated from stool samples of ICU hospitalized patients in China. C. difficile strains were routinely grown under anaerobic conditions in a Bactron III-2 anaerobic chamber in 3.7% Brain Heart Infusion (BHI) (Cat#53286, Sigma) supplemented with 0.05% yeast extract and 0.01% L-cysteine (BHIS) broth or on BHIS agar plates. Multi-locus sequence typing (MLST) of C. difficile strains was performed using a previously established method (Griffiths et al., 2010). Capillary PCR ribotyping was performed on Xy06 and Xy07 against the database of the Dutch National Reference Laboratory, a center recognized by the Europe Center for Disease Prevention and Control for the typing of C. difficile (Fawley et al., 2015),
2.2 Whole genome sequencing
Whole genome sequences of Xy06 and Xy07 were obtained using paired-end libraries and the sequencing-by-synthesis Illumina Hiseq 3000 platform. Approximately 200 Mb of clean data for each strain was obtained. Reads were assembled using Velvet (Zerbino and Birney, 2008), and contigs of 500 bp were scaffolded with SSPACE (Boetzer et al., 2011). This Whole Genome Shotgun project for Xy06 and Xy07 has been deposited at DDBJ/ENA/GenBank under the accession numbers JANFNF000000000 and JANFVQ000000000, respectively.
2.3 Phylogenetic analysis of A-B+ C. difficile strains
A literature search of Pubmed and Google Scholar databases with the search terms “toxin A-B+ C. difficile strain” was performed on August 6th, 2021, to identify previously isolated and sequenced A-B+ C. difficile strains. The appropriate genome sequence or accession number for each strain was sourced from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). Additional information on the characteristics of the genomes was also gathered from the GenBank entries. To produce a phylogenetic tree, the sequence files or accession numbers were uploaded to the Type Strain Genome Server (TYGS) (Meier-Kolthoff and Göker, 2019). TYGS compared the genomes using the Genome BLAST Distance Phylogeny approach (GBDP) (Wu and Wu, 2013) and generated a minimum evolution tree. The tree data file was visualized using an online tool (ETE Toolkit Phylogenetic tree (newick) viewer).
2.4 Prophage analysis
Putative prophages were detected in the bacterial genome using PHASTER (upgrade 6) (Arndt et al., 2016), which screens for regions with high similarity to a database of phage genes. The software assigned prophage regions a score that determines whether they are considered complete (>90), questionable (70-90), or incomplete (<70). The sequences of predicted complete Xy06 prophages were aligned with sequences of previously isolated phages using ClustalW in MegaX software (version 11.0.13) on default settings. A Maximum-likelihood phylogenetic analysis was performed using default parameters and 100 bootstrap replicates. PaLoc gene sequences for tcdA, tcdB, tcdR, tcdE, and tcdC were downloaded from the GenBank entry for the reference strain CD630 (AM180355.1), and CdtLoc gene sequences for cdtR, cdtA, and cdtB were downloaded from the GenBank entry for the reference strain R20291 (FN545816.1). PaLoc and CdtLoc sequences were aligned against the putative prophage sequences using the online BLAST+ function of the Galaxy Web server (https://test.galaxyproject.org/).
2.5 Genome comparison
Blast Ring Image Generator (BRIG) (Alikhan et al., 2011) was used to compare non-toxigenic strains and the reference strain CD630. The upper, middle, and minimum identity percentage thresholds were set to 100%, 80%, and 50%, respectively. To identify transposons in Xy06, transposon sequences were mined from GenBank files of various strains using the boundaries of each element as previously reported (Brouwer et al., 2011). BLASTn comparisons between transposon sequences and the Xy06 genome were performed online through the National Center for Biotechnology Information (NCBI, https://blast.ncbi.nlm.nih.gov/Blast.cgi). The locations of alignments between the transposon sequences and the Xy06 genome were annotated on a GenBank file of the Xy06 genome so that BRIG software could illustrate the putative transposon-like regions.
2.6 Analysis of toxin loci
C. difficile genomes were downloaded from GenBank, and then the previously annotated pathogenicity locus (PaLoc) and surrounding genes of each strain were color-coded in Artemis (Rutherford et al., 2000). Upstream and downstream genes from PaLoc were identified in the genome annotations based on previous work describing the conserved cdu and cdd genes in these areas (Braun et al., 1996b). Color-coded annotations were imported into EasyFig software (version 2.2.5) (Sullivan et al., 2011) to generate BLAST comparisons. The binary toxin locus (CdtLoc) was illustrated in the same manner.
To generate alignments of PaLoc genes, the sequences of PaLoc genes were mined from the genome using Artemis and aligned using an online T-COFFEE aligner (Di Tommaso et al., 2011) or the online MPI Bioinformatics Toolkit T-COFFEE server (Gabler et al., 2020). The alignment of the CdtLoc of CDT-negative (CDT-) strains was performed by mining these sequences from the appropriate GenBank files before aligning the sequences using MAFFT (Katoh and Standley, 2013) on the MPI Bioinformatics Toolkit server (Gabler et al., 2020).
A phylogenetic tree of TcdB amino acid sequences was generated based on previous work (Shen et al., 2020a) modifications. TcdB amino acid sequences were mined from GenBank (see Supplementary Material for details) and aligned using Muscle Algorithm in MegaX software (Kumar et al., 2018). A Neighbor-joining tree was then constructed in MegaX with 1000 bootstrap replicates.
2.7 Toxin A and toxin B production determined by ELISA
Toxin production was measured at four different time points (12, 24, 48 and 72 hours post-inoculation). Single colonies of the strains were initially cultured in BHIS medium for 18-24 hours, and cultures (0.5% v/v) were inoculated into fresh TY medium (3% w/v tryptose, 2% w/v yeast extract, 0.1% w/v thioglycollate, pH 7.4), different strain cultures at each time-point were adjusted to the same OD600 value after incubation. Cultures at each time point were centrifuged at 12,000 × g for 10 min at 4°C. Each supernatant was filtered through a 0.45-μm membrane filter. Toxin production was measured by ELISA. 96-well plates were coated with 50 μl per well of anti-TcdA (Novus Biologicals, USA) or anti-TcdB (A1, Gene Tex, USA) antibody at a concentration of 0.5 μg/ml and plates were kept at 4°C overnight. The coated plates were washed with PBST (washing buffer, 1×PBS+0.05% Tween 20) and blocked with 60 μl per well of blocking buffer (PBS+5% dry milk) for 2 hours. After being washed with PBST, the plates were incubated with 50 μl of supernatants/well collected at 12, 24, 48, and 72 hours post-inoculation in TY medium at room temperature for 1.5 hours. After being washed with PBST, the plates were further incubated with 50 μl of HRP-Chicken anti-Tcd A antibody (1: 5000 dilution, Gallus Immunotech, USA) or 50 μl HRP-Chicken anti-TcdB antibody (1: 2500 dilution, Gallus Immunotech, USA) per well at 37°C for 1 hour. The plate was washed again, and each well was supplemented with 50 μl TMB substrate and was incubated for 20-30 minutes. 50 μl of stop solution (2 N of H2SO4) was added to each well and mixed well. The optical density of each well was determined immediately using a spectrometer (Biotek Synergy HT Multi-Mode Microplate Reader).
2.8 Cytotoxicity assay
The murine CT26 cells cultured in RPMI 1640 (Gibco) with 10% (vol/vol) FBS (Gibco), 10 mM L-glutamine (Gibco), and 100 μg/mL penicillin/streptomycin (Gibco) were seeded in 96-well plates and were cultured to more than 80% confluence followed by addition of 16 μl of C. difficile culture supernatants collected at 72 hours post-inoculation in TY medium. Cell rounding was visualized by phase-contrast microscopy every two hours. Approximately 100 cells from 5 random fields were counted, and the percentage of rounded cells was determined. The experiments were repeated twice, and triplicate wells were assessed for cell rounding in each experiment.
2.9 C. difficile spore preparation
C. difficile culture was streaked on pre-reduced BHIS agar medium and incubated under anaerobic conditions for 48 hours. One colony was scraped up and suspended in 5 ml of Columbia Broth (CB) in a sterile 15 ml plastic tube and incubated for 48 hours. 50 μl of culture was then inoculated into 20 ml CB in a 50 ml tube and incubated for 24 hours. The entire 20 ml culture was inoculated into 500 ml of Clospore medium (Special Peptone Mix (SPM; Oxoid; LP0072), 10 g/L; Yeast extract, 10 g/L; (NH4)2SO4, 0.6 g/L; MgSO4.7H2O, 0.12 g/L; CaCl2.2H2O, 0.08 g/L; K2CO3, 3.48 g/L; KH2PO4, 2.6 g/L; pH 7.9± 0.1) in a 1 L bottle for 7-10 days of incubation. The spore suspensions were centrifuged at 10,000×g for 10 minutes, and the pellets were washed five times each with sterilized H2O. The final pellet was resuspended in 10 ml of sterilized H2O for stock. The suspension was then layered on top of a 10 ml bed volume of 50% (wt/vol) sucrose in water. The gradient was centrifuged at 3,200 × g for 20 min. During the centrifugation, vegetative cells and debris were collected at the interface while spores migrated through the 50% sucrose bed as pellets. After centrifugation, the spore pellet was washed five times to remove the sucrose and suspended in water. All spore preparations were >99% free of vegetative cells and debris, and all spores appeared phase bright. Before the adherence assay, spore suspensions were heated at 60°C for 20 minutes to activate the remaining vegetative cells. Spore concentration was determined as previously described (Sorg and Dineen, 2009).
2.10 Measurement of sporulation rate
C. difficile strains were cultured to mid-log phase in BHIS medium supplemented with 0.1% taurocholate (Sigma) and 0.5% fructose at 37 °C in an anaerobic chamber. A mixture of 70:30 sporulation medium (70% SMC medium and 30% BHIS medium including 63 g Bacto peptone, 3.5 g protease peptone, 11.1 g BHI medium, 1.5 g yeast extract, 1.06 g Tris base, 0.7 g NH4SO4) was prepared. Cultures were subsequently diluted to an optical density at OD600 of 0.05 in 70:30 medium. Approximately 24 hours after the start of the stationary phase (T24), 1-2 ml of samples were taken from the cultures.
To measure ethanol-resistant spore formation, 500 µl of samples from the sporulation medium were removed from the anaerobic chamber and mixed 1:1 with 95% ethanol for 15 min to kill vegetative cells. Samples were then returned to the anaerobic chamber. 100 µl of ethanol-treated cultures were mixed with 100 µl of 10% taurocholate and were plated onto BHIS agar to induce germination of C. difficile spores. Ethanol-resistant CFU/ml was determined after incubation for 24 h. Ethanol-resistant CFU/ml was divided by the total CFU/ml of the non-ethanol-treated cultures. A minimum of three biological replicates were performed for each strain.
2.11 Determination of spore germination
The initiation of spore germination was monitored aerobically at OD600. C. difficile spore germination was initiated by suspending spores in the germination solution [(10 mM Tris (pH 7.5), 150 mM NaCl, 100 mM glycine, and 10 mM taurocholic acid)]. Spores were heat-shocked at 65°C for 30 minutes and then placed on ice. Next, 5 µl of spores were diluted into 995 µl of buffer with or without germinant and mixed, and the change in optical density at 600nm (OD600) was monitored. The ratio of the OD600 at time X (Tx) to the OD600 at time zero (T0) was plotted against time. Germination rates were determined using the slopes of the linear portions of the germination plots. Data are reported as the averages from three independent experiments with standard deviations.
2.12 Determination of the spore colony-forming efficiency
Spore suspensions were serially diluted and plated on BHIS agar supplemented with 0.1%(w/v) taurocholic acid (TA). Plates were incubated anaerobically at 37°C for 24 h and colony counts of the starting suspension were determined.
The number of phase-bright spores/ml of starting suspension was counted directly by phase-contrast microscopy using a hemocytometer counting chamber (Bright-Line). The germination efficiency was defined as the percentage of total spores that gave rise to colonies on BHIS agar with 0.1% (w/v) TA, calculated by c.f.u. per ml colony count/c.f.u. per ml direct count by microscopy × 100%.
2.13 Ability of C. difficile spores to adhere to HCT-8 cells
Human gut epithelial HCT-8 cells were cultured in RPMI 1640 (Gibco) with 10% (vol/vol) FBS, 10 mM L-glutamine, and 100 μg/mL penicillin/streptomycin. Following five days of incubation, HCT-8 cells (5 ×105 cells per ml in a 6-well plate) were co-cultured with 100 μl of 5×107 spores/ml in sterile deionized water and incubated for 100 minutes at 37°C in the anaerobic chamber. After incubation, the cell-spore mixture was washed with 1× PBS via centrifugation at 800× g for 1 minute to remove any unattached spores. This washing step was performed three times, and the supernatants from each step were collected to enumerate any spores not adhering to the cells. The spores in the supernatant were enumerated on pre-reduced BHI plus 0.1% taurocholic acid (w/v). Controls included PBS incubated with spores and RPMI incubated with spores. All cell culture assays were performed in triplicates. The percentage of spore adherence was calculated by using the following formula (initial CFU/ml - eluted CFU/ml)/initial CFU/ml.
2.14 Evaluation of C. difficile strain pathogenicity in the mouse model of CDI
Six-week-old C57BL/6 female mice were purchased from Charles River Laboratories, MA. All animal experiments were approved by the institutional committee for animal care and performed at the University of South Florida. Forty mice were divided into four groups. Group 1 (n=10) was challenged with spores of C. difficile R20291. Group 2 (n=10) was challenged with spores of C. difficile CD630E, and Group 3 (n=10) was challenged with spores of C. difficile Xy06. Group 4 (n=10) was challenged with spores of C. difficile Xy07. All mice were given drinking water containing a mixture of five antibiotics including kanamycin (0.4 mg/ml), gentamycin (0.035 mg/ml), colistin (0.042 mg/ml), metronidazole (0.215 mg/ml) and vancomycin (0.045 mg/ml) for five days, and then received autoclaved water for two days, followed by a single dose of clindamycin (10 mg/kg) intraperitoneal injection before challenge day (day 0). On the challenge day (day 0), mice in each group were challenged with 106 C. difficile spores by gavage. Mice were monitored daily for a week after challenge with C. difficile spores for weight changes, survival, diarrhea, and other disease symptoms. Diarrhea was defined as wet tails along with loose or watery feces. The percentage of diarrhea was defined as the number of mice that developed diarrhea divided by the total number of mice in each group. The number of recorded dead mice included those that died after infection and those that were euthanized if weight loss was greater than 20%.
2.15 Statistical analysis
Animal survival was analyzed by Kaplan–Meier survival analysis with a log-rank test of significance. When comparing results for two groups, a Student’s unpaired t-test was used to assess significance. When comparing more than two groups, one-way analysis of variance (ANOVA) with post hoc analysis by Bonferroni tests was used. The results are expressed as the means ± standard errors of the means. Differences were considered significant if p < 0.05 (*). All statistical analyses were performed using GraphPad Prism.
3 Results
3.1 Ribotype and genomic characteristics of strain Xy06
To place the Xy06 strain in a phylogenetic context, we performed capillary PCR ribotyping and whole genome sequencing. Xy06 was determined to be an RT017 strain by capillary PCR ribotyping. Analysis of the draft genome sequence of Xy06 using a 7-gene multi-locus sequencing scheme confirmed the strain to be ST37, similar to other RT017 isolates. To analyze the Xy06 genome, we compiled sequenced A-B+ C. difficile strains from the literature for comparison (Supplementary Table 1). Strain Xy06 contains a 4,194,266 bp genome similar in size to previously reported A-B+ C. difficile genomes (Supplementary Table 1). The Xy06 genome has a GC content of 28.5%, in line with the 28-30% GC content typically observed in C. difficile genomes. The NCBI Prokaryotic Genome Annotation Pipeline (PGAP) predicted 48 tRNAs and 11 rRNAs.
3.2 Genomic comparison between Xy06 and the sequenced A-B+ C. difficile strains as well as the reference strain CD630 with a focus on PaLoc region
Genomes of all A-B+ C. difficile strains in Supplementary Table 1 were uploaded to the Type Strain Genome Server (TYGS) (Meier-Kolthoff and Göker, 2019) to generate a minimum evolution phylogenetic tree (Figure 1). The results indicate that Xy06 is most closely related to strains CDT4, DSM29627, M68, and CD161. Xy06, CD161, and CDT4 were all isolated from China, while DSM29627 and M68 were isolated in France and Ireland. Although not all the included strains have been ribotyped, Xy06 is more closely related to fellow RT017 strains (1470, M68, DSM 29627, CF5) than non-RT017 strains (88.64, SUC36, ES130, WA151, 173070). G89 and CD060, which are two ST81 strains, cluster with the ST37 strains used in this analysis (Xy06, M68, CDT4, CD161, DSM 29627, 1470, and CF5). A few prior studies have established that ST81 and ST37 strains are closely related (Wang et al., 2018b; Su et al., 2021a), which is reflected in our analysis.
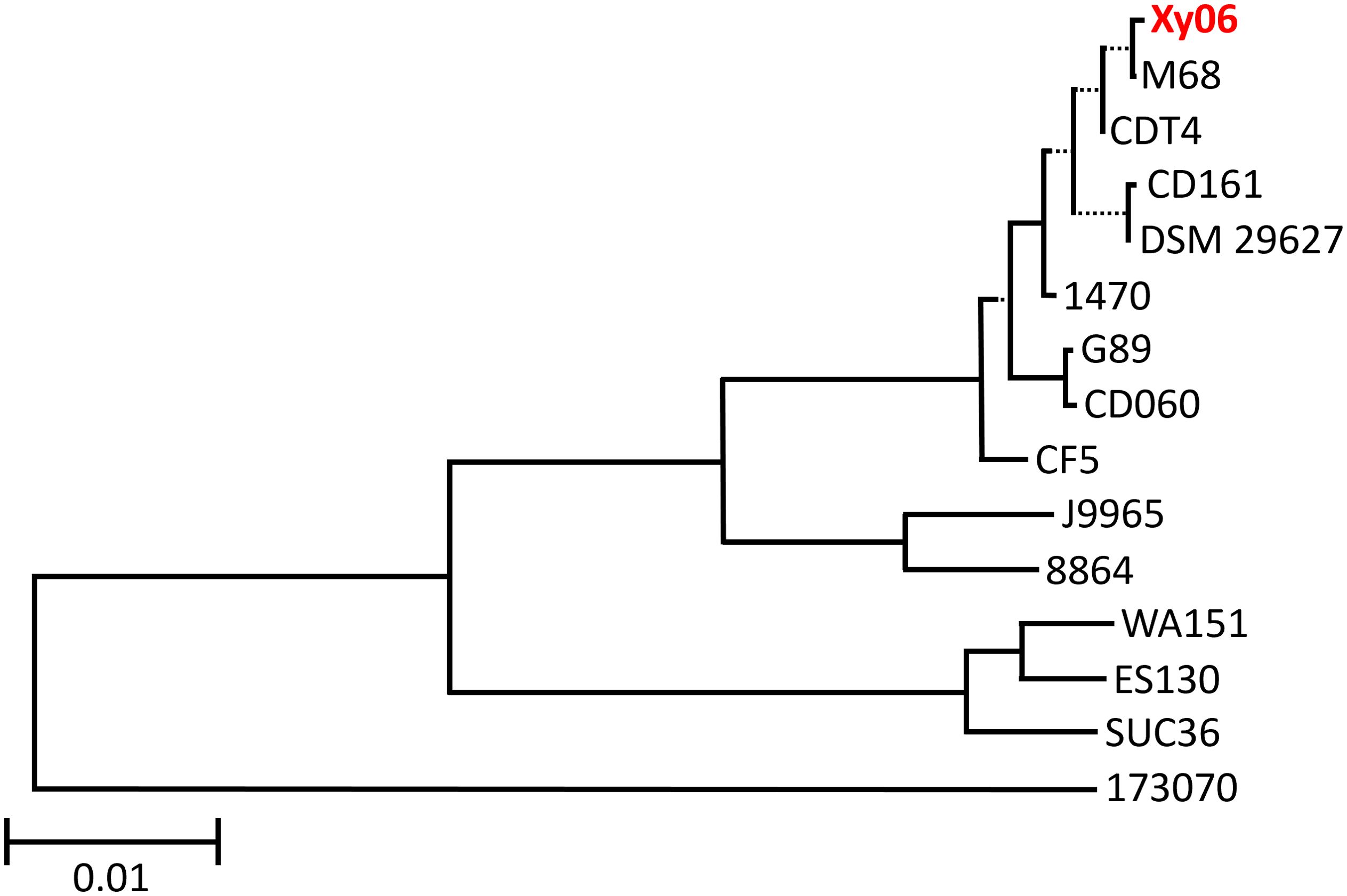
Figure 1. Phylogenetic tree of A-B+ C. difficile strains. A minimum evolution phylogenetic tree was generated using the Type (Strain) Genome Server. Xy06 is indicated in red. The scale bar indicates 0.01 nucleotide substitutions per site. Dotted lines separate select branches for easier viewing and do not indicate evolutionary distance.
In general, the pathogenicity locus (PaLoc) of C. difficile comprises a 19.6 kb region encoding five genes, including tcdA and tcdB. However, variant PaLocs have been noted in a minority of strains (Monot et al., 2015). Other genes in the locus include tcdR, tcdC, and tcdE. Specifically, tcdR is involved in the positive regulation of toxin transcription, tcdC may negatively regulate toxin transcription, and tcdE may facilitate toxin secretion (Braun et al., 1996a; Monot et al., 2015).
We examined how the PaLoc region and surrounding genes of strain Xy06 compared to other A-B+ C. difficile strains and the A+B+ reference strain CD630 (Figure 2). Upstream of the PaLoc region, C. difficile genomes typically encode cdu1 and cdu2, while downstream genes include cdd2, cdd3, and cdd4 (Braun et al., 1996b). Xy06 is an exception to this trend, as the cdd2-4 genes are not encoded directly downstream of the PaLoc region, but rather are ~3Mbp upstream of the other PaLoc genes. A handful of other A-B+ strains (173070, ES130, and WA151) are similar in that the usual downstream genes are not adjacent to the PaLoc region. On another note, the upstream cdu2 and cdu1 genes, while intact in strain Xy06, are noted to be either absent in strain 173070 or show a deletion in cdu2 in strains ES130, SUC36, and WA151.
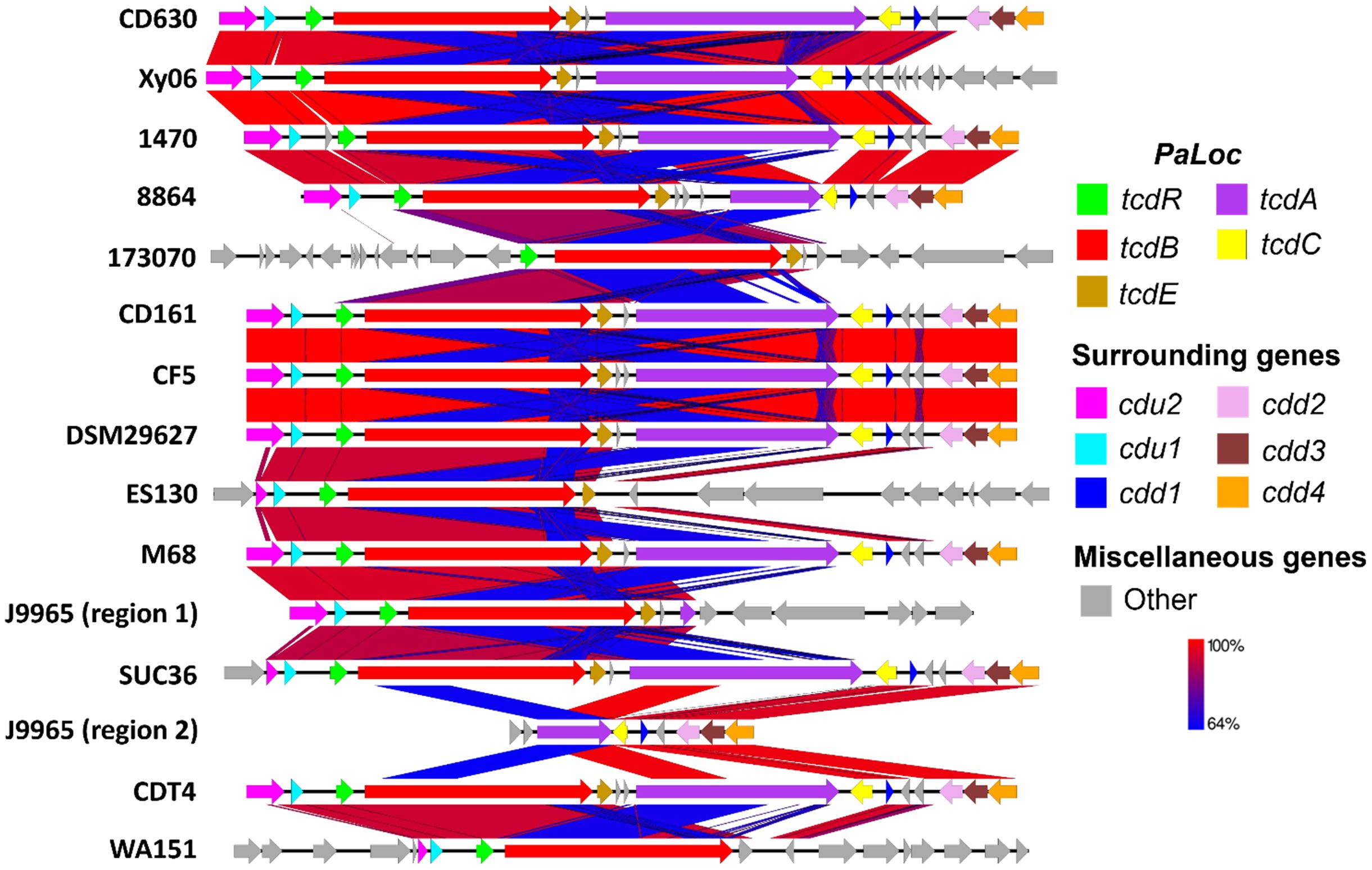
Figure 2. Comparative sequence analysis of PaLoc region. C. difficile typically encodes the genes cdu2 (pink) and cdu1 (light blue) upstream of PaLoc and cdd2 (pink), cdd3 (maroon), and cdd4 (orange) downstream of PaLoc, so these were used as boundaries for visualizing the genome below. PaLoc-adjacent genes were visualized for strains lacking the above surrounding genes (e.g., 173070). A-B+ C. difficile strains display an atypical organization of their PaLoc region and surrounding genes, such as deletions in tcdA (e.g., Xy06) or multiple genes (e.g., 173070, ES130, J9965). Blue BLAST alignments indicate regions with relatively lower (64%) shared identity, while red regions indicate high similarity.
3.2.1 Variations in tcdA
The tcdA gene of Xy06, like the tcdA genes of the other A-B+ strains, is visually shorter than that of CD630 (Figure 2). First, we aligned the tcdA sequence of Xy06 and other strains using the T-coffee algorithm on an online server (Di Tommaso et al., 2011). Second, we determined the length of each tcdA gene using the online ENDMEMO DNA/RNA Length Calculator tool (Table 1). It was determined the Xy06 tcdA exhibits a ~1.8 kB deletion relative to the wild-type CD630 tcdA. Strains 1470, CD161, CDT4, CF5, DSM29627, and M68 have deletions of virtually identical sizes. Upon translating the DNA sequence of the above strains’ tcdA gene, we detected a premature stop codon at AA position 47. These findings align with a prior study that reported these mutations in strain 1470 (Von Eichel-Streiber et al., 1999).
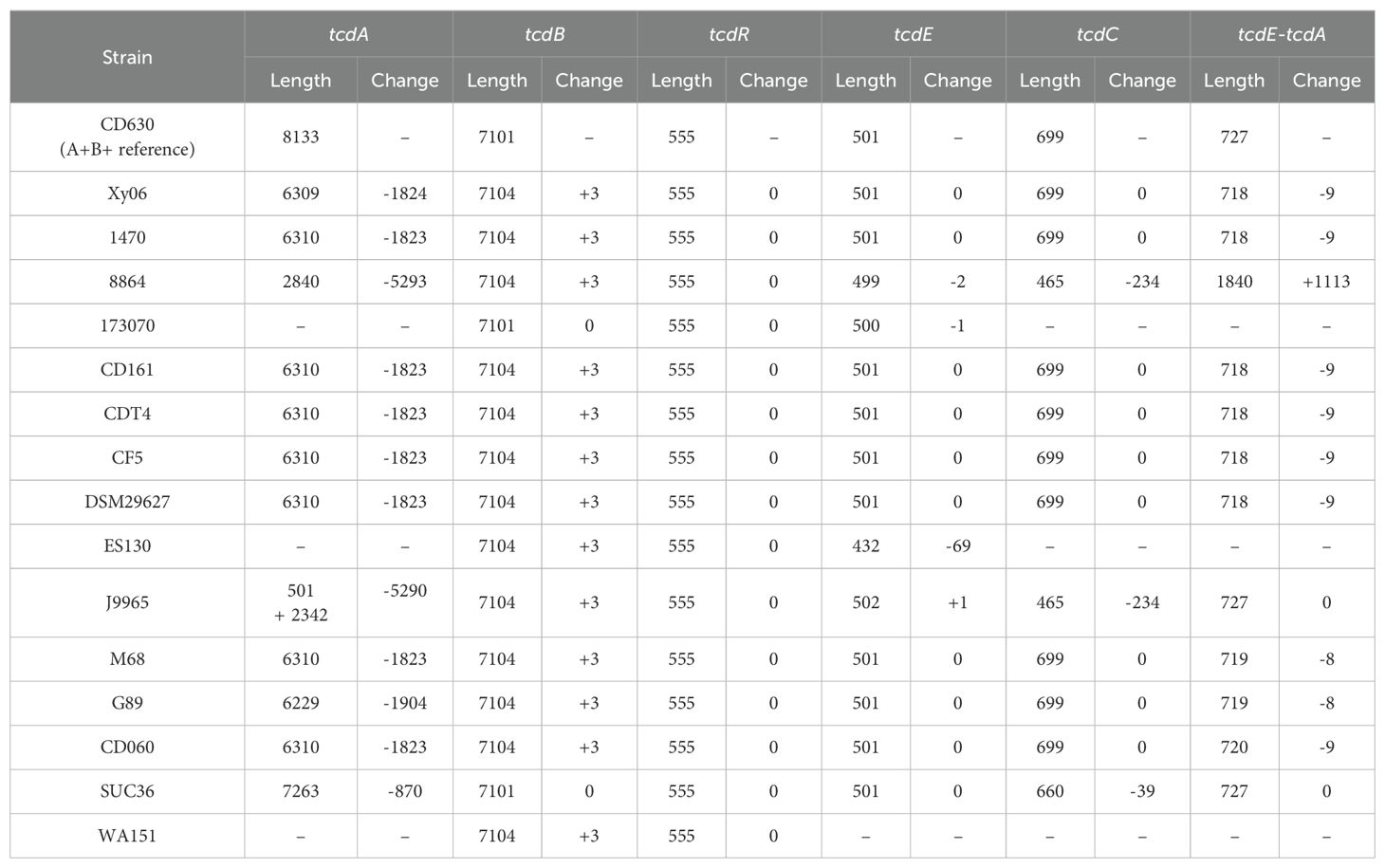
Table 1. Size of PaLoc genes in reference strain and A-B+ C. difficile strains.
Strain 8864 was found to have a ~5.3 kB deletion in tcdA, which is somewhat smaller than the previously reported figure of ~5.9 kB (Soehn et al., 1998). Translating the gene also indicated the presence of a stop codon at AA 699 before the end of the polypeptide sequence. It has also been noted that 8864 encodes a ~1kb insertion between tcdE and tcdA (Soehn et al., 1998). Our calculations in Table 1 confirm this result and demonstrate that no other A-B+ strain in our analysis has this feature. SUC36 displays the smallest deletion (<1 Kb) of the analyzed strains. Finally, strains 173070, ES130, and WA151 exhibit deletions of the entire tcdA and tcdC genes.
Another unique A-B+ strain is J9965. While Xy06 and the other strains contain a single PaLoc region, J9965 exhibits a split PaLoc. The first study to report on the J9965 (Rupnik et al., 2003) classified the strain in toxinotype XVII, and it appears that their PCR experiment successfully amplified the first, smaller tcdA fragment (J9965 region 1, Figure 2). The second, larger fragment that we report (J9965 region 2, Figure 2) was not detected by the prior study. The researchers noted that their amplified tcdA gene was ~1.9kb smaller than the tcdA of 8864, which is similar to the difference between the 8864 tcdA and J9965 tcdA fragment 1 lengths we calculated using sequencing data (~2.3kB, Table 1). Regarding the formation of the J9965 tcdA fragment 2, it is possible that a past recombination event split the PaLoc region at the tcdA gene, thereby abolishing tcdA expression.
3.2.2 Variations in tcdB
TcdB production is essential for the virulence of C. difficile. All A-B+ strains except for those in toxinotype XVI (strain SUC36) have been reported to contain a hybrid tcdB that is homologous to TcsL of C. sordellii (Chaves-Olarte et al., 2003). In line with this observation, strain Xy06 displays 83.68% similarity over 99% coverage between its tcdB and the tcsL toxin gene. Based on the T-coffee alignment of the tcdB genes of A-B+ strains as well as the calculated lengths of the tcdB genes (Table 1), we confirmed that the tcdB gene has a consistent size across A-B+ strains. A previous study classified TcdB into eight subtypes (Shen et al., 2020b), while another study demonstrated that certain subtypes may be more damaging than others in disease models (Pan et al., 2021). To determine the TcdB subtype of Xy06 and how it compares with other A-B+ strains as well as the hypervirulent R20291 strain, a neighbor-joining phylogenetic tree of TcdB amino acid sequences was constructed with annotated TcdB subtypes based on a previous subtyping of C. difficile TcdB (Figure 3) (Shen et al., 2020b). Just as Xy06 tcdA was similar to the tcdA of strains 1470, CD161, CDT4, CF5, DSM29627, and M68, the TcdB sequences were also highly similar between these strains as evidenced by the cluster of TcdB3 strains and Xy06 on the phylogenetic tree. The results suggest that Xy06 encodes a TcdB3 subtype toxin B, distinct from the TcdB1 subtype of CD630 and the TcdB2 subtype of R20291. Interestingly, the two ST81 strains used in this analysis, G89 and CD060, also encode TcdB3 toxins that are identical to that encoded by Xy06. Studies so far suggest that ST81 strains’ increased presence in healthcare facilities is, in part, due to their increased sporulation, motility, and antibiotic resistance (Wang et al., 2018b; Su et al., 2021a). Certain ST81 strains may express less tcdB at the mRNA level (Wang et al., 2018b), but no major mutations have been noted in ST81 TcdB relative to ST37 TcdB (Wang et al., 2018b; Su et al., 2021a). Our analysis shows that the G89 and CD060 encode the same TcdB sequence as Xy06 and other ST37 strains in our dataset, further suggesting that differences in virulence between ST37 and ST81 A-B+ strains is due to factors other than TcdB.
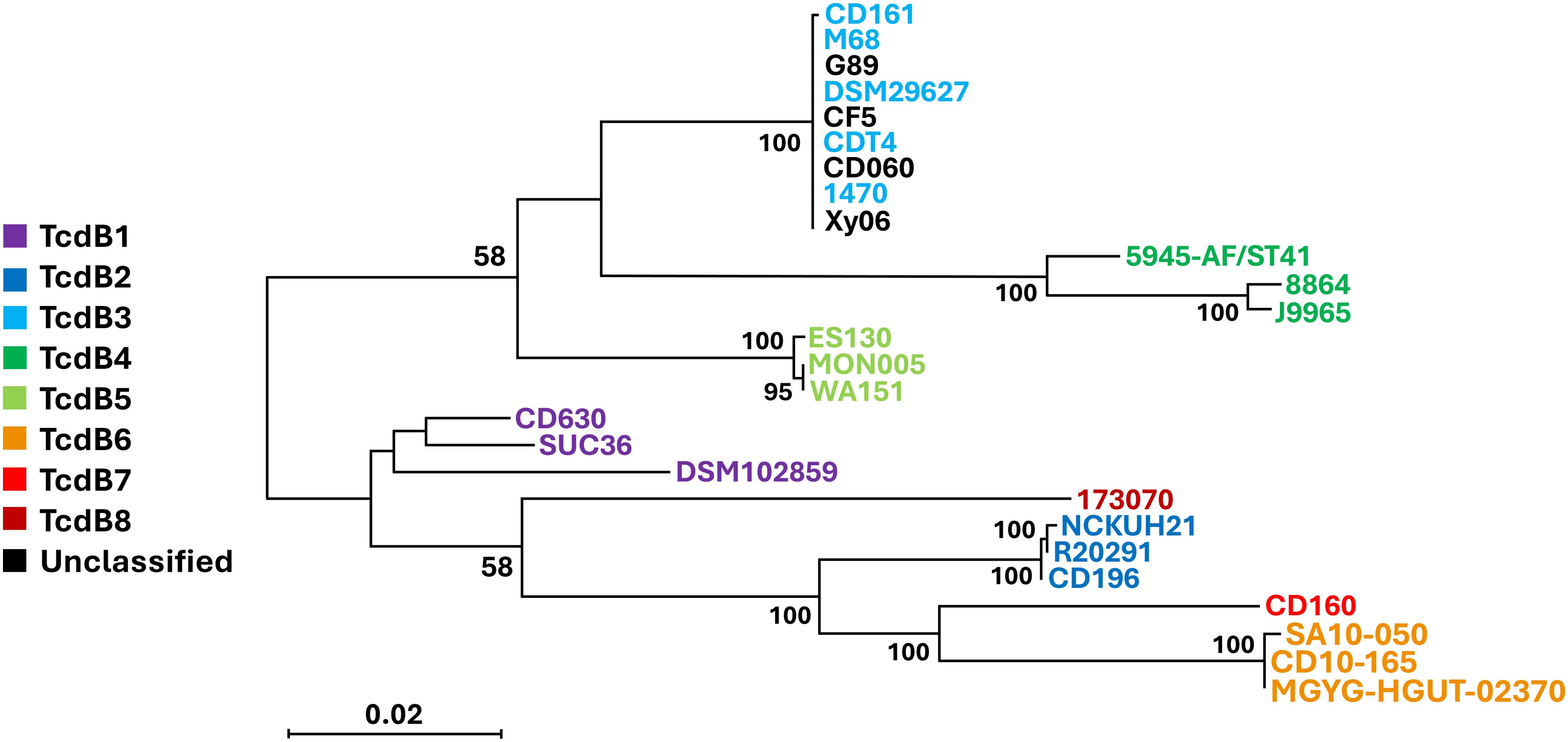
Figure 3. Phylogenetic tree of A-B+ C. difficile TcdB subtypes. A neighbor-joining phylogenetic tree of TcdB amino acid sequences from the A-B+ strains examined in this study and other strains representing the various subtypes of TcdB. Xy06 TcdB is clustered with several A-B+ strains that encode TcdB3 (light blue). The scale bar indicates 0.020 amino acid substitutions per site. Bootstrap values of 50 or greater are displayed.
3.2.3 Variations in tcdE, tcdR, and tcdC
The T-coffee alignment (Supplementary Figure 1) showed highly similar tcdE sequences across the strains surveyed. One exception is ES130, which displays a 73 bp deletion at the 5’ end of the gene and a 4bp insertion at the 3’ end relative to the other A-B+ strains analyzed.
Finally, tcdC and tcdR were also generally well conserved among A-B+ strains. TcdR showed no significant deletions or additions in the strains analyzed (Supplementary Figure 2). However, some strains did display variations in tcdC (Supplementary Figure 3). J9965 and 8864 have identical 234 bp deletions in their tcdC genes, whereas SUC36 has a smaller 39 bp deletion (Supplementary Figure 3, Table 1).
3.3 Xy06 is a robust TcdB producer
TcdA and TcdB production of Xy06 was evaluated in comparison with strains R20291 (ST1), CD630E (ST54), and the Xy07 strain that was isolated from a patient at the same institute as Xy06. Whole genome sequencing showed that Xy07 belongs to ST54. Consistent with this, Xy07 was determined to be an RT012 strain by capillary PCR ribotyping. Xy06 strain did not produce detectable TcdA by ELISA, while R20291, CD630E, and Xy07 produced both TcdA and TcdB. The highest concentrations of both toxins were detected at 48 h or 72 h post-inoculation. Of the strains tested, R20291 showed the highest level of TcdA production (> 100 ng/ml, Figure 4A). Of note, Xy06 showed the highest level of TcdB production (331 ± 18.50 ng/ml) at 48 hours post-inoculation (Figures 4B, C).
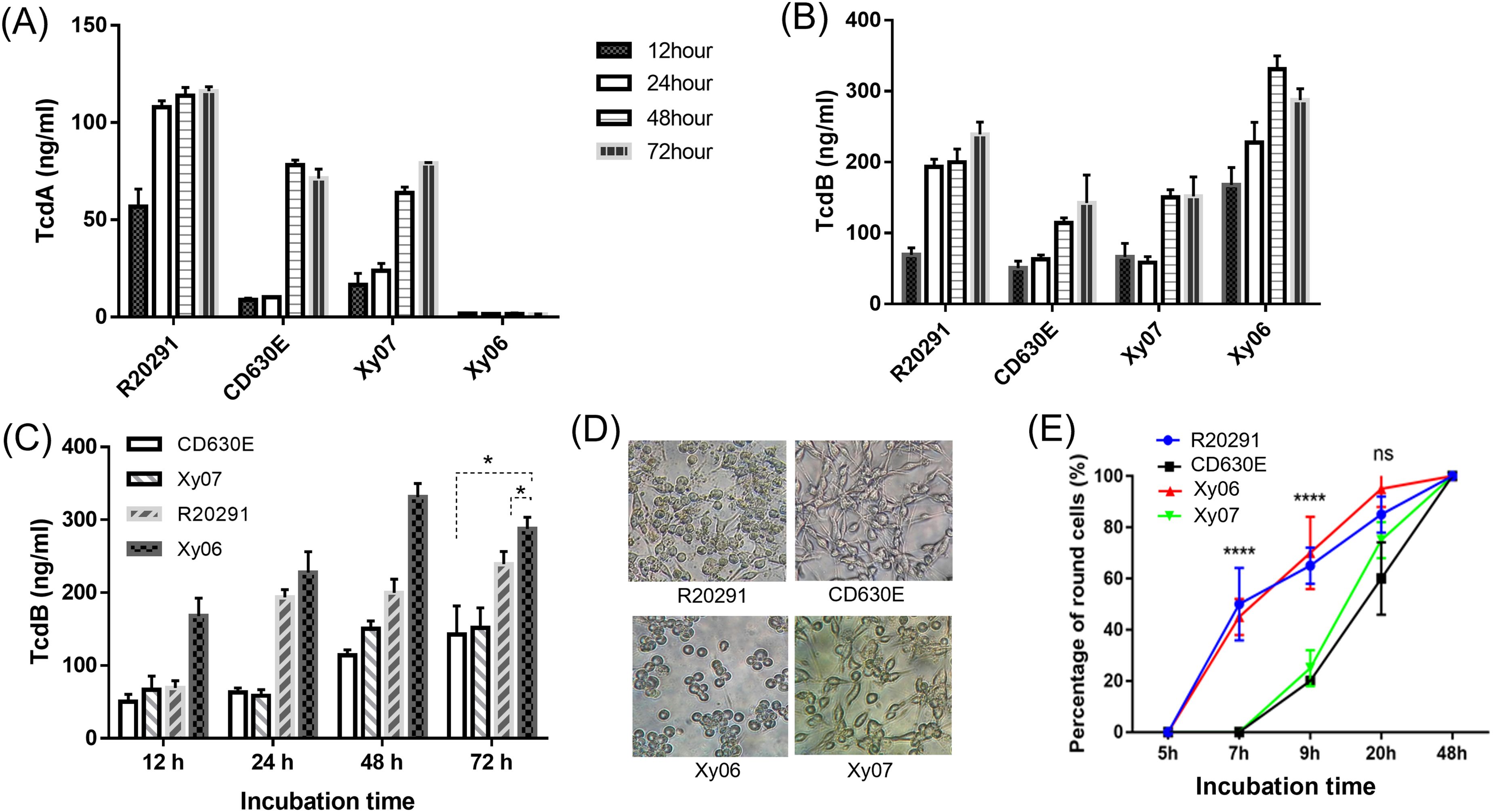
Figure 4. Toxin production in strains Xy06 and Xy07. TcdA (A) and TcdB (B) concentrations of C. difficile culture supernatants at different time points. (C) TcdB concentrations of C. difficile culture supernatants were compared at the same time point. *P < 0.05 (t-test). (D) Cytotoxicity of C. difficile culture supernatants. C. difficile culture supernatants (16 μl) collected at 72 hours post-inoculation in TY medium were added to CT26 cells seeded in 96-well plates. CT26 cell rounding was visualized by phase-contrast microscopy (40X) at 20 hours post-treatment. (E) Percentage of round cells at different time points (****P<0.0001, one-way ANOVA). Data are presented as mean ± SEM (n = triplicate). ns- not significant.
Even though Xy06 only produces TcdB, its culture supernatant induced comparable cell rounding to the culture supernatant from R20291 (A+B+) strain (Figure 4D), with significantly higher cell cytotoxicity compared with CD630E and Xy07 at 7 hours and 9 hours post-inoculation (one-way ANOVA, P<0.0001) (Figure 4E). Significant cell rounding was already evident at 7 hours post-inoculation for cells exposed to culture supernatants of Xy06 (ca. 45%) and R20291 (ca. 50%), whereas limited toxicity was observed for the two ST54 strains at this time point. Nearly complete cell rounding (95%) was visualized at 20 h post-inoculation with Xy06 culture supernatant. Thus, Xy06 culture supernatant induced a faster rounding of CT26 cells compared to CD630E and Xy07 (Figure 4E). Taken together, our data demonstrate that Xy06 is a robust TcdB producer capable of inducing cytopathic effects similar to RT027 strains.
3.4 Xy-06 shows strong binding to human gut epithelial cells
The adhesion and persistence of C. difficile spores in the gut are the root cause of CDI and recurrence (Castro-Cordova et al., 2021). We also determined the binding capability of spores to HCT-8 human gut epithelial cells. Of the strains tested, Xy06 spores showed comparable adherence efficiency to CD630E spores (p=0.8011), and the adherence efficacy of Xy06 spores was significantly higher than R20291 spores (unpaired t-test, p=0.0097) (Figure 5).
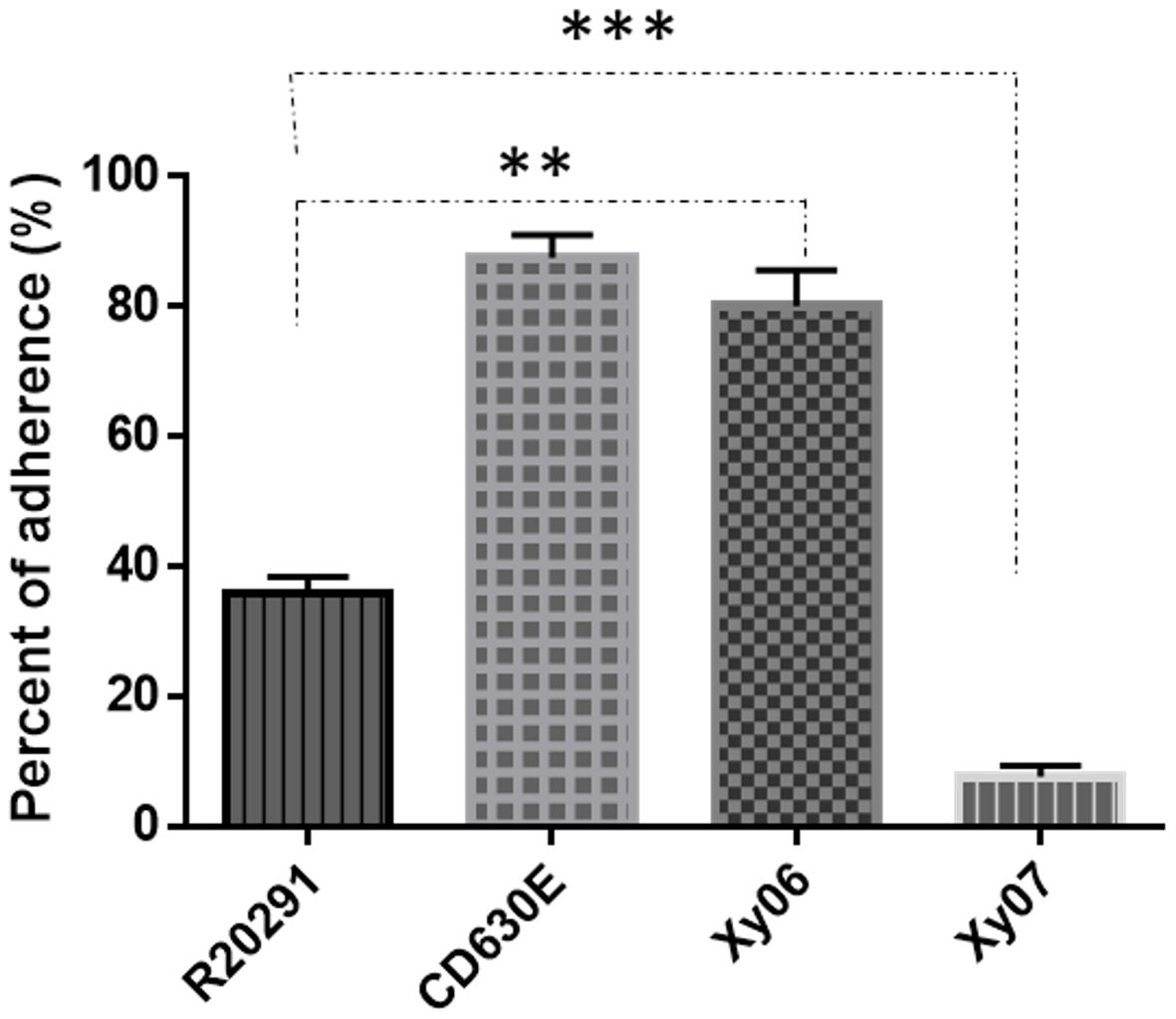
Figure 5. Adhesion of C. difficile spores to HCT-8 cells. HCT-8 cells were incubated with spores for 100 minutes under anaerobic conditions, and the relative number of adherent spores was calculated. Data are from three biological replicates and reported as mean ± SEM. **p=0.0097 (Xy06 VS R20291); p=0.8011 (Xy06 VS CD630E); *** p<0.0001 (R20291 VS Xy07).
3.5 Sporulation and germination capabilities of Xy06
Sporulation and germination play critical roles in C. difficile pathogenesis and transmission. Sporulation and germination capabilities of Xy06 were also determined in comparison with strains R20291, CD630E, and Xy07. Xy06 is significantly more efficient in sporulation (p=0.0065) (Figure 6A) and germination (p=0.0001) (Figure 6B) than CD630E, significantly less efficient in sporulation than R20291 (P=0.0398) (Figure 6A) while comparable with R20291 in germination (P=0.0714). In addition, Xy06 is less efficient in sporulation (ns, p=0.0666) while significantly more efficient in germination than Xy07 (p=0.0021) (Figure 6B). To further assess the germination efficiency of C. difficile Xy06, Xy07, R20291 and CD630E, the colony-forming efficiency of spores was determined. Xy06 spores are significantly more efficient in forming colonies than CD630E spores but comparable with R20291 and Xy07 spores (Figure 6C). Our data indicate that Xy06 readily sporulates and germinates.
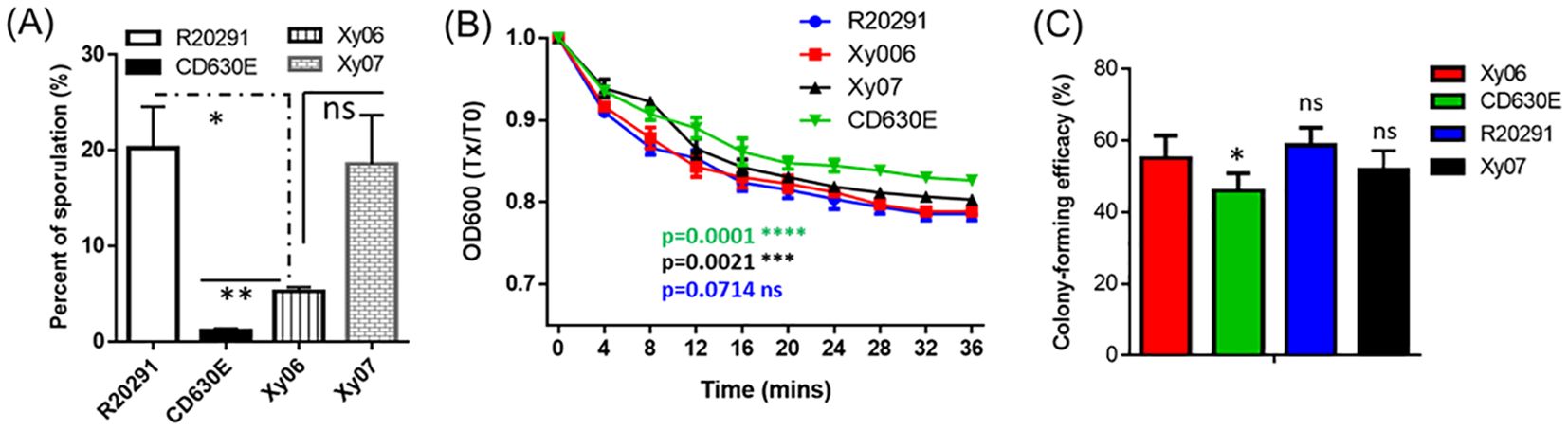
Figure 6. Sporulation and germination rates of C. difficile strains. (A) Sporulation. *P<0.05; **P<0.01. (B) Germination: Purified C. difficile spores were suspended in buffer supplemented with taurocholic acid and glycine to induce germination. Germination was monitored by plotting the ratio of the OD600 at a given time to the OD600 at time zero. Data are from three biological replicates and reported as mean ± SEM. ****p=0.0001(CD630E vs Xy06); ***p=0.0021 (Xy07 vs Xy06); ns: p=0.0714 (R20291 vs Xy06). (C) Spore colony-forming efficiency. Spore suspensions were serially diluted and plated on BHIS agar supplemented with 0.1%(w/v) taurocholic acid (TA). The spore colony-forming efficiency was defined as the percentage of total spores that gave rise to colonies on BHIS agar with 0.1% (w/v) TA, calculated by c.f.u. per ml colony count/c.f.u. per ml direct count by microscopy × 100%. Data are from three biological replicates and reported as mean ± SEM. * p=0.0276 (CD630E vs Xy06); ns: no significance (R20291, Xy07 vs Xy06). ns- not significant.
3.6 Xy06 induces strong symptoms in a mouse model of CDI
The pathogenicity of Xy06 in vivo was evaluated in the mouse model of CDI in comparison with strains R20291, CD630E, and Xy07. C. difficile R20291 challenged group showed a mortality rate of 55% (Figure 7B) and a diarrhea rate of 100% (Figure 7C). The Xy06 challenged group also presented a high death rate (30%) and a diarrhea rate (70%), which were higher than those of the CD630E challenged group ((10% death rate (p=0.1164 between Xy06 and CD630E groups) and 55% diarrhea rate, Figure 7C) and were also significantly higher than those of the Xy07 challenged mice ((5% death rate (p=0.0362 between Xy06 and Xy07 groups) and 30% diarrhea rate, Figure 7C). The Xy06-challenged group lost significantly more weight at post-infection day 2 compared to the CD630E-challenged group (Figure 7A). The R20291-challenged group lost significantly more weight at post-infection days 2 and 3 than the Xy06-challenged group (p=0.0015 and p=0.0076, respectively). Taken together, Xy-06 was more virulent than strain CD630E and Xy07, while less virulent with no significance than strain R20291 in mortality (p=0.1556 between Xy06 and R20291 groups) in the mouse model of CDI.
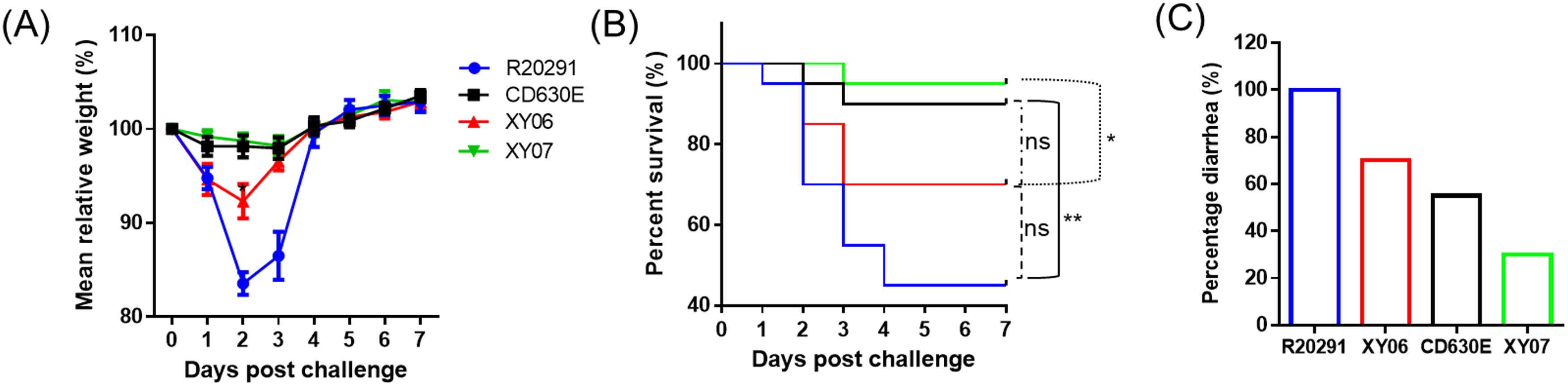
Figure 7. Virulence of Xy06 in the mouse model of CDI. Four groups of mice (n = 20) were challenged with 106 spores of Xy06, Xy07, CD630E and R20291, respectively. Mice were monitored for one week for weight changes (A), survival (B), and diarrhea (C). Animal survival was analyzed by Kaplan–Meier survival analysis with a log-rank test of significance. The mean relative weight of mice was analyzed for significance using a Student’s t-test. Xy06 challenged group lost significantly more weight at day 2 post-infection compared to CD630E challenged group (p=0.0145). R20291 challenged group lost significantly more weight at days 2 and 3 post-infection than Xy06 challenged group (p=0.0015 and p=0.0076, respectively) (A). (*p < 0.05, **p < 0.01 and **p < 0.01). ns- not significant.
4 Discussion
Though initially identified in Asia, RT017/ST37 strains have also been identified in other countries such as Canada, Argentina, Australia, Israel, South Africa, and throughout Europe (Cairns et al., 2017). Strikingly, the proportion of RT017/ST37 is much higher in China than other areas of the world (Wu et al., 2022). Several publications reported that infections with the strains of type ST37/RT017 are the leading epidemic healthcare-associated CDIs in China (Yan et al., 2013; Jin et al., 2017; Yang et al., 2017), but factors contributing to their success as a pathogen in this region are still unknown. Although RT027 strains cause outbreaks in the Western world and are associated with increased morbidity and mortality, RT027 cases in China were only occasionally reported, and no significant outbreaks and death cases in China have been documented (Cheng et al., 2016; Jia et al., 2016; Wu et al., 2022). There are some indications that RT017 strains might exhibit clinical characteristics similar to hypervirulent RT 027 strains (Goorhuis et al., 2011).
Here, we provide experimental evidence supporting these indications. In particular, Xy06 showed a higher production of TcdB than CD630E and Xy07 (ST54) clinical strains under laboratory conditions. Notably, Xy06 produced more TcdB and at a faster rate than R20291 (highest TcdB production at 48 hours for Xy06 and at 72 hours for R20291 and other strains). In the cytotoxicity assay, Xy06 culture supernatant also induced a high level of cell cytotoxicity similar to R20291. At present, the reason for the high toxin expression is unknown. Regulation of toxin expression is complex. In some studies, mutations within the tcdC gene have been postulated to affect its function as a negative regulator (Matamouros et al., 2007), but others have not found such a relation (Cartman et al., 2012). In this study, we did not find any deletion or mutation of tcdC that might inactivate TcdC and lead to overexpression of tcdA and tcdB.
TcdA and TcdB share 49% identity and 63% similarity and share a similar domain structure (Pruitt and Lacy, 2012). The relative role of each toxin in the pathophysiology of CDI has long been a matter of debate, though TcdB is considered to be the more potent cytotoxin. The sequence of PaLoc comparison showed that the Xy06 genome carries a truncation of the tcdA gene and a functional tcdB gene, which codes for a hybrid TcdB (TcdB3) that is homologous to TcsL of C. sordellii. This hybrid tcdB gene seems highly conserved in RT017 C. difficile strains, and TcdB3 has been reported to target different sets of Rho GTPases in mammalian cells in comparison with TcdB from non-RT017 strains (Chaves-Olarte et al., 1999; Genth et al., 2014). However, a recent study showed that TcdB1 (from representative strain CD630), TcdB2 (from representative strain R20291), and TcdB3 exhibited comparable potency in a direct toxin challenge by intraperitoneal injection in mice (Shen et al., 2020b), though these TcdB variants induced different pathological effects and are highly diverse in their receptor preference (Pan et al., 2021). Even though Xy06 produces TcdB faster and in larger quantity than R20291 and Xy06 spores show stronger binding to HCT-8 cells than R20291 spores at least in vitro, it is less virulent, though not significant, than R20291 in the mouse model of CDI, indicating the complexed interplays among virulence factors and colonization, especially TcdA, TcdB and CDT in vivo that lead to the disease symptoms. In this regard, one of the limitations of this study is the missing data on C. difficile numbers and toxin levels in fecal samples of the infected mice, which may indicate the toxin production and C. difficile colonization in vivo.
Notably, our results showed that the Xy06 genome clusters closely with those of other RT017 strains (Figure 1), and the PaLoc genes (tcdA, tcdB, tcdC, tcdR, and tcdE) are highly conserved among all RT017 strains (Figure 3 and Supplementary Figures 1, Supplementary Figure 2, Supplementary Figure 3). This suggests that the Xy06 virulence characteristics, such as the strong adherence to HCT-8 cells, ability to form spores and germinate, and robust toxin production may be representative of other RT017 strains.
C. difficile genomes can harbor multiple prophages (Heuler et al., 2021). Some C. difficile phages, such as phiCD119 and phiCD38-2, have been shown to regulate toxin expression, while the phage phiSemix9p1 encodes the entire binary toxin locus (Fortier, 2018). Given the potential that phages may influence C. difficile virulence, we searched for putative prophages in the Xy06 genome and prophage-encoded toxin genes. In Xy06, PHASTER predicted eight incomplete prophage-like regions and two putative complete prophages (Supplementary Table 2). The first complete prophage (sequence 2 in Supplementary Table 2) was predicted to have a length of 44.5 Kb and a GC content of 29.4%. Meanwhile, the second complete prophage (sequence 4 in Supplementary Table 2) was predicted to have a length of 50 Kb and a GC content of 27.8%. These statistics of both prophages are in agreement with the length and GC% ranges observed in other C. difficile bacteriophages (Heuler et al., 2021). We sought to determine if either of the complete prophages (Prophage 2, Prophage 4) were related to previously sequenced C. difficile phages implicated in virulence mechanisms. A Maximum-likelihood phylogenetic analysis shows that both Prophage 2 and Prophage 4 of strain Xy06 are most closely related to phage JD032. JD032 infection has been shown to downregulate a hemolysin, the sporulation-related sigma factor sigH, and two surface-proteins of C. difficile related to adhesion (Cwp2, Cwp66) (Li et al., 2020). The authors of this study hypothesized that the initiation of JD032 infection could attenuate the virulence of its host strain, which raises the possibility of a similar relationship between strain Xy06 and its prophages. Also, like JD032, the prophages of Xy06 do not appear to encode either PaLoc genes or CdtLoc genes based on BLAST analysis.
Several conjugative transposons (CTns) and transposable elements (Tns) have been identified in C. difficile strains, such as CD630 (Brouwer et al., 2011). CTns and Tns can significantly impact the virulence of strains that encode them. For example, the Tn5398 element encoded in CD630 confers tetracycline and erythromycin resistance (Mullany et al., 1995). The strong similarity between transposons of various C. difficile strains suggests that bacteria frequently exchange these regions through horizontal gene transfer (Brouwer et al., 2011). To identify CTns and Tns in Xy06, transposon sequences were mined from GenBank files of various strains using the boundaries of each element as previously reported (Brouwer et al., 2011). These were aligned with the Xy06 genome using NCBI BLAST, and the results were reported in Supplementary Table 3. The sequences of CTn5 and Tn6106 produced strong alignments with regions of the Xy06 genome that are at least 90% the size of the reference transposon sequences and shared at least 98% identity. CTn5-like elements in C. difficile strains such as R20291 and QCD-23M63 encode putative sigma factors that could potentially impact the transcription profile of CTn5-encoding strains (Brouwer et al., 2011). Tn6106, unlike CTn5, has not been shown to be excisable, but it does encode putative recombinases and a putative sigma factor among other genes (Brouwer et al., 2011). Additional six elements (CTn1, CTn2, CTn3, Tn6073, Tn6107, and Tn6110) aligned with sequences of the Xy06 genome also showed high shared identity (at least 85%) but were shorter than the reference transposon sequence (50-69% query coverage). Four transposons (CTn6, Tn5398, Tn6103, and Tn6104) displayed poor quality alignments because of low query cover (below 50%). CTn4 and Tn6105 did not produce any significant alignment according to the BLAST algorithm. It will be interesting to investigate the exact roles of these CTn and Tns in Xy06 physiology and fitness in the future.
In summary, Xy06 is more virulent than strains CD630E and Xy07, and is comparable to strain R20291 in virulence. Xy06 carries two putative prophages and several mobilizable elements, which might contribute to its increased virulence and fitness.
Data availability statement
Data will be available from the corresponding authors once requested. The genomes of C. difficile strains Xy06 and Xy07 presented in this study were deposited in the GenBank repository (https://www.ncbi.nlm.nih.gov/genbank/), accession numbers JANFNF000000000 and JANFVQ000000000, respectively.
Ethics statement
The animal study was approved by University of South Florida Institutional Animal Care and use Committee (IACUC #IS00011981). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
CL: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. JH: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. DZ: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. XM: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. SC: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. CH: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. SW: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. ZP: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. WS: Writing – original draft, Writing – review & editing. AW: Writing – original draft, Writing – review & editing. XS: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by the University of South Florida Startup Fund and the Anthony Gagliardi Foundation to XS, and Xiangya Sinobioway Health Research Fund to CL (No. xywm2015I11).
Acknowledgments
We would like to thank other team members in Sun Lab at the University of South Florida for their critical comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1412408/full#supplementary-material
References
Alikhan, N.-F., Petty, N. K., Zakour, N. L. B., Beatson, S. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 1–10. doi: 10.1186/1471-2164-12-402
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Braun, V., Hundsberger, T., Leukel, P., Sauerborn, M., Von Eichel-Streiber, C. (1996a). Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 181, 29–38. doi: 10.1016/S0378-1119(96)00398-8
Braun, V., Hundsberger, T., Leukel, P., Sauerborn, M., Von Eichel-Streiber, C. J. G. (1996b). Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 181, 29–38. doi: 10.1016/S0378-1119(96)00398-8
Brouwer, M. S., Warburton, P. J., Roberts, A. P., Mullany, P., Allan, E. J. P. O. (2011). Genetic organisation, mobility and predicted functions of genes on integrated, mobile genetic elements in sequenced strains of Clostridium difficile. PloS One 6, e23014. doi: 10.1371/journal.pone.0023014
Cairns, M. D., Preston, M. D., Hall, C. L., Gerding, D. N., Hawkey, P. M., Kato, H., et al. (2017). Comparative genome analysis and global phylogeny of the toxin variant clostridium difficile PCR ribotype 017 reveals the evolution of two independent sublineages. J. Clin. Microbiol. 55, 865–876. doi: 10.1128/JCM.01296-16
Cartman, S. T., Kelly, M. L., Heeg, D., Heap, J. T., Minton, N. P. (2012). Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl. Environ. Microbiol. 78, 4683–4690. doi: 10.1128/AEM.00249-12
Castro-Cordova, P., Mora-Uribe, P., Reyes-Ramirez, R., Cofre-Araneda, G., Orozco-Aguilar, J., Brito-Silva, C., et al. (2021). Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat. Commun. 12. doi: 10.1038/s41467-021-21355-5
Chaves-Olarte, E., Freer, E., Parra, A., Guzmán-Verri, C., Moreno, E., Thelestam, M.J.J.O.B.C. (2003). R-Ras glucosylation and transient RhoA activation determine the cytopathic effect produced by toxin B variants from toxin A-negative strains of Clostridium difficile. J. Biol. Chem. 278, 7956–7963. doi: 10.1074/jbc.M209244200
Chaves-Olarte, E., Low, P., Freer, E., Norlin, T., Weidmann, M., Von Eichel-Streiber, C., et al. (1999). A novel cytotoxin from Clostridium difficile serogroup F is a functional hybrid between two other large clostridial cytotoxins. J. Biol. Chem. 274, 11046–11052. doi: 10.1074/jbc.274.16.11046
Cheng, J. W., Xiao, M., Kudinha, T., Xu, Z. P., Hou, X., Sun, L. Y., et al. (2016). The first two clostridium difficile ribotype 027/ST1 isolates identified in beijing, China-an emerging problem or a neglected threat? Sci. Rep. 6, 18834. doi: 10.1038/srep18834
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J.-M., et al. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39.suppl_2 39, W13–W17. doi: 10.1093/nar/gkr245
Du, P. C., Cao, B., Wang, J., Li, W. G., Jia, H. B., Zhang, W., et al. (2014). Sequence Variation in tcdA and tcdB of Clostridium difficile: ST37 with Truncated tcdA Is a Potential Epidemic Strain in China. J. Clin. Microbiol. 52, 3264–3270. doi: 10.1128/JCM.03487-13
Fawley, W. N., Knetsch, C. W., Maccannell, D. R., Harmanus, C., Du, T., Mulvey, M. R., et al. (2015). Development and validation of an internationally-standardized, high-resolution capillary gel-based electrophoresis PCR-ribotyping protocol for Clostridium difficile. PloS One 10, e0118150. doi: 10.1371/journal.pone.0118150
Fortier, L.-C. (2018). Bacteriophages contribute to shaping Clostridioides (Clostridium) difficile species. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02033
Gabler, F., Nam, S. Z., Till, S., Mirdita, M., Steinegger, M., Soding, J., et al. (2020). Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinf. 72, e108. doi: 10.1002/cpbi.108
Genth, H., Pauillac, S., Schelle, I., Bouvet, P., Bouchier, C., Varela-Chavez, C., et al. (2014). Haemorrhagic toxin and lethal toxin from Clostridium sordellii strain vpi9048: molecular characterization and comparative analysis of substrate specificity of the large clostridial glucosylating toxins. Cell Microbiol. 16, 1706–1721. doi: 10.1111/cmi.2014.16.issue-11
Goorhuis, A., Debast, S. B., Dutilh, J. C., Van Kinschot, C. M., Harmanus, C., Cannegieter, S. C., et al. (2011). Type-specific risk factors and outcome in an outbreak with 2 different Clostridium difficile types simultaneously in 1 hospital. Clin. Infect. Dis. 53, 860–869. doi: 10.1093/cid/cir549
Griffiths, D., Fawley, W., Kachrimanidou, M., Bowden, R., Crook, D. W., Fung, R., et al. (2010). Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 48, 770–778. doi: 10.1128/JCM.01796-09
Hawkey, P. M., Marriott, C., Liu, W. E., Jian, Z. J., Gao, Q., Ling, T. K. W., et al. (2013). Molecular epidemiology of clostridium difficile infection in a major chinese hospital: an underrecognized problem in asia? J. Clin. Microbiol. 51, 3308–3313. doi: 10.1128/JCM.00587-13
Heuler, J., Fortier, L. C., Sun, X. (2021). Clostridioides difficile phage biology and application. FEMS Microbiol. Rev. 45. doi: 10.1093/femsre/fuab012
Imwattana, K., Knight, D. R., Kullin, B., Collins, D. A., Putsathit, P., Kiratisin, P., et al. (2019). Clostridium difficile ribotype 017 - characterization, evolution and epidemiology of the dominant strain in Asia. Emerg. Microbes Infect. 8, 796–807. doi: 10.1080/22221751.2019.1621670
Imwattana, K., Putsathit, P., Collins, D. A., Leepattarakit, T., Kiratisin, P., Riley, T. V., et al. (2022). Global evolutionary dynamics and resistome analysis of Clostridioides difficile ribotype 017. Microb. Genom 8. doi: 10.1099/mgen.0.000792
Jia, H., Du, P., Yang, H., Zhang, Y., Wang, J., Zhang, W., et al. (2016). Nosocomial transmission of Clostridium difficile ribotype 027 in a Chinese hospital 2012-2014, traced by whole genome sequencing. BMC Genomics 17, 405. doi: 10.1186/s12864-016-2708-0
Jin, D. Z., Luo, Y., Huang, C., Cai, J., Ye, J. L., Zheng, Y., et al. (2017). Molecular epidemiology of clostridium difficile infection in hospitalized patients in eastern China. J. Clin. Microbiol. 55, 801–810. doi: 10.1128/JCM.01898-16
Katoh, K., Standley, D.M.J.M.B. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, J., Kim, Y., Pai, H. (2016). Clinical characteristics and treatment outcomes of clostridium difficile infections by PCR ribotype 017 and 018 strains. PloS One 11, e0168849. doi: 10.1371/journal.pone.0168849
Kim, J., Pai, H., Seo, M. R., Kang, J. O. (2012). Clinical and microbiologic characteristics of tcdA-negative variant Clostridium difficile infections. BMC Infect. Dis. 12, 109. doi: 10.1186/1471-2334-12-109
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, T., Zhang, Y., Dong, K., Kuo, C.-J., Li, C., Zhu, Y.-Q., et al. (2020). Isolation and characterization of the novel phage JD032 and global transcriptomic response during JD032 infection of Clostridioides difficile ribotype 078. MSystems 5, e00017–e00020. doi: 10.1128/msystems.00017-20
Martin-Verstraete, I., Peltier, J., Dupuy, B. (2016). The regulatory networks that control clostridium difficile toxin synthesis. Toxins (Basel) 8. doi: 10.3390/toxins8050153
Matamouros, S., England, P., Dupuy, B. (2007). Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol. Microbiol. 64, 1274–1288. doi: 10.1111/j.1365-2958.2007.05739.x
Meier-Kolthoff, J. P., Göker, M. J. N. C. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-10210-3
Monot, M., Eckert, C., Lemire, A., Hamiot, A., Dubois, T., Tessier, C., et al. (2015). Clostridium difficile: New Insights into the Evolution of the Pathogenicity Locus. Sci. Rep. 5, 15023. doi: 10.1038/srep15023
Mullany, P., Wilks, M., Tabaqchali, S. (1995). Transfer of macrolide-lincosamide-streptogramin B (MLS) resistance in Clostridium difficile is linked to a gene homologous with toxin A and is mediated by a conjugative transposon, Tn5398. J. Antimicrobial Chemotherapy 35, 305–315. doi: 10.1093/jac/35.2.305
Pan, Z., Zhang, Y., Luo, J., Li, D., Zhou, Y., He, L., et al. (2021). Functional analyses of epidemic Clostridioides difficile toxin B variants reveal their divergence in utilizing receptors and inducing pathology. PloS Pathog. 17, e1009197. doi: 10.1371/journal.ppat.1009197
Pruitt, R. N., Lacy, D. B. (2012). Toward a structural understanding of Clostridium difficile toxins A and B. Front. Cell Infect. Microbiol. 2, 28. doi: 10.3389/fcimb.2012.00028
Rupnik, M., Kato, N., Grabnar, M., Kato, H. (2003). New types of toxin A-negative, toxin B-positive strains among Clostridium difficile isolates from Asia. J. Clin. Microbiol. 41, 1118–1125. doi: 10.1128/JCM.41.3.1118-1125.2003
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M.-A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Senoh, M., Kato, H., Fukuda, T., Niikawa, A., Hori, Y., Hagiya, H., et al. (2015). Predominance of PCR-ribotypes, 018 (smz) and 369 (trf) of Clostridium difficile in Japan: a potential relationship with other global circulating strains? J. Med. Microbiol. 64, 1226–1236. doi: 10.1099/jmm.0.000149
Shen, E., Zhu, K., Li, D., Pan, Z., Luo, Y., Bian, Q., et al. (2020a). Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B. Commun. Biol. 3, 347. doi: 10.1038/s42003-020-1078-y
Shen, E., Zhu, K., Li, D., Pan, Z., Luo, Y., Bian, Q., et al. (2020b). Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B. Commun. Biol. 3, 347. doi: 10.1038/s42003-020-1078-y
Soehn, F., Wagenknecht-Wiesner, A., Leukel, P., Kohl, M., Weidmann, M., Von Eichel-Streiber, C., et al. (1998). Genetic rearrangements in the pathogenicity locus of Clostridium difficile strain 8864–implications for transcription, expression and enzymatic activity of. Mol. Gen. Genet. MGG 258, 222–232. doi: 10.1007/s004380050726
Sorg, J. A., Dineen, S. S. (2009). Laboratory maintenance of Clostridium difficile. Curr. Protoc. Microbiol. 9, Unit9A 1. doi: 10.1002/9780471729259.mc09a01s12
Su, T., Chen, W., Wang, D., Cui, Y., Ni, Q., Jiang, C., et al. (2021a). Complete genome sequencing and comparative phenotypic analysis reveal the discrepancy between Clostridioides difficile ST81 and ST37 isolates. Front. Microbiol. 12, 776892. doi: 10.3389/fmicb.2021.776892
Su, T., Chen, W., Wang, D., Cui, Y., Ni, Q., Jiang, C., et al. (2021b). Complete genome sequencing and comparative phenotypic analysis reveal the discrepancy between clostridioides difficile ST81 and ST37 isolates. Front. Microbiol. 12, 776892. doi: 10.3389/fmicb.2021.776892
Sullivan, M. J., Petty, N. K., Beatson, S. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Von Eichel-Streiber, C., Zec-Pirnat, I., Grabnar, M., Rupnik, M.J.F.M.L. (1999). A nonsense mutation abrogates production of a functional enterotoxin A in Clostridium difficile toxinotype VIII strains of serogroups F and X. FEMS Microbiol. Lett. 178, 163–168. doi: 10.1111/fml.1999.178.issue-1
Wang, B., Peng, W., Zhang, P., Su, J. (2018a). The characteristics of Clostridium difficile ST81, a new PCR ribotype of toxin A- B+ strain with high-level fluoroquinolones resistance and higher sporulation ability than ST37/PCR ribotype 017. FEMS Microbiol. Lett. 365. doi: 10.1093/femsle/fny168
Wang, B., Peng, W., Zhang, P., Su, J. (2018b). The characteristics of Clostridium difficile ST81, a new PCR ribotype of toxin A-B+ strain with high-level fluoroquinolones resistance and higher sporulation ability than ST37/PCR ribotype 017. FEMS Microbiol. Lett. 365, fny168. doi: 10.1093/femsle/fny168
Wu, S., Wu, H. J. B. B. (2013). More powerful significant testing for time course gene expression data using functional principal component analysis approaches. BMC Bioinf. 14, 1–13. doi: 10.1186/1471-2105-14-6
Wu, Y., Wang, Y. Y., Bai, L. L., Zhang, W. Z., Li, G. W., Lu, J. X. (2022). A narrative review of Clostridioides difficile infection in China. Anaerobe 74, 102540. doi: 10.1016/j.anaerobe.2022.102540
Yan, Q., Zhang, J., Chen, C., Zhou, H., Du, P., Cui, Z., et al. (2013). Multilocus sequence typing (MLST) analysis of 104 Clostridium difficile strains isolated from China. Epidemiol. Infect. 141, 195–199. doi: 10.1017/S0950268812000453
Yang, J., Zhang, X., Liu, X., Cai, L., Feng, P., Wang, X., et al. (2017). Antimicrobial susceptibility of Clostridium difficile isolates from ICU colonized patients revealed alert to ST-37 (RT 017) isolates. Diagn. Microbiol. Infect. Dis. 89, 161–163. doi: 10.1016/j.diagmicrobio.2017.06.021
Yang, Z., Huang, Q., Qin, J., Zhang, X., Jian, Y., Lv, H., et al. (2020). Molecular epidemiology and risk factors of clostridium difficile ST81 infection in a teaching hospital in eastern China. Front. Cell Infect. Microbiol. 10, 578098. doi: 10.3389/fcimb.2020.578098
Keywords: Clostridioides difficile, pathogenicity locus (PaLoc), ST37, RT017, virulence, toxin
Citation: Li C, Heuler J, Zhu D, Meng X, Chakraborty S, Harmanus C, Wang S, Peng Z, Smits WK, Wu A and Sun X (2024) Genomic and phenotypic characterization of a Clostridioides difficile strain of the epidemic ST37 type from China. Front. Cell. Infect. Microbiol. 14:1412408. doi: 10.3389/fcimb.2024.1412408
Received: 04 April 2024; Accepted: 23 September 2024;
Published: 18 October 2024.
Edited by:
Ziad A. Memish, Alfaisal University, Saudi ArabiaReviewed by:
Alexey V. Rakov, Central Research Institute of Epidemiology (CRIE), RussiaDaniel Paredes-Sabja, Texas A and M University, United States
Copyright © 2024 Li, Heuler, Zhu, Meng, Chakraborty, Harmanus, Wang, Peng, Smits, Wu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingmin Sun, c3VuNUB1c2YuZWR1; Chunhui Li, bGljaHVuaHVpQGNzdS5lZHUuY24=
†These authors have contributed equally to this work