
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 19 July 2024
Sec. Parasite and Host
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1405198
 Miriam Rodi1
Miriam Rodi1 Katarzyna Kawecka1Laura Stephan1
Katarzyna Kawecka1Laura Stephan1 Lilith Berner1Martha Salinas Medina1Albert Lalremruata1,2Tamirat Gebru Woldearegai1,2Pierre Blaise Matsiegui3Mirjam Groger4,5
Lilith Berner1Martha Salinas Medina1Albert Lalremruata1,2Tamirat Gebru Woldearegai1,2Pierre Blaise Matsiegui3Mirjam Groger4,5 Rella Zoleko Manego4,5,6Dorothea Ekoka Mbassi4,5,6
Rella Zoleko Manego4,5,6Dorothea Ekoka Mbassi4,5,6 Ghyslain Mombo-Ngoma6,7,8
Ghyslain Mombo-Ngoma6,7,8 Selidji Todagbe Agnandji1,6Michael Ramharter4,5,6
Selidji Todagbe Agnandji1,6Michael Ramharter4,5,6 Benjamin Mordmüller1,6,9Juliana Inoue1
Benjamin Mordmüller1,6,9Juliana Inoue1 Jana Held1,2,6*
Jana Held1,2,6*Introduction: Plasmodium malariae is the most common non-falciparum species in sub-Saharan Africa. Despite this, data on its genetic diversity is scarce. Therefore, we aimed to establish a P. malariae genotyping approach based on size polymorphic regions that can be easily applied in molecular epidemiological studies.
Methods: Four potential genotyping markers, Pm02, Pm09, P. malariae thrombospondin-related anonymous protein (pmtrap), and P. malariae merozoite surface protein fragment 2 (pmmsp1 F2) were amplified via nested PCR and analysed using automated capillary gel electrophoresis.
Results: We observed the highest allelic diversity for pmtrap (MOI = 1.61) and pmmsp1 F2 (He = 0.81). Further applying the two markers pmtrap and pmmsp1 F2 on a different sample set of 21 P. malariae positive individuals followed up over one week, we saw a high consistency in their performance. The results show a large complexity and high dynamics of P. malariae infections in the asymptomatic Gabonese study population.
Discussion: We successfully implemented a new genotyping panel for P. malariae consisting of only two markers: pmtrap and pmmsp1 F2. It can be easily applied in other settings to investigate the genotype diversity of P. malariae populations, providing further important data on the molecular epidemiology of this parasite species.
Plasmodium malariae is the second most abundant Plasmodium species in sub-Saharan Africa causing malaria in humans (Garnham, 1966; Coatney, 1971; Sutherland, 2016; Nundu et al., 2021; Culleton et al., 2023; Miezan et al., 2023). It is detected worldwide in many tropical and subtropical regions and is often found in co-infections with other malaria parasites, mostly P. falciparum (Zhou et al., 1998; Herman et al., 2023; Nguiffo-Nguete et al., 2023). Over the past decades a worldwide decline in malaria cases and deaths has been reported by the World Health Organization (WHO) (Organization, 2023). However, this decrease in case numbers has not been observed for P. malariae. On the contrary, there are many endemic countries reporting an increase in P. malariae prevalence (Cao et al., 2016; Yman et al., 2019; Agonhossou et al., 2022; Nainggolan et al., 2022). Due to improvements and increased usage of molecular diagnostics, the number of P. malariae infections has been shown to be substantially higher than previously described (Woldearegai et al., 2019; Mbama Ntabi et al., 2022; Nguiffo-Nguete et al., 2023). However, treatment as well as epidemiological studies and vaccine development are mainly directed towards P. falciparum and more recently also P. vivax, while other species are almost entirely neglected (Groger et al., 2022). A deeper understanding of the biology and epidemiology of the non-falciparum species would contribute to our understanding of transmission dynamics which is a prerequisite for achieving malaria elimination.
P. malariae causes a more benign form of malaria characterized by comparably mild symptoms and low parasitemia. However, serious complications have also been reported, mainly including severe anemia (Langford et al., 2015) and acute kidney injury (Ehrich and Eke, 2007). Infections are described as more chronic, persisting in the human host for a long time, indicating that P. malariae is successfully adapted to its human host.
A powerful tool to further characterize a parasite population and its transmission dynamics is the usage of genotyping markers. With this approach, diverse genetic regions are used to distinguish parasite genotypes. For P. malariae two genotyping marker panels have been identified previously (Bruce et al., 2007; Mathema et al., 2020). Both approaches focus on microsatellite markers, that represent genomic repetitive regions displaying a high genetic diversity. Few other P. malariae genes have been investigated additionally regarding their variability (Guimarães et al., 2015; Lalremruata et al., 2017; Srisutham et al., 2018; Putaporntip et al., 2022). In the present study, we aimed to establish a panel with a minimum number of genotyping markers for P. malariae yet with a high discriminative power. We assessed four markers from previous studies - two microsatellites, Pm02 and Pm09, as well as P. malariae thrombospondin-related anonymous protein (pmtrap), and P. malariae merozoite surface protein 1 fragment 2 (pmmsp1 F2) - the most promising candidate markers based on published diversity and successful amplification (Bruce et al., 2007; Srisutham et al., 2018; Mathema et al., 2020). We optimized the workflow, confirmed their diversity in a large set of P. malariae samples from Gabon, a malaria-endemic country in sub-Saharan Africa, and analysed dynamics and complexity of infections, by following-up asymptomatically infected individuals from Gabon in a tight schedule over one week.
A total of 410 P. malariae samples from two different study populations were investigated in this study. Study 1 was a cross-sectional study, aiming to characterize Plasmodium infections in rural areas in Fougamou and surrounding villages in Gabon in February/March 2016 (Manego et al., 2017; Groger et al., 2018; Woldearegai et al., 2019). A total of 840 individuals aged from 1-96 years were included in the original study, from which 193 participants were positive by 18S rRNA qPCR for P. malariae. Of these, 95 samples were used here, selected according to low Cq values.
Study 2 was conducted in 2019/2020 in Lambaréné, Gabon, assessing safety and efficacy of ivermectin against P. falciparum infections in asymptomatic adults (Ekoka Mbassi et al., 2023); samples were collected throughout the year. As ivermectin at the given dose showed similar activity as placebo against Plasmodia, individuals are regarded as not treated for this study. Out of 49 participants, 21 were positive for P. malariae at inclusion. The following 15 timepoints were investigated: Screening (SCR), after 8 (H8), 16 (H16), 24 (H24), 32 (H32), 40 (H40), 48 (H48), 56 (H56), 64 (H64), 72 (H72), 96 (H96), 120 (H120) hours, day 6 (D6), day 7 (D7). On day 7 a complete treatment of artemether-lumefantrine was given, and additional sampling was done on day 14 (D14).
Both studies and the corresponding experimental protocols were approved by the Institutional Ethics Committee of CERMEL (CEI-007/2014, CEI/CERMEL 006/2019). Study 2 was registered with the Pan-African Clinical Trials Registry (PACTR201908520097051). All methods were carried out in accordance with relevant guidelines and regulations. Informed consent was obtained from all adult participants or legal guardians.
Blood samples were stabilized in RNAlater prior to extraction. DNA was extracted either manually using the QIAamp DNA mini blood kit (QIAGEN) or automated in the QIAsymphony (QIAGEN) or in the KingFisher™ Flex Purification System (Thermo Fisher Scientific) with the sbeadex blood kit (LGC), according to the manufacturer’s protocols. Plasmodium species were determined by qPCR as described previously targeting the 18S rRNA of the different Plasmodium species (Groger et al., 2018; Woldearegai et al., 2019; Ekoka Mbassi et al., 2023).
We chose four loci distributed on different regions of the P. malariae genome, namely Pm02, Pm09, pmtrap, and pmmsp1 F2, based on high genetic diversity published in other P. malariae populations (Bruce et al., 2007; Guimarães et al., 2015; Srisutham et al., 2018). Pm09 is located on chromosome 1 and pmmsp1 F2 on chromosome 7. Pm02 and pmtrap are both located on chromosome 12. The two microsatellite markers show the following repeat sizes: Pm02 has a repeat unit of 4 bp (CATA) and Pm09 of 17 bp (GCAAAATAACAAAAAGA). Pmtrap shows different patterns of 12 bp repeat units (CCAGAGGATAGA; CCAGAGAATAGA; CCAGAGAATAGT).
Each of the four length-polymorphic markers was amplified by nested PCR. In all PCR runs the following controls were used: non-template control (H2O), negative control (human whole blood from a malaria naïve person), positive control (P. malariae mono infection).
Optimized cycling conditions and primer sequences for Pm02, Pm09, pmtrap and pmmsp1 F2 are shown in Supplementary Table S1. New outer (Pm02 and Pm09 forward) and inner (pmmsp1 F2 and pmtrap) primers were designed according to the amplicon length needed for the analysis method used, which is described below. Primer design was based on the P. malariae reference genome PmUG01 sequences (LT594622 – LT594635). Samples that were negative in two consecutive runs, were repeated with 5 more cycles, to exclude low DNA content as a source of error.
Before further analysis, the amplicon size and concentration were measured using automated capillary gel electrophoresis (QIAxcel, QIAGEN). Specific conditions and descriptions are listed in Supplementary Table S2 and Supplementary File S1.
Verification of automated capillary gel electrophoresis is described in Supplementary File S1.
We assessed the performance and diversity of the P. malariae genotyping markers along 95 P. malariae positive samples from study 1. Whereas samples from study 2 were used to analyze the performance of the markers over time. The maximum multiplicity of infection (MOI) was counted based on the highest number of alleles per marker in one sample. The mean MOI for each marker was calculated as followed: . To estimate the allele frequency for each detected allele size, we divided the number of alleles detected by the total number of PCR positive samples. The expected heterozygosity (He) was calculated using the following formula:
where p corresponds to the ith of k alleles and N to the number of positive samples.
Based on literature and in silico analysis, we chose four different loci Pm02, Pm09, pmtrap, and pmmsp1 F2.
The allelic diversity of the markers was assessed on a set of 95 samples from study 1. The number of allele groups detected in the 95 samples was highest for pmmsp1 F2 and Pm02, with 15 distinct groups detected each (Figure 1). While 11 allele groups were detected for pmtrap, Pm09 resulted in 5 distinct allele groups, which was the lowest number detected. Overall, the allele frequency of groups was unevenly distributed for all four markers, with mostly one major allele group. For pmmsp1 F2 seven minor allele groups were identified, each appearing only once.
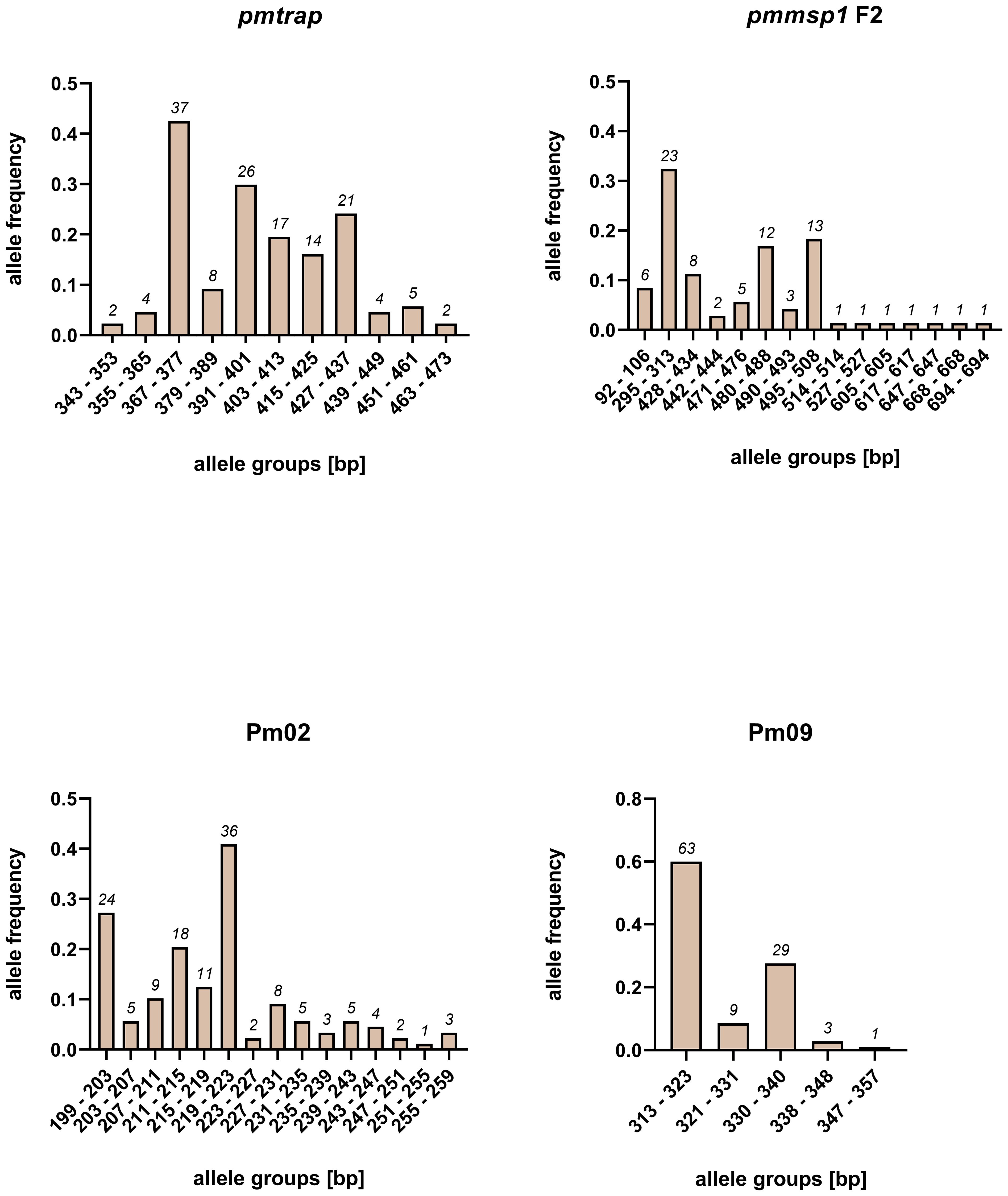
Figure 1 Allele frequencies of four P. malariae markers pmtrap, pmmsp1 F2, Pm02 and Pm09, arranged in different allele groups according to the fragment size. Frequencies and sizes include alleles from multiclonal infections. In total 95 samples were screened.
An overall comparison of allele characteristics showed that pmtrap and pmmsp1 F2 performed best among the four markers when aiming at displaying diversity (Table 1). The highest expected heterozygosity (He) of 0.81 was calculated for pmmsp1 F2 while pmtrap had the highest mean MOI of 1.61. Pm02 performed comparably well, with a mean MOI of 1.55 and an He of 0.68. Despite the high resolution of the chosen QIAxcel method, the differentiation of the allele groups was challenging for the marker Pm02, as it has a repeat size unit of only 4 bp, which falls within the maximum resolution of 3-5 bp that can be achieved with the underlying method. Pm09 was the least diverse locus, with the lowest He (0.56) as well as a low MOI (1.19). Amplification success exceeded 90% for three out of the four markers, while pmmsp1 F2 exhibited a lower performance, with a PCR positivity rate of only 76%. The gene is known for its polymorphisms; therefore, it is likely that for some of the samples the primer regions were too diverse to anneal without mismatches. Indeed, we found one mismatch in the reverse primer region of 1/12 generated pmmsp1 F2 sequences. We do not see any effect of the Cq value on the amplification success of the samples.
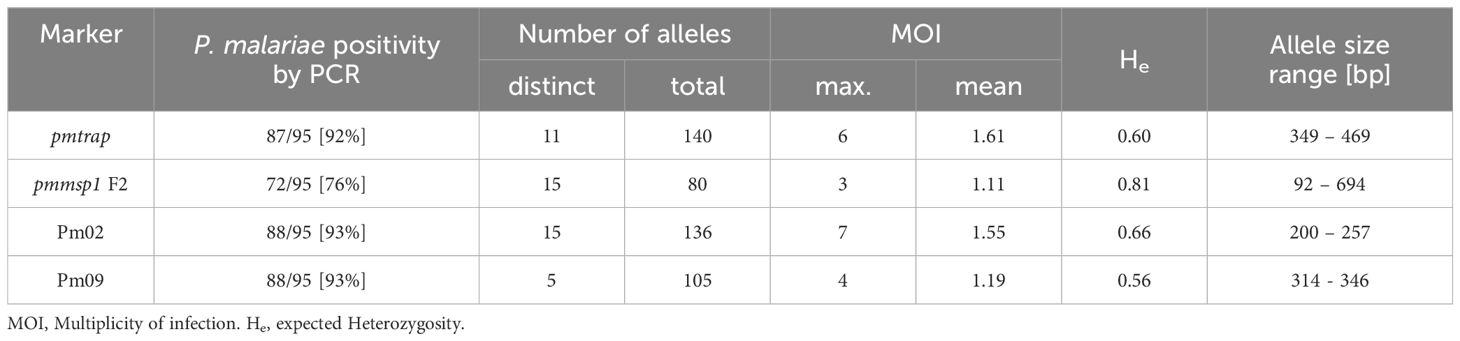
Table 1 Allele characteristics of four P. malariae markers based on size polymorphisms.
In addition to the P. malariae reference genome obtained from Uganda, we identified sequences from regions outside of Africa within the NCBI database that align to the four markers explored with samples from study 1. We also considered P. brasilianum sequences. 32 sequences were retrieved for pmtrap and 50 sequences for pmmsp1. All 32 pmtrap sequences are derived from samples collected in Southeast Asia. 35/50 pmmsp1 sequences originate from Southeast Asia as well and the remaining 15/50 from South America. For Pm02 and Pm09, one and two published sequences respectively aligned to our sequences, all three originating from South America.
When comparing the sequences from pmtrap and pmmsp1 F2 to the above-mentioned sequences, we found four new genotypes using pmtrap (accession numbers: OR829576-OR829579) and five new genotypes using pmmsp1 F2 (accession numbers: OR829569-OR829571, OR829572, OR829573), among our samples that were not published before. For Pm09 two new genotypes (accession numbers: OR829566, OR829568) were found, each displaying a new repeat unit motif of 17 bp (GAAGAGCAAAATAACAA) and 8 bp (AACAAACA). For Pm02 seven new genotypes were found (accession numbers: OR829554, OR829557, OR829558, OR829560-OR829564), one with a new repeat unit of 6 bp (CCACAC). New genotypes are defined by a new number of repeat units for pmtrap, Pm02 and Pm09 or an amplicon size not reported before for pmmsp1 F2. The genotype derived from the reference genome was found among our sequences in all four markers.
We investigated which combination of the four markers gives the highest diversity compared to using a single marker (Supplementary Figure S1). This was assessed on the 95 samples from study 1. Hereby the number of samples with either 1 (i.e., MOI=1) or more than 1 genotype (i.e., MOI>1), was used as a benchmark to evaluate the different combinations. The more samples with an MOI>1, the better the marker combination reflects the allelic diversity of the sample set. In addition, the PCR failure rate, i.e., number of samples that were not amplified, was used for comparison. The number of negative samples was highest (at least 7/95 samples) when the markers were used separately than in any other combination (less than 6/95 samples). All combinations of two markers enhanced diversity by either reducing the number of negative samples or increasing the number of samples with multiple genotypes.
For any combination of 3 markers, the multiplicity is even higher, with at least 33/95 samples showing a MOI>1. Combining all four markers decreased the number of negative samples to 3/95 and increased samples with an MOI>1 to 40/95. Although this combination reflects diversity best, we did not select it for further investigation. We excluded Pm09 from the combinations because of its low heterozygosity. Pm02 on the other hand was diverse enough, however the analysis of the generated amplicons was challenging and time consuming as of the small repeat size of 4 bp. Instead, we chose the combination of pmtrap and pmmsp1 F2, identifying 61/95 samples with one genotype only (Figure 2). While 5/95 samples were not amplified by any of the two marker, 29/95 samples were carrying two or more genotypes simultaneously.
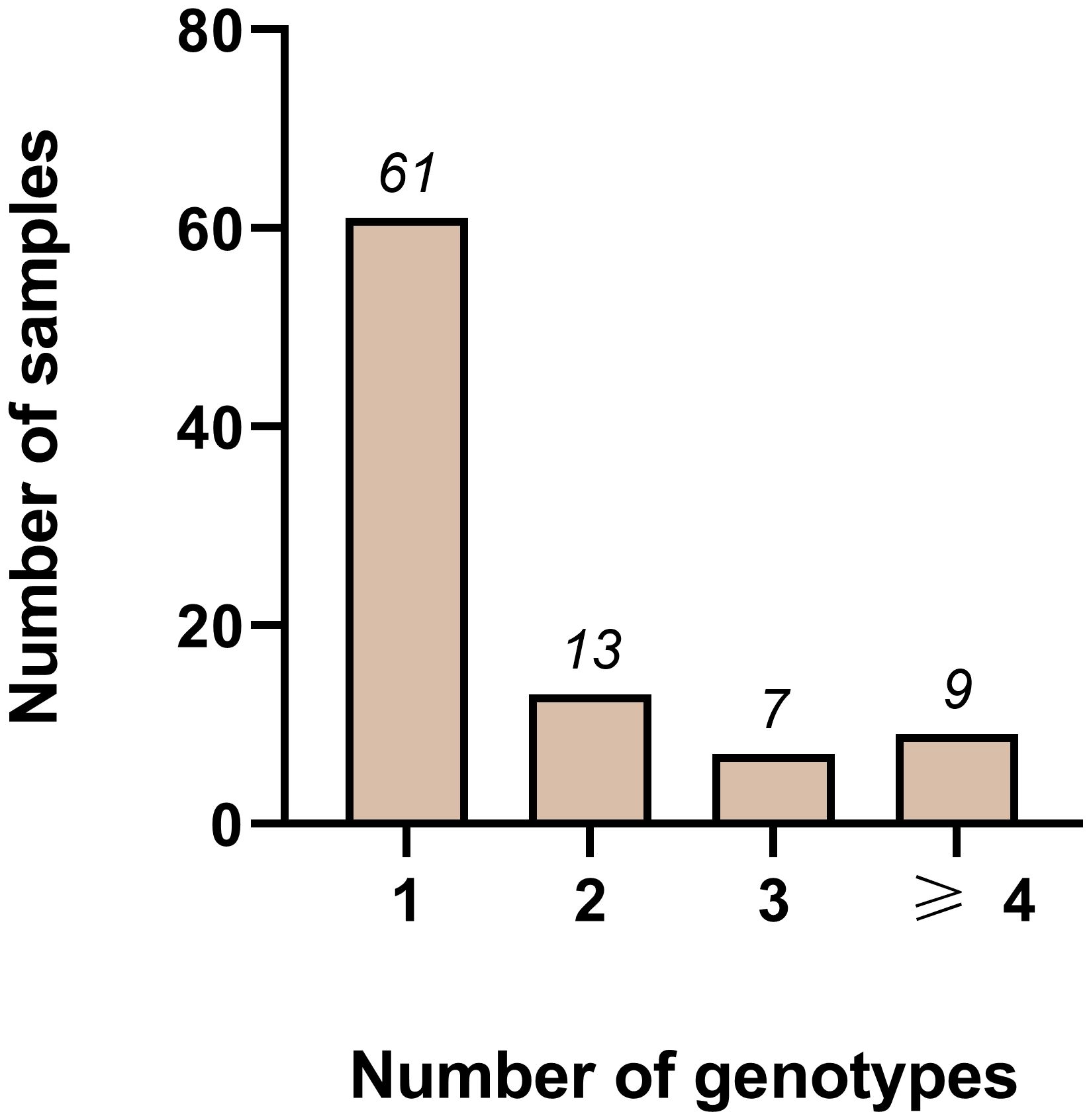
Figure 2 Distribution of different genotypes measured using the two markers pmmsp1 F2 and pmtrap in combination among 95 samples from Gabon. Out of 95 samples, 61 were infected with one genotype only. Whereas 13, 7 and 9 samples were infected with two, three or more than three genotypes respectively. In total 5/95 samples failed to amplify.
We further applied the two markers (pmmsp1 F2 and pmtrap) on a unique sample set from study 2, consisting of 21 P. malariae positive individuals that were followed for 14 days. In total, 15/21 individuals were positive for both markers by PCR while 6/21 were positive for only one marker respectively (two for pmtrap and four for pmmsp1 F2).
Overall individuals showed different patterns throughout the course of the infection (for an overview of all 21 individuals see Supplementary Table S3): 2/15 individuals showed only one genotype for both markers over all sampling timepoints (individual i.01 in Figure 3) and 11/15 individuals were infected with multiple genotypes according to both markers, one was even detected with 8 genotypes using pmtrap (individual i.02 in Figure 3). In all 11 mentioned individuals the maximum MOI was higher for pmtrap than pmmsp1 F2. The maximum MOI measured with pmtrap and pmmsp1 F2 is depicted in Figure 3 for individuals i.01 – i.03 as an example for different genotype patterns detected. The remaining individuals can be seen in Supplementary Figure S2 (individuals i.04 – i.15). In 2/15 cases the maximum MOI was higher for pmmsp1 F2 and in two other cases it was similar. Overall, the different genotypes were detected very stable along the 15 timepoints. Some genotypes were fluctuating over time, detected at some timepoints and undetected in others (individual i.03 in Figure 3). This shows the high dynamics and complexity of P. malariae infections. 17/21 individuals were negative on day 14 (D14), as per protocol participants were treated with artemether-lumefantrine latest on day 7.
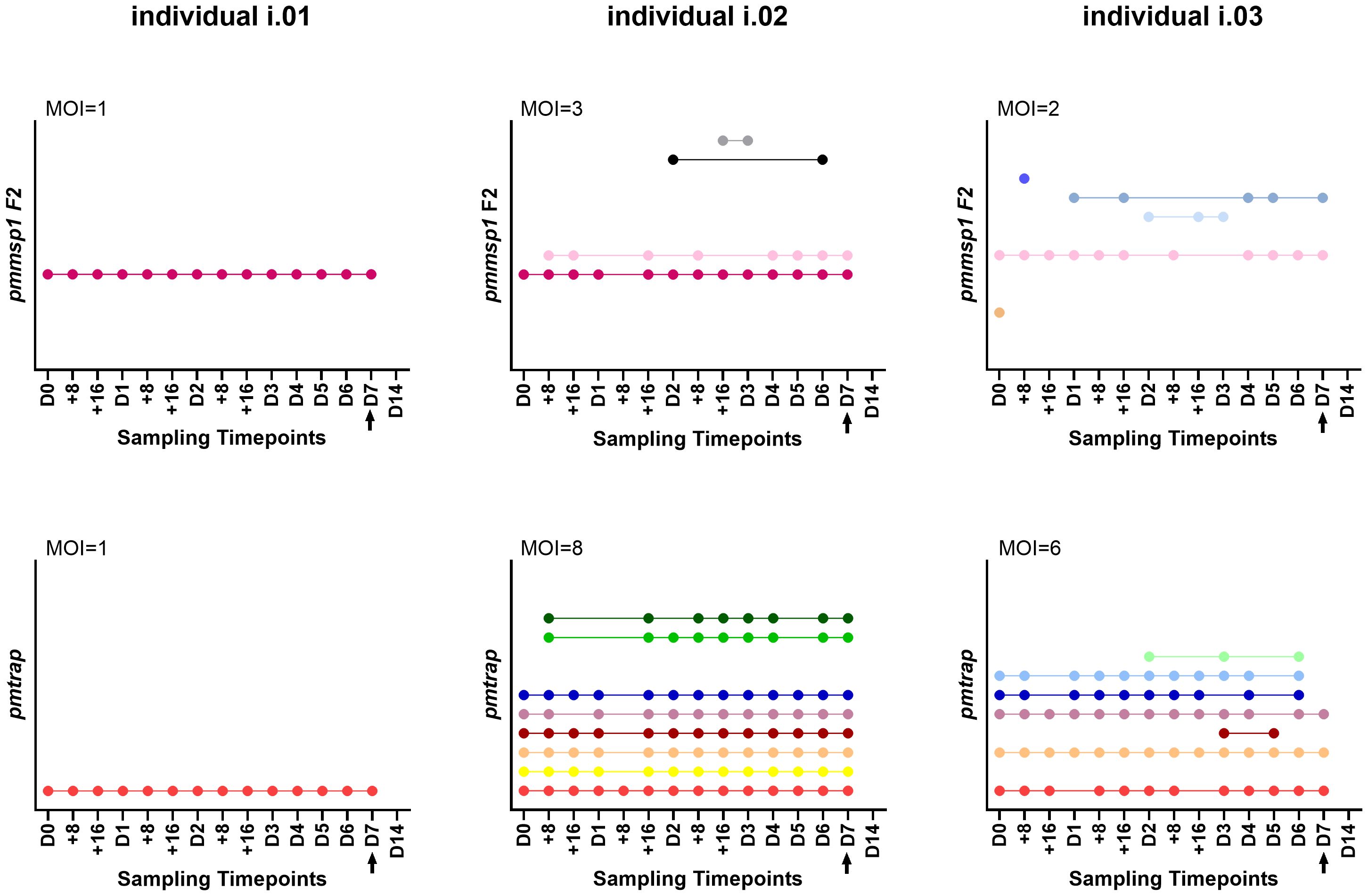
Figure 3 Three individuals i.01, i.02 and i.03 were genotyped at 15 timepoints using the markers pmmsp1 F2 (upper row) and pmtrap (lower row). Each color represents a different genotype for the respective marker. The arrow indicates treatment with artemether-lumefantrine on day 7. The remaining 18 individuals are shown in Supplementary Figure S2. MOI, multiplicity of infection, referring to the maximum MOI measured among the 15 timepoints.
The number of P. malariae infections in endemic countries has been shown to be substantially higher than previously acknowledged. Nevertheless, there are many open questions regarding the genetic epidemiology of this species. In the present study, we assessed four genotyping markers and selected two of them for establishment of a genotyping panel for P. malariae with a minimum number of markers and a high discriminative power.
First, we analysed the individual performance of the four markers using a sample set of 95 P. malariae infected individuals from Gabon. Our analysis showed that pmtrap, pmmsp1 F2 and Pm02 are the most diverse among the chosen loci in the population studied. They showed the highest number of distinct alleles - 11, 15 and 15, respectively -, whereas only 5 distinct alleles were found with Pm09 in the 95 samples. The mean MOI was highest for pmtrap (1.61), showing that this marker can very well reflect the multiplicity of P. malariae infections. The expected heterozygosity was highest for pmmsp1 F2 (0.81), demonstrating that this marker can detect a wide variety of different P. malariae genotypes. In terms of theses parameters, Pm02 performs comparatively well. However, this marker seems unsuitable for the fragment analysis using automated capillary gel electrophoresis with a resolution of 3-5 bp. The repeat size of only 4 bp is too small to reliably distinguish single alleles in infections with multiple genotypes, a limitation of this method. Similar results were found in another study on P. vivax genotyping, where automated capillary gel electrophoresis also reached its limit in distinguishing alleles that are close to each other in size (Manrique et al., 2015). The observed differences in the respective PCR efficiency of each marker can be attributed to various influencing factors. Low DNA concentrations in the sample are most likely the major reason why some samples were not amplified at all. We found a mismatch in the primer region in one of the pmmsp1 F2 sequences. This further underlines the high rate of polymorphisms in this region and might have led to the low amplification success for this marker. If primer binding is not efficient, less DNA is amplified. PCR conditions were thoroughly optimized to increase PCR efficiency to a maximum.
Overall, the range of heterozygosity (0.56 – 0.80) is comparable to other published marker panels by Bruce et al. (0.331 – 0.839) and Mathema et al. (0.530 – 0.922); the mean MOI for the four markers (1.11 – 1.61) is even slightly higher (Bruce et al., 2007; Mathema et al., 2020). However, both parameters depend on the characteristics of the investigated population, as geographical origin, clinical status, participants age, or seasonality. The samples used here for the validation of genetic diversity are from one geographical region (Lambaréné and surrounding villages, Gabon) and were collected within two months (study 1). The same is true for the P. malariae sample set from Myanmar of Mathema et al. In contrast Bruce et al. tested samples from diverse geographical origin (Thailand, Malawi, and Gambia), different clinical status and transmission seasons. Therefore, a direct comparison of the different marker panels using different settings can be misleading.
A notable observation for the pmmsp1 F2 marker was that comparatively more amplicons with a small size were detected and that the MOI was lower than for the other three markers. This effect of preferential amplification of shorter over longer amplicons has been seen in P. falciparum genotyping before, targeting the msp1 locus as well (Messerli et al., 2017). It can lead to an underestimation of the MOI. However, we saw no evidence of preferential amplification of smaller fragments for the pmmsp1 F2 locus (Supplementary File S2). Also, it is unlikely that a low parasite density of the minor genotype leads to these findings in infections with multiple genotypes, as the other three markers have a higher MOI. Another explanation for the seen effect could be mutations in the primer region of the bigger alleles.
We optimized our genotyping approach based on automated capillary gel electrophoresis. To validate the method, we sequenced amplicons from samples that were infected with one genotype only and compared the amplicon lengths from both methods. The high concordance observed for all four markers shows that our chosen analysis method is very reliable (Supplementary File S3). This approach has been proven efficient in previous Plasmodium genotyping approaches as well (Manrique et al., 2015; Nguyen et al., 2019; Tadele et al., 2022). General advantages of the method include the fast and high throughput, as well as the straightforward analysis. The high deviation of up to a maximum of 21 bp seen for pmmsp1 F2 can be explained by the lower resolution of 20 bp given by the used method. As the allele size range was bigger for this marker (92 – 694 bp), a method with a lower resolution (20 bp) had to be used.
Previous studies on P. malariae genotyping have been carried out in samples from South America and/or Southeast Asia (Guimarães et al., 2015; Srisutham et al., 2018; Putaporntip et al., 2022). The pmtrap and pmmsp1 sequences generated here originate from Gabon and therefore represent a valuable set of genotypes from Africa. A comparison of spatial dynamics of P. malariae genotypes reveals that some genotypes appear in all three continents mentioned: for pmtrap 3/10 identified genotypes have been found in Southeast Asia as well as Africa; for pmmsp1 F2 4/25 genotypes have been identified either in Southeast Asia and South America (2 genotypes), in South America and Africa (1 genotype) or in all three continents (1 genotype). However, most genotypes are shared among samples from the same region, forming spatial genotype clusters. As most of the available sequences are from Southeast Asia, the biggest cluster was identified from this region for both markers (4/10 using pmtrap and 14/25 using pmmsp1 F2). Another genotype cluster exclusively seen in samples from South America, consists of three genotypes and was identified using pmmsp1 F2. We found three new genotypes using both markers in our samples from Gabon, Africa, that were not seen in the sequencing data from any other continent before.
In the end, a combination of the two markers pmmsp1 F2 and pmtrap resulted in the panel with the best discriminative power. While pmmsp1 F2 can detect a high variety of diverse genotypes, pmtrap is able to best cover the multiplicity of P. malariae infections. In combination the two markers provide a powerful tool for studies on genetic diversity of P. malariae.
Based on the results from the sample set of the two-weekly follow-up study, both markers demonstrated a very consistent performance over time. Genotypes were detected consistently throughout the different sampling timepoints, which shows the method`s effectiveness. We detected up to 8 genotypes in one individual at the same timepoint. This surprisingly high number of circulating P. malariae genotypes in an asymptomatic population is particularly interesting. In a previous study from Gabon including symptomatic individuals, P. malariae was the species with the highest number of genotypes (Lalremruata et al., 2017). To which extend these findings are related to the clinical status needs further investigation.
Throughout the follow-up period, fluctuations of genotypes were evident, indicating that not all genotypes are present at every sampling occasion. This observation is likely because the amount of DNA from these minor genotypes approaches the limit of detection, so that they are detectable at one time point but not at the other. However, it is also possible that some of the appearing genotypes are new infections. The 72-hour cycle of P. malariae could not be seen in the pattern of detected genotypes from our data.
We followed the P. malariae infections for a limited time, providing valuable insights into their dynamics. As P. malariae infections are known for their longevity and to persist in the human host for even decades (Ciusa et al., 2022), studying infections over longer periods would provide even more information on the biology of this species. However, the high complexity of asymptomatic P. malariae infections with multiple genotypes and variable detection, shown by our findings, makes longitudinal investigations challenging to interpret since a genotype may not be detected on one sampling timepoint but be present in another. This could be eventually addressed with next generation sequencing (NGS) approaches that can increase sensitivity and the probability to detect minor clones, potentially adding further insights into P. malariae genotypes, infection dynamics and biology (Gruenberg et al., 2019; Wamae et al., 2022).
Some individuals in our study were still PCR-positive on day 14, seven days after treatment with artemether-lumefantrine. We do not think that these are treatment failures, but rather residual DNA that can still be detected with our highly sensitive PCR. Similar results were also found for P. falciparum (Inoue et al., 2024). In addition, no parasites were detected microscopically at those timepoints. However, in order to be certain that these are no treatment failures and that the individuals turn PCR negative later, follow-up samples from later timepoints would have been required.
We were able to successfully implement a new P. malariae genotyping panel consisting of only two markers, pmtrap and pmmsp1 F2. This set of markers will add a valuable tool to characterize the genetic diversity of P. malariae infections from different regions and to better elucidate the transmission dynamics of this neglected malarial parasite species. It can be implemented in many settings as its usage is straightforward, having a short turnaround time as only two markers are required.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OR829554-OR829580.
The studies involving humans were approved by Institutional Ethics Committee of CERMEL. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
MRo: Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. KK: Investigation, Methodology, Validation, Writing – review & editing. LS: Investigation, Writing – review & editing. LB: Investigation, Writing – review & editing. MS: Investigation, Methodology, Writing – review & editing. AL: Methodology, Writing – review & editing. TW: Resources, Writing – review & editing. PB: Resources, Writing – review & editing. MG: Resources, Writing – review & editing. RZ: Resources, Writing – review & editing. DE: Resources, Writing – review & editing. GM-N: Resources, Writing – review & editing. SA: Resources, Writing – review & editing. MRa: Resources, Writing – review & editing. BM: Resources, Writing – review & editing. JI: Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing. JH: Conceptualization, Project Administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We acknowledge support by Open Access Publishing Fund of University of Tübingen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor KM declared a past co-authorship with the author JH.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1405198/full#supplementary-material
Agonhossou, R., Akoton, R., Dossou, Y. A., Avokpaho, E., Mbama, D. N. J., Boussougou-Sambe, T. S., et al. (2022). Surveillance of Plasmodium malariae infection among inhabitants of rural areas in Ouidah–Kpomasse–Tori Bossito health district, Benin. Parasitol. Res. 121, 275–286. doi: 10.1007/s00436-021-07398-z
Bruce, C. M., Macheso, A., Galinski, R. M., Barnwell, W. J. (2007). Characterization and application of multiple genetic markers forPlasmodium malariae. Parasitology 134, 637–650. doi: 10.1017/S0031182006001958
Cao, Y., Wang, W., Liu, Y., Cotter, C., Zhou, H., Zhu, G., et al. (2016). The increasing importance of Plasmodium ovale and Plasmodium malariae in a malaria elimination setting: an observational study of imported cases in Jiangsu Province, China 2011–2014. Malaria J. 15. doi: 10.1186/s12936-016-1504-2
Ciusa, G., Guida Marascia, F., Virruso, R., Angheben, A., Guaraldi, G., Cascio, A. (2022). Clinical recrudescence of chronic untreated. IDCases 27, e01456. doi: 10.1016/j.idcr.2022.e01456
Coatney, G. R., Collins, W. E., Warren, M., Contacos, P. G. (1971). The primate malarias. (National Institute of Allergy and Infectious Diseases (U.S.)).
Culleton, R., Pain, A., Snounou, G. (2023). Plasmodium malariae: the persisting mysteries of a persistent parasite. Trends Parasitol. 39, 113–125. doi: 10.1016/j.pt.2022.11.008
Ehrich, J. H. H., Eke, F. U. (2007). Malaria-induced renal damage: facts and myths. Pediatr. Nephrol. 22, 626–637. doi: 10.1007/s00467-006-0332-y
Ekoka Mbassi, D., Mombo-Ngoma, G., Held, J., Okwu, D. G., Ndzebe-Ndoumba, W., Kalkman, L. C., et al. (2023). Efficacy and safety of ivermectin for the treatment of Plasmodium falciparum infections in asymptomatic male and female Gabonese adults - a pilot randomized, double-blind, placebo-controlled single-centre phase Ib/IIa clinical trial. EBioMedicine 97, 104814. doi: 10.1016/j.ebiom.2023.104814
Groger, M., Tona Lutete, G., Mombo-Ngoma, G., Ntamabyaliro, N. Y., Kahunu Mesia, G., Muena Mujobu, T. B., et al. (2022). Effectiveness of pyronaridine-artesunate against Plasmodium malariae, Plasmodium ovale spp, and mixed-Plasmodium infections: a post-hoc analysis of the CANTAM-Pyramax trial. Lancet Microbe 3, e598–e605. doi: 10.1016/S2666-5247(22)00092-1
Groger, M., Veletzky, L., Lalremruata, A., Cattaneo, C., Mischlinger, J., Zoleko-Manego, R., et al. (2018). Prospective Clinical Trial Assessing Species-Specific Efficacy of Artemether-Lumefantrine for the Treatment of Plasmodium malariae, Plasmodium ovale, and Mixed Plasmodium Malaria in Gabon. Antimicrobial Agents Chemotherapy 62, AAC.01758–17. doi: 10.1128/AAC.01758-17
Gruenberg, M., LERCH, A., BECK, H. P., FELGER, I. (2019). Amplicon deep sequencing improves Plasmodium falciparum genotyping in clinical trials of antimalarial drugs. Sci. Rep. 9, 17790. doi: 10.1038/s41598-019-54203-0
Guimarães, L. O., Wunderlich, G., Alves, J. M. P., Bueno, M. G., Röhe, F., Catão-Dias, J. L., et al. (2015). Merozoite surface protein-1 genetic diversity in Plasmodium malariae and Plasmodium brasilianum from Brazil. BMC Infect. Dis. 15. doi: 10.1186/s12879-015-1238-8
Herman, C., Leonard, C. M., Uhomoibhi, P., Maire, M., Moss, D., Inyang, U., et al. (2023). Non-falciparum malaria infection and IgG seroprevalence among children under 15 years in Nigeri. Nat. Commun. 14, 1360. doi: 10.1038/s41467-023-37010-0
Inoue, J., Galys, A., Rodi, M., Ekoka Mbassi, D., Mombo-Ngoma, G., Adegnika, A. A., et al. (2024). Dynamics of Plasmodium species and genotype diversity in adults with asymptomatic infection in Gabon. Int. J. Infect. Dis. 143, 107013. doi: 10.1016/j.ijid.2024.107013
Lalremruata, A., Jeyaraj, S., Engleitner, T., Joanny, F., Lang, A., Bélard, S., et al. (2017). Species and genotype diversity of Plasmodium in malaria patients from Gabon analysed by next generation sequencing. Malaria J. 16. doi: 10.1186/s12936-017-2044-0
Langford, S., Douglas, N. M., Lampah, D. A., Simpson, J. A., Kenangalem, E., Sugiarto, P., et al. (2015). Plasmodium malariae infection associated with a high burden of anemia: A hospital-based surveillance study. PloS Negl. Trop. Dis. 9, e0004195. doi: 10.1371/journal.pntd.0004195
Manego, R. Z., Mombo-Ngoma, G., Witte, M., Held, J., Gmeiner, M., Gebru, T., et al. (2017). Demography, maternal health and the epidemiology of malaria and other major infectious diseases in the rural department Tsamba-Magotsi, Ngounie Province, in central African Gabon. BMC Public Health 17, 130. doi: 10.1186/s12889-017-4045-x
Manrique, P., Hoshi, M., Fasabi, M., Nolasco, O., Yori, P., Calderón, M., et al. (2015). Assessment of an automated capillary system for Plasmodium vivax microsatellite genotyping. Malar J. 14, 326. doi: 10.1186/s12936-015-0842-9
Mathema, V. B., Nakeesathit, S., Pagornrat, W., Smithuis, F., White, N. J., Dondorp, A. M., et al. (2020). Polymorphic markers for identification of parasite population in Plasmodium malariae. Malaria J. 19. doi: 10.1186/s12936-020-3122-2
Mbama Ntabi, J. D., Lissom, A., Djontu, J. C., Diafouka-Kietela, S., Vouvoungui, C., Boumpoutou, R. K., et al. (2022). Prevalence of non-Plasmodium falciparum species in southern districts of Brazzaville in The Republic of the Congo. Parasites Vectors 15. doi: 10.1186/s13071-022-05312-9
Messerli, C., Hofmann, N. E., Beck, H. P., Felger, I. (2017). Critical evaluation of molecular monitoring in malaria drug efficacy trials and pitfalls of length-polymorphic markers. Antimicrob. Agents Chemother. 61. doi: 10.1128/AAC.01500-16
Miezan, A. J. S., Gnagne, A. P., Bedia-Tanoh, A. V., Kone, E. G. M., Konate-Toure, A. A., Angora, K. E., et al. (2023). Molecular epidemiology of non-falciparum Plasmodium infections in three different areas of the Ivory Coast. Malar J. 22, 211. doi: 10.1186/s12936-023-04639-7
Nainggolan, I. R. A., Syafutri, R. D., Sinambela, M. N., Devina, C., Hasibuan, B. S., Chuangchaiya, S., et al. (2022). The presence of Plasmodium malariae and Plasmodium knowlesi in near malaria elimination setting in western Indonesia. Malaria J. 21. doi: 10.1186/s12936-022-04335-y
Nguiffo-Nguete, D., Nongley Nkemngo, F., Ndo, C., Agbor, J.-P., Boussougou-Sambe, S. T., Salako Djogbénou, L., et al. (2023). Plasmodium malariae contributes to high levels of malaria transmission in a forest–savannah transition area in Cameroon. Parasites Vectors 16. doi: 10.1186/s13071-022-05635-7
Nguyen, T. T., Nzigou Mombo, B., Lalremruata, A., Koehne, E., Zoleko Manego, R., Dimessa Mbadinga, L. B., et al. (2019). DNA recovery from archived RDTs for genetic characterization of Plasmodium falciparum in a routine setting in Lambaréné, Gabon. Malar J. 18, 336. doi: 10.1186/s12936-019-2972-y
Nundu, S. S., Culleton, R., Simpson, S. V., Arima, H., Muyembe, J. J., Mita, T., et al. (2021). Malaria parasite species composition of Plasmodium infections among asymptomatic and symptomatic school-age children in rural and urban areas of Kinshasa, Democratic Republic of Congo. Malar J. 20, 389. doi: 10.1186/s12936-021-03919-4
Putaporntip, C., Kuamsab, N., Rojrung, R., Seethamchai, S., Jongwutiwes, S. (2022). Structural organization and sequence diversity of the complete nucleotide sequence encoding the Plasmodium malariae merozoite surface protein-1. Sci. Rep. 12. doi: 10.1038/s41598-022-19049-z
Srisutham, S., Saralamba, N., Sriprawat, K., Mayxay, M., Smithuis, F., Nosten, F., et al. (2018). Genetic diversity of three surface protein genes in Plasmodium malariae from three Asian countries. Malaria J. 17. doi: 10.1186/s12936-018-2176-x
Sutherland, C. J. (2016). Persistent parasitism: the adaptive biology of malariae and ovale malaria. Trends Parasitol. 32, 808–819. doi: 10.1016/j.pt.2016.07.001
Tadele, G., Jaiteh, F. K., Oboh, M., Oriero, E., Dugassa, S., Amambua-Ngwa, A., et al. (2022). Low genetic diversity of Plasmodium falciparum merozoite surface protein 1 and 2 and multiplicity of infections in western Ethiopia following effective malaria interventions. Malar J. 21, 383. doi: 10.1186/s12936-022-04394-1
Wamae, K., Ndwiga, L., Kharabora, O., Kimenyi, K., Osoti, V., De Laurent, Z., et al. (2022). Targeted amplicon deep sequencing of ama1 and mdr1 to track within-host. Wellcome Open Res. 7, 95. doi: 10.12688/wellcomeopenres
Woldearegai, T. G., Lalremruata, A., Nguyen, T. T., Gmeiner, M., Veletzky, L., Tazemda-Kuitsouc, G. B., et al. (2019). Characterization of Plasmodium infections among inhabitants of rural areas in Gabon. Sci. Rep. 9. doi: 10.1038/s41598-019-46194-9
Yman, V., Wandell, G., Mutemi, D. D., Miglar, A., Asghar, M., Hammar, U., et al. (2019). Persistent transmission of Plasmodium malariae and Plasmodium ovale species in an area of declining Plasmodium falciparum transmission in eastern Tanzania. PloS Negl. Trop. Dis. 13, e0007414. doi: 10.1371/journal.pntd.0007414
Zhou, M., Liu, Q., Wongsrichanalai, C., Suwonkerd, W., Panart, K., Prajakwong, S., et al. (1998). High prevalence of Plasmodium malariae and Plasmodium ovale in malaria patients along the Thai-Myanmar border, as revealed by acridine orange staining and PCR-based diagnoses. Trop. Med. Int. Health 3, 304–312. doi: 10.1046/j.1365-3156.1998.00223.x
Keywords: Plasmodium malariae, genotyping, size polymorphism, pmtrap, pmmsp1 F2
Citation: Rodi M, Kawecka K, Stephan L, Berner L, Medina MS, Lalremruata A, Woldearegai TG, Matsiegui PB, Groger M, Manego RZ, Ekoka Mbassi D, Mombo-Ngoma G, Agnandji ST, Ramharter M, Mordmüller B, Inoue J and Held J (2024) Genetic diversity of Plasmodium malariae in sub-Saharan Africa: a two-marker genotyping approach for molecular epidemiological studies. Front. Cell. Infect. Microbiol. 14:1405198. doi: 10.3389/fcimb.2024.1405198
Received: 22 March 2024; Accepted: 09 July 2024;
Published: 19 July 2024.
Edited by:
Kai Matuschewski, Humboldt University of Berlin, GermanyReviewed by:
Dalma Maria Banic, Oswaldo Cruz Foundation (Fiocruz), BrazilCopyright © 2024 Rodi, Kawecka, Stephan, Berner, Medina, Lalremruata, Woldearegai, Matsiegui, Groger, Manego, Ekoka Mbassi, Mombo-Ngoma, Agnandji, Ramharter, Mordmüller, Inoue and Held. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jana Held, amFuYS5oZWxkQHVuaS10dWViaW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.