 Aswath P. Chandrasekar
Aswath P. Chandrasekar Mark Maynes3,4
Mark Maynes3,4 Andrew D. Badley
Andrew D. Badley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 05 March 2024
Sec. Virus and Host
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1354502
This article is part of the Research TopicHIV Latency: Novel insights into the viral reservoir and therapeutic strategiesView all 5 articles
HIV cure still remains an elusive target. The “Shock and Kill” strategy which aims to reactivate HIV from latently infected cells and subsequently kill them through virally induced apoptosis or immune mediated clearance, is the subject of widespread investigation. NF-κB is a ubiquitous transcription factor which serves as a point of confluence for a number of intracellular signaling pathways and is also a crucial regulator of HIV transcription. Due to its relatively lower side effect profile and proven role in HIV transcription, the non-canonical NF-κB pathway has emerged as an attractive target for HIV reactivation, as a first step towards eradication. A comprehensive review examining this pathway in the setting of HIV and its potential utility to cure efforts is currently lacking. This review aims to summarize non-canonical NF-κB signaling and the importance of this pathway in HIV shock-and-kill efforts.
The HIV latent reservoir refers to the pool of integrated-but-transcriptionally-silent HIV pro-viral DNA that persists indefinitely in vivo (Siliciano and Greene, 2011). The latent reservoir begins to seed within days of acute HIV infection. Combination anti-retroviral therapy (ART) can suppress viral replication and plasma viremia to undetectable levels; however, treatment interruption and ensuing reservoir reactivation allow for viral rebound and the return of plasma viremia (Vanhamel et al., 2019). The exact mechanisms underlying this partial and selective transcriptional silencing are poorly understood, and it has been observed that multiple factors contribute to determining the size of the reservoir including the efficacy of the host immune response, the level of plasma viremia in acute infection and the interval between infection and anti-HIV therapy initiation. Additionally, it has now become apparent that even under the cover of suppressive ART (which is capable of reducing plasma viremia to undetectable levels), the HIV reservoir remains dynamic with low-level replication and reservoir seeding through clonal expansion (Yeh et al., 2021; Hosmane et al., 2017; Bui et al., 2017; Bachmann et al., 2019; Woldemeskel et al., 2020).
A cure for HIV may be achieved either as a “sterilizing cure”, which is the complete eradication of the virus; or a “functional cure”, achieving the absence of viremia without ART. Within this paradigm, the “shock and kill” theory is currently under investigation at various levels as a feasible sterilizing or functional cure strategy. Briefly, shock and kill therapy aims to therapeutically reactivate latent HIV and subsequently clear it through a combination of immune mechanisms, the cytopathic effects of viral reactivation, and drugs that selectively eradicate reactivating cells (Kim et al., 2018; Chandrasekar and Badley, 2022). Importantly, identifying a feasible shock agent that potently activates HIV remains the major objective. Furthermore, learning the various mechanisms that drive HIV replication is central to the shock and kill strategy. An understanding of key transcriptional regulators is necessary to identify viable drug candidates that may reactivate the latent reservoir. The Nuclear Factor Kappa Light Chain Enhancer of B Cells (NF-κB) signaling pathway has long been recognized as a major driver of HIV replication and its relationship to HIV transcription has been extensively studied (Hiscott et al., 2001). Between the canonical and non-canonical pathways that the NF-κB family can signal through, studies are now beginning to identify the non-canonical NF-κB pathway as a potential target for HIV cure efforts. This review aims to examine the non-canonical NF-κB pathway in depth and provide a comprehensive resource that would help inform future investigations.
The Nuclear Factor Kappa Light Chain Enhancer of B Cells (NF-κB) complex constitutes one of the key transcription regulatory factor families, and is present in almost all cells, across most living higher organisms (Gilmore, 2006). The NF-κB family, defined by the presence of a conserved homology domain known as “Rel”, includes five individual Rel containing proteins – NF-KB1, NF-KB2, RelA, RelB and c-Rel (O'Dea and Hoffmann, 2010). These proteins interact with each other to form dimers that are capable of binding to DNA and activating transcription.
The NF-κB1 and NF-κB2 proteins in their inactive states are termed p105 and p100, respectively. Following activation or as a result of translational arrest, these proteins are transformed to their active, shorter conformations – p50 and p52 through protease mediated cleavage. The p50 and p52 proteins can form dimers either with each other, with themselves or with the RelA (p65), RelB and c-Rel members of the family and exert downstream effects (Gilmore, 2006; O'Dea and Hoffmann, 2010; Hoffmann and Baltimore, 2006). Of the possible dimer combinations, the combinations of p50:p50, p52:p52 and p50:p52, while capable of binding to DNA, are not known to exert transcriptional regulation. Additionally, the RelA : RelB, c-Rel : RelB and RelB : RelB dimers are incapable of binding to DNA. The remaining possible combinations are all capable of initiating transcription (O'Dea and Hoffmann, 2010).
NF-κB1 and 2 are, at baseline, in an inactive state; maintained as such, by their association with a group of regulatory, inhibitory proteins known as Inhibitory Kappa B (IκB). This family can further be subclassified into the typical IκB proteins: IκBα, IκBβ and IκBϵ, and the atypical IκB proteins: BCL-3, IκBξ and IκBNS (Tam and Sen, 2001; Whiteside et al., 1997; Nolan et al., 1993; Yu et al., 2020). These proteins bind to the NF-κB precursor proteins p105 and p100 and prevent their cleavage into their active forms. The Inhibitory Kappa B Kinase complex (IKK), consisting of the enzymes IKKα, IKKβ and IKKγ (also called NF-κB essential modifier (NEMO)), bind, phosphorylate and cause the degradation of the IκB proteins, allowing for p52 or p50 release and downstream signaling (Scheidereit, 2006).
Through exogenous or endogenous initiators, the NF-κB signaling cascade may be activated, leading to the regulation of cellular transcription and the up or down regulation of protein production. There may be numerous cascades through which NF-κB signaling may occur, but broadly, two major signaling pathways exist: a canonical NF-κB (cNF-κB) pathway and non-canonical NF-κB (ncNF-κB) pathway (Gilmore, 2006; O'Dea and Hoffmann, 2010; Hoffmann and Baltimore, 2006; Pomerantz and Baltimore, 2002). The cNF-κB pathway involves NF-κB1 protein in a p50:RelA (p65) heterodimer that is capable of binding to DNA and inducing transcriptional regulation and is largely the most constitutively active NF-κB signaling cascade. A comprehensive review of the mechanics and effects of the cNF-κB cascade is beyond the scope of this article, [reviewed in detail in (Yu et al., 2020)], but briefly, canonical NF-κB signaling may be initiated through multiple mechanisms including T cell receptors, B-cell receptors, cytokine receptors, and innate pattern recognition receptors. Canonical signaling promotes immune cell activation, the production of pro-inflammatory cytokines, angiogenesis and leads to immune recruitment. Dysregulated cNF-κB signaling has been described as a pathogenic mechanism in autoimmune diseases and malignancy (Yu et al., 2020). The ncNF-κB pathway involves the NF-κB2 protein in a p52:RelB heterodimer that is examined in detail below.
The Non-canonical NF-κB pathway culminates in transcriptional regulation by the NF-κB2 protein, in a p52:RelB heterodimer. At baseline, the p100 protein restricts RelB activity, functioning like a IκB protein. Following proteolysis to p52 and dimerization with RelB, the complex undergoes nuclear translocation and regulates transcription (Sun, 2011). The central component to ncNF-κB signaling is the NF-κB inducing kinase (NIK), a MAP3K like protein, which is a potent and specific inducer of p100 processing. NIK activation leads to downstream phosphorylation, ubiquitination, and activation of the ncNF-κB cascade through IKKα activation, and potentiation of IKKα-p100 binding (Senftleben et al., 2001; Xiao et al., 2004; Xiao et al., 2001). At cellular homeostasis, p100 is conjugated to the SUMO1 protein in a post-translational SUMOylating process, mediated through the Ubc9 enzyme, a process critical to normal p100 processing (Vatsyayan et al., 2008).
The activation the ncNF-κB cascade may be initiated by receptor-ligand interaction on the cell surface. Specifically, signaling through tumor necrosis factor (TNF) superfamily members and their receptors such as: CD40, B-cell activating factor (BAFFR), Lymphotoxin β Receptor (LTβR), Receptor activator for NF-κB (RANK), TNFR2, Fibroblast growth factor inducible factor (FN14), CD27, CD30, or OX40 (CD134) have been shown to activate the ncNF-κB. Additionally, signaling through the macrophage colony stimulating factor receptor (MCSFR) or membrane attack complexes (MACs) have also been shown to activate the ncNF-κB. Activation of the retinoic acid inducible gene 1 (RIG-1) by viral pathogens has also been shown to activate the ncNF-κB (Sun, 2011; Sun, 2017).
ncNF-κB activation through cell surface receptor-ligand interactions are primarily dependent on the recruitment and degradation of the TNF receptor associated factors (TRAF) 2 and 3. TRAF2 and TRAF3, in conjunction with the cellular inhibitor of apoptosis-1&2 (cIAP1 & cIAP2) proteins, function as the main negative regulators of ncNF-κB signaling (Vallabhapurapu et al., 2008; Zarnegar et al., 2008). In the inactivated state, TRAF3 is bound to NIK leading to its ubiquitination and proteasomal degradation, preventing p52 processing. TRAF3-NIK binding is mediated by the cIAP protein in a TRAF2-dependent manner. Following receptor ligation, activated cIAP mediates TRAF3 degradation, which allows for the release and accumulation of NIK (Sun, 2011; 2017). This process may be positively or negatively regulated in vivo.
Positive regulation of the cascade may be mediated by receptor ligand interaction or by intracellular proteins. Increased ligand-receptor interaction would likely lead to increased signaling. Intracellularly, NIK activation may also be positively regulated by cytoplasmic proteins such as Zfp91, which leads to NIK stabilization and downstream p100 processing (Jin et al., 2010). Positive regulation of NIK may also occur through MALT-1, which promotes TRAF3 ubiquitination (Sun, 2011), or BCL-10, which promotes NIK phosphorylation (Bhattacharyya et al., 2010). Additionally, independent activation of IKKα by proteins such as STAT3 have also been shown to induce p100 processing. Another key regulatory step in the ncNF-κB cascade is the NIK induced processing of p100 which is mediated through the binding of βTRcP to p100 (following its phosphorylation at serine residues 866 and 870) and subsequent ubiquitination and degradation (Yu et al., 2020). At the nuclear level, p100 degradation may be mediated by the Fbw7 protein (Fukushima et al., 2012).
The ncNF-κB cascade is negatively regulated at various steps in the pathway. As mentioned above, at baseline, the TRAF3-TRAF2-cIAP complex prevents the intracellular accumulation and activation of NIK; however, even following receptor-ligand binding, the TRAF3 ubiquitination process may be inhibited by the cytoplasmic deubiquitinase belonging to the OUT family, such as A20 and OTUD7B, attenuating the pathway (Hu et al., 2013; Pujari et al., 2013). NIK induced IKKα activation may independently lead to NIK destabilization by IKKα in a negative feedback mechanism (Razani et al., 2010). Intracytoplasmic proteins such as NLRP12 (Allen et al., 2012) and Tank binding kinase (TBK1) may lead to NIK degradation (Jin et al., 2012). In neural tissues, TRIM9 has been shown to inhibit NIK mediated p100 processing (Shi et al., 2014). IKKα may also be independently inhibited by specific microRNAs.
Ultimately, the accumulation of stable NIK leads to IKKα activation and p100 processing. The released p52 protein associates with RelB and translocates to the nucleus where it regulates transcription, binding to specific κB sites. It has been suggested that there exists similar DNA-binding specificity of canonical and non-canonical NF-κB members (Britanova et al., 2008).
A summary of the non-canonical pathway and the involved receptors and regulators is provided in Figure 1.
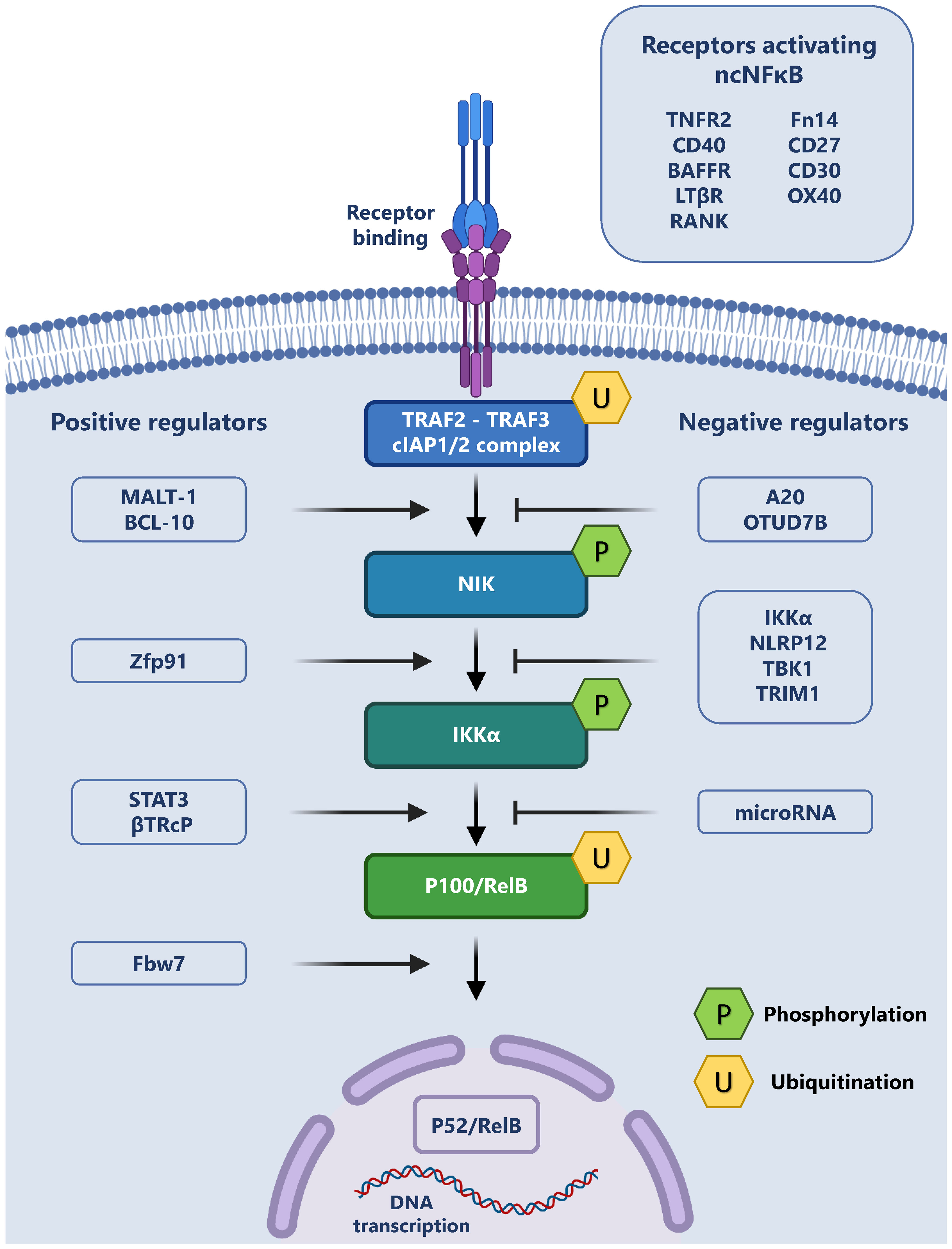
Figure 1 The Non-canonical NF-κB pathway and its regulation: The non-canonical signaling pathway may be initiated through the binding of a variety of exogenous ligands to their cognate receptors (top right). This ligand receptor interaction leads to the recruitment of TRAF2, TRAF3 and cIAP1/2, forming a complex, which allows for the release and phosphorylation of NIK. Phospho-NIK is able to in turn phosphorylate IKKα, which leads to the ubiquitination of the p100/RelB complex and p52 production. P52 subsequently translocates to the nucleus and regulates transcription. This cascade may be positively regulated (left) or negatively regulated (right) by a variety of intracellular proteins.
While at the population level, there is wide variance to sequences into which HIV integrates, some regions have been identified as preferential sites of integration, and having bearing on HIV persistence, (Reviewed in (Hughes and Coffin, 2016)). These include regions with transcription-associated histone modifications and regions that correspond to specific mutations in HIV integrase (Hughes and Coffin, 2016; Wang et al., 2007; Serrao et al., 2014). Serving as a major transcriptional regulator, the role of NF-κB in HIV transcription during active infection has been extensively studied (reviewed in (Hiscott et al., 2001)) and is essential for efficient replication. Additionally, it has been demonstrated that the long terminal repeat (LTR) of the HIV genome harbors two independent NF-κB binding sites that are essential for normal HIV transcription and replication, and cells with lower levels of NF-κB may facilitate the establishment of HIV latency (Hiscott et al., 2001; Jiang and Dandekar, 2015; Kwon et al., 1998; Alcamí et al., 1995). CD4+ T cells (which harbor the vast majority of the latent reservoir) are described to have very low levels of NF-κB activity at baseline, and this is mostly mediated through the non-canonical p50 pathway (Hiscott et al., 2001). It is widely accepted that memory CD4+ T Cells harbor the vast majority of the latent reservoir and therefore, understanding of the infection-induced modifications to the regulators of the ncNF-κB pathway becomes essential to inform cure studies and will be examined here, and are summarized in Table 1.
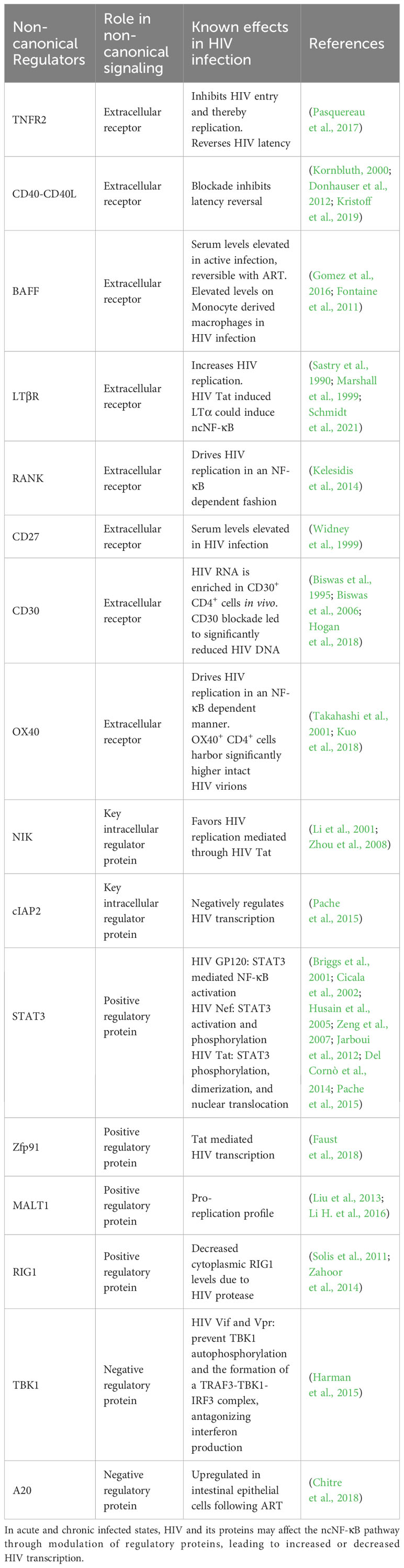
Table 1 The known effects of non-canonical NF-κB regulators in HIV infection.
As mentioned above, non-canonical signaling may be initiated through receptor ligand interactions at the cell surface. Chronic infection may up or downregulate the expression of these receptors, leading to potential pathogenic ramifications downstream.
Tumor Necrosis Factor Receptor 2 (TNFR2) stimulation activates both the canonical and non-canonical NF-κB pathways. TNF-mediated stimulation has been shown to reactivate HIV in in-vitro models of latency and in ex vivo studies in combination with other latency reversal agents but has been seen to be associated with significant toxicity to bystander cells. TNFR2 signaling may also control HIV replication through inhibition of HIV entry through CD4 downregulation (Pasquereau et al., 2017).
CD40-CD40L binding in active HIV infection contributes to HIV control through the regulation of chemokine secretion, antibody production and immune effector function. The levels of plasma and surface expression of CD40 have been observed to vary in acute, chronic, and treated infection (Kornbluth, 2000; Donhauser et al., 2012). With respect to shock and kill strategies, it has been observed that CD40L-CD40R blockade was sufficient to significantly reduce HIV latency reversal and protein production in a myeloid dendritic cell induced model (Kristoff et al., 2019).
B-cell activating factor (BAFF) is primarily produced on the surface of antigen-presenting cells of myeloid lineage such as monocytes and dendritic cells. HIV has been demonstrated to independently upregulate BAFF expression in monocyte derived macrophages (Gomez et al., 2016). In HIV infection, serum BAFF levels have been seen to increase steadily over the duration of infection, and in animal models, this increase was reversible with ART (Fontaine et al., 2011). The BAFF/BAFFR axis, while primarily involved in B-Cell regulation, has been demonstrated to influence T cell activation and proliferation (Huard et al., 2001; Ye et al., 2004). In HIV infection, therefore, BAFF upregulation on the surface of dendritic cells and B-Cells as well as secreted BAFF in the microenvironment could exert pro-transcriptional activity and drive HIV replication.
Lymphotoxin β Receptor (LTβR) signaling is known to increase HIV replication alone and in the presence of TNFα (Marshall et al., 1999). It has been established that Naïve T cells are the major producers of LTβ in lymphoid tissues and that HIV-associated CD4+ T cell depletion leads to dysregulation of normal immune architecture, concurrent with depleted LTβR signaling, an effect that was seen to be reversible with ART (Zeng et al., 2012). HIV Tat has also been shown to induce the production of Lymphotoxin α (LTα), which may also signal through the LTβR and drive non-canonical NF-κB activation. However, considering the observed off-target effect of therapeutic targeting of this receptor, further research is necessary to improve its feasibility as a safe target for latency-reversal therapy (Sastry et al., 1990; Schmidt et al., 2021).
Receptor activator for NF-κB (RANK), RANK ligand (RANKL) and its soluble receptor Osteoprotegerin (OPG) may be up or down regulated during active HIV infection or following ART which has been extensively reviewed elsewhere (Kelesidis et al., 2014). RANK-RANKL signaling has been shown to drive HIV replication in an NF-κB dependent fashion in acutely and chronically infected T cells (Kelesidis et al., 2014).
Fibroblast growth factor inducible factor (FN14) is a transient receptor to the TNF-like weak inducer of apoptosis (TWEAK/TWK) protein that is expressed on the cell surface in response to cellular injury (Winkles, 2008). The levels of soluble TWEAK in the plasma of treated and untreated HIV-infected individuals were found to be lower than in uninfected controls (Beltrán et al., 2014). FN14 expression in γδ T Cell subsets has been shown to be IL-21dependent (Vermijlen et al., 2007). Plasma IL-21 levels have been shown to be decreased in chronic HIV infection, an effect that is reversed in the presence of ART. Additionally, Elite controllers (patients exhibiting spontaneous immune control of HIV) have been shown to have higher circulating levels of IL-21. However, it has been seen that infected individuals may harbor higher levels of circulating IL-21 positive CD4+ T cells (Pallikkuth et al., 2012). While it is yet to be clearly established that FN14 expression is significantly dysregulated in HIV infection, the concurrent downregulation of TWEAK and cytokines such as IL-21 which may stimulate FN14 production suggest that FN14 signaling may represent a feasible therapeutic target for latency reversal.
CD30 levels in the plasma of HIV infected individuals have been found to be directly related to disease progression (Biswas et al., 1995; Biswas et al., 2006). It was recently demonstrated that in HIV infection, CD4+ T cells express higher levels of CD30 on their surface, regardless of ART status, whereas levels of soluble CD30 were only elevated in the viremic group. HIV RNA was seen to be enriched in the CD30 positive CD4+ T cell subsets from the blood of both untreated and treated patients, and also coincided with magnitude higher levels of HIV DNA. mRNA levels in the gut associated lymphoid tissue was also seen to co-localize with CD30 expression. The same study also utilized a clinically relevant anti-CD30 antibody, brentuximab vedotin, to treat ex-vivo PBMCs from HIV infected individuals which was seen to reduce the total amount of HIV-1 DNA (Hogan et al., 2018).
OX40 (CD134) ligation by its cognate ligand has been described to drive HIV replication in an NF-κB driven manner (Takahashi et al., 2001). In the setting of ART, OX40-expressing cells were observed to harbor significantly higher HIV DNA copy numbers and higher levels of clonally expanded HIV DNA. Intact proviruses were also seen to be enriched in the OX40-positive cells in four of the five subjects analyzed (Kuo et al., 2018).
NF-κB inducing kinase (NIK), the primary protein involved in the ncNF-κB pathway, has been demonstrated to favor HIV replication. HIV Tat has been described to facilitate NIK mediated IKKβ activation, and it has been observed that knockdown of NIK leads to the inhibition of Tat driven HIV transcription (Zhou et al., 2008; Li et al., 2001).
Cellular inhibitor of apoptosis 2 (cIAP2) another key regulatory protein in the ncNF-κB cascade was identified to be able to directly inhibit HIV transcription through the inhibition of the ncNF-κB signaling pathway (Pache et al., 2015).
Signal transducer and activator of transcription 3 (STAT3) has been described to be activated and phosphorylated by different HIV proteins. HIV Gp120 has been described to drive STAT3-mediated NF-κB activation and cytokine production (Cicala et al., 2002; Del Cornò et al., 2014). HIV Nef has been described to activate and phosphorylate STAT3 in Macrophages, dendritic cells, and podocytes (Briggs et al., 2001; Husain et al., 2005). HIV Tat has been shown to cause STAT3 phosphorylation, dimerization, and nuclear translocation (Zeng et al., 2007; Jarboui et al., 2012).
Zinc finger protein 91 (Zfp91), which has been shown to associate with NIK and lead to p100 processing, was found to be involved in Tat mediated transcription, identified by siRNA mediated knockdown of Zfp91 leading to decreased Tat mediated HIV transcription (Faust et al., 2018).
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), a Para-caspase that has been shown to promote TRAF3 ubiquitination has been demonstrated to favor a pro-replication profile, through the degradation of the MCPIP1 RNAse. It was demonstrated that MALT1 inhibition induced significant HIV-infected cell death and significantly impacted the level of HIV post reactivation with an LRA (Liu et al., 2013; Li H. et al., 2016).
Retinoic acid-inducible gene 1 (RIG1) is a positive regulator of the ncNF-κB pathway. In HIV infection, RIG1 activity has been demonstrated to be antagonized by HIV protease, with infection leading to decreased cytoplasmic RIG1 levels (Solis et al., 2011). On the contrary, it has also been described that HIV Vpr may acutely induce increased expression of RIG1 mRNA in human monocyte-derived macrophages (Zahoor et al., 2014).
TANK-binding kinase 1 (TBK1) is a negative regulator of the ncNF-κB cascade, and it has been demonstrated that HIV Vif and Vpr may bind to TBK1, preventing its autophosphorylation and the formation of a TRAF3-TBK1-IRF3 complex that is necessary for interferon production in human dendritic cells and macrophages (Harman et al., 2015). It is therefore plausible that HIV interactions with TBK1 may also influence TBK1-TRAF3 mediated NF-κB signaling, though this is yet to be established.
A20, an important negative regulator of NF-κB, is downregulated in intestinal epithelial cells during HIV infection due to the effects of interferon alpha, rendering cells more susceptible to cytokine mediated cell death. This downregulation was seen to be reversed following ART therapy in HIV infection (Chitre et al., 2018). It is possible that a similar upregulation could exist in cells that harbor the latent reservoir, allowing for transcriptional silence. Importantly, there has been evidence that the PKC agonist Prostratin upregulates A20 when used for latency reversal, leading directly to NF-κB inhibition, an effect that may be abrogated when co-administered with HMBA (Chen et al., 2016).
As evidenced above, HIV-induced modulation to the regulators of ncNF-κB signaling plays a significant role in developing strategies to target HIV for latency reversal, either through the potentiation of positive regulatory effects or the inhibition of the negative inhibitory effects.
Current latency reversal agents that are under investigation for “shock and kill” include, but are not limited to, Histone deacetylase inhibitors (HDACis), Bromodomain inhibitors, DNA methyltransferase inhibitors, proteosome inhibitors, Protein kinase C (PKC) agonists, WNT inhibitors and second mitochondria-derived activator of caspases (SMAC) mimetics. Nearly all of these agents have been shown to reactive latency invitro or in vivo with minimal effect on depleting total viral reservoir size (Kim et al., 2018). It has been shown that many of these agents have effects on the canonical NF-κB pathway. However, due to the generally proinflammatory nature of NF-κB and the significantly varied downstream effects of the canonical pathway, there is cause for concern with regards to off-target toxicity (Pache et al., 2015). In this scenario, selective agents targeting the ncNF-κB pathway would be preferred to mitigate toxicity risks.
SMAC mimetics are a group of drugs that have been shown to selectively target and inhibit cIAPs, allowing for downstream ncNF-κB signaling. The SMAC mimetics SBI-0637142 and LCL161 have been demonstrated to drive HIV reactivation through non-canonical signaling in cell lines. Additionally, in combination with HDAC inhibitors, they were able to reactivate latent HIV from ex vivo CD4+ T cells (Pache et al., 2015). Subsequently, a bivalent SMAC mimetic, Ciapavir, was shown to reactivate HIV in an in vivo mouse model, at the bone marrow level, in four out of the six mice treated with the agent, in the setting of suppressive ART (Pache et al., 2020). Debio 1143, another IAP antagonist, was shown to reactivate HIV from latency through the potentiation of the ncNF-κB pathway (Bobardt et al., 2019).
The SMAC mimetic AZD5582 which functions through the ncNF-κB pathway has been the focus of multiple recent studies and was recently demonstrated to reactivate HIV in a mouse model and in SIV infected macaques (Nixon et al., 2020). It is of note that minimal off-target effects were observed in these studies. The effect of AZD5582 was seen to be potentiated by crotonylation, allowing for superior latency reversal along with significant increases in p52 protein levels (Li et al., 2021). The effects of AZD5582 were also seen to be synergistically enhanced by selective and pan BET domain inhibition in a cell-line model of latency, however this was not seen to consistently result in HIV latency reversal in ex-vivo primary CD4+ T cells (Falcinelli et al., 2022). A recent study, examined the combination of AZD5582 with the DEAD-box polypeptide 3 (DDX3) inhibitor FH1321. It was observed that AZD5582 alone and in combination with DDX3 inhibition resulted in robust HIV reactivation in in-vitro Jurkat models and resulted in reservoir depletion in ex-vivo studies using PBMCs from HIV infected individuals (Jansen et al., 2023).
Interestingly, the latency reversal effects of AZD5582 differed between SIV-infected, ART-suppressed infant rhesus macaques and adult macaques, revealing lower levels of on-ART plasma viremia. In the same study, transcriptomic profiling in the infant macaques revealed that the expression of the ncNF-κB signaling genes RELB and NFKB2 were not significantly increased, contrary to prior observations in adults (Bricker et al., 2022).
Combination therapy with AZD5582 and a cocktail of 3 HIVxCD3 DART molecules (having human A32, 7B2, or PGT145 anti-HIV-1 envelope (Env) specificities) in a SHIV infected macaque model failed to result in an observable decrease in the viral reservoir, possibly as a result of poor latency reversal, with none of the infected animals demonstrating detectable viremia. It was suggested that lower pre-ART viral loads, and low pre-intervention reservoir sizes may have affected the potency of latency reversal in this study (Dashti et al., 2020). A more recent study examined AZD5582 with or without the IL-15 superagonist, N-803, in combination with SIV Env-specific Rhesus monoclonal antibodies (RhmAbs). N-803 is a potent LRA (discussed separately below), which was seen to enhance AZD5582 driven latency reversal. The combination of the RhmAbs with AZD5582 ± N-803, was observed to cause differential SIV-DNA depletion in CD4+ T cells based on anatomic location. However, significant decreases in “total body” SIV-DNA in CD4+ T cells (graphed as the sum of all SIV-DNA results from blood, lymph nodes, bone marrow and gastrointestinal tract) were observed following treatment with RhmAbs + AZD5582 ± N-803. Once again, it was observed that the magnitude of latency reversal was directly associated with pre-ART viral loads and the post-ART SIV-DNA CD4+ T cell reservoir, suggesting that viral reservoir size may be a crucial determinant of the efficacy of AZD5582 (Dashti et al., 2023).
PKC agonists such as bryostatin and prostratin are amongst the most investigated class of drugs for HIV latency reversal, both in vivo and invitro. Studies have indicated induction of cNF-κB and HIV latency reversal by PKC agonists (Bullen et al., 2014; Jiang and Dandekar, 2015; Kim et al., 2018; French et al., 2020). PKC agonism by PMA in cell lines was seen to result in the activation of the non-canonical NF-κB pathway via the recruitment of RelB to the APOBEC3B promoter, increasing the expression of APOBEC3B, a protein that is becoming increasingly recognized as a dynamic modulator of HIV replication (Gillick et al., 2013; Leonard et al., 2015; Bandarra et al., 2021). Prostratin was also seen to upregulate APOBEC3B in primary CD4+ T cells, but the involvement of RelB was not defined (Sung and Rice, 2006).
The IL-15 super agonists have been under study as a potential latency-reversal agent that has been shown to reactivate HIV and prime latently infected cells for clearance by immune effectors. Notably, IL-15 was seen to increase gene sets involved in TNF signaling via NF-κB and in STAT3 signaling in bulk CD4+ T cells (Jones et al., 2016; McBrien et al., 2020). Additionally, IL-15 was also seen to increase the expression of NF-κB2 in bulk CD4+ T cells (extended data (McBrien et al., 2020)). As mentioned previously, STAT3 can independently activate p100 processing and lead to ncNF-κB signaling. This mechanism remains to be described in the context of HIV latency reversal by these agents.
Proteasome inhibitors such as Bortezomib and Ixazomib are now under study as latency reversal agents via the activation of the NF-κB pathway (Miller et al., 2013; Natesampillai et al., 2018; Li et al., 2019; Timmons et al., 2020; Alto et al., 2021; Cummins et al., 2021), and they have recently been studied in the clinical setting (Cummins et al., 2021). The generation of p52 has been described as proteasome dependent with studies demonstrating that inhibition of the proteasome results in decreased p52 protein (Heusch et al., 1999). It has also been very well established that ncNF-κB inhibition is crucial to the efficacy of proteasome inhibitors in multiple myeloma (Chauhan et al., 2011; Dash et al., 2020). It is still to be elucidated whether the ncNF-κB pathway may play a similar role for HIV latency reversal by proteasome inhibitors.
CD40 targeted antibodies are increasingly becoming relevant in oncology. Agonists lead to observable T cell activation and anti-tumor potentiation induced by dendritic cells, and it has been well established that CD40-signaling can potentiate the ncNF-κB pathway and that a CD40-blockade inhibits dendritic-cell induced HIV latency reversal. Targeted antibodies to potentiate the effects of CD40 on ncNF-κB activity therefore represents a feasible strategy to achieve latency reversal, but the concomitant effects on the canonical pathway need to be considered (Hostager and Bishop, 2013; Kristoff et al., 2019; Vonderheide, 2020).
OX40 agonistic antibodies have now become increasingly relevant in cancer therapy, and OX40 agonism has been shown to boost HIV replication (Takahashi et al., 2001; Linch et al., 2015). As mentioned above, OX40, MALT1 and BCL-10 are all positive regulators of the non-canonical pathway. OX40 agonism has been seen to recruit and activate MALT-1 (Linch et al., 2015; Israël and Bornancin, 2018). Additionally, OX40-expressing CD4+ T cells were preferentially enriched for clonally expanded HIV, and some subjects harbored higher quantities of intact Proviral DNA (Kuo et al., 2018). As mentioned above, MALT-1 favors a pro-transcriptional profile. Interestingly, another protein, NEDD4-binding protein 1 (N4BP1), was seen to inhibit HIV replication, a function that was antagonized by MALT1. MALT1-mediated N4BP1 degradation was seen to facilitate the reactivation of latent HIV proviruses (Yamasoba et al., 2019). Targeted efforts to boost OX40 signaling or MALT1 potentiation, either in an OX40-dependent or independent manner may therefore represent feasible latency reversal agents not only through the non-canonical NF-κB pathway but also through other protein interactions.
RIG-1, a positive regulator of ncNF-κB, was targeted through the RIG-1 agonist acitretin which increased HIV replication in in-vitro and ex vivo models, and lead to preferential HIV infected cell apoptosis. However, a second study failed to reproduce similar results, suggesting further research is necessary to examine RIG-1 as a shock agent (Li P. et al., 2016; Garcia-Vidal et al., 2017). The interactions of these agents with the nc-NF-kB pathway has been summarized in Table 2.
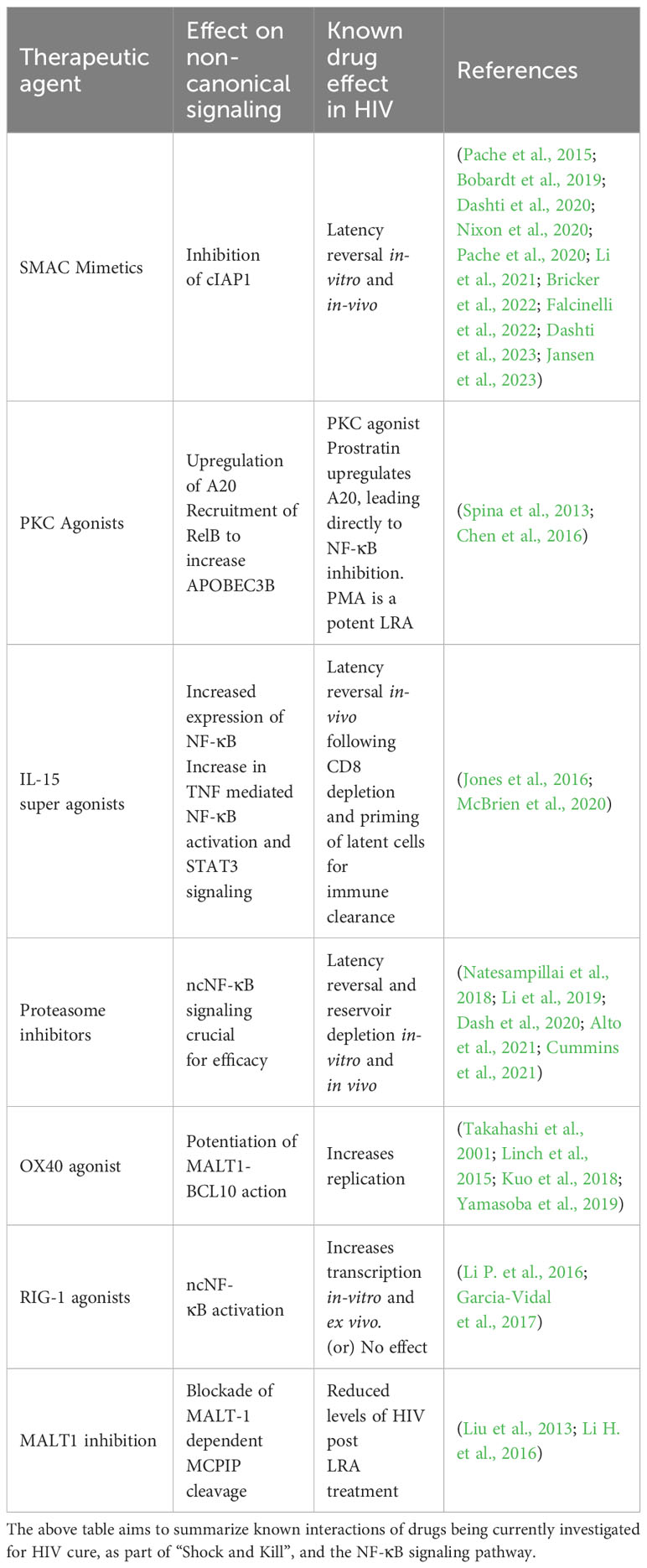
Table 2 Shock and kill agents and their known effects on the non-canonical pathway.
Current ART therapy is incapable of achieving either a sterilizing or a functional cure of HIV. The current “shock and kill” strategies to eradicate HIV may involve the ncNF-κB pathway. As further studies in these fields progress, the role of this pathway may become better defined. Efforts to identify specific agonists of this pathway would significantly enhance “shock and kill” efforts and may ultimately contribute to the cure of HIV.
AC: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. MM: Writing – original draft, Writing – review & editing. AB: Funding acquisition, Resources, Supervision, Visualization, Writing – review & editing, Conceptualization.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Portions of this work were funded through grants awarded to AB, (grants AI110173 and AI120698) from the National Institute of Allergy and Infectious Diseases of the NIH and the Mayo Clinic Foundation.
AB is supported by grants from NIAID grants AI110173 and AI120698 Amfar #109593 and Mayo Clinic HH Sheikh Khalifa Bin Zayed Al-Nahyan Named Professorship of Infectious Diseases. AB is a paid consultant for Abbvie, Gilead, Freedom Tunnel, Pinetree therapeutics Primmune, Immunome, MarPam, Rion, Symbiosis, NexImmune and Flambeau Diagnostics, is a paid member of the DSMB for Corvus Pharmaceuticals, Equilium, CSL Behring, and Excision Biotherapeutics, has received fees for speaking for Reach MD, Peer Voice, and Medscape, owns equity for scientific advisory work in Tier 1 Bio, Zentalis, Rion, and Nference, and is founder and President of Splissen therapeutics, and Member of the Board of Attivare.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The opinions expressed are solely of the authors and do not necessarily represent the opinions of the funding organization(s).
Alcamí, J., Lera Laín, T., Folgueira, L., Pedraza, M. A., Jacqué, J. M., Bachelerie, F., et al. (1995). Absolute dependence on kappa B responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. EMBO J. 14, 1552–1560. doi: 10.1002/j.1460-2075.1995.tb07141.x
Allen, I. C., Wilson, J. E., Schneider, M., Lich, J. D., Roberts, R. A., Arthur, J. C., et al. (2012). NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36, 742–754. doi: 10.1016/j.immuni.2012.03.012
Alto, A., Natesampillai, S., Chandrasekar, A. P., Krogman, A., Misra, A., Shweta, F., et al. (2021). The combination of venetoclax and ixazomib selectively and efficiently kills HIV-infected cell lines but has unacceptable toxicity in primary cell models. J. Virol. 95. doi: 10.1128/jvi.00138-21
Bachmann, N., von Siebenthal, C., Vongrad, V., Turk, T., Neumann, K., Beerenwinkel, N., et al. (2019). Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat. Commun. 10, 3193. doi: 10.1038/s41467-019-10884-9
Bandarra, S., Miyagi, E., Ribeiro, A.C., Gonçalves, J., Strebel, K., Barahona, I. (2021). APOBEC3B potently restricts HIV-2 but not HIV-1 in a vif-dependent manner. J. Virol. 95, e0117021. doi: 10.1128/jvi.01170-21
Beltrán, L. M., Muñoz Hernández, R., de Pablo Bernal, R. S., García Morillo, J. S., Egido, J., Noval, M. L., et al. (2014). Reduced sTWEAK and increased sCD163 levels in HIV-infected patients: modulation by antiretroviral treatment, HIV replication and HCV co-infection. PloS One 9, e90541. doi: 10.1371/journal.pone.0090541
Bhattacharyya, S., Borthakur, A., Dudeja, P. K., Tobacman, J. K. (2010). Lipopolysaccharide-induced activation of NF-κB non-canonical pathway requires BCL10 serine 138 and NIK phosphorylations. Exp. Cell Res. 316, 3317–3327. doi: 10.1016/j.yexcr.2010.05.004
Biswas, P., Smith, C. A., Goletti, D., Hardy, E. C., Jackson, R. W., Fauci, A. S. (1995). Cross-linking of CD30 induces HIV expression in chronically infected T cells. Immunity 2, 587–596. doi: 10.1016/1074-7613(95)90003-9
Biswas, P., Cozzi-Lepri, A., Delfanti, F., Galli, A., Colangeli, V., Moioli, M. C., et al. (2006). Significant link between sCD30 changes and HIV viremia in patients treated with HAART. J. Med. Virol. 78, 1513–1519. doi: 10.1002/jmv.20733
Bobardt, M., Kuo, J., Chatterji, U., Chanda, S., Little, S. J., Wiedemann, N., et al. (2019). The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PloS One 14, e0211746. doi: 10.1371/journal.pone.0211746
Bricker, K. M., Obregon-Perko, V., Williams, B., Oliver, D., Uddin, F., Neja, M., et al. (2022). Altered response pattern following AZD5582 treatment of SIV-infected, ART-suppressed rhesus macaque infants. J. Virol. 96, e0169921. doi: 10.1128/jvi.01699-21
Briggs, S. D., Scholtz, B., Jacque, J. M., Swingler, S., Stevenson, M., Smithgall, T.E. (2001). HIV-1 Nef promotes survival of myeloid cells by a Stat3-dependent pathway. J. Biol. Chem. 276, 25605–25611. doi: 10.1074/jbc.M103244200
Britanova, L. V., Makeev, V. J., Kuprash, D. V. (2008). In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem. Biophys. Res. Commun. 365, 583–588. doi: 10.1016/j.bbrc.2007.10.200
Bui, J. K., Sobolewski, M. D., Keele, B. F., Spindler, J., Musick, A., Wiegand, A., et al. (2017). Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PloS Pathog. 13, e1006283. doi: 10.1371/journal.ppat.1006283
Bullen, C. K., Laird, G. M., Durand, C. M., Siliciano, J. D., Siliciano, R. F. (2014). New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency. vivo. Nat. Med. 20, 425–429. doi: 10.1038/nm.3489
Chandrasekar, A. P., Badley, A. D. (2022). Prime, shock and kill: BCL-2 inhibition for HIV cure. Front. Immunol. 13, 1033609. doi: 10.3389/fimmu.2022.1033609
Chauhan, D., Tian, Z., Zhou, B., Kuhn, D., Orlowski, R., Raje, N., et al. (2011). In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 17, 5311–5321. doi: 10.1158/1078-0432.Ccr-11-0476
Chen, D., Wang, H., Aweya, J. J., Chen, Y., Chen, M., Wu, X., et al. (2016). HMBA enhances prostratin-induced activation of latent HIV-1 via suppressing the expression of negative feedback regulator A20/TNFAIP3 in NF-κB signaling. BioMed. Res. Int. 2016, 5173205. doi: 10.1155/2016/5173205
Chitre, A. S., Kattah, M. G., Rosli, Y. Y., Pao, M., Deswal, M., Deeks, S. G., et al. (2018). A20 upregulation during treated HIV disease is associated with intestinal epithelial cell recovery and function. PloS Pathog. 14, e1006806. doi: 10.1371/journal.ppat.1006806
Cicala, C., Arthos, J., Selig, S.M., Dennis, G., Jr., Hosack, D.A., Van Ryk, D., et al. (2002). HIV envelope induces a cascade of cell signals in non-proliferating target cells that favor virus replication. Proc. Natl. Acad. Sci. U.S.A. 99, 9380–9385. doi: 10.1073/pnas.142287999
Cummins, N. W., Baker, J., Chakraborty, R., Dean, P. G., Garcia-Rivera, E., Krogman, A., et al. (2021). Single center, open label dose escalating trial evaluating once weekly oral ixazomib in ART-suppressed, HIV positive adults and effects on HIV reservoir size. vivo. EClinicalMedicine 42, 101225. doi: 10.1016/j.eclinm.2021.101225
Dash, A. B., Zhang, J., Shen, L., Li, B., Berg, D., Lin, J., et al. (2020). Clinical benefit of ixazomib plus lenalidomide-dexamethasone in myeloma patients with non-canonical NF-κB pathway activation. Eur. J. Haematol. 105, 274–285. doi: 10.1111/ejh.13435
Dashti, A., Waller, C., Mavigner, M., Schoof, N., Bar, K. J., Shaw, G. M., et al. (2020). SMAC mimetic plus triple-combination bispecific HIVxCD3 retargeting molecules in SHIV.C.CH505-infected, antiretroviral therapy-suppressed rhesus macaques. J. Virol. 94. doi: 10.1128/jvi.00793-20
Dashti, A., Sukkestad, S., Horner, A. M., Neja, M., Siddiqi, Z., Waller, C., et al. (2023). AZD5582 plus SIV-specific antibodies reduce lymph node viral reservoirs in antiretroviral therapy-suppressed macaques. Nat. Med. 29, 2535–2546. doi: 10.1038/s41591-023-02570-7
Del Cornò, M., Donninelli, G., Varano, B., Da Sacco, L., Masotti, A., Gessani, S. (2014). HIV-1 gp120 activates the STAT3/interleukin-6 axis in primary human monocyte-derived dendritic cells. J. Virol. 88, 11045–11055. doi: 10.1128/jvi.00307-14
Donhauser, N., Pritschet, K., Helm, M., Harrer, T., Schuster, P., Ries, M., et al. (2012). Chronic immune activation in HIV-1 infection contributes to reduced interferon alpha production via enhanced CD40:CD40 ligand interaction. PloS One 7, e33925. doi: 10.1371/journal.pone.0033925
Falcinelli, S. D., Peterson, J. J., Turner, A. W., Irlbeck, D., Read, J., Raines, S. L., et al (2022). Combined noncanonical NF-κB agonism and targeted BET bromodomain inhibition reverse HIV latency ex vivo. J. Clin. Invest. 132. doi: 10.1172/jci157281
Faust, T. B., Li, Y., Bacon, C. W., Jang, G. M., Weiss, A., Jayaraman, B., et al. (2018). The HIV-1 Tat protein recruits a ubiquitin ligase to reorganize the 7SK snRNP for transcriptional activation. Elife 7. doi: 10.7554/eLife.31879
Fontaine, J., Chagnon-Choquet, J., Valcke, H. S., Poudrier, J., Roger, M. (2011). High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood 117, 145–155. doi: 10.1182/blood-2010-08-301887
French, A. J., Natesampillai, S., Krogman, A., Correia, C., Peterson, K. L., Alto, A., et al. (2020). Reactivating latent HIV with PKC agonists induces resistance to apoptosis and is associated with phosphorylation and activation of BCL2. PloS Pathog. 16, e1008906. doi: 10.1371/journal.ppat.1008906
Fukushima, H., Matsumoto, A., Inuzuka, H., Zhai, B., Lau, A. W., Wan, L., et al. (2012). SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep. 1, 434–443. doi: 10.1016/j.celrep.2012.04.002
Garcia-Vidal, E., Castellví, M., Pujantell, M., Badia, R., Jou, A., Gomez, L., et al. (2017). Evaluation of the innate immune modulator acitretin as a strategy to clear the HIV reservoir. Antimicrob. Agents Chemother. 61. doi: 10.1128/aac.01368-17
Gillick, K., Pollpeter, D., Phalora, P., Kim, E. Y., Wolinsky, S. M., Malim, M. H. (2013). Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 87, 1508–1517. doi: 10.1128/jvi.02587-12
Gilmore, T. D. (2006). Introduction to NF-κB: players, pathways, perspectives. Oncogene 25, 6680–6684. doi: 10.1038/sj.onc.1209954
Gomez, A. M., Ouellet, M., Deshiere, A., Breton, Y., Tremblay, M. J. (2016). HIV-1-mediated BAFF secretion in macrophages does not require endosomal TLRs, type-I IFN, and nef, but depends on the cellular phenotype status. J. Immunol. 196, 3806–3817. doi: 10.4049/jimmunol.1501249
Harman, A. N., Nasr, N., Feetham, A., Galoyan, A., Alshehri, A. A., Rambukwelle, D., et al. (2015). HIV blocks interferon induction in human dendritic cells and macrophages by dysregulation of TBK1. J. Virol. 89, 6575–6584. doi: 10.1128/jvi.00889-15
Heusch, M., Lin, L., Geleziunas, R., Greene, W. C. (1999). The generation of nfkb2 p52: mechanism and efficiency. Oncogene 18, 6201–6208. doi: 10.1038/sj.onc.1203022
Hiscott, J., Kwon, H., Génin, P. (2001). Hostile takeovers: viral appropriation of the NF-kappaB pathway. J. Clin. Invest. 107, 143–151. doi: 10.1172/jci11918
Hoffmann, A., Baltimore, D. (2006). Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 210, 171–186. doi: 10.1111/j.0105-2896.2006.00375.x
Hogan, L. E., Vasquez, J., Hobbs, K. S., Hanhauser, E., Aguilar-Rodriguez, B., Hussien, R., et al. (2018). Increased HIV-1 transcriptional activity and infectious burden in peripheral blood and gut-associated CD4+ T cells expressing CD30. PloS Pathog. 14, e1006856. doi: 10.1371/journal.ppat.1006856
Hosmane, N. N., Kwon, K. J., Bruner, K. M., Capoferri, A. A., Beg, S., Rosenbloom, D. I., et al. (2017). Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 214, 959–972. doi: 10.1084/jem.20170193
Hostager, B. S., Bishop, G. A. (2013). CD40-mediated activation of the NF-κB2 pathway. Front. Immunol. 4. doi: 10.3389/fimmu.2013.00376
Hu, H., Brittain, G. C., Chang, J. H., Puebla-Osorio, N., Jin, J., Zal, A., et al. (2013). OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 494, 371–374. doi: 10.1038/nature11831
Huard, B., Schneider, P., Mauri, D., Tschopp, J., French, L. E. (2001). T cell costimulation by the TNF ligand BAFF. J. Immunol. 167, 6225–6231. doi: 10.4049/jimmunol.167.11.6225
Hughes, S. H., Coffin, J. M. (2016). What integration sites tell us about HIV persistence. Cell Host Microbe 19, 588–598. doi: 10.1016/j.chom.2016.04.010
Husain, M., D'Agati, V. D., He, J. C., Klotman, M. E., Klotman, P. E. (2005). HIV-1 Nef induces dedifferentiation of podocytes in vivo: a characteristic feature of HIVAN. . AIDS 19, 1975–1980. doi: 10.1097/01.aids.0000191918.42110.27
Israël, L., Bornancin, F. (2018). Ways and waves of MALT1 paracaspase activation. Cell Mol. Immunol. 15, 8–11. doi: 10.1038/cmi.2017.77
Jansen, J., Kroeze, S., Man, S., Andreini, M., Bakker, J. W., Zamperini, C., et al. (2023). Noncanonical-NF-κB activation and DDX3 inhibition reduces the HIV-1 reservoir by elimination of latently infected cells ex-vivo. Microbiol. Spectr. 12, e0318023.
Jarboui, M. A., Bidoia, C., Woods, E., Roe, B., Wynne, K., Elia, G., et al. (2012). Nucleolar protein trafficking in response to HIV-1 Tat: rewiring the nucleolus. PloS One 7, e48702. doi: 10.1371/journal.pone.0048702
Jiang, G., Dandekar, S. (2015). Targeting NF-κB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retroviruses 31, 4–12. doi: 10.1089/aid.2014.0199
Jin, J., Xiao, Y., Chang, J.H., Yu, J., Hu, H., Starr, R., et al. (2012). The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-κB signaling. Nat. Immunol. 13, 1101–1109. doi: 10.1038/ni.2423
Jin, X., Jin, H. R., Jung, H. S., Lee, S. J., Lee, J. H., Lee, J. J.. (2010). An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J. Biol. Chem. 285, 30539–30547. doi: 10.1074/jbc.M110.129551
Jones, R. B., Mueller, S., O'Connor, R., Rimpel, K., Sloan, D. D., Karel, D., et al. (2016). A subset of latency-reversing agents expose HIV-infected resting CD4+ T-cells to recognition by cytotoxic T-lymphocytes. PloS Pathog. 12, e1005545. doi: 10.1371/journal.ppat.1005545
Kelesidis, T., Currier, J. S., Yang, O. O., Brown, T. T. (2014). Role of RANKL-RANK/osteoprotegerin pathway in cardiovascular and bone disease associated with HIV infection. AIDS Rev. 16, 123–133.
Kim, Y., Anderson, J. L., Lewin, S. R. (2018). Getting the "Kill" into "Shock and kill": strategies to eliminate latent HIV. Cell Host Microbe 23, 14–26. doi: 10.1016/j.chom.2017.12.004
Kornbluth, R. S. (2000). The emerging role of CD40 ligand in HIV infection. J. Leukoc. Biol. 68, 373–382. doi: 10.1189/jlb.68.3.373
Kristoff, J., Palma, M. L., Garcia-Bates, T. M., Shen, C., Sluis-Cremer, N., Gupta, P., et al. (2019). Type 1-programmed dendritic cells drive antigen-specific latency reversal and immune elimination of persistent HIV-1. EBioMedicine 43, 295–306. doi: 10.1016/j.ebiom.2019.03.077
Kuo, H. H., Ahmad, R., Lee, G. Q., Gao, C., Chen, H. R., Ouyang, Z., et al. (2018). Anti-apoptotic protein BIRC5 maintains survival of HIV-1-infected CD4(+) T cells. Immunity 48, 1183–1194.e1185. doi: 10.1016/j.immuni.2018.04.004
Kwon, H., Pelletier, N., DeLuca, C., Genin, P., Cisternas, S., Lin, R., et al. (1998). Inducible expression of IkappaBalpha repressor mutants interferes with NF-kappaB activity and HIV-1 replication in Jurkat T cells. J. Biol. Chem. 273, 7431–7440. doi: 10.1074/jbc.273.13.7431
Leonard, B., McCann, J. L., Starrett, G. J., Kosyakovsky, L., Luengas, E. M., Molan, A. M., et al. (2015). The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 75, 4538–4547. doi: 10.1158/0008-5472.Can-15-2171-t
Li, D., Dewey, M. G., Wang, L., Falcinelli, S. D., Wong, L. M., Tang, Y., et al. (2021). Crotonylation sensitizes IAPi-induced disruption of latent HIV by enhancing p100 cleavage into p52. iScience 103649.
Li, H., He, H., Gong, L., Fu, M., Wang, T. T. (2016). Short communication: preferential killing of HIV latently infected CD4(+) T cells by MALT1 inhibitor. AIDS Res. Hum. Retroviruses 32, 174–177. doi: 10.1089/aid.2015.0343
Li, P., Kaiser, P., Lampiris, H. W., Kim, P., Yukl, S. A., Havlir, D. V., et al. (2016). Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 22, 807–811. doi: 10.1038/nm.4124
Li, X., Josef, J., Marasco, W. A. (2001). Hiv-1 Tat can substantially enhance the capacity of NIK to induce IkappaB degradation. Biochem. Biophys. Res. Commun. 286, 587–594. doi: 10.1006/bbrc.2001.5442
Li, Z., Wu, J., Chavez, L., Hoh, R., Deeks, S. G., Pillai, S. K., et al. (2019). Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PloS Pathog. 15, e1007498. doi: 10.1371/journal.ppat.1007498
Linch, S. N., McNamara, M. J., Redmond, W. L. (2015). OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front. Oncol. 5. doi: 10.3389/fonc.2015.00034
Liu, S., Qiu, C., Miao, R., Zhou, J., Lee, A., Liu, B., et al. (2013). MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proc. Natl. Acad. Sci. U.S.A. 110, 19083–19088. doi: 10.1073/pnas.1316208110
Marshall, W. L., Brinkman, B. M., Ambrose, C. M., Pesavento, P. A., Uglialoro, A. M., Teng, E., et al. (1999). Signaling through the lymphotoxin-beta receptor stimulates HIV-1 replication alone and in cooperation with soluble or membrane-bound TNF-alpha. J. Immunol. 162, 6016–6023. doi: 10.4049/jimmunol.162.10.6016
McBrien, J. B., Mavigner, M., Franchitti, L., Smith, S. A., White, E., Tharp, G. K., et al. (2020). Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8(+) cells. Nature 578, 154–159. doi: 10.1038/s41586-020-1946-0
Miller, L. K., Kobayashi, Y., Chen, C. C., Russnak, T. A., Ron, Y., Dougherty, J. P. (2013). Proteasome inhibitors act as bifunctional antagonists of human immunodeficiency virus type 1 latency and replication. Retrovirology 10, 120. doi: 10.1186/1742-4690-10-120
Natesampillai, S., Cummins, N. W., Nie, Z., Sampath, R., Baker, J. V., Henry, K., et al. (2018). HIV protease-generated casp8p41, when bound and inactivated by bcl2, is degraded by the proteasome. J. Virol. 92. doi: 10.1128/jvi.00037-18
Nixon, C. C., Mavigner, M., Sampey, G. C., Brooks, A. D., Spagnuolo, R. A., Irlbeck, D. M., et al. (2020). Systemic HIV and SIV latency reversal via non-canonical NF-κB signaling in vivo. Nature 578, 160–165. doi: 10.1038/s41586-020-1951-3
Nolan, G. P., Fujita, T., Bhatia, K., Huppi, C., Liou, H.C., Scott, M. L., et al. (1993). The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol. Cell Biol. 13, 3557–3566. doi: 10.1128/mcb.13.6.3557-3566.1993
O'Dea, E., Hoffmann, A. (2010). The regulatory logic of the NF-kappaB signaling system. Cold Spring Harbor Perspect. Biol. 2, a000216. doi: 10.1101/cshperspect.a000216
Pache, L., Dutra, M. S., Spivak, A. M., Marlett, J. M., Murry, J. P., Hwang, Y., et al. (2015). BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by smac mimetics to promote reversal of viral latency. Cell Host Microbe 18, 345–353. doi: 10.1016/j.chom.2015.08.009
Pache, L., Marsden, M. D., Teriete, P., Portillo, A. J., Heimann, D., Kim, J. T., et al. (2020). Pharmacological activation of non-canonical NF-κB signaling activates latent HIV-1 reservoirs. In Vivo. Cell Rep. Med. 1, 100037. doi: 10.1016/j.xcrm.2020.100037
Pallikkuth, S., Parmigiani, A., Pahwa, S. (2012). The role of interleukin-21 in HIV infection. Cytokine Growth Factor Rev. 23, 173–180. doi: 10.1016/j.cytogfr.2012.05.004
Pasquereau, S., Kumar, A., Herbein, G. (2017). Targeting TNF and TNF receptor pathway in HIV-1 infection: from immune activation to viral reservoirs. Viruses 9. doi: 10.3390/v9040064
Pomerantz, J. L., Baltimore, D. (2002). Two pathways to NF-kappaB. Mol. Cell 10, 693–695. doi: 10.1016/s1097-2765(02)00697-4
Pujari, R., Hunte, R., Khan, W. N., Shembade, N. (2013). A20-mediated negative regulation of canonical NF-κB signaling pathway. Immunol. Res. 57, 166–171. doi: 10.1007/s12026-013-8463-2
Razani, B., Zarnegar, B., Ytterberg, A. J., Shiba, T., Dempsey, P. W., Ware, C. F., et al. (2010). Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal 3, ra41. doi: 10.1126/scisignal.2000778
Sastry, K. J., Reddy, H. R., Pandita, R., Totpal, K., Aggarwal, B. B. (1990). HIV-1 tat gene induces tumor necrosis factor-beta (lymphotoxin) in a human B-lymphoblastoid cell line. J. Biol. Chem. 265, 20091–20093. doi: 10.1016/S0021-9258(17)30473-8
Scheidereit, C. (2006). IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25, 6685–6705. doi: 10.1038/sj.onc.1209934
Schmidt, M., Weidemann, A., Poser, C., Bigot, A., von Maltzahn, J. (2021). Stimulation of non-canonical NF-κB through lymphotoxin-β-receptor impairs myogenic differentiation and regeneration of skeletal muscle. Front. Cell Dev. Biol. 9. doi: 10.3389/fcell.2021.721543
Senftleben, U., Cao, Y., Xiao, G., Greten, F. R., Krähn, G., Bonizzi, G., et al. (2001). Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293, 1495–1499. doi: 10.1126/science.1062677
Serrao, E., Krishnan, L., Shun, M. C., Li, X., Cherepanov, P., Engelman, A., et al. (2014). Integrase residues that determine nucleotide preferences at sites of HIV-1 integration: implications for the mechanism of target DNA binding. Nucleic Acids Res. 42, 5164–5176. doi: 10.1093/nar/gku136
Shi, M., Cho, H., Inn, K. S., Yang, A., Zhao, Z., Liang, Q., et al. (2014). Negative regulation of NF-κB activity by brain-specific TRIpartite Motif protein 9. Nat. Commun. 5, 4820. doi: 10.1038/ncomms5820
Siliciano, R. F., Greene, W. C. (2011). HIV latency. Cold Spring Harb. Perspect. Med. 1, a007096. doi: 10.1101/cshperspect.a007096
Solis, M., Nakhaei, P., Jalalirad, M., Lacoste, J., Douville, R., Arguello, M., et al. (2011). RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 85, 1224–1236. doi: 10.1128/jvi.01635-10
Spina, C. A., Anderson, J., Archin, N. M., Bosque, A., Chan, J., Famiglietti, M., et al. (2013). An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PloS Pathog. 9, e1003834. doi: 10.1371/journal.ppat.1003834
Sun, S. C. (2011). Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85. doi: 10.1038/cr.2010.177
Sun, S. C. (2017). The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558. doi: 10.1038/nri.2017.52
Sung, T. L., Rice, A. P. (2006). Effects of prostratin on Cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 3, 66. doi: 10.1186/1742-4690-3-66
Takahashi, Y., Tanaka, Y., Yamashita, A., Koyanagi, Y., Nakamura, M., Yamamoto, N. (2001). OX40 stimulation by gp34/OX40 ligand enhances productive human immunodeficiency virus type 1 infection. J. Virol. 75, 6748–6757. doi: 10.1128/jvi.75.15.6748-6757.2001
Tam, W. F., Sen, R. (2001). IkappaB family members function by different mechanisms. J. Biol. Chem. 276, 7701–7704. doi: 10.1074/jbc.C000916200
Timmons, A., Fray, E., Kumar, M., Wu, F., Dai, W., Bullen, C. K., et al. (2020). HSF1 inhibition attenuates HIV-1 latency reversal mediated by several candidate LRAs. In Vitro Ex Vivo. Proc. Natl. Acad. Sci. U.S.A. 117, 15763–15771. doi: 10.1073/pnas.1916290117
Vallabhapurapu, S., Matsuzawa, A., Zhang, W., Tseng, P. H., Keats, J. J., Wang, H., et al. (2008). Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 9, 1364–1370. doi: 10.1038/ni.1678
Vanhamel, J., Bruggemans, A., Debyser, Z. (2019). Establishment of latent HIV-1 reservoirs: what do we really know? J. Virus Erad 5, 3–9. doi: 10.1016/S2055-6640(20)30275-2
Vatsyayan, J., Qing, G., Xiao, G., Hu, J. (2008). SUMO1 modification of NF-kappaB2/p100 is essential for stimuli-induced p100 phosphorylation and processing. EMBO Rep. 9, 885–890. doi: 10.1038/embor.2008.122
Vermijlen, D., Ellis, P., Langford, C., Klein, A., Engel, R., Willimann, K., et al. (2007). Distinct cytokine-driven responses of activated blood gammadelta T cells: insights into unconventional T cell pleiotropy. J. Immunol. 178, 4304–4314. doi: 10.4049/jimmunol.178.7.4304
Vonderheide, R. H. (2020). CD40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med. 71, 47–58. doi: 10.1146/annurev-med-062518-045435
Wang, G. P., Ciuffi, A., Leipzig, J., Berry, C. C., Bushman, F. D. (2007). HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 17, 1186–1194. doi: 10.1101/gr.6286907
Whiteside, S. T., Epinat, J. C., Rice, N. R., Israël, A. (1997). I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. EMBO J. 16, 1413–1426. doi: 10.1093/emboj/16.6.1413
Widney, D., Gundapp, G., Said, J. W., van der Meijden, M., Bonavida, B., Demidem, A., et al. (1999). Aberrant expression of CD27 and soluble CD27 (sCD27) in HIV infection and in AIDS-associated lymphoma. Clin. Immunol. 93, 114–123. doi: 10.1006/clim.1999.4782
Winkles, J. A. (2008). The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 7, 411–425. doi: 10.1038/nrd2488
Woldemeskel, B. A., Kwaa, A. K., Blankson, J. N. (2020). Viral reservoirs in elite controllers of HIV-1 infection: Implications for HIV cure strategies. EBioMedicine 62, 103118. doi: 10.1016/j.ebiom.2020.103118
Xiao, G., Fong, A., Sun, S. C. (2004). Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J. Biol. Chem. 279, 30099–30105. doi: 10.1074/jbc.M401428200
Xiao, G., Harhaj, E. W., Sun, S. C. (2001). NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7, 401–409. doi: 10.1016/s1097-2765(01)00187-3
Yamasoba, D., Sato, K., Ichinose, T., Imamura, T., Koepke, L., Joas, S., et al. (2019). N4BP1 restricts HIV-1 and its inactivation by MALT1 promotes viral reactivation. Nat. Microbiol. 4, 1532–1544. doi: 10.1038/s41564-019-0460-3
Ye, Q., Wang, L., Wells, A. D., Tao, R., Han, R., Davidson, A., et al. (2004). BAFF binding to T cell-expressed BAFF-R costimulates T cell proliferation and alloresponses. Eur. J. Immunol. 34, 2750–2759. doi: 10.1002/eji.200425198
Yeh, Y. J., Yang, K., Razmi, A., Ho, Y. C. (2021). The clonal expansion dynamics of the HIV-1 reservoir: mechanisms of integration site-dependent proliferation and HIV-1 persistence. Viruses 13. doi: 10.3390/v13091858
Yu, H., Lin, L., Zhang, Z., Zhang, H., Hu, H. (2020). Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct. Target. Ther. 5, 209. doi: 10.1038/s41392-020-00312-6
Zahoor, M. A., Xue, G., Sato, H., Murakami, T., Takeshima, S. N., Aida, Y. (2014). HIV-1 Vpr induces interferon-stimulated genes in human monocyte-derived macrophages. PloS One 9, e106418. doi: 10.1371/journal.pone.0106418
Zarnegar, B. J., Wang, Y., Mahoney, D. J., Dempsey, P. W., Cheung, H. H., He, J., et al. (2008). Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378. doi: 10.1038/ni.1676
Zeng, Y., Zhang, X., Huang, Z., Cheng, L., Yao, S., Qin, D., et al. (2007). Intracellular Tat of human immunodeficiency virus type 1 activates lytic cycle replication of Kaposi's sarcoma-associated herpesvirus: role of JAK/STAT signaling. J. Virol. 81, 2401–2417. doi: 10.1128/jvi.02024-06
Zeng, M., Paiardini, M., Engram, J. C., Beilman, G. J., Chipman, J. G., Schacker, T. W., et al. (2012). Critical role of CD4 T cells in maintaining lymphoid tissue structure for immune cell homeostasis and reconstitution. Blood 120, 1856–1867. doi: 10.1182/blood-2012-03-418624
Keywords: HIV, NFkappa B signaling pathway, shock and kill strategies, HIV cure strategies, latency reversal agents
Citation: Chandrasekar AP, Maynes M and Badley AD (2024) Dynamic modulation of the non-canonical NF-κB signaling pathway for HIV shock and kill. Front. Cell. Infect. Microbiol. 14:1354502. doi: 10.3389/fcimb.2024.1354502
Received: 12 December 2023; Accepted: 16 February 2024;
Published: 05 March 2024.
Edited by:
Ana Carolina Soares De Oliveira, University of São Paulo, BrazilReviewed by:
Guochun Jiang, University of North Carolina at Chapel Hill, United StatesCopyright © 2024 Chandrasekar, Maynes and Badley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew D. Badley, YmFkbGV5LmFuZHJld0BtYXlvLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.