 Kumaragurubaran Karthik1
Kumaragurubaran Karthik1 Saraswathi Subramanian2
Saraswathi Subramanian2 Michael Vinoli Priyadharshini2
Michael Vinoli Priyadharshini2 Ayyaru Jawahar2
Ayyaru Jawahar2 Subbaiyan Anbazhagan3Ramaiyan Selvaraju Kathiravan2
Subbaiyan Anbazhagan3Ramaiyan Selvaraju Kathiravan2 Prasad Thomas4
Prasad Thomas4 Ramasamy Parthiban Aravindh Babu2Krishnaswamy Gopalan Tirumurugaan2
Ramasamy Parthiban Aravindh Babu2Krishnaswamy Gopalan Tirumurugaan2 Gopal Dhinakar Raj5*
Gopal Dhinakar Raj5*- 1Department of Veterinary Microbiology, Veterinary College and Research Institute, Tamil Nadu Veterinary and Animal Sciences University, Udumalpet, India
- 2Translational Research Platform for Veterinary Biologicals, Tamil Nadu Veterinary and Animal Sciences University (TANUVAS), Chennai, India
- 3Indian Council of Medical Research (ICMR)-National Animal Resource Facility for Biomedical Research, Hyderabad, Telangana, India
- 4Division of Bacteriology and Mycology, Indian Council of Agricultural Research (ICAR)- India Veterinary Research Institute, Bareilly, Uttar Pradesh, India
- 5Department of Animal Biotechnology, Madras Veterinary College, Tamil Nadu Veterinary and Animal Sciences University (TANUVAS), Chennai, India
Introduction: Mycobacterium orygis, a member of MTBC has been identified in higher numbers in the recent years from animals of South Asia. Comparative genomics of this important zoonotic pathogen is not available which can provide data on the molecular difference between other MTBC members. Hence, the present study was carried out to isolate, whole genome sequence M. orygis from different animal species (cattle, buffalo and deer) and to identify molecular marker for the differentiation of M. orygis from other MTBC members.
Methods: Isolation and whole genome sequencing of M. orygis was carried out for 9 samples (4 cattle, 4 deer and 1 buffalo) died due to tuberculosis. Comparative genomics employing 53 genomes (44 from database and 9 newly sequenced) was performed to identify SNPs, spoligotype, pangenome structure, and region of difference.
Results: M. orygis was isolated from water buffalo and sambar deer which is the first of its kind report worldwide. Comparative pangenomics of all M. orygis strains worldwide (n= 53) showed a closed pangenome structure which is also reported for the first time. Pairwise SNP between TANUVAS_2, TANUVAS_4, TANUVAS_5, TANUVAS_7 and NIRTAH144 was less than 15 indicating that the same M. orygis strain may be the cause for infection. Region of difference prediction showed absence of RD7, RD8, RD9, RD10, RD12, RD301, RD315 in all the M. orygis analyzed. SNPs in virulence gene, PE35 was found to be unique to M. orygis which can be used as marker for identification.
Conclusion: The present study is yet another supportive evidence that M. orygis is more prevalent among animals in South Asia and the zoonotic potential of this organism needs to be evaluated.
1 Introduction
Members of the Mycobacterium tuberculosis complex (MTBC) cause tuberculosis in humans and various animal species. M. bovis was traditionally thought to be the major infectious zoonotic pathogen causing animal tuberculosis worldwide. Later, it was reported that another member of MTBC, M. orygis, was found in higher numbers in cases of animal tuberculosis in South Asia (Thapa et al., 2022). M. orygis has been isolated from different animal species such as cattle, bison, spotted deer, rhesus monkey, free-ranging rhinoceros, black buck, and African buffalo (Thapa et al., 2015; Thapa et al., 2016; Rahim et al., 2017; Refaya et al., 2019; Sharma et al., 2023). Human cases of tuberculosis due to M. orygis have been documented in New Zealand (Dawson et al., 2012), Australia (Lavender et al., 2013), the United States of America (Marcos et al., 2017), the United Kingdom (Lipworth et al., 2019), Norway (Eldholm et al., 2021), the Netherlands, and recently India (Sumanth et al., 2023). There is growing evidence that the number of animal tuberculosis cases in India caused by M. orygis is higher than cases caused by other MTBC members.
The biology, epidemiology, and genomics of M. orygis are poorly understood. There is a difference of opinion among researchers on the primary host of M. orygis. It was previously reported that the organism was an animal-adapted MTBC member that mainly affects wild animals (van Ingen et al., 2012), while a recent hypothesis states that cattle are the primary host, and spill-over events can lead to infection in wild animals and humans (Rahim et al., 2017; Brites et al., 2018). The sudden rise in the number of M. orygis infections in South Asia may be due to the lack of assays that can clearly discriminate the members of MTBC, leading to incorrect reporting as M. bovis or M. tuberculosis. Tools such as multilocus sequence typing (MLST), spoligotyping, region of difference (RDs) analysis, and single nucleotide polymorphisms (SNPs) can be used for the discrimination of MTBC members. Recently, RDs specific to M. orygis were reported, although only 32 genomes were used for analysis, and further validation was required to use these RDs as specific markers for the differentiation of M. orygis (Bespiatykh et al., 2021).
Next-generation sequencing and analysis have provided a platform for clear differentiation of bacterial strains. Recently, a number of reports documented the isolation and whole genome sequencing of M. orygis from domestic and captive wild animals (Refaya et al., 2022; Sharma et al., 2023). Still, there is no comprehensive study comprising the isolation and whole genome sequencing (WGS) of M. orygis from different animal species. Hence, in the present study, the WGS of M. orygis from cattle, water buffalo, sambar deer, and spotted deer was carried out. Comparative genomics of M. orygis was carried out in this study using publicly available data to identify a marker for the discrimination of M. orygis from other MTBC members.
2 Methods
2.1 Sample collection
Lung and lymph node samples were collected from nine dead animals (cattle= 4, buffalo= 1, deer= 4) that were suspected as having had tuberculosis (Table 1).

Table 1 Details of the samples processed for isolation of Mycobacterium spp.
Cattle and buffalo were maintained in an organized farm in Chengalpattu district, Tamil Nadu, India. Post-mortem examination of dead cattle and buffalo was done to identify the cause of death. It should be mentioned that three cattle and one buffalo were tested to be reactors for tuberculosis by a single intradermal comparative cervical tuberculin (SICCT) test earlier in 2021. Free-ranging deer in Guindy National Park Forest area in Chennai that had died from unknown causes were presented for post-mortem at Madras Veterinary College, Chennai. Post-mortem examination of the dead animals revealed numerous nodules in the lungs and lymph nodes, while one cattle (ID 1046) had numerous nodules in the liver. All the collected samples were processed in the BACTEC MGIT 960 system for isolation of Mycobacterium spp. All the samples were initially decontaminated using sodium hydroxide-NALC. After inoculation of the samples, MGIT tubes were incubated in the BACTEC instrument for 45 days before declaring the sample as negative for the presence of Mycobacterium spp. MGIT tubes with growth units/turbidity were used for acid-fast staining, and acid-fast positive samples were further used for isolation on an LJ solid medium. Simultaneously, acid-fast positive MGIT cultures were used for molecular confirmation. DNA was extracted from 1.0 ml of BACTEC MGIT culture fluid using a MagGenome Xpress DNA kit following the manufacturer’s instructions. PCR for the amplification of Mycobacterium tuberculosis complex (MTBC) was performed using primers targeting the IS6110 region (Miller et al., 1997). Differentiation among M. tuberculosis, M. bovis, and M. orygis was performed using primers targeting the RD12 region (Duffy et al., 2020).
2.2 Whole genome sequencing, assembly, and annotation of M. orygis
All of the nine M. orygis isolates were used for whole genome sequencing. DNA samples extracted using the MagGenome Xpress DNA kit were submitted to MedGenome Labs Ltd., Bengaluru, Karnataka, for whole genome sequencing using Illumina HiseqX technology. The quality of raw reads were tested with the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). For de novo genome assembly, Unicycler version 0.4.8.0 was used with default options. Normal bridging mode and spades error correction were used to create error-free genome assembly. Annotation was carried out with Prokka 1.14.5 and the Rapid Annotations using Subsystem Technology (RAST) annotation pipeline (Aziz et al., 2008; Tatusova et al., 2016; Wick et al., 2017). The virulence factors of the M. orygis genomes (n=9) were predicted with the VFDB: virulence factor database (Liu B. et al., 2022). Accessibility of the database was carried out with the ABRicate tool available in the Galaxy server (Galaxy Version 1.0.1). For this, a minimum of 75% DNA identity and 80% DNA coverage was used (Seemann, 2016). All the genome generated in this study was submitted to the NCBI SRA database with the bio project number PRJNA785380.
2.3 Whole genome phylogeny
The whole genome sequence assemblies of M. orygis (n=9) isolated in this study were used along with 45 genome assemblies of M. orygis and other MTBC isolates (n=40) retrieved from the NCBI database (Supplementary Table 1). All the genome assemblies were aligned using the Reference Sequence Alignment Based Phylogeny Builder (REALPHY) (https://realphy.unibas.ch/realphy/) pipeline employing M. tuberculosis H37Rv as the reference genome for the genome alignment (Bertels et al., 2014). Whole genome phylogeny was constructed with the maximum likelihood (ML) method and substitution model (GTR+G). Bootstrap replicates 1000 was set to obtain a reliable tree. The Galaxy server-based IQTREE program was used to infer the phylogeny relatedness (Minh et al., 2020). The treefile format from IQTREE was visualized by using Interactive Tree of Life (iTOL). The tree was rooted with mid-point rooting, and tree branches were annotated with host, isolation source, country and year of isolation, respectively (Letunic and Bork, 2021).
2.4 Pangenome analysis of M. orygis
The pan genomic analysis of M. orygis (n= 53) was carried out with Roary and Panaroo pan genome pipeline (Page et al., 2015). M. orygis strain 115(1)C was not used for the pangenome analysis due to poor quality of genome. Genome assemblies of M. orygis were annotated with Prokka. The GFF outputs from Prokka were used for Roary and Panaroo pan genomic pipeline. Pangenomic tools screen and sort out every isolate based on gene presence or absence (Page et al., 2015; Tonkin-Hill et al., 2020). The CSV output from Roary and Panaroo was used to visualize the pangenome with FriPan (http://drpowell.github.io/FriPan/), and open and closeness of the genome were calculated using PanGP. Fripan is an interactive web tool to analyze and group bacterial strains based on the accessory genomes. Script roary2fripan (https://github.com/kwongj/roary2fripan) was used to convert the CSV file of Roary to FriPan formats. Rtab binary file from Roary was used for the PanGP analysis. The complete pan-genome profile analysis was carried out using the Heaps law (y = Apan x Bpan +Cpan) formula. The PanGP tool works based on the DG algorithm to calculate the genome diversity of the population (Zhao et al., 2014). The results of Roary and Panaroo were compared to indicate the effectiveness of pipelines. Core genome phylogeny was constructed using the Panaroo generated core genome alignment. Panaroo identified and aligned the conserved orthologous genes from available genomes. A further maximum likelihood tree was constructed with 1000 bootstraps by using the iqtree webserver. Interactive web tool iTol was used for the visualization of core genome phylogeny.
2.5 Genotyping
Spoligotyping is a spacer oligonucleotide typing which is widely used for genotyping Mycobacterium spp. Lorikeet is the tool for digital spoligotyping of mycobacterial genomes from Illumina reads. The WGS raw reads from M. orygis strains (n=53) were subjected to lorikeet spoligotyping to determine the lineage of strains (Cohen et al., 2015). Multilocus sequence typing is sequence-based genotyping, utilizing multiple housekeeping genes to classify the bacterial strains. Sequence types and allelic profiles of M. orygis genomes were identified using galaxy server-based MLST (version 2.22.0) tool. Available mycobacteria pubMLST schemes with a minimum DNA identity of 95% and DNA coverage of 10% were used for determining similar allelic profiles and sequence typing (Seemann, 2016).
2.6 Region of difference analysis
MTBC species are classified based on the SNPs and large polymorphisms present in the region of difference (RD). Presence or absence of RD in the NGS sequence reads can be used to predict the lineage of strains (Bespiatykh et al., 2021). RDscan (https://github.com/dbespiatykh/RDscan) was used to determine the deletion regions (RDs) in all the M. orygis used in the study (n= 55). Sequence reads of the genomes were extracted and downloaded through the Galaxy Europe server. RDscan script was executed with snakemake python script in Ubuntu 20.04.
2.7 Single nucleotide polymorphism analysis
The M. orygis genomes from NCBI and our study were subjected to SNP analysis. Sequence reads were directly extracted from the NCBI to the galaxy server. Sequence reads were trimmed and quality was assessed with trimmometric and FastQC tools. Mapping and variant calling was performed with Snippy 4.6.0 software. The software uses BWA for read mapping and variant calling with the M. tuberculosis (H37Rv) reference genome (Seemann, 2020). The VCF file obtained from Snippy was merged and filtered with the TB variant filter tool with region, snv, and depth. The TB variant report was also prepared with Galaxy server tools and analyzed for SNPs and indels. The genes predicted with SNPs were evaluated for the ortholog analysis using the EggNOG webserver. Fasta sequences of SNP-containing genes were extracted and classified for the subsystems (Huerta-Cepas et al., 2016).
3 Results
A total of nine MGIT cultures showed acid-fast positive bacilli, and Mycobacterium-specific colonies were observed on LJ slant. All the nine isolates showed 445 bp product size for IS6100 PCR, depicting the isolates as MTBC. RD12 region-based PCR showed amplification of 264 bp confirming the isolates as M. orygis (Figure 1). M. orygis-positive isolates were recovered from samples of four cattle, four deer, and one buffalo. The features of all the nine whole genomes carried out in this study are provided in Supplementary Table 2.
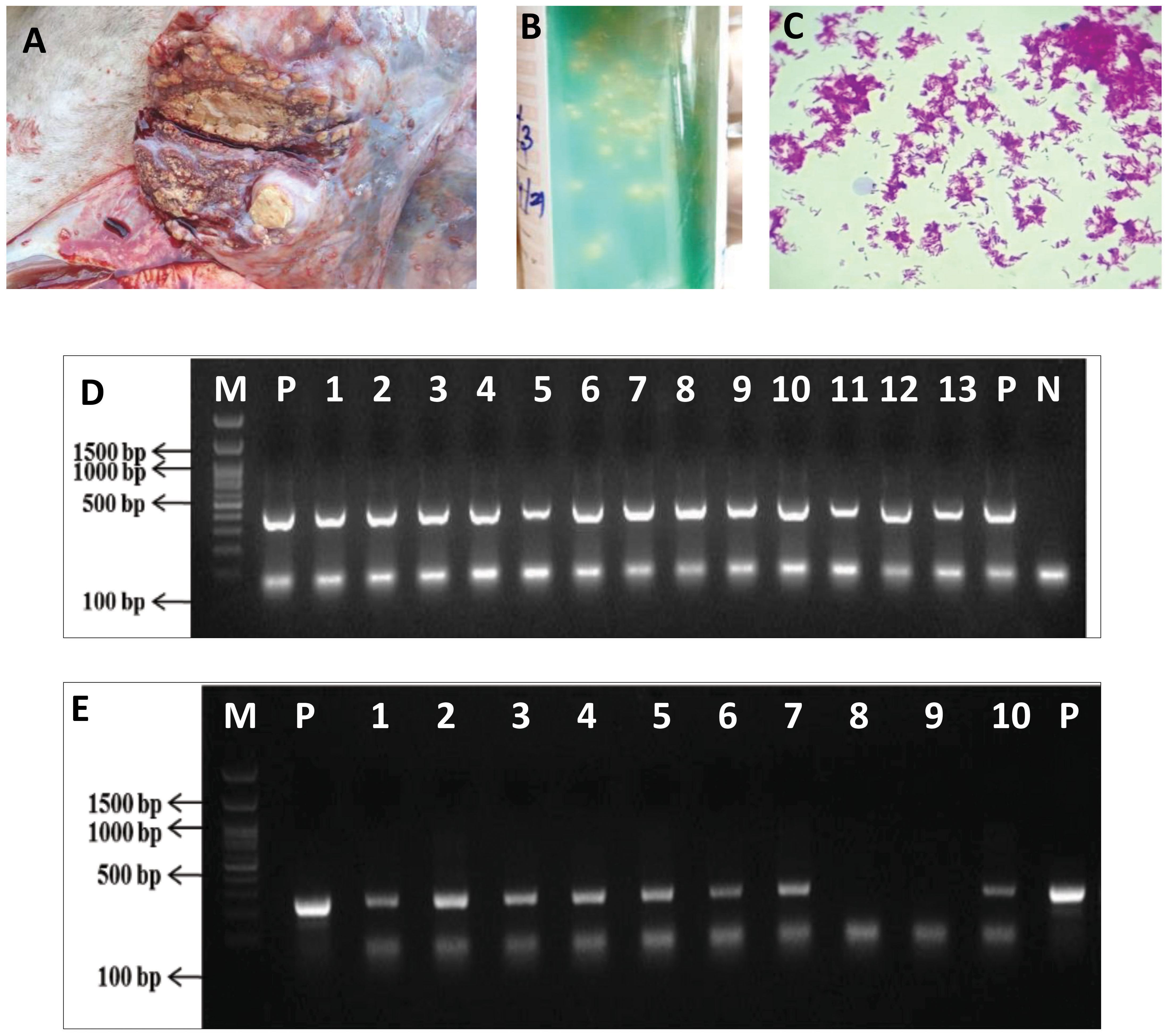
Figure 1 Isolation and confirmation of M. orygis from clinical samples. (A) Caseous nodule in the lung of cattle (ID Jx25). (B) Mycobacterium colonies on LJ slant. (C) Acid-fast positive bacilli from LJ slant. (D) IS6110 PCR for confirmation of MTBC. Lane P—Positive control (M. bovis AN5), Lane N—Negative control, Lanes 1 to 9—M. orygis TANUVAS_1, TANUVAS_2, and TANUVAS_4 to TANUVAS_10. Lanes 10 and 11—M. bovis BCG strain. Lanes 12 and 13—M. tuberculosis. (E) RD12 PCR for confirmation of M. orygis. Lane P—positive control (M. orygis). Lanes 1 to 7—M. orygis TANUVAS_1, TANUVAS_2, and TANUVAS_4 to TANUVAS_8. Lane 8—M. tuberculosis. Lane 9—M. bovis BCG. Lane 10—M. orygis TANUVAS_9.
All M. orygis strains used in the analysis clearly separated from the other members of the Mycobacterium tuberculosis complex (MTBC) based on whole genome phylogeny (Figure 2). All M. orygis strains recovered in the study were clustered together. Strains TANUVAS_2, TANUVAS_4, TANUVAS_5, TANUVAS_7, and NIRTAH144 were genetically more closely related. Similarly, TANUVAS_9 and TANUVAS_10 were more closely related, while TANUVAS_6 was related closely to NIRTKL036, and TANUVAS_1 was related to NIRTKL276. There was no geographical distinction or host-based distinction based on whole genome phylogeny.
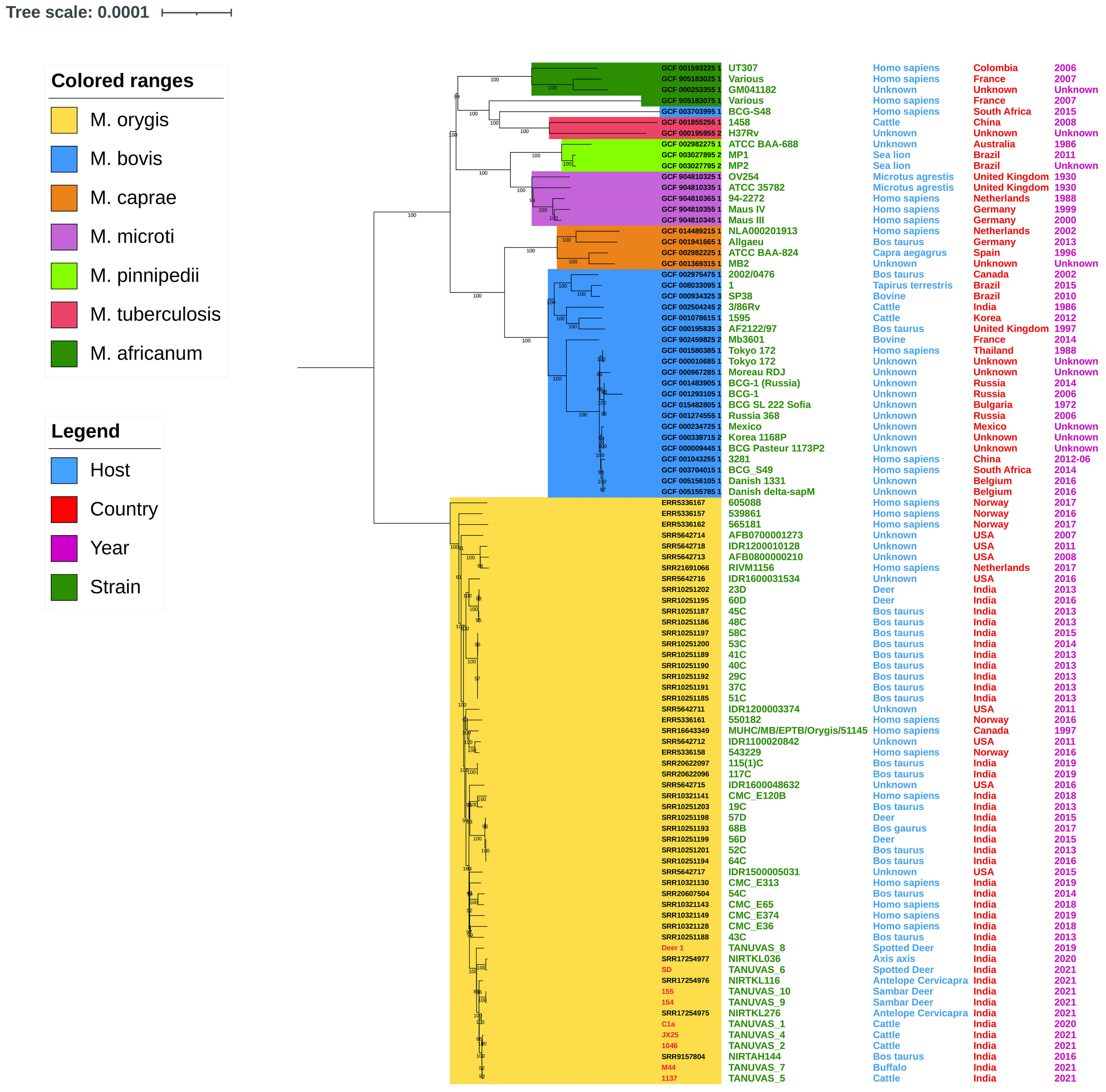
Figure 2 Whole genome phylogeny of MTBC strains (n= 94). Colored ranges indicate different MTBC members, and host, country, and year of isolation are marked adjacent to the strains. M. orygis forms a separate cluster from the rest of the MTBC members.
In the present study, Pairwise SNP were below 100 when compared between strains isolated from Tamil Nadu, India (present study or earlier report). Comparison of strains from other parts of India with Tamil Nadu strains showed SNPs between 100 and 200. Similarly, strains from Norway, USA, and the Netherlands showed higher SNPs (120–1064) when compared with Indian strains (Supplementary Table 3). A maximum of 1064 SNPs was found between strains 565181 and TANUVAS_4. Strains TANUVAS_2, TANUVAS_4, TANUVAS_5, TANUVAS_7, and NIRTAH144 had SNPs less than 10, while other strains from this study had SNPs more than 25. Among the 10 M. orygis strains isolated in the study, all strains except TANUVAS_6 were predicted with 66 virulence genes. Gene mgtC was not predicted in TANUVAS_6, while the clpP gene was not predicted in any the 10 M. orygis strains.
3.1 Typing
Based on MLST, all the M. orygis strains used in the study belonged to ST215 except strain IDR1600048632, which was untyped. Spoligotyping using SpolPred tool showed that all the M. orygis strains had a similar binary pattern (1100000001111000000000001111111101001101111), except strains MUHC/MB/EPTB/Orygis/51145 (1100000000000000000000000000000000000101111), 565181 (1000000001110000000000001111111111001101111), and 43C (1100000001111000000000000110000000000001111) (Supplementary Table 4). There was no spoligotype international type (SIT) assigned for the binary pattern. On further manual checking of the spacer regions, spacer 2 and 3 was found in all M. orygis, while spacer 33 was absent in MUHC/MB/EPTB/Orygis/51145, 52C, 68B, 64C, and 43C. Hence, all the M. orygis strains except MUHC/MB/EPTB/Orygis/51145, 52C, 68B, 64C, and 43C were assigned with SIT 587.
3.2 Pangenome analysis of M. orygis
Pangenome analysis of the 53 M. orygis strains showed a total of 3976 genes, 3031 core genes, 724 soft core genes, 156 shell genes, and 65 cloud genes (accessory genome). The Bpan value of the pan genome is 0 showing that it is a “closed” pangenome. The pangenome curve shows a plateau indicating the “closed” pangenome structure. There was no addition of new genes upon addition of new genome to the pangenome analysis (Figure 3). There was no clear discrimination of strains based on geographical location or year of isolation (Supplementary Figure 1). Clustering of M. orygis in core genome phylogeny was similar to whole genome phylogeny (Supplementary Figure 2).
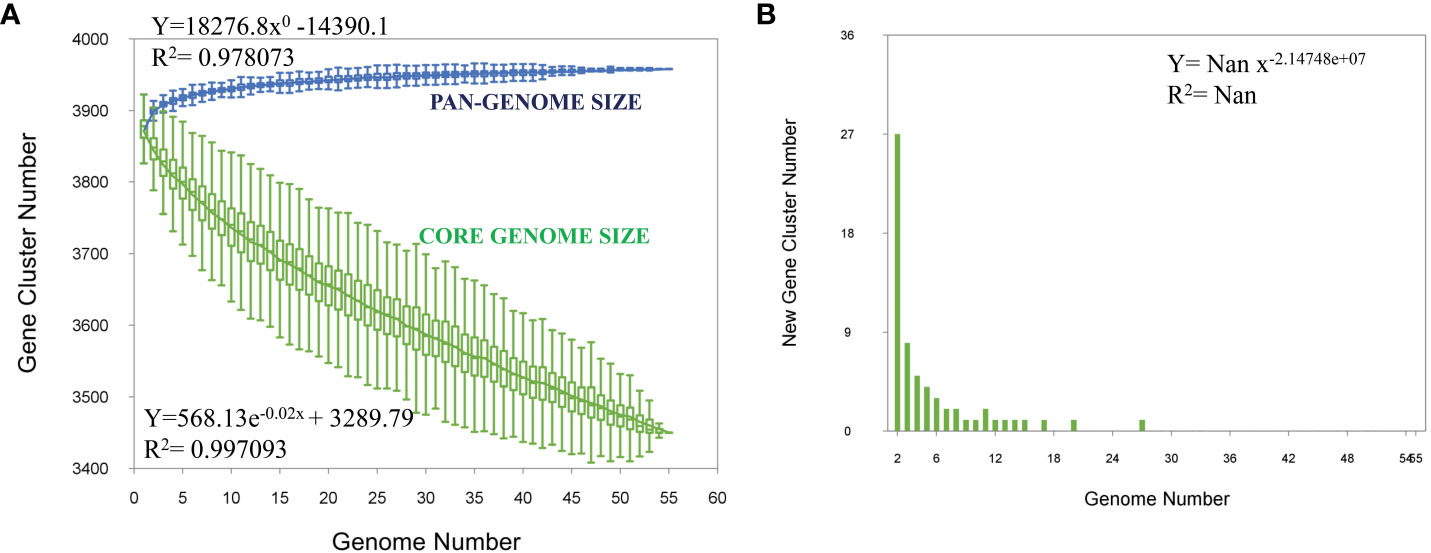
Figure 3 Pangenome analysis. (A) Core and pangenome analysis for 53 M. orygis strains analyzed. Total number of core (green) and pan (blue) genome are plotted. The pangenome curve has attained a plateau, indicating that M. orygis has a closed pangenome structure. (B) Strain specific gene analysis for M. orygis strains. Number of new genes added to the gene pool on addition of new genome is plotted.
3.3 SNP analysis
A total of 8696 SNPs were predicted when the M. orygis genome was compared with the reference genome M. tuberculosis H37Rv. In total, 66 SNPs were predicted to affect the genes heavily. These SNPs belong to start lost, stop gained, stop lost, and stop lost; splice region variant. Multiple intragenic variant SNPs were observed in the tRNA(ile) region. A total of 1700 non-synonymous SNPs were predicted and were used for identifying the COG functional classification. Several genes (n= 439) were not predicted with functional category; 105 genes belonged to the energy production and conversion categories (C); 115 genes belonged to lipid metabolism (I); 92 genes belonged to amino acid metabolism and transport (E); 98 genes belonged to replication, recombination, and repair (L); 46 genes belonged to secretion, motility, and chemotaxis (N); 73 genes belonged to inorganic ion transport and metabolism (P); 86 genes belonged to secondary metabolites biosynthesis, transport, and catabolism (Q); 13 genes belonged to intracellular trafficking, secretion, and vesicular transport (U); and 34 genes belonged to defense mechanism (V) (Supplementary Table 5).
The SNPs present in the virulence factors were analyzed, and the majority of the genes were shown to share the synonymous and non-synonymous mutations. A total of 378 SNPs were present in the 165 genes, among which 238 missense and 138 synonymous SNPs were recorded. Secretion system (PPE4, PE35) and cell surface components (fad13, MRA2983) genes gained stop codons, whereas mps1 lost the stop codon. All the M. orygis strains analyzed gained a stop codon in the PE35 gene before the actual stop codon (Supplementary Figure 3). Initiator codon variant was noticed in the copper uptake gene (ctpV). The genes categorized as regulation shared non-synonymous SNPs. These non-synonymous SNP and stop codons may cause a high or moderate impact on the expression of virulence factor-associated genes (Supplementary Table 6).
3.4 Region of difference
A total of 79 regions were predicted by RDscan in all the M. orygis strains, except RIVM1156 where there were no RDs predicted. RD7, RD8, RD9, RD10, RD12, RD236a, RD301, RD315 were not present in any of the M. orygis (n= 53) analyzed. RD12oryx, RDcan, and RDoryx_4 were not predicted in all M. orygis strains except CMC_E120B. RDOryx_1 and RD-Sur1 were predicted only in IDR1200010128, AFB0800000210, 550182, AFB0700001273, IDR1200003374, 565181, IDR1200010128, and 543229 (Figure 4).
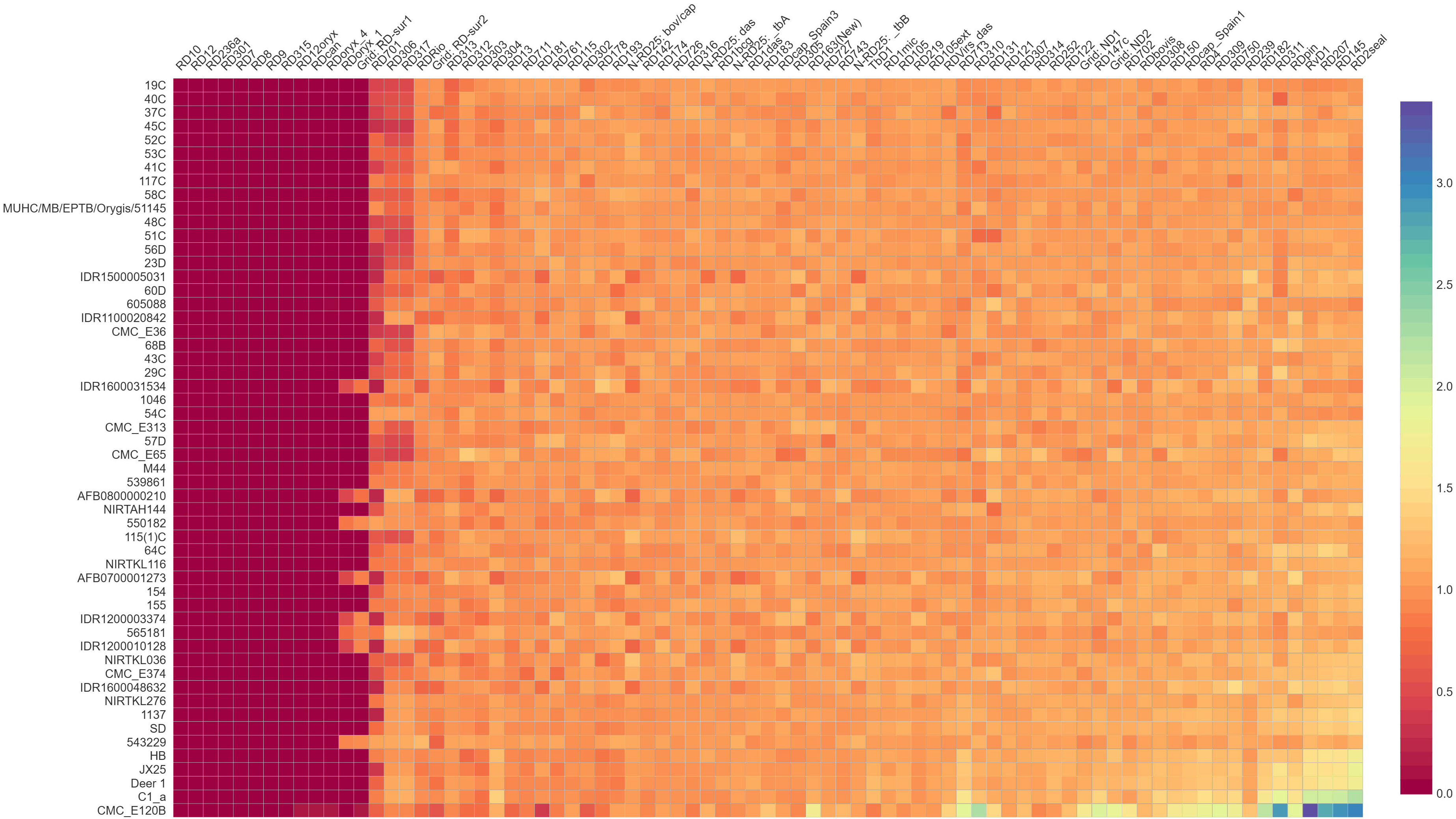
Figure 4 Region of difference among M. orygis strains. Values obtained from RDscan are plotted in a heat map depiction. Dark violet color indicates absence of the region (RDscan value= 0), while orange/yellow color indicates presence of the region (RDscan value >0.5 up to 3). RD10, RD12, RD236a, RD7, RD8, RD12oryx, RD301, and RD315 are absent in all the M. orygis genome analyzed.
4 Discussion
Studies from India have shown the presence of M. orygis in humans (Duffy et al., 2020) and animal species such as cattle, spotted deer, black buck, and Indian bison (Refaya et al., 2022; Sharma et al., 2023), suggesting that there is need for a nationwide surveillance of this pathogen to elucidate its role in tuberculosis. Isolation of M. orygis but not M. bovis in this study correlates with earlier reports from India. To the best of our knowledge, this report on the isolation and whole genome sequencing of M. orygis from water buffalo (Bubalus bubalis) and sambar deer (Cervus unicolor) is the first of its kind.
The whole genome phylogeny shows three major clades of Mycobacterium spp. affecting animals: M. orygis forming a separate clade (I), M. bovis and M. caprae together forming the second clade, and M. microti and M. pinnipedii forming the third clade, as reported by Brites et al. (2018). Strains TANUVAS_2, TANUVAS_4, TANUVAS_5, TANUVAS_7, and NIRTAH144 were isolated from same geographical location, which may be the reason for genetic relatedness. There may be possibility that same M. orygis strain might be the reason for disease in the same geographical location (Tamil Nadu) since SNP cut-off for disease transmission among the affected animals was 3–14 (Walker et al., 2013). Similarly, 0–6 SNPs between M. orygis strains were reported earlier in human patients, suggesting the possible role of single source infection (Lipworth et al., 2019).
Pangenome analysis for M. orygis has not been reported previously, and hence it has been attempted in this study. The pangenome of M. orygis was found to be closed, which was similar to the closed nature of M. bovis reported previously (Ceres et al., 2022). The genome structure of M. orygis is compact, and genes that are essential for survival are present. Since a fewer number of accessory genomes are present, the chances for genetic exchange are lower. Pangenome analysis of M. orygis using the Roary pan-genome pipeline showed an open pangenome structure (date not shown), while the Panaroo pipeline showed a closed structure. Similarly, Reis and Cunha (2021) reported an open pangenome structure of M. bovis using Roary, while Ceres et al. (2022) reported closed a pangenome by using Panaroo. Hence, in the present study, the results obtained from Panaroo were used.
Similar to this study, M. orygis was assigned with spoligotype SIT587, while SIT701 and orphan types were also reported (Gey van Pittius et al., 2012; Riojas et al., 2018). Since only nine M. orygis strains were available, new spoligotype patterns predicted for strains like MUHC/MB/EPTB/Orygis/51145 using bioinformatics could not be confirmed using hybridization assay, which is a limitation of the study. Subsequently, Mycobacterium spp. has less genetic diversity, and MLST and spoligotyping may not be useful for genotyping or construction of phylogeny; instead, large sequence polymorphisms (LSPs) or RDs can be used (Bespiatykh et al., 2021). RD301 and RD315, which were reported to be uniquely absent in M. orygis (Bespiatykh et al., 2021), were absent in all the M. orygis genomes tested in the study. Contradictory to earlier findings (Bespiatykh et al., 2021; Liu Z. et al., 2022), RD236a was absent in all the M. orygis genome analyzed. As reported earlier, RD8 (comprising of Rv3617, Rv3618, Rv3619c, Rv3620c, Rv3621c, Rv3622c, and Rv3623 affected genes) is absent in all M. orygis, and RD236a corresponds to the Rv3617 (ephA) gene. Since the Rv3617 gene is common in both RDs, RD236a might not have been predicted in the M. orygis genome. Blast analysis of the M. orygis genome used in this study also showed the absence of Rv3617.
Prediction of RDoryx_1 in 7/32 M. orygis at other regions of the genome was reported earlier (Bespiatykh et al., 2021), and that may be the reason for the prediction of RDoryx_1 in eight strains in this study. RDoryx_1 comprises eight gene deletions, while RD-Sur1 has 15 gene deletions that includes the eight genes of RDoryx_1 (Liu Z. et al., 2022). Since the genes for RDoryx_1 were predicted in eight strains and similar genes correspond to the RDSur-1 region, RDSur-1 might have been predicted in these eight strains. Bespiatykh et al. (2021) analyzed 32 M. orygis strains, while in the present study, 54 strains were used for RD analysis. Based on both the studies, RD301 and RD315 were specifically absent in all the M. orygis analyzed. The same was confirmed by amplifying the RD301 region using 14 M. orygis genomes (nine isolated in this study and five isolates available in the laboratory), which showed no amplification by PCR. The same region had specific PCR amplification for M. tuberculosis and M. bovis BCG (Supplementary Figure 4). There are reports regarding the misidentification of M. orygis as M. africanum since better diagnostic tools are not available to differentiate the MTBC. Since MTBC members are more closely related, a better diagnostic tool is essential to identify M. orygis (Rahim et al., 2007; Islam et al., 2023). Hence, RD301 or RD315 can be used as a marker for identifying or discriminating M. orygis from other members of MTBC. Since only a limited number of M. orygis genomes were available while drafting the manuscript, analysis with additional datasets from different geographical regions would help to improve confidence regarding the RDs.
A higher number of synonymous SNPs was identified in the M. orygis. Non-synonymous SNPs were identified in the genes related to the lipid and energy metabolism. Similar types of non-synonymous SNPs were detected in the M. bovis strains originating from Spain and France (Hauer et al., 2019; Perea et al., 2021). Non-synonymous SNPs may affect the structure and arrangement of lipid and protein virulence factors. SNPs in these genes may be involved in the evolution, persistence, and transmission of M. orygis in wild and domestic animals of tropical countries including India. Further, the impact of SNPs on the phenotypic nature of M. orygis needs to be studied to understand the pathogenesis of this bacterium in humans and animals.
The genes PPE4 and PE35 are linked with the ESX1 and ESX3 secretion system of mycobacterial species. These genes gained a stop codon in M. orygis, which will alter the amino acid sequences of the protein. PE/PPE gene products were related to bacterial virulence and involved in host–pathogen interactions such as invasion, immune regulation, and intracellular survival (Qian et al., 2020). PPE4 carry out iron or zinc acquisition from the cells and regulate mycobactin utilization (Tufariello et al., 2016). PE35 is a proline-rich protein and is encoded in RD1 region. PE35 is important for M. tuberculosis infection and cellular-level response. Due to the partial loss of RD1 in the BCG strain, it is used to differentiate vaccinated and infected humans (Jiang et al., 2016). Polymorphism in this gene region affects intracellular survival and immune regulation during infection and could be employed as a marker in future diagnostic tools.
5 Conclusion
This study supports that hypothesis that M. orygis is the most prevalent Mycobacterium spp. in animals in India. The study is also the first to report M. orygis in water buffalo and sambar deer. Compared to spoligotyping and MLST, RD-based analysis discriminates M. orygis from other MTBC members, and RD301 and RD315 as validated by PCR can be used as diagnostic markers for confirmation of the presence of M. orygis. Similarly, SNPs in the PE35 gene can be used as molecular markers for the identification of M. orygis.
Data availability statement
All the genomes generated in this study have been submitted tothe NCBI SRA database with the bio project number PRJNA785380.
Ethics statement
The study does not involve animal trials, and samples receivedat the laboratory were used for isolation, hence, this study does notrequire approval for conducting animal trials.
Author contributions
KK: Conceptualization, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SS: Data curation, Investigation, Methodology, Writing – review & editing. MV: Data curation, Investigation, Methodology, Writing – review & editing. AJ: Data curation, Methodology, Writing – review & editing. SA: Methodology, Software, Writing – original draft, Writing – review & editing. RK: Methodology, Resources, Writing – review & editing. PT: Software, Validation, Writing – review & editing. RA: Data curation, Supervision, Writing – review & editing. KGT: Funding acquisition, Resources, Validation, Writing – review & editing. GD: Funding acquisition, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research project has been implemented with financial contributions from the Department of Biotechnology, Government of India under the DBT Network Program on Bovine Tuberculosis Control: Mycobacterial Diseases in Animals Network (MyDAN) Program (Scheme Code No.22270) and Department of Biotechnology, Government of India under the Translational Research Platform for Veterinary Biologicals- Phase III (BT/TRPVB/TANUVAS/2011- Phase III).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1302393/full#supplementary-material
Supplementary Table 3 | Pairwise SNP distance of M. orygis genomes used in the study.
Supplementary Table 4 | Spoliogotyping results of the M. orygis genomes.
Supplementary Table 5 | COG categorization of functional genes that possess missense mutation.
Supplementary Figure 1 | Pangenome-based phylogeny and multidimensional scaling. A. Pangenome-based phylogeny. B. Multidimensional scaling of M. orygis genome. No major categorization based on place, year, or host of isolation.
Supplementary Figure 2 | Core genome phylogeny of MTBC isolates used in this study.
Supplementary Figure 3 | Amino acid sequence alignment of PE35 gene showing stop codon (gained) marked as *. The first sequence is the M. tuberculosis reference sequence to which all the M. orygis is compared.
Supplementary Figure 4 | Representative gel electrophoresis image of PCR for RD307 and RD315. A. M. tuberculosis DNA. Lane 1—RD301, Lane 2—RD315. B. M. bovis BCG and M. orygis DNA. Lanes 1 and 2—M. bovis BCG and M. orygis tested for RD301 region respectively. Lanes 3 and 4—M. bovis BCG and M. orygis tested for RD315 region respectively.
References
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 1–15. doi: 10.1186/1471-2164-9-75
Bertels, F., Silander, O. K., Pachkov, M., Rainey, P. B., Van Nimwegen, E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31 (5), 1077–1088. doi: 10.1093/molbev/msu088
Bespiatykh, D., Bespyatykh, J., Mokrousov, I., Shitikov, E. (2021). A comprehensive map of mycobacterium tuberculosis complex regions of difference. mSphere 6 (4), e0053521. doi: 10.1128/mSphere.00535-21
Brites, D., Loiseau, C., Menardo, F., Borrell, S., Boniotti, M. B., Warren, R., et al. (2018). A new phylogenetic framework for the animal-adapted mycobacterium tuberculosis complex. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02820
Ceres, K. M., Stanhope, M. J., Gröhn, Y. T. (2022). A critical evaluation of Mycobacterium bovis pangenomics, with reference to its utility in outbreak investigation. Microbial Genome 8 (6), mgen000839. doi: 10.1099/mgen.0.000839
Cohen, K. A., Abeel, T., Manson McGuire, A., Desjardins, C. A., Munsamy, V., Shea, T. P., et al. (2015). Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PloS Med. 12 (9), e1001880. doi: 10.1371/journal.pmed.1001880
Dawson, K. L., Bell, A., Kawakami, R. P., Coley, K., Yates, G., Collins, D. M. (2012). Transmission of Mycobacterium orygis (M. tuberculosis Complex Species) from a Tuberculosis Patient to a Dairy Cow in New Zealand. J. Clin. Microbiol. 50 (9), 3136–3138. doi: 10.1128/JCM.01652-12
Duffy, S. C., Srinivasan, S., Schilling, M. A., Stuber, T., Danchuk, S. N., Michael, J. S., et al. (2020). Reconsidering Mycobacterium bovis as a proxy for zoonotic tuberculosis: a molecular epidemiological surveillance study. Lancet Microbe 1 (2), e66–e73. doi: 10.1016/S2666-5247(20)30038-0
Eldholm, V., Rønning, J. O., Mengshoel, A. T., Arnesen, T. (2021). Import and transmission of Mycobacterium orygis and Mycobacterium africanum, Norway. BMC Infect. Dis. 21 (1), 562. doi: 10.1186/s12879-021-06269-3
Gey van Pittius, N. C., van Helden, P. D., Warren, R. M. (2012). Characterization of mycobacterium orygis. Emerging Infect. Dis. 18 (10), 1708–1709. doi: 10.3201/eid1810.120569
Hauer, A., Michelet, L., Cochard, T., Branger, M., Nunez, J., Boschiroli, M. L., et al. (2019). Accurate phylogenetic relationships among mycobacterium bovis strains circulating in France based on whole genome sequencing and single nucleotide polymorphism analysis. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00955
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44 (D1), D286–D293. doi: 10.1093/nar/gkv1248
Islam, M. R., Sharma, M. K., KhunKhun, R., Shandro, C., Sekirov, I., Tyrrell, G. J., et al. (2023). Whole genome sequencing-based identification of human tuberculosis caused by animal-lineage Mycobacterium orygis. J. Clin. Microbiol. 25, e0026023. doi: 10.1128/jcm.00260-23
Jiang, Y., Wei, J., Liu, H., Li, G., Guo, Q., Qiu, Y., et al. (2016). Polymorphisms in the PE35 and PPE68 antigens in Mycobacterium tuberculosis strains may affect strain virulence and reflect ongoing immune evasion. Mol. Med. Rep. 13 (1), 947–954. doi: 10.3892/mmr.2015.4589
Lavender, C. J., Globan, M., Kelly, H., Brown, L. K., Sievers, A., Fyfe, J. A. M., et al. (2013). Epidemiology and control of tuberculosis in Victoria, a low-burden state in south-eastern Australia 2005-10. Int. J. Tuberculosis Lung Dis. 17 (6), 752–758. doi: 10.5588/ijtld.12.0791
Letunic, I., Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (1), W293–W296. doi: 10.1093/nar/gkab301
Lipworth, S., Jajou, R., de Neeling, A., Bradley, P., van der Hoek, W., Maphalala, G., et al. (2019). SNP-IT tool for identifying subspecies and associated lineages of mycobacterium tuberculosis complex. Emerging Infect. Dis. 25 (3), 482–488. doi: 10.3201/eid2503.180894
Liu, B., Zheng, D., Zhou, S., Chen, L., Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50 (D1), D912–D917. doi: 10.1093/nar/gkab1107
Liu, Z., Jiang, Z., Wu, W., Xu, X., Ma, Y., Guo, X., et al. (2022). Identification of region of difference and H37Rv-related deletion in Mycobacterium tuberculosis complex by structural variant detection and genome assembly. Front. Microbiol. 13, 7. doi: 10.3389/fmicb.2022.984582
Marcos, L. A., Spitzer, E. D., Mahapatra, R., Ma, Y., Halse, T. A., Shea, J., et al. (2017). Mycobacterium orygis lymphadenitis in New York, USA. Emerging Infect. Dis. 23 (10), 1749–1751. doi: 10.3201/eid2310.170490
Miller, J., Jenny, A., Rhgyan, J., Saari, D., Saurez, D. (1997). Detection of Mycobacterium bovis in formalin-fixed, paraffin-embedded tissues of cattle and elk by PCR amplification of an IS6110 sequence specific for M. tuberculosis complex organisms. J. Vet. Diagn. Invest. 9, 244–249. doi: 10.1177/104063879700900304
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Haeseler, A. V., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37 (5), 1530–1534. doi: 10.1093/molbev/msaa015
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31 (22), 3691–3693. doi: 10.1093/bioinformatics/btv421
Perea, C., Ciaravino, G., Stuber, T., Thacker, T. C., Robbe-Austerman, S., Allepuz, A., et al. (2021). Whole-genome SNP analysis identifies putative mycobacterium bovis transmission clusters in livestock and wildlife in Catalonia, Spain. Microorganisms 9 (8), 1629. doi: 10.3390/microorganisms9081629
Qian, J., Chen, R., Wang, H., Zhang, X. (2020). Role of the PE/PPE family in host–pathogen interactions and prospects for anti-tuberculosis vaccine and diagnostic tool design. Front. Cell. infection Microbiol. 10, 594288. doi: 10.3389/fcimb.2020.594288
Rahim, Z., Möllers, M., te Koppele-Vije, A., de Beer, J., Zaman, K., Matin, M. A., et al. (2007). Characterization of Mycobacterium africanum subtype I among cows in a dairy farm in Bangladesh using Spoligotyping. Southeast Asian J. Trop. Med. Public Health 38, 706–713.
Rahim, Z., Thapa, J., Fukushima, Y., van der Zanden, A. G. M., Gordon, S. V., Suzuki, Y., et al. (2017). Tuberculosis caused by Mycobacterium orygis in dairy cattle and captured monkeys in Bangladesh: a new scenario of Tuberculosis in South Asia. Transboundary Emerging Dis. 64, 1965–1969. doi: 10.1111/tbed.12596
Refaya, A. K., Kumar, N., Raj, D., Veerasamy, M., Balaji, S., Shanmugam, S., et al. (2019). Whole-genome sequencing of a mycobacterium orygis strain isolated from cattle in Chennai India. Microbiol. Resource Announcement 8 (40), e01080-19. doi: 10.1128/MRA.01080-19
Refaya, A. K., Ramanujam, H., Ramalingam, M., Rao, G. V. S., Ravikumar, D., Sangamithrai, D., et al. (2022). Tuberculosis caused by Mycobacterium orygis in wild ungulates in Chennai, South India. Transboundary Emerging Dis. 69 (5), e3327–e3333. doi: 10.1111/tbed.14613
Reis, A. C., Cunha, M. V. (2021). The open pan-genome architecture and virulence landscape of Mycobacterium bovis. Microbial Genome 7 (10), 664. doi: 10.1099/mgen.0.000664
Riojas, M. A., McGough, K. J., Rider-Riojas, C. J., Rastogi, N., Hazbón, M. H. (2018). Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycobacterium tuberculosis. Int. J. Systematic Evolutionary Microbiol. 68 (1), 324–332. doi: 10.1099/ijsem.0.002507
Seemann, T. (2016). ABRicate: mass screening of contigs for antibiotic resistance genes (San Francisco, CA: GitHub).
Seemann, T. (2020). Snippy: rapid haploid variant calling and core genome alignment (San Francisco, CA: GitHub). Available at: https://github.com/tseemann/snippy.
Sharma, M., Mathesh, K., Dandapat, P., Mariappan, A. K., Kumar, R., Kumari, S., et al. (2023). Emergence of mycobacterium orygis-associated tuberculosis in wild ruminants, India. Emerging Infect. Dis. 29 (3), 661–663. doi: 10.3201/eid2903.221228
Sumanth, L. J., Suresh, C. R., Venkatesan, M., Manesh, A., Behr, M. A., Kapur, V., et al. (2023). Clinical features of human tuberculosis due to Mycobacterium orygis in Southern India. J. Clin. Tuberculosis Other Mycobacterial Dis. 32, 100372. doi: 10.1016/j.jctube.2023.100372
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Thapa, J., Gordon, S. V., Nakajima, C., Suzuki, Y. (2022). Threat from Mycobacterium orygis-associated tuberculosis in south Asia. Lancet Microbe 3 (9), e641–e642. doi: 10.1016/S2666-5247(22)00149-5
Thapa, J., Nakajima, C., Maharjan, B., Poudell, A., Suzuki, Y. (2015). Molecular characterization of Mycobacterium orygis isolates from wild animals of Nepal. Japanese J. Veterinary Res. 63 (3), 151–158.
Thapa, J., Paudel, S., Sadaula, A., Shah, Y., Maharjan, B., Kaufman, G. E., et al. (2016). Mycobacterium orygis: associated tuberculosis in free-ranging rhinoceros, Nepal. Emerging Infect. Dis. 22 (3), 3. doi: 10.3201/eid2203.151929
Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21, 180. doi: 10.1186/s13059-020-02090-4
Tufariello, J. M., Chapman, J. R., Kerantzas, C. A., Wong, K. W., Vilchèze, C., Vilchèze, C., et al. (2016). Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc. Natl. Acad. Sci. USA 113 (3), E348–E357. doi: 10.1073/pnas.1523321113
van Ingen, J., Rahim, Z., Mulder, A., Boeree, M. J., Simeone, R., Brosch, R., et al. (2012). Characterization of Mycobacterium orygis as M. tuberculosis complex subspecies. Emerging Infect. Dis. 18, 653–655. doi: 10.3201/eid1804.110888
Walker, T. M., Ip, C. L. C., Harrell, R. H., Evans, J. T., Kapatai, G., Dedicoat, M. J., et al. (2013). Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 13, 137–146. doi: 10.1016/S1473-3099(12)70277-3
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Keywords: Mycobacterium orygis, buffalo, sambar deer, India, comparative genomics, region of difference, SNP
Citation: Karthik K, Subramanian S, Vinoli Priyadharshini M, Jawahar A, Anbazhagan S, Kathiravan RS, Thomas P, Babu RPA, Gopalan Tirumurugaan K and Raj GD (2023) Whole genome sequencing and comparative genomics of Mycobacterium orygis isolated from different animal hosts to identify specific diagnostic markers. Front. Cell. Infect. Microbiol. 13:1302393. doi: 10.3389/fcimb.2023.1302393
Received: 26 September 2023; Accepted: 22 November 2023;
Published: 22 December 2023.
Edited by:
Rui Miguel Gil Da Costa, Federal University of Maranhão, BrazilReviewed by:
Jeewan Thapa, Hokkaido University, JapanMarcelo Andrade, Federal University of Maranhão, Brazil
Copyright © 2023 Karthik, Subramanian, Vinoli Priyadharshini, Jawahar, Anbazhagan, Kathiravan, Thomas, Babu, Gopalan Tirumurugaan and Raj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gopal Dhinakar Raj, ZGlydHJwdmJAZ21haWwuY29t