
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 12 December 2023
Sec. Intestinal Microbiome
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1274690
This article is part of the Research Topic Human Microbiome and COVID-19 View all 7 articles
 Ruyue Fan1,2†
Ruyue Fan1,2† Shuai Liu3,4*†Na Sun1,2Ying Yang1,2Xia Deng5
Shuai Liu3,4*†Na Sun1,2Ying Yang1,2Xia Deng5 Bin Hu1,2Changhua Sun1,2Chengli Wen1,2Hui Li1,2Dong Cheng1,2Chuanjun Huang3,4Peibin Hou1,2*Tianliang Zhang1,2*
Bin Hu1,2Changhua Sun1,2Chengli Wen1,2Hui Li1,2Dong Cheng1,2Chuanjun Huang3,4Peibin Hou1,2*Tianliang Zhang1,2*Background: Human gut microbiota play a crucial role in the immune response of the host to respiratory viral infection. However, evidence regarding the association between the gut microbiome, host immune responses, and disease severity in coronavirus disease 2019 (COVID-19) remains insufficient.
Methods: To better comprehend the interactions between the host and gut microbiota in COVID-19, we conducted 16S rRNA sequencing and characterized the gut microbiome compositions in stool samples from 40 COVID-19 patients and 33 non-pneumonia controls. We assessed several hematological parameters to determine the immune status.
Results: We found that the gut microbial composition was significantly changed in COVID-19 patients, which was characterized by increased opportunistic pathogens and decreased commensal bacteria. The frequency of prevalent opportunistic pathogens Enterococcus and Lactobacillus increased, especially in severe patients; yet the abundance of butyrate-producing bacteria, Faecalibacterium, Roseburia, and Anaerostipes, decreased significantly, and Faecalibacterium prausnitzii might help discriminate severe patients from moderate patients and non-pneumonia people. Furthermore, we then obtained a correlation map between the clinical characteristics of COVID-19 and severity-related gut microbiota. We observed a notable correlation between the abundance of Enterococcus faecium and abnormal neutrophil or lymphocyte percentage in all COVID-19 patients. Faecalibacterium was positively correlated with lymphocyte counts, while negatively correlated with neutrophil percentage.
Conclusion: These results suggested that the gut microbiome could have a potential function in regulating host immune responses and impacting the severity or consequences of diseases.
The coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is still a health problem worldwide. Patients with COVID-19 can be identified by typical respiratory symptoms; however, common gastrointestinal (GI) symptoms have also been demonstrated to be significant features of the symptomatology associated with COVID-19, such as anorexia, vomiting, and abdominal pain (Zuo et al., 2020; Hayashi et al., 2021). Moreover, gastrointestinal symptoms have been reported to not only appear in patients with COVID-19 but also occur prior to the onset of typical respiratory symptoms (Schmulson et al., 2020). Certain studies note a substantial involvement of the GI tract after SARS-CoV-2 infection, such as the capability of the virus to infect and replicate in human small intestine enterocytes (Lamers et al., 2020), the positive detection of surviving virus particles in stool samples (Merola et al., 2020), and gut microbiota dysbiosis occurring in patients with COVID-19 (Zuo et al., 2020). Previous research has indicated that SARS-CoV-2 enters the human body via the angiotensin-converting enzyme 2 (ACE2) receptor, which has significant expression in epithelial cells of the lung and intestine (Lucas et al., 2020). Given the high distribution of ACE2 receptors in the intestinal mucosa, the GI tract becomes a crucial biological niche for the expansion of pathogens into the respiratory and cardiovascular systems.
The dysregulated inflammatory response of the host is the key to determining disease severity and progression (Lucas et al., 2020; Cao et al., 2021). When the virus enters a cell, it has the potential to trigger innate and adaptive host immune responses, which are essential parts of the defense against viral infections (Schultze and Aschenbrenner, 2021; Sette and Crotty, 2021). The dysregulated inflammatory responses could contribute to tissue degradation and the development of the disease (Nasrollahi et al., 2023). These include the overproduction of inflammatory cytokines, overactivation of neutrophils and macrophages, T lymphopenia, and dysregulated T- and B-lymphocyte function, when there is preexisting immunity to the virus (Wilk et al., 2020; Schultze and Aschenbrenner, 2021; Sette and Crotty, 2021). In the gut, the local immune disorders were closely associated with gut bacterial dysbiosis and instability, which further exacerbated the impairment of gut health.
Human intestinal microbial communities play a crucial role in maintaining host homeostasis through various mechanisms such as immunomodulation, nutrient metabolism, and structural protection against pathogens. The GI tract is recognized as the major immunological organ, and its resident microbiota are known to be involved in modulating the host immune system and our entire physiology (Donaldson et al., 2016; Schirmer et al., 2016). Respiratory viral infection has been found to result in notable alterations in the gut microbiome, even in the absence of identifiable virions inside the GI tract (Lu et al., 2017; Hanada et al., 2018; Libertucci and Young, 2019). Several studies also described a possible connection between COVID-19 severity and gut microbiota composition (Dhar and Mohanty, 2020; Yeoh et al., 2021; Zuo et al., 2021). The relative abundance of Clostridium innocuum, Alistipes finegoldii, and Ruthenibacterium lactatiformans was positively correlated with inflammatory biomarkers and tended to increase with COVID-19 progression (Schult et al., 2022). COVID-19 severity was correlated with an increased abundance of Bacteroides but a reduced abundance of Roseburia (Hazan et al., 2022). Additionally, a negative correlation between disease severity and the abundance of bacteria associated with tryptophan metabolism and maintenance of immune homeostasis was observed in COVID-19 patients (Reinold et al., 2021). Nevertheless, data revealing the holistic interplay between the gut microbiota and host immune responses to SARS-CoV-2 infection still remain limited. There is still substantial work to be done to address the direct causality between gut microbiota and COVID-19 deterioration or prognosis.
Here, we conducted a characterization of the gut microbiome and plasma inflammatory cytokines among 18 moderate and 22 severe patients with COVID-19 during hospitalization and 33 individuals with no symptoms or recovery from mild infection. Furthermore, we analyzed the diversity and composition of gut microbiota and uncovered the possible links between gut microbiota and host systemic immune responses. This will provide valuable insight into the function of the gut microbiota and shed light on the development of gut microbiota-based treatment for COVID-19.
Hospitalized patients with COVID-19 were recruited during the peak SARS-CoV-2 season of 2022–2023 in the Department of Pulmonary and Critical Care Medicine of Shandong Provincial Hospital. All patients were diagnosed with pneumonia by chest computed tomography scans. The throat swab samples were tested by reversing real-time polymerase chain reaction (RT-PCR) to detect the presence of SARS-CoV-2. Referring to the New Coronavirus Pneumonia Diagnosis and Treatment Program (10th edition) published by the National Health Commission of China, severe pneumonia was defined as dyspnea, respiratory frequency ≥30/min, blood oxygen saturation ≤93%, partial pressure of arterial oxygen (PaO2)-to-fraction of inspired oxygen (FiO2) ratio ≤300, and/or lung infiltrates >50% within 24–48 h, respiratory failure, septic shock, and/or multiple organ dysfunction or failure. Moderate pneumonia was defined as fever or respiratory tract infection symptoms with imaging indicating pneumonia. Patients were classified as “non-pneumonia” if there were no radiographic indications of pneumonia with minor clinical symptoms. Blood and stool samples from in-hospital patients were collected, flash-frozen, and stored in liquid nitrogen at −80°C until use.
A total of 73 stool samples were collected from COVID-19 patients at admission and donors recovering from mild infections or asymptomatic individuals. All samples were stored at −80°C immediately until processing. According to the manufacturer’s instructions for the QIAamp Fast DNA Stool Mini Kit (Qiagen, Germany), stool DNA was extracted. Approximately 200 mg of stool samples was used for DNA extraction and sequencing libraries. PCR was performed to amplify the V3–V4 hypervariable region of the 16S rRNA gene in all samples, following the guidelines provided in the 16S Metagenomic Sequencing Library Preparation guide. The amplicon primers were as follows: forward primer, 5′-AGAGTTTGATCCTGGCTCAG-3′; and reverse primer, 5′-GNTACCTTGTTACGACTT-3′. The PCR conditions were as follows: initial denaturation at 95°C for 3 min; 30 cycles of denaturation at 95°C for 30 s, annealing at 56°C for 30 s, extension at 72°C for 3 min; at 72°C for 10 min; hold at 4°C. The PCR products that had been purified were combined in equal numbers and subsequently utilized to generate the amplicon library for single-molecule real-time (SMRT) sequencing analysis using the PacBio sequencing platform (Novogene Co., Ltd., Beijing, China). The reads obtained from the platform were subjected to processing using SMRT software to eliminate the unqualified reads (including excessively long or short reads, chimeras, and inferior-quality reads). Subsequently, qualified sequences were clustered and divided into operational taxonomic units (OTUs) based on a threshold of 97% similarity, as described by Liu et al. (Zhang et al., 2021). Raw sequence data generated for this study are available in the Sequence Read Archive under BioProject accession PRJNA1004732.
Sequence analysis was conducted using Uparse software (Uparse v7.0.1001) (Edgar, 2013). Sequences with ≥97% similarity were allocated to identical OTUs. The representative sequence of each OTU was subjected to a screening process for further annotation. For every representative sequence, the SSUrRNA Database of the Silva Database was used based on the Mothur algorithm to annotate taxonomic information (Quast et al., 2013). Prior to analyzing alpha-diversity and beta-diversity, the original OTU table was rarefied to 5,599 reads (the smallest number of bacterial reads among samples) to allow for comparable comparisons. The alpha-diversity indices, including observed species, Chao1, Shannon, Simpson, and ACE, were determined by rarefaction analysis using Mothur software (version 1.21.1). The indices in our samples were computed using QIIME (version 1.9.1) and visualized using R software (version 2.15.3). Beta-diversity analysis was used to assess variations of samples in species complexity, and the QIIME software (version 1.9.1) was utilized to quantify beta-diversity on both weighted and unweighted UniFrac (Lozupone et al., 2011). Principal coordinate analysis (PCoA) and analysis of similarities (ANOSIM) based on the Bray–Curtis similarity were used to assess the statistical significance of the diversity index variation among the samples. The Kruskal–Wallis rank-sum test was used to examine the alterations and disparities between samples. The construction of PCoA was performed using the WGCNA package, stat packages, and ggplot2 package in R software (version 2.15.3). A p-value <0.05 was considered significant, and 0.05 ≤ p < 0.10 was considered a tendency. Linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed to define COVID-19-related microbial characteristics.
Tests for blood cells, inflammation biomarkers, and albumin of COVID-19 patients were performed using an automated hematology and biochemical analyzer. The full whole blood cell subsets were analyzed using the CytoFLEX S Flow Cytometer (Beckman Coulter, Brea, CA, USA) with FlowJo V10.7.2 software. A correlation matrix between gut microbes from the moderate and severe patients and their clinical features of host values was then generated using Spearman’s rank correlation coefficient implemented in R version 4.2.3. Spearman’s correlations were calculated using the function corr.test of the R package psych, and the correlation heatmap was drowned using the R package ggplot2. Student’s t-test was performed to determine the significance analysis of the means between groups. *p < 0.05, **p < 0.01, and ***p < 0.001 represent significant differences.
COVID-19 patients were recruited between 2022 and 2023 (n = 40). Out of our cohort of 73 subjects, 18 were classified as “moderate”, 22 as “severe”, and 33 as “non-pneumonia” based on the criteria outlined in the Materials and Methods section. The three groups were comparable in terms of sex. All COVID-19 patients enrolled had at least one comorbidity, with pneumonia disease being the most prevalent. The superior respiratory support strategy was chosen and summarized. Among all patients, 85% of patients (34 of 40) needed supplemental oxygen. In the severe group, 40.91% of patients (9 of 22) required superior respiratory support (such as mechanical ventilatory support). The hospital mortality rate in the severe cases of pneumonitis was 9.09% (2 of 22). The chest computed tomography from severe patients who eventually progressed to death showed confluent and predominantly patchy ground-glass opacities with pronounced peripheral distribution and partial consolidation (Supplementary Figures 1A,B). The severe group had a significantly higher percentage of neutrophil counts and C-reactive protein (CRP) and human interleukin (IL)-6 levels whereas substantially reduced lymphocyte counts and PaO2/FiO2 ratio levels compared to the moderate group. The characteristics of cohorts are listed in Table 1.
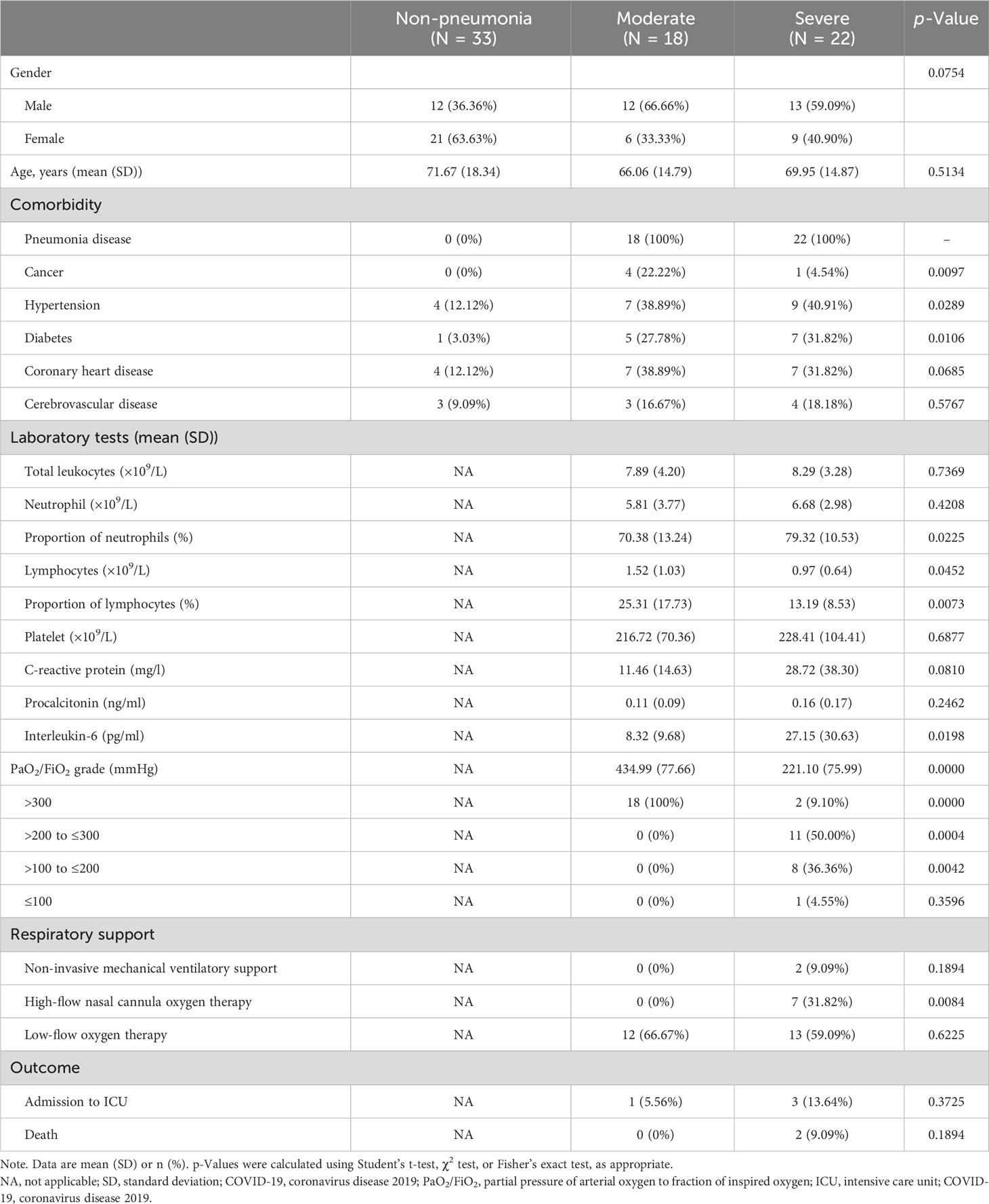
Table 1 Demographics of the COVID-19 and non-pneumonia cohorts.
To investigate the distribution of bacterial taxa, the composition of the gut microbes at the phylum level was subjected to visualization. The results showed that in patients and controls, Firmicutes, Bacteroidetes, and Proteobacteria were dominant communities. Particularly, the phylum Tenericutes was relatively more abundant in severe COVID-19 patients than in non-pneumonia individuals (mean <0.01% vs. 0.8%; p < 0.01, Mann–Whitney test) (Figure 1A, Supplementary Figure 2A). The corresponding heatmap of the top 35 abundant genera is shown in Figure 1B and Supplementary Figure 2B. From these results, it was found that most of the members of the top 35 genera were classified as phyla Firmicutes, Bacteroidetes, and Proteobacteria. Moreover, the dominant genera were different between samples; Blautia was dominant in the severe groups, while unidentified Prevotellaceae dominated in the moderate group. The differences of microbial communities at the genus level were further analyzed, and the higher enrichment of Enterococcus was observed in the COVID-19 patients group compared with the control group (M vs. N, p = 0.038; S vs. N, p = 0.001) (Figures 2A, B). Moreover, 4 and 15 bacterial genera (e.g., Romboutsia and Roseburia) were abundant in the moderate and severe COVID-19 groups, and 15 bacterial genera (e.g., Faecalibacterium and Dialister) were decreased in the severe COVID-19 group compared with the control group. In addition, the significant depletion of the genera Faecalibacterium, Dialister, and Roseburia was found in the severe COVID-19 patients compared with the moderate group (Figure 2C). To better identify bacteria with value as potential COVID-19 indicators, bacterial communities at the species level were evaluated. The abundance of several bacteria showed obvious differences between the COVID-19 patients and non-pneumonia groups (Supplementary Figures 3A,B). Particularly, Enterococcus faecium was enriched in the severe group (S vs. N, p = 0.001), while Bacteroides plebeius and Clostridium celatum were decreased compared with the control group (Supplementary Figure 3B). Furthermore, the abundance of four bacterial species, including Faecalibacterium prausnitzii, Anaerostipes hadrus, Roseburia faecis, and Coprococcus comes, was obviously lower in the severe group compared to the moderate group (Supplementary Figure 3C). These results demonstrated that Enterococcus was significantly elevated in the COVID-19 group, which indicated that Enterococcus had an obvious effect in distinguishing COVID-19 and non-pneumonia groups and could be used as a potential candidate for aiding in the diagnosis and prediction of the disease severity of COVID-19.
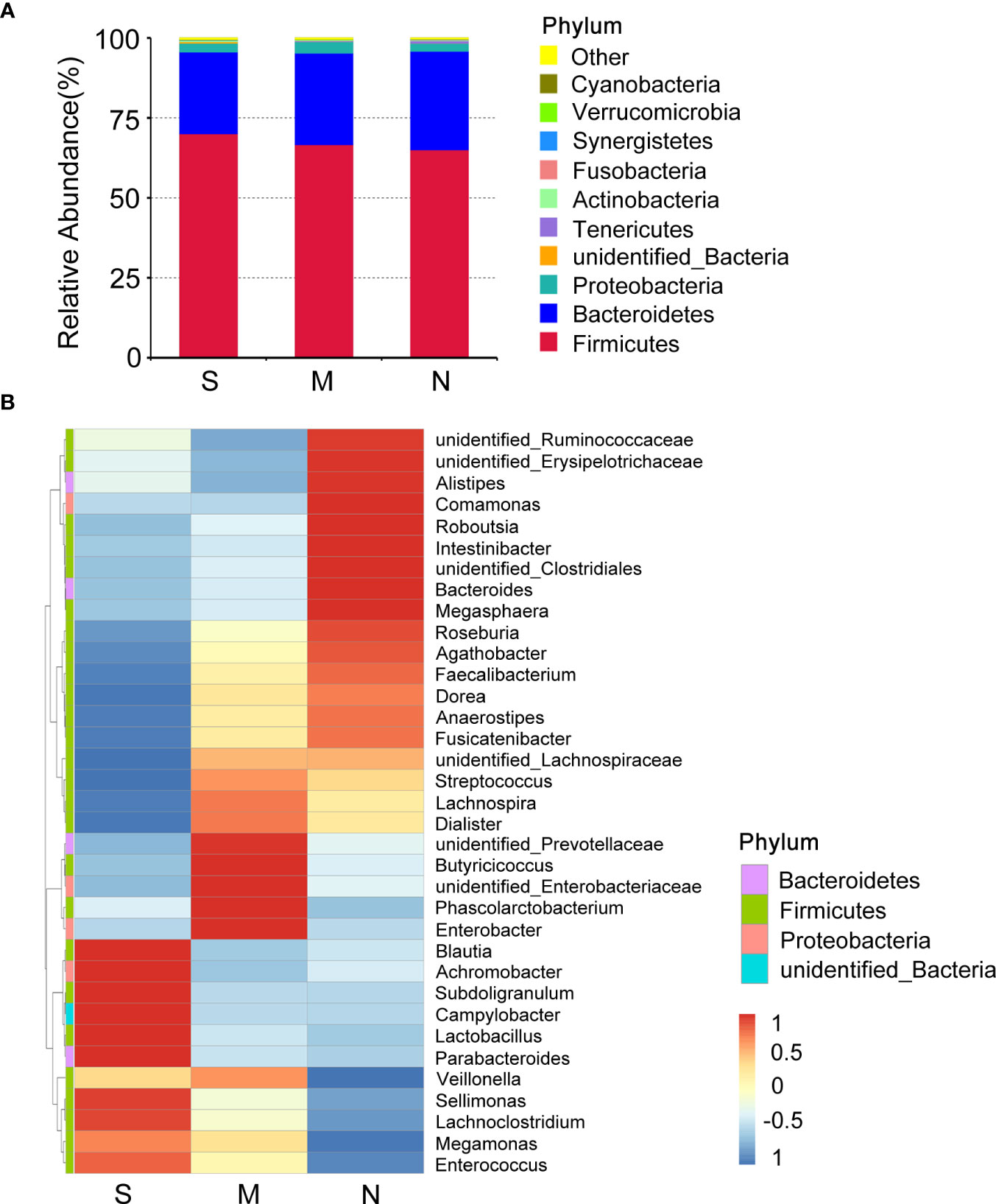
Figure 1 Composition differences in gut microbiota between coronavirus disease 2019 (COVID-19) patients and non-pneumonia individuals. (A) Top 10 phyla in stool samples from patients with severe and moderate COVID-19 and non-pneumonia individuals. The dominant phyla were Firmicutes and Bacteroidetes in all groups. The x-axis indicates group names (S: severe; M, moderate; N, non-pneumonia individuals). The y-axis indicates relative abundance. “Others” indicates the sum of the relative abundance of all phyla except the top 10. “unidentified_Bacteria” represents OTUs that could not be assigned at the phylum level. (B) Heatmap of the relative abundance of the top 35 most abundant genera among the three groups. The values corresponding to the heatmap are Z-values obtained by standardizing the relative abundance of the species in each row. The hierarchical taxon clustering on the left was based on the Euclidian distance. The left vertical bars correspond to the phylum level classification of each genus; the top 35 genera belonged to three phyla. Blautia is dominant in the severe groups.
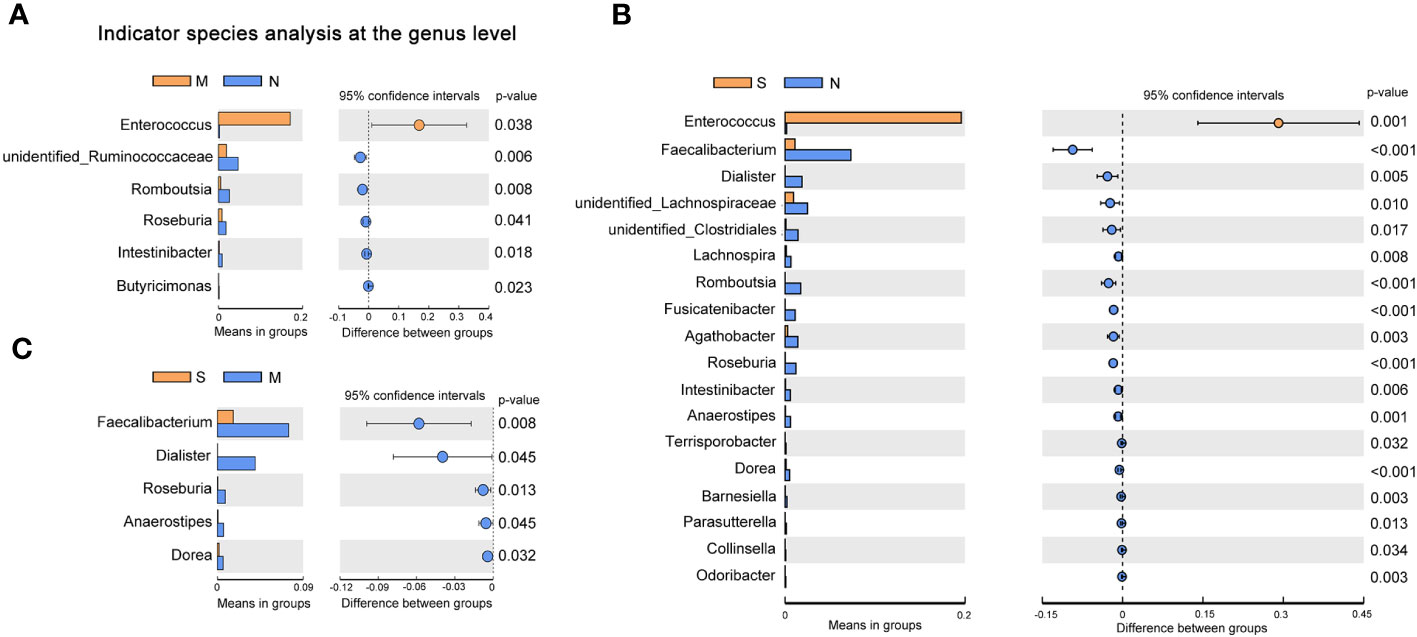
Figure 2 Changes of gut microbiota composition in the patients with coronavirus disease 2019 (COVID-19). Differentially bacterial species among the patients with severe, moderate COVID-19, and non-pneumonia individuals. operational taxonomic units (OTUs) and taxon differences are shown with p-values less than 0.05. (A) The differences between the moderate and non-pneumonia groups. (B) The differences between the severe and non-pneumonia groups. (C) The differences between the moderate and severe groups.
Next, a diversity analysis was performed to gain a deeper understanding of the species richness and microbiome composition of each group. It was found that compared with the non-pneumonia group, the alpha-diversity indices (ACE, Chao1, Shannon, and Simpson) were significantly reduced in the moderate and severe groups (p < 0.05). However, no significant difference was observed between the moderate and severe patients (Figures 3A–D). This indicated that the uniformity of bacterial community was lower in COVID-19 cases than in non-pneumonia cases but was similar in the moderate and severe groups. The rank abundance curve reflected the similarity in structure across all microbial communities, as seen by the same patterns observed in both the frequency and abundance of grades within the same group of samples, and the control group had higher species richness and uniformity than the COVID-19 groups (Figure 3E). To further demonstrate microbiome space among the three groups, the beta-diversity analysis was performed. PCoA was used to analyze two classic beta-diversity indices, and the separate clusters of the COVID-19 and control groups at the species level were confirmed. The PCoA plot, which was generated using unweighted UniFrac distances, revealed a significant difference between the non-pneumonia group and the other two groups, and PCo1 and PCo2 accounted for 13.6% and 12.99% of the total analysis results, respectively (Figure 4A). Similarly, weighted UniFrac distance-based bacterial structure analysis showed that the samples in the non-pneumonia group were discriminated against those in the moderate and severe groups. PCo1 and PCo2 explained 41.89% and 21.67% of the variation, respectively (Figure 4B). These results indicated different bacterial communities in the COVID-19 groups compared to the non-pneumonia group. The significant difference between the two indices (unweighted and weighted UniFrac distances) among the three groups was shown using the Wilcoxon rank-sum test (Figures 4C, D). According to the distance index ranking, ANOSIM provided confirmation that the distance between groups was considerably larger than the distance within groups, demonstrating a major difference in the microbiome structure across various groups (Supplementary Figures 4A–C).
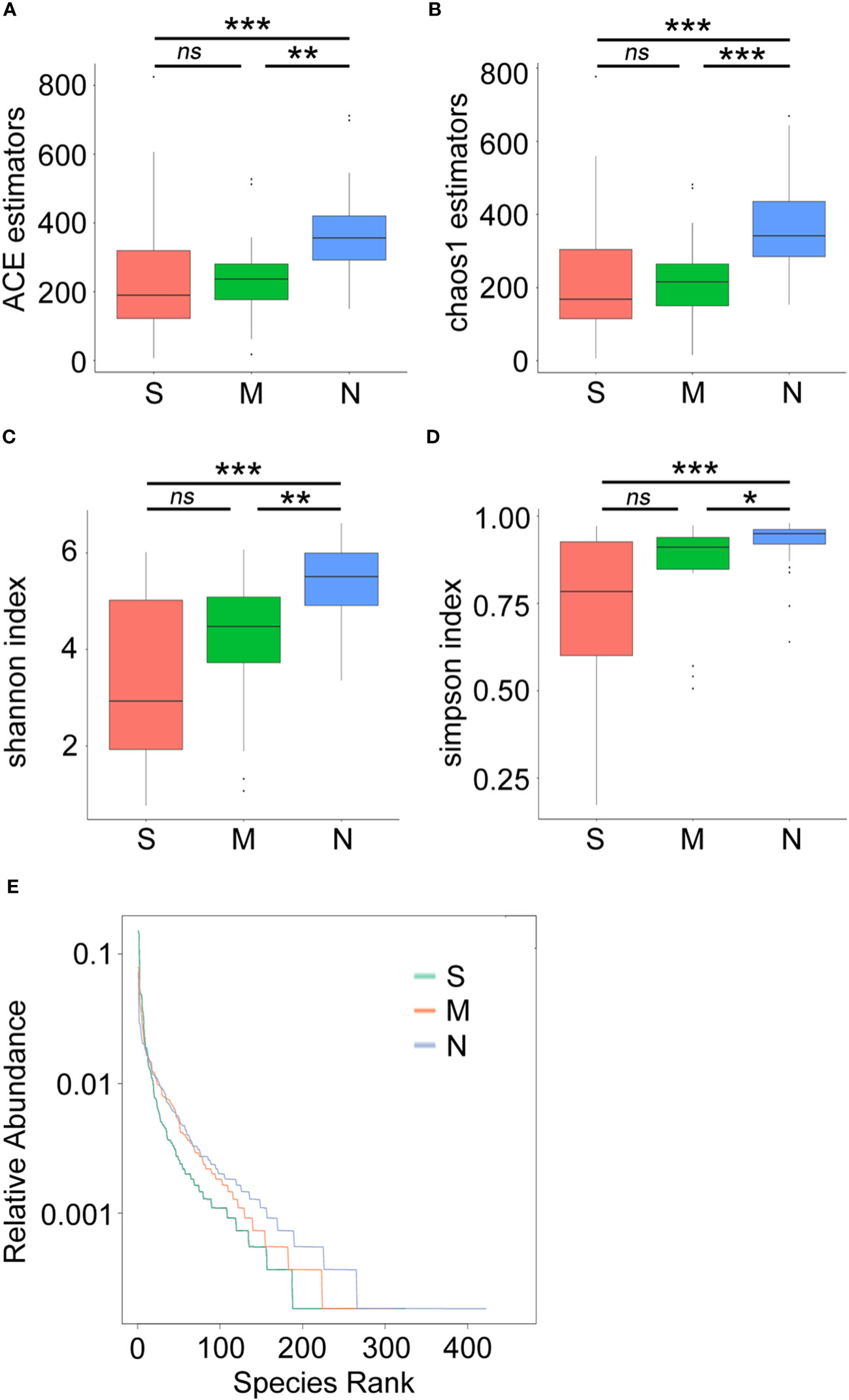
Figure 3 Alpha-diversity analysis of microbial community composition in stool samples from patients with coronavirus disease 2019 (COVID-19) and non-pneumonia individuals. The boxplots represent the minimum, maximum, and median of the sample values obtained from the richness estimators ACE (A) and Chao1 (B) and diversity indices Shannon (C) and Simpson (D). The rank abundance curve represents richness of bacterial community (E) *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
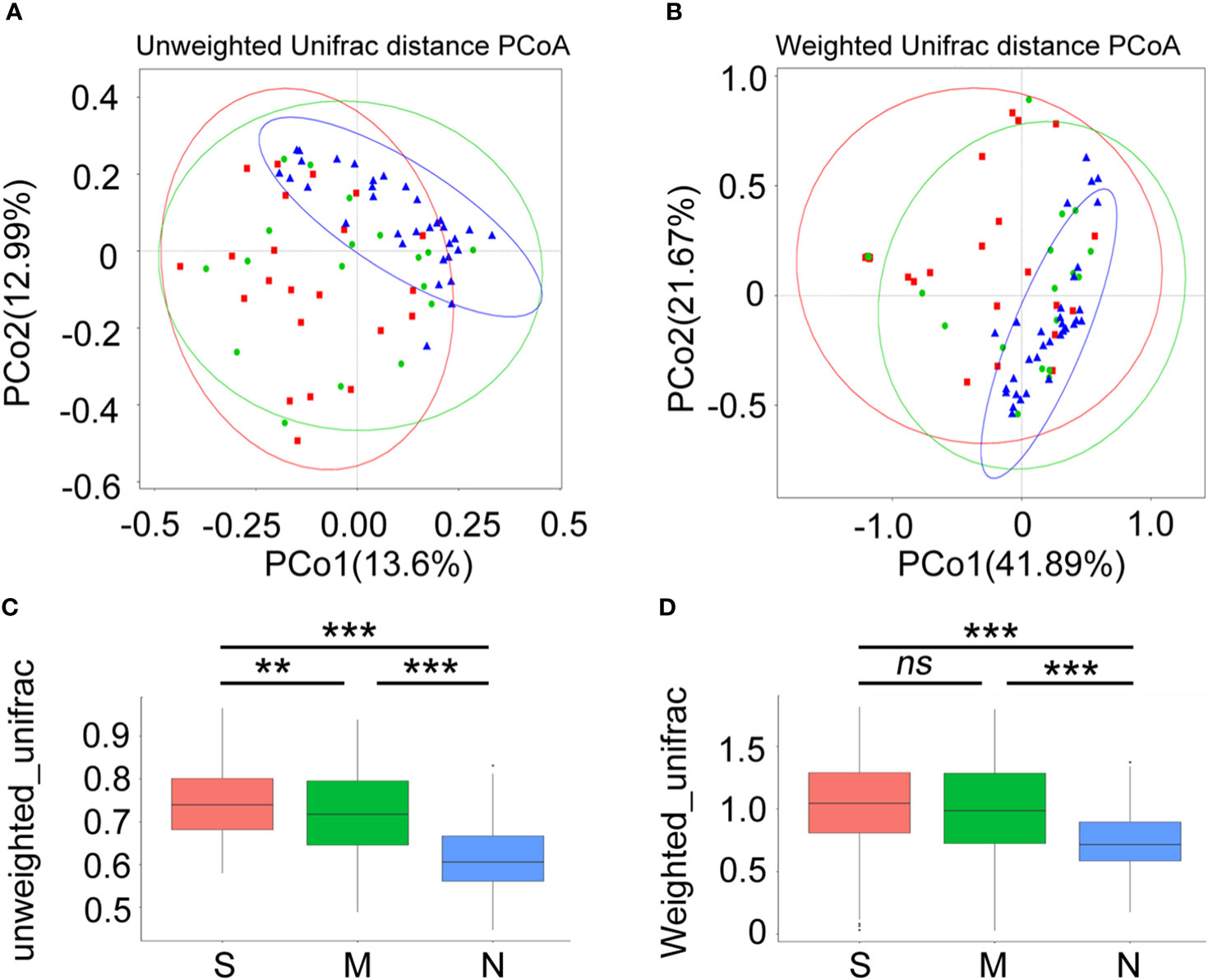
Figure 4 Beta-diversity analysis of gut microbiota composition of coronavirus disease 2019 (COVID-19) patients and non-pneumonia individuals. Principal coordinate analysis (PCoA) of gut microbiota based on unweighted UniFrac distance (A) and weighted UniFrac distance (B) in three groups. Blue dots refer to the non-pneumonia group. Red and blue dots refer to the severe and moderate groups, respectively. The ellipses represent the 68% confidence interval for each time point. The samples from COVID-19 patients and non-pneumonia individuals showed clustering distributions. (C, D) The boxplots show the extent of gut microbiota compositional changes among patients with severe, moderate COVID-19 and non-pneumonia individuals. **p < 0.01, ***p < 0.001, ns, not significant.
In order to identify bacterial taxa (at taxonomic levels) that were differentially abundant among the three groups, we used LEfSe analysis, at an LDA score >4. The results showed that the family Enterococcaceae, genus Enterococcus, and two species (Parabacteroides merdae and E. faecium) were abundant in the severe COVID-19 group, while the genus Dialister was enriched in the moderate group (Figure 5A). In the non-pneumonia group, three domain families (Lachnospiraceae, Peptostreptococcaceae, and Ruminococcaceae) and two genera (Faecalibacterium and Romboutsia) were detected. In addition, the cladogram analysis of taxa also showed a significant difference in gut microbiota among the three groups, and the severe group was characterized by taxa in the family Enterococcaceae (Figure 5B). Of note is that the two severe cases eventually progressed to death, and the abundance of Enterococcus and Lactobacillales accounted for 87% and 54% of their gut microbiota, respectively (Supplementary Figures 1C,D).
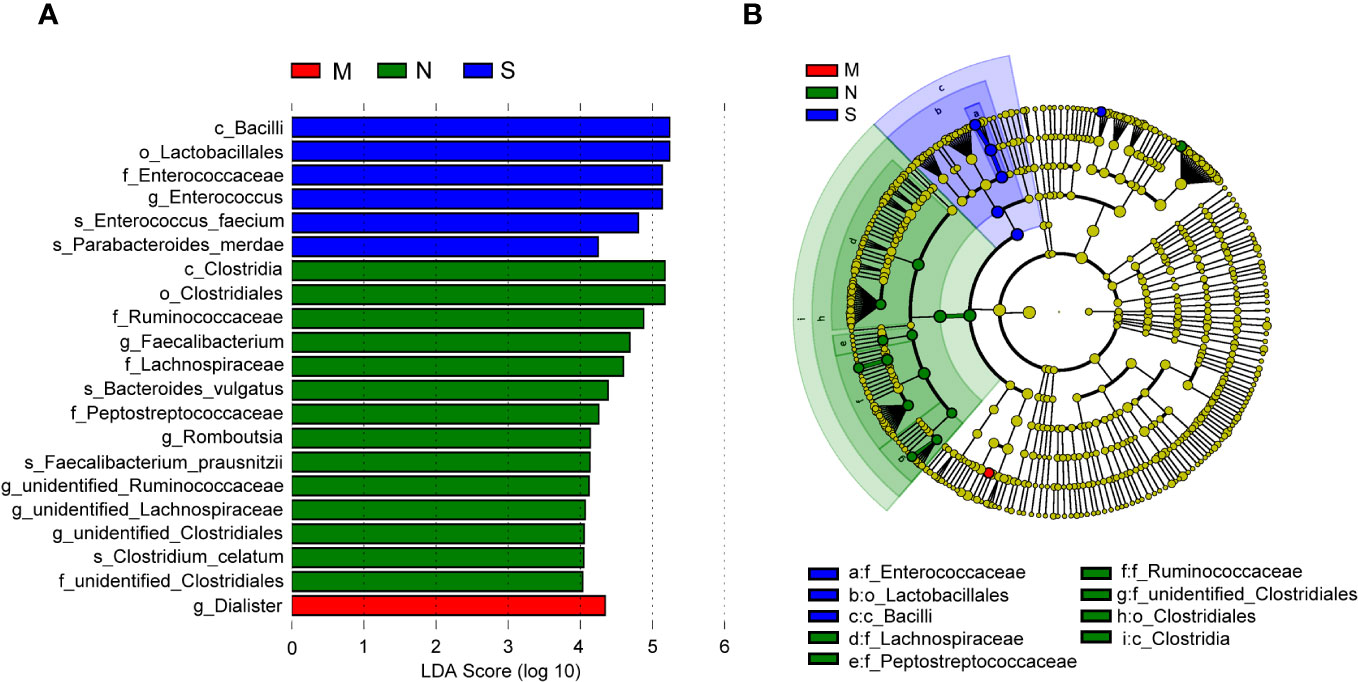
Figure 5 Linear discriminant analysis effect size (LEfSe) analysis of bacterial species. (A) The bar graph shows the linear discriminant analysis (LDA) scores calculated for characteristics at the operational taxonomic unit (OTU) levels. Blue and red bars refer to the severe and moderate groups, respectively. The red ones refer to the non-pneumonia group. (B) The cladogram shows the relative abundance of OTUs. Green and blue (light and dark green or blue) areas represent the non-pneumonia and severe groups, respectively. Green, blue, and red nodes in the branches represent bacterial species that played an important role in their groups. Yellow nodes represent bacterial species that did not play an important role in both groups.
To explore the interplay of the gut microbiota with certain specific blood biomarkers or clinical parameters that reflect the host immune response, we first investigated the distribution of bacterial tax in all COVID-19 patients. The results showed that at the phylum level, Firmicutes and Bacteroidetes were dominant communities (Supplementary Figure 5A). We further assessed the correlation between specific species enriched or depleted in COVID-19 patients and inflammatory indicators. The results are listed in Supplementary Table 1. At the species level, we found that Phascolarctobacterium faecium, Bacteroides coprocola, Lactobacillus reuteri, Blautia massiliensis, and Lactobacillus mucosae were positively correlated with lymphocyte counts or lymphocyte percentage, while E. faecium was negatively correlated with lymphocyte percentage, which suggested a potential supplementary function of these bacteria in the lymphoid reactions to SARS-CoV-2 infection. E. faecium, B. coprocola, and Coprococcus eutactus were positively correlated with neutrophil percentage or neutrophil counts, while Streptococcus salivarius was negatively correlated with neutrophil percentage (Figure 6), which suggested that these bacteria were closely involved in neutrophil-mediated inflammatory responses. Moreover, Eubacterium hallii and Enterococcus faecalis were positively correlated with CRP; L. reuteri, L. mucosae, and Lactobacillus fermentum were positively correlated with procalcitonin (PCT). Due to many COVID‐19 patients developing hypoalbuminemia at admission, we further analyzed the correlation between gut microbes and albumin. B. plebeius was found to be positively correlated with albumin, while Lactobacillus salivarius and Bacteroides fragilis were inversely related to albumin, indicating a potential connection between gut microbes and potential organ injury in COVID-19 patients. We used LEfSe analysis, at an LDA score >4. The results showed that the genus Faecalibacterium was abundant in the moderate COVID-19 group, while species E. faecium was enriched in the severe group (Supplementary Figure 5B). We also selected and analyzed some differentially abundant gut microbiota between two groups in LDA. At the genus level, Faecalibacterium was positively correlated with lymphocyte counts while negatively correlated with neutrophil percentage. We further evaluated the association with immunological indicators at the species level and found that E. faecium was positively correlated with neutrophil percentage while negatively correlated with lymphocyte percentage (Supplementary Figure 5C). Overall, these data suggested an association between gut microbiota composition and aberrant immune responses.
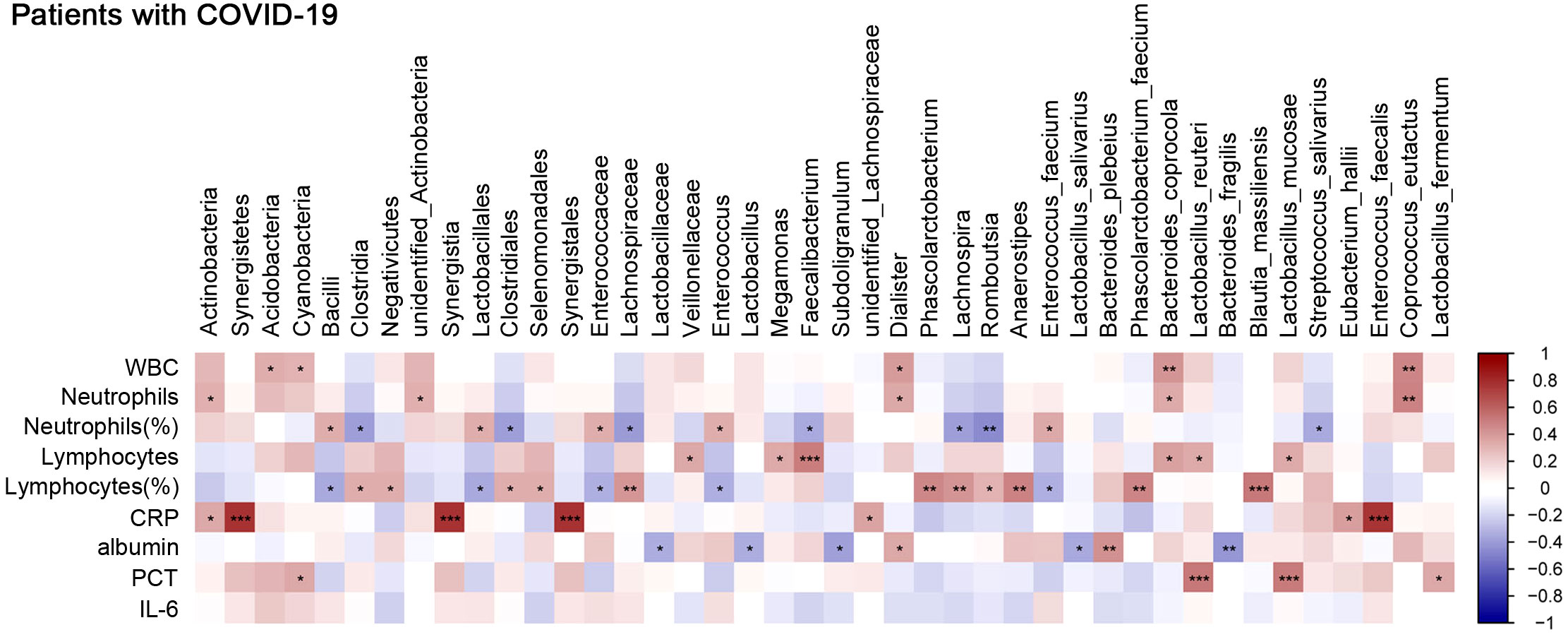
Figure 6 Correlation between gut microbes and clinical parameters that reflect host immune response. Analysis of the correlation between gut microbes and clinical traits. Red bars indicate positive associations, while blue bars indicate negative associations. The color key indicates the association strength and direction in terms of the t-test. Pearson’s correlation coefficient and p-value are used for plotting. Black asterisks indicate associations with p-value. *p < 0.05, **p < 0.01, and ***p < 0.001 represent significant differences.
Since late 2019, the SARS-CoV-2 strains, including the Delta and Omicron mutation types, have caused complicated multisystem diseases. In addition to different initial respiratory symptoms, gastrointestinal symptoms, including diarrhea, nausea, and vomiting, are common clinical manifestations. The ACE2 receptor modulates innate immunity and affects gut microbiota composition (Perlot and Penninger, 2013). SARS-CoV-2 infection is initiated in human intestinal epithelial cells by the binding of the viral spike protein to ACE2, thereby disrupting intestinal immune homeostasis in the host and resulting in the adverse consequences of COVID-19. Growing evidence suggests that the crosstalk between lung immunity and gut microbiota, which is called the gut–lung axis, is actively involved in the protective and pathologic immune responses to SARS-CoV-2 (de Oliveira et al., 2021; Rastogi et al., 2022). Therefore, changes in intestinal microbiota communities may significantly affect disease severity and progression as well as post-COVID-19 syndrome development (Mahooti et al., 2020; Ancona et al., 2023).
A previous study on the gut microbiota of hospitalized patients with COVID-19 revealed significant changes in microbiota communities, with an increased presence of opportunistic microorganisms and a decrease in beneficial microbes (Zuo et al., 2020). Studies have reported that after SARS-CoV-2 infection, the abundance of Faecalibacterium, Coprococcus, Ruminococcus, and Roseburia was decreased (Tao et al., 2020; Gaibani et al., 2021), whereas that of Enterococcus, Rothia, and Lactobacillus was increased (Tao et al., 2020; Wu et al., 2021). In the present study, we observed that the microbial relative abundance at the taxonomic level was associated with COVID-19 severity. Furthermore, bacterial diversity was decreased in the gut microbiota of patients with COVID-19, particularly in the severe group. Although we observed no significant differences in alpha-diversity between the moderate and severe groups, there were obvious changes in bacterial composition. This finding may be attributed to similar comorbidities or antibiotic usage between both cohorts. Moreover, we observed a consistent decrease in the alpha-diversity of the gut microbiota of patients with COVID-19 compared with that of the healthy population. This can be attributed to direct infection and the indirect effects of SARS-CoV-2. First, healthy gut microbiota may be vital for balancing the optimal immune responses against harmful excessive inflammatory responses. However, SARS-CoV-2 directly binds to ACE2 in intestinal epithelial cells and induces gut microbiota imbalance, further decreasing floral diversity in the intestine and the abundance of short-chain fatty acid-producing bacteria and increasing the abundance of opportunistic bacteria belonging to the family Enterobacteriaceae. Second, SARS-CoV-2 infection can affect the gut–lung axis and alter the functional and metabolic characteristics of the gut microbiota, further changing the abundance and diversity of the gut microbiota in the gastrointestinal tract (Zhang et al., 2023).
Notably, in the present study, the abundance of Enterococcus was increased in the severe group compared with the moderate and non-pneumonia groups. Enterococcus is a typical opportunistic pathogen. Some studies have revealed that enterococci may be associated with the development of severe illnesses. For example, Enterococcus may contribute to the development of cytokine storms in some patients with COVID-19 by activating Toll-like receptors (Gago et al., 2022; Righi et al., 2022). Furthermore, the excessive proliferation of Enterococcus leads to subsequent intestinal dysbiosis, intestinal epithelial barrier injury, and secondary infections (Bäumler and Sperandio, 2016; Hanada et al., 2018). E. faecium is an anaerobic opportunistic pathogen with extensive antibiotic resistance. It often causes severe healthcare-related infections, particularly in critically ill patients admitted to the intensive care unit (Fiore et al., 2019). In the present study, compared with the non-pneumonia group, the abundance of E. faecium was increased in the severe group. Moreover, there was a correlation between E. faecium and neutrophil or lymphocyte percentage in all patients with COVID-19. Collectively, these results suggest that Enterococcus has an obvious effect on distinguishing the COVID-19 and non-pneumonia groups and can be used as a potential candidate for diagnosing and predicting the severity of COVID-19.
Lactobacillus can produce lactic acid to regulate immunity and maintain intestinal barrier function (Hou et al., 2018; Wu et al., 2020). Tang et al. have reported that the abundance of Lactobacillus is below or close to the lower limit of the normal range in critically ill patients (Tang et al., 2020). Furthermore, another study has reported that hypoalbuminemia is associated with COVID-19 outcomes (Huang et al., 2020). This can be owing to increased capillary permeability resulting from a systemic inflammatory response, leading to albumin leakage into the interstitial space, poor nutritional status, and impaired liver function. In the present study, we observed that the relative abundance of Lactobacillus was dominant in severe patients and negatively correlated with albumin levels in patients with COVID-19; this suggests that Lactobacillus is associated with liver function maintenance. Furthermore, the abundance of L. reuteri, L. mucosae, and L. fermentum was positively correlated with the abnormal PCT concentration in the plasma of patients with COVID-19. The increase in Lactobacillus may exacerbate inflammatory responses against viral pneumonia and is positively correlated with disease severity. Therefore, additional studies are warranted to determine the dynamics of beneficial intestinal bacteria under the stress of viral infections.
Of note, in patients with severe COVID-19 who eventually died, we observed that the relative abundance of Lactobacillus and Enterococcus was dominant; furthermore, the percentage of neutrophils was higher and that of lymphocytes was lower. Lung lobe-based analysis revealed the typical imaging features of multiple ground-glass opacities with consolidation, which may be attributed to the ability of the intestinal microbiota to affect lung disease progression via the gut–lung axis. Many studies have suggested that gut dysbiosis plays a vital role in the pathogenesis of sepsis and acute respiratory distress syndrome (ARDS) (Chakraborty et al., 2022). The decrease in microbial diversity and accumulation of opportunistic microorganisms can lead to dysbiosis; this, in turn, may lead to immune dysregulation and even ARDS, similar to the manifestations observed in severe COVID-19 (Trompette et al., 2014; Anand and Mande, 2018). Therefore, additional studies on the relationship among inflammatory responses, gut microbiota, and COVID-19 severity are warranted.
Butyrate plays an important role in inhibiting the growth of opportunistic pathogens, maintaining the integration of the intestinal barrier, and activating or enhancing adaptive immune responses (Louis and Flint, 2009; Park et al., 2015). Studies have confirmed that the enrichment of the butyrate-producing genus Faecalibacterium is negatively correlated with disease severity (Li et al., 2021; Yeoh et al., 2021) and that the abundance of F. prausnitzii, which favors an anti-inflammatory microenvironment, is inversely correlated with COVID-19 severity (Zuo et al., 2020). Our study results are consistent with these findings. We observed that the abundance of Faecalibacterium, Roseburia, and Anaerostipes was decreased in patients with severe COVID-19. Of note, butyrate-producing bacteria such as Faecalibacterium, Anaerostipes, and Lachnospiraceae were positively correlated with several lymphoid-related markers, including lymphocyte count and lymphocyte percentage; this indicates a potential interaction between the gut microbiota and lymphocyte regulation. Indeed, previous studies have demonstrated that butyrate or butyrate-producing bacteria can modulate the quantity and function of regulatory T cells while also activating T helper cells (Park et al., 2015; Zhu et al., 2018). Therefore, the decrease in butyrate-producing bacteria may exacerbate inflammatory responses against viral pneumonia and exhibit a positive correlation with disease severity. We inferred that the changes in the abundance of Faecalibacterium can help accurately distinguish between critical and general patients and that microbial characterization and classification may be powerful tools to predict disease severity.
The present study has some limitations. First, we did not conduct metabolomic, transcriptomic, or proteomic studies or analyze their relationship with changes in the gut microbiota. Determining how gut bacteria affect host intestinal immune responses through microbial metabolites is vital. Second, several factors, including diet, may affect the human gut microbiota. However, data on lifestyle and diet were lacking; nevertheless, all patients had a similar diet, at least during hospitalization, and their meals were managed by the hospital. Third, stool samples were obtained from the patients with COVID-19 after antibiotic treatment and short-term hormone therapy; this may have affected the intestinal bacteria. Fortunately, all patients received the same class of antibiotics (moxifloxacin or third-generation cephalosporin). Therefore, species comparison between the moderate and severe groups was conducted under the same conditions.
Only one stool sample was collected from each patient within 24 h of admission, and the dynamic changes in gut microbiota were not elucidated. Incorporating these dynamic data of patients after discharge and expanding the clinical information of the non-pneumonia cohort may provide additional power to our study. Furthermore, future studies with larger sample sizes and more advanced techniques are warranted to confirm these findings and investigate the mechanisms underlying the observed associations.
In the present study, we used 16S rRNA sequencing to characterize the gut microbiome compositions. The gut microbial composition was significantly changed in COVID-19 patients, and the opportunistic pathogens were increased, yet the commensal bacteria were decreased. Furthermore, a correlation map between clinical hematological parameters and gut microbiota was obtained from COVID-19 patients, demonstrating that gut microbiota could have a function in regulating host immune responses and possibly impacting the severity and consequences of diseases. Still, more in-depth research is warranted to understand the immune-related pathogenesis of COVID-19 in the future.
The data presented in the study are deposited in the NCBI repository, accession number PRJNA1004732.
The studies involving humans were approved by Shandong Provincial Hospital Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
RF: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Software, Validation, Writing – original draft, Writing – review & editing. SL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing. NS: Conceptualization, Data curation, Project administration, Writing – review & editing. YY: Data curation, Methodology, Resources, Writing – review & editing. XD: Project administration, Validation, Writing – review & editing. BH: Data curation, Formal Analysis, Project administration, Writing – review & editing. CS: Conceptualization, Methodology, Writing – review & editing. CW: Data curation, Formal Analysis, Writing – review & editing. HL: Data curation, Project administration, Writing – original draft. DC: Investigation, Software, Supervision, Writing – review & editing. CH: Formal Analysis, Resources, Validation, Visualization, Writing – review & editing. PH: Formal Analysis, Project administration, Writing – review & editing. TZ: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Natural Science Foundation of China (No.82200014), Natural Science Foundation of Shandong Province (ZR2022QH216).
The authors thank Xuefeng Pan and Weimin Liu for their helpful discussions and technical support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1274690/full#supplementary-material
Anand, S., Mande, S. S. (2018). Diet, microbiota and gut-lung connection. Front. Microbiol. 9, 2147. doi: 10.3389/fmicb.2018.02147
Ancona, G., Alagna, L., Alteri, C., Palomba, E., Tonizzo, A., Pastena, A., et al. (2023). Gut and airway microbiota dysbiosis and their role in COVID-19 and long-COVID. Front. Immunol. 14. doi: 10.3389/fimmu.2023.1080043
Bäumler, A. J., Sperandio, V. (2016). Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535 (7610), 85–93. doi: 10.1038/nature18849
Cao, Y., Xu, X., Kitanovski, S., Song, L., Wang, J., Hao, P., et al. (2021). Comprehensive comparison of RNA-seq data of SARS-coV-2, SARS-coV and MERS-coV infections: alternative entry routes and innate immune responses. Front. Immunol. 12. doi: 10.3389/fimmu.2021.656433
Chakraborty, C., Sharma, A. R., Bhattacharya, M., Dhama, K., Lee, S. S. (2022). Altered gut microbiota patterns in COVID-19: Markers for inflammation and disease severity. World J. Gastroenterol. 28 (25), 2802–2822. doi: 10.3748/wjg.v28.i25.2802
de Oliveira, G. L. V., Oliveira, C. N. S., Pinzan, C. F., de Salis, L. V. V., Cardoso, C. R. B. (2021). Microbiota modulation of the gut-lung axis in COVID-19. Front. Immunol. 12. doi: 10.3389/fimmu.2021.635471
Dhar, D., Mohanty, A. (2020). Gut microbiota and Covid-19- possible link and implications. Virus Res. 285, 198018. doi: 10.1016/j.virusres.2020.198018
Donaldson, G. P., Lee, S. M., Mazmanian, S. K. (2016). Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14 (1), 20–32. doi: 10.1038/nrmicro3552
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 (10), 996–998. doi: 10.1038/nmeth.2604
Fiore, E., Van Tyne, D., Gilmore, M. S. (2019). Pathogenicity of enterococci. Microbiol. Spectr. 7 (4). doi: 10.1128/microbiolspec.GPP3-0053-2018
Gago, J., Filardo, T. D., Conderino, S., Magaziner, S. J., Dubrovskaya, Y., Inglima, K., et al. (2022). Pathogen species is associated with mortality in nosocomial bloodstream infection in patients with COVID-19. Open Forum Infect. Dis. 9 (6), ofac083. doi: 10.1093/ofid/ofac083
Gaibani, P., D'Amico, F., Bartoletti, M., Lombardo, D., Rampelli, S., Fornaro, G., et al. (2021). The gut microbiota of critically ill patients with COVID-19. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.670424
Hanada, S., Pirzadeh, M., Carver, K. Y., Deng, J. C. (2018). Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front. Immunol. 9, 2640. doi: 10.3389/fimmu.2018.02640
Hayashi, Y., Wagatsuma, K., Nojima, M., Yamakawa, T., Ichimiya, T., Yokoyama, Y., et al. (2021). The characteristics of gastrointestinal symptoms in patients with severe COVID-19: a systematic review and meta-analysis. J. Gastroenterol. 56 (5), 409–420. doi: 10.1007/s00535-021-01778-z
Hazan, S., Stollman, N., Bozkurt, H. S., Dave, S., Papoutsis, A. J., Daniels, J., et al. (2022). Lost microbes of COVID-19: Bifidobacterium, Faecalibacterium depletion and decreased microbiome diversity associated with SARS-CoV-2 infection severity. BMJ Open Gastroenterol. 9 (1), e000871. doi: 10.1136/bmjgast-2022-000871
Hou, Q., Ye, L., Liu, H., Huang, L., Yang, Q., Turner, J. R., et al. (2018). Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 25 (9), 1657–1670. doi: 10.1038/s41418-018-0070-2
Huang, J., Cheng, A., Kumar, R., Fang, Y., Chen, G., Zhu, Y., et al. (2020). Hypoalbuminemia predicts the outcome of COVID-19 independent of age and co-morbidity. J. Med. Virol. 92 (10), 2152–2158. doi: 10.1002/jmv.26003
Lamers, M. M., Beumer, J., van der Vaart, J., Knoops, K., Puschhof, J., Breugem, T. I., et al. (2020). SARS-CoV-2 productively infects human gut enterocytes. Science 369 (6499), 50–54. doi: 10.1126/science.abc1669
Li, S., Yang, S., Zhou, Y., Disoma, C., Dong, Z., Du, A., et al. (2021). Microbiome profiling using shotgun metagenomic sequencing identified unique microorganisms in COVID-19 patients with altered gut microbiota. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.712081
Libertucci, J., Young, V. B. (2019). The role of the microbiota in infectious diseases. Nat. Microbiol. 4 (1), 35–45. doi: 10.1038/s41564-018-0278-4
Louis, P., Flint, H. J. (2009). Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 294 (1), 1–8. doi: 10.1111/j.1574-6968.2009.01514.x
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. Isme. J. 5 (2), 169–172. doi: 10.1038/ismej.2010.133
Lu, H. F., Li, A., Zhang, T., Ren, Z. G., He, K. X., Zhang, H., et al. (2017). Disordered oropharyngeal microbial communities in H7N9 patients with or without secondary bacterial lung infection. Emerg. Microbes Infect. 6 (12), e112. doi: 10.1038/emi.2017.101
Lucas, C., Wong, P., Klein, J., Castro, T. B. R., Silva, J., Sundaram, M., et al. (2020). Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584 (7821), 463–469. doi: 10.1038/s41586-020-2588-y
Mahooti, M., Miri, S. M., Abdolalipour, E., Ghaemi, A. (2020). The immunomodulatory effects of probiotics on respiratory viral infections: A hint for COVID-19 treatment? Microb. Pathog. 148, 104452. doi: 10.1016/j.micpath.2020.104452
Merola, E., Armelao, F., de Pretis, G. (2020). Prevalence of gastrointestinal symptoms in coronavirus disease 2019: a meta-analysis. Acta Gastroenterol. Belg. 83 (4), 603–615.
Nasrollahi, H., Talepoor, A. G., Saleh, Z., Eshkevar Vakili, M., Heydarinezhad, P., Karami, N., et al. (2023). Immune responses in mildly versus critically ill COVID-19 patients. Front. Immunol. 14. doi: 10.3389/fimmu.2023.1077236
Park, J., Kim, M., Kang, S. G., Jannasch, A. H., Cooper, B., Patterson, J., et al. (2015). Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol. 8 (1), 80–93. doi: 10.1038/mi.2014.44
Perlot, T., Penninger, J. M. (2013). ACE2 - from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect. 15 (13), 866–873. doi: 10.1016/j.micinf.2013.08.003
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi: 10.1093/nar/gks1219
Rastogi, S., Mohanty, S., Sharma, S., Tripathi, P. (2022). Possible role of gut microbes and host's immune response in gut-lung homeostasis. Front. Immunol. 13. doi: 10.3389/fimmu.2022.954339
Reinold, J., Farahpour, F., Fehring, C., Dolff, S., Konik, M., Korth, J., et al. (2021). A pro-inflammatory gut microbiome characterizes SARS-coV-2 infected patients and a reduction in the connectivity of an anti-inflammatory bacterial network associates with severe COVID-19. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.747816
Righi, E., Lambertenghi, L., Gorska, A., Sciammarella, C., Ivaldi, F., Mirandola, M., et al. (2022). Impact of COVID-19 and antibiotic treatments on gut microbiome: A role for enterococcus spp. Biomedicines 10 (11). doi: 10.3390/biomedicines10112786
Schirmer, M., Smeekens, S. P., Vlamakis, H., Jaeger, M., Oosting, M., Franzosa, E. A., et al. (2016). Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 167 (4), 1125–1136.e1128. doi: 10.1016/j.cell.2016.10.020
Schmulson, M., Dávalos, M. F., Berumen, J. (2020). Beware: Gastrointestinal symptoms can be a manifestation of COVID-19. Rev. Gastroenterol. Mex. (Engl. Ed). 85 (3), 282–287. doi: 10.1016/j.rgmx.2020.04.001
Schult, D., Reitmeier, S., Koyumdzhieva, P., Lahmer, T., Middelhoff, M., Erber, J., et al. (2022). Gut bacterial dysbiosis and instability is associated with the onset of complications and mortality in COVID-19. Gut. Microbes 14 (1), 2031840. doi: 10.1080/19490976.2022.2031840
Schultze, J. L., Aschenbrenner, A. C. (2021). COVID-19 and the human innate immune system. Cell 184 (7), 1671–1692. doi: 10.1016/j.cell.2021.02.029
Sette, A., Crotty, S. (2021). Adaptive immunity to SARS-coV-2 and COVID-19. Cell 184 (4), 861–880. doi: 10.1016/j.cell.2021.01.007
Tang, L., Gu, S., Gong, Y., Li, B., Lu, H., Li, Q., et al. (2020). Clinical significance of the correlation between changes in the major intestinal bacteria species and COVID-19 severity. Eng. (Beijing). 6 (10), 1178–1184. doi: 10.1016/j.eng.2020.05.013
Tao, W., Zhang, G., Wang, X., Guo, M., Zeng, W., Xu, Z., et al. (2020). Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med. Microecol. 5, 100023. doi: 10.1016/j.medmic.2020.100023
Trompette, A., Gollwitzer, E. S., Yadava, K., Sichelstiel, A. K., Sprenger, N., Ngom-Bru, C., et al. (2014). Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 20 (2), 159–166. doi: 10.1038/nm.3444
Wilk, A. J., Rustagi, A., Zhao, N. Q., Roque, J., Martínez-Colón, G. J., McKechnie, J. L., et al. (2020). A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26 (7), 1070–1076. doi: 10.1038/s41591-020-0944-y
Wu, Y., Cheng, X., Jiang, G., Tang, H., Ming, S., Tang, L., et al. (2021). Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms. Microbiomes. 7 (1), 61. doi: 10.1038/s41522-021-00232-5
Wu, H., Xie, S., Miao, J., Li, Y., Wang, Z., Wang, M., et al. (2020). Lactobacillus reuteri maintains intestinal epithelial regeneration and repairs damaged intestinal mucosa. Gut. Microbes 11 (4), 997–1014. doi: 10.1080/19490976.2020.1734423
Yeoh, Y. K., Zuo, T., Lui, G. C., Zhang, F., Liu, Q., Li, A. Y., et al. (2021). Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 70 (4), 698–706. doi: 10.1136/gutjnl-2020-323020
Zhang, J. J., Cao, Y. Y., Tan, G., Dong, X., Wang, B. C., Lin, J., et al. (2021). Clinical, radiological, and laboratory characteristics and risk factors for severity and mortality of 289 hospitalized COVID-19 patients. Allergy 76 (2), 533–550. doi: 10.1111/all.14496
Zhang, F., Lau, R. I., Liu, Q., Su, Q., Chan, F. K. L., Ng, S. C. (2023). Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 20 (5), 323–337. doi: 10.1038/s41575-022-00698-4
Zhu, C., Song, K., Shen, Z., Quan, Y., Tan, B., Luo, W., et al. (2018). Roseburia intestinalis inhibits interleukin−17 excretion and promotes regulatory T cells differentiation in colitis. Mol. Med. Rep. 17 (6), 7567–7574. doi: 10.3892/mmr.2018.8833
Zuo, T., Wu, X., Wen, W., Lan, P. (2021). Gut microbiome alterations in COVID-19. Genomics Proteomics Bioinf. 19 (5), 679–688. doi: 10.1016/j.gpb.2021.09.004
Keywords: gut microbiota, COVID-19, host immune responses, disease severity, opportunistic pathogens
Citation: Fan R, Liu S, Sun N, Yang Y, Deng X, Hu B, Sun C, Wen C, Li H, Cheng D, Huang C, Hou P and Zhang T (2023) Gut microbiota composition is associated with disease severity and host immune responses in COVID-19. Front. Cell. Infect. Microbiol. 13:1274690. doi: 10.3389/fcimb.2023.1274690
Received: 08 August 2023; Accepted: 21 November 2023;
Published: 12 December 2023.
Edited by:
Kundlik Gadhave, Johns Hopkins University, United StatesReviewed by:
Manish Kumar, University of California, San Diego, United StatesCopyright © 2023 Fan, Liu, Sun, Yang, Deng, Hu, Sun, Wen, Li, Cheng, Huang, Hou and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuai Liu, bGl1c2h1YWlfc2FtQDE2My5jb20=; Peibin Hou, aG91cGVpYmluQDE2My5jb20=; Tianliang Zhang, c2R6dGxAdG9tLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.