
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 14 November 2023
Sec. Molecular Bacterial Pathogenesis
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1274573
This article is part of the Research Topic Epidemiology of Antimicrobial Resistance and Virulence Factors of Emerging and Re-emerging bacteria View all 11 articles
 Claudio Neidhöfer1,2*†
Claudio Neidhöfer1,2*† Marcel Neuenhoff3†
Marcel Neuenhoff3† Robert Jozič1
Robert Jozič1 Brenda Atangcho1,4Sandra Unsleber5Ulrike Neder5
Brenda Atangcho1,4Sandra Unsleber5Ulrike Neder5 Silke Grumaz5
Silke Grumaz5 Marijo Parčina1
Marijo Parčina1Background: Bloodstream infections (BSIs) remain a significant cause of mortality worldwide. Causative pathogens are routinely identified and susceptibility tested but only very rarely investigated for their resistance genes, virulence factors, and clonality. Our aim was to gain insight into the clonality patterns of different species causing BSI and the clinical relevance of distinct virulence genes.
Methods: For this study, we whole-genome-sequenced over 400 randomly selected important pathogens isolated from blood cultures in our diagnostic department between 2016 and 2021. Genomic data on virulence factors, resistance genes, and clonality were cross-linked with in-vitro data and demographic and clinical information.
Results: The investigation yielded extensive and informative data on the distribution of genes implicated in BSI as well as on the clonality of isolates across various species.
Conclusion: Associations between survival outcomes and the presence of specific genes must be interpreted with caution, and conducting replication studies with larger sample sizes for each species appears mandatory. Likewise, a deeper knowledge of virulence and host factors will aid in the interpretation of results and might lead to more targeted therapeutic and preventive measures. Monitoring transmission dynamics more efficiently holds promise to serve as a valuable tool in preventing in particular BSI caused by nosocomial pathogens.
Bloodstream infections (BSIs) pose a substantial global health threat, leading to increased morbidity and mortality rates (McNamara et al., 2018). While identifying the causative pathogen and its antibiotic susceptibility remains a clinical priority, exploring additional factors such as genetic relatedness, virulence genes, and antibiotic resistance genes can significantly enhance patient outcomes and alleviate the overall burden (Wren, 2000; Leavis et al., 2006; Wyres et al., 2020; Allen et al., 2021). Whole-genome sequencing (WGS) studies have revolutionized our understanding of pathogen identification, antibiotic resistance, and epidemiology (Wren, 2000). Insights into clonality patterns can guide interventions to curtail the dissemination of specific strains and inform targeted prevention strategies (Wren, 2000; Leavis et al., 2006; Wyres et al., 2020). Furthermore, studying pathogen clonality provides valuable insights into the evolutionary dynamics of BSI pathogens, facilitating the prediction of future trends in antibiotic resistance and virulence (Wren, 2000; Allen et al., 2021).
Virulence factors play a crucial role in the colonization, invasion, and evasion of the host immune system by pathogens. Understanding the specific virulence factors associated with BSI-causing bacteria provides insights into disease mechanisms, severity, and potential therapeutic targets (Wren, 2000; L Thomas and Lee, 2012; Wyres et al., 2020). In-depth investigations of bacterial virulence factors associated with BSI have yielded significant findings concerning pathogenesis and host–pathogen interactions (Wren, 2000; Leavis et al., 2006; L Thomas and Lee, 2012; Wyres et al., 2020). Studying resistance genes and correlating them with phenotypical susceptibility are of paramount importance for addressing key research questions in the field of antimicrobial resistance leading to a comprehensive understanding of the interplay between genotype and phenotype (Wren, 2000; L Thomas and Lee, 2012; Mahfouz et al., 2020; Allen et al., 2021). It enables the validation and verification of resistance mechanisms and provides a more accurate assessment of the clinical implications of specific genetic variants.
In this study, we conducted a comprehensive whole-genome sequencing of over 400 randomly selected common BSI-causing pathogens to elucidate clonality patterns and assess the clinical significance of virulence and resistance genes, including their association with mortality. Our study findings advance the understanding of BSI pathogenesis and hold implications for more targeted therapeutic interventions.
For this study, over 400 bacterial isolates of common pathogens detected in blood cultures between January 2019 and December 2021 in our microbiological diagnostic unit, which services a tertiary referral and maximum care hospital and other hospitals in the area, were randomly selected for the genera Acinetobacter, Bacteroides, Citrobacter, Enterobacter, and Serratia and the species Enterococcus faecalis, Enterococcus faecium, Escherichia coli, Klebsiella pneumonia, Proteus mirabilis, Pseudomonas aeruginosa, Staphylococcus aureus, Stenotrophomonas maltophilia, Streptococcus pneumoniae, and Streptococcus pyogenes from cryo-storage. These were thawed, subcultured twice on Columbia 5% sheep blood agar (Becton Dickinson, Heidelberg, Germany), and inspected by two experienced operators prior to sequencing. Isolate information was complemented by accessible laboratory information on phenotypical susceptibility, growth of additional bacteria in the same blood culture, and routine diagnostic resistance gene detection, as well as accessible patient information on age, sex, hospitalization, 30-day mortality, and outcome. We constructed a database that was password-protected and accessible by only three operators who ensured that all patient data were fully de-identified prior to analysis. Unless specifically mentioned or reported, the minimum inhibitory concentrations (MICs) were determined by the VITEK 2 system (bioMérieux, Marcy-l’Etoile, France). Isolate susceptibility was inferred based on EUCAST Cl. Br. Tables v. 13.0.
DNA isolation, library preparation, sequencing, and sequence assembly were carried out by Noscendo GmbH, Germany. Genomic DNA was prepared from pellets obtained from 5 ml of culture in a brain heart infusion (BHI) medium (Becton Dickinson, Heidelberg, Germany). Cell pellets were prepared, shipped on dry ice, and stored at −80°C until further processing. DNA was isolated with the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions with a Vortex Genie 2 device equipped with an SI-H524 horizontal tube holder (Scientific Industries, Bohemia, NY, USA) to perform mechanical cell disruption for 10 min. DNA concentration was measured with the Qubit 1X dsDNA Assay-Kit on a Qubit 2.0 instrument (Thermo Fisher Scientific, Waltham, MA, USA), and size distribution was checked with the Agilent Genomic DNA 50 kb Kit on a 5200 Fragment Analyzer System (Agilent Technologies, Santa Clara, CA, USA). The libraries were prepared using the Ligation Sequencing Kit SQK-LSK109 with Native Barcoding Expansion 1–12 (PCR-free) EXP-NBD104 and Native Barcoding Expansion 13–24 (PCR-free) EXP-NBD114 according to the manufacturer’s instructions (Oxford Nanopore Technologies, Oxford, UK) together with NEB Blunt/TA Ligase Master Mix and NEBNext Companion Module for Oxford Nanopore Technologies Ligation Sequencing (New England Biolabs, MA, USA). Then, the libraries were prepared, pooled in equimolar ratio, quality checked, loaded on a MinION Flow Cell (R9.4.1), and finally sequenced on a MinION benchtop sequencer (Mk1B). For each batch preparation, a pellet of a 1.5-ml overnight culture (BHI medium) of DSM 1576–Escherichia coli was used as an internal quality control during the whole process, starting with DNA isolation.
After sequencing, fast5 data were basecalled using the Oxford Nanopore Technologies neural-network-based basecalling software Guppy (version 5.0.7) applying a high accuracy mode (config dna_r9.4.1_450bps_hac.cfg). Fastq statistics on read length distributions, expected coverages, and N50 values were calculated. Following closely the manual of the tool Trycycler (Wick et al., 2021) (https://github.com/rrwick/Trycycler, version 0.4.2), long-read consensus assemblies were produced from multiple input assemblies of the same input data set by using the following assemblers: Flye (https://github.com/fenderglass/Flye, version 2.8.3), Miniasm and Minipolish (https://github.com/rrwick/Minipolish, version 0.1.3), and Raven (https://github.com/lbcb-sci/raven, version 1.4.0). Before assembly, reads below a length of 1,000 bp were removed using the tool Filtlong (https://github.com/rrwick/Filtlong, version 0.2.0), at most removing 5% of the original data set. Nine subsamples were created, providing a minimum read depth of 100×. In cases in which average coverages were below 100×, the minimum read depth after subsampling was lowered to 50×. Subsampled data sets were then forwarded to the assemblers, producing nine assemblies in total, three of every assembler. The Trycycler then clustered similar contigs to detect spurious, incomplete, or misassembled contigs. After manual inspection, the conspicuous clusters were removed. The remaining clusters were further processed to result in circular chromosomes and plasmids. Consensus sequences were polished using Medaka (https://github.com/nanoporetech/medaka, version 1.4.3) to increase base accuracy and minimize assembly and sequencing errors.
Assembly quality was finally evaluated by Busco (https://busco.ezlab.org/, https://gitlab.com/ezlab/busco/-/releases/5.2.1, version 5.2.1). Furthermore, reads were mapped to the consensus sequence by using Minimap2 (https://github.com/lh3/minimap2, version 2.18) and Flye-samtools to get coverage values of every consensus base. Assemblies having a Busco completeness value above 95% and every consensus base covered with at least 5× coverage were regarded as high quality. All isolates with valid results and sufficient coverage were included in the analysis.
Detection of antimicrobial resistance (AMR) genes, stress response genes, and virulence factors was detected with AMRFinderPlus (3.11.4) “–plus” option/NCBI reference gene database (2023-02-23.1) (Feldgarden et al., 2021; Feldgarden et al., 2022). If species were present in the list of curated organisms, the “–organism” option was used to ignore universal species mutations and resistance genes and to screen for known point mutations. Only results with identity and coverage >90% were included. For further analysis, assemblies were imported into bactopia (Shen, ; Ewels et al., 2020; Petit and Read, 2020). FastANI (v1.33) was used to calculate the average nucleotide identity within species (Jain et al., 2018). MLST types were determined with the PubMLST database using MLST (2.23.0, https://github.com/tseemann/mlst) (Jolley et al., 2018). For Enterobacterales, plasmids were detected using plasmidFinder (2.1.6) (Camacho et al., 2009; Carattoli et al., 2014).
Strain-specific tools were used to obtain more in-depth information: E. coli—ECTyper (1.0.0) (Iguchi et al., 2015), K. pneumoniae—Kleborate (2.3.2) (Lam et al., 2021), P. aeruginosa—pasty (1.0.2, https://github.com/rpetit3/pasty) (Camacho et al., 2009; Thrane et al., 2016), and S. pneumoniae—Seroba (1.0.2) (Epping et al., 2018).
Phylogenetic trees and ANI values in supplementary data were created with Anvi’o v7.1 (Eren et al., 2021) using “anvi’o pangenomics workflow” (Edgar, 2004; Hyatt et al., 2010; Buchfink et al., 2015; Pritchard et al., 2016; Chan and Lowe, 2019). All detected resistance genes, stress response genes, and virulence factors for each species can be found in the Supplementary Material. (Sequence names are mentioned yellow for findings with shared gene symbol but multiple possible alleles. These were merged for our purposes if the genes belong to the same subclass.)
All data relevant to the study are included in the article or uploaded as supplementary information. Due to collaboration agreements, genomes have not been uploaded to any publicly accessible platform but can be shared upon reasonable request.
The Ethics Committee of the University Hospital Bonn confirmed that no ethics approval was required for this study.
The 364 isolates for which genomes were available with satisfactory coverage and quality belonged to 364 different patients of which the majority were men (60.16%). Detailed information on patients and isolates is listed in Table 1. In 55 cases (15.11%), the pathogen under study was not the only one isolated from the respective blood culture. In 7, there were 2 additional pathogens, and in another three, 3. Fourteen isolates were methicillin-resistant S. aureus (MRSA), 10 were vancomycin-resistant enterococci (VRE), and 26 were gram-negative rods falling into the German guideline classification of multidrug-resistant gram-negative rods on the basis of resistance against three (3MRGN) or four (4MRGN) of the following antibiotic groups: acylureidopenicillins, third- and fourth-generation cephalosporins, carbapenems, and fluoroquinolones.
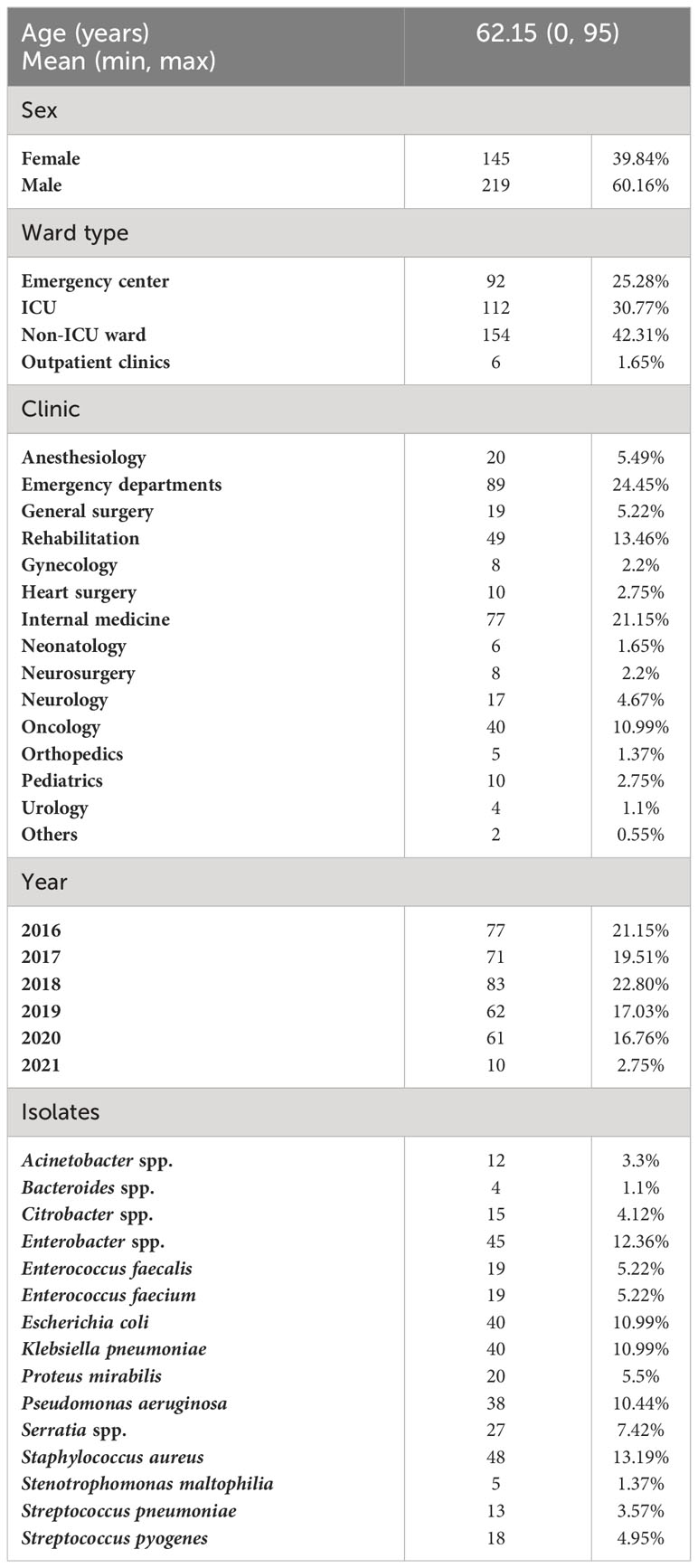
Table 1 Patient and isolate information.
The most frequently detected AMR genes were fosA (107), oqxA (73), and emrD (70). These were, however, only detected in Enterobacterales and P. aeruginosa that made up a large part of all isolates. An overview of detected virulence, stress response, and AMR genes can be found in the Supplementary Material. Figure 1 displays to what degree the average nucleotide identity could vary across different pathogens and the number of virulence genes, AMR genes, stress response genes, and plasmids that different pathogens carried. The 30-day and 90-day mortality did not correlate with the number of resistance, AMR, or stress response genes but with patient age (rpb = 0.22, n = 248, p = 0.001 and rpb = 0.25, n = 201, p = <0.001, respectively).
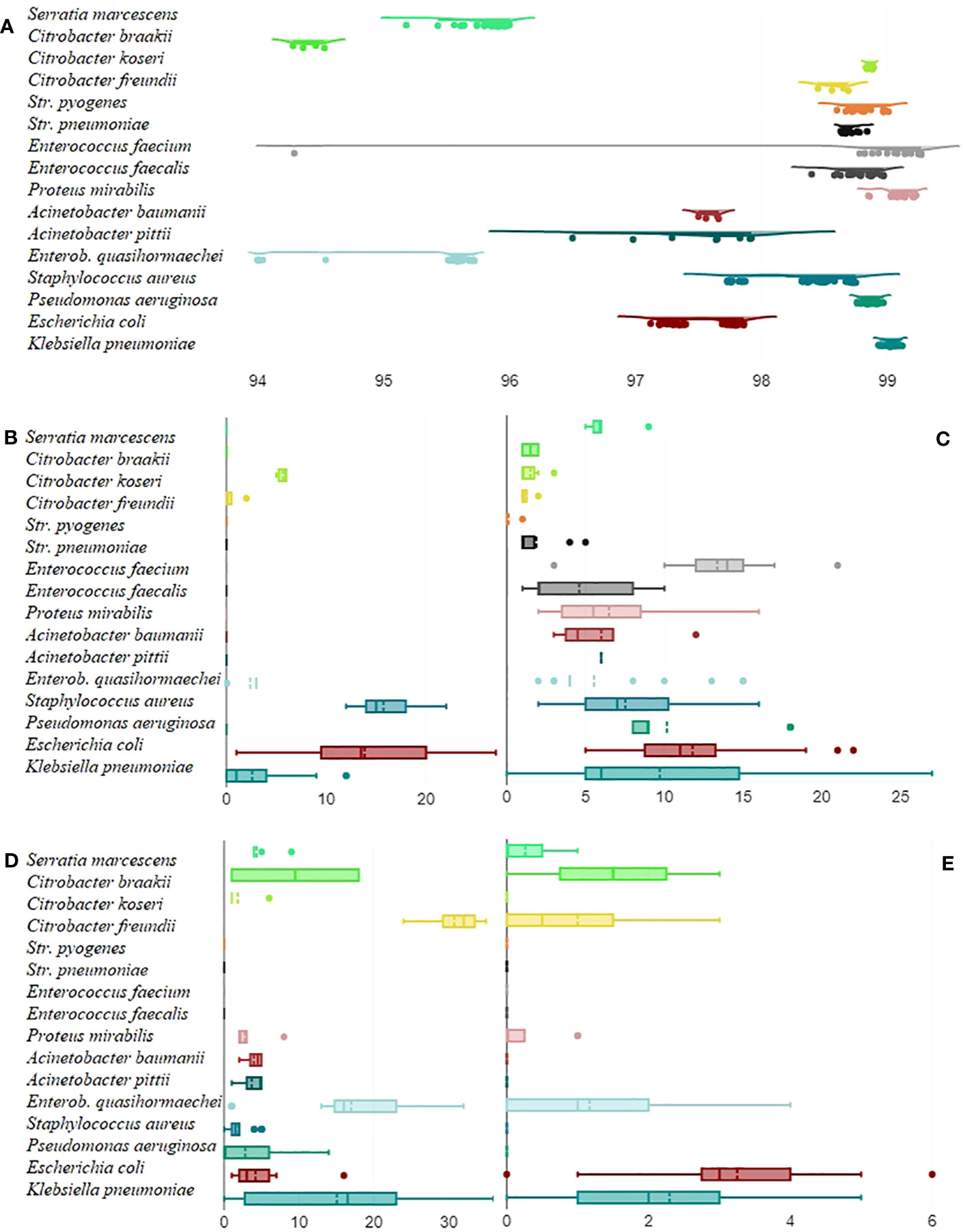
Figure 1 Mean average nucleotide identity by pathogen. For each isolate, the average nucleotide identity it had to all the other isolates within the same group in percent (x-axis) was calculated and displayed as a raincloud plot (A). Boxplots on the number (x-axis) of virulence genes (B), AMR genes (C), stress response genes (D), and plasmids (E) that pathogens carried.
The majority of the isolates originated from patients in intensive care units (ICUs) with eight cases, followed by the emergency center (EC) with four cases. None of the isolates were identified as VRE either phenotypically or genotypically. There were 11 unique multilocus sequence typing (MLST) types represented once and four types represented twice. The isolates showed an average nucleotide identity of 98.78% to each other, ranging from 98.40% to 98.97%. Notably, patients infected with E. faecalis isolates encoding dfrF, gyrA_S83I, and parC_S80I exhibited significantly lower 30-day (all p = 0.015) and 90-day survival rates (all p = 0.047), although the assumptions for the χ2 test were not met due to low cell frequencies. Out of all the isolates, only two showed higher MICs for trimethoprim/sulfamethoxazole than the threshold of ≤10, and both of these isolates tested positive for the dfrG gene.
Out of the E. faecium isolates, 10 were classified as VRE, but their presence was not associated with a lower 30-day or 90-day survival rate. Among these isolates, the MLST types of seven could not be determined, while six belonged to ST117 and three to ST80. Five ST117 isolates and two ST80 isolates were identified as VRE. No significant differences were observed in the 30-day and 90-day survival rates based on the presence or absence of specific genes. However, a positive correlation was found between survival and younger age (rpb = 0.71, p = 0.001). The isolates displayed an average nucleotide identity of 98.84% to each other, with a range of 94.29% to 99.27%.
Fourteen isolates were MRSA, of which all were mecA-positive. Two isolates were resistant to tetracycline, while tet(k) was only encoded by one of them and by one phenotypically susceptible isolate; tet(38) was encoded by both resistant ones but also by all but one of those susceptible. Out of four that were resistant to rifampicin, only one was found to have genes conferring resistance to it (rboB_H481Y, rboB_L466S, and rpoB_S486L). Resistance to levofloxacin well matched the presence of resistance genes gyrA_S84L, parC_E84G, parC_E84K, parC_S80F, and/or parE_P585S. While all isolates were susceptible to linezolid, in nine, 23S_C2220T was found. Out of 23 resistant to erythromycin, 11 encoded erm(A), 4 erm(C), 2 erm(T), and another 2 msr(A) and mph(C); in 4, no macrolide resistance genes were detected. For 40 isolates, information on the 30-day survival could be retrieved, as well as for 37 on 90-day survival and 30-day and 90-day outcomes. The only significant difference that was found was that of a lower 30-day survival in patients with S. aureus isolates that encoded splE (p = 0.045; Cramér’s V 0.32) but without meeting χ2 test assumptions due to low cell frequencies and without significant Fisher exact test (p = 0.072). Isolates had an average nucleotide identity to each other of 98.36, ranging from 97.73 to 98.75 (see also Figure A1 in the Supplementary Material). The two most represented MLST types were 225 (10) and 22 (9).
In 11 out of 13 cases in which S. pneumoniae grew, blood cultures were collected in the EC. The available data were insufficient to perform statistical analyses pertaining to hypotheses involving 30-day and 90-day outcomes and survival rates. Only three isolates had MICs to penicillin sufficiently high to be considered susceptible only at increased dosage (I). Of these, two encoded pbp1a and pbp2b. Two out of three isolates resistant to erythromycin encoded erm(B). Three out of four isolates resistant to tetracycline encoded tet(M). Isolates had an average nucleotide identity to each other of 98.71, ranging from 98.63 to 98.84. Isolates belonged to 10 different serotypes, the most frequent being 8, with 16F and 17F having two isolates each, and these isolates had 13 different MLST types.
In 12 out of 18 cases in which S. pyogenes grew, blood cultures were collected in the EC. Here, too, the available data were insufficient to perform statistical analyses pertaining to hypotheses involving 30-day and 90-day outcomes and survival rates. Both isolates resistant to tetracycline carried tet(M). Isolates had an average nucleotide identity to each other of 98.82, ranging from 98.60 to 99.01. The two most represented MLST types were ST28 (5) and ST39 (4).
The four B. fragilis isolates had an average nucleotide identity of 98.97 to each other that ranged from 98.91 to 99.03. One MLST type could not be determined, while the remaining were ST40, ST67, and ST129. The only isolate resistant to clindamycin was also the only one to encode mef(A), as well as the only isolate to have an increased MIC to meropenem was the only one to encode cfxA.
The mean nucleotide identity of E. coli isolates ranged from 96.74 to 98.42 (see also Figure A2 in the Supplementary Material) with an average of 97.63, with MLST type ST69 being the most frequent (6). Four isolates belonged to ST12, ST73, and ST131 each, and two isolates to ST88 and ST95 each. Two isolates could not be assigned and the remaining isolates all belonged to different MLST types. The most represented O-antigen types were O4, O6, and O25, with four isolates each, and the most represented H-antigen type was H4 (9). The majority of isolates carried one or more plasmids with the most frequent being IncFIB(APoo1918) (32), Col156 (17), IncFll (14), and IncFlA (13). The significant differences found were only that of a lower 30-day and 90-day survival in patients with isolates encoding IncFll(pSE11) and p0111 (p = 0.001, Cramér’s V = 0.69) but without χ2 test assumptions being met due to low cell frequencies.
For three K. pneumoniae isolates, the MLST type could not be determined, and another three belonged to ST147. Six MLST types were represented twice (ST14, ST15, ST20, ST78, ST661, and ST3328), and another 22 just once. With Kleborate, the missing MLSTs were identified as ST307, ST223-1LV, and ST1013-1LV. Isolates had an average nucleotide identity of 99.03 to each other, ranging from 98.93 to 99.12 (see also Figure A3 in the Supplementary Material), and carried an average of 2.4 plasmids, ranging from two isolates carrying none to three carrying five. The number of encoded stress tolerance, AMR, and virulence genes varied from none to 36, from none to 27, and from none to 12, respectively, with only one isolate encoding neither of each. significant differences found were that of a lower 30-day and 90-day survival in patients with isolates encoding iucA, iucB, and iucC (p = 0.039, Cramér’s V = 0.35) and iroB and iroN (p = 0.039, Cramér’s V = 0.35), but all violating χ2 test assumptions due to low cell frequencies, and for aac(3)-IId, aph(3′)-VI, gyrA_D87G, and phoQ_R16C (p = 0.046, Cramér’s V = 0.34) and blaSHV, dfrA1, iucC, and ompK36_D135DGD (p = 0.039, Cramér’s V = 0.35), additionally without significant Fisher exact test. The four isolates encoding blaOXA-48 and the three isolates encoding blaNDM-1 were all correctly identified as such in routine diagnostics.
The sequenced Enterobacter isolates were all reported in routine diagnostics as Enterobacter cloacae complex isolates, but WGS revealed these to be E. quasihormaechei (31), E. quasiroggenkampii (4), E. sichuanensis (3), E. cloacae (2), E. chengduensis (2), and E. wuhouensis (1). Isolates either encoded no virulence genes or three (iroB, iroC, iroN), with all isolates encoding three being E. quasihormaechei isolates. The number of carried AMR and stress response genes varied significantly among the isolates, ranging from 1 to 15 for AMR genes and 1 to 35 for stress response genes. Isolates encoded between none and four plasmids, the most frequent being IncFII(pECLA) (12), IncFIB(pECLA) (10), and Col440I (8). The one blaVIM-1-positive isolate was phenotypically susceptible to imipenem and meropenem and was, hence, not investigated for carrying carbapenemases in routine diagnostics. MLST type could not be determined in 11 cases; in seven, ST50 was identified, and in three, ST118 was identified. Other MLST types were only represented once or twice. No significant differences were found regarding the 30-day and 90-day survival and the presence or absence of investigated genes.
Four Citrobacter isolates were C. freundii, another four were C. braakii, and seven were C. koseri. The average nucleotide identity of C. koseri (98.88) and C. freundii (98.59) isolates was higher than that of C. braakii (94.41) isolates, despite C. freundii isolates belonging to four different MLST types. Among all eight C. freundii and C. braakii isolates, there was only one isolate carrying virulence genes (ybtP and ybtQ), whereas all C. koseri isolates encoded the same virulence factors (iucA, iucB, iucC, ybtP, and ybtQ) with the exception of senB, which was only encoded by four. Isolates encoded between one and three AMR genes. The most significant variation was observed in the number of stress response genes encoded by the isolates. Two C. braakii isolates and six C. koseri isolates only encoded a single stress response gene, which was fief, while the remaining C. koseri isolates encoded six, the remaining C. braakii isolates encoded 17, and C. freundii isolates encoded between 24 and 35. Isolates carried from none to four plasmids with not a single plasmid carried by more than one isolate. The only two Citrobacter isolates encoding qnrB38 were the only ones to have MICs to moxifloxacin above the lowest measurable level.
Proteus mirabilis isolates had an average nucleotide identity of 99.12 to each other, ranging from 98.83 to 99.25. The significant differences found were that of a lower 30-day survival when isolates encoded aph(3′)-la (p = 0.028; Cramér’s V 0.63); however, χ2 test assumptions were violated due to low cell frequencies and the Fisher exact test was not significant (p = 0.091). All isolates carrying blaTEM-1 were resistant to ampicillin and all but one isolate to ampicillin/sulbactam. All isolates resistant to trimethoprim/sulfamethoxazole carried dfrA1 and in addition either sul1 or sul2. Four isolates carried an IncQ1 plasmid.
More than half of the isolates were isolated from patients in the ICU, but isolates only had an average nucleotide identity to each other of 95.79, ranging from 95.18 to 96.00 (see also Figure A4 in the Supplementary Material). No significant differences were found in the 30-day and 90-day survival linked to the presence or absence of certain genes, age, or ward type. Four isolates carried a pSM22 plasmid and another three isolates either Col440I, IncX5, or IncX6. The IncX5 carrying isolate carried a blaVIM-1 alongside it that was neither specifically investigated nor detected in routine diagnostics. That isolate was resistant to piperacillin–tazobactam, tested cephalosporins, and imipenem but had a MIC to meropenem of 0.19 as determined by the gradient strip test (Liofilchem, Roseto degli Abruzzi, Italy). It was also the only one to be sul1-positive although it was just as susceptible to trimethoprim/sulfamethoxazole as all the other isolates. Isolates carried four or five out of 10 stress response genes and either five or six out of 13 AMR genes. The most frequent AMR genes were ssmE, smdB, smdA, sdeY, sdeA (27 each), smfY, sdeB (26 each), aac(6′) (25), blaSRT (22), and tet(41) (21).
Among the isolates of the A. baumannii complex, four were A. baumannii isolates and eight were A. pittii. Among the A. baumannii isolates, the MLST types represented were ST1, ST40, and ST213, with one isolate unassigned and with an average nucleotide identity to each other ranging from 97.54 to 97.69 and from 96.52 to 98.03 among the A. pittii isolates. No virulence genes were identified; however, between one and five stress tolerance genes were identified in all isolates and either five or six AMR genes in A. pittii and between 3 and 12 in A. baumannii. In both species, the most frequently encoded stress tolerance genes were nreB (12), clpK (6), yfdX2 (5), trxLHR (5), and hdeD-GI (5), while the most frequent AMR genes were amvA (12) and ant(3″)-IIa (10). The available data were insufficient to perform statistical analyses pertaining to hypotheses involving 30-day and 90-day outcomes and survival rates.
The average nucleotide identity of P. aeruginosa isolates was 98.85 and ranged from 98.66 to 98.94. For 11 isolates, no MLST type could be determined, while three belonged to ST234, ST253, and ST823 each, two to ST308, ST316, and ST446 each, and all the remaining isolates to different MLST types. Isolates belonged mainly to serogroups O11 (13), O6 (9), O1 (5), and O10 (4). No virulence genes were detected, and stress response genes were only detected in less than half of the isolates in which they ranged from 2 to 14 in number. The most frequently encoded were merE (14), merT (12), merR (12), merP (12), merD (11), and merA (11). No isolate had fewer than 8 AMR genes with some carrying up to 18. By far, the most frequent were mexE (38), mexA (38), fosA (38), aph(3′)-llb (38), mexX (37), catB7 (37), and crpP (29). Thirty-five isolates carried blaOXA genes and four blaVIM (three blaVIM-2 and one blaVIM-11). Five isolates were classified as 4MRGN and three as 3MRGN. One blaVIM-2 encoding isolate did not previously classify as 4MRGN as it had below-resistant MICs to piperacillin–tazobactam and cefepime and the blaVIM-2 gene was neither investigated nor detected in routine diagnostics. It was resistant to imipenem, meropenem, ciprofloxacin, and ceftazidime. No significant differences were found in the 30-day and 90-day survival linked to the presence or absence of certain genes. Isolates exhibited a distinct clustering pattern, roughly partitioning into three distinct branches. Notably, one of these branches encompassed all but one isolate from patients with negative 30-day survival (see Figure A5 in the Supplementary Material) (p = 0.038).
Stenotrophomonas maltophilia isolates only had an average nucleotide identity of 93.15 to each other, ranging from 92.09 to 94.17. One MLST type could not be determined, while all remaining were of different ones: ST4, ST23, ST224, and ST233. Only one did not have any stress response genes, the others had from three to nine, and all isolates carried either seven or eight antimicrobial resistance genes. Those shared among all isolates were aph(6), emrA, emrB, emrC, and smeF.
The present study aimed to conduct a comprehensive analysis of blood culture isolates’ genomes, with a retrospective correlation between the detected virulence genes, resistance genes, and stress tolerance genes with the phenotypical susceptibilities and patient outcomes. The findings of this investigation yielded extensive and informative data on the distribution of genes implicated in bloodstream infections across various species. However, the interpretation of these results necessitates careful consideration due to several noteworthy factors. The intriguing observed associations between survival outcomes and the presence of specific resistance genes warrant caution, particularly given the limited knowledge regarding many of the identified virulence factors. The χ2 test assumptions were persistently violated due to the relatively small sample sizes in the groups of patients with negative 30-day and 90-day survival available for each species, making it imperative to validate and strengthen these associations by replicating the study with larger sample sizes for different species. One key consideration is the possibility that host factors may play a more significant role than pathogen factors in determining the outcomes of bloodstream infections and the inherent difficulty in fully accounting for such factors in studies of this nature (Hekker et al., 2000; Casadevall and Pirofski, 2001; Tseng et al., 2002; Beck et al., 2004; Tseng et al., 2005; Newman et al., 2017).
For example, research on splE in S. aureus still aims to uncover its precise functions and mechanisms. It seems that splE plays a significant role in S. aureus pathogenesis by promoting immune evasion through the degradation of immune components and facilitating tissue invasion by breaking down extracellular matrix proteins (Stach et al., 2018). Confirming its role in survival would further emphasize the importance of understanding splE for developing effective strategies against staphylococcal infections. The IncFll(pSE11) and p0111 plasmids that were linked to lower survival in E. coli are known for disseminating AMR genes (Tasleem Jan and Tiwari, 2017; Wang et al., 2022), and their likely correlation with factors such as prolonged hospitalizations cannot be excluded to confound results, emphasizing that interpretation of the results warrants considered caution. The lower survival in aph(3′)-la encoding P. mirabilis isolate is probably the most questionable finding, because aside from resistance to aminoglycosides, it is not reported to contribute to virulence (Shaw et al., 1993), and aminoglycosides are infrequently used in our setting.
The study highlights important differences in genetic variation across different species and might be representative to some degree for these pathogens involved in BSI, while it is certainly important to exercise caution in generalizing these findings or applying them to other settings. The identity and coverage values for E. faecium ST117 and E. coli ST131 were both 100% for their exact alleles. Notably, ECO 131 has been highlighted in several studies as a rapidly expanding multidrug-resistant pathogen with high receptivity and the potential to develop resistance to last-resort antibiotics such as carbapenems and colistin (Pitout and Finn, 2020; Li et al., 2021; Taati Moghadam et al., 2021; Brumwell et al., 2023). Serotypes H4 and O25 were found to match all isolates in our investigation, consistent with previous studies (Brumwell et al., 2023), but none of the isolates exhibited the antimicrobial MDR profile of 3MRGN or 4MRGN. Enterococcus faecium ST117 was previously identified as a rising factor in vancomycin-resistant enterococci (VRE) (Weber et al., 2020), and five out of six isolates in our study were found to carry vanB genes. A comparison with this recent comprehensive study involving 120 isolates revealed that all the genes mentioned in the study (msrC, efmA, erm(B), dfrG, aac(6′)-Ii, gyrA, parC, and pbp5) matched those present in our isolates, except for efmA, which was absent in the NCBI database used. The notable clustering of ST50 E. quasihormaechei isolates calls for focused investigations, given the escalating local and global issue of sewage-related clonal colonization in hospital sanitary facilities (Babouee Flury et al., 2016; Kehl et al., 2022; Stokes et al., 2022). Similarly, the recurring identification of ST147 K. pneumoniae and ST235 and ST823 P. aeruginosa isolates, even in wards geographically distant from those assumed to have a contamination source in our setting (Kehl et al., 2022; Neidhöfer et al., 2023a), urges us to implement transmission dynamics monitoring networks (Bohl et al., 2022; Ko et al., 2022; Neidhöfer et al., 2023b).
Conducting replication studies with larger sample sizes for each species is imperative to cautiously interpret associations between survival outcomes and the presence of specific genes, emphasizing the need for further research in this area. A deeper understanding of virulence and host factors will not only aid in the interpretation of results but also pave the way for the development of more targeted therapeutic and preventive measures, thus enhancing patient outcomes. In addition, implementing more efficient transmission dynamics monitoring holds significant promise as a valuable tool in preventing bloodstream infections caused by certain pathogens, highlighting the importance of establishing robust surveillance systems.
All data and original contributions presented in the study are included in the article/Supplementary Material, except for genomes that cannot be readily made available because of binding collaboration agreements. These can, however, be made available upon request directed to the corresponding author.
The studies involving humans were approved by the Ethics Committee of the University Hospital Bonn. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
CN: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. MN: Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Data curation. RJ: Data curation, Formal Analysis, Methodology, Project administration, Writing – review & editing. BA: Data curation, Writing – review & editing. SU: Data curation, Methodology, Software, Writing – original draft, Writing – review & editing. UN: Data curation, Methodology, Software, Writing – original draft, Writing – review & editing. SG: Data curation, Methodology, Software, Writing – original draft, Writing – review & editing. MP: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
SG is coinventor of one pending international patent on diagnostic algorithms and chief scientific officer and cofounder of Noscendo GmbH in Duisburg, Germany. SU and UN are employed by Noscendo GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1274573/full#supplementary-material
Allen, J. P., Snitkin, E., Pincus, N. B., Hauser, A. R. (2021). Forest and trees: exploring bacterial virulence with genome-wide association studies and machine learning. Trends Microbiol. 29 (7), 621–633. doi: 10.1016/j.tim.2020.12.002
Babouee Flury, B., Ellington, M. J., Hopkins, K. L., Turton, J. F., Doumith, M., Loy, R., et al. (2016). Association of novel nonsynonymous single nucleotide polymorphisms in ampD with cephalosporin resistance and phylogenetic variations in ampC, ampR, ompF, and ompC in Enterobacter cloacae isolates that are highly resistant to carbapenems. Antimicrobial Agents chemotherapy 60 (4), 2383–2390. doi: 10.1128/AAC.02835-15
Beck, M. A., Handy, J., Levander, O. A. (2004). Host nutritional status: the neglected virulence factor. Trends Microbiol. 12 (9), 417–423. doi: 10.1016/j.tim.2004.07.007
Bohl, J. A., Lay, S., Chea, S., Ahyong, V., Parker, D. M., Gallagher, S., et al. (2022). Discovering disease-causing pathogens in resource-scarce Southeast Asia using a global metagenomic pathogen monitoring system. Proc. Natl. Acad. Sci. 119, 11, e2115285119. doi: 10.1073/pnas.2115285119
Brumwell, A., Sutton, G., Lantos, P. M., Hoffman, K., Ruffin, F., Brinkac, L., et al. (2023). Escherichia coli ST131 associated with increased mortality in bloodstream infections from urinary tract source. J. Clin. Microbiol., e00199–e00123. doi: 10.1128/jcm.00199-23
Buchfink, B., Xie, C., Huson, D. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12 (1), 59–60. doi: 10.1038/nmeth.3176
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58 (7), 3895–3903. doi: 10.1128/aac.02412-14
Casadevall, A., Pirofski, L.-a. (2001). Host-pathogen interactions: the attributes of virulence. J. Infect. Dis. 184 (3), 337–344. doi: 10.1086/322044
Chan, P. P., Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Epping, L., van Tonder, A. J., Gladstone, R. A., The Global Pneumococcal Sequencing Consortium, Bentley, S. D., Page, A. J., et al. (2018). SeroBA: rapid high- throughput serotyping of Streptococcus pneumoniae from whole genome sequence data. Microbial Genomics 4 (7):e000186. doi: 10.1099/mgen.0.000186
Eren, A. M., Kiefl, E., Shaiber, A., Veseli, I., Miller, S. E., Schechter, M. S., et al. (2021). Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6 (1), 3–6. doi: 10.1038/s41564-020-00834-3
Ewels, P., Peltzer, A., Fillinger, S., Patel, H., Alneberg, J., Wilm, A., et al. (2020). Nahnsen S The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38 (3), 276–278. doi: 10.1038/s41587-020-0439-x
Feldgarden, M., Brover, V., Fedorov, B., Haft, D. H., Prasad, A. B., Klimke, W. (2022). Curation of the AMRFinderPlus databases: applications, functionality and impact. Microb. Genom. 8 (6), mgen000832. doi: 10.1099/mgen.0.000832
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J. G., Haendiges, J., Haft, D. H., et al. (2021). AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11 (1), 12728. doi: 10.1038/s41598-021-91456-0
Hekker, T. A., Groeneveld, A. B., Simoons-Smit, A. M., de Man, P., Connell, H., MacLaren, D. M. (2000). Role of bacterial virulence factors and host factors in the outcome of Escherichia coli bacteraemia. Eur. J. Clin. Microbiol. Infect. Dis. 19, 312–316. doi: 10.1007/s100960050483
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Iguchi, A., Iyoda, S., Kikuchi, T., Ogura, Y., Katsura, K., Ohnishi, M., et al. (2015). A complete view of the genetic diversity of the Escherichia coli O-antigen biosynthesis gene cluster. DNA Res. 22 (1), 101–107. doi: 10.1093/dnares/dsu043
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., Aluru, S. (2018). Aluru S High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9 (1), 5114. doi: 10.1038/s41467-018-07641-9
Jolley, K. A., Bray, J. E., Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Kehl, K., Schallenberg, A., Szekat, C., Albert, C., Sib, E., Exner, M., et al. (2022). Dissemination of carbapenem resistant bacteria from hospital wastewater into the environment. Sci. Total Environ. 806, 151339. doi: 10.1016/j.scitotenv.2021.151339
Ko, K. K. K., Chng, K. R., Nagarajan, N. (2022). Metagenomics-enabled microbial surveillance. Nat. Microbiol. 7 (4), 486–496. doi: 10.1038/s41564-022-01089-w
Lam, M. M. C., Wick, R. R., Watts, S. C., Cerdeira, L. T., Wyres, K. L., Holt, K. E. (2021). A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12, 4188. doi: 10.1038/s41467-021-24448-325
Leavis, H. L., Bonten, M. J. M., Willems, R. J. L. (2006). Identification of high-risk enterococcal clonal complexes: global dispersion and antibiotic resistance. Curr. Opin. Microbiol. 9 (5), 454–460. doi: 10.1016/j.mib.2006.07.001
Li, D., Wyrsch, E. R., Elankumaran, P., Dolejska, M., Marenda, M. S., Browning, G. F., et al. (2021). Genomic comparisons of Escherichia coli ST131 from Australia. Microbial Genomics 7 (12), 000721. doi: 10.1099/mgen.0.000721
Thomas, C. L., Lee, S. W. (2012). Knowing is half the battle: Targeting virulence factors of group A streptococcus for vaccine and therapeutics. Curr. Drug Targets 13 (3), 308–322. doi: 10.2174/138945012799424679
Mahfouz, N., Ferreira, I., Beisken, S., von Haeseler, A., Posch, A. E. (2020). Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: a systematic review. J. Antimicrobial Chemotherapy 75 (11), 3099–3108. doi: 10.1093/jac/dkaa257
McNamara, J. F., Righi, E., Wright, H., Hartel, G. F., Harris, P. N. A., Paterson, D. L. (2018). Long-term morbidity and mortality following bloodstream infection: a systematic literature review. J. Infection 77 (1), 1–8. doi: 10.1016/j.jinf.2018.03.005
Neidhöfer, C., Sib, E., Neuenhoff, M., Schwengers, O., Dummin, T., Buechler, C., et al. (2023a). Hospital sanitary facilities on wards with high antibiotic exposure play an important role in maintaining a reservoir of resistant pathogens, even over many years. Antimicrobial Resistance Infection Control 12 (1), 1–17. doi: 10.1186/s13756-023-01236-w
Neidhöfer, C., Sib, E., Benhsain, A. H., Mutschnik-Raab, C., Schwabe, A., Wollkopf, A., et al. (2023b). Examining different analysis protocols targeting hospital sanitary facility microbiomes. Microorganisms 11 (1), 185. doi: 10.3390/microorganisms11010185
Newman, J. W., Floyd, R. V., Fothergill, J. L. (2017). The contribution of Pseudomonas aeruginosa virulence factors and host factors in the establishment of urinary tract infections. FEMS Microbiol. Lett. 364 (15). doi: 10.1093/femsle/fnx124
Petit, R. A., III, Read, T. D. (2020). Bactopia: a flexible pipeline for complete analysis of bacterial genomes. mSystems. 5 (4), e00190–20. doi: 10.1128/mSystems.00190-2024
Pitout, J. D. D., Finn, T. J. (2020). The evolutionary puzzle of Escherichia coli ST131. Infection Genet. Evol. 81, 104265. doi: 10.1016/j.meegid.2020.104265
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Shaw, K. J., Rather, P. N., Hare, R. S., Miller, G. H. (1993). Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiological Rev. 57 (1), 138–163. doi: 10.1128/mr.57.1.138-163.1993
Stach, N., Kalinska, M., Zdzalik, M., Kitel, R., Karim, A., Serwin, K., et al. (2018). Unique substrate specificity of SplE serine protease from Staphylococcus aureus. Structure 26 (4), 572–579. doi: 10.1016/j.str.2018.02.008
Stokes, W., Peirano, G., Matsumara, Y., Nobrega, D., Pitout, J. D. D. (2022). Population-based surveillance of Enterobacter cloacae complex causing blood stream infections in a centralized Canadian region. Eur. J. Clin. Microbiol. Infect. Dis. 41, 119–125. doi: 10.1007/s10096-021-04309-z
Taati Moghadam, M., Mirzaei, M., Fazel Tehrani Moghaddam, M., Babakhani, S., Yeganeh, O., Asgharzadeh, S., et al. (2021). The challenge of global emergence of novel colistin-resistant Escherichia coli ST131. Microbial Drug resistance 27 (11), 1513–1524. doi: 10.1089/mdr.2020.0505
Hemlata, J. A. T., Tiwari, A. (2017). The ever changing face of antibiotic resistance: Prevailing problems and preventive measures. Curr. Drug Metab. 18 (1), 69–77. doi: 10.2174/1389200217666161014163324
Thrane, S. W., Taylor, V. L., Lund, O., Lam, J. S., Jelsbak, L. (2016). Application of whole- genome sequencing data for O-specific antigen analysis and in silico serotyping of pseudomonas aeruginosa isolates. J. Clin. Microbiol. 54 (7), 1782–1788. doi: 10.1128/JCM.00349-16
Tseng, C.-C., Wu, J. J., Liu, H. L., Sung, J. M., Huang, J. J. (2002). Roles of host and bacterial virulence factors in the development of upper urinary tract infection caused by Escherichia coli. Am. J. Kidney Dis. 39 (4), 744–752. doi: 10.1053/ajkd.2002.32992
Tseng, C.-C., Wu, J. J., Wang, M. C., Hor, L. I., Ko, Y. H., Huang, J. J. (2005). Host and bacterial virulence factors predisposing to emphysematous pyelonephritis. Am. J. Kidney Dis. 46 (3), 432–439. doi: 10.1053/j.ajkd.2005.05.019
Wang, M., Jiang, L., Wei, J., Zhu, H., Zhang, J., Liu, Z., et al. (2022). Similarities of P1-like phage plasmids and their role in the dissemination of bla CTX-M-55. Microbiol. Spectr. 10 (5), e01410-22. doi: 10.1128/spectrum.01410-22
Weber, A., Maechler, F., Schwab, F., Gastmeier, P., Kola, A. (2020). Increase of vancomycin-resistant Enterococcus faecium strain type ST117 CT71 at Charité - Universitätsmedizin Berlin, 2008 to 2018. Antimicrob. Resist. Infect. Control 9 (1), 109. doi: 10.1186/s13756-020-00754-1
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Méric, G., Vezina, B., et al. (2021). Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 22, 266. doi: 10.1186/s13059-021-02483-z
Wren, B. W. (2000). Microbial genome analysis: insights into virulence, host adaptation and evolution. Nat. Rev. Genet. 1 (1), 30–39. doi: 10.1038/35049551
Keywords: bloodstream infections, whole-genome sequencing, clonality patterns, virulence factors, resistance genes, genotype-phenotype correlation, genomic characterization of BSI, molecular epidemiology of BSI
Citation: Neidhöfer C, Neuenhoff M, Jozič R, Atangcho B, Unsleber S, Neder U, Grumaz S and Parčina M (2023) Exploring clonality and virulence gene associations in bloodstream infections using whole-genome sequencing and clinical data. Front. Cell. Infect. Microbiol. 13:1274573. doi: 10.3389/fcimb.2023.1274573
Received: 08 August 2023; Accepted: 18 September 2023;
Published: 14 November 2023.
Edited by:
Manuel Gerardo Ballesteros Monrreal, University of Sonora, MexicoReviewed by:
Edwin Barrios-Villa, Universidad de Sonora, MexicoCopyright © 2023 Neidhöfer, Neuenhoff, Jozič, Atangcho, Unsleber, Neder, Grumaz and Parčina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Neidhöfer, Y2xhdWRpby5uZWlkaG9lZmVyQHVrYm9ubi5kZQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.