 Shahrzad Shoraka
Shahrzad Shoraka Amali E. Samarasinghe
Amali E. Samarasinghe Amir Ghaemi
Amir Ghaemi Seyed Reza Mohebbi
Seyed Reza Mohebbi- 1Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran
- 2Department of Microbiology and Microbial Biotechnology, Faculty of Life Sciences and Biotechnology, Shahid Beheshti University, Tehran, Iran
- 3Division of Pulmonology, Allergy and Immunology, Department of Pediatrics, College of Medicine, University of Tennessee Health Science Center, Memphis, TN, United States
- 4Children’s Foundation Research Institute, Memphis, TN, United States
- 5Department of Virology, Pasteur Institute of Iran, Tehran, Iran
- 6Gastroenterology and Liver Diseases Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran
Since December 2019, the world has been facing viral pandemic called COVID-19 (Coronavirus disease 2019) caused by a new beta-coronavirus named severe acute respiratory syndrome coronavirus-2, or SARS-CoV-2. COVID-19 patients may present with a wide range of symptoms, from asymptomatic to requiring intensive care support. The severe form of COVID-19 is often marked by an altered immune response and cytokine storm. Advanced age, age-related and underlying diseases, including metabolic syndromes, appear to contribute to increased COVID-19 severity and mortality suggesting a role for mitochondria in disease pathogenesis. Furthermore, since the immune system is associated with mitochondria and its damage-related molecular patterns (mtDAMPs), the host mitochondrial system may play an important role during viral infections. Viruses have evolved to modulate the immune system and mitochondrial function for survival and proliferation, which in turn could lead to cellular stress and contribute to disease progression. Recent studies have focused on the possible roles of mitochondria in SARS-CoV-2 infection. It has been suggested that mitochondrial hijacking by SARS-CoV-2 could be a key factor in COVID-19 pathogenesis. In this review, we discuss the roles of mitochondria in viral infections including SARS-CoV-2 infection based on past and present knowledge. Paying attention to the role of mitochondria in SARS-CoV-2 infection will help to better understand the pathophysiology of COVID-19 and to achieve effective methods of prevention, diagnosis, and treatment.
Highlights
● Mitochondria are the “powerhouse” of the cell and have several other important functions. Multiple mitochondrial functions make them essential to the cell, so when a virus “hijacks” mitochondrial function, this allows it to control the entire cell.
● Studies have suggested that host mitochondria are hijacked by the SARS-CoV-2 based on in silico predictions. Emerging evidence also suggested that SARS-CoV-2 hijacks the mitochondria of immune cells, proliferate in mitochondrial structures, and impairs mitochondrial functions. The data indicate that patients with COVID-19 have a population of T cells with mitochondrial dysfunction, as well as mitochondrial markers in monocytes have altered.
● The interaction of several SARS-CoV-2 proteins with mitochondrial components components may occur.
● Lung tissue studies in COVID-19 patients have shown that mitochondrial function is altered following infection.
● Monitoring mitochondrial function of immune cells in the blood could be a non-invasive marker to diagnose and predict the prognosis of COVID-19.
● Understanding the cause of more severity and mortality in people with underlying diseases, particularly metabolic and age-related diseases, could help improve the management of the COVID-19 pandemic.
● There are various strategies with pharmacological agents and lifestyle interventions to target mitochondria that could be promising to prevent SARS-CoV-2 infection or reduce disease progression. However, there is little empirical evidence of their potential.
● Given the important roles of mitochondria in viral infection, mitochondria could be considered a therapeutic target for COVID-19 disease.
Introduction
The new member of the coronavirus family, like other members, is enveloped virus containing a positive single-stranded RNA [(+) ssRNA] genome. During the last two decades, and before the current COVID-19 pandemic, coronaviruses also caused two epidemic diseases, SARS (severe acute respiratory syndrome) in 2003 and MERS (the Middle East respiratory syndrome) in 2012. SARS-CoV-2 like the SARS-CoV and MERS-CoV, belongs to the beta group of coronaviruses and, similar to SARS-CoV, belongs to the B lineage of betacoronaviruses (Eriani and Martin, 2022). The SARS-CoV-2 genome encodes 4 structural proteins (spike (S), envelope (E), membrane (M), nucleocapsid protein (N)) and 16 nonstructural proteins (NSP1-16) that are important for replication, infection, and triggering host immune response (Bai et al., 2022).
SARS-CoV-2 invades the host cell by attaching its surface spike protein to cell receptors such as angiotensin-converting enzyme 2 (ACE2), a receptor expressed in a variety of organs (Kuppusamy et al., 2021). Most COVID-19 patients present with respiratory symptoms, although clinical manifestations of extra-respiratory symptoms have also been reported. Also, COVID-19 patients may show a wide range of symptoms, from asymptomatic to severe disease (Shoraka et al., 2021). Although the main cause of severe COVID-19 is not clearly understood, it appears to correlate with host immune-viral interactions, excessive and/or impaired inflammatory response and cytokine storm play important roles in this regard (Zheng and Gao, 2023).
Mitochondria
The mitochondrion is an organelle in the cytoplasm that function as the energy wheelhouse of eukaryotic cells. Mitochondrial DNA (mtDNA) is a 16.5 kb extra-chromosomal circular double-stranded DNA. The number of mtDNA copies in a cell varies depending on the cell type. mtDNA copy number could also change in response to physiological signals and some diseases (Hummel et al., 2023).
Mitochondrial perform numerous other cellular functions in addition to cell energy production by performing oxidative phosphorylation (OXPHOS), serving as the main site of reactive oxygen species (ROS) generation as a byproduct of the electron transfer chain (ETC), and controlling cellular processes involved in metabolism and immune responses (Kuznetsov et al., 2022).
Role of mitochondria in viral infection
The prominent role of mitochondria in the outcome of viral infection has been demonstrated in the clinical phenotype of patients with inherited mitochondrial defects (Kruk et al., 2019). In 2015, Thakar et al. reported that decreased efficacy of the influenza vaccine is associated with decreased mitochondrial biogenesis (Thakar et al., 2015). Mitochondrial dysfunction has been also considered not only in HIV infection but also in antiviral drug therapies (Ganta and Chaubey, 2019). Thus, impaired mitochondrial function could affect both the host response to the virus and the development of an effective response to vaccine or treatment (Nunn et al., 2020).
The role of mitochondria in the immune system of mammals has been substantially proven. Many studies show that mitochondria and mitochondrial damage-related molecular patterns (mtDAMPs), such as mtDNA, cardiolipin, N-formyl peptides, and cytochrome c, have an important role in the host immune response against viral infections (Garg et al., 2022). Since the host immune response against viruses is significantly based on mitochondrial function, it is not surprising that viruses modulate mitochondrial function to promote their proliferation and survival (Tiku et al., 2020). In the following section, mitochondrial roles in viral infection will be discussed briefly.
Mitochondrial antiviral signaling (MAVS)
The innate immune response is the first line of host defense against viruses and begins with the identification of pathogen-related molecular patterns (PAMPs) or DAMPs by pattern recognition receptors (PRRs). This leads to activation of some signaling pathways and subsequently, induction of interferons (IFNs), pro-inflammatory cytokines, and other antiviral effector genes. Among PRRs, RIG-I-like receptors (RLRs) and MDA5 have been identified as cytosolic viral RNA sensors in mammalian cells. After viral RNA binding, RLRs translocate to mitochondrial adaptor protein MAVS (mitochondrial antiviral signaling) in the mitochondrial membrane and activate MAVS to induce the formation of oligomers in a prion-like manner. The formation of the MAVS complex is modulated by mitochondrial translocase of outer membrane 70 (Tom70) and 20 (Tom20). This pathway can either activate type I IFN transcription and stimulate interferon-stimulated genes (ISGs) or release NF-κB to activate the transcription of pro-inflammatory cytokines. While both pathways converge on antiviral immunity, overstimulation of the second pathway may lead to cytokine storms and viral persistence in infected cells (Fu et al., 2021; Sharma et al., 2021). During RNA virus infection, MAVS also appears to play a major role in activating NLRP3 inflammasome. Inflammasome activation often leads to programmed pro-inflammatory cell death known as pyroptosis with heightened secretion of interleukin-1β (IL-1β) (Paik et al., 2021).
The MAVS pathway is targeted by many viruses. Some DNA or RNA viruses have evolved mechanisms targeting mitochondria to escape IFN-I-mediated host immune responses (Iessi et al., 2020). Human Rhinovirus C encodes a protease that could cleave MAVS to inhibit RLR signaling (Pang et al., 2017). The influenza virus protein PB1-F2 binds to and disrupts MAVS and reduces the IFN response (Varga et al., 2012). Similarly, the ORF-9b protein in SARS-CoV restricts host IFN responses by degrading MAVS. ORF-9b leads to viral escape by manipulating mitochondrial function (Shi et al., 2014). Also, SARS-CoV ORF-7a and ORF-8b can translocate to mitochondria and alter MAVS and mitochondrial function (Ganji and Reddy, 2021). These data suggest that blocking innate immune signaling through mitochondrial manipulation could be one of the viral evasion strategies (Ferreira et al., 2016).
Studies have demonstrated that SARS-CoV-2 RNA could activate RIG-I/MAVS signaling pathway (Wu et al., 2021). Downregulation of MAVS has also been confirmed in SARS-CoV-2-infected CaCo-2 (human colon epithelial carcinoma) cells (Bojkova et al., 2020). Studies suggest that SARS-CoV-2 uses several conserved virulence genes to antagonize the IFN response (Wu et al., 2021; Znaidia et al., 2022) (See Table 1).
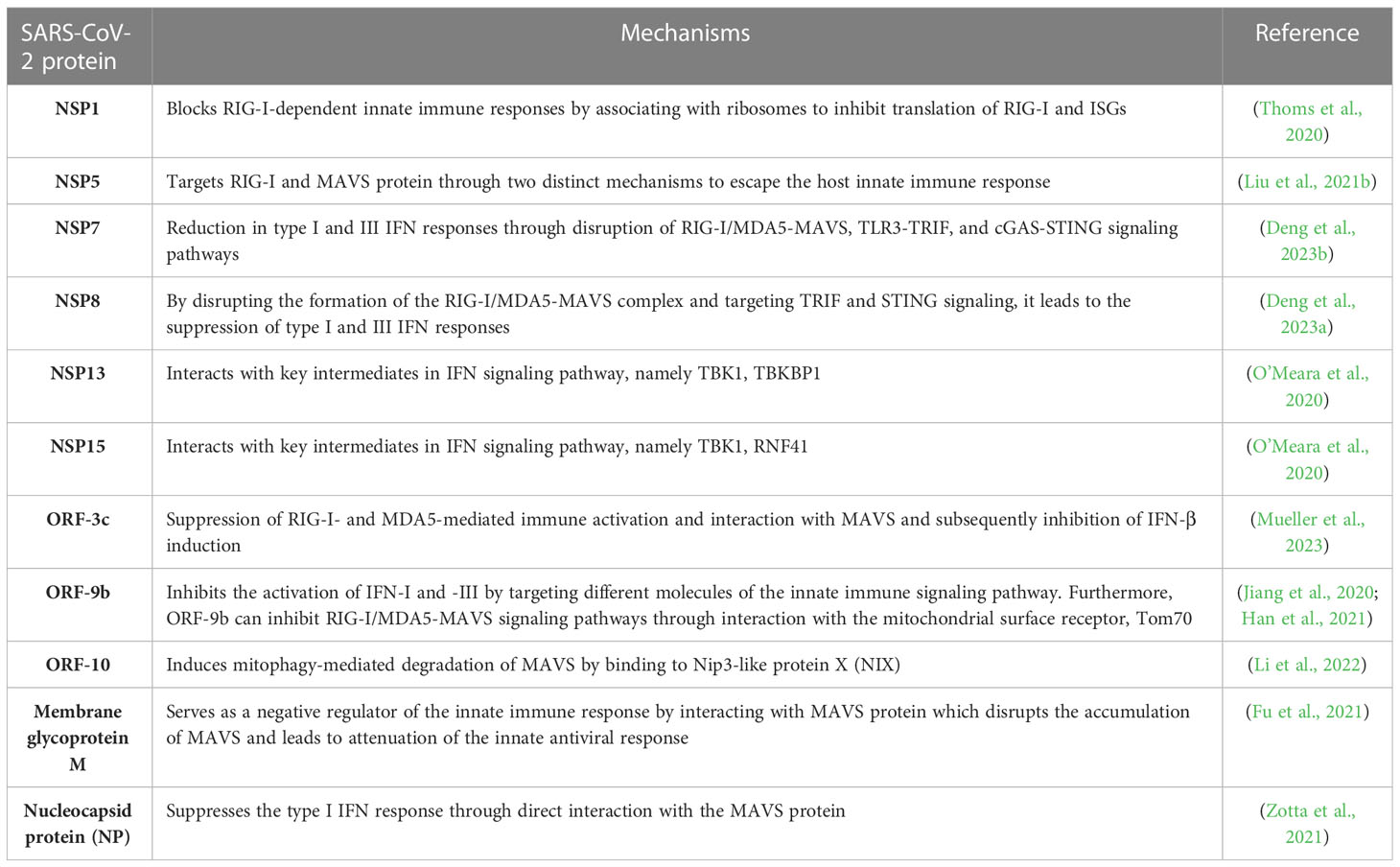
Table 1 Interaction of SARS-CoV-2 proteins with host immune response components and modulation of MAVS signaling.
Mitochondrial dynamics
Mitochondria are also dynamic organelles that modulate and adapt in response to environmental stresses. Mitochondrial dynamics include fusion and fission events, mitochondrial autophagy (mitophagy), and biogenesis. DRP1 and FIS1 proteins play key roles in mitochondrial fission (a process by which mitochondria divide and are essential for cell growth and division) and sometimes occur during mitochondrial damage. Fission increases ROS production and also facilitates mitophagy and mitochondrial distribution. Mitochondrial fusion, wherein they elongate, maximizes oxidative capacity in response to stress. MFN1, MFN2, and OPA1 proteins are involved in the fusion process (Giacomello et al., 2020; Al Ojaimi et al., 2022). In contrast to fission that occurs during nutrient-replete states resulting in nutrient storage and reduced bioenergetics efficiency, mitochondrial fusion is promoted in nutrient-deficient conditions and increases the bioenergetics efficiency of mitochondria (Schrepfer and Scorrano, 2016; Giacomello et al., 2020).
Changes in metabolic or physiological conditions, such as infection, induce mitochondrial morphological alterations that have functional consequences which significantly affect the innate immune response (Banoth and Cassel, 2018). The close association between RLR signaling and mitochondrial dynamics has been confirmed. Efficient RLR signaling requires MAVS interaction with MFN1, while mitochondrial fission factor (MFF1) or DRP1 as a fusion inhibitor reduces virus-induced NF-κB and interferon regulatory transcription factor 3 (IRF-3) activation. After depletion of DRP1 and FIS1 in cells, RLR signaling increases, and elongation of the mitochondrial network enhances the endoplasmic reticulum (ER)-mitochondria interaction during viral infection and increases the association of MAVS to enhance RLR signaling (Castanier et al., 2010; Kim et al., 2018).
Therefore, the lack of the drp1 gene leads to improved antiviral responses during infection through mitochondrial elongation, while inhibition of MFN1 and OPA1 leads to mitochondrial degradation and decreased antiviral response (Kim et al., 2018; Lai et al., 2018). Also, drp1 knockdown targets NLRP3 inflammasome accumulation and activates caspase-1 and IL-1β (Park et al., 2015).
Viruses are able to manipulate the function of cellular factors affecting mitochondrial dynamics such as DRP1, MFN, and OPA1 pushing mitochondria toward fusion or fission. Viral infections may lead to mitochondrial elongation, potentially increasing antiviral signals (Castanier et al., 2010). Also, DRP1 is usually targeted by viruses. For example, HIV protein gp120 could impair mitochondrial fission. Mitochondrial fission is important for clearance of defective mitochondria (Fields et al., 2016). Conversely, some viruses, such as hepatitis B virus, alter mitochondrial dynamics toward fission and mitophagy thus leading to viral persistence (Kim et al., 2013). The ORF-9b protein of SARS-CoV in addition to degrading MAVS, could alter mitochondrial dynamics and modulate host IFN responses. ORF-9b protein causes DRP1 degradation and mitochondrial elongation through physical interaction with DRP1 (Shi et al., 2014).
Studies of lung tissue in patients with COVID-19 show that the mitochondrial dynamic balance is disturbed, thus resulting in oxidative stress, pro-inflammatory status, cytokine production, and cell death (Tay et al., 2020). An early study on SARS-CoV-2 infection proposed an increase in fusion, leading to mitochondrial elongation, preventing apoptosis, and creating an intracellular environment suitable for virus propagation in infected cells (Holder and Reddy, 2021). A recent study also showed that mitochondria were strongly fused in SARS-CoV-2 positive human placenta (Gabanella et al., 2022).
Mitochondrial DNA
Mitochondrial DNA damage increases the mtDNA copy numbers, seemingly as a compensatory mechanism. The number of mtDNA copies is a reflection of the loss of mtDNA integrity and a marker for mitochondrial function and oxidative stress (Riou et al., 2020).
In addition, the ratio of the mitochondrial genome to the nuclear genome (mt/n) may change during oxidative stress. The initial response to increased oxidative stress could be an adaptive response in which the mt/n ratio increases as a result of heightened mitochondrial biogenesis. While prolonged oxidative stress may lead to a decrease in mt/n ratio with mitochondrial dysfunction due to damage to mitochondrial DNA and proteins. The inflammatory response could be triggered by the accumulation of damaged mtDNA in the cell (Malik and Czajka, 2013). In some conditions, mtDNA could leak into the cytoplasm and extracellular space triggering inflammation. Various factors could contribute to mtDNA release, for example, the stress induced by the absence of TFAM (a transcription factor that binds to mtDNA) leads to improper packaging of mtDNA and ultimately its release into cytosol (Rongvaux, 2018). Some immune pathways, such as IL-1β signaling, leading to mitochondrial stress and mtDNA release (Cheng et al., 2020). In addition, many viruses can cause mitochondrial stress. As reported for Herpesvirus 1 (HSV-1), cell-free mtDNA (cf-mtDNA) targets inflammatory and antiviral responses. This signaling mechanism is conserved in many viral infections (Corcoran et al., 2009). Metabolic stress due to HIV replication has a role in mitochondrial damage and mtDNA leakage (Pernas et al., 2017). Studies showed that positive single-stranded RNA viruses produce a strong inflammatory response that involves the release of mtDNA out of the cell (Storci et al., 2020a). Also in a process termed NETosis (neutrophil extracellular trap), neutrophils release their nuclear and mitochondrial DNA into the surrounding tissue when stimulated by virus infection (Cao et al., 2022).
In addition, mtDNA is a circular loop that contains a significant number of CpG islands evolved from prokaryotic symbiosis. Therefore extracellular mtDNA could target various pro-inflammatory signaling pathways (Comish et al., 2022). Toll-like receptor-9 (TLR-9) induces NF-κB-dependent pro-inflammatory signaling by binding to mtDNA. Also, the detection of mtDNA in the cytosol by cGAS involves activation of stimulator of interferon genes (STING) (located in the endoplasmic reticulum) and induction of IFN response. The activated cGAS-STING pathway also induces NF-κB pro-inflammatory signaling. Activation of mtDNA-dependent inflammasome also leads to the production of pro-inflammatory IL-1 and IL-8 (Riley and Tait, 2020).
In recent studies, a significant association between serum/plasma mtDNA levels and various diseases such as trauma or viral and bacterial infections has been demonstrated (Cossarizza et al., 2011). These findings suggest that cell-free mtDNA is a marker for cell death and pathogen-induced disease severity (Gambardella et al., 2019). The presence of cell-free mtDNA in the plasma of COVID-19 patients has been reported, and there is a significant relationship between the high level of mtDNA and the disease severity (Edinger et al., 2022; Leonard et al., 2022). However, the source and mechanism of releasing mtDNA in response to SARS-CoV-2 needs further investigation. The ability of mtDNA to activate TLR-9, cGAS and inflammasomes has made circulating mtDNA a candidate for the pro-inflammatory response in COVID-19 patients, especially in the elderly (Storci et al., 2020b).
In addition to mitochondrial DNA, other mitochondrial DAMPs have also been investigated in COVID-19 (Garg et al., 2022). Mitochondrial N-formylmethionine peptides contribute to neutrophil hyperactivation in COVID-19 patients (Kuley et al., 2023). Higher production of anti-cardiolipin antibodies in patients with COVID-19 may also be related to DAMP activity of mitochondrial membrane cardiolipin (Bhowal et al., 2023). Although studies on mitochondrial RNAs such as mitochondrial long non-coding RNAs (mt-lncRNAs) and mitochondrial small RNAs (mt-sRNAs) are limited, it has been suggested that double-stranded mitochondrial RNAs (mtRNA) triggers antiviral signaling. Cytosolic mitochondrial RNA is recognized by RIG-I or MDA5 and ultimately leads to immune activation. A recent study showed that the expression of mt-sRNAs may change during COVID-19 recovery, suggesting these relatively unknown RNAs may be good candidates for further studies (Pozzi, 2022; Marchi et al., 2023).
Activation of the cGAS-STING pathway by released mtDNA
In addition to the MAVS pathway, mtDNA -since it acts as a DAMP- could also be involved in the immune response. As mentioned, released mtDNA could activate both the NF-κB signaling pathway and the cGAS-STING pathway. Therefore, cell-free mtDNA can activate innate immune signaling pathways (Mills et al., 2017) (See Figure 1).
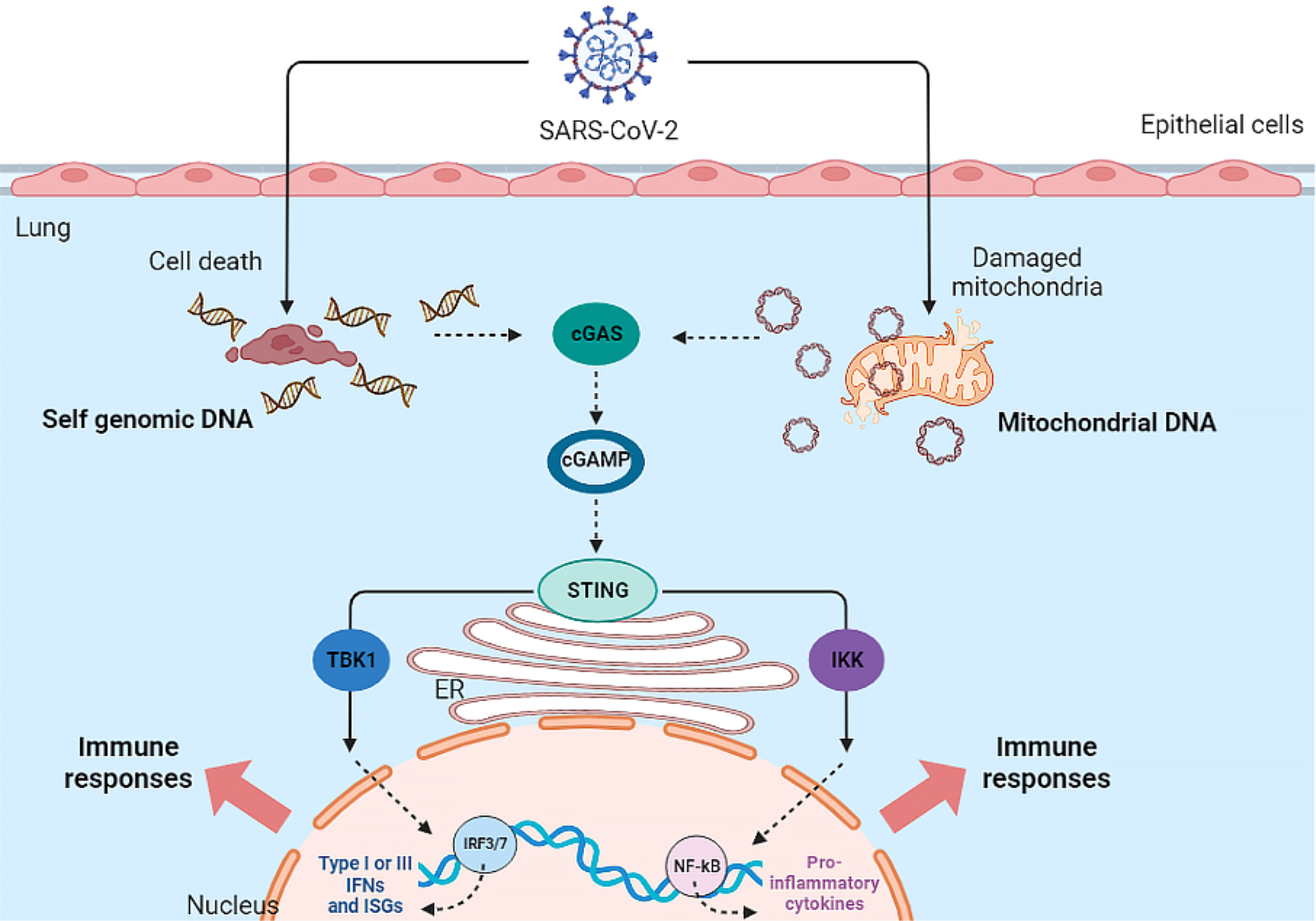
Figure 1 Activation of cGAS-STING Pathway by SARS-CoV-2 infection. The SARS-CoV-2 virus indirectly activates the cGAS-STING pathway and ultimately the inflammatory response in the host cell.
The cGAS-STING pathway is activated by mtDNA in various cells such as macrophages, dendritic cells (DCs), endothelia, and epithelia, resulting in the production of type I IFN and the release of inflammatory cytokines. In addition, the cGAS-STING signaling pathway interacts with apoptosis, necrosis, pyroptosis, and also autophagy (Ma et al., 2020). To inhibit or limit cGAS activation, viruses use various strategies to destroy or reduce cGAS. Dengue virus NS2B3 protease cleaves cGAS in order to suppress its activation (Bhattacharya et al., 2023). Papain-like protease (PL-pro) in HCoV-NL63 coronavirus disrupts STING oligomerization and ultimately leads to virus escape from the innate immune system (Sun et al., 2012). Manipulation of the cGAS-STING pathway by PLpro has also been reported in SARS-CoV, resulting in the regulation of the host NF-κB and IFN pathways (Ma and Damania, 2016).
Recent studies have attributed a delayed cGAS-STING response to SARS-CoV-2 infection. At the onset of SARS-CoV-2 infection, the virus ORF-9b protein inhibits the induction of type I and III IFNs by inhibiting the cGAS-STING signaling pathway. ORF-9b exerts its inhibitory effect by interacting with STING and inhibiting TBK1 phosphorylation (Han et al., 2021). Studies have shown that other SARS-CoV-2 proteins, ORF-3a, ORF-10, NSP7, NSP8, PLpro and 3CL protease, can also inhibit the STING pathway and the subsequent interferon response, leading to viral escape from innate immunity (Rui et al., 2021; Han et al., 2022; Deng et al., 2023b; Deng et al., 2023a). However, in the secondary phase of COVID-19, damaged host DNA could lead to STING over-activation, release of IFN-β, and cytokine storm following IRF-3 and NF-κB activation (Berthelot and Lioté, 2020).
Mitochondria-induced inflammasome activation
Inflammasomes are macromolecular immune complexes that are activated in response to PAMPs and DAMPs and lead to the activation, maturation, and release of pro-inflammatory cytokines. Inflammasomes play a dual role in viral infection. Activated inflammasomes provide inflammatory caspases to induce pyroptosis and cytokine production, leading to the elimination of virus-infected cells. However, impaired inflammasome activation could lead to a cytokine storm and hyperinflammation (Wang et al., 2023). Several molecular and cellular events have been shown to activate inflammasomes. Many studies have suggested the role of ROS, mitochondrial dysfunction, and mtDNA in NLRP3 inflammasome activation (Kelley et al., 2019). MAVS also physically interacts with the NLRP3, and it has been suggested that MAVS is required for the NLRP3 inflammasome activation by viral infections (Franchi et al., 2014). NLRP3 inflammasome activity is negatively regulated by autophagy (Zhou et al., 2011). In addition, free DNA or mtDNA can activate the AIM2 and NLRP3 inflammasomes (Uresti-Rivera and García-Hernández, 2023).
Aberrant inflammasome activation has been reported in COVID-19 patients (Aymonnier et al., 2022). SARS-CoV-2 activates inflammasomes through several pathways. SARS-CoV-2 infection can lead to increased expression and synthesis of the NLRP3 and IL-1β by upregulating the NF-κB pathway. SARS-CoV-2 proteins including S, E, and NSP2 stimulate NLRP3 transcription Also, viral N protein directly interacts with NLRP3 to induce inflammasome formation and subsequent inflammatory response. On the other hand, the inflammasomes can be activated in a way related to dsDNA released from infected epithelial cells and mtDNA released from damaged mitochondria. Mitochondrial ROS and lysosomal degradation further activate the NLRP3 inflammasome (Dutta et al., 2022; Deng C-H. et al., 2023).
Mitochondrial apoptosis pathway
Cellular apoptosis is a final resort in defense against invading pathogens. Programmed cell death eliminates the infected cell and limits the production of viruses. In addition, activation of antiviral transcription and cell death are necessary to control viral infection and provide long-term immunity (Orzalli and Kagan, 2017). The three main pathways for activating the caspase and apoptosis signals in mammalian cells include; the extrinsic pathway, intrinsic pathway (mitochondrial pathway), and cell death mediated by granzyme (Murthy et al., 2020). Mitochondria are directly and indirectly involved in apoptosis (Ghosh and Sil, 2021).
The intrinsic pathway is activated in response to cellular stress and requires mitochondrial outer membrane permeability. This permeability is induced by BAX/BAK proteins which are in the cytoplasm inactivated by BCL-2 protein at a steady state. In response to apoptotic stimulation, BAX/BAK is transported into the mitochondria and undergoes conformational changes, releasing cytochrome c from mitochondria and activation of downstream caspases. Caspase-9 is activated and leads to cell death via cleavage of caspase-3 and caspase-7 (Orzalli and Kagan, 2017).
Disruption of mitochondrial fusion/fission machinery can also lead to mitochondrial destruction and the initiation of apoptotic processes (Scaini et al., 2017). Loss of mitochondrial membrane potential (MMP, Δψm) leads to an imbalance between outer and inner membrane potentials, arresting normal cellular biosynthetic functions and bioenergetics within the cell. MMP changes occur during the pathogenesis of viral proteins, toxins, and pro-oxidants. Long-term loss of MMP leads to serious irreparable cell damage. Therefore, any viral agent that affects MMP has a major effect on cellular fate, either by inducing or inhibiting cell death (Reshi et al., 2014).
On the other hand, mitochondrial apoptosis could inhibit inflammatory signaling initiated by mtDNA. The role of mtDNA in initiating cGAS-STING mediated IFN response and inhibition of this inflammatory response by activation of caspase-9 has been demonstrated. Mitochondria have a dual capacity; on the one hand, using mtDNA to induce an IFN response, and on the other hand, with the help of caspase, it inactivates this response (Murthy et al., 2020). The mechanism of reducing the inflammatory response by the cascade of apoptotic caspases has recently been identified. Caspase-3 prevents the overproduction of type I IFN during viral infection by cleavage of cGAS, IRF-3, and MAVS. By inhibiting caspases, mitochondrial permeability leads to activation of cGAS-STING and transcription of NF-κB. The cytokines induced by NF-κB could lead to macrophage activation (Murthy et al., 2020; Riley and Tait, 2020). Viruses utilize a variety of strategies to prevent apoptosis; such as inducing the expression of survival genes such as anti-apoptotic BCL-2 proteins. (+)ssRNA viruses appear to activate mitophagy to remove damaged mitochondria and thus prevent apoptosis. SARS-CoV has increased the survival of infected cells by inhibiting apoptosis through mitochondrial fusion (Prasun, 2020). Conversely, SARS-CoV 3a protein could activate mitochondrial cell death pathways (Padhan et al., 2008). Also, MERS-CoV effectively activates extrinsic and intrinsic apoptotic pathways in T cells (Chu et al., 2016).
SARS-CoV-2 infection in lung epithelial cells activates caspase-8 and also targets apoptosis and the processing of inflammatory cytokines such as IL-1β into the active form (Li et al., 2020). SARS-CoV-2 ORF-3a and ORF-7b induces apoptosis in vitro (Ren et al., 2020; Yang et al., 2021). A recent study demonstrated that SARS-CoV-2 could induce both extrinsic and intrinsic apoptotic pathways (Liu et al., 2021a).
Autophagy and mitophagy
Autophagy is responsible for the selective removal of dysfunctional organelles, intracellular pathogens, and misplaced proteins, as well as the regulation of the immune response. The autophagy process is induced by Atg genes and is regulated by several signaling pathways such as AMPK, MAPK/ERK, and PI3K/Akt. mTOR is a negative regulator of autophagy. The dual role of autophagy in the formation and regulation of NETs has been proven. Thus autophagy is closely related to the host inflammatory response. Autophagy has a protective role in reducing the excessive release of cytokines in ARDS (Cheng et al., 2020; Cicco et al., 2020).
During viral infections, TLR activation induces autophagy and enhances IFN production, while negative regulation of autophagy helps terminate TLR signaling. Studies also show the induction of STING-dependent autophagy during viral infections. Some coronaviruses negatively regulate the IFN response through autophagic degradation of key molecules in the IFN signaling cascade. Manipulation of the autophagy process by various viruses has been reported (García-Pérez et al., 2020). SARS-CoV ORF-9b protein could induce autophagy and activate NF-κB (Shi et al., 2014).
The selective destruction of mitochondria by the process of autophagy is called mitophagy. Under normal conditions, damaged mitochondria are removed from the cell by mitophagy. The proteins involved in this pathway are PINK1 and PARKIN. Studies confirm that mice without the parkin or pink1 genes show higher levels of pro-inflammatory IFN-β and IL-6 than wild-type mice. An increase in cell-free mtDNA has also been observed in mice without parkin (Riley and Tait, 2020).
Mitophagy is also manipulated by various factors including viral infections. Some viruses directly or indirectly control the mitophagy process with different strategies. This promotes infection and reduces the host innate immune responses. Viral infections inhibit inflammasome activation by inducing mitophagy. In the absence of mitophagy, mitochondrial accumulation could increase MAVS levels and thus enhance its antiviral signaling pathway. Therefore, mitophagy induced by viral infection leads to decreased MAVS signaling and IFN-I response (Chen T. et al., 2023). (+)ssRNA viruses could activate mitophagy to remove damaged mitochondria and prevent apoptosis. SARS-CoV targets mitophagy with the help of ORF-9b protein (Gatti et al., 2020).
SARS-CoV-2 NSP8 induces mitophagy and incomplete autophagy in vivo (Zong et al., 2023). SARS-CoV-2 ORF-3c impairs autophagy in vivo (Mozzi et al., 2023). Also, SARS-CoV-2 ORF-7a initiates autophagy and restricts autophagosome-lysosome fusion to promote virus replication (Hou et al., 2023). A 2023 study showed that both S1 and RBD proteins, by inhibiting mitophagy and increasing mtROS, lead to IL-18 expression and inflammasome NLRP3 activation in vivo and in vitro (Liang et al., 2023).
ROS and antioxidant defense system
ROS are produced during mitochondrial oxidative metabolism or in response to ultraviolet (UV) radiation, xenobiotics, bacterial invasion, and viral infection. Mitochondria are thought to play a major role in the generation of intracellular ROS in most cell types. ROS participates in cellular signaling as secondary messengers and could regulate hormone action, cytokines, apoptosis, and immunomodulation (Reshi et al., 2014; El-Amine et al., 2018). ROS have a role in innate immunity as defense mechanism as well as in cell types involved in the acquired immune response (Yang et al., 2013).
Oxidative damage to DNA, proteins, and lipids is associated with increased ROS production, mitochondrial dysfunction, and eventually aging and cell death. Continuous production of ROS by mitochondria throughout cell life causes age-related chronic oxidative stress which leads to oxidative modification or deletion of bases especially in mtDNA (Giorgi et al., 2018). Overproduction of ROS and deficiency in antioxidant defense systems lead to oxidative stress, a condition that may contribute to various human diseases and is common during viral invasion (Reshi et al., 2018).
The main target of ROS which are produced inside a cell during viral infection is the mtDNA. Because mitochondrial ATP production requires proteins from the nuclear and mitochondrial genomes, damage to the mtDNA by ROS disrupts oxidative ATP production, although ROS have other cellular targets (Shokolenko et al., 2009; Reshi et al., 2018). Oxidative stress caused by the virus through the production of ROS is important for the life cycle and pathogenicity of the virus (Bermano et al., 2021). Oxidative stress could reduce the function of the host immune system. Almost all viruses including RNA viruses cause cell death by producing oxidative stress in infected cells. In hepatitis C virus infection and HIV, oxidative stress always plays a significant role in pathogenesis (Couret and Chang, 2016; Ivanov et al., 2018; Reshi et al., 2018). Respiratory viral infections are associated with inhibition of Nrf2 antioxidant pathways and/or activation of NF-κB signaling, leading to inflammation and oxidative stress. Several respiratory viruses cause the formation of dysregulated ROS as a result of increased inflammatory cells at the site of infection. Furthermore, viral infections may disrupt antioxidant mechanisms, leading to an imbalanced status and consequent oxidative cell damage (Komaravelli and Casola, 2014). Several studies suggested that the onset of severe lung injury in SARS-CoV patients depends on the activation of the oxidative stress system, which is coupled with innate immunity and activates transcription factors such as NF-κB, leading to an intensified pro-inflammatory host response (Delgado-Roche and Mesta, 2020). Lin et al. showed that SARS-CoV 3CLpro caused a significant increase in ROS production in HL-CZ cells. They also suggested that the ROS-activated NF-κB signaling pathway, induced by SARS-CoV 3CLpro, may be a key player in the pathophysiology of SARS-CoV (Lin et al., 2006).
Biopsy of COVID-19 patients demonstrated decreased expression of genes related to the Nrf2 pathway, which may be associated with viral oxidative cell damage (Olagnier et al., 2020). Also, a recent study demonstrated that serum ROS levels were significantly higher in COVID-19 patients compared to healthy controls and in ICU patients compared to non-ICU patients (Ahmed et al., 2023). The high neutrophil-to-lymphocyte ratio described in the severe form of COVID-19 is associated with excessive levels of ROS. This increases the cascade of biological events that drive the pathological host response (Laforge et al., 2020). A recent study reported that the release of TNF-α during a cytokine storm intensified ROS production through a positive feedback loop. Production of TNF-α-mediated ROS may be an important contributing factor to the observed damage to distal tissue such as the brain in COVID-19 patients (Wichmann et al., 2020). ROS storms may also occur in response to and/or lead to the breakdown of the antioxidant system during COVID-19. In support of this argument, a recent study showed that SARS-COV-2 leads to change in the intracellular/extracellular redox balance in the host immune cells, and the antioxidant defense was inhibited in the upper respiratory tract (Tavassolifar et al., 2023). Another study suggested that the cytokine storm in severe COVID-19 patients leads to a decrease in the antioxidant defense of endothelial cells through downregulation of the Nrf2 transcription factor (Rodrigues et al., 2023). Damaged mitochondria increase ROS and production of pro-inflammatory cytokine, along with the release of mtDNA leading to cell death, inflammation, and tissue damage. Taken together, all of these events can cause oxidative stress, hyperferritinemia, blood clotting, and thrombosis which are all reported in severe COVID-19 (Iessi et al., 2020).
Mitochondrial metabolism
Mitochondria known for their vital role in cell bioenergy and metabolism, are central sites for metabolism including the TCA (Krebs) cycle, fatty acid β-oxidation (FAO), and OXPHOS (Garaude, 2019).
Studies have shown that many immune signaling pathways are highly integrated with cellular metabolism, which not only provides fuel for active cells but also provides guidance for deciding cellular fate. These studies have led to a new field of research called “immunometabolism”. Thus, mitochondrial metabolism is one of the important factors in innate and acquired immunity (Weinberg et al., 2015), since mitochondria can modulate metabolic and physiological states in different types of immune cells (de Souza Breda et al., 2019).
Viruses hijack host cell metabolism and cause alterations in cellular and physiological functions (Thaker et al., 2019). The function of central metabolic pathways, such as OXPHOS is often altered by viruses to provide energy, biosynthetic resources, or evade the immune system to facilitate their replication (Qu et al., 2019b). Reprogramming of various aspects of host central carbon metabolism has been reported by both DNA and RNA viruses, including increased glycolysis, and increased pentose phosphate activity to support nucleotide production, amino acid production, and lipid synthesis (Thaker et al., 2019). Also, since changes in amino acid metabolism are closely related to inflammatory and immune responses, amino acid metabolism can play a role in controlling pathogen infection and regulating inflammation. Arginine (Arg) has been proposed to be a key amino acid for viral replication of many DNA and RNA viruses (Tomé, 2021). Hijacking of host lipid metabolism by some viruses to improve viral replication has also been suggested (Gong et al., 2023).
Biochemical manifestations specific to SARS-CoV-2 infection include an acute inflammatory condition with cytokine storm and oxidative stress with devastating downstream effects such as chronic hypoxia, acidosis, hypercoagulation, and changes in aerobic glycolytic metabolism (Shenoy, 2020). Transcriptome data analysis showed an increase in the expression of genes associated with OXPHOS in both PBMC and in bronchoalveolar lavage fluid (BALF) of COVID-19 patients (Gardinassi et al., 2020). Ajaz et al. evaluated mitochondrial function in PBMC of COVID-19 patients. ATP-dependent respiration and maximal respiration represent a modified mitochondrial functional response to SARS-CoV-2 infection. This study confirmed mitochondrial dysfunction and metabolic alterations with increased glycolysis in PBMC of SARS-CoV-2 infected patients (Ajaz et al., 2021). Another study confirmed that the SARS-CoV-2 induces glycolysis and one-carbon metabolism and further supports viral RNA and protein expression, proliferation, and cytopathic effect (Zhang et al., 2021). SARS-CoV-2 spike protein subunits in human pulmonary microvascular endothelial cells showed changes in mitochondrial metabolism, indicating a change from aerobic to anaerobic metabolism (Zekri-Nechar et al., 2022). SARS-CoV-2 ORF-3c alters mitochondrial metabolism, causing a shift from glucose oxidation to fatty acids and increased oxidative phosphorylation (Mozzi et al., 2023). Also, COVID-19 patients have shown alterations in both lipid and amino acid metabolisms compared to healthy controls (Masoodi et al., 2022). Lipid metabolism is altered in COVID-19 patients through direct cellular infection as well as systemic inflammatory response. Elevated long-chain polyunsaturated fatty acid (PUFA) levels, in the absence of elevated long-chain acylcarnitines, which may reflect impaired mitochondrial fatty acid oxidation, suggest a possible role for phospholipase A2 in COVID-19 (Casari et al., 2021).
Thus, the immune‐metabolic phenotype, including mitochondrial targeting could be used to understand the pathogenesis of the disease and possible treatments (Thompson et al., 2020). Figure 2 summarizes the changes in mitochondria-related events in SARS-CoV-2 infection.
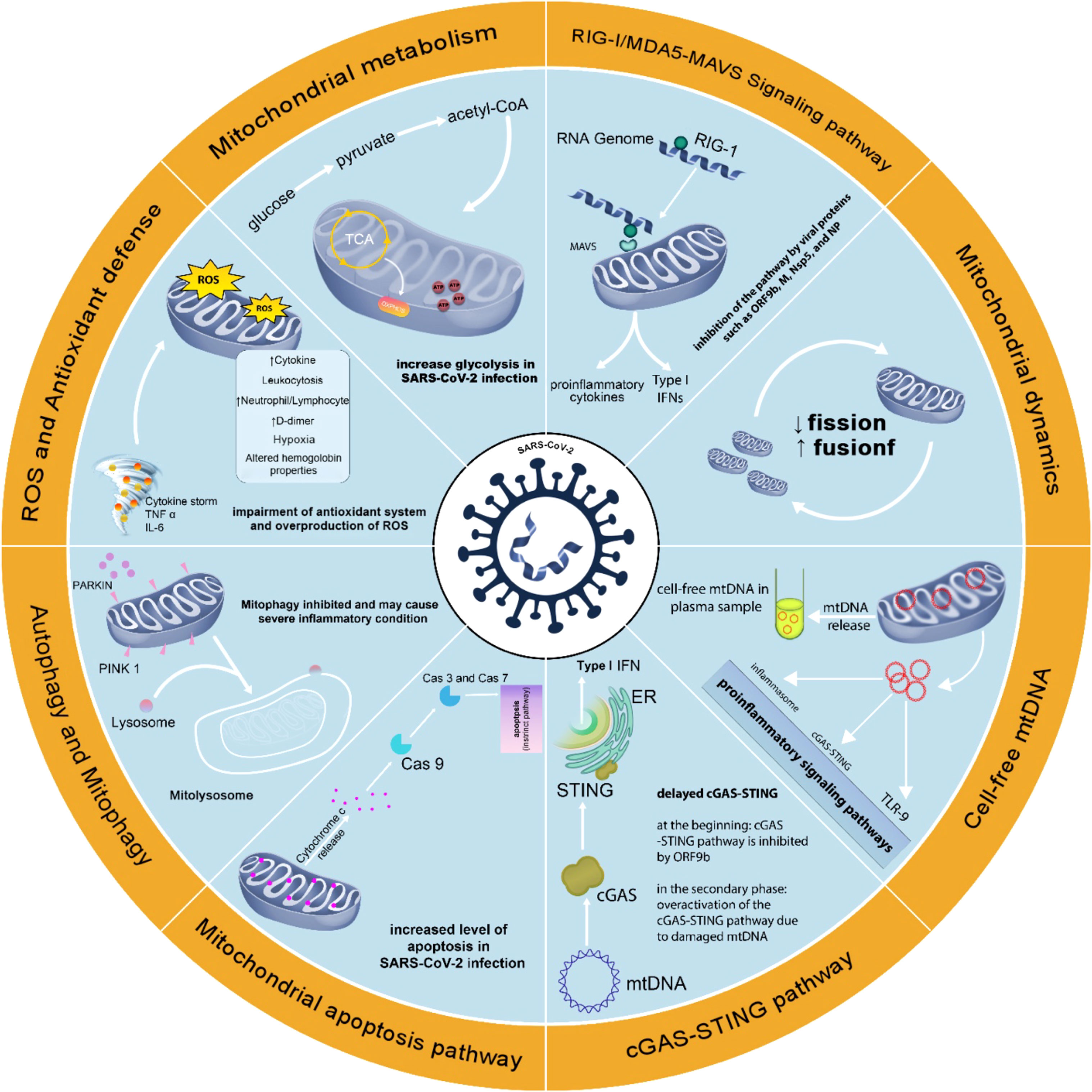
Figure 2 Changes in mitochondria-related events in SARS-CoV-2 infection. Changes in mitochondrial function in SARS-CoV-2 infection support the role of this organelle in COVID-19. Attention to mitochondrial function in SARS-CoV-2 infection may help better understand the pathophysiology of COVID-19 disease and achieve effective methods of prevention, diagnosis, and treatment.
Mitochondria-associated membranes
Mitochondria-associated membranes (MAMs), the points of contact between the ER and mitochondria, have been implicated in autophagy, apoptosis, mitochondrial dynamics, and activation of NLRP3 inflammasome (Namgaladze et al., 2019). In addition, recent studies have shown that some viruses exploit multiple ER membrane machineries to facilitate different stages of their life cycle (Woo et al., 2023). A 2023 study showed that hepatitis C virus replication requires the integrity of MAMs (Duponchel et al., 2023). A recent study by Jiao et al., showed that SARS-CoV-2 NSP6 induced ER stress which ultimately led to lysosomal degradation of STING and decreased IFN production (Jiao et al., 2023).
Age-related mitochondrial dysfunction
There is a link between mitochondrial dysfunction and aging, age-related disorders, and metabolic syndromes. Age-related mitochondrial dysfunction could also be due to an imbalance of mitochondrial dynamics, autophagy and mitophagy, and biogenesis that result in the accumulation of damaged mitochondria (López-Lluch, 2017).
Immunosenescence has been described as age-related deterioration of the immune system. Accumulation of mitochondrial damage combined with reduction of the efficiency in energy production affects the capacity of the immune system through reducing the capacity to respond to viral infections by lower IFN-I release. Mitochondrial dysfunction releases damage signals to the cytosol, which leads to the release of inflammatory cytokines and the activation of the inflammasome, leading to chronic inflammation that is associated with aging and age-related diseases (Ayala et al., 2020).
Indeed, the decay of mitochondrial function is key to the progression of aging and age-related diseases. Mitochondrial dysfunction is also implicated in deteriorating immune systems as a key factor in inflammation, higher susceptibility to viral infections, and T cell immunodeficiency found in the elderly and age-related diseases (McGuire, 2019).
The COVID-19 severity is associated with impaired immune function found in the elderly, and more recently this aged immunity has been thought to intensify COVID-19. In addition, many age-related diseases such as metabolic syndromes, type 2 diabetes, obesity, hypertension, and cardiovascular disease exacerbate COVID-19 (Zheng et al., 2020). Age-related mitochondrial dynamic dysfunction has been reported in many studies. Therefore, the accumulation of inefficient mitochondria may be one of the causes of high COVID-19 mortality in the elderly, mainly due to mitochondrial dysfunction in senescent macrophages and lymphocytes that intensify the inflammatory response (Ayala et al., 2020).
Therefore, the study of mitochondrial immunity against SARS-CoV-2 could provide insight into why older people may have difficulty coping with COVID-19 (Ganji and Reddy, 2021).
Cell-free mitochondrial DNA as a non-invasive biomarker
It is not unreasonable to expect that local and systemic changes following cell injury and disease are rapidly reflected in the cellular and molecular components of the blood (Hsiao and Hoppel, 2018). Because viruses can directly or indirectly modulate mitochondrial function in different cell types, the occurrence of mitochondrial alterations in blood cells could be a reflection of tissue mitochondrial changes (Cruz and Kang, 2018; Nunn et al., 2020).
Changes in somatic mtDNA have been considered as one of the primary aims in diagnosis and predicting various types of diseases, including infectious diseases (Qu et al., 2019a). One of the advantages of circulating mtDNA is the ability to be used as a liquid biopsy (González-Masiá et al., 2013). In recent years, mtDNA content has been considered a biomarker of mitochondrial function. mtDNA is also studied for disease-related changes in blood samples and other body fluids (Rosa et al., 2020). Among the advantages of mtDNA compared to nDNA are shorter length, easier and cheaper screening, high number of copies, and found in various fluids (Scozzi et al., 2021).
A recent study evaluated cf-mtDNA levels in the plasma of COVID-19 patients. Results showed plasma MT-CYTB levels had similar AUCs for predicting patient mortality compared with LDH, ferritin, and D-dimer. This study suggested high levels of circulating mtDNA as a poor biomarker for the prognosis of COVID-19 (Scozzi et al., 2021).
Although circulating mtDNA is one of the most common mtDAMPs evaluated as biomarker, other mitochondrial DAMPs may also be novel biomarker candidates (Shen et al., 2022). However, studies on mitochondria as a biomarker for the diagnosis or prognosis of COVID-19 are very limited.
Mitochondrial targeted therapy for the treatment of COVID-19
Therapeutically targeting mitochondrial dysfunction might represent a successful strategy for disease (Thompson et al., 2020). As mitochondrial dysfunction has been shown to play an important role in inhibiting the antiviral response in infected cells. It seems that restoring mitochondrial function and creating new healthy mitochondria could enhance cellular resistance to infection-induced stress by regulating bioenergy and the innate immune response (Li et al., 2019). Therapies that improve mitochondrial function and inhibit inflammation may be some of the more effective treatments for COVID-19 (Holder and Reddy, 2021). Decreased inefficient mitochondria could diminish the inflammatory response to SARS-CoV-2 infection (Zheng et al., 2020). Also, a 2023 study reported that lower peripheral levels of mtDNA were associated with lower cellular immunity due to COVID-19 vaccination, suggesting that maintaining mitochondrial integrity is also essential for adequate immune responses to vaccination (Ikezaki et al., 2023). Possible therapeutic strategies for COVID-19 based on mitochondria as classified in Figure 3 are reviewed in this section.
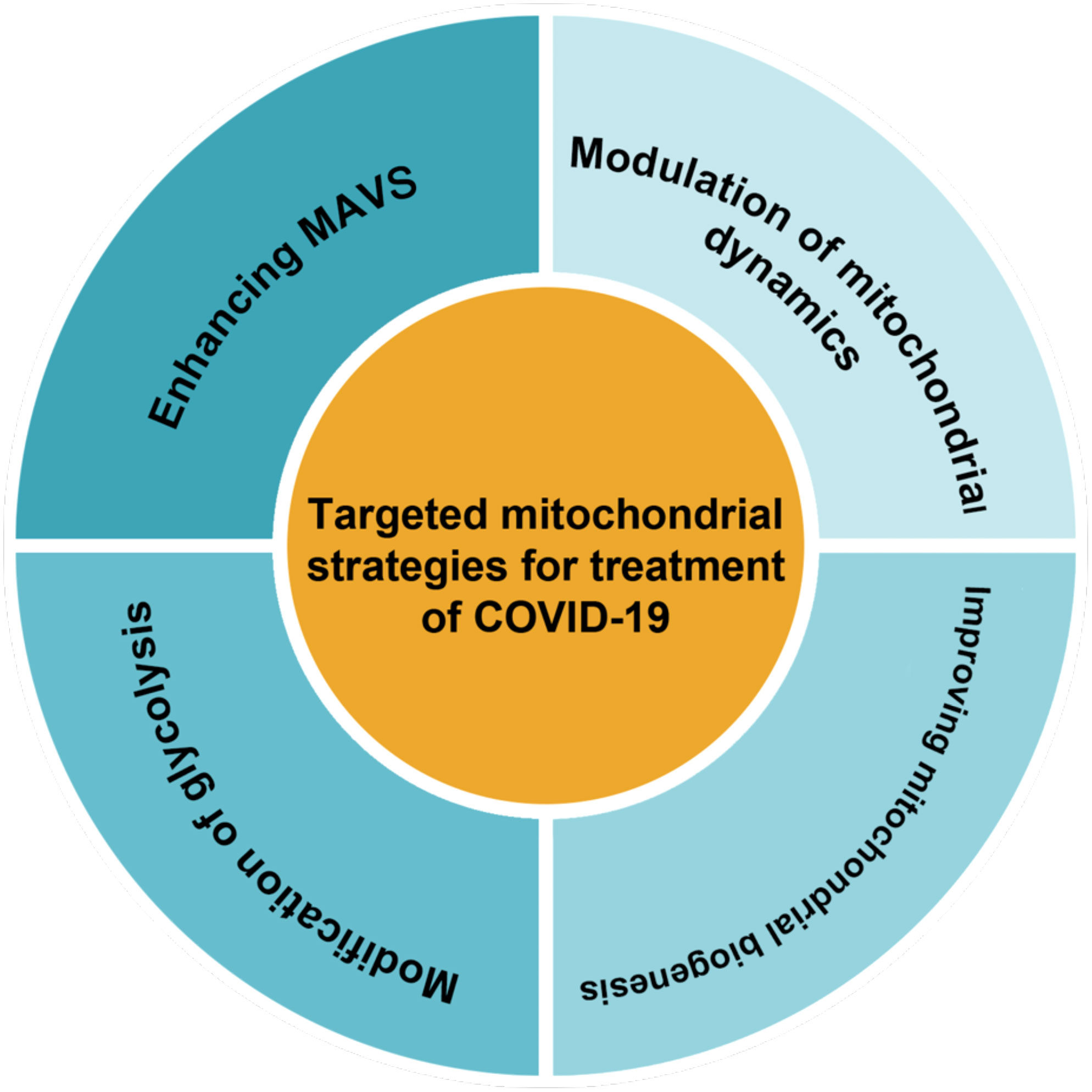
Figure 3 There are several methods for classifying targeted mitochondrial strategies.
Modification of glycolysis
Metabolic phenotypes resulting from viral infection often reflect metabolic changes observed in cancer cells, such as positive regulation of nutrient intake and anabolism to support viral replication or rapid cell growth, respectively (Thaker et al., 2019). Recently targeted metabolic reprogramming of immune cells or cancerous cells in the treatment of various diseases such as cancers has been considered (Castro et al., 2019; Faubert et al., 2020). In addition, studies have been performed on targeted metabolic reprogramming to improve the effectiveness of viral therapy (Kennedy et al., 2020). The metabolic manipulation by SARS-CoV-2 stimulates an enhanced inflammatory response that helps symptoms of COVID-19. Targeting the mitochondrial metabolic pathways could help define new strategies for COVID-19 treatment (Ajaz et al., 2021). An enhanced glycolytic pathway upon SARS-CoV-2 infection is one example of viral infection-induced metabolic reprogramming. A study reported the use of a known glycolytic inhibitor, 2-deoxy-d-glucose (2-DG), to effectively counter SARS-CoV-2 replication in host cells (Bhatt et al., 2022).
Enhancing MAVS
Improving the MAVS pathway may be a possible treatment. Babajani et al. proposed a hypothesis for targeted therapy of COVID-19 with overexpressed MAVS protein from manipulated mesenchymal stem cells that express viral S protein to boost the innate immune response and the production of appropriate IFNs (Babajani et al., 2021).
Modulation of mitochondrial dynamics
A delay or absence of types I and III IFN responses with strong cytokine production was observed in COVID-19. Because there is a negative feedback loop between autophagy and the IFN-I response, inhibition or activation of autophagy is important depending on the stage of the SARS-CoV-2 infection. Inhibition of autophagy in the first stage of COVID-19 may lead to the regulation of the interferon antiviral response and inhibition of viral replication. Activation of autophagy in the late stage of the disease could lead to the removal of ROS, damaged organelles and reduce inflammation and balance the immune response. For these reasons, autophagy-related therapeutics should be selected according to the COVID-19 timeline (García-Pérez et al., 2020).
Improving mitochondrial biogenesis
Oxidative stress affects mitochondrial functions such as biogenesis and antioxidant defense mechanisms (Gao and Chen, 2022). The use of antioxidant compounds in SARS-CoV-2 infection is also recommended. These compounds scavenge ROS in a variety of ways and could therefore prevent the pathophysiological consequences of COVID-19 disease (Laforge et al., 2020; Camp et al., 2021). While ROS scavengers prevent the virus from entering the host cell, an effective dosage of ROS inducers could modify the reduction–oxidation (redox) potential in host cells to induce oxidative stress, resulting in the degradation of nascent RNA. It would largely affect viral RNA. Furthermore, the ROS causes cytotoxicity in infected cells as their DNA repair mechanism would be compromised (Nadhan et al., 2021). Oxidative stress and persistent pathogenesis in SARS-CoV-2 are almost certainly related. The cytokine storms and free radical storms should be considered in COVID-19 (Delgado-Roche and Mesta, 2020; Wu, 2020; de Oliveira et al., 2021).
Mitochondria and long-COVID
Post-COVID-19 syndrome, or “long-COVID”, is a term that describes various conditions, such as inflammation and the consequences of organ damage that persist for a long time after SARS-CoV-2 infection. Although the mechanism underlying the long-COVID is not yet elucidated, the role of mitochondrial dysfunction and the subsequent immune response has recently received attention. It has been suggested that some post-COVID complications are related to mitochondrial dysfunction (Chen T-H. et al., 2023). The long-COVID status could be caused by damage related to the host response to initial severe infection, such as cytokine storms, and could lead to oxidative stress and oxidative and inflammatory damage. This suggests that antioxidant therapies may also be useful in the treatment of long-COVID (Wood et al., 2020; Chen T-H. et al., 2023).
Conclusion
Manipulation of host mitochondria by SARS-CoV-2 plays a significant role in disease progression through modulation of various functions. The evidence indicates that SARS-CoV-2 infection could lead to modulation/disruption of the MAVS pathway, mitochondrial dynamics, apoptosis, autophagy and mitochondrial metabolism. Further research into the molecular mechanisms that hijack mitochondria in SARS-CoV-2 infection to suppress the immune system will provide insight into new treatment and prevention. Also, host mitochondria may be the key to effective and/or ineffective response in individuals, especially the elderly, to the COVID-19 vaccine. Improving or maintaining mitochondrial function may prevent dysfunction of mitochondria following viral infection. Targeted drug interventions that are effective in enhancing mitochondrial function may improve innate immunity against SARS-CoV-2 infection. In addition, evaluation of mitochondrial status and related markers in accessible tissues such as blood in response to SARS-CoV-2 may provide perspectives to predicting susceptibility to and the severity of COVID-19. High mtDNA copy number allows assessment of changes in different samples, which is also usually indicative of the mitochondrial biogenesis. Also, the small size of mtDNA provides an easy and cheap analysis method. However, the lack of studies on the role of other mitochondrial DAMPs in SARS-CoV-2 infection is noticeable.
Author contributions
SRM: article decision, planning, and writing. SS: literature search and reviewing and article writing. AES: manuscript revision. AG: article reviewing. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, S. A., Alahmadi, Y. M., Abdou, Y. A. (2023). The impact of serum levels of reactive oxygen and nitrogen species on the disease severity of COVID-19. Int. J. Mol. Sci. 24 (10), 8973. doi: 10.3390/ijms24108973
Ajaz, S., McPhail, M. J., Singh, K. K., Mujib, S., Trovato, F. M., Napoli, S., et al. (2021). Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiology-Cell Physiol. 320 (1), C57–C65. doi: 10.1152/ajpcell.00426.2020
Al Ojaimi, M., Salah, A., El-Hattab, A. W. (2022). Mitochondrial fission and fusion: molecular mechanisms, biological functions, and related disorders. Membranes 12 (9), 893. doi: 10.3390/membranes12090893
Ayala, DJM-F, Navas, P., López-Lluch, G. (2020). Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 111147, 111147-60. doi: 10.1016/j.exger.2020.111147
Aymonnier, K., Ng, J., Fredenburgh, L. E., Zambrano-Vera, K., Münzer, P., Gutch, S., et al. (2022). Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Advances. 6 (7), 2001–2013. doi: 10.1182/bloodadvances.2021005949
Babajani, A., Hosseini-Monfared, P., Abbaspour, S., Jamshidi, E., Niknejad, H. (2021). Targeted mitochondrial therapy with over-expressed MAVS protein from mesenchymal stem cells: A new therapeutic approach for COVID-19. Front. Cell Dev. Biol. 9, 1475. doi: 10.3389/fcell.2021.695362
Bai, C., Zhong, Q., Gao, G. F. (2022). Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 65 (2), 280–294. doi: 10.1007/s11427-021-1964-4
Banoth, B., Cassel, S. L. (2018). Mitochondria in innate immune signaling. Trans. Res. 202, 52–68. doi: 10.1016/j.trsl.2018.07.014
Bermano, G., Méplan, C., Mercer, D. K., Hesketh, J. E. (2021). Selenium and viral infection: are there lessons for COVID-19? Br. J. Nutr. 125 (6), 618–627. doi: 10.1017/S0007114520003128
Berthelot, J.-M., Lioté, F. (2020). COVID-19 as a STING disorder with delayed over-secretion of interferon-beta. EBioMedicine 56, 102801. doi: 10.1016/j.ebiom.2020.102801
Bhatt, A. N., Kumar, A., Rai, Y., Kumari, N., Vedagiri, D., Harshan, K. H., et al. (2022). Glycolytic inhibitor 2-deoxy-d-glucose attenuates SARS-CoV-2 multiplication in host cells and weakens the infective potential of progeny virions. Life Sci. 295, 120411. doi: 10.1016/j.lfs.2022.120411
Bhattacharya, M., Bhowmik, D., Tian, Y., He, H., Zhu, F., Yin, Q. (2023). The Dengue virus protease NS2B3 cleaves cyclic GMP-AMP synthase to suppress cGAS activation. J. Biol. Chem. 299 (3), 102986-96. doi: 10.1016/j.jbc.2023.102986
Bhowal, C., Ghosh, S., Ghatak, D., De, R. (2023). Pathophysiological involvement of host mitochondria in SARS-CoV-2 infection that causes COVID-19: a comprehensive evidential insight. Mol. Cell Biochem. 478 (6), 1325–1343. doi: 10.1007/s11010-022-04593-z
Bojkova, D., Klann, K., Koch, B., Widera, M., Krause, D., Ciesek, S., et al. (2020). Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583 (7816), 469–472. doi: 10.1038/s41586-020-2332-7
Camp, O. G., Bai, D., Gonullu, D. C., Nayak, N., Abu-Soud, H. M. (2021). Melatonin interferes with COVID-19 at several distinct ROS-related steps. J. Inorg Biochem. 111546, 111546-54. doi: 10.1016/j.jinorgbio.2021.111546
Cao, Z., Zhao, M., Sun, H., Hu, L., Chen, Y., Fan, Z. (2022). Roles of mitochondria in neutrophils. Front. Immunol. 13, 934444. doi: 10.3389/fimmu.2022.934444
Casari, I., Manfredi, M., Metharom, P., Falasca, M. (2021). Dissecting lipid metabolism alterations in SARS-CoV-2. Prog. Lipid Res. 82, 101092. doi: 10.1016/j.plipres.2021.101092
Castanier, C., Garcin, D., Vazquez, A., Arnoult, D. (2010). Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 11 (2), 133–138. doi: 10.1038/embor.2009.258
Castro, I., Sampaio-Marques, B., Ludovico, P. (2019). Targeting metabolic reprogramming in acute myeloid leukemia. Cells 8 (9), 967. doi: 10.3390/cells8090967
Chen, T.-H., Chang, C.-J., Hung, P.-H. (2023). Possible pathogenesis and prevention of long COVID: SARS-CoV-2-induced mitochondrial disorder. Int. J. Mol. Sci. 24 (9), 8034. doi: 10.3390/ijms24098034
Chen, T., Tu, S., Ding, L., Jin, M., Chen, H., Zhou, H. (2023). The role of autophagy in viral infections. J. BioMed. Sci. 30 (1), 1–25. doi: 10.1186/s12929-023-00899-2
Cheng, Z., Dai, T., He, X., Zhang, Z., Xie, F., Wang, S., et al. (2020). The interactions between cGAS-STING pathway and pathogens. Signal Transduction Targeted Ther. 5 (1), 1–15. doi: 10.1038/s41392-020-0198-7
Chu, H., Zhou, J., Wong, B. H.-Y., Li, C., Chan, J. F.-W., Cheng, Z.-S., et al. (2016). Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J. Infect. diseases. 213 (6), 904–914. doi: 10.1093/infdis/jiv380
Cicco, S., Cicco, G., Racanelli, V., Vacca, A. (2020). Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm. 2020. doi: 10.1155/2020/7527953
Comish, P. B., Liu, M.-M., Huebinger, R., Carlson, D., Kang, R., Tang, D. (2022). The cGAS-STING pathway connects mitochondrial damage to inflammation in burn-induced acute lung injury in rat. Burns 48 (1), 168–175. doi: 10.1016/j.burns.2021.04.007
Corcoran, J. A., Saffran, H. A., Duguay, B. A., Smiley, J. R. (2009). Herpes simplex virus UL12. 5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol. 83 (6), 2601–2610. doi: 10.1128/JVI.02087-08
Cossarizza, A., Pinti, M., Nasi, M., Gibellini, L., Manzini, S., Roat, E., et al. (2011). Increased plasma levels of extracellular mitochondrial DNA during HIV infection: a new role for mitochondrial damage-associated molecular patterns during inflammation. Mitochondrion 11 (5), 750–755. doi: 10.1016/j.mito.2011.06.005
Couret, J., Chang, T. L. (2016). Reactive oxygen species in HIV infection. EC Microbiol. 3 (6), 597.
Cruz, C. S. D., Kang, M.-J. (2018). Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 41, 37–44. doi: 10.1016/j.mito.2017.12.001
Delgado-Roche, L., Mesta, F. (2020). Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 51 (5), 384–387. doi: 10.1016/j.arcmed.2020.04.019
Deng, C.-H., Li, T.-Q., Zhang, W., Zhao, Q., Wang, Y. (2023). Targeting inflammasome activation in viral infection: A therapeutic solution? Viruses 15 (7), 1451. doi: 10.3390/v15071451
Deng, J., Zheng, S. N., Xiao, Y., Nan, M. L., Zhang, J., Han, L., et al. (2023a). SARS-CoV-2 NSP8 suppresses type I and III IFN responses by modulating the RIG-I/MDA5, TRIF, and STING signaling pathways. J. Med. Virol. 95 (4), e28680. doi: 10.1002/jmv.28680
Deng, J., Zheng, Y., Zheng, S. N., Nan, M. L., Han, L., Zhang, J., et al. (2023b). SARS-CoV-2 NSP7 inhibits type I and III IFN production by targeting the RIG-I/MDA5, TRIF, and STING signaling pathways. J. Med. Virol. 95 (3), e28561. doi: 10.1002/jmv.28561
de Oliveira, A. A., Priviero, F., Lima, V. V., Webb, R. C., Nunes, K. P. (2021). COVID-19 and ROS storm: what is the forecast for hypertension. Am. J. Hypertens. 34 (8), 779. doi: 10.1093/ajh/hpab085
de Souza Breda, C. N., Davanzo, G. G., Basso, P. J., Câmara, N. O. S., Moraes-Vieira, P. M. M. (2019). Mitochondria as central hub of the immune system. Redox Biol. 26, 101255. doi: 10.1016/j.redox.2019.101255
Duponchel, S., Monnier, L., Molle, J., Bendridi, N., Alam, M. R., Gaballah, A., et al. (2023). Hepatitis C virus replication requires integrity of mitochondria-associated ER membranes. JHEP Rep. 5 (3), 100647. doi: 10.1016/j.jhepr.2022.100647
Dutta, D., Liu, J., Xiong, H. (2022). NLRP3 inflammasome activation and SARS-CoV-2-mediated hyperinflammation, cytokine storm and neurological syndromes. Int. J. Physiol. Pathophysiol Pharmacol. 14 (3), 138.
Edinger, F., Edinger, S., Koch, C., Markmann, M., Hecker, M., Sander, M., et al. (2022). Peak Plasma Levels of mtDNA Serve as a Predictive Biomarker for COVID-19 in-Hospital Mortality. J. Clin. Med. 11 (23), 7161. doi: 10.3390/jcm11237161
El-Amine, R., Germini, D., Zakharova, V. V., Tsfasman, T., Sheval, E. V., Louzada, R. A., et al. (2018). HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 15, 97–108. doi: 10.1016/j.redox.2017.11.024
Eriani, G., Martin, F. (2022). Viral and cellular translation during SARS-CoV-2 infection. FEBS Open Bio. 12 (9), 1584–1601. doi: 10.1002/2211-5463.13413
Faubert, B., Solmonson, A., DeBerardinis, R. J. (2020). Metabolic reprogramming and cancer progression. Science 368 (6487), eaaw5473-83. doi: 10.1126/science.aaw5473
Ferreira, A. R., Magalhaes, A. C., Camões, F., Gouveia, A., Vieira, M., Kagan, J. C., et al. (2016). Hepatitis C virus NS 3-4A inhibits the peroxisomal MAVS-dependent antiviral signalling response. J. Cell Mol. Med. 20 (4), 750–757. doi: 10.1111/jcmm.12801
Fields, J. A., Serger, E., Campos, S., Divakaruni, A. S., Kim, C., Smith, K., et al. (2016). HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol. Dis. 86, 154–169. doi: 10.1016/j.nbd.2015.11.015
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R., Ozkurede, U., Kim, Y.-G., Chakrabarti, A., et al. (2014). Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 193 (8), 4214–4222. doi: 10.4049/jimmunol.1400582
Fu, Y.-Z., Wang, S.-Y., Zheng, Z.-Q., Huang, Y., Li, W.-W., Xu, Z.-S., et al. (2021). SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol. Immunol. 18 (3), 613–620. doi: 10.1038/s41423-020-00571-x
Gabanella, F., Barbato, C., Corbi, N., Fiore, M., Petrella, C., de Vincentiis, M., et al. (2022). Exploring mitochondrial localization of SARS-CoV-2 RNA by padlock assay: A pilot study in human placenta. Int. J. Mol. Sci. 23 (4), 2100. doi: 10.3390/ijms23042100
Gambardella, S., Limanaqi, F., Ferese, R., Biagioni, F., Campopiano, R., Centonze, D., et al. (2019). ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front. Immunol. 10, 1064. doi: 10.3389/fimmu.2019.01064
Ganji, R., Reddy, P. H. (2021). Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front. Aging Neurosci. 12, 502. doi: 10.3389/fnagi.2020.614650
Ganta, K. K., Chaubey, B. (2019). Mitochondrial dysfunctions in HIV infection and antiviral drug treatment. Expert Opin. Drug Metab. Toxicol. 15 (12), 1043–1052. doi: 10.1080/17425255.2019.1692814
Gao, J., Chen, J. (2022). Clinical studies on curcumin in improving mitochondrial biogenesis. MEDS Clin. Med. 3 (7), 33–38. doi: 10.23977/medsc.2022.030706
Garaude, J. (2019). Reprogramming of mitochondrial metabolism by innate immunity. Curr. Opin. Immunol. 56, 17–23. doi: 10.1016/j.coi.2018.09.010
García-Pérez, B. E., González-Rojas, J. A., Salazar, M. I., Torres-Torres, C., Castrejón-Jiménez, N. S. (2020). Taming the autophagy as a strategy for treating COVID-19. Cells 9 (12), 2679. doi: 10.3390/cells9122679
Gardinassi, L. G., Souza, C. O., Sales-Campos, H., Fonseca, S. G. (2020). Immune and metabolic signatures of COVID-19 revealed by transcriptomics data reuse. Front. Immunol. 11, 1636. doi: 10.3389/fimmu.2020.01636
Garg, M., Johri, S., Chakraborty, K. (2022). Immunomodulatory role of mitochondrial DAMPs: a missing link in pathology? FEBS J. doi: 10.1111/febs.16563
Gatti, P., Ilamathi, H. S., Todkar, K., Germain, M. (2020). Mitochondria targeted viral replication and survival strategies—prospective on SARS-CoV-2. Front. Pharmacol. 11. doi: 10.3389/fphar.2020.578599
Ghosh, N., Sil, P. C. (2021). “Mitochondria and apoptosis,” in Mitochondrial Physiology and Vegetal Molecules (Academic Press), 127–149.
Giacomello, M., Pyakurel, A., Glytsou, C., Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21 (4), 204–224. doi: 10.1038/s41580-020-0210-7
Giorgi, C., Marchi, S., Simoes, I. C., Ren, Z., Morciano, G., Perrone, M., et al. (2018). Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell Mol. Biol. 340, 209–344. doi: 10.1016/bs.ircmb.2018.05.006
Gong, Z., Yan, Z., Liu, W., Luo, B. (2023). Oncogenic viruses and host lipid metabolism: a new perspective. J. Gen. Virol. 104 (6), 001861. doi: 10.1099/jgv.0.001861
González-Masiá, J. A., García-Olmo, D., García-Olmo, D. C. (2013). Circulating nucleic acids in plasma and serum (CNAPS): applications in oncology. Onco Targets Ther. 6, 819. doi: 10.2147/OTT.S44668
Han, L., Zheng, Y., Deng, J., Nan, M. L., Xiao, Y., Zhuang, M. W., et al. (2022). SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J. Med. Virol. 94 (11), 5174–5188. doi: 10.1002/jmv.27965
Han, L., Zhuang, M. W., Deng, J., Zheng, Y., Zhang, J., Nan, M. L., et al. (2021). SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J. Med. Virol. 93 (9), 5376–5389. doi: 10.1002/jmv.27050
Holder, K., Reddy, P. H. (2021). The COVID-19 effect on the immune system and mitochondrial dynamics in diabetes, obesity, and dementia. Neuroscientist. 27 (4), 331–339. doi: 10.1177/1073858420960443
Hou, P., Wang, X., Wang, H., Wang, T., Yu, Z., Xu, C., et al. (2023). The ORF7a protein of SARS-CoV-2 initiates autophagy and limits autophagosome-lysosome fusion via degradation of SNAP29 to promote virus replication. Autophagy 19 (2), 551–569. doi: 10.1080/15548627.2022.2084686
Hsiao, C.-P., Hoppel, C. (2018). Analyzing mitochondrial function in human peripheral blood mononuclear cells. Anal. Biochem. 549, 12–20. doi: 10.1016/j.ab.2018.03.003
Hummel, E., Piovesan, K., Berg, F., Herpertz, S., Kessler, H., Kumsta, R., et al. (2023). Mitochondrial DNA as a marker for treatment-response in post-traumatic stress disorder. Psychoneuroendocrinology 148, 105993. doi: 10.1016/j.psyneuen.2022.105993
Iessi, E., Cittadini, C., Anticoli, S., Fecchi, K., Matarrese, P., Ruggieri, A. (2020). Sex differences in antiviral immunity in SARS-CoV-2 infection: Mitochondria and mitomiR come into view. Acta Physiol. (Oxf). 231(2), e13571. doi: 10.1111/apha.13571
Ikezaki, H., Nomura, H., Shimono, N. (2023). Impact of peripheral mitochondrial DNA level on immune response after COVID-19 vaccination. Iscience. 26 (7), 107094-106. doi: 10.1016/j.isci.2023.107094
Ivanov, A. V., Khomich, O. A., Bartosch, B. (2018). Oxidative stress in hepatitis C infection. Liver. 1, 1–13. doi: 10.1016/B978-0-12-803951-9.00001-X
Jiang, H.-w., Zhang, H.-n., Meng, Q.-f., Xie, J., Li, Y., Chen, H., et al. (2020). SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol. Immunol. 17 (9), 998–1000. doi: 10.1038/s41423-020-0514-8
Jiao, P., Fan, W., Ma, X., Lin, R., Zhao, Y., Li, Y., et al. (2023). SARS-CoV-2 nonstructural protein 6 triggers endoplasmic reticulum stress-induced autophagy to degrade STING1. Autophagy, 1–19. doi: 10.1080/15548627.2023.2238579
Kelley, N., Jeltema, D., Duan, Y., He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20 (13), 3328. doi: 10.3390/ijms20133328
Kennedy, B. E., Sadek, M., Gujar, S. A. (2020). Targeted metabolic reprogramming to improve the efficacy of oncolytic virus therapy. Mol. Ther. 28 (6), 1417–1421. doi: 10.1016/j.ymthe.2020.03.014
Kim, S.-J., Ahn, D.-G., Syed, G. H., Siddiqui, A. (2018). The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 41, 21–27. doi: 10.1016/j.mito.2017.11.007
Kim, S.-J., Khan, M., Quan, J., Till, A., Subramani, S., Siddiqui, A. (2013). Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PloS Pathog. 9 (12), e1003722. doi: 10.1371/journal.ppat.1003722
Komaravelli, N., Casola, A. (2014). Respiratory viral infections and subversion of cellular antioxidant defenses. Journal of pharmacogenomics & pharmacoproteomics 5 (4), 1000141-61. doi: 10.4172/2153-0645.1000141
Kruk, S. K., Pacheco, S. E., Koenig, M. K., Bergerson, J. R., Gordon-Lipkin, E., McGuire, P. J. (2019). Vulnerability of pediatric patients with mitochondrial disease to vaccine-preventable diseases. J. Allergy Clin. Immunology: In Practice. 7 (7), 2415–8. e3. doi: 10.1016/j.jaip.2019.03.046
Kuley, R., Duvvuri, B., Wallin, J. J., Bui, N., Adona, M. V., O’Connor, N. G., et al. (2023). Mitochondrial N-formyl methionine peptides contribute to exaggerated neutrophil activation in patients with COVID-19. Virulence 14 (1), 2218077. doi: 10.1080/21505594.2023.2218077
Kuppusamy, M., Wankhar, W., Gurugubelli, K. R., Mahadevappa, V. H., Lepcha, L., kumar Choudhary, A. (2021). Angiotensin-converting enzyme 2 (ACE2): COVID 19 gate way to multiple organ failure syndromes. Respir. Physiol. Neurobiol. 283, 103548. doi: 10.1016/j.resp.2020.103548
Kuznetsov, A. V., Margreiter, R., Ausserlechner, M. J., Hagenbuchner, J. (2022). The complex interplay between mitochondria, ROS and entire cellular metabolism. Antioxidants 11 (10), 1995. doi: 10.3390/antiox11101995
Laforge, M., Elbim, C., Frère, C., Hémadi, M., Massaad, C., Nuss, P., et al. (2020). Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 20 (9), 515–516. doi: 10.1038/s41577-020-0407-1
Lai, J.-H., Luo, S.-F., Ho, L.-J. (2018). Operation of mitochondrial machinery in viral infection-induced immune responses. Biochem. Pharmacol. 156, 348–356. doi: 10.1016/j.bcp.2018.08.044
Leonard, A., Rolfsen, M., Ix, J., Odish, M., Coufal, N., Lam, M., et al. (2022). Circulating, cell-free mitochondrial DNA marks disease severity in COVID-19. B45. PREDICTING COVID SEVERITY AND OUTCOMES: Am. Thorac. Society;, A2927–A292A.
Li, C., Cheung, M. K., Han, S., Zhang, Z., Chen, L., Chen, J., et al. (2019). Mesenchymal stem cells and their mitochondrial transfer: a double-edged sword. Biosci. Rep. 39 (5), BSR20182417-26. doi: 10.1042/BSR20182417
Li, X., Hou, P., Ma, W., Wang, X., Wang, H., Yu, Z., et al. (2022). SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol. Immunol. 19 (1), 67–78. doi: 10.1038/s41423-021-00807-4
Li, S., Zhang, Y., Guan, Z., Li, H., Ye, M., Chen, X., et al. (2020). SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal transduction targeted Ther. 5 (1), 1–10. doi: 10.1038/s41392-020-00334-0
Liang, S., Bao, C., Yang, Z., Liu, S., Sun, Y., Cao, W., et al. (2023). SARS-CoV-2 spike protein induces IL-18-mediated cardiopulmonary inflammation via reduced mitophagy. Signal Transduction Targeted Ther. 8 (1), 108. doi: 10.1038/s41392-023-01368-w
Lin, C.-W., Lin, K.-H., Hsieh, T.-H., Shiu, S.-Y., Li, J.-Y. (2006). Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 46 (3), 375–380. doi: 10.1111/j.1574-695X.2006.00045.x
Liu, Y., Garron, T. M., Chang, Q., Su, Z., Zhou, C., Qiu, Y., et al. (2021a). Cell-type apoptosis in lung during SARS-CoV-2 infection. Pathogens 10 (5), 509. doi: 10.3390/pathogens10050509
Liu, Y., Qin, C., Rao, Y., Ngo, C., Feng, J. J., Zhao, J., et al. (2021b). SARS-CoV-2 Nsp5 demonstrates two distinct mechanisms targeting RIG-I and MAVS to evade the innate immune response. Mbio 12 (5), e02335–e02321. doi: 10.1128/mBio.02335-21
López-Lluch, G. (2017). Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech. Ageing Dev. 162, 108–121. doi: 10.1016/j.mad.2016.12.005
Ma, Z., Damania, B. (2016). The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19 (2), 150–158. doi: 10.1016/j.chom.2016.01.010
Ma, R., Ortiz Serrano, T. P., Davis, J., Prigge, A. D., Ridge, K. M. (2020). The cGAS-STING pathway: The role of self-DNA sensing in inflammatory lung disease. FASEB J. 34 (10), 13156–13170. doi: 10.1096/fj.202001607R
Malik, A. N., Czajka, A. (2013). Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 13 (5), 481–492. doi: 10.1016/j.mito.2012.10.011
Marchi, S., Guilbaud, E., Tait, S. W., Yamazaki, T., Galluzzi, L. (2023). Mitochondrial control of inflammation. Nat. Rev. Immunol. 23 (3), 159–173. doi: 10.1038/s41577-022-00760-x
Masoodi, M., Peschka, M., Schmiedel, S., Haddad, M., Frye, M., Maas, C., et al. (2022). Disturbed lipid and amino acid metabolisms in COVID-19 patients. J. Mol. Med. 100 (4), 555–568. doi: 10.1007/s00109-022-02177-4
McGuire, P. J. (2019). Mitochondrial dysfunction and the aging immune system. Biology 8 (2), 26. doi: 10.3390/biology8020026
Mills, E. L., Kelly, B., O'Neill, L. A. (2017). Mitochondria are the powerhouses of immunity. Nat. Immunol. 18 (5), 488–498. doi: 10.1038/ni.3704
Mozzi, A., Oldani, M., Forcella, M. E., Vantaggiato, C., Cappelletti, G., Pontremoli, C., et al. (2023). SARS-CoV-2 ORF3c impairs mitochondrial respiratory metabolism, oxidative stress, and autophagic flux. Iscience. 26 (7), 107118-144. doi: 10.1016/j.isci.2023.107118
Mueller, M., Herrmann, A., Fujita, S., Kolberg, E. J., Kruth, C., Stange, A., et al. (2023). SARS-CoV-2 ORF3c suppresses immune activation by inhibiting innate sensing. bioRxiv. 2023-02. doi: 10.1101/2023.02.27.530232
Murthy, A. M., Robinson, N., Kumar, S. (2020). Crosstalk between cGAS–STING signaling and cell death. Cell Death Differ. 27 (11), 2989–3003. doi: 10.1038/s41418-020-00624-8
Nadhan, R., Patra, D., Krishnan, N., Rajan, A., Gopala, S., Ravi, D., et al. (2021). Perspectives on mechanistic implications of ROS inducers for targeting viral infections. Eur. J. Pharmacol. 890, 173621. doi: 10.1016/j.ejphar.2020.173621
Namgaladze, D., Khodzhaeva, V., Brüne, B. (2019). ER-mitochondria communication in cells of the innate immune system. Cells 8 (9), 1088. doi: 10.3390/cells8091088
Nunn, A. V., Guy, G. W., Brysch, W., Botchway, S. W., Frasch, W., Calabrese, E. J., et al. (2020). SARS-CoV-2 and mitochondrial health: implications of lifestyle and ageing. Immun. Ageing. 17 (1), 1–21. doi: 10.1186/s12979-020-00204-x
Olagnier, D., Farahani, E., Thyrsted, J., Blay-Cadanet, J., Herengt, A., Idorn, M., et al. (2020). SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 11 (1), 1–12. doi: 10.1038/s41467-020-18764-3
O’Meara, M. J., Guo, J. Z., Swaney, D. L., Tummino, T. A., Hüttenhain, R. (2020). A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. BioRxiv, 123. doi: 10.1101/2020.03.22.002386
Orzalli, M. H., Kagan, J. C. (2017). Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol. 27 (11), 800–809. doi: 10.1016/j.tcb.2017.05.007
Padhan, K., Minakshi, R., Towheed, M. A. B., Jameel, S. (2008). Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J. Gen. Virol. 89 (8), 1960–1969. doi: 10.1099/vir.0.83665-0
Paik, S., Kim, J. K., Silwal, P., Sasakawa, C., Jo, E.-K. (2021). An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 18 (5), 1141–1160. doi: 10.1038/s41423-021-00670-3
Pang, L.-l., Yuan, X.-h., Shao, C.-s., Li, M.-z., Wang, Y., Wang, H.-m., et al. (2017). The suppression of innate immune response by human rhinovirus C. Biochem. Biophys. Res. Commun. 490 (1), 22–28. doi: 10.1016/j.bbrc.2017.05.169
Park, S., Won, J.-H., Hwang, I., Hong, S., Lee, H. K., Yu, J.-W. (2015). Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 5 (1), 1–14. doi: 10.1038/srep15489
Pernas, B., Rego-Pérez, I., Tabernilla, A., Balboa, V., Relaño, S., Grandal, M., et al. (2017). Plasma mitochondrial DNA levels are inversely associated with HIV-RNA levels and directly with CD4 counts: potential role as a biomarker of HIV replication. J. Antimicrob. Chemother. 72 (11), 3159–3162. doi: 10.1093/jac/dkx272
Pozzi, A. (2022). COVID-19 and mitochondrial non-coding RNAs: new insights from published data. Front. Physiol. 12, 2586. doi: 10.3389/fphys.2021.805005
Prasun, P. (2020). Letter to the editor: COVID-19, mitochondria, and interferon. J. Interferon Cytokine Res. 40 (9), 466–467. doi: 10.1089/jir.2020.0112
Qu, C., Zhang, S., Li, Y., Wang, Y., Peppelenbosch, M. P., Pan, Q. (2019a). Mitochondria in the biology, pathogenesis, and treatment of hepatitis virus infections. Rev. Med. Virol. 29 (5), e2075. doi: 10.1002/rmv.2075
Qu, C., Zhang, S., Wang, W., Li, M., Wang, Y., van der Heijde-Mulder, M., et al. (2019b). Mitochondrial electron transport chain complex III sustains hepatitis E virus replication and represents an antiviral target. FASEB J. 33 (1), 1008–1019. doi: 10.1096/fj.201800620R
Ren, Y., Shu, T., Wu, D., Mu, J., Wang, C., Huang, M., et al. (2020). The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 17 (8), 881–883. doi: 10.1038/s41423-020-0485-9
Reshi, M. L., Su, Y.-C., Hong, J.-R. (2014). RNA viruses: ROS-mediated cell death. Int. J. Cell Biol. 2014. doi: 10.1155/2014/467452
Reshi, L., Wang, H.-V., Hong, J.-R. (2018). Modulation of mitochondria during viral infections. Mitoch Dis. 2018, 443. doi: 10.5772/intechopen.73036
Riley, J. S., Tait, S. W. (2020). Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21 (4), e49799. doi: 10.15252/embr.201949799
Riou, M., Alfatni, A., Charles, A.-L., Andrès, E., Pistea, C., Charloux, A., et al. (2020). New insights into the implication of mitochondrial dysfunction in tissue, peripheral blood mononuclear cells, and platelets during lung diseases. J. Clin. Med. 9 (5), 1253. doi: 10.3390/jcm9051253
Rodrigues, D., MaChado, M. R., Alves, J. V., Fraga-Silva, T. F., Martins, R. B., Campos, L. C., et al. (2023). Cytokine storm in individuals with severe COVID-19 decreases endothelial cell antioxidant defense via downregulation of the Nrf2 transcriptional factor. Am. J. Physiology-Heart Circulatory Physiol. 325 (2), H252–HH63. doi: 10.1152/ajpheart.00096.2023
Rongvaux, A. (2018). Innate immunity and tolerance toward mitochondria. Mitochondrion 41, 14–20. doi: 10.1016/j.mito.2017.10.007
Rosa, H. S., Ajaz, S., Gnudi, L., Malik, A. N. (2020). A case for measuring both cellular and cell-free mitochondrial DNA as a disease biomarker in human blood. FASEB J. 34 (9), 12278–12288. doi: 10.1096/fj.202000959RR
Rui, Y., Su, J., Shen, S., Hu, Y., Huang, D., Zheng, W., et al. (2021). Unique and complementary suppression of cGAS-STING and RNA sensing-triggered innate immune responses by SARS-CoV-2 proteins. Signal transduction targeted Ther. 6 (1), 123. doi: 10.1038/s41392-021-00515-5
Scaini, G., Fries, G., Valvassori, S., Zeni, C., Zunta-Soares, G., Berk, M., et al. (2017). Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Trans. Psychiatry 7 (5), e1111–e111e. doi: 10.1038/tp.2017.83
Schrepfer, E., Scorrano, L. (2016). Mitofusins, from mitochondria to metabolism. Mol. Cell. 61 (5), 683–694. doi: 10.1016/j.molcel.2016.02.022
Scozzi, D., Cano, M., Ma, L., Zhou, D., Zhu, J. H., O’Halloran, J. A., et al. (2021). Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 6 (4), e143299-115. doi: 10.1172/jci.insight.143299
Sharma, A., Kontodimas, K., Bosmann, M. (2021). The MAVS immune recognition pathway in viral infection and sepsis. Antioxidants Redox Signaling. 35 (16), 1376-92 doi: 10.1089/ars.2021.0167
Shen, Y., Chen, L., Chen, J., Qin, J., Wang, T., Wen, F. (2022). Mitochondrial damage-associated molecular patterns in chronic obstructive pulmonary disease: Pathogenetic mechanism and therapeutic target. J. Trans. Internal Med. 0). doi: 10.2478/jtim-2022-0019
Shenoy, S. (2020). Coronavirus (Covid-19) sepsis: revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflammation Res. 69, 1077–85. doi: 10.1007/s00011-020-01389-z
Shi, C.-S., Qi, H.-Y., Boularan, C., Huang, N.-N., Abu-Asab, M., Shelhamer, J. H., et al. (2014). SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193 (6), 3080–3089. doi: 10.4049/jimmunol.1303196
Shokolenko, I., Venediktova, N., Bochkareva, A., Wilson, G. L., Alexeyev, M. F. (2009). Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 37 (8), 2539–2548. doi: 10.1093/nar/gkp100
Shoraka, S., Ferreira, M. L. B., Mohebbi, S. R., Ghaemi, A. (2021). SARS-CoV-2 infection and Guillain-Barré syndrome: a review on potential pathogenic mechanisms. Front. Immunol. 12, 674922. doi: 10.3389/fimmu.2021.674922
Storci, G., Bonifazi, F., Garagnani, P., Olivieri, F., Bonafè, M. (2020a). How studies on inflamm-aging may help to understand and combat COVID-19 pandemic. doi: 10.20944/preprints202004.0158.v1
Storci, G., Bonifazi, F., Garagnani, P., Olivieri, F., Bonafè, M. (2020b). The role of extracellular DNA in COVID-19: Clues from inflamm-aging. Ageing Res. Rev. 101234, 101234-41. doi: 10.1016/j.arr.2020.101234
Sun, L., Xing, Y., Chen, X., Zheng, Y., Yang, Y., Nichols, D. B., et al. (2012). Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PloS One 7 (2), e30802. doi: 10.1371/journal.pone.0030802
Tavassolifar, M. J., Aghdaei, H. A., Sadatpour, O., Maleknia, S., Fayazzadeh, S., Mohebbi, S. R., et al. (2023). New insights into extracellular and intracellular redox status in COVID-19 patients. Redox Biol. 59, 102563. doi: 10.1016/j.redox.2022.102563
Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A., Ng, L. F. (2020). The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20 (6), 363–374. doi: 10.1038/s41577-020-0311-8
Thakar, J., Mohanty, S., West, A. P., Joshi, S. R., Ueda, I., Wilson, J., et al. (2015). Aging-dependent alterations in gene expression and a mitochondrial signature of responsiveness to human influenza vaccination. Aging (Albany NY). 7 (1), 38. doi: 10.18632/aging.100720
Thaker, S. K., Ch’ng, J., Christofk, H. R. (2019). Viral hijacking of cellular metabolism. BMC Biol. 17 (1), 1–15. doi: 10.1186/s12915-019-0678-9
Thompson, E. A., Cascino, K., Ordonez, A. A., Zhou, W., Vaghasia, A., Hamacher-Brady, A., et al. (2020). Mitochondrial induced T cell apoptosis and aberrant myeloid metabolic programs define distinct immune cell subsets during acute and recovered SARS-CoV-2 infection. MedRxiv. doi: 10.1101/2020.09.10.20186064
Thoms, M., Buschauer, R., Ameismeier, M., Koepke, L., Denk, T., Hirschenberger, M., et al. (2020). Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369 (6508), 1249–1255. doi: 10.1126/science.abc8665
Tiku, V., Tan, M.-W., Dikic, I. (2020). Mitochondrial functions in infection and immunity. Trends Cell Biol. 30 (4), 263–275. doi: 10.1016/j.tcb.2020.01.006
Tomé, D. (2021). Amino acid metabolism and signalling pathways: potential targets in the control of infection and immunity. Eur. J. Clin. Nutr. 75 (9), 1319–1327. doi: 10.1038/s41430-021-00943-0
Uresti-Rivera, E., García-Hernández, M. (2023). Potential role of AIM2 inflammasome in SARS-CoV-2 infection. Scand. J. Immunol. 97 (2), e13239. doi: 10.1111/sji.13239
Varga, Z. T., Grant, A., Manicassamy, B., Palese, P. (2012). Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 86 (16), 8359–8366. doi: 10.1128/JVI.01122-12
Wang, M., Yu, F., Chang, W., Zhang, Y., Zhang, L., Li, P. (2023). Inflammasomes: a rising star on the horizon of COVID-19 pathophysiology. Front. Immunol. 14, 1185233. doi: 10.3389/fimmu.2023.1185233
Weinberg, S. E., Sena, L. A., Chandel, N. S. (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42 (3), 406–417. doi: 10.1016/j.immuni.2015.02.002
Wichmann, D., Sperhake, J.-P., Lütgehetmann, M., Steurer, S., Edler, C., Heinemann, A., et al. (2020). Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann. Intern. Med. 173 (4), 268–277. doi: 10.7326/M20-2003
Woo, T.-T., Williams, J. M., Tsai, B. (2023). How host ER membrane chaperones and morphogenic proteins support virus infection. J. Cell Sci. 136 (13), jcs2611213. doi: 10.1242/jcs.261121
Wood, E., Hall, K. H., Tate, W. (2020). Role of mitochondria, oxidative stress and the response to antioxidants in myalgic encephalomyelitis/chronic fatigue syndrome: a possible approach to SARS-CoV-2 ‘long-haulers’? Chronic Dis. Trans. Med. 7 (1), 14–26. doi: 10.1016/j.cdtm.2020.11.002
Wu, J. (2020). Tackle the free radicals damage in COVID-19. Nitric. Oxide 102, 39–41. doi: 10.1016/j.niox.2020.06.002
Wu, J., Shi, Y., Pan, X., Wu, S., Hou, R., Zhang, Y., et al. (2021). SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 34 (7), 108761. doi: 10.1016/j.celrep.2021.108761
Yang, Y., Bazhin, A. V., Werner, J., Karakhanova, S. (2013). Reactive oxygen species in the immune system. Int. Rev. Immunol. 32 (3), 249–270. doi: 10.3109/08830185.2012.755176
Yang, R., Zhao, Q., Rao, J., Zeng, F., Yuan, S., Ji, M., et al. (2021). SARS-CoV-2 accessory protein ORF7b mediates tumor necrosis factor-α-induced apoptosis in cells. Front. Microbiol. 12, 654709. doi: 10.3389/fmicb.2021.654709
Zekri-Nechar, K., Zamorano-León, J. J., Reche, C., Giner, M., López-de-Andrés, A., Jiménez-García, R., et al. (2022). Spike protein subunits of SARS-CoV-2 alter mitochondrial metabolism in human pulmonary microvascular endothelial cells: involvement of factor Xa. Dis. Markers 2022. doi: 10.1155/2022/1118195
Zhang, Y., Guo, R., Kim, S. H., Shah, H., Zhang, S., Liang, J. H., et al. (2021). SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 12 (1), 1–11. doi: 10.1038/s41467-021-21903-z
Zheng, Y., Gao, C. (2023). Phase separation: the robust modulator of innate antiviral signaling and SARS-CoV-2 infection. Pathogens 12 (2), 243. doi: 10.3390/pathogens12020243
Zheng, Z., Peng, F., Xu, B., Zhao, J., Liu, H., Peng, J., et al. (2020). Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 81 (2), e16–e25. doi: 10.1016/j.jinf.2020.04.021
Zhou, R., Yazdi, A. S., Menu, P., Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469 (7329), 221–225. doi: 10.1038/nature09663
Znaidia, M., Demeret, C., van der Werf, S., Komarova, A. V. (2022). Characterization of SARS-CoV-2 evasion: interferon pathway and therapeutic options. Viruses 14 (6), 1247. doi: 10.3390/v14061247
Zong, S., Wu, Y., Li, W., You, Q., Peng, Q., Wang, C., et al. (2023). SARS-CoV-2 Nsp8 induces mitophagy by damaging mitochondria. Virol. Sin. 38 (4), 520–530. doi: 10.1016/j.virs.2023.05.003
Keywords: coronavirus, SARS-CoV-2, COVID-19, mitochondria, viral infection
Citation: Shoraka S, Samarasinghe AE, Ghaemi A and Mohebbi SR (2023) Host mitochondria: more than an organelle in SARS-CoV-2 infection. Front. Cell. Infect. Microbiol. 13:1228275. doi: 10.3389/fcimb.2023.1228275
Received: 24 May 2023; Accepted: 07 August 2023;
Published: 25 August 2023.
Edited by:
Sounak Ghosh Roy, Ph.D., Henry M Jackson Foundation for the Advancement of Military Medicine (HJF), United StatesReviewed by:
Juan C. Hernandez, Cooperative University of Colombia, ColombiaBhaskar Saha, University of New Mexico, United States
Rishi Bhardwaj, Yale University, United States
Copyright © 2023 Shoraka, Samarasinghe, Ghaemi and Mohebbi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed Reza Mohebbi, c3IubW9oZWJiaUBzYm11LmFjLmly
†ORCID: Seyed Reza Mohebbi, orcid.org/0000-0002-7020-7889