 Assaf Katz1
Assaf Katz1 Lorena Porte2
Lorena Porte2 Thomas Weitzel2,3
Thomas Weitzel2,3 Carmen Varela2Cristina Muñoz-Rehbein4Juan A. Ugalde4
Carmen Varela2Cristina Muñoz-Rehbein4Juan A. Ugalde4 Christopher Grim5
Christopher Grim5 Narjol González-Escalona5
Narjol González-Escalona5 Carlos J. Blondel6*
Carlos J. Blondel6* Verónica Bravo7*
Verónica Bravo7*- 1Programa de Biología Celular y Molecular, ICBM, Facultad de Medicina, Universidad de Chile, Santiago, Chile
- 2Laboratorio Clínico, Clínica Alemana de Santiago, Facultad de Medicina, Clínica Alemana, Universidad del Desarrollo, Santiago, Chile
- 3Instituto de Ciencias e Innovación en Medicina (ICIM), Facultad de Medicina Clínica Alemana, Universidad del Desarrollo, Santiago, Chile
- 4Center for Bioinformatics and Integrative Biology, Facultad de Ciencias de la Vida, Universidad Andrés Bello, Santiago, Chile
- 5Center for Food Safety and Applied Nutrition, Food and Drug Administration, College Park, MD, United States
- 6Instituto de Ciencias Biomédicas, Facultad de Medicina y Facultad de Ciencias de la Vida, Universidad Andrés Bello, Santiago, Chile
- 7Centro de Investigaciones Biomédicas y Aplicadas (CIBAP), Escuela de Medicina, Facultad de Ciencias Médicas, Universidad de Santiago de Chile, Santiago, Chile
Campylobacter is the leading cause of bacterial gastroenteritis worldwide and an emerging and neglected pathogen in South America. This zoonotic pathogen colonizes the gastrointestinal tract of a wide range of mammals and birds, with poultry as the most important reservoir for human infections. Apart from its high morbidity rates, the emergence of resistant strains is of global concern. The aims of this work were to determine genetic diversity, presence of antimicrobial resistance determinants and virulence potential of Campylobacter spp. isolated from patients with acute gastrointestinal disease at ‘Clinica Alemana’, Santiago de Chile. The study considered the isolation of Campylobacter spp., from stool samples during a 20-month period (January 2020 to September 2021). We sequenced (NextSeq, Illumina) and performed an in-depth analysis of the genome sequences of 88 Campylobacter jejuni and 2 Campylobacter coli strains isolated from clinical samples in Chile. We identified a high genetic diversity among C. jejuni strains and the emergence of prevalent clonal complexes, which were not identified in our previous reports. While ~40% of strains harbored a mutation in the gyrA gene associated with fluoroquinolone resistance, no macrolide-resistance determinants were detected. Interestingly, gene clusters encoding virulence factors such as the T6SS or genes associated with long-term sequelae such as Guillain-Barré syndrome showed lineage-relatedness. In addition, our analysis revealed a high degree of variability regarding the presence of fT3SS and T6SS effector proteins in comparison to type strains 81-176, F38011, and NCTC 11168 and 488. Our study provides important insights into the molecular epidemiology of this emerging foodborne pathogen. In addition, the differences observed regarding the repertoire of fT3SS and T6SS effector proteins could have an impact on the pathogenic potential and transmissibility of these Latin American isolates, posing another challenge in characterizing the infection dynamics of this emergent and neglected bacterial pathogen.
1 Introduction
Campylobacter spp. is one of the most frequent causes of human bacterial gastrointestinal infections worldwide (Acheson and Allos, 2001; Kaakoush et al., 2015). Of the 31 species, which so far have been described (Costa and Iraola, 2019), Campylobacter jejuni and Campylobacter coli are the most prevalent species isolated from humans (Butzler, 2004). Infections are mainly associated with the consumption of contaminated raw or undercooked poultry (Humphrey et al., 2007). Campylobacteriosis typically manifests as self-limiting diarrhea; however, in some cases, it might cause long-term sequelae such as Miller Fisher and Guillain-Barré syndrome (GBS) (Finsterer, 2022). Antimicrobial therapy, recommended for prolonged or severe manifestations, is hampered by increasing resistance to different groups of antimicrobials in isolates from human and animal sources worldwide (Rivera et al., 2011; Tang et al., 2017; World Health Organization E, 2017).
In contrast to other gastrointestinal pathogens, Campylobacter does not rely solely on classical virulence factors such as toxins and adhesins (Elmi et al., 2021). Virulence is mainly based on other phenotypical characteristics such as motility, metabolic potential, and the modification of surface structures (capsule and lipooligosaccharides (LOS)) (Kreling et al., 2020). In addition, recent studies on C. jejuni virulence highlighted the importance of protein secretion systems such as the flagellar Type III Secretion System (fT3SS) and the Type VI Secretion System (T6SS) to C. jejuni virulence (Lertpiriyapong et al., 2012; Bleumink-Pluym et al., 2013; Agnetti et al., 2019; Gabbert et al., 2023). Flagellar Type III Secretion System (fT3SS) comprises an MS ring and C ring surrounding the system core in the inner membrane and a rod and hook structure that crosses the periplasm and connects to the flagellar filament extended towards the exterior of the bacterial cell (Burnham and Hendrixson, 2018). C. jejuni translocates at least ten effector proteins, contributing in vitro to host-cell adhesion and invasion and in vivo to colonization and immune modulation of infected chickens (Konkel et al., 1999; Barrero-Tobon and Hendrixson, 2012). Notably, a recent bioinformatic analysis predicted 57 putative fT3SS effector candidates encoded in the type strains F38011, NCTC 11168, and 81-176 (Gabbert et al., 2023), suggesting that the total number of effectors translocated by C. jejuni is more extensive than previously estimated.
Some C. jejuni strains also encode for a T6SS (Robinson et al., 2021) which is a multiprotein contractile injection system that delivers effector proteins into target cells (Ho et al., 2014; Zoued et al., 2014; Coulthurst, 2019). While some T6SS effectors target only bacteria, others target bacterial and eukaryotic cells (Durand et al., 2014; Alcoforado Diniz et al., 2015; Hachani et al., 2016). By employing the T6SS against bacterial rivals, several strains have been demonstrated competitive advantages in various environments, such as the plant surface or the intestinal epithelium (Bernal et al., 2018; Wu et al., 2018; Allsopp et al., 2020). Other enteric pathogens such as Salmonella, Shigella, and Vibrio use the T6SS to colonize the gut (Sana et al., 2017; Chassaing and Cascales, 2018), while some of the Bacteroides strains of the gut microbiota use their T6SSs to antagonize certain pathogenic species (Coyne and Comstock, 2019). In C. jejuni, the T6SS contribute to a variety of in vitro and in vivo phenotypes, ranging from host cell adherence, cellular invasion, resistance to bile salts or oxidative stress, and interbacterial competition to the ability to colonize both mouse and chicken in vivo infection models efficiently (Lertpiriyapong et al., 2012; Bleumink-Pluym et al., 2013; Liaw et al., 2019). No T6SS effector protein has been fully characterized for Campylobacter, but recent bioinformatic analyzes of the genome of C. jejuni strain 488 have predicted the presence of at least 15 effector candidates (Robinson et al., 2021; Robinson et al., 2022).
Despite the importance of Campylobacter as an emerging enteric pathogen, its prevalence in low and middle-income countries is widely unknown (Platts-Mills and Kosek, 2014; Kaakoush et al., 2015; Collado et al., 2018; Levican et al., 2019; Collado, 2020). Recent reports in the Latin American region have shown a high prevalence of human campylobacteriosis (Porte et al., 2023), including significant outbreaks causing long-term autoimmune sequelae such as GBS (Ramos et al., 2021; Quino et al., 2022). The absence of widely globally distributed lineages in some areas of Latin American countries such as Peru (Pascoe et al., 2020) emphasizes the need to gain a deeper insight into the molecular epidemiology and characteristics of the predominance of particular genotypes in the region. In addition, the variability of virulence and pathogenic potential of Latin American isolates is vastly unknown, which affects our understanding of the dynamics of this neglected bacterial pathogen.
In this context and continuing with our prior efforts to understand the genomic epidemiology of Campylobacter spp in Chile, this work provides a genetic characterization of clinical Campylobacter strains from patients with gastrointestinal disease in Santiago. In addition to the most prevalent lineages, we described Clonal Complexes (CCs) not previously identified in the city as well as virulence factors over-represented in some lineages. In addition, our analysis revealed that Chilean clinical strains harbor distinct combinations of T3SS and T6SS effector protein candidates, previously identified in C. jejuni reference strains 81-176, F38011, NCTC 11168 and 488.
Our work increases our understanding of the genomic epidemiology of this emerging pathogen in Chile. It also highlights the variability of the repertoire of T3SS and T6SS effector proteins encoded in these clinical strains, which may have important implications for the environmental fitness, transmission, and pathogenic potential of these strains.
2 Materials and methods
2.1 Bacterial cultures and DNA extraction
Campylobacter strains derived from clinical stool samples obtained from patients with gastrointestinal disease, were tested for bacterial pathogens at ‘Clínica Alemana’, a private non-for-profit healthcare center in Santiago, Chile, between January 2020 and September 2021 (Porte et al., 2016). Briefly, isolates were initially cultivated on CASA agar (BioMerieux®) and identified by MALDI-TOF mass spectrometry (VITEK MS, BioMerieux®). Then, each isolate was stored at -80°C in cryotubes (Cryobank, Mast Group Ltd.). For DNA extraction, strains were thawed and cultivated overnight on Mueller-Hinton 5% sheep blood agar plates at 42°C under microaerophilic conditions (Biomerieux®). Genomic DNA was extracted using the DNeasy blood and tissue kit (Qiagen®) following the manufacturer´s protocol.
2.2 Genome data set, sequencing, and annotation
From a total of 116 strains isolated in the period mentioned above, we randomly chose 90 Campylobacter spp isolates for sequencing. The genome sequence dataset analyzed in this study is listed in Supplementary Table S1. Sequencing libraries were prepared with the Nextera DNA Prep library kit and DNA/RNA UD sequencing indexes (Illumina, San Diego, CA USA), with 100-200 ng of input DNA. Sequencing libraries were prepared on a Sciclone G3 NGSx iQ liquid handling workstation (Perkin Elmer, Waltham, MA USA). Genomes were sequenced on a NextSeq 2000 (Illumina, San Diego, CA USA), using a P2 300 cycle (2 × 150-bp paired-end) chemistry sequencing, according to the manufacturer’s instructions, aiming for a genome coverage of 60x-600x. Default parameters were used for all software unless otherwise specified. The Illumina paired reads were analyzed with the CLC Genomics Workbench v9.5.2 (Qiagen), assessed for quality (Q>30) with the quality control tool, and trimmed (adapter trimming, quality trimming, and length trimming) with the trim sequences tool. The trimmed data was de novo assembled using CLC Genomics Workbench and a minimum contig size threshold of 500 bp.
A subset of strains was chosen for long-read sequencing (Supplementary Table S1). The selected 7 strains were: CFSAN122719 (SAMN31838813), CFSAN122739 (SAMN31838830), CFSAN122748 (SAMN31838839), CFSAN122778 (SAMN31838866), CFSAN122786 (SAMN31838874), CFSAN122787 (SAMN31838875), and CFSAN122822 (SAMN31838909). These strains were selected according to their virulence factors distribution pattern. Thus, strains CFSAN122719 and CFSAN122786 were closed to confirm the presence of pVir plasmid and strains CFSAN122739, CFSAN122748, CFSAN122778 and CFSAN122787 to determine the presence/absence of genes encoding structural and effectors proteins of the T6SS. Those isolates belong to different STs. Finally, strain CFSAN122822 was chosen because it belongs to the specie C. coli which has been poorly characterized in the country.
Long reads were generated on a GridION sequencer (Nanopore Technologies, Oxford, UK). Sequencing libraries were prepared using the rapid barcoding sequencing kit (SQK-RBK004). Seven libraries were pooled and run in a FLO-MIN106 (R9.4.1) flow cell for 48 hours, according to the manufacturer’s instructions. Base-calling was performed using default settings (MinKNOW v22.10.5, Guppy v6.3.8). Sequencing outputs was 2.50 Gb (quality score, 11.29; N50, 3,405 bp; total reads, 150.727), for an estimated average genome coverage of 30 to 50×. Reads of <2,000 bp and a quality score of <7 were discarded for downstream analysis. The short-read whole-genome sequence for these strains were generated as described earlier using a NextSeq sequencer. The final complete genome sequences (comprising the chromosome and plasmid, when present) were obtained using a previously described pipeline (Maguire et al., 2021). Unicycler software v0.4.8 (Wick et al., 2017) was utilized to perform hybrid assembly and short and long-read sequencing output and resulted in completely or nearly completely closed and circular genomes.
The genomes were annotated using Prokka (Seemann, 2014) with the genomes of C. coli aerotolerant strain OR12 (accID GCF_002024185.1) and C. jejuni strain NCTC 11168 (accID GCF_000009085.1) as references and by forcing GenBank compliance.
2.3 MLST and cgMLST analysis
For MLST and cgMLST, sequencing reads were mapped against an MLST scheme based on seven housekeeping genes and 637 core and 958 accessory gene cgMLST scheme (Cody et al., 2017), using Ridom SeqSphere software (Münster, Germany). A phylogenetic comparison of the 90 Campylobacter genome sequences was performed using a neighbor-joining (NJ) tree based on a distance matrix of the core genomes of all the strains. STs and clonal complexes (CCs) were determined after automated allele submission to the PubMLST server for Campylobacter MLST (https://pubmlst.org/campylobacter/) (Jolley et al., 2018). Further annotations and visualizations of trees were performed using iTOL v.6 (Letunic and Bork, 2019).
2.4 Virulome and resistome analysis
The bacterial virulome was determined by genome hits against the genes of the virulence factor database (VFDB) (Liu et al., 2019) using the minimum length coverage of 60% aligned to the reference and 85% of identity. Antimicrobial resistance determinants were screened in each Campylobacter genome using ABRicate (Seemann, 2020), which includes the Resfinder (Florensa et al., 2022), CARD (Alcock et al., 2022), ARG-ANNOT (Gupta et al., 2014) and NCBI ARRGD (Feldgarden et al., 2019) databases. gyrA and 23S RNA genes point mutations were determined using Resfinder (Florensa et al., 2022). BLAST analysis was performed as previously described (Bravo et al., 2021) and genetic clusters were visualized using EasyFig (Sullivan et al., 2011). In both virulome and resistome analysis, short-reads assemblies of each strain were used. To predict the source of the Campylobacter strains, first cgMLST profiles were assigned using chewBBACA (Silva et al., 2018) using the Campylobacter pubMLST database (Cody et al., 2017). The resulting cgMLST profile was processed using aiSource (Arning et al., 2021) to predict source attribution of the isolate. For the analysis of the distribution of T3SS and T6SS effector candidates, the nucleotidic sequences of genes coding for each previously described effector protein (Gabbert et al., 2023) were used as baits in BLAST analysis, as described above. Hierarchical clustering analysis was performed using the versatile matrix visualization and analysis software Morpheus using a presence/absence matrix of each effector candidate (Table S2). The hierarchical clustering recursively merges objects based on their pair-wise distance and this results in dendrogram where profiles which are more similar will cluster together in closer branches (one minus Pearson correlation, average linkage method) (https://software.broadinstitute.org/morpheus).
2.5 Comparative genomic and sequence analysis
Genome nucleotide sequences were visualized with the Artemis 18.1 software (Carver et al., 2012) and analyzed with the Artemis Comparison Tool (ACT) release 6 (Carver et al., 2005), the multiple aligner Mauve (Darling et al., 2010) and EasyFig v2.2.2 software (Sullivan et al., 2011). Putative effectors were analyzed with the structure-based homology tool HHpred (Zimmermann et al., 2018). For the pangenome analysis of pVir plasmids, we performed long-read sequencing of CFSAN122719 and CFSAN122748 strains, following the protocol described above. Nucleotide plasmid sequences were compared to the pVir plasmid nucleotide sequence of C. jejuni 81-176. For that comparison we used Roary (Page et al., 2015), setting a minimum percentage identity of 90% for BLASTp.
For the identification of novel T6SS effector candidates, the amino acid sequence of each ORF was analyzed with the Bastion6 pipeline (Wang et al., 2018), the DeepTMHTMM software (Hallgren et al., 2022) to predict putative transmembrane domains, the structure-based homology tool HHpred (Zimmermann et al., 2018) and the PROSITE, NCBI-CDD, Motif-finder, and Pfam databases to identify putative conserved functional domains (Kanehisa, 2002; Sigrist et al., 2012; Finn et al., 2014; Lu et al., 2020). For this analysis, long-read assemblies were used.
3 Results
3.1 cgMLST analysis and phylogenetic distribution of Chilean C. jejuni strains
The study included a total of 90 clinical Campylobacter strains of which 88 (97.8%) were C. jejuni and 2 (2.2%) were C. coli. The cgMLST analysis showed a high degree of diversity within the 90 strains analyzed. These strains belong to 49 sequence types (STs), identified within 22 clonal complexes (CCs) (Figure 1A). More than 50% of isolates (n=53) belonged to the worldwide prevalent lineages CC-353, CC-21, CC-22, and CC-48 (Figure 1A; Supplementary Table S3). ST-475 belonging to CC-48, was the most frequent STs identified in this study (n=12, 13.3%). Based on previous reports we cataloged all CCs based on the most probable isolation source. CC-21 was the most abundant and diverse among host-generalist clonal complexes (Dearlove et al., 2016) accounting for 22 isolates of 5 different STs with a predominance of ST-883 (6 isolates) and ST-50 (8 isolates) (Figure 1A; Supplementary Table S3). CC-353, a chicken-associated complex (Sheppard et al., 2011), was the most prevalent host-specialist CC (representing 11.1% of the isolates; n=10), followed by the cattle-associated CC-22 (Epping et al., 2021) (7.8%; n=7). In agreement with these observations, a source attribution analysis confirmed that the most prevalent CCs found in this study (CC-21, CC-353- CC-22) are related to known animal sources (Figure 2).
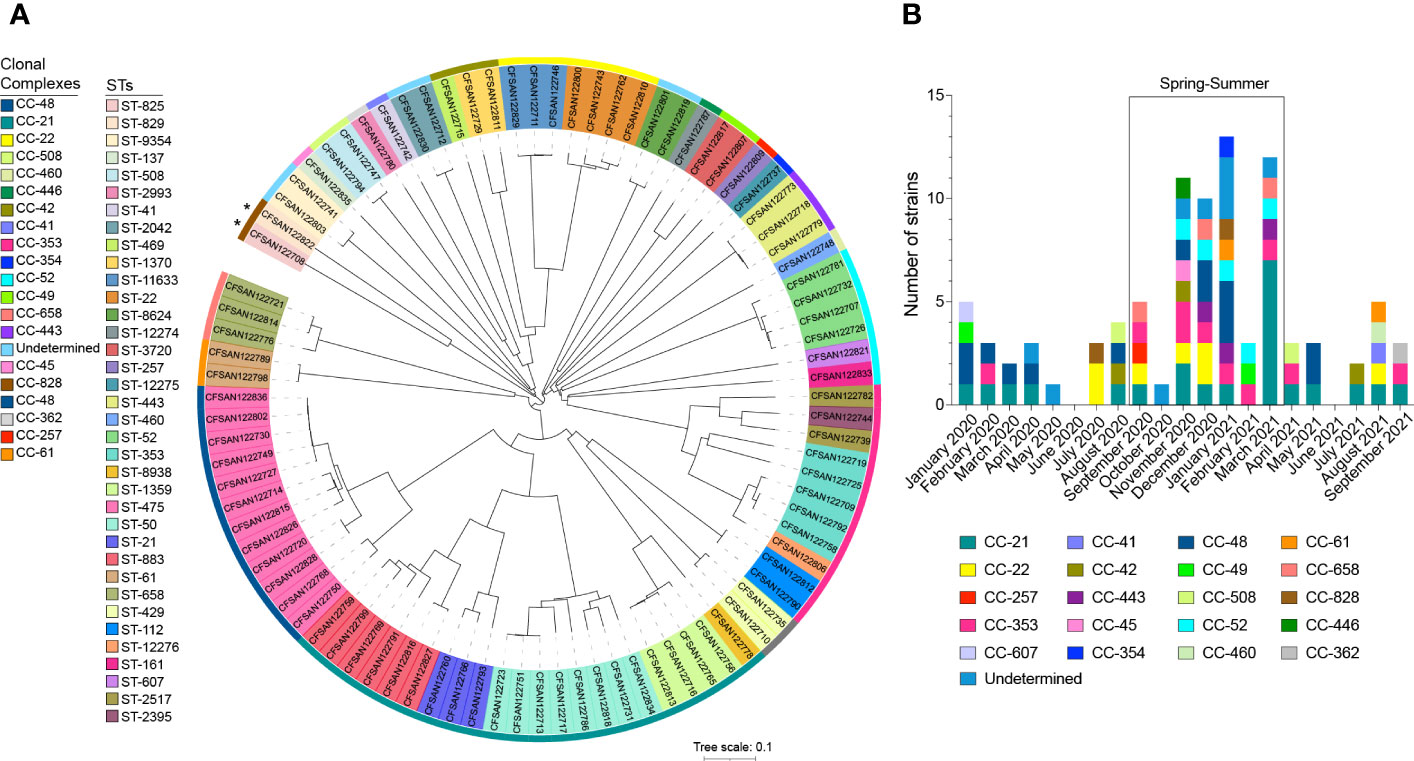
Figure 1 Phylogenetic Analysis and distribution of CC over time. (A) cgMLST phylogenetic analysis of 90 clinical Campylobacter strains isolated from patients with gastrointestinal disease in Santiago, Chile. Neighbor joining (NJ) tree was constructed with RIDOM SeqSphere+ using Campylobacter jejuni subsp. jejuni NCTC 11168 as reference genome. The outer colored strip represents the different clonal complexes described in the study. The colored background of each strain name denotes STs. iTOL (Letunic and Bork, 2019) was used for tree visualization. Asterisks denotes C coli isolates. (B) Monthly distribution of CCs in clinical Campylobacter strains collected in a 20 months period (January 2020 to September 2021) in Santiago, Chile. Colored bars represent each clonal complex described in the study.
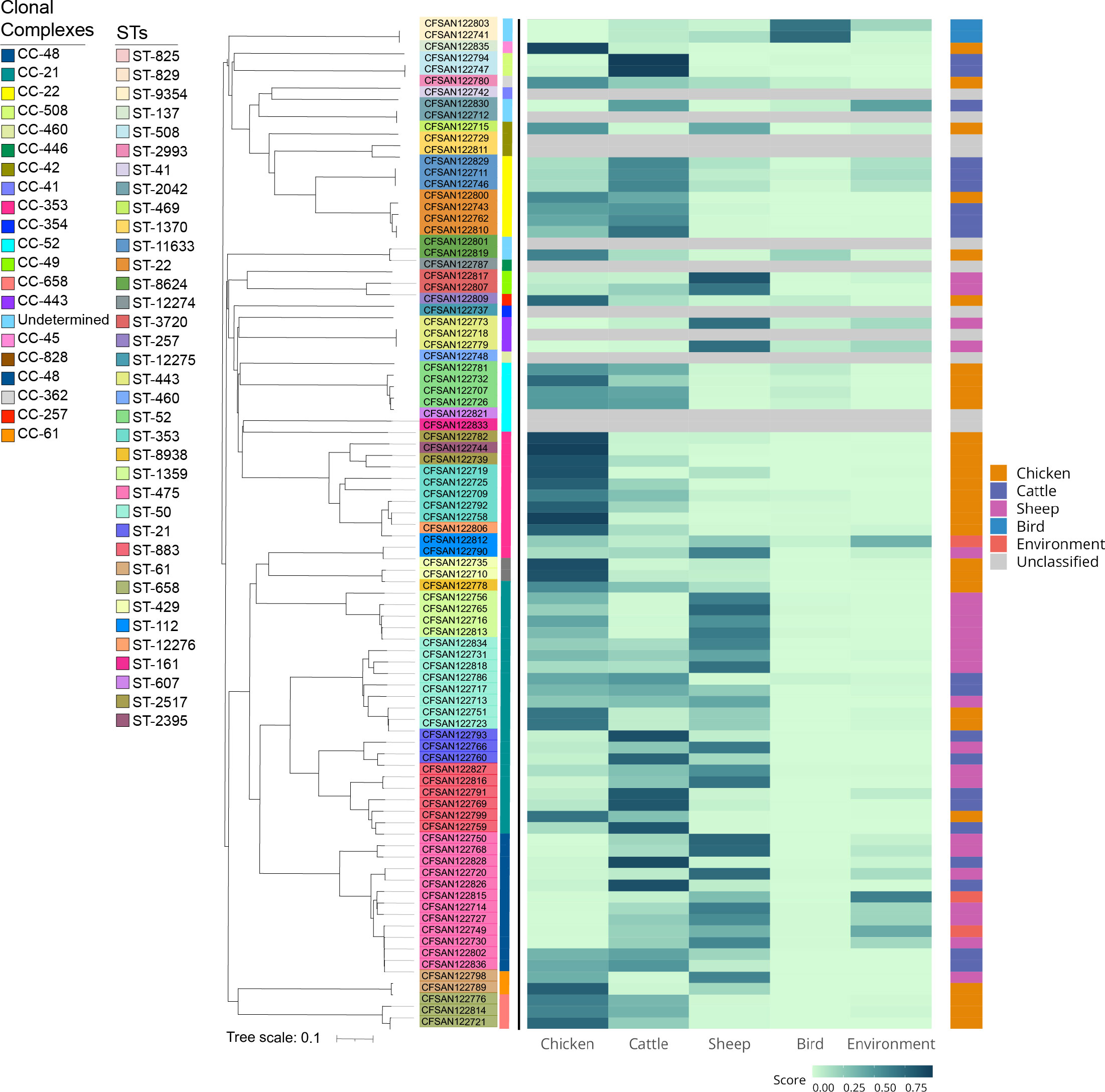
Figure 2 Source attribution of clinical Campylobacter isolates. The source of each isolate was predicted based on cgMLST profiles processed using aiSource (Arning et al., 2021). The figure shows a heat map based on the probability of attribution to the tested sources for each strain ordered based on the NJ tree. The colored bar on the right indicates the predicted source while the bar in the left denotes the CC for each strain.
Several studies have reported that campylobacteriosis cases follow a distinctive seasonality, with peak cases during the spring and summer (Nylen et al., 2002). Most strains (62.2%; n=56) were isolated during those seasons (September to March in Chile) (Figure 1B). The most prevalent strains followed a similar trend. While strains of CC-353 and CC-48 were mostly isolated during the spring-summer period, CC-21 was present over the whole period of study with a peak in summer (Figure 1B).
3.2 Resistome analysis of Chilean C. jejuni strains clinical strains
Antimicrobial determinants in the genome sequences of the 90 Campylobacter strains were identified using Abricate software and Resfinder (Camacho et al., 2009; Zankari et al., 2017; Bortolaia et al., 2020; Seemann, 2020). None of the analyzed isolates encoded for the mutations A2058C and A2059G in the 23S RNA gene, which are associated with macrolide resistance (Tang et al., 2017) (Figure 3A). The C257T point mutation in the quinolone resistance-determining regions (QRDR) of the gyrA gene, which confers class-wide resistance to fluoroquinolones, was detected in 36.7% of isolates (n=33) (Porte et al., 2023). As shown in Figure 3A, all ST-1359 strains harbored this mutation, while all the isolates from the most common ST (ST-475) are predicted to be sensitive to this group of antimicrobials. The analysis of genomes grouped by month of isolation, revealed an increase in the isolates predicted to be resistant to fluoroquinolones in April/May, both for strains isolated in 2020 and 2021 (Figure 3B).
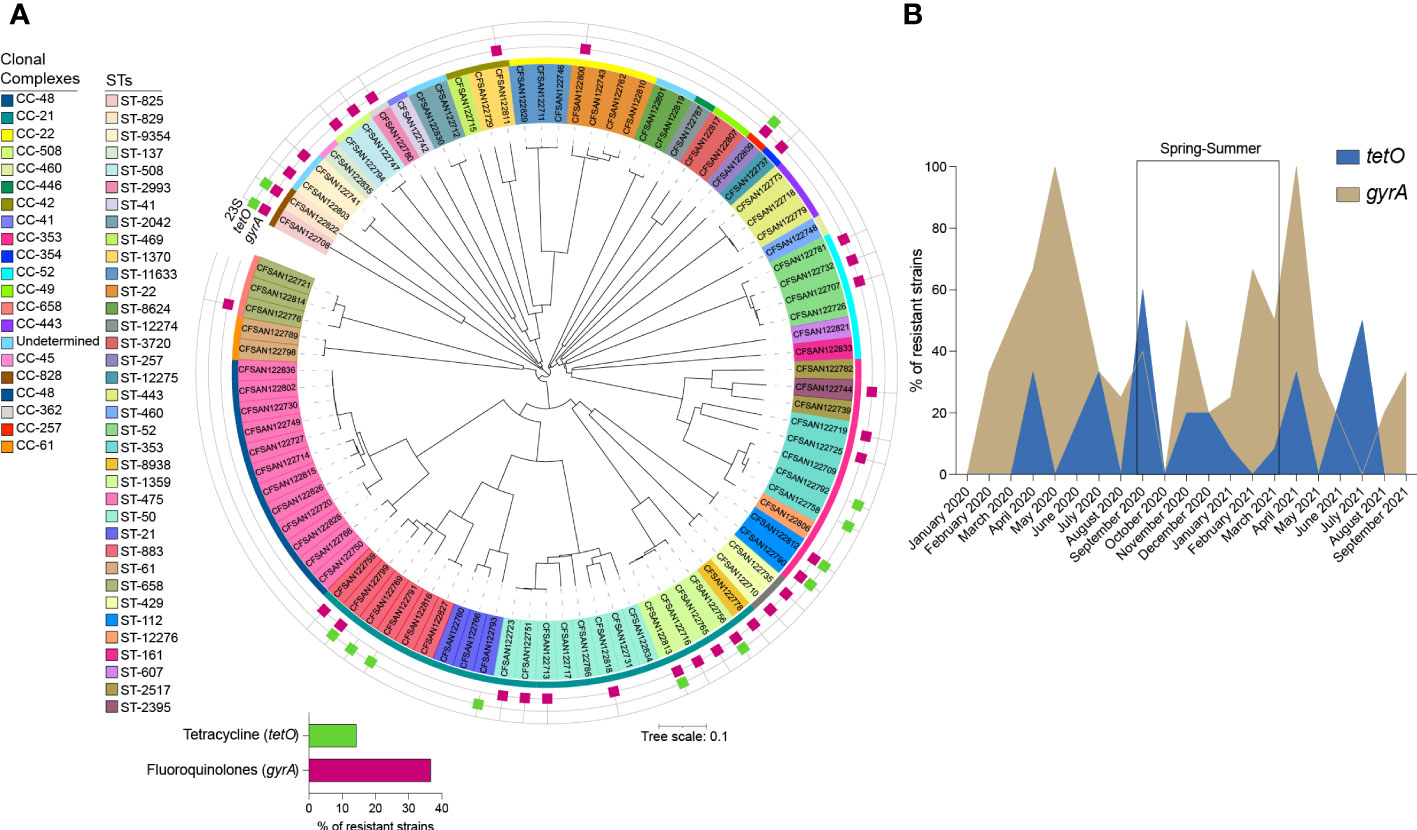
Figure 3 Presence of genetic resistance determinants amongst clinical Campylobacter strains collected in Santiago, Chile. (A) NJ tree indicating the presence of resistance determinants among Campylobacter spp isolates. Resistance determinants are denoted by colored squares surrounding the tree and grouped according to the spectrum of action: Fluoroquinolones (magenta), macrolides, not detected (light blue), and tetracycline (pea). (B) Monthly distribution of fluoroquinolones (khaki) and tetracycline (royal blue) determinants.
The tetO gene, which encodes for the ribosomal protection protein involved in tetracycline resistance, was present in 14.4% of the analyzed genomes (n=13). ß-lactamase encoding genes (blaOXA-61, blaOXA-193, blaOXA-450, and blaOXA-605), were also prevalent among the Campylobacter genomes (Supplementary Table S4). The analysis of the genome sequence of a predicted multi-resistant strain (strain CFSAN122769), revealed the presence of a DNA fragment that encodes genes conferring resistance to aminoglycosides (aph(2″)-If, aac(6’)-Im, ant (6)-Ia), and tetracycline (tetO). Additionally, multidrug efflux pump genes cmeABC were detected in all the C. jejuni isolates.
3.3 Analysis of the distribution of virulence-related genes in Chilean C. jejuni strains
The distribution of virulence determinants is shown in Figure 4; Supplementary Table S5. Genes were classified according to their associated functions, including capsule synthesis, toxins production, LOS synthesis, fT3SS, T4SS, and T6SS.
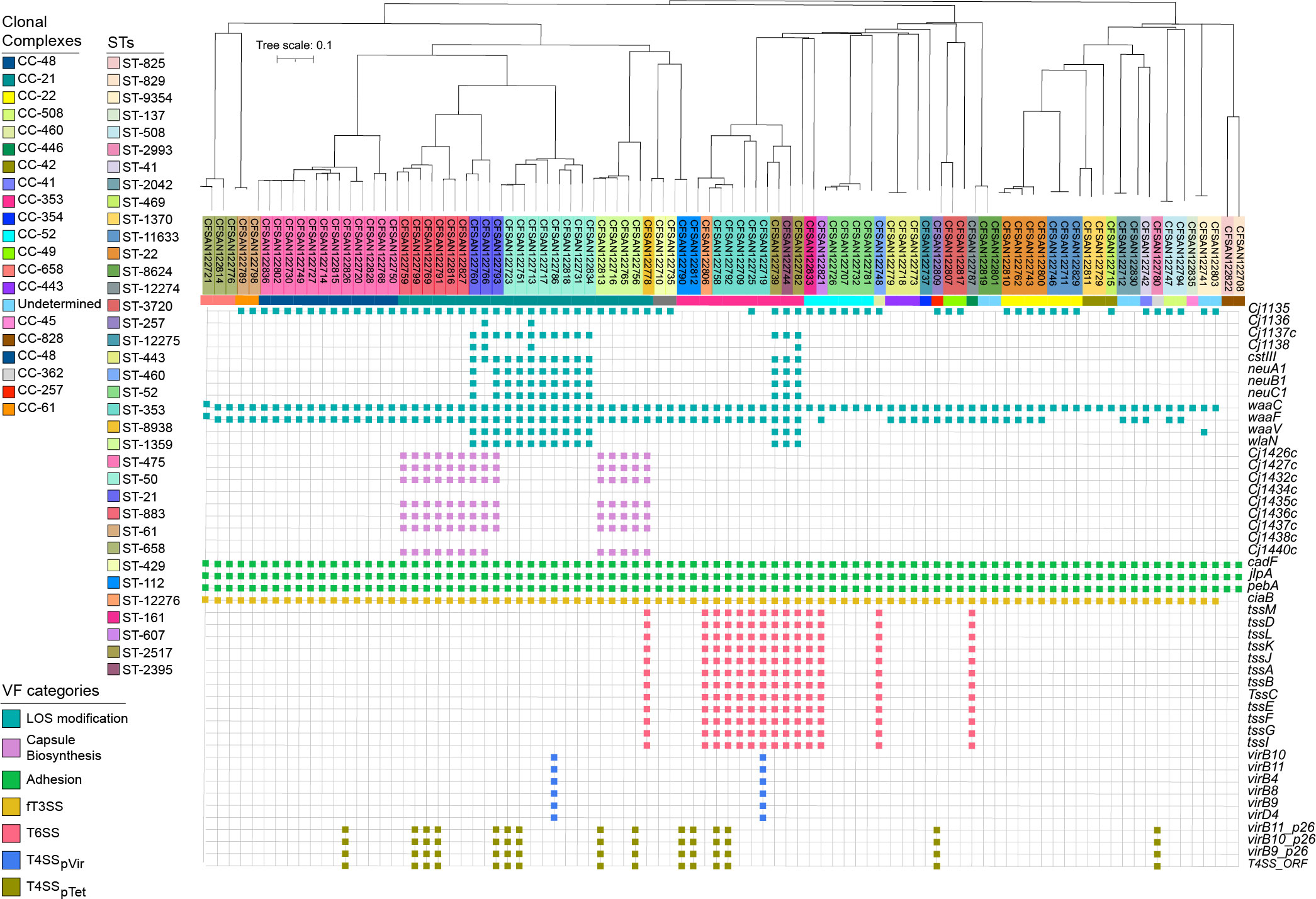
Figure 4 Virulome analysis of clinical Campylobacter strains isolated from patients with gastroenteritis. The colored strip represents the CCs identified in this group of strains. Virulence determinants were divided into seven different categories and highlighted by colored squares: LOS modification (teal), capsule modification (lavender), adhesion (spring green), invasion (golden), T6SS (pale red), T4SSpVir (azure) and T4SSpTet (olive).
Adhesion associated genes (cadF, jlpA, pebA) as well as the cytotoxin-encoding cluster cdtABC were detected in all the analyzed genome sequences (Figure 4). Genes involved in capsule biosynthesis (Cj1426c, Cj1427c, Cj1432c, Cj1434c, Cj1435c, Cj1436c, Cj1437c, Cj1438c, Cj1440c) were only present in strains belonging to CC-21, particularly in all isolates from ST-883, ST-21, and ST-1359. Additionally, cstIII, neuABC and wlaN, which have been involved in molecular mimicry (Linton et al., 2000; Linton et al., 2002; Culebro et al., 2018), were detected in 100% of the strains belonging to ST-50 and ST-21.
Regarding the fT3SS, in each of the strains we detected the genes encoding the major structural components such as the flagellar basal body (flgB and flgC), hook (flgE2), filament (flaA and flaB), as well as genes encoding essential components for flagellar protein export (flhB and fliI) (Supplementary Table S5). This suggests that each of the strains encode a functional fT3SS. To determine the presence and distribution of the known and putative fT3SS effectors among Chilean strains, we generated a local database with the genome assemblies of genome sequences of C. jejuni from our previous (Bravo et al., 2020b; Bravo et al., 2020a) and current work (total of 171 strains). Our analysis also included the C. jejuni type strains F38011, NCTC 11168, and 81-176. As shown in Figure 5, there is a differential distribution of an important group of effector proteins in the Chilean isolates. Hierarchical clustering analysis revealed five subgroups with distinct repertoires of effector protein candidates (Figure 5). Interestingly, most Chilean clinical strains cluster with type strain F38011, while only a few clustered with type strains NCTC 11168 and 81-176. In addition, two subgroups (II and V) did not cluster with any type strain.
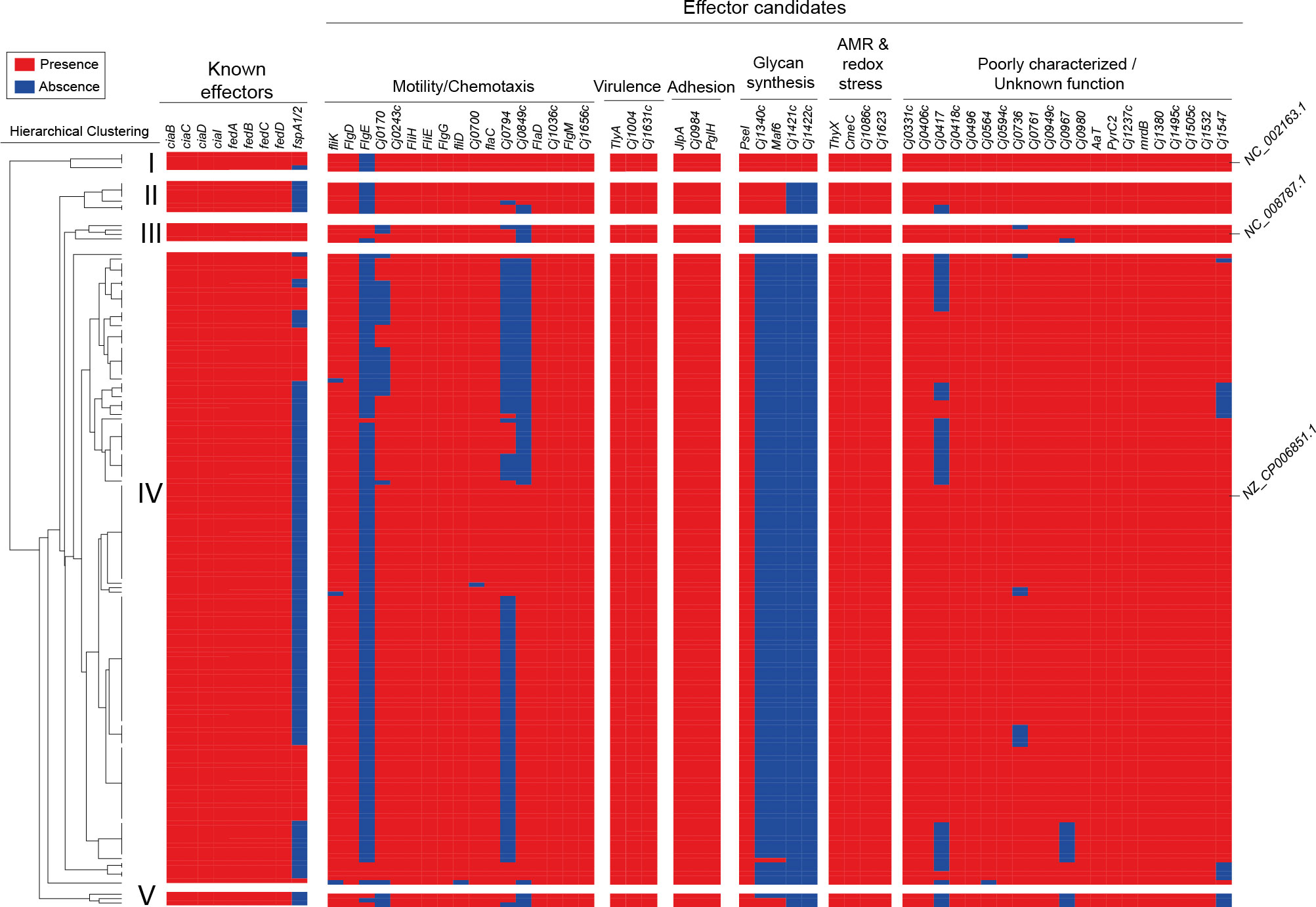
Figure 5 Distribution and hierarchical clustering analysis of T3SS components in clinical C. jejuni strains. Clustering analysis was performed using the versatile matrix visualization and analysis software Morpheus (one minus Pearson correlation, average linkage method). The red and blue boxes represent the presence and absence of individual genes, respectively.
Every Chilean isolate harbored previously described effectors, except for FspA1 and FspA2 that were absent in several isolates (Figure 5). The largest diversity was observed in terms of the presence of the effector candidates identified by Gabbert et al. (Gabbert et al., 2023), especially in the motility/chemotaxis category. Predicted effectors related to glycan synthesis were absent in most Chilean isolates (Figure 5). These results highlight the diversity regarding fT3SS effectors in these Chilean strains.
Another protein secretion system linked to C. jejuni virulence is the T4SS. This system is a membrane-associated complex used by bacteria to deliver molecules to several target cells in a contact-dependent manner and is usually encoded in the pTet family plasmids of Campylobacter (Bacon et al., 2000; Marasini et al., 2018). In the current study, this system was associated with several STs, most of them belonging to CC-21 and CC-353 (Figure 4). However, not all isolates of these STs encoded this secretion system. Additionally, we identified two strains harboring the pVir plasmid, associated with increased invasion of epithelial cells in C. jejuni 81-176 strain (Cody et al., 2012). While the presence of this plasmid is low among clinical isolates worldwide (Panzenhagen et al., 2021), this is the first report of its presence in clinical strains from Chile. Sequence-based analysis showed that these plasmids were more similar to pVir isolated from Japan (strain THJ065) (Supplementary Figure S1). The main differences between pVir isolated from Chilean strains and strain 81-176 is a set of hypothetical proteins and the DinJ-YafQ Type II toxin-antitoxin (TA) module in the Chilean strains (Supplementary Table S6).
As mentioned above, the T6SS has emerged as a novel fitness and virulence factor in C. jejuni, contributing to host colonization and resistance to oxidative stress (Liaw et al., 2019). Our bioinformatic analysis identified putative T6SS gene clusters (based on the detection of each of the major T6SS structural components) in 14 C. jejuni strains (~16%), more than 2-fold increase compared to our previous study from the same health center (Bravo et al., 2021) (Supplementary Table S5). Interestingly, 64% (n=9) of the T6SS positive strains of this study belonged to the CC-353 clonal complex, which is generally attributed to a chicken source.
To determine and compare this distribution with other countries, we analyzed the databases of van Vliet et al. and Robinson et al. and included the CC and ST of every C. jejuni strain from Chile sequenced to date (van Vliet et al., 2021; Robinson et al., 2022). The analysis corroborated the global representation of CC-464, CC-353, CC-573, and CC-403 (Figure 6A). However, an analysis based on the country of origin revealed important differences between Latin American countries such as Chile and Brazil and countries from the Northern Hemisphere, such as the UK and the United States. While in the UK the two most frequent T6SS positive CCs are CC-464 (32.6%) and CC-353 (23.6%), in Chile and Brazil, only the CC-353 is found at a high frequency (58.3% and 59.4%, respectively), with very few strains from CC-464 found only in Chile (4.2%). CC-464 is also not frequent in strains from the United States and CC-353 is present only in 28,8% of strains (Figure 6B).
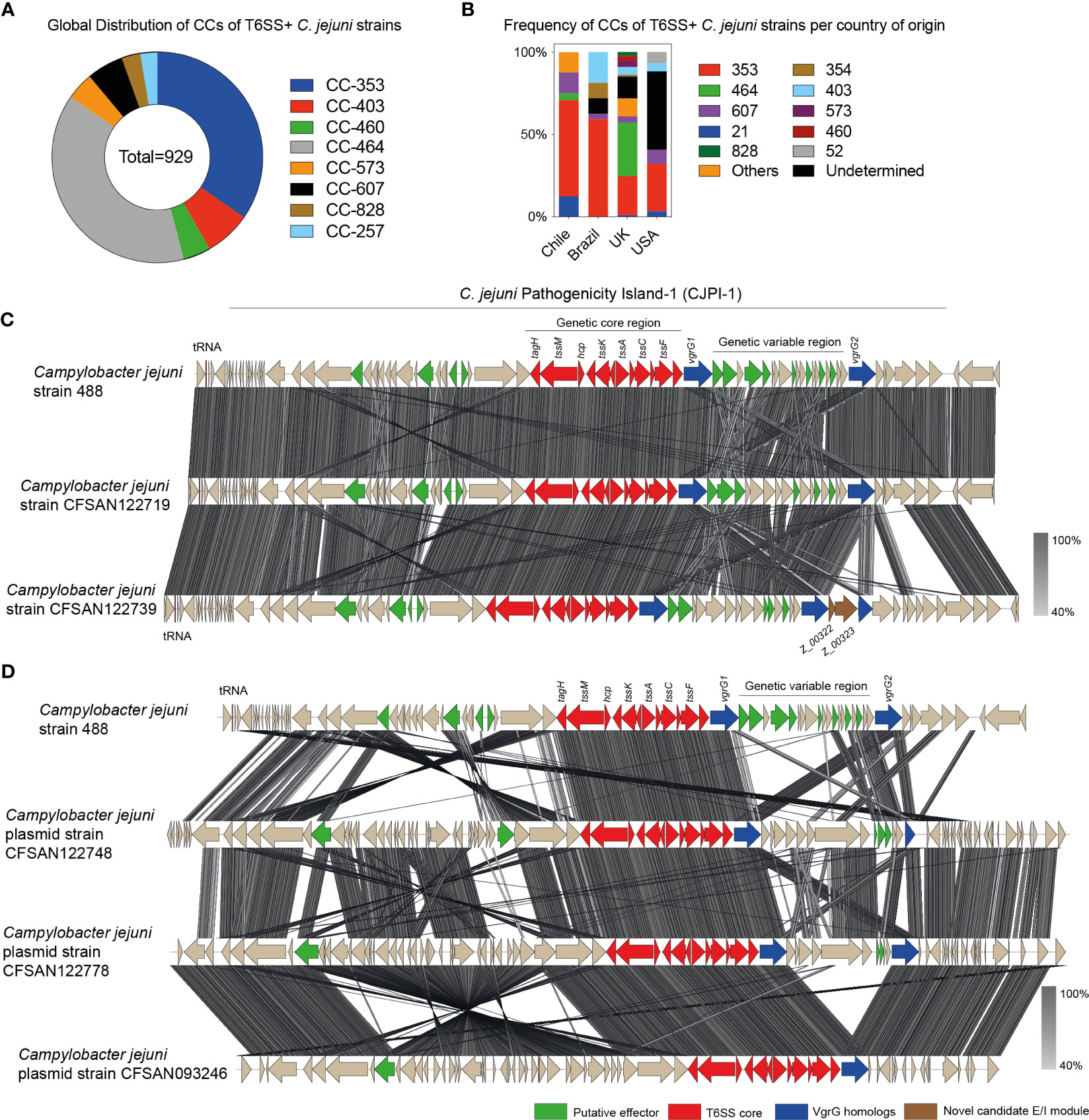
Figure 6 Clonal complex distribution and genomic comparative analysis of C jejuni encoding T6SSs. (A) Analysis of the global distribution of CC of T6SS positive C jejuni strains. (B) Frequency analysis of the CCs of T6SS positive C jejuni strains per country of origin. (C) and (D) Comparison of the T6SS gene cluster in C jejuni 488 with the T6SS gene clusters encoded in either the chromosome C or plasmids (D) from clinical strains isolated from Chile. tBLASTx alignments were performed and visualized using Easyfig. Genes encoding different T6SS-related components are highlighted in color.
To gain insight into the genomic context of the putative T6SS gene clusters, we performed a comparative genomic analysis between the T6SS gene cluster of C. jejuni strain 488 and the T6SS gene clusters of 4 C. jejuni strains with closed or near-finished genomes (genomes with contigs large enough to contain complete T6SS gene clusters) (strains CFSAN122719, CFSAN122739, CFSAN122748 and CFSAN122778). As shown in Figure 6C there was a high degree of synteny between the chromosomally encoded T6SS clusters. This synteny was also observed in the variable region described initially in strain 488 and located between two copies of the vgrG gene (Figure 6C). Strain CFSAN122739 harbored an additional copy of vgrG and several additional ORFs flanking these gene encoding diverse hypothetical proteins. Our analysis showed that the T6SS gene clusters of strains CFSAN122719 and CFSAN122739 are located in the chromosome within the CjiE3 conjugative element (just as in the 488 strain). Interestingly, the T6SS gene clusters of strains CFSAN122748 and CFSAN122778 were encoded in distinct plasmids (Figure 6D) and were similar to the plasmid of strain CFSAN093246 previously identified in the same clinic (Bravo et al., 2021), although with difference in the genetic variable region of the T6SS gene cluster. A recent bioinformatic survey identified that Campylobacter T6SS gene clusters could be classified into three distinct cluster organizations (types I, II, and III). Analysis of T6SS genetic organization revealed that each of the T6SS gene clusters identified belonged to the T6SS group I-a, the same T6SS group as the T6SS of the Brazilian type strain 488 (Robinson et al., 2021; Robinson et al., 2022). In addition, we analyzed each ORF of the chromosomally encoded T6SS gene clusters in strains CFSAN122719 and CFSAN122739 and found a novel putative effector/immunity module (ORFs Z_00322 and Z_00323) in the variable region of the T6SS gene cluster of strain CFSAN122739 (Figure 6C). ORF Z_00322 encodes a putative membrane protein with a transmembrane domain. It is predicted to be co-transcribed with ORF Z_00323 that possesses low homology to N-acetylmuramoyl-L-alanine amidases (Supplementary Figure S2), suggesting that it could target the peptidoglycan of target cells contributing to the antibacterial activity of this T6SS.
To further characterize the T6SS of the Chilean clinical strains, we performed a bioinformatic screen for the presence and absence of previously described putative effectors and immunity proteins in each genome from our Chilean database (Robinson et al., 2021; Robinson et al., 2022). As shown in Figure 7, the hierarchical clustering analysis revealed the presence of 5 major subgroups (I to V) with different profiles of T6SS effector and immunity encoding genes. The C. jejuni strains from the present study clustered in the different subgroups, mostly in subgroup 1. Notably, the C. jejuni strain 488, in which most of the bioinformatic analysis of the T6SS have been performed, clustered in a different group (V), highlighting that the Chilean clinical strains harbor different repertoires of T6SS effector proteins in comparison to the type 488 strain. The most conserved effector candidates between strain 488 and the Chilean clinical strains where the CJ488_00948c (PAAR-like), CJ488_00957c (Lipase) and CJ488_00962 (Lisozyme) while the least conserved effectors where CJ488_00980 (Tox-Rease), CJ488_00982 (Tox-Rease) and CJ488_00990 (PAAR-like). The differential distribution of putative T6SS effector candidates suggest that these clinical strains might differ in their ability to interact with host-cells and/or compete with the host microbiota.
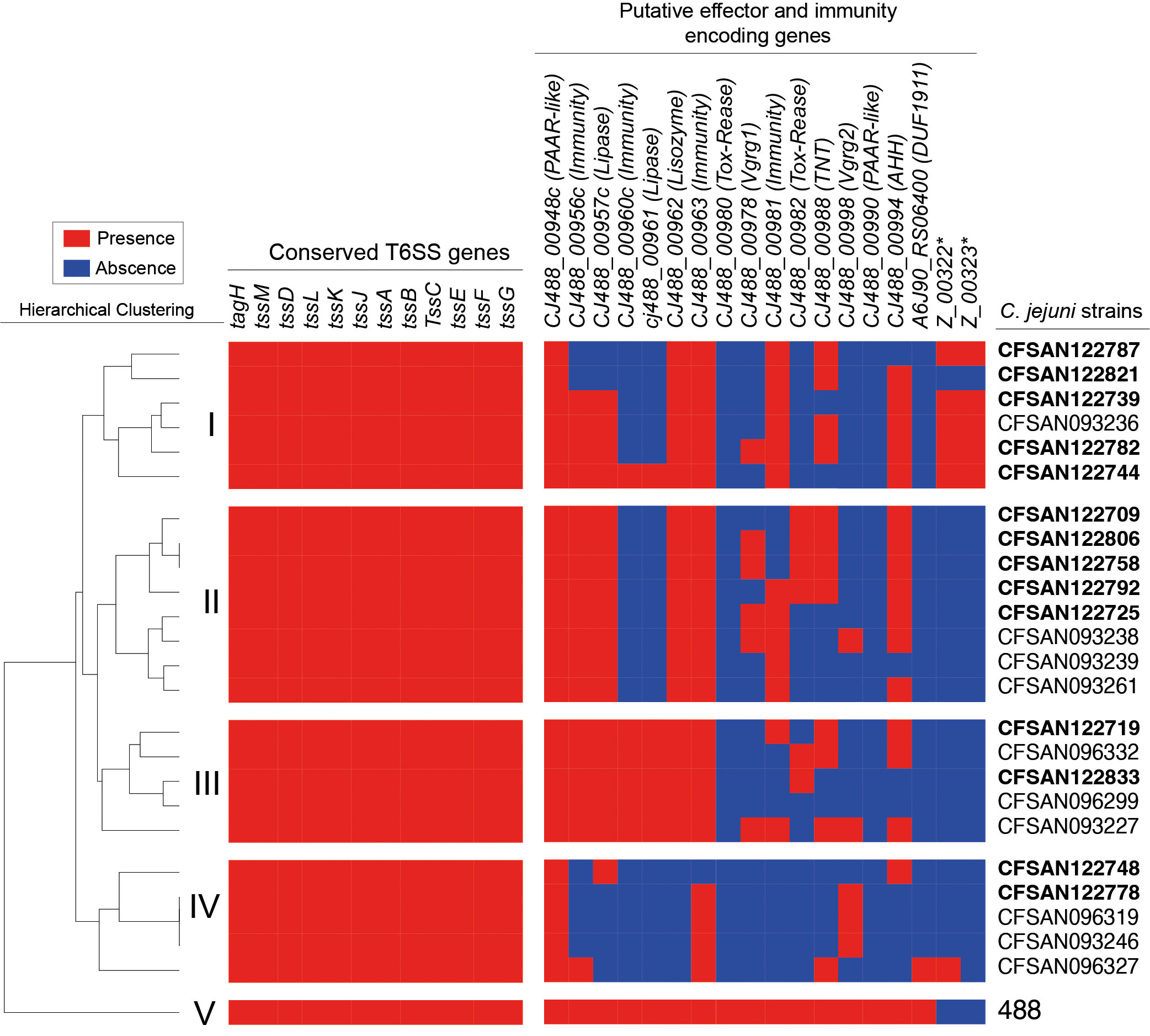
Figure 7 Distribution and hierarchical clustering analysis of T6SS components in C. jejuni strains. Clustering analysis was performed using the versatile matrix visualization and analysis software Morpheus (one minus Pearson correlation, average linkage method). The red and blue boxes represent the presence and absence of individual genes, respectively. Asterisks highlight the novel E/I module identified in this work.
4 Discussion
4.1 ST-475 is the major sequence type among Chilean C. jejuni strains
The presented Chilean data is consistent with international studies, reporting a predominance of C. jejuni as the most prevalent species causing human campylobacteriosis (Acheson and Allos, 2001; World Health Organization, Food and Agriculture Organization of the United Nations, World Organisation for Animal Health, 2013; Kaakoush et al., 2015; EFSA, 2019). However, we found an important decline of C. coli within our study site from >10% during 2017-2019 (Bravo et al., 2021) to 2.2% in the present study. This difference might be related to the reduced mobility of the urban population of Santiago during the SARS CoV-2 pandemic, since higher prevalence of C. coli have been reported from rural environments (Roux et al., 2013). Remarkably, we did not identify emerging Campylobacter species, as reported in other South American countries such as Peru, where C. concisus, C. upsaliensis, and Candidatus Campylobacter infans were described recently (Garcia Bardales et al., 2022; Parker et al., 2022).
In contrast to reports from other regions of South America (Pascoe et al., 2020), the most common CCs identified in Chile have a rather global distribution (CC-21, CC-353, CC-48, CC-22) (Cobo-Díaz et al., 2021). Interestingly, we found an increased frequency and higher genetic diversity of CC-353 compared to our previous study (Bravo et al., 2021). Additionally, CC-22, which was not identified in previous studies in Santiago, emerged as a frequent lineage (Supplementary Figure S3A). The presence of this and other predominant CCs (CC-41, CC-48, CC-353, CC-21 among others) was previously reported in southern Chile (Collado et al., 2018). The presence of genetically related lineages in these geographically distant regions (approx. 800 km) might indicate common food-associated sources (e.g., poultry or cattle); such relatedness is supported by common phenotypic properties including antimicrobial resistance and virulence factors.
In Chile there is limited information about Campylobacter sources and their relationship with certain lineages However, recent genetic signatures demonstrated an association of allelic variants to host-specific lineages (Berthenet et al., 2019; Cody et al., 2019). In the current study we identified a relevant number of strains belonging to host-generalist lineages (CC-21 and CC-48), suggesting that they are capable of colonizing various animal reservoirs. Even though CC-48 has also been associated with a host-generalist lifestyle, most of the isolates within this CC belonged to the ST-475. Méric et al., 2018 performed a deep nucleotide and amino acid sequence analysis of an extensive collection of Campylobacter spp. isolates from multiple origins and determined an association between this ST and its high prevalence in human hosts (Méric et al., 2018). This could indicate that asymptomatic human carriers could play a role as reservoir in Chile or that there is no sufficient data about Campylobacter isolated from other animal sources. In fact, the source attribution analysis showed that most of the isolates from this ST are related to either environmental or ovine sources (Figure 2). This result is unexpected due to the low consumption of ovine-derived products in Santiago and the absence of reports associating Campylobacter cases to environmental transmission in the country. These data suggest the need of further epidemiological studies and active national surveillance to determine such potential reservoirs of this and other STs in Chile.
4.2 Resistome analysis predicts a high susceptibility to macrolides in clinical strains
This study identified 25 distinct antimicrobial resistance determinants and point mutations within the Chilean isolates. Even though a low percentage of strains are predicted to be resistant to aminoglycosides, we found diverse genetic determinants associated with this resistance, which agrees with worldwide observations (Cobo-Díaz et al., 2021). The high percentage of isolates harboring the C257T point mutation in the QRDR of gyrA confirmed the global trends regarding fluoroquinolones resistance in Campylobacter spp (World Health Organization E, 2017; Gomes et al., 2020). However, the presence of the gyrA mutation decreased compared to the findings of our previous work (Bravo et al., 2021). A similar trend was observed for the mutations and genetic markers related to macrolides resistance, which is the group of choice for treatment of human campylobacteriosis (Tang et al., 2017) (Supplementary Figure S3B). Although we lack information regarding antimicrobials used in animal production, these trends might result from a more restricted veterinary usage of these drugs in the last years.
4.3 Lineage-relatedness of LOS and capsular modifications genes
The virulome analysis revealed that all isolates harbored genes related to adherence and invasion (cadF, jlpA, pebA, ciaB), which is in accordance with previous reports (Redondo et al., 2019; Truccollo et al., 2021). Interestingly, virulence factors associated with lipooligosaccharide (LOS) and capsular modifications were lineage-related, i.e., wlaN, neuA1, neuB1, neuBC1, cj1136, and cstIII were only identified in strains belonging to CC-21 and CC-353, particularly ST-50, ST-21, ST-2395, and ST-2517. Guirado et al. had previously reported similar findings in C. jejuni strains isolated from different sources (Guirado et al., 2020), showing a high prevalence of wlaN and cgtB (related to GBS (Guirado et al., 2020)) (both involved in LOS sialylation) in CC-21 strains. The fact that these genetic determinants were found in high-prevalent lineages reinforces the importance of epidemiological surveillance in order to better respond to outbreaks.
4.4 First report of the pVir plasmid C. jejuni strains from Chile
Our study firstly reports the presence of the pVir plasmid in clinical strains from Chile having a high similarity to pVir plasmids from Japan strains. Although this plasmid is infrequent in clinical isolates worldwide and its role in pathogenesis is unclear, its presence has been associated to the occurrence of bloody stools in patients with campylobacteriosis (Tracz et al., 2005). Given its possible role in the clinical outcome, it is important to follow-up the dynamics of the bacterial population carrying this plasmid in Chile and South America.
4.5 Chilean clinical strains harbor different repertories of fT3SS and T6SS effector protein candidates
In addition to its role in bacterial motility, the fT3SS is one of the major virulence factors of C. jejuni (Gabbert et al., 2023). Our analysis showed the presence of the fT3SS in every strain analyzed (Supplementary Table S5), highlighting its importance for C. jejuni. Notably, we identified important differences regarding the presence of known and putative T3SS effector proteins in C. jejuni clinical strains isolated from Chile. One of the major differences was the absence of FspA homologs in most Chilean strains. FspA proteins are secreted by the fT3SS, and FspA1 induces apoptosis to host-cells in vitro (Poly et al., 2007). Therefore, the absence of these proteins may impact the virulence of these strains. Besides known fT3SS effector genes, we also detected important differences regarding the presence of 57 recently identified T3SS effector candidates (Gabbert et al., 2023). Based on the presence/absence of these putative effectors, the hierarchical analysis revealed five major subgroups with distinct effector repertoires (Figure 5). Different fT3SS effector repertories could translate to different infection outcomes due to differences in the ability of these strains to invade and colonize the gut. In addition, some of the most conserved effectors of the fT3SS (Figure 5) are poorly characterized highlighting the need to dissect the contribution of these effectors to the pathogenesis of Campylobacter.
We also observed significant differences regarding the presence of the T6SS and the distribution of its putative effector proteins. T6SS has emerged as a potential new virulence factor of Campylobacter spp (Liaw et al., 2019; Ungureanu et al., 2019), and was detected more frequently compared to our previous work (Bravo et al., 2021). Notably, the presence of T6SS was associated with strains of the chicken-specific CC-353. A lineage-relatedness was also reported by Rokney et al. with a high prevalence of T6SS in CC-353, CC-460, CC-607, and CC-446 (Rokney et al., 2018) and by Van Vliet in CC-464, CC- 353, CC-573 and CC-403 (van Vliet et al., 2021). Even though there is no evidence that this system is related to more severe clinical outcomes (Agnetti et al., 2019), its presence in C. jejuni strains from chicken-associated clonal complexes suggests that it might play a role in the increased ability to colonize the gastrointestinal tract of humans and chickens.
Regarding the presence and distribution of T6SS effector proteins, we observed significant differences between the Chilean clinical strains and type strain 488. The hierarchical analysis identified four distinct effector protein profiles, different from strain 488. Differences that may impact the ability of these clinical strains to compete with other bacteria during gut colonization in both chicken and humans. One group (IV) lacked most of the predicted effectors for this strain. We identified a putative new E/I module within the variable region of the T6SS gene cluster of strain CFSAN122739 (Figure 6). This module was present in most strains of subgroup I of the hierarchical analysis (Figure 7), suggesting that at least some clinical strains could harbor novel effectors beyond the repertoire predicted for the type 488 strain. The differences observed for both fT3SS and T6SS effector protein candidates could account for strain-dependent differences in pathogenicity and the ability of these strains to survive in the environment or colonize different hosts.
5 Final remarks
In conclusion, the study demonstrated the emergence of certain clones of C. jejuni, not previously identified in the Central region of Chile, and the presence of lineage-related virulence factors that might be relevant for transmission and/or clinical outcomes. More genomic studies are necessary to gain further information on the transmission dynamics of pathogenic Campylobacter spp. to humans and the potential emergence of new virulence and antimicrobial resistance markers in Chile and other Latin American countries.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
AK: Methodology, Formal analysis, Investigation, Visualization, Resources, Data curation, Writing-Original Draft and review and editing. LP: Investigation, Visualization, review and editing final manuscript. TW: Investigation, Visualization, review and editing final manuscript. CV: Investigation, review and editing final manuscript. CM-R: Formal analysis, Investigation, Visualization, review and editing final manuscript. JU: Formal analysis, Investigation, Visualization, review and editing final manuscript. NG-E: Methodology, Formal analysis, Investigation, Visualization, Resources, Data curation, review and editing final manuscript. CG: Methodology, Formal analysis, Investigation, Visualization, Resources, Data curation, review and editing final manuscript. CB: Methodology, Formal analysis, Investigation, Visualization, Resources, Data curation, Funding acquisition, Writing-Original Draft and review and editing. VB: Conceptualization, Methodology, Formal analysis, Investigation, Visualization, Resources, Data curation, Writing-Original Draft and review and editing, Funding acquisition, Supervision and Project administration. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by DICYT grant 022001BZ USACH from VB. CB was supported by FONDECYT grant 1201805, ECOS-ANID ECOS200037 and HHMI-Gulbenkian International Research Scholar Grant #55008749. JU and CM-R were supported by FONDECYT grant 1221209. NG-E and CG were supported by FDA Foods Program Intramural Funds. AK was supported by FONDECYT grant 1191074.
Acknowledgments
Universidad de Santiago de Chile, USACH. Proyecto Dicyt 022001BZ, Vicerrectoría de Investigación, Desarrollo e Innovación.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1208825/full#supplementary-material
Supplementary Figure 1 | Blast-based dendrograms of clinical Campylobacter strains harboring the pVir plasmid.
Supplementary Figure 2 | Analysis and sequence alignment of the putative T6SS effector protein Z_00323 by the structure-based homology tool HHpred.
Supplementary Figure 3 | Comparison of CCs distribution and antimicrobial resistance determinants in two periods of study in clinical Campylobacter strains isolated in Santiago, Chile. (A). Comparison of clonal complexes distribution amongst clinical Campylobacter strains in two periods of study (2017-2019 vs. 2020-2021). Colored bars represent the different CCs described in both periods of study. (B). Comparison of fluoroquinolones (azure), macrolides (pea) and tetracycline (yellow) resistance levels in the studied periods (2017-2019 vs. 2020-2021).
References
Acheson, D., Allos, B. M. (2001). Campylobacter jejuni infections: update on emerging issues and trends. Clin. Infect. Diseases 32 (8), 1201–1206. doi: 10.1086/319760
Agnetti, J., Seth-Smith, H. M. B., Ursich, S., Reist, J., Basler, M., Nickel, C., et al. (2019). Clinical impact of the type VI secretion system on virulence of campylobacter species during infection. BMC Infect. Dis. 19 (1), 237. doi: 10.1186/s12879-019-3858-x
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2022). CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51 (D1), D690–D699. doi: 10.1093/nar/gkac920
Alcoforado Diniz, J., Liu, Y., Coulthurst, S. J. (2015). Molecular weaponry: diverse effectors delivered by the type VI secretion system. Cell. Microbiol. 17 (12), 1742–1751. doi: 10.1111/cmi.12532
Allsopp, L. P., Bernal, P., Nolan, L. M., Filloux, A. (2020). Causalities of war: the connection between type VI secretion system and microbiota. Cell. Microbiol. 22 (3), e13153. doi: 10.1111/cmi.13153
Arning, N., Sheppard, S. K., Clifton, D. A., Wilson, D. J. (2021). Machine learning to predict the source of campylobacteriosis using whole genome data. Bioinformatics 17 (10), e1009436. doi: 10.1101/2021.02.23.432443
Bacon, D. J., Alm, R. A., Burr, D. H., Hu, L., Kopecko, D. J., Ewing, C. P., et al. (2000). Involvement of a plasmid in virulence of Campylobacter jejuni 81-176. Infect. Immun. 68 (8), 4384–4390. doi: 10.1128/IAI.68.8.4384-4390.2000
Barrero-Tobon, A. M., Hendrixson, D. R. (2012). Identification and analysis of flagellar coexpressed determinants (Feds) of Campylobacter jejuni involved in colonization: flagellar coexpressed determinants of C. jejuni. Mol. Microbiol. 84 (2), 352–369. doi: 10.1111/j.1365-2958.2012.08027.x
Bernal, P., Llamas, M. A., Filloux, A. (2018). Type VI secretion systems in plant-associated bacteria. Environ. Microbiol. 20 (1), 1–15. doi: 10.1111/1462-2920.13956
Berthenet, E., Thépault, A., Chemaly, M., Rivoal, K., Ducournau, A., Buissonnière, A., et al. (2019). Source attribution of Campylobacter jejuni shows variable importance of chicken and ruminants reservoirs in non-invasive and invasive French clinical isolates. Sci. Rep. 9 (1), 8098. doi: 10.1038/s41598-019-44454-2
Bleumink-Pluym, N. M. C., van Alphen, L. B., Bouwman, L. I., Wösten, M. M. S. M., van Putten, J. P. M. (2013). Identification of a functional type VI secretion system in Campylobacter jejuni conferring capsule polysaccharide sensitive cytotoxicity. PLoS Pathog. 9 (5), e1003393. doi: 10.1371/journal.ppat.1003393
Bortolaia, V., Kaas, R. S., Ruppe, E., Roberts, M. C., Schwarz, S., Cattoir, V., et al. (2020). ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75 (12), 3491–3500. doi: 10.1093/jac/dkaa345
Bravo, V., Katz, A., Porte, L., Weitzel, T., Varela, C., Gonzalez-Escalona, N., et al. (2021). Genomic analysis of the diversity, antimicrobial resistance and virulence potential of clinical Campylobacter jejuni and campylobacter coli strains from Chile. PLoS Negl. Trop. Dis. 15 (2), e0009207. doi: 10.1371/journal.pntd.0009207
Bravo, V., Porte, L., Weitzel, T., Varela, C., Blondel, C. J., Gonzalez-Escalona, N. (2020a). Complete genome sequences of 17 clinical Campylobacter jejuni strains from Chile. Microbiol. Resour. Announc. 9 (30), e00535–e00520. doi: 10.1128/MRA.00535-20
Bravo, V., Varela, C., Porte, L., Weitzel, T., Kastanis, G. J., Balkey, M., et al. (2020b). Whole-genome sequences of 51 Campylobacter jejuni and 12 campylobacter coli clinical isolates from Chile. Microbiol. Resour. Announc. 9 (18), MRA.00072-20, e00072–20. doi: 10.1128/MRA.00072-20
Burnham, P. M., Hendrixson, D. R. (2018). Campylobacter jejuni: collective components promoting a successful enteric lifestyle. Nat. Rev. Microbiol. 16 (9), 551–565. doi: 10.1038/s41579-018-0037-9
Butzler, J. P. (2004). Campylobacter, from obscurity to celebrity. Clin. Microbiol. Infection 10 (10), 868–876. doi: 10.1111/j.1469-0691.2004.00983.x
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., McQuillan, J. A. (2012). Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 28 (4), 464–469. doi: 10.1093/bioinformatics/btr703
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., Parkhill, J. (2005). ACT: the Artemis comparison tool. Bioinformatics. 21 (16), 3422–3423. doi: 10.1093/bioinformatics/bti553
Chassaing, B., Cascales, E. (2018). Antibacterial weapons: targeted destruction in the microbiota. Trends Microbiol. 26 (4), 329–338. doi: 10.1016/j.tim.2018.01.006
Cobo-Díaz, J. F., González del Río, P., Álvarez-Ordóñez, A. (2021). Whole resistome analysis in Campylobacter jejuni and c. coli genomes available in public repositories. Front. Microbiol. 12, 662144. doi: 10.3389/fmicb.2021.662144
Cody, A. J., Bray, J. E., Jolley, K. A., McCarthy, N. D., Maiden, M. C. J. (2017). Core genome multilocus sequence typing scheme for stable, comparative analyses of Campylobacter jejuni and c. coli human disease isolates. J. Clin. Microbiol. 55 (7), 2086–2097. doi: 10.1128/JCM.00080-17
Cody, A. J., Maiden, M. C., Strachan, N. J., McCarthy, N. D. (2019) A systematic review of source attribution of human campylobacteriosis using multilocus sequence typing. Euro Surveill. 24 (43), 1800696. doi: 10.2807/1560-7917.ES.2019.24.43.1800696
Cody, A. J., McCarthy, N. M., Wimalarathna, H. L., Colles, F. M., Clark, L., Bowler, I. C. J. W., et al. (2012). A longitudinal 6-year study of the molecular epidemiology of clinical campylobacter isolates in Oxfordshire, united kingdom. J. Clin. Microbiol. 50 (10), 3193–3201. doi: 10.1128/JCM.01086-12
Collado, L. (2020). Diagnóstico microbiológico y vigilancia epidemiológica de la campilobacteriosis en Chile: situación actual y desafíos futuros. Rev. Chil Infectol 37 (3). doi: 10.4067/s0716-10182020000300244
Collado, L., Muñoz, N., Porte, L., Ochoa, S., Varela, C., Muñoz, I. (2018). Genetic diversity and clonal characteristics of ciprofloxacin-resistant Campylobacter jejuni isolated from Chilean patients with gastroenteritis. Infection Genet. Evolution 58, 290–293. doi: 10.1016/j.meegid.2017.12.026
Costa, D., Iraola, G. (2019). Pathogenomics of emerging Campylobacter species. Clin. Microbiol. Rev. 32 (4), e00072–e00018. doi: 10.1128/CMR.00072-18
Coulthurst, S. (2019). The type VI secretion system: a versatile bacterial weapon. Microbiology. 165 (5), 503–515. doi: 10.1099/mic.0.000789
Coyne, M. J., Comstock, L. E. (2019). Type VI secretion systems and the gut microbiota. Microbiol. Spectr. 7 (2), 7. doi: 10.1128/9781683670285.ch27
Culebro, A., Machado, M. P., Carriço, J. A., Rossi, M. (2018). Origin, evolution, and distribution of the molecular machinery for biosynthesis of sialylated lipooligosaccharide structures in campylobacter coli. Sci. Rep. 8 (1), 3028. doi: 10.1038/s41598-018-21438-2
Darling, A. E., Mau, B., Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5 (6), e11147. doi: 10.1371/journal.pone.0011147
Dearlove, B. L., Cody, A. J., Pascoe, B., Méric, G., Wilson, D. J., Sheppard, S. K. (2016). Rapid host switching in generalist campylobacter strains erodes the signal for tracing human infections. ISME J. 10 (3), 721–729. doi: 10.1038/ismej.2015.149
Durand, E., Cambillau, C., Cascales, E., Journet, L. (2014). VgrG, tae, tle, and beyond: the versatile arsenal of type VI secretion effectors. Trends Microbiol. 22 (9), 498–507. doi: 10.1016/j.tim.2014.06.004
EFSA (2019). The European union one health 2018 zoonoses report. EFSA J. 17 (12), e05926. doi: 10.2903/j.efsa.2019.5926
Elmi, A., Nasher, F., Dorrell, N., Wren, B., Gundogdu, O. (2021). Revisiting Campylobacter jejuni virulence and fitness factors: role in sensing, adapting, and competing. Front. Cell Infect. Microbiol. 10, 607704. doi: 10.3389/fcimb.2020.607704
Epping, L., Walther, B., Piro, R. M., Knüver, M. T., Huber, C., Thürmer, A., et al. (2021). Genome-wide insights into population structure and host specificity of Campylobacter jejuni. Sci. Rep. 11 (1), 10358. doi: 10.1038/s41598-021-89683-6
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019). Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63 (11), e00483–e00419. doi: 10.1128/AAC.00483-19
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucl. Acids Res. 42 (D1), D222–D230. doi: 10.1093/nar/gkt1223
Finsterer, J. (2022). Triggers of Guillain–Barré syndrome: Campylobacter jejuni predominates. IJMS. 23 (22), 14222. doi: 10.3390/ijms232214222
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., Aarestrup, F. M. (2022). ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 8 (1), 000748. doi: 10.1099/mgen.0.000748
Gabbert, A. D., Mydosh, J. L., Talukdar, P. K., Gloss, L. M., McDermott, J. E., Cooper, K. K., et al. (2023). The missing pieces: the role of secretion systems in Campylobacter jejuni virulence. Biomolecules. 13 (1), 135. doi: 10.3390/biom13010135
Garcia Bardales, P. F., Schiaffino, F., Huynh, S., Paredes Olortegui, M., Peñataro Yori, P., Pinedo Vasquez, T., et al. (2022). Candidatus campylobacter infans” detection is not associated with diarrhea in children under the age of 2 in Peru. PLoS Negl. Trop. Dis. 16 (10), e0010869. doi: 10.1371/journal.pntd.0010869
Gomes, C. N., Frazão, M. R., Passaglia, J., Duque, S. S., Medeiros, M. I. C., Falcão, J. P. (2020). Molecular epidemiology and resistance profile of Campylobacter jejuni and Campylobacter coli strains isolated from different sources in Brazil. Microbial Drug Resistance 26 (12), 1516–1525. doi: 10.1089/mdr.2019.0266
Guirado, P., Paytubi, S., Miró, E., Iglesias-Torrens, Y., Navarro, F., Cerdà-Cuéllar, M., et al. (2020). Differential distribution of the wlaN and cgtB genes, associated with Guillain-Barré syndrome, in Campylobacter jejuni isolates from humans, broiler chickens, and wild birds. Microorganisms. 8 (3), 325. doi: 10.3390/microorganisms8030325
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58 (1), 212–220. doi: 10.1128/AAC.01310-13
Hachani, A., Wood, T. E., Filloux, A. (2016). Type VI secretion and anti-host effectors. Curr. Opin. Microbiol. 29, 81–93. doi: 10.1016/j.mib.2015.11.006
Hallgren, J., Tsirigos, K. D., Pedersen, M. D., Almagro Armenteros, J. J., Marcatili, P., Nielsen, H., et al. (2022). DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. Bioinformatics. Available at: http://biorxiv.org/lookup/doi/10.1101/2022.04.08.487609.
Ho, B. T., Dong, T. G., Mekalanos, J. J. (2014). A view to a kill: the bacterial type VI secretion system. Cell Host Microbe 15 (1), 9–21. doi: 10.1016/j.chom.2013.11.008
Humphrey, T., O’Brien, S., Madsen, M. (2007). Campylobacters as zoonotic pathogens: a food production perspective. Int. J. Food Microbiol. 117 (3), 237–257. doi: 10.1016/j.ijfoodmicro.2007.01.006
Jolley, K. A., Bray, J. E., Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Kaakoush, N. O., Castaño-Rodríguez, N., Mitchell, H. M., Man, S. M. (2015). Global epidemiology of campylobacter infection. Clin. Microbiol. Rev. 28 (3), 687–720. doi: 10.1128/CMR.00006-15
Kanehisa, M. (2002). The KEGG databases at GenomeNet. Nucleic Acids Res. 30 (1), 42–46. doi: 10.1093/nar/30.1.42
Konkel, M. E., Kim, B. J., Rivera-Amill, V., Garvis, S. G. (1999). Bacterial secreted proteins are required for the internaliztion of Campylobacter jejuni into cultured mammalian cells. Mol. Microbiol. 32 (4), 691–701. doi: 10.1046/j.1365-2958.1999.01376.x
Kreling, V., Falcone, F. H., Kehrenberg, C., Hensel, A. (2020). Campylobacter sp.: pathogenicity factors and prevention methods–new molecular targets for innovative antivirulence drugs? Appl. Microbiol. Biotechnol. 104 (24), 10409–10436. doi: 10.1007/s00253-020-10974-5
Lertpiriyapong, K., Gamazon, E. R., Feng, Y., Park, D. S., Pang, J., Botka, G., et al. (2012). Campylobacter jejuni type VI secretion system: roles in adaptation to deoxycholic acid, host cell adherence, invasion, and In vivo colonization. PLoS One 7 (8), e42842. doi: 10.1371/journal.pone.0042842
Letunic, I., Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47 (W1), W256–W259. doi: 10.1093/nar/gkz239
Levican, A., Ramos-Tapia, I., Briceño, I., Guerra, F., Mena, B., Varela, C., et al. (2019). Genomic analysis of Chilean strains of Campylobacter jejuni from human faeces. BioMed. Res. Int. 2019, 1–12. doi: 10.1155/2019/1902732
Liaw, J., Hong, G., Davies, C., Elmi, A., Sima, F., Stratakos, A., et al. (2019). The Campylobacter jejuni type VI secretion system enhances the oxidative stress response and host colonization. Front. Microbiol. 10, 2864. doi: 10.3389/fmicb.2019.02864
Linton, D., Gilbert, M., Hitchen, P. G., Dell, A., Morris, H. R., Wakarchuk, W. W., et al. (2002). Phase variation of a β-1,3 galactosyltransferase involved in generation of the ganglioside GM1-like lipo-oligosaccharide of Campylobacter jejuni: c. jejuni LOS phase variation. Mol. Microbiol. 37 (3), 501–514. doi: 10.1046/j.1365-2958.2000.02020.x
Linton, D., Karlyshev, A. V., Hitchen, P. G., Morris, H. R., Dell, A., Gregson, N. A., et al. (2000). Multiple n-acetyl neuraminic acid synthetase (neuB) genes in Campylobacter jejuni: identification and characterization of the gene involved in sialylation of lipo-oligosaccharide. Mol. Microbiol. 35 (5), 1120–1134. doi: 10.1046/j.1365-2958.2000.01780.x
Liu, B., Zheng, D., Jin, Q., Chen, L., Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47 (D1), D687–D692. doi: 10.1093/nar/gky1080
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48 (D1), D265–D268. doi: 10.1093/nar/gkz991
Maguire, M., Khan, A. S., Adesiyun, A. A., Georges, K., Gonzalez-Escalona, N. (2021). Closed genome sequence of a salmonella enterica serotype senftenberg strain carrying the mcr-9 gene isolated from broken chicken eggshells in Trinidad and Tobago. Microbiol. Resour. Announc. 10 (21), e01465–e01420. doi: 10.1128/MRA.01465-20
Marasini, D., Karki, A. B., Buchheim, M. A., Fakhr, M. K. (2018). Phylogenetic relatedness among plasmids harbored by Campylobacter jejuni and campylobacter coli isolated from retail meats. Front. Microbiol. 9, 2167. doi: 10.3389/fmicb.2018.02167
Méric, G., McNally, A., Pessia, A., Mourkas, E., Pascoe, B., Mageiros, L., et al. (2018). Convergent amino acid signatures in polyphyletic Campylobacter jejuni subpopulations suggest human niche tropism. Genome Biol. Evolution 10 (3), 763–774. doi: 10.1093/gbe/evy026
Nylen, G., Dunstan, F., Palmer, S. R., Andersson, Y., Bager, F., Cowden, J., et al. (2002). The seasonal distribution of campylobacter infection in nine European countries and new Zealand. Epidemiol. Infect. 128 (3), 383–390. doi: 10.1017/S0950268802006830
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 31 (22), 3691–3693. doi: 10.1093/bioinformatics/btv421
Panzenhagen, P., Portes, A. B., dos Santos, A. M. P., Duque S da, S., Conte Junior, C. A. (2021). The distribution of Campylobacter jejuni virulence genes in genomes worldwide derived from the NCBI pathogen detection database. Genes. 12 (10), 1538. doi: 10.3390/genes12101538
Parker, C. T., Schiaffino, F., Huynh, S., Paredes Olortegui, M., Peñataro Yori, P., Garcia Bardales, P. F., et al. (2022). Shotgun metagenomics of fecal samples from children in Peru reveals frequent complex co-infections with multiple campylobacter species. PloS Negl. Trop. Dis. 16 (10), e0010815. doi: 10.1371/journal.pntd.0010815
Pascoe, B., Schiaffino, F., Murray, S., Meric, G., Bayliss, S. C., Hitchings, M. D., et al. (2020). Genomic epidemiology of Campylobacter jejuni associated with asymptomatic paediatric infection in the Peruvian Amazon. PLoS Negl. Trop. Dis. 14 (8), e0008533. doi: 10.1371/journal.pntd.0008533
Platts-Mills, J. A., Kosek, M. (2014). Update on the burden of campylobacter in developing countries. Curr. Opin. Infect. Dis. 27 (5), 444–450. doi: 10.1097/QCO.0000000000000091
Poly, F., Ewing, C., Goon, S., Hickey, T. E., Rockabrand, D., Majam, G., et al. (2007). Heterogeneity of a Campylobacter jejuni protein that is secreted through the flagellar filament. Infect. Immun. 75 (8), 3859–3867. doi: 10.1128/IAI.00159-07
Porte, L., Pérez, C., Barbé, M., Varela, C., Vollrath, V., Legarraga, P., et al. (2023). Campylobacter spp. prevalence in Santiago, Chile: a study based on molecular detection in clinical stool samples from 2014 to 2019. Pathogens. 12 (3), 504. doi: 10.3390/pathogens12030504
Porte, L., Varela, C., Haecker, T., Morales, S., Weitzel, T. (2016). Impact of changing from staining to culture techniques on detection rates of campylobacter spp. in routine stool samples in Chile. BMC Infect. Dis. 16 (1), 196. doi: 10.1186/s12879-016-1546-7
Quino, W., Caro-Castro, J., Mestanza, O., Hurtado, V., Zamudio, M. L., Cruz-Gonzales, G., et al. (2022). Emergence and molecular epidemiology of Campylobacter jejuni ST-2993 associated with a Large outbreak of Guillain-Barré syndrome in Peru. Microbiol. Spectr. 10 (5), e01187–e01122. doi: 10.1128/spectrum.01187-22
Ramos, A. P., Leonhard, S. E., Halstead, S. K., Cuba, M. A., Castañeda, C. C., Dioses, J. A., et al. (2021). Guillain-Barré Syndrome outbreak in Peru 2019 associated with Campylobacter jejuni infection. Neurol. Neuroimmunol. Neuroinflamm. 8 (2), e952. doi: 10.1212/NXI.0000000000000952
Redondo, N., Carroll, A., McNamara, E. (2019). Molecular characterization of campylobacter causing human clinical infection using whole-genome sequencing: virulence, antimicrobial resistance and phylogeny in Ireland. PLoS One 14 (7), e0219088. doi: 10.1371/journal.pone.0219088
Rivera, F. N., Bustos, B. R., Montenegro, H. S., Sandoval, M. M., Castillo, N. J., Fernández, J. H., et al. (2011). [Genotyping and antibacterial resistance of campylobacter spp strains isolated in children and in free range poultry]. Rev. Chil. Infectol. 28 (6), 555–5862.
Robinson, L., Liaw, J., Omole, Z., Corcionivoschi, N., Hachani, A., Gundogdu, O. (2022). In silico investigation of the genus campylobacter type VI secretion system reveals genetic diversity in organization and putative effectors: this article is part of the “Genomics, epidemiology and evolution of campylobacter, helicobacter and related organisms” collection. Microbial Genomics 8 (10), mgen000898. doi: 10.1099/mgen.0.000898
Robinson, L., Liaw, J., Omole, Z., Xia, D., van Vliet, A. H. M., Corcionivoschi, N., et al. (2021). Bioinformatic analysis of the Campylobacter jejuni type VI secretion system and effector prediction. Front. Microbiol. 12, 694824. doi: 10.3389/fmicb.2021.694824
Rokney, A., Valinsky, L., Moran-Gilad, J., Vranckx, K., Agmon, V., Weinberger, M. (2018). Genomic epidemiology of Campylobacter jejuni transmission in Israel. Front. Microbiol. 9, 2432. doi: 10.3389/fmicb.2018.02432
Roux, F., Sproston, E., Rotariu, O., MacRae, M., Sheppard, S. K., Bessell, P., et al. (2013). Elucidating the aetiology of human campylobacter coli infections. Alter, T.PloS One 8 (5), e64504. doi: 10.1371/journal.pone.0064504
Sana, T. G., Lugo, K. A., Monack, D. M. (2017). T6SS: the bacterial “fight club” in the host gut. PloS Pathog. 13 (6), e1006325. doi: 10.1371/journal.ppat.1006325
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068–2069. doi: 10.1093/bioinformatics/btu153
Seemann, T. (2020) Tseemann/abricate. Available at: https://github.com/tseemann/abricate.
Sheppard, S. K., Colles, F. M., McCARTHY, N. D., Strachan, N. J. C., Ogden, I. D., Forbes, K. J., et al. (2011). Niche segregation and genetic structure of Campylobacter jejuni populations from wild and agricultural host species. Mol. Ecology 20 (16), 3484–3490. doi: 10.1111/j.1365-294X.2011.05179.x
Sigrist, C. J. A., De Castro, E., Cerutti, L., Cuche, B. A., Hulo, N., Bridge, A., et al. (2012). New and continuing developments at PROSITE. Nucleic Acids Res. 41 (D1), D344–D347. doi: 10.1093/nar/gks1067
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., et al. (2018). chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 4 (3), e000166. doi: 10.1099/mgen.0.000166
Sullivan, M. J., Petty, N. K., Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics. 27 (7), 1009–1010. doi: 10.1093/bioinformatics/btr039
Tang, Y., Fang, L., Xu, C., Zhang, Q. (2017). Antibiotic resistance trends and mechanisms in the foodborne pathogen, campylobacter. Anim. Health Res. Rev. 18 (2), 87–98. doi: 10.1017/S1466252317000135
Tracz, D. M., Keelan, M., Ahmed-Bentley, J., Gibreel, A., Kowalewska-Grochowska, K., Taylor, D. E. (2005). pVir and bloody diarrhea in Campylobacter jejuni enteritis. Emerg. Infect. Dis. 11 (6), 839–843. doi: 10.3201/eid1106.041052
Truccollo, B., Whyte, P., Burgess, C., Bolton, D. (2021). Genetic characterisation of a subset of Campylobacter jejuni isolates from clinical and poultry sources in Ireland. PloS One 16 (3), e0246843. doi: 10.1371/journal.pone.0246843
Ungureanu, V. A., AStratakos, A. c. h., Gundogdu, O., Stef, L., Pet, I., Pet, E., et al. (2019). Virulence of a T6SS Campylobacter jejuni chicken isolate from north Romania. BMC Res. Notes 12 (1), 180. doi: 10.1186/s13104-019-4201-8
van Vliet, A. H. M., Charity, O. J., Reuter, M. (2021). A campylobacter integrative and conjugative element with a CRISPR-Cas9 system targeting competing plasmids: a history of plasmid warfare? Microbial Genomics 7 (11), 000729. doi: 10.1099/mgen.0.000729
Wang, J., Yang, B., Leier, A., Marquez-Lago, T. T., Hayashida, M., Rocker, A., et al. (2018). Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics. 34 (15), 2546–2555. doi: 10.1093/bioinformatics/bty155
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13 (6), e1005595. doi: 10.1371/journal.pcbi.1005595
World Health Organization E (2017). Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics 7.
World Health Organization, Food and Agriculture Organization of the United Nations & World Organisation for Animal Health (2013). The global view of campylobacteriosis: report of an expert consultation (Utrecht, Netherlands: World Health Organization). Available at: https://apps.who.int/iris/handle/10665/80751.
Wu, C. F., Smith, D. A., Lai, E. M., Chang, J. H. (2018). “The agrobacterium type VI secretion system: a contractile nanomachine for interbacterial competition,” in Agrobacterium biology 418, 215–231. doi: 10.1007/82_2018_99.
Zankari, E., Allesøe, R., Joensen, K. G., Cavaco, L. M., Lund, O., Aarestrup, F. M. (2017). PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72 (10), 2764–2768. doi: 10.1093/jac/dkx217
Zimmermann, L., Stephens, A., Nam, S. Z., Rau, D., Kübler, J., Lozajic, M., et al. (2018). A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430 (15), 2237–2243. doi: 10.1016/j.jmb.2017.12.007
Keywords: molecular epidemiology, sequence analysis, campylobacteriosis, clonal complexes, antimicrobial resistance, T6SS, fT3SS
Citation: Katz A, Porte L, Weitzel T, Varela C, Muñoz-Rehbein C, Ugalde JA, Grim C, González-Escalona N, Blondel CJ and Bravo V (2023) Whole-genome sequencing reveals changes in genomic diversity and distinctive repertoires of T3SS and T6SS effector candidates in Chilean clinical Campylobacter strains. Front. Cell. Infect. Microbiol. 13:1208825. doi: 10.3389/fcimb.2023.1208825
Received: 19 April 2023; Accepted: 16 June 2023;
Published: 13 July 2023.
Edited by:
Rachel Leite Ribeiro, Fluminense Federal University, BrazilReviewed by:
Marja-Liisa Hänninen, University of Helsinki, FinlandRoberto Rosales-Reyes, National Autonomous University of Mexico, Mexico
Stephanie S.R. Souza, University at Albany, United States
Copyright © 2023 Katz, Porte, Weitzel, Varela, Muñoz-Rehbein, Ugalde, Grim, González-Escalona, Blondel and Bravo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Verónica Bravo, dmVyb25pY2EuYnJhdm9AdXNhY2guY2w=; Carlos J. Blondel, Y2FybG9zLmJsb25kZWxAdW5hYi5jbA==