 Junya Yamagishi
Junya Yamagishi Onur Ceylan
Onur Ceylan Xuenan Xuan
Xuenan Xuan Ferda Sevinc
Ferda Sevinc- 1International Institute for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 2Global Station for Zoonosis Control, GI-CoRE, Hokkaido University, Sapporo, Japan
- 3Department of Parasitology, Faculty of Veterinary Medicine, University of Selcuk, Konya, Türkiye
- 4National Research Center for Protozoan Diseases, Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan
Ovine babesiosis, caused by Babesia ovis, is an acute, lethal, and endemic disease worldwide and causes a huge economic loss to animal industry. Pathogen genome sequences can be utilized for selecting diagnostic markers, drug targets, and antigens for vaccine development; however, those for B. ovis have not been available so far. In this study, we obtained a draft genome sequence for B. ovis isolated from an infected sheep in Turkey. The genome size was 7.81 Mbp with 3,419 protein-coding genes. It consisted of 41 contigs, and the N50 was 526 Kbp. There were 259 orthologs identified among eight Babesia spp., Plasmodium falciparum, and Toxoplasma gondii. A phylogeny was estimated on the basis of the orthologs, which showed B. ovis to be closest to B. bovis. There were 43 ves genes identified using hmm model as well. They formed a discriminating cluster to other ves multigene family of Babesia spp. but showed certain similarities to those of B. bovis, B. caballi, and Babesia sp. Xinjiang, which is consistent with the phylogeny. Comparative genomics among B. ovis and B. bovis elucidated uniquely evolved genes in these species, which may account for the adaptation.
Background
Babesia ovis is a tick-borne intraerythrocytic apicomplexan parasite. It is one of the causative agents of ovine babesiosis in small ruminants. The disease is a worldwide epidemic, especially in Europe, Africa, and Asia. The manifestation is characterized by fever, hemolytic anemia, hemoglobinuria, and icterus (Yeruham et al., 1998; Ahmed et al., 2006; Fakhar et al., 2012; Ranjbar-Bahadori et al., 2012; Sevinc et al., 2013; Fanelli, 2021). Interestingly, it is highly virulent in sheep but milder in goats (Yeruham et al., 1998). However, correlation and etiology among geographic distribution, phenotype, and genotype still need to be elucidated owing to the paucity of genome information. Current countermeasures rely on pharmacotherapy of the affected animals and vector control by acaricides, which poses risks of resistance in both pathogen and vector. Therefore, effective vaccines are also desired but have yet to be available so far. Attenuation by successive blood passages in splenectomized lambs was conducted but was not successful (Sevinc et al., 2014). Latent infection and recurrence after splenectomy were reported (Habela et al., 1990). These observations might provide clues to the difficulty in vaccine development. Potential vaccine antigens designated as B. ovis–secreted antigen 1 and B. ovis–secreted antigen 2 have been nominated by immune screening (Sevinc et al., 2015a; Sevinc et al., 2015b). Genetic diversity among the species is mainly examined using 18S rRNA genes, and the resolution is not high enough to reflect the geographic diversity (Tumwebaze et al., 2020; Galon et al., 2022a; Galon et al., 2022b). Whole genome sequence for B. ovis is promising in enhancing vaccine development and optimizing genotyping markers as well.
Whole genome sequence is one of the fundamental information in biology, and the genomes of following Babesia spp. are publicly available: B. bigemina, B. bovis, B. caballi, B. divergens B. microti, B. ovata, and Babesia sp. Xinjiang (Brayton et al., 2007; Cornillot et al., 2012; Ma et al., 2013; Jackson et al., 2014; Guan et al., 2016; Yamagishi et al., 2017). In terms of disease control, they contribute to the development of diagnostic methods and vaccines, screening of drug targets, and identification of drug-resistant genes. They are also indispensable to understand the evolution and adaptation of the parasites. By comparative genomics among Babesia spp., it is demonstrated that there is an expansion of ves genes. In B. bovis, it has ves1α and ves1β gene that code Variant Erythrocyte Surface Antigen 1a (VESA1a) and 1b (VESA1b) (Allred, 2019), respectively. According to the genome analysis, there are more than 104 ves genes including other groups of ves, ves1γ, and ves2 (Brayton et al., 2007). VESA1a and VESA1b are expressed as a set and form a heterodimer that is involved in cell adhesion and pathogenicity (Allred et al., 1993; O’Connor and Allred, 2000; Pedroni et al., 2013). In other Babesia spp., they also have species specific ves multigene families, namely, ves1a, ves1b, and ves2 of B. bigemina (Jackson et al., 2014) and B. ovata (Yamagishi et al., 2017), ves1 of B. divergens (Jackson et al., 2014) and ves1 of Babesia sp. Xinjiang (Guan et al., 2016), although their function is not well demonstrated (O’Connor and Allred, 2000; Pedroni et al., 2013). In contrast, it is known that there are species-specific gene expansions such as smorf, mtm type A and B in B. bovis, and mfs in B. bigemina and B. ovata (Brayton et al., 2007; Yamagishi et al., 2017; Hakimi et al., 2020; Hakimi et al., 2022). The smorf genes form a multigene family of B. bovis that codes small open reading frame protein and locate proximal to the ves1α and ves1β (Brayton et al., 2007). Their function is not clear (Ferreri et al., 2012), but the involvement in the transcriptional regulation of the ves genes is suggested (Allred, 2019). The mtm type A and B genes of B. bovis are group of genes with eight or more predicted multi-transmembrane domains (Hakimi et al., 2020). They are exported to the surface of infected red blood cells (RBCs) (Hakimi et al., 2022). One of the mtm type B is proven to confer drug resistance, and others are speculated to be involved in nutrition uptake (Hakimi et al., 2020; Hakimi et al., 2022). The mfs genes code Major Facilitator Superfamily proteins with eight or more predicted multi-transmembrane domains. They widely exist in Babesia spp., but the expansion is only found in B. bigemina and B. ovata (Yamagishi et al., 2017; Hakimi et al., 2020). Their function has not yet been elucidated.
In this study, we assembled a whole genome sequence of B. ovis obtained from infected sheep in the field. Comparative genomics among B. ovis and related Babesia spp. revealed a diversity of multiplied genes that may be involved in host switch and adaptation through the evolution.
Materials and methods
Genomic DNA extraction, library construction, and sequencing
Babesia ovis–infected blood sample was obtained from the natural case (50% parasitemia levels) located in an area in the Central Anatolian Region of Turkey in which ovine babesiosis is enzootic in 2015. The parasite was monitored by a microscopic examination of Giemsa-stained thin blood smears and verified by the PCR analysis. B. ovis–infected blood was washed five times with phosphate-buffered saline (PBS) by centrifugation at 2,000 rpm for 15 min to separate RBCs from other cells, and, then, infected RBCs were diluted 10 times with 0.1% saponin (in PBS) to lyse. It was kept at room temperature for 5 min and centrifuged at 4,000 rpm for 20 min to collect merozoites. The merozoite pellet was diluted with PBS, and DNA extraction was performed by using Phenol Chloroform Isoamyl Alcohol (Sigma P2069) (Green and Sambrook, 2012). MinION libraries were constructed with the Ligation Sequencing Kit 1D, LSK108 (Oxford Nanopore Technologies), and then sequenced with three FLO-MIN106 flow cells. A PacBio library was sequenced using the PacBio RS II sequencer with two SMRT cells. An Illumina library was constructed with a TruSeq DNA PCR-Free kit (Illumina), and 300-bp paired-end sequencing was performed using a MiSeq sequencer (Illumina). Reads with low quality were removed using Trimmomatic v0.36 based on the following parameters: LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 MINLEN:36.
RNAseq analysis
Infected blood from the same sheep was dissolved into a PAXgene blood RNA tube, and RNA was purified using the PAXgene Blood RNA Kit (QIAGEN). The quality and quantity of the purified RNA were validated with Bioanalyzer (Agilent). Library construction for RNAseq was performed as per the instruction manual with the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina), and 300-bp paired-end sequencing was performed using a MiSeq sequencer (Illumina).
De novo genome assembly
Obtained fast5 data were base-called to fastq using poretools 0.6.0 (Loman and Quinlan, 2014) or albacore.1.2.6. The fastq derived from MinION and Pacbio preassembled reads and Illumina paired-end reads were assembled using SPAdes-3.11.1 with -k 127 option (Bankevich et al., 2012). The resulting scaffolds were examined to subtract possible host contamination and artifacts based on the following criteria. First, scaffolds with equal or more than 1,000 bp were selected. Second, redundancy among the scaffolds was excluded if more than 90% of the length of each scaffold had more than 80% identity against the other scaffolds. The identity was evaluated by BLASTN (Altschul et al., 1990). Third, scaffolds were excluded if the region showing the highest identity against B. bovis or B. bigemina was less than 10% of each scaffold. The identity was evaluated by BLASTX (Altschul et al., 1990) using B. bovis (Brayton et al., 2007), B. bigemina (Jackson et al., 2014), and Ovis aries (Archibald et al., 2010) coding sequences and non-redundant protein database provided by National Center for Biotechnology Information (NCBI). Finally, sequencing depth was calculated using BamDeal (https://github.com/BGI-shenzhen/BamDeal) with aligned reads made by bowtie2 version 2.3.4.1 (Langmead and Salzberg, 2012) for the Illumina reads and BWA version 0.7.17 (Li and Durbin, 2010) for the nanopore and PacBio reads, and, then, scaffolds with too low (less than 10) or too high (more than 100) coverage were excluded using the Illumina reads. Scaffolds corresponding to both apicoplast and mitochondria genomes were additionally specified on the basis of sequence similarity. The qualified scaffolds were polished using the Illumina reads with Pilon with 15 iterations until a plateau was reached (Walker et al., 2014). To eliminate redundancy, further quality filtering was performed using Redundans (Pryszcz and Gabaldón, 2016). The qualified contigs were validated using BlobTools v1.1.1 (Laetsch and Blaxter, 2017). The translated amino acids sequences of each contigs were examined using Diamond version 2.1.6 (Buchfink et al., 2021) against NCBI non-redundant protein (nr) database and then subjected to BlobTools as inputs. Integrity of the assembly was validated using BUSCO using the apicomplexa_odb10 dataset (Manni et al., 2021).
Gene model estimation and functional annotation
The gene model was estimated by AUGUSTUS version 3.1.0 (Stanke et al., 2008). Trained parameters for AUGUSTUS were acquired by webAugustus (Hoff and Stanke, 2013) using the B. bovis gene model obtained from PiroplasmaDB version 5.1 (Ma et al., 2013). The RNAseq data were mapped onto the B. ovis–assembled draft genome with Tophat2 (Trapnell et al., 2009). The first round of AUGUSTUS was performed to obtain an exon–exon junction database. The second round of AUGUSTUS was performed using the database to obtain the final gene model. Our previous report describes the procedure in detail (Yamagishi et al., 2017). Functional annotation of B. ovata genes, including gene ontology (GO) terms, was conducted by Blast2GO (Conesa et al., 2005). tRNA and rRNA genes in the genome were predicted by tRNAscan (Lowe and Eddy, 1997) and RNAmmer 1.2 web servers (Lagesen et al., 2007) with default parameters, respectively. Gene model in the apicoplast and mitochondria genomes was estimated by MiGAP considering their bacterial origin.
Acquisition of publicly available sequences and gene annotation
For the comparative genomic analysis among apicomplexan parasites, we used genome assembly and annotation released in PiroplasmaDB-37 for B. bovis T2Bo strain, PiroplasmaDB-36 for B. bigemina BOND strain, PiroplasmaDB-40 for B. ovata Miyake strain, PiroplasmaDB-61 for B. caballi USDA-D6B2 strain, PiroplasmaDB-40 for B. microti RI strain, PiroplasmaDB-46 for B. divergens 1802A strain, PiroplasmaDB-55 for Babesia sp. Xinjiang, ToxoDB-40 for the T. gondii ME49 strain, and PlasmoDB-56 for the P. falciparum 3D7 strain.
Phylogenetic analyses
Orthologous genes conserved among these parasites were specified using OMA version 2.5.0 with the default parameters (Altenhoff et al., 2015). The orthologs coded in the B. ovis genome and all the nine parasites above were selected. Amino acid sequences of the orthologs were aligned using MAFFT with the default parameters (Katoh et al., 2002). The resulting gaps were trimmed, and the remaining sequences were concatenated. A phylogenetic tree by the maximum likelihood method with 100 times of bootstrap replications was constructed using MEGA version 10.0.5 (Kumar et al., 2018)
Synteny analyses
Synteny between B. ovis and B. bovis genomes were examined using MCScanX (Wang et al., 2012). The identity between the amino acid sequences of the genes was calculated using BLASTP, and gene pairs with e-values less than 1e-100 were selected and then the synteny was visualized by AccuSyn (https://accusyn.usask.ca/). Structural diversity among apicoplast genomes of B. ovis (BaOVIS_AP_20230606), B. bovis (NC_011395.1), and Babesia sp. Xinjiang (KX881914.1) was examined by dotplot using YASS (Noe and Kucherov, 2005). Structural diversity among mitochondrial genomes of B. ovis (BaOVIS_MT_20230606), B. bovis (EU075182.1), and Babesia sp. Xinjiang (KX698108.1) was examined in the same way.
Uniquely gained and lost genes in B. ovis
The uniquely gained genes were defined as the B. ovis genes that have no orthologs in either species, as shown below. The first set was closely related to B. bovis and Babesia sp. Xinjiang. The other set was wider range of Babesia spp., B. bovis, B. bigemina, B. ovata, B. caballi, B. divergens, and Babesia sp. Xinjiang. The uniquely lost genes were defined as the genes conserved in Babesia spp. mentioned above but lack in B. ovis. Those in B. bovis were retrieved as representatives. GO enrichment analysis for both gained and lost genes was performed using agriGO (Tian et al., 2017).
Identification of multigene families
The ves and smorf multigene families in the B. ovis genome were searched using hidden Markov model. The training dataset was constructed on the basis of the public genomes and annotations mentioned above. In detail, ves1α, ves1β, and smorf in B. bovis were retrieved from the gff by searching “variant erythrocyte surface antigen-1, alpha,” “variant erythrocyte surface antigen-1, beta,” and “smorf,” respectively. Babesia divergens ves1 genes were retrieved from the gff by searching “variant erythrocyte surface antigen.” Babesia bigemina ves1a, ves1b, ves1ba, and ves2 were retrieved from the website by Sanger institute (https://www.sanger.ac.uk/resources/downloads/protozoa/babesia-bigemina.html). Babesia sp. Xinjiang ves was obtained from GenBank sequence MBFZ00000000.1 (Guan et al., 2016). Hidden Markov models for each gene set were constructed, and, then, ves-related genes in B. ovis were identified using HMMER version 3.3.2 (Mistry et al., 2013). The genes in B. ovis and the abovementioned ves and smorf genes in B. bovis, B. divergens, B. bigemina, and Babesia sp. Xinjiang were aligned using BLASTP (Altschul et al., 1990). Those with more than 200-bit score were selected, and mutual similarity was visualized with Gephi using a Fruchterman–Reingold layout (Bastian et al., 2009).
The mtm multigene family in B. ovis was examined using an algorithm predicting transmembrane domain, TMHMM version 2.0c (Krogh et al., 2001), if there were eight or more predicted transmembrane domains and at least one TM domain average per 100 amino acids. Those in B. caballi, B. divergens, and Babesia sp. Xinjiang identified with the same method. Those in B. bovis and B. bigemina were retrieved from the previous study (Hakimi et al., 2020). Mutual similarity among them was examined and visualized as mentioned above.
To find other multigene families, the genes in B. ovis were aligned to themselves using BLASTP. Those with more than 200-bit score were selected, and mutual similarity was visualized with Gephi using a Fruchterman–Reingold layout (Bastian et al., 2009). Clusters were assigned by Gephi function, and their functions were estimated using BLASTP with nr database.
Results
De novo assembly of B. ovis genome
Using MiSeq, we obtained 1.9 million paired-end reads consisting of 1.1 Gbp. In parallel, a total of 196,077 reads and 295,660 subreads, consisting of 0.6 and 1.8 Gbp, were obtained using MinION FLO-MIN106 and PacBio RS II, respectively.
Using SPAdes assembler, these reads were assembled into 2,920 scaffolds comprising approximately 9.2 Mbp. Following subtraction based on redundancy, possible host contamination, and coverage, 80 scaffolds were qualified. Sequentially, we conducted error correction using the Illumina reads with Pilon. In 15 iterations, the modification reached a plateau resulting in the correction of substitution of 353 nucleotides, 11 insertions, and six deletions, respectively. To eliminate redundancy, further quality filtering was performed using Redundans (Pryszcz and Gabaldón, 2016), and 39 redundant contigs were removed. Coverage, guanine-cytosine (GC) contents, and coding sequences of the qualified contigs were validated using BlobTools (Laetsch and Blaxter, 2017) and confirmed that the coverage and GC contents were consistent except for apicoplast and mitochondria contigs. In addition, the majority of the amino acid sequences encoded by each contig were related to Apicomplexan parasite genes (Figure S1), suggesting that there was no migration of sequences other than B. ovis in the qualified contigs. The sequencing depth was 27.1, 9.3, and 1.9 for the Illumina, Nanopore and PacBio reads, respectively. Finally, the draft genome consisting of 7,811,629 bp was successfully obtained and designated as Selcuk assembly (Table 1). The longest scaffolds, N50, and L50 were 1,533,616 bp, 525,571 bp, and the fifth longest contig out of the 41 scaffolds, respectively. The integrity of the assembly was validated by BUSCO, and percentages of completeness, fragmented, and missing core genes were 96.6%, 0.9%, and 2.5%, respectively.
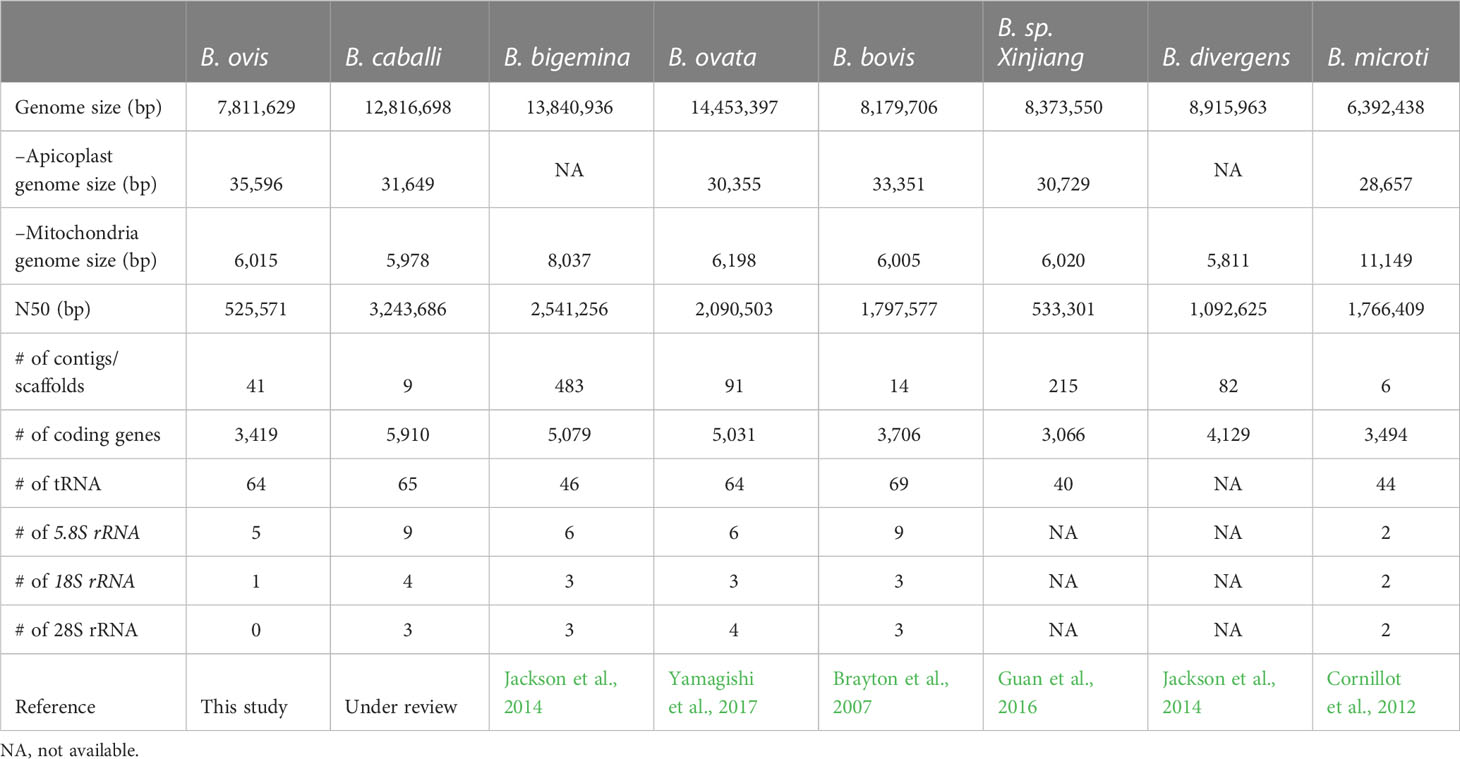
Table 1 Comparative analysis of genome and gene among representative Babesia spp.
Gene model estimation and functional annotation
In this study, we applied AUGUSTUS, which provides a more reliable gene model estimation supported by experimentally obtained transcriptome (Stanke et al., 2008). A total of 3.8-Mbp paired-end reads were obtained by RNAseq. Of these, 10.7% were successfully mapped to the assembled draft genome using Tophat2. Considering that the blood specimen included host sheep cells together with B. ovis–infected RBC, the mapped ratio was reasonable. Two scaffolds had high sequence similarity with significant e-value (“0.0” for both) against the apicoplast and mitochondria genomes of B. bovis, respectively, and, then, MiGAP was utilized for gene modeling, considering that they evolved from bacterial symbionts. Collectively, 3,419 coding regions (CDS) were predicted (Table 1). It corresponded to the number of CDS in B. bovis. tRNAs, 5.8s rRNA, and 18S rRNA were estimated in 64, five, and one locus, respectively. Functional annotation of the CDS was performed with Blast2GO. On the basis of the estimation, 2,772 CDS were functionally annotated, whereas the remaining 647 CDS were then simply assigned as hypothetical proteins.
Phylogenetic and synteny analyses
To create an orthology-based phylogenetic tree, we identified 259 single-copy orthologous genes among B. bovis, B. bigemina, B. caballi, B. microti, B. divergens, B. ovata, Babesia sp. Xinjiang, P. falciparum, T. gondii, and B. ovis using OMA. Their amino acid sequences were aligned; then, gaps were removed and concatenated; and a phylogenetic tree was constructed (Figure 1). This genome-wide analysis revealed that B. ovis was phylogenetically most close to B. bovis.
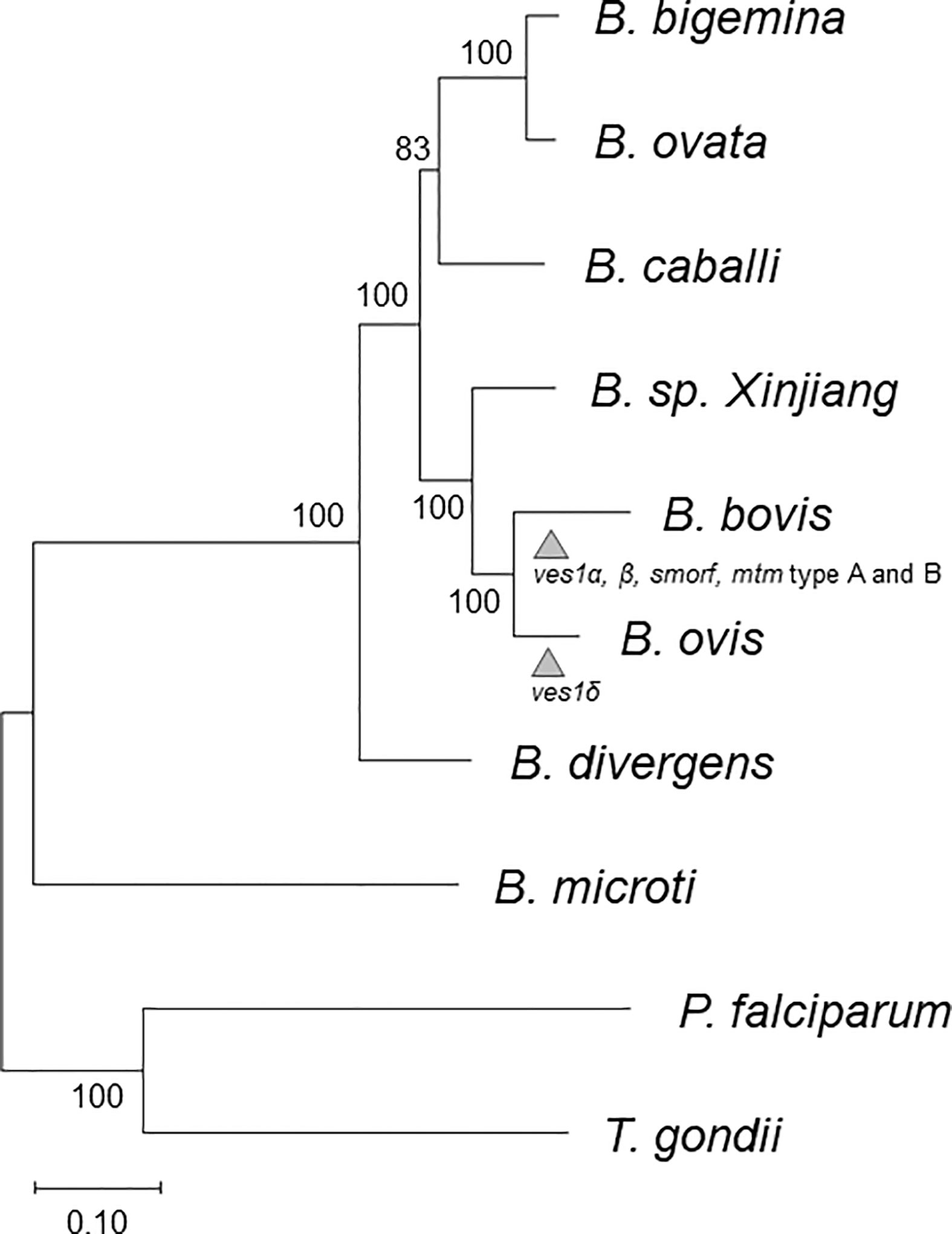
Figure 1 Phylogenetic tree based on 259 orthologs among B. caballi, B. bovis, B. bigemina, B. ovata, B. microti, B. divergens, B. sp. Xinjiang, P. falciparum, T. gondii, and B. ovis. The arrowheads represent estimated points for expansion of each gene family. The numbers at the branches represent bootstrap values.
We then examined synteny between B. ovis and the closest Babesia species, B. bovis (Figure 2). It implied that synteny blocks among them were generally conserved although better genome assembly is needed to conclude this. In addition, the contig 17 of the B. ovis genome diverges from chromosomes 2 and 3 of the B. bovis genome, indicating the presence of structural recombination. The synteny of apicoplast and mitochondrial genome among B. ovis, B. bovis, and Babesia sp. Xinjiang was examined as well. Dotplot analyses were applicable due to high sequence identity among them, and their synteny was well conserved (Figures S2, S3).
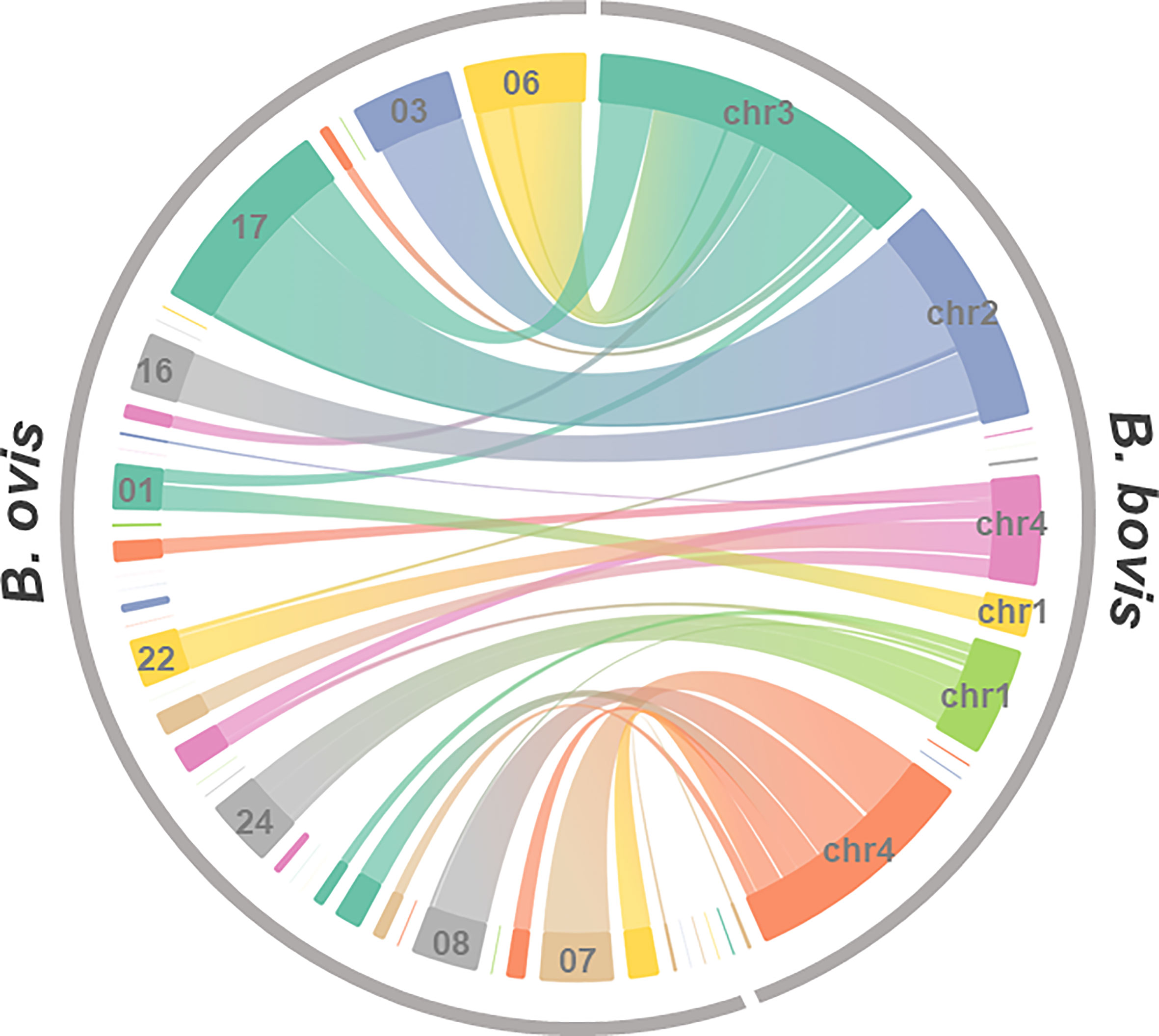
Figure 2 Genome synteny between B. ovis and B. ovata. Synteny blocks identified by amino acid sequence similarity of genes of both species were linked.
Uniquely gained and lost genes in B. ovis
We have identified 281 uniquely gained genes compared to closely related Babesia spp., B. bovis, and Babesia sp. Xinjiang. To evaluate systematic gain in function, we analyzed the 281 genes using gene ontology; however, there was no enrichment compared to the whole ontology in B. ovis. We also identified 19 uniquely gained genes in comparison to wider Babesia spp. However, there was no specific enrichment as well. We have identified 229 and 57 uniquely lost genes against a close and wide range of Babesia spp., but there was not any enrichment as well.
Multiple-gene families
Using hmm model, 43 ves1-like genes were identified in B. ovis (Table S1). Mutual homologies among the B. ovis ves1-like genes, smorf, and ves of B. bovis, B. bigemina, B. caballi, B. divergens, and Babesia sp. Xinjiang were visualized. It demonstrated that the B. ovis ves1-like genes were differentiated from ves1α and ves1β of B. bovis (Figure 3). A part of Babesia sp. Xinjiang ves and B. ovis ves1-like genes had similarity; however, they clustered separately from each other.
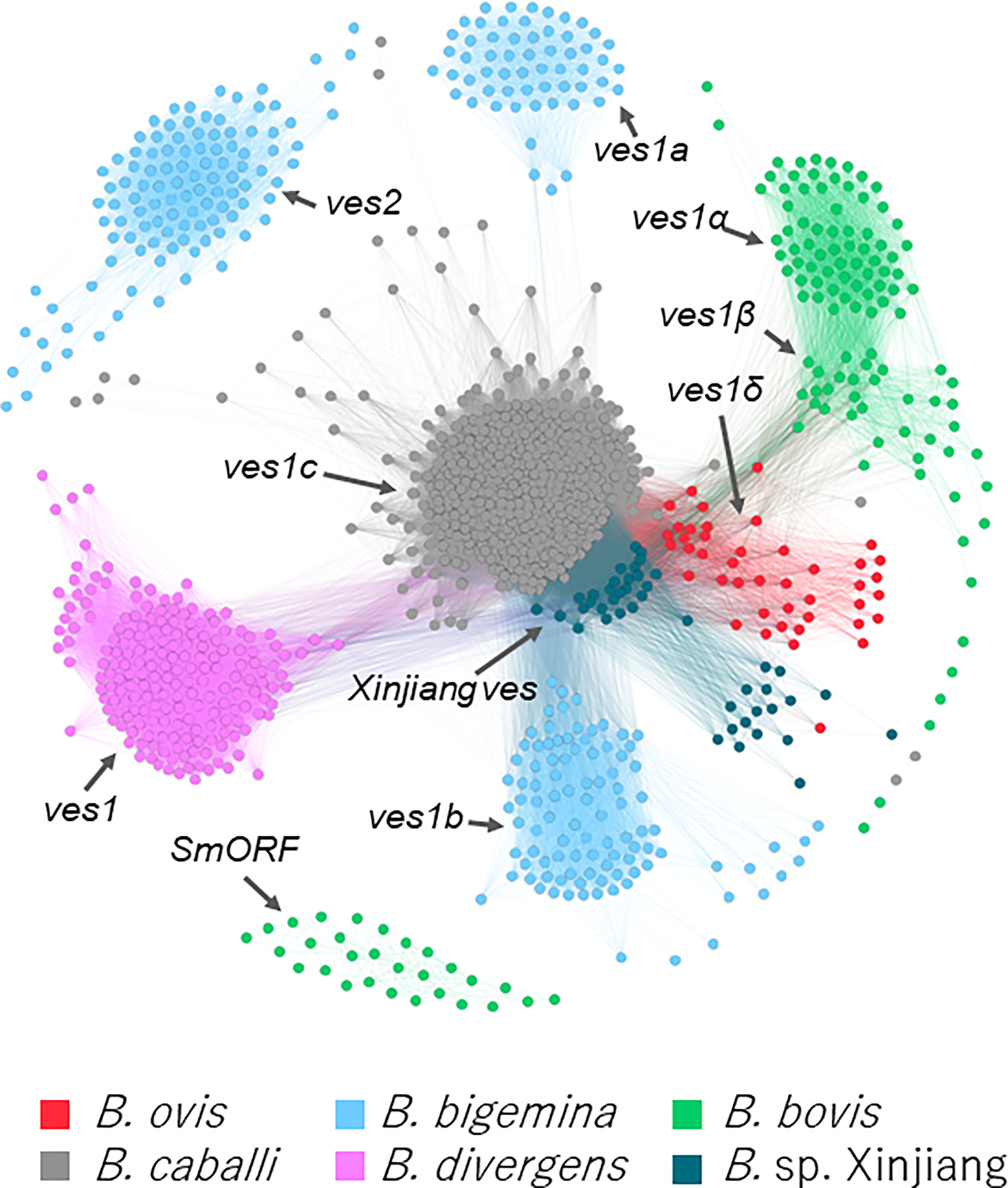
Figure 3 Clustering based on sequence of ves -like genes and smorf in B. bigemina, B. caballi, B. divergens, B. bovis, Babesia sp. Xinjiang, and B. ovis. Each node represents a protein-coding gene in the five parasites. Edges represent the similarity between connected nodes.
Genes that encode proteins with multi-transmembrane domains were examined, and 60 genes with eight or more predicted transmembrane domains were identified (Figure S4). It is known that B. bovis and B. bigemina have unique duplicated genes with the multi-transmembrane domains, mtm and msf, respectively. However, B. ovis did not have such duplicated genes.
Other multigene families in B. ovis were examined using a similarity search among B. ovis genes (Figure S5). The biggest cluster was cluster 1, consisting of 16 genes annotated as DEAD box ATP-dependent RNA helicase, followed by clusters 2 and 3 consisting of 10 and 8 genes, respectively. They were annotated as 26S protease regulatory subunit 8 and T-complex protein 1, respectively.
Regarding the expanded B. ovis ves1-like genes, those number could be under estimated because the redundancy of the assembled contigs was enough removed. On the other hand, it is also less likely that the expansion of the mtm family genes had been overlooked at all. Because the expanded mtm genes of B. bovis and B. bigemina are scattered over the contigs regardless of the size of contig, however, there is no expansion of mtm in the B. ovis genome. It is true that gene expansion and contraction can only be fully concluded by the chromosome level of the genome; however, it is possible to qualitatively assert the presence or absence of expansions.
Discussion
Hybrid scheme using long reads and short reads is the current standard for de novo genome assembly (Segerman, 2020). We obtained approximately 7.81-Mbp genome sequence consisting of 41 contigs using the approach. The BUSCO analysis revealed that it contained almost all genes in the contigs. However, the number of contigs was much larger than the known chromosome number in Babesia spp., which ranges from four or five (Ray et al., 1992; Jones et al., 1997; Depoix et al., 2002; Cornillot et al., 2013). The similar fragmented assembly is reported in B. ovata (Table 1). In contrast, the hybrid assembly in B. caballi gives almost complete assembly. One of the potential factors discriminating the results among them is with or without the sub-cloning step. Through long-term in vitro culture, quasi-species can be derived from recombination among chromosomes and accumulated, which can hamper assembly (unpublished data). Indeed, we obtained better assemblies by sub-cloning in B. bovis (data not shown). In B. ovis, the source of genome DNA was obtained from neither isolated clone nor sub-clone but from wildly infected sheep; therefore, heterogeneity in the genome DNA might be retained in the population.
The B. ovis genome sequence coded 3,419 protein-coding genes (Table 1). Previous studies using 18S rRNA genes suggest that B. ovis is the closest to B. bovis, followed by Babesia sp. Xinjiang, although bootstrap values are not definitive (Oosthuizen et al., 2008). This was consistent with our phylogenetic tree described on the basis of 259 homologs among Apicomplexan parasites and demonstrated more precisely (Figure 1). The number of genes in B. ovis was comparable to those in B. bovis and Babesia sp. Xinjiang as well. There were some gained and lost genes compared to B. bovis and Babesia sp. Xinjiang; however, there was no ontology terms that were found to be enriched in this set of genes. These lines of evidence suggest that fundamental biological flamework is conserved among them.
It is known that every Babesia sp. except B. microti has ves multigene family (Brayton et al., 2007; Cornillot et al., 2012; Jackson et al., 2014; Guan et al., 2016; Yamagishi et al., 2017). In B. ovis, we identified 43 ves1-like genes using hmm model (Figure 3, Table S1). They made a cluster that was discriminated from other species specific ves clusters; therefore, it should be appropriate to name them ves1δ. Without ves1δ of the B. ovis genome, ves1α and ves1β of B. bovis are almost separated from other clusters; however, ves1δ filled the gap, suggesting that they evolved continuously, shared the same ancestral genes, and differentiated along with the speciation (Figure 3). Similarly, smorf and mtm A type and B type of B. bovis seem to appear after the speciation between B. ovis and B. bovis. It is speculated that smorf is involved in transcriptional regulation for ves1α and ves1β (Allred, 2019). The fact that smorf, ves1α, and ves1β emerge simultaneously in B. bovis but absent in B. ovis supported the hypothesis. Other well-described multigene families in B. bovis are mtm A type and B type (Figure S4). It is known that one of the mtm is involved in drug resistance, and it is speculated that mtm family genes are involved in nutrition uptake (Hakimi et al., 2020; Hakimi et al., 2022). It is interesting to estimate the origin of smorf and mtm family genes; however, their homologs are not identified among any Babesia spp., including the most closely related B. ovis (Figures 3, S4). It is possible to speculate that they were acquired by horizontal gene transfer or that their evolution was too fast to trace back to their ancestral genes. However, more pieces of evidence are required to clarify the role of these factors. On the basis of the comparative genomics among B. ovis and related Babesia spp., the uniqueness of B. bovis was uncovered. Considering that both B. ovis and Babesia sp. Xinjiang infect the sheep Ovis, it is possible to speculate that adaptation from Ovis to Bos required differentiation of ves1α and ves1β from ancestors of ves1δ and expansion of smorf and mtm (Figure 1). Regarding the expanded B. ovis ves1δ, those number could be underestimated because the redundancy of the assembled contigs was enough removed. On the other hand, it is also less likely that the expansion of the mtm family genes had been overlooked at all. Because the expanded mtm genes of B. bovis and B. bigemina are scattered over the contigs regardless of the size of contig, however, there is no expansion of mtm in the B. ovis genome. It is true that gene expansion and contraction can only be fully concluded by the chromosome level of the genome; however, it is possible to qualitatively assert the presence or absence of expansions.
Whole genome sequencing of pathogens provides much more data than conventional single-gene sequencing, which makes significant contributions to the advances in the diagnosis and prevention of diseases (Akoniyon et al., 2022). Pathogen genome sequences provide invaluable and large-scale data for the selection of new diagnostic and drug targets and the identification of antigens to be used in vaccine development studies. Today, although genome sequence information is available for many Babesia species including B. microti, B. bigemina, B. bovis, B. divergens, B. ovata, Babesia sp. Xinjiang, and B. caballi, this information is not yet available for B. ovis. This study introduces the B. ovis whole genome sequence, which is 7.81 Mbp in size and contains 3,419 protein-coding genes. In the future, it is strongly anticipated that these data will contribute to increasing novel vaccine and drug targets in the prevention of B. ovis–induced babesiosis.
Data availability statement
The genome sequence and annotation presented in the study are deposited in the NCBI GenBank, accession number BLIY00000000 (https://www.ncbi.nlm.nih.gov/nuccore/BLIY00000000). Corresponding BioProject and Biosample ID are PRJDB7132 (https://www.ncbi.nlm.nih.gov/bioproject/PRJDB7132) and SAMD00128968 (https://www.ncbi.nlm.nih.gov/biosample/SAMD00128968), respectively. Illumina, Nanopore and PacBioreads are available from SRA under the following accession numbers, DRR489302-DRR489304 (https://ddbj.nig.ac.jp/resource/sra-run/DRR489302- https://ddbj.nig.ac.jp/resource/sra-run/DRR489304), respectively.
Ethics statement
The animal study was reviewed and approved by the Ethics Committee of Selcuk University Faculty of Veterinary Medicine. The samples were obtained as part of the diagnosis of livestock and the informed consent was obtained orally. The infected blood samples were collected in the project supported by the Scientific and Technological Research Council of Turkey (Project No. 113O336). Animal experiments performed following the rules of the Ethics Committee of Selcuk University Faculty of Veterinary Medicine (No. 2013/003).
Author contributions
JY, XX, and FS conceived and designed the study. OC and FS conducted sample collection. JY, OC, and FS performed the experiments. JY, OC, XX, and FS conducted the literature search, performed data extraction and analysis, and interpreted the results. JY drafted and wrote the manuscript. OC, XX, and FS critically reviewed the manuscript for important intellectual content and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by a Grant-in-Aid for Scientific Research (18KK0188) and the JSPS Core-to-Core program, both from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, as well as a grant from the Strategic International Collaborative Research Project (JPJ008837) promoted by the Ministry of Agriculture, Forestry, and Fisheries of Japan.
Acknowledgments
We thank Ms. Naoko Kawai for conducting sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1194608/full#supplementary-material
Supplementary Figure 1 | Profile of the 41 assembled contigs of B. ovis by BlobTools. It was confirmed that no contigs were found to have low sequence depth or deviate from the average GC% except apicoplast and mitochondrial genome.
Supplementary Figure 2 | Dotplot among apicoplast genomes of B. ovis. B. bovis and Babesia. sp. Xinjiang.
Supplementary Figure 3 | Dotplot among mitochondrial genomes of B. ovis. B. bovis and Babesia. sp. Xinjiang.
Supplementary Figure 4 | Clustering based on sequence of genes with more than eight multi-transmembrane domains in B. bigemina, B. caballi, B. divergens, B. bovis, Babesia sp. Xinjiang, and B. ovis. Each node and edge represent a protein-coding gene and similarity between connected nodes, respectively.
Supplementary Figure 5 | Clustering based on sequence in B. ovis genes. Each node and edge represent a protein-coding gene and similarity between connected nodes, respectively. The clusters were colored if they consisted of five or more genes.
References
Ahmed, J. S., Luo, J., Schnittger, L., Seitzer, U., Jongejan, F., Yin, H. (2006). Phylogenetic position of small-ruminant infecting piroplasms. Ann. New York Acad. Sci. 1081, 498–504. doi: 10.1196/ANNALS.1373.074
Akoniyon, O. P., Adewumi, T. S., Maharaj, L., Oyegoke, O. O., Roux, A., Adeleke, M. A., et al. (2022). Whole genome sequencing contributions and challenges in disease reduction focused on malaria. Biology 11 (4), 587. doi: 10.3390/BIOLOGY11040587
Allred, D. R. (2019). Variable and variant protein multigene families in Babesia bovis persistence. Pathog. (Basel Switzerland) 8 (2), 76. doi: 10.3390/pathogens8020076
Allred, D. R., Hines, S. A., Ahrens, K. P. (1993). Isolate-specific parasite antigens of the Babesia bovis-infected erythrocyte surface. Mol. Biochem. Parasitol. 60 (1), 121–132. doi: 10.1016/0166-6851(93)90035-V
Altenhoff, A. M., Škunca, N., Glover, N., Train, C.-M., Sueki, A., Piližota, I., et al. (2015). The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res. 43 (Database issue), D240–D249. doi: 10.1093/nar/gku1158
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi: 10.1016/S0022-2836(05)80360-2
Archibald, A. L., Cockett, N. E., Dalrymple, B. P., Faraut, T., Kijas, J. W., Maddox, J. F., et al. (2010). The sheep genome reference sequence: A work in progress. Anim. Genet. 41 (5), 449–453. doi: 10.1111/j.1365-2052.2010.02100.x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Bastian, M., Heymann, S., Jacomy, M. (2009) Gephi: An Open Source Software for Exploring and Manipulating Networks. Available at: http://www.aaai.org/ocs/index.php/ICWSM/09/paper/view/154.
Brayton, K. a, Lau, A. O. T., Herndon, D. R., Hannick, L., Kappmeyer, L. S., Berens, S. J., et al. (2007). Genome sequence of Babesia bovis and comparative analysis of apicomplexan hemoprotozoa. PloS Pathog. 3 (10), 1401–1413. doi: 10.1371/journal.ppat.0030148
Buchfink, B., Reuter, K., Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18 (4), 366–368. doi: 10.1038/s41592-021-01101-x
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinf. (Oxford England) 21 (18), 3674–3676. doi: 10.1093/bioinformatics/bti610
Cornillot, E., Dassouli, A., Garg, A., Pachikara, N., Randazzo, S., Depoix, D., et al. (2013). Whole genome mapping and re-organization of the nuclear and mitochondrial genomes of Babesia microti isolates. PloS One 8 (9), e72657. doi: 10.1371/journal.pone.0072657
Cornillot, E., Hadj-Kaddour, K., Dassouli, A., Noel, B., Ranwez, V., Vacherie, B., et al. (2012). Sequencing of the smallest Apicomplexan genome from the human pathogen Babesia microti. Nucleic Acids Res. 40 (18), 9102–9114. doi: 10.1093/nar/gks700
Depoix, D., Carcy, B., Jumas-Bilak, E., Pages, M., Precigout, E., Schetters, T. P. M., et al. (2002). Chromosome number, genome size and polymorphism of European and South African isolates of large Babesia parasites that infect dogs. Parasitology 125 (4), 313–321. doi: 10.1017/S0031182002002202
Fakhar, M., Hajihasani, A., Maroufi, S., Alizadeh, H., Shirzad, H., Piri, F., et al. (2012). An epidemiological survey on bovine and ovine babesiosis in Kurdistan Province, western Iran. Trop. Anim. Health Production 44 (2), 319–322. doi: 10.1007/S11250-011-0023-Y
Fanelli, A. (2021). A historical review of Babesia spp. associated with deer in Europe: Babesia divergens/Babesia divergens-like, Babesia capreoli, Babesia venatorum, Babesia cf. odocoilei. Veterinary Parasitol. 294, 109433. doi: 10.1016/j.vetpar.2021.109433
Ferreri, L. M., Brayton, K. A., Sondgeroth, K. S., Lau, A. O. T., Suarez, C. E., McElwain, T. F. (2012). Expression and strain variation of the novel “small open reading frame” (smorf) multigene family in Babesia bovis. Int. J. Parasitol. 42 (2), 131–138. doi: 10.1016/J.IJPARA.2011.10.004
Galon, E. M., Ybañez, R. H., Macalanda, A. M., Estabillo, G. R., Montano, M. T. R., Veedor, M. D., et al. (2022a). First molecular identification of babesia, theileria, and anaplasma in goats from the Philippines. Pathogens 11 (10), 1109. doi: 10.3390/pathogens11101109
Galon, E. M., Zafar, I., Ji, S., Li, H., Ma, Z., Xuan, X. (2022b). Molecular reports of ruminant babesia in Southeast Asia. Pathogens 11 (8), 915. doi: 10.3390/pathogens11080915
Green, M. R., Sambrook, J. (2012). Molecular Cloning: A Laboratory Manual, 4th ed. (New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbor).
Guan, G., Korhonen, P. K., Young, N. D., Koehler, A. V., Wang, T., Li, Y., et al. (2016). Genomic resources for a unique, low-virulence Babesia taxon from China. Parasites Vectors 9 (1), 1–8. doi: 10.1186/s13071-016-1846-1
Habela, M., Reina, D., Nieto, C., Navarrete, I. (1990). Antibody response and duration of latent infection in sheep following experimental infection with Babesia ovis. Veterinary Parasitol. 35 (1–2), 1–10. doi: 10.1016/0304-4017(90)90111-N
Hakimi, H., Templeton, T. J., Sakaguchi, M., Yamagishi, J., Miyazaki, S., Yahata, K., et al. (2020). Novel Babesia bovis exported proteins that modify properties of infected red blood cells. PloS Pathog. 16 (10), e1008917. doi: 10.1371/journal.ppat.1008917
Hakimi, H., Yamagishi, J., Kawazu, S. I., Asada, M. (2022). Advances in understanding red blood cell modifications by Babesia. PloS Pathog. 18 (9), e1010770. doi: 10.1371/journal.ppat.1010770
Hoff, K. J., Stanke, M. (2013). WebAUGUSTUS–a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41 (Web Server issue), W123–W128. doi: 10.1093/nar/gkt418
Jackson, A. P., Otto, T. D., Darby, A., Ramaprasad, A., Xia, D., Echaide, I. E., et al. (2014). The evolutionary dynamics of variant antigen genes in Babesia reveal a history of genomic innovation underlying host-parasite interaction. Nucleic Acids Res. 42 (11), 7113–7131. doi: 10.1093/nar/gku322
Jones, S. H., Lew, A. E., Jorgensen, W. K., Barker, S. C. (1997). Babesia bovis: Genome size, number of chromosomes and telomeric probe hybridisation. Int. J. Parasitol. 27 (12), 1569–1573. doi: 10.1016/S0020-7519(97)00099-4
Katoh, K., Misawa, K., Kuma, K., Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30 (14), 3059–3066. doi: 10.1093/nar/gkf436
Krogh, A., Larsson, B., von Heijne, G., Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305 (3), 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi: 10.1093/molbev/msy096
Laetsch, D. R., Blaxter, M. L. (2017). BlobTools: Interrogation of genome assemblies. F1000Research 6, 1287. doi: 10.12688/f1000research.12232.1
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H.-H., Rognes, T., Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35 (9), 3100–3108. doi: 10.1093/nar/gkm160
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinf. (Oxford England) 26 (5), 589–595. doi: 10.1093/BIOINFORMATICS/BTP698
Loman, N. J., Quinlan, A. R. (2014). Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics 30 (23), 3399–3401. doi: 10.1093/bioinformatics/btu555
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 (5), 955–964. doi: 10.1093/nar/25.5.955
Ma, C., Gunther, S., Cooke, B., Coppel, R. L. (2013). Geneious plugins for the access of PlasmoDB and PiroplasmaDB databases. Parasitol. Int. 62 (2), 134–136. doi: 10.1016/j.parint.2012.11.003
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., Zdobnov, E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38 (10), 4647–4654. doi: 10.1093/MOLBEV/MSAB199
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41 (12), e121. doi: 10.1093/NAR/GKT263
Noe, L., Kucherov, G. (2005). YASS: enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 33 (Web Server issue), W540–W543. doi: 10.1093/nar/gki478
O’Connor, R. M., Allred, D. R. (2000). Selection of Babesia bovis-infected erythrocytes for adhesion to endothelial cells coselects for altered variant erythrocyte surface antigen isoforms. J. Immunol. (Baltimore Md.: 1950) 164 (4), 2037–2045. doi: 10.4049/jimmunol.164.4.2037
Oosthuizen, M. C., Zweygarth, E., Collins, N. E., Troskie, M., Penzhorn, B. L. (2008). Identification of a novel Babesia sp. from a sable antelope (Hippotragus niger Harris 1838). J. Clin. Microbiol. 46 (7), 2247–2251. doi: 10.1128/JCM.00167-08
Pedroni, M. J., Sondgeroth, K. S., Gallego-Lopez, G. M., Echaide, I., Lau, A. O. T. (2013). Comparative transcriptome analysis of geographically distinct virulent and attenuated Babesia bovis strains reveals similar gene expression changes through attenuation. BMC Genomics 14, 763. doi: 10.1186/1471-2164-14-763
Pryszcz, L. P., Gabaldón, T. (2016). Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44 (12), e113. doi: 10.1093/NAR/GKW294
Ranjbar-Bahadori, S., Eckert, B., Omidian, Z., Shirazi, N. S., Shayan, P. (2012). Babesia ovis as the main causative agent of sheep babesiosis in Iran. Parasitol. Res. 110 (4), 1531–1536. doi: 10.1007/S00436-011-2658-Z
Ray, B. K., Bailey, C. W., Jensen, J. B., Andrew Carson, C. (1992). Chromosomes of Babesia bovis and Babesia bigemina. Mol. Biochem. Parasitol. 52 (1), 123–126. doi: 10.1016/0166-6851(92)90041-H
Segerman, B. (2020). The most frequently used sequencing technologies and assembly methods in different time segments of the bacterial surveillance and RefSeq genome databases. Front. Cell. Infection Microbiol. 10, 527102. doi: 10.3389/FCIMB.2020.527102
Sevinc, F., Cao, S., Xuan, X., Sevinc, M., Ceylan, O. (2015a). Identification and expression of Babesia ovis secreted antigen 1 and evaluation of its diagnostic potential in an enzyme-linked immunosorbent assay. J. Clin. Microbiol. 53 (5), 1531–1536. doi: 10.1128/JCM.03219-14
Sevinc, F., Cao, S., Zhou, M., Sevinc, M., Ceylan, O., Xuan, X. (2015b). A new immunoreactive recombinant protein designated as rBoSA2 from Babesia ovis: Its molecular characterization, subcellular localization and antibody recognition by infected sheep. Veterinary Parasitol. 214 (1–2), 213–218. doi: 10.1016/J.VETPAR.2015.09.022
Sevinc, F., Sevinc, M., Ekici, O. D., Yildiz, R., Isik, N., Aydogdu, U. (2013). Babesia ovis infections: detailed clinical and laboratory observations in the pre- and post-treatment periods of 97 field cases. Veterinary Parasitol. 191 (1–2), 35–43. doi: 10.1016/J.VETPAR.2012.07.025
Sevinc, F., Sevinc, M., Koc, Y., Alkan, F., Ekici, O. D., Yildiz, R., et al. (2014). The effect of 12 successive blood passages on the virulence of Babesia ovis in splenectomized lambs: A preliminary study. Small Ruminant Res. 116 (1), 66–70. doi: 10.1016/j.smallrumres.2013.10.010
Stanke, M., Diekhans, M., Baertsch, R., Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinf. (Oxford England) 24 (5), 637–644. doi: 10.1093/bioinformatics/btn013
Tian, T., Liu, Y., Yan, H., You, Q., Yi, X., Du, Z., et al. (2017). agriGO v2.0: a GO analysis toolkit for the agricultural community 2017 update. Nucleic Acids Res. 45 (W1), W122–W129. doi: 10.1093/NAR/GKX382
Trapnell, C., Pachter, L., Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinf. (Oxford England) 25 (9), 1105–1111. doi: 10.1093/bioinformatics/btp120
Tumwebaze, M. A., Byamukama, B., Tayebwa, D. S., Byaruhanga, J., Angwe, M. K., Galon, E. M., et al. (2020). First molecular detection of babesia ovis, theileria spp., anaplasma spp., and ehrlichia ruminantium in goats from Western Uganda. Pathogens 9 (11), 1–16. doi: 10.3390/pathogens9110895
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49–e49. doi: 10.1093/NAR/GKR1293
Yamagishi, J., Asada, M., Hakimi, H., Tanaka, T. Q., Sugimoto, C., Kawazu, S. (2017). Whole-genome assembly of Babesia ovata and comparative genomics between closely related pathogens. BMC Genomics 18 (1), 832. doi: 10.1186/s12864-017-4230-4
Keywords: ovine babesiosis, Babesia ovis, comparative genomics, multigene families, apicomplexa
Citation: Yamagishi J, Ceylan O, Xuan X and Sevinc F (2023) Whole genome sequence and diversity in multigene families of Babesia ovis. Front. Cell. Infect. Microbiol. 13:1194608. doi: 10.3389/fcimb.2023.1194608
Received: 27 March 2023; Accepted: 04 July 2023;
Published: 01 August 2023.
Edited by:
Haiyan Gong, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Amit Sinha, New England Biolabs, United StatesMingming Liu, Hubei University of Arts and Science, China
Copyright © 2023 Yamagishi, Ceylan, Xuan and Sevinc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junya Yamagishi, anVueWFAY3pjLmhva3VkYWkuYWMuanA=; Ferda Sevinc, ZnNldmluY0BzZWxjdWsuZWR1LnRy