 Alessandro de Sa Pinheiro1,2
Alessandro de Sa Pinheiro1,2 James W. Kazura
James W. Kazura Alvin H. Schmaier
Alvin H. Schmaier
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 10 August 2023
Sec. Parasite and Host
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1184896
This article is part of the Research Topic Kallikrein-Kinin System: Insights into a Multifunctional System View all 5 articles
Malaria is a parasitic disease of global health significance and a leading cause of death in children living in endemic regions. Although various Plasmodium species are responsible for the disease, Plasmodium falciparum infection accounts for most severe cases of the disease in humans. The mechanisms of cerebral malaria pathogenesis have been studied extensively in humans and animal malaria models; however, it is far from being fully understood. Recent discoveries indicate a potential role of bradykinin and the kallikrein kinin system in the pathogenesis of cerebral malaria. The aim of this review is to highlight how bradykinin is formed in cerebral malaria and how it may impact cerebral blood-brain barrier function. Areas of interest in this context include Plasmodium parasite enzymes that directly generate bradykinin from plasma protein precursors, cytoadhesion of P. falciparum infected red blood cells to brain endothelial cells, and endothelial cell blood-brain barrier disruption.
Malaria continues to be a major cause of morbidity and mortality in tropical and sub-tropical areas of the world. Life-threatening clinical malaria syndromes – severe anemia, respiratory distress accompanied by metabolic acidosis, cerebral malaria (CM) and combinations therein – primarily affect children and pregnant women with Plasmodium falciparum (Pf) infection. In 2021 there was an estimated 619,000 malaria deaths world-wide; 593,000 of these deaths occurred in sub-Saharan Africa (WHO, 2022). Children younger than 3-5 years with little prior exposure to Pf blood stage infection and minimal naturally acquired immunity to clinical malaria are the major age group at risk of death from CM. Most deaths from Plasmodium falciparum (Pf) infection are due to cerebral malaria.
Cerebral malaria is a neurological complication caused by Pf blood stage infection. The World Health Organization has defined 3 clinical criteria for the diagnosis of CM: i) a peripheral blood smear or rapid diagnostic test positive for asexual stage Pf parasites; ii) exclusion of any other etiology of neurologic dysfunction; and iii) coma and seizures (W. H. O, 2012). Magnetic resonance imaging studies of children and adults with CM indicate that vasogenic edema resulting from breakdown of the blood-brain barrier (BBB) underlies progressive brain swelling that can lead to fatal brainstem herniation (Seydel et al., 2015; Mohanty et al., 2017). The exact pathophysiological mechanisms underlying cerebral edema with hemorrhage are not known. Systemic inflammation, activation of coagulation and the cytokine cascade, neutrophil degranulation, and cytoadherence of Pf infected red blood cells (iRBCs) to brain vascular endothelium are collectively thought to initiate the breakdown of the BBB (Moxon et al., 2009; Moxon et al., 2020; Patel et al., 2020; Guenther et al., 2021). Fundoscopic examination of the eye has revealed that distinctive pathologic features of retinal blood vessels, e.g., macular and peripheral whitening due to ischemia associated with iRBC cytoadhesion, are associated with high risk of fatal CM relative to CM cases without these retinal changes (Beare, 2023; Wilson et al., 2023). Seminal studies conducted in Africa and Southeast Asia indicate that prompt treatment with parenteral artesunate reduces the death rate of CM relative to quinine, the previous standard of care (Dondorp et al., 2010). Nevertheless, 10 percent of children with severe malaria of any clinical phenotype die despite appropriate treatment (Gething et al., 2016; Maitland, 2016).
Children who recover from CM and other severe malaria syndromes may experience long-term neurocognitive and motor deficits (John et al., 2008; Idro et al., 2016). Persistent elevation of plasma biomarkers of inflammation, such as C-reactive protein, following clinical recovery have been associated with these deficits (Conroy et al., 2021). However, there is minimal understanding of the etiology and prevention of long-term neurologic sequelae in children who survive severe malaria.
The proximate molecular pathways underlying progressive brain edema in CM are not known. Pathologic examination of brains of children with fatal CM shows extensive intravascular sequestration of iRBCs with vascular thrombosis, perivascular hemorrhage, and infiltration of adjacent brain parenchyma with monocyte/macrophages, hemozoin (a parasite byproduct of hemoglobin degradation), and CD8+ T cells (Jensen et al., 2020; Moxon et al., 2020; Riggle et al., 2020). Immunostaining studies of brain slices indicate that platelet and neutrophil activation have occurred in vivo (Amulic et al., 2020). Relative to children with non-life threatening uncomplicated malaria, iRBCs isolated from the blood of children with CM express variants of Pf erythrocyte protein-1 (PfEMP1) on the iRBC surface that bind to intercellular adhesion molecule 1 (ICAM-1) and endothelial protein C receptor (EPCR) on the brain endothelial cell (EC) luminal surface (Kessler et al., 2017; Sahu et al., 2021; Ramachandran and Sharma, 2022). Sequestration of iRBCs in cerebral microvessels produces mechanical obstruction to blood flow and metabolic disturbances in the EC and adjacent brain tissue. Infected RBC binding triggers EC signaling pathways that lead to reorganization of endothelium tight junction complexes and leakiness of the BBB (van der Heyde et al., 2006; Wassmer and Grau, 2017). Disruption of BBB and plasma leakage are hallmark features in CM pathology, as demonstrated in murine experimental cerebral malaria (ECM) by measuring extravasation of impermeable dyes into the brains of infected mice where local hemorrhage is observed (Renia et al., 2012).
In this review, we ask the question, what is the evidence for bradykinin, a biologic peptide with vasogenic properties, as an etiologic agent in the pathogenesis of cerebral malaria? This review is a summary of human, animal, and in vitro data addressing this question. Our focus is the generation of bradykinin in CM and its possible vascular effects on infected patients.
Bradykinin (BK) is a 9 amino acid peptide (RPPGFSPFR) liberated by cleavage of high or low molecular weight kininogen (HK and LK) by plasma kallikrein (PKa), tissue kallikrein, and/or activated factor XII (FXIIa). The in vivo half-life of BK is less than 1 minute as various plasma proteases rapidly degrade the peptide (Cicardi and Zuraw, 2018; Pinheiro et al., 2022). Related peptides with similar biological effects on ECs, i.e., the capacity to increase vascular permeability and local angioedema, include Lys BK, a decapeptide with an amino acid sequence identical to BK with lysine at position 10 formed by tissue kallikreins and desArg9BK, an octapeptide with an amino acid sequence identical to BK with Arg lacking at position 9 formed by several BK carboxypeptidases (Pinheiro et al., 2022). Lys BK is liberated from LK by tissue kallikrein. In general, tissue kallikreins do not participate in cleaving HK within the intravascular compartment. PKa and FXIIa of the plasma contact activation system are present as enzymatically inactive zymogens [denoted respectively as prekallikrein (PK) and factor XII (FXII)]. Negatively charged biological molecules in plasma, such as polyphosphates, cell lysates, collagen, lipids, aggregated proteins and artificial products, support FXII auto-activation to FXIIa (Colman and Schmaier, 1997) (Figure 1, on the right). Small amounts of FXIIa catalytically hydrolyze plasma PK to PKa which, in turn, produces more FXIIa in reciprocal activation leading to amplification of activation. Both PKa and FXIIa cleave HK or LK between a Lys-Arg at the N-terminus of BK and Arg-Ser at the C-terminus to liberate the intact peptide (Schmaier, 2016). HK also binds to EC, platelets, and neutrophils to serve as a receptor for PK (Gustafson et al., 1986; Schmaier et al., 1988).
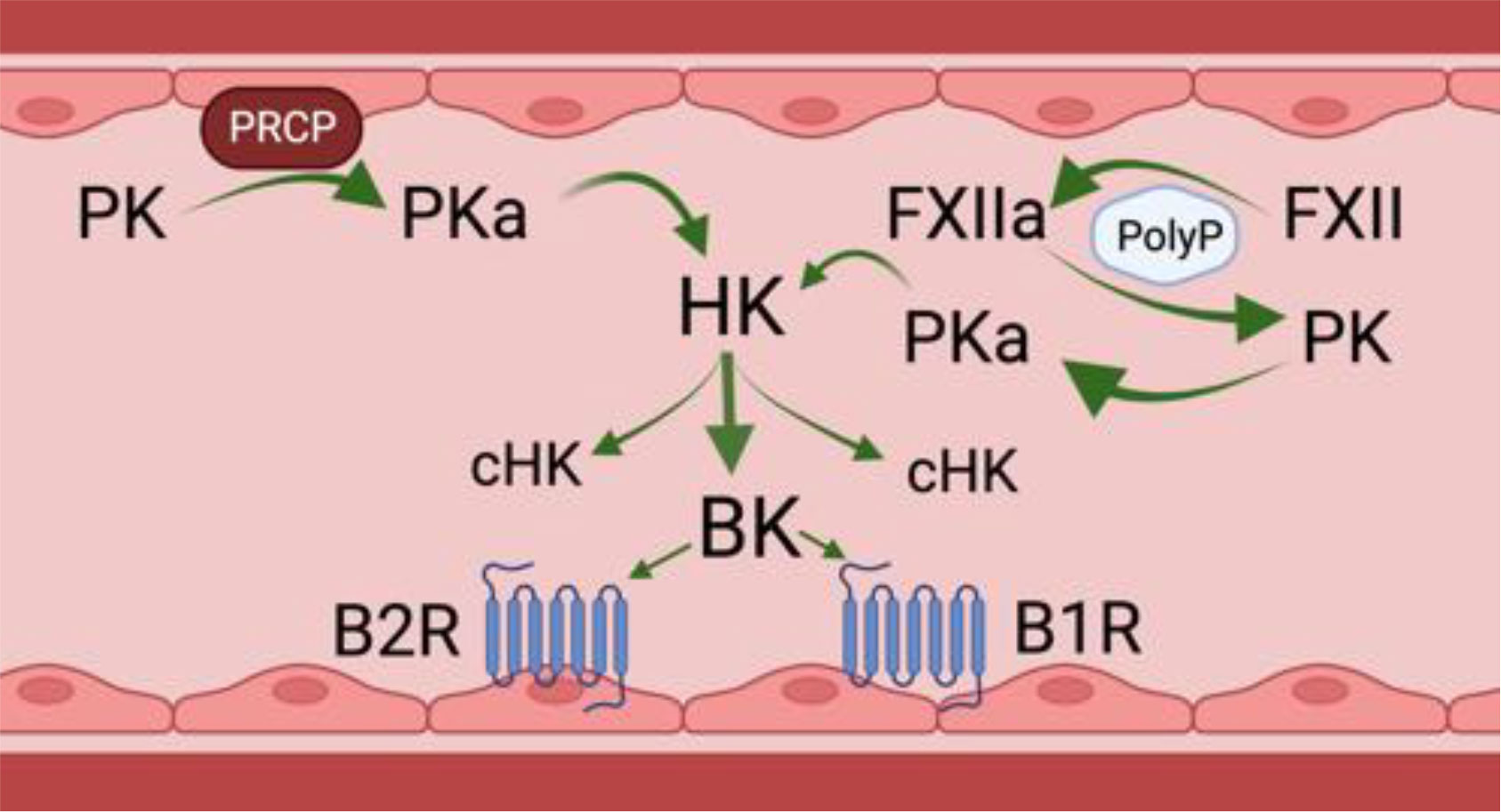
Figure 1 Physiologic bradykinin forming pathways in the intravascular compartment. PK, prekallikrein; PKa, plasma kallikrein; PRCP, prolylcarboxypeptidase; FXII, factor XII; FXIIa, activated factor XII; HK, high molecular weight kininogen; cHK, cleaved HK or bradykinin-free HK; BK, bradykinin; B2R, bradykinin B2; B1R, bradykinin B1 receptor. There are two physiologic mechanisms for BK formation in the intravascular compartment. The first is on the left of the figure. It shows that on a negatively activated surface, e.g., PolyP, factor XII autoactivates into an enzyme FXIIa. FXIIa then activates zymogen PK to plasma PKa). A second mechanism to form PKa is on the right. PK bound to endothelial cells by HK is proteolyzed by a membrane expressed PRCP. PKa formed by any mechanism cleaves the HK it is bound to, liberating BK and leaving a residual bradykinin-free HK or cHK. Formed BK binds to its endothelial cell constitutive receptor, the B2R to stimulate endothelial cell activation. In inflammatory states, a second receptor, termed the B1R also is expressed to mediate BK’s effect on endothelial cell biology. Figure made with Biorender.
In addition to contact activation, there is another pathway that leads to PK activation on the vessel wall, Endothelial cell prolylcarboxypeptidase (PRCP) is a serine protease that also hydrolyzes PK to PKa when bound to HK on the cells to liberate BK (Pinheiro et al., 2022) (Figure 1, on the left). This pathway is independent of FXII. On ECs the degree of PK activation is regulated inversely by the ambient concentration of C1 inhibitor (C1INH) and directly by PRCP (Kaplan and Joseph, 2017; Merkulova et al., 2023). FXII binds ECs, but in the absence of PK and HK, it does not autoactivate to FXIIa. Even in the presence of high concentrations of C1INH, the degree of inhibition of cleavage of HK and BK liberation by forming plasma kallikrein is only 50% (Kaplan and Joseph, 2017; Merkulova et al., 2023). This statement implies that some BK is always being formed on EC regardless of the C1INH concentration.
Angiotensin converting enzyme (ACE) is the major BK degrading enzyme in the intravascular compartment. In total, there are 13 plasma and tissue BK peptidases (Pinheiro et al., 2022). Carboxypeptidases degrade BK and Lys-BK to DesArg9BK and LysDesArg9BK, respectively, and are ligands for the BK G-protein coupled B2 receptor (B2R) and B1 receptor (B1R), respectively (Figure 1). The B2R is the constitutive receptor in the intravascular compartment. The B1R is expressed in inflammatory states or as a response to injury. BK binding to B2R on the EC surface stimulates the release of nitric oxide and prostacyclin, followed by loss of BBB function, increased vascular permeability, and vasodilation due to decreased smooth muscle cell tone. It is not known if the BK signaling pathway for loss of BBB function is the same for vasodilation.
The archetype contact activation system disease state is hereditary angioedema, an inflammatory disorder manifested by localized edema in skin, soft tissue, bowel, and the larynx. This condition has been associated with defects or deficiency of C1 inhibitor, FXII, plasminogen, angiopoietin 1, HK, heparan sulfate, and myoferlin (Schmaier et al., 1986; Kaplan and Joseph, 2017; Schmaier, 2019; Kaplan et al., 2022). Since BK formation leads to tissue edema with associated microvessel thrombosis, we hypothesize that proteins of the plasma contact activation system and BK participate in CM pathogenesis by mediating breakdown of the BBB. In addition, we propose that BK itself may impact malaria parasite growth and transmission to mosquito vectors. The following is a presentation of what is known about the participation of FXII and the KKS in BK formation in malaria. We will describe published evidence suggesting that: 1) BK is generated during malaria infection; 2) BK is a potential causal agent of vascular leakage in humans and experimental animals with severe malaria, including CM; 3) A cysteine protease expressed by Pf parasites directly generates BK from HK; 4) BK has direct anti-plasmodium activity. A schematic of potential pathways by which BK may contribute to CM pathogenesis is shown in Figure 2.
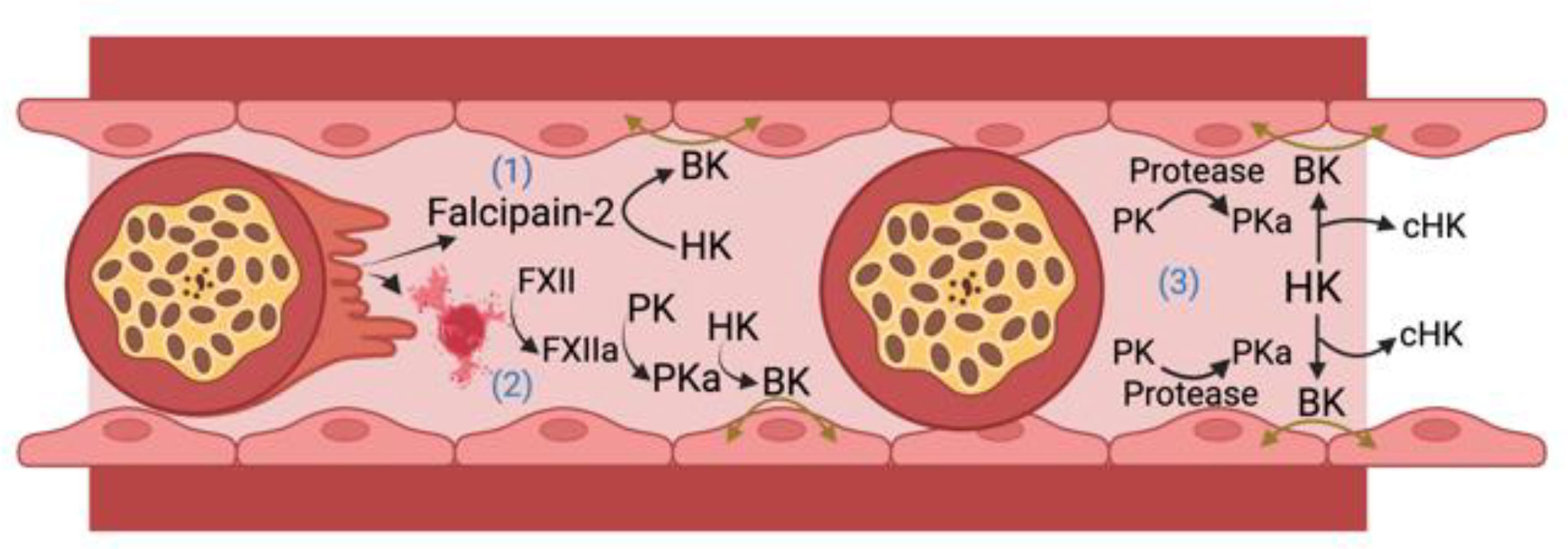
Figure 2 Bradykinin (BK) formation in cerebral malaria. HK, high molecular weight kininogen; PK, plasma prekallikrein; PKa, plasma kallikrein; FXII, factor XII; FXIIa, activated FXII; cHK, cleaved (bradykinin free) high molecular weight kininogen. There are 3 ways iRBCs may lead to BK formation. 1) Falcipain-2 itself released from Plasmodium falciparum cleaves HK to liberate BK; 2) The ruptured membranes or microparticles/exosomes from P. falciparum infected RBCs or parasites themselves support FXII autoactivation leading to PKa formation that liberates BK from HK; 3) Occluding P. falciparum infected red blood cells upregulate an endothelial cell PK activator (Protease) that leads to more PKa activation with cleavage of HK liberating BK to produce edema in the vessel wall. Figure made with Biorender.
Observational studies of human malaria conducted in the first half of 20th century suggested that increased vascular permeability and capillary leakage contribute to the pathogenesis of severe malaria. Estimated changes in intravascular versus extravascular plasma distribution in patients undergoing malaria-induced “fever therapy” for the treatment of neurosyphilis were among the first to suggest that Pf blood stage infection leads to vascular leakiness (O'Leary, 1928; Wagner-Jauregg and Bruetsch, 1946; Verhave, 2013; Kraft et al., 2019). Notably, this form of therapy for neurosyphilis, used in the pre-antibiotic era before the discovery and widespread availability of penicillin, did not undergo rigorous randomized clinical trials. From today’s vantage point, these historical therapies appear neither safe nor ethical.
In a report describing 9 malaria naïve neurosyphilis patients inoculated intravenously with Pf or P. vivax iRBCs, one of the four patients infected with Pf developed hypotension and decreased consciousness concomitant with high parasitemia and decreased intravascular plasma volume measured by dilution of Evans blue dye relative to the baseline value before iRBC infection (Feldman and Murphy, 1945). Recovery ensued two to three days later following transfusion of whole blood and plasma. The authors suggested that vascular leakage was a component of the acute decrease in plasma volume. In contrast, studies of American soldiers with uncomplicated non-life threatening malaria had no evidence of plasma vascular leakage measured by radio-iodinated serum albumin dilution (Malloy et al., 1967). More recently, Davis et al. (1992) measured vascular endothelium permeability by trans-capillary escape of radiolabeled albumin and the urinary albumin/creatinine ratio in adult Thai patients with severe malaria and patients with uncomplicated malaria. The former two measures of capillary leakage were higher in patients with severe malaria than patients with uncomplicated malaria. Severe malaria patients also had significantly greater trans-capillary escape of radio-labeled albumin than those with uncomplicated malaria. Another study of 22 Thai adults with severe malaria, including 6 with CM, reported that FXII and PK levels were significantly decreased at presentation, consistent with prior activation of the intrinsic pathway of coagulation (Clemens et al., 1994). The C1 inhibitor activity/antigen ratio, a biomarker for prior contact activation, was also decreased.
With respect to primate malaria, a series of studies conducted in the mid to late 1960s showed that BK-like activity measured by increased skin vascular permeability in guinea pigs was increased, and the BK precursor protein, bradykininogen (now referred to as HK), decreased in plasma of rhesus monkeys (Macaca mulatta) infected with P. knowlesi relative to uninfected controls (Tella and Maegraith, 1966; Maegraith and Tella, 1968; Maegraith and Onabanjo, 1969a; Maegraith and Onabanjo, 1969b; Maegraith and Onabanjo, 1970). In 1969, Colman et al. (Colman et al., 1969) and Maegraith and Onabanjo (1969a) showed that the kininogenase (now referred to as plasma PK) increased permeability in skin capillaries of guinea pigs. Onabanjo and Maegraith subsequently observed that brain capillaries of P. knowlesi infected monkeys occluded by iRBCs had elevated plasma PK, increased EC permeability, and vasodilation (Onabanjo and Maegraith, 1970a). Injection of plasma PKa fractions containing pontamine blue dye into the brain increased EC permeability along with lymphocyte and eosinophil infiltration and RBC diapedesis (Onabanjo and Maegraith, 1970b). Increased plasma PKa also was associated with a fall in plasma bradykininogen (HK) in infected animals (Onabanjo and Maegraith, 1970c). Finally, severe hypotension in rhesus monkeys infected with P. knowlesi was found to be accompanied by PK activation, cleavage of HK, and high BK levels (Skirrow et al., 1964).
The above observations in primate malaria are similar to those reported in studies of mice with experimental CM following infection with Plasmodium berghei (NK65 strain) (Ohtomo and Katori, 1972). Plasma HK levels were markedly reduced and kinin levels increased concomitant with progressive severity of neurologic dysfunction. Further, treatment of P. berghei ANKA infected mice with captopril, an ACE inhibitor, increased BK levels and directly modulated CD8+ T cell responses (Silva-Filho et al., 2013; Silva-Filho et al., 2017). These combined studies indicate that malaria infection in rodents and primates was associated with plasma proteins that contribute to the generation of BK.
Plasmodium falciparum iRBCs internalize plasma HK and actively process it through cysteine proteases to liberate BK and its analogs to the extracellular space (Bagnaresi et al., 2012). This process may contribute to activation of the kallikrein-kinin system in ECs. Based on this precedent, investigations examined whether kinins generated in the conditioned medium of Plasmodium falciparum could modulate iRBC cytoadhesion as well as barrier function (Silva et al., 2019). The conditioned media of Pf cultures stimulated B2R and B1R to increase iRBC cytoadhesion to brain microvascular endothelial cells. These activities were blocked by antagonists to B2R (HOE-140) and B1R (DALBK). The same results were observed when cells were treated with BK alone. Bradykinin or conditioned medium of Pf cultures stimulation of brain EC monolayers impaired tight junctions with loss of ZO-1 and β-catenin expression on cell surface, resulting in changes in EC function that allowed for increased permeability of bovine serum albumin tracer (Silva et al., 2019).
Kininogens such as HK have been known to be substrates and inhibitors of cysteine proteases for over 40 years. We showed that HK inhibits platelet calpain 2 with high affinity, and molar excess calpain 2 destroys HK (Schmaier et al., 1986; Bradford et al., 1990). Further, in stochiometric ratios (1:1) of calpain:HK, BK is liberated from HK (Higashiyama et al., 1986). These data serve as a guide to how falcipains, a Pf cysteine protease, cleaved HK to liberate BK. It has been reported that the Pf aminopeptidase P in food vacuoles hydrolyzes BK (Ragheb et al., 2009). Plasmodium chabaudi and Pf iRBCs also internalize and process plasma HK that subsequently release the vasoactive kinins, Lys-BK, BK, and desArg9BK (Bagnaresi et al., 2012). Furthermore, recombinant falcipain-2 and -3 proteolyze HK to liberate functional kinins that induced ileum contraction and activated the B2R and B1R as measured by calcium mobilization. These activities were blocked with antagonists to the B2R (HOE140) and B1R [DesArg9(Leu8)] and, independently, E64 protease. In other studies, both falcipain-2 and falcipain-3 generated BK (Cotrin et al., 2013). Both enzymes generated Met-Lys-BK, Lys-BK, and BK from the substrate Abz-MISLMKRPPGFSPFRSSRI-NH2. BK formed by any means stimulates nitric oxide production, induces vasodilatation, activates EC, enhances microvascular permeability, and modulates cellular metabolism.
The role of BK in malaria depends on its local or systemic production, the correlate BK receptor to which it binds and signals, and Plasmodium species involved. Bradykinin has been reported to have anti-plasmodial activity against P. gallinaceum, a malarial strain that infects chickens (Silva et al., 2015).
In other studies, the addition of BK to P. falciparum schizont cultures inhibited parasite growth similarly to captopril, an ACE inhibitor (Saraiva et al., 2011; Silva et al., 2016). ACE connects the kallikrein/kinin system (KKS) to the renin-angiotensin (RAS) and its inhibition leads to increase in both BK and Ang- (Moxon et al., 2009; W. H. O, 2012; Seydel et al., 2015; Mohanty et al., 2017; Santos et al., 2018; Moxon et al., 2020; Guenther et al., 2021; WHO, 2022) levels. In fact, the effect of BK was prevented by HOE-140 or A779, blockers of B2 or MAS receptors, respectively. Also, BK-induced inhibition of PKA activity in erythrocytes was sensitive to both pharmacological treatments, suggesting a coordinated effect between RAS and KKS. Co-immunoprecipitation assays demonstrated a possible association between B2R and Mas in the RBC membrane. B2R and Mas heterodimers have been demonstrated in the EC surface membrane as well. However, if this mechanism occurs during malaria infection remains to be determined (Abadir et al., 2006).
Published data reviewed here suggest that BK may be generated and contribute to the pathogenesis of vasogenic edema in CM in several ways (Figure 2). Falcipain-2 from the parasite itself cleaves HK and liberates BK. Further, malaria iRBCs and their microparticles released into the circulation provide negatively charged surfaces allowing for FXII autoactivation with activation of PK to plasma PKa leading to cleavage of HK and liberation of BK. Lastly, iRBC occlusion of microvessels in the brain could perturb the EC membrane and allow for vessel wall associated proteases such as prolylcarboxypeptidase to activate PK to produce plasma PKa to cleave HK with BK liberation that also contributes to reciprocal activation of FXII. Future investigations need to show causality of the proteins of the factor XII and kallikrein/kinin system in CM. These approaches should be done with global and selective gene deleted mice along with the use of precise pharmacologic agents already developed to treat diseases like hereditary angioedema (Abdulkarim and Craig, 2023; Wilkerson and Moellman, 2023). More in depth studies of the role(s) of contact activation and kallikrein-kinin system in CM may provide justification to test these drugs as adjunctive therapeutics to anti-malarial drugs. Finally, future studies of cerebral malaria, other severe malaria syndromes, and uncomplicated malaria that should include measurements of BK levels and activation of the kallikrein-kinin system before and after treatment with antimalarial drugs.
All authors contributed to the manuscript. AP wrote the first draft. The other authors worked together to complete the manuscript. All authors have seen the final version of the manuscript.
This work was aupported in part by National Institutes of Health grant numbers HL157405, HL144113, HL143402, CA223301, and AI130131.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abadir, P. M., Periasamy, A., Carey, R. M., Siragy, H. M. (2006). Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 48 (2), 316–322. doi: 10.1161/01.HYP.0000228997.88162.a8
Abdulkarim, A., Craig, T. J. (2023). “Hereditary Angioedema,” in StatPearls (Treasure Island (FL) ineligible companies, StatPearl’s Publishing).
Amulic, B., Moxon, C. A., Cunnington, A. J. (2020). A more granular view of neutrophils in malaria. Trends Parasitol. 36 (6), 501–503. doi: 10.1016/j.pt.2020.03.003
Bagnaresi, P., Barros, N. M., Assis, D. M., Melo, P. M., Fonseca, R. G., Juliano, M. A., et al. (2012). Intracellular proteolysis of kininogen by malaria parasites promotes release of active kinins. Malar J. 11, 156. doi: 10.1186/1475-2875-11-156
Beare, N. A. V. (2023). Cerebral malaria-using the retina to study the brain. Eye (Lond). doi: 10.1038/s41433-023-02432-z
Bradford, H. N., Schmaier, A. H., Colman, R. W. (1990). Kinetics of inhibition of platelet calpain II by human kininogens. Biochem. J. 270 (1), 83–90. doi: 10.1042/bj2700083
Cicardi, M., Zuraw, B. L. (2018). Angioedema due to bradykinin dysregulation. J. Allergy Clin. Immunol. Pract. 6 (4), 1132–1141. doi: 10.1016/j.jaip.2018.04.022
Clemens, R., Pramoolsinsap, C., Lorenz, R., Pukrittayakamee, S., Bock, H. L., White, N. J. (1994). Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br. J. Haematol 87 (1), 100–105. doi: 10.1111/j.1365-2141.1994.tb04877.x
Colman, R. W., Mattler, L., Sherry, S. (1969). Studies on the prekallikrein (kallikreinogen)–kallikrein enzyme system of human plasma. I. Isolation and purification of plasma kallikreins. J. Clin. Invest. 48 (1), 11–22. doi: 10.1172/JCI105959
Colman, R. W., Schmaier, A. H. (1997). Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 90 (10), 3819–3843.
Conroy, A. L., Opoka, R. O., BangIrana, P., Namazzi, R., Okullo, A. E., Georgieff, M. K., et al. (2021). Parenteral artemisinins are associated with reduced mortality and neurologic deficits and improved long-term behavioral outcomes in children with severe malaria. BMC Med. 19 (1), 168. doi: 10.1186/s12916-021-02033-1
Cotrin, S. S., Gouvea, I. E., Melo, P. M., Bagnaresi, P., Assis, D. M., Araujo, M. S., et al. (2013). Substrate specificity studies of the cysteine peptidases falcipain-2 and falcipain-3 from Plasmodium falciparum and demonstration of their kininogenase activity. Mol. Biochem. Parasitol. 187 (2), 111–116. doi: 10.1016/j.molbiopara.2013.01.002
Davis, T. M., Suputtamongkol, Y., Spencer, J. L., Ford, S., Chienkul, N., Schulenburg, W. E., et al. (1992). Measures of capillary permeability in acute falciparum malaria: relation to severity of infection and treatment. Clin. Infect. Dis. 15 (2), 256–266. doi: 10.1093/clinids/15.2.256
Dondorp, A. M., Fanello, C. I., Hendriksen, I. C., Gomes, E., Seni, A., Chhaganlal, K. D., et al. (2010). Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet 376 (9753), 1647–1657. doi: 10.1016/S0140-6736(10)61924-1
Feldman, H. A., Murphy, F. D. (1945). The effect of alternations in blood volume on the anemia and hypoproteinemia of human malaria. J. Clin. Invest. 24, 780–792.
Gething, P. W., Casey, D. C., Weiss, D. J., Bisanzio, D., Bhatt, S., Cameron, E., et al. (2016). Mapping Plasmodium falciparum Mortality in Africa between 1990 and 2015. N Engl. J. Med. 375 (25), 2435–2445. doi: 10.1056/NEJMoa1606701
Guenther, G., Muller, D., Moyo, D., Postels, D. (2021). Pediatric cerebral malaria. Curr. Trop. Med. Rep. 8 (2), 69–80. doi: 10.1007/s40475-021-00227-4
Gustafson, E. J., Schutsky, D., Knight, L. C., Schmaier, A. H. (1986). High molecular weight kininogen binds to unstimulated platelets. J. Clin. Invest. 78 (1), 310–318. doi: 10.1172/JCI112567
Higashiyama, S., Ishiguro, H., Ohkubo, I., Fujimoto, S., Matsuda, T., Sasaki, M. (1986). Kinin release from kininogens by calpains. Life Sci. 39 (18), 1639–1644. doi: 10.1016/0024-3205(86)90160-8
Idro, R., Kakooza-Mwesige, A., Asea, B., Ssebyala, K., BangIrana, P., Opoka, R. O., et al. (2016). Cerebral malaria is associated with long-term mental health disorders: a cross sectional survey of a long-term cohort. Malar J. 15, 184. doi: 10.1186/s12936-016-1233-6
Jensen, A. R., Adams, Y., Hviid, L. (2020). Cerebral Plasmodium falciparum malaria: The role of PfEMP1 in its pathogenesis and immunity, and PfEMP1-based vaccines to prevent it. Immunol. Rev. 293 (1), 230–252. doi: 10.1111/imr.12807
John, C. C., BangIrana, P., Byarugaba, J., Opoka, R. O., Idro, R., Jurek, A. M., et al. (2008). Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics 122 (1), e92–e99. doi: 10.1542/peds.2007-3709
Kaplan, A. P., Joseph, K. (2017). Pathogenesis of hereditary angioedema: The role of the bradykinin-forming cascade. Immunol. Allergy Clin. North Am. 37 (3), 513–525. doi: 10.1016/j.iac.2017.04.001
Kaplan, A. P., Joseph, K., Ghebrehiwet, B. (2022). The complex role of kininogens in hereditary angioedema. Front. Allergy 3. doi: 10.3389/falgy.2022.952753
Kessler, A., Dankwa, S., Bernabeu, M., Harawa, V., Danziger, S. A., Duffy, F., et al. (2017). Linking EPCR-binding pfEMP1 to brain swelling in pediatric cerebral malaria. Cell Host Microbe 22 (5), 601–614 e5. doi: 10.1016/j.chom.2017.09.009
Kraft, S. A., Duenas, D. M., Kublin, J. G., Shipman, K. J., Murphy, S. C., Shah, S. K. (2019). Exploring ethical concerns about human challenge studies: A qualitative study of controlled human malaria infection study participants' Motivations and attitudes. J. Empir Res. Hum. Res. Ethics 14 (1), 49–60. doi: 10.1177/1556264618820219
Maegraith, B. G., Onabanjo, A. O. (1969a). Isolation and estimation of pure monkey serum kallikrein. J. Physiol. 205 (2), 9P–10P.
Maegraith, B. G., Onabanjo, A. O. (1969b). The involvement of histamine in malaria. Br. J. Pharmacol. 37 (2), 535P–536P.
Maegraith, B. G., Onabanjo, A. O. (1970). The effects of histamine in malaria. Br. J. Pharmacol. 39 (4), 755–764. doi: 10.1111/j.1476-5381.1970.tb09902.x
Maegraith, B. G., Tella, A. (1968). Kallidin–a probable factor in the pathogenesis of malaria. Br. J. Pharmacol. 34 (1), 235P.
Maitland, K. (2016). Severe malaria in african children - the need for continuing investment. N Engl. J. Med. 375 (25), 2416–2417. doi: 10.1056/NEJMp1613528
Malloy, J. P., Brooks, M. H., Barry, K. G. (1967). Pathophysiology of acute falciparum malaria. II. Fluid compartmentalization. Am. J. Med. 43 (5), 745–750. doi: 10.1016/0002-9343(67)90116-7
Merkulova, A. A., Abdalian, S., Silbak, S., Pinheiro, A. S., Schmaier, A. H. C1 inhibitor and prolylcarboxypeptidase modulate prekallikrein activation on endothelial cells. J Allergy Clin. Immunol. (2023) S0091-6749 (23), 00856–4. doi: 10.1016/j.jaci.2023.06.017
Mohanty, S., Benjamin, L. A., Majhi, M., Panda, P., Kampondeni, S., Sahu, P. K., et al. (2017). Magnetic resonance imaging of cerebral malaria patients reveals distinct pathogenetic processes in different parts of the brain. mSphere 2 (3), e00193-17. doi: 10.1128/mSphere.00193-17
Moxon, C. A., Gibbins, M. P., McGuinness, D., Milner, D. A., Jr., Marti, M. (2020). New insights into malaria pathogenesis. Annu. Rev. Pathol. 15, 315–343. doi: 10.1146/annurev-pathmechdis-012419-032640
Moxon, C. A., Heyderman, R. S., Wassmer, S. C. (2009). Dysregulation of coagulation in cerebral malaria. Mol. Biochem. Parasitol. 166 (2), 99–108. doi: 10.1016/j.molbiopara.2009.03.006
O'Leary, P. A. (1928). Treatment of neurosyphilis by malaria-fourth annual report. Cal West Med. 29 (3), 160–163.
Ohtomo, H., Katori, M. (1972). Reliable evidence of involvement of the kinin system in mouse malaria. Jpn J. Pharmacol. 22 (4), 493–509. doi: 10.1254/jjp.22.493
Onabanjo, A. O., Maegraith, B. G. (1970a). Inflammatory changes in small blood vessels induced by kallikrein (kininogenase) in the blood of Macaca mulatta infected with Plasmodium knowlesi. Ann. Trop. Med. Parasitol. 64 (2), 227–236. doi: 10.1080/00034983.1970.11686685
Onabanjo, A. O., Maegraith, B. G. (1970b). Pathological lesions produced in the brain by kllikrein (kininogenase) in Macaca mulatta infected with Plasmodium knowlesi. Ann. Trop. Med. Parasitol. 64 (2), 237–242. doi: 10.1080/00034983.1970.11686686
Onabanjo, A. O., Maegraith, B. G. (1970c). Kallikrein as a pathogenic agent in Plasmodium knowlesi infection in Macaca mulatta. Br. J. Exp. Pathol. 51 (5), 523–533.
Patel, H., Dunican, C., Cunnington, A. J. (2020). Predictors of outcome in childhood Plasmodium falciparum malaria. Virulence 11 (1), 199–221. doi: 10.1080/21505594.2020.1726570
Pinheiro, A. S., Silbak, S., Schmaier, A. H. (2022). Bradykinin - An elusive peptide in measuring and understanding. Res. Pract. Thromb. Haemost. 6 (2), e12673. doi: 10.1002/rth2.12673
Ragheb, D., Bompiani, K., Dalal, S., Klemba, M. (2009). Evidence for catalytic roles for Plasmodium falciparum aminopeptidase P in the food vacuole and cytosol. J. Biol. Chem. 284 (37), 24806–24815. doi: 10.1074/jbc.M109.018424
Ramachandran, A., Sharma, A. (2022). Dissecting the mechanisms of pathogenesis in cerebral malaria. PLoS Pathog. 18 (11), e1010919. doi: 10.1371/journal.ppat.1010919
Renia, L., Howland, S. W., Claser, C., Charlotte Gruner, A., Suwanarusk, R., Hui Teo, T., et al. (2012). Cerebral malaria: mysteries at the blood-brain barrier. Virulence 3 (2), 193–201. doi: 10.4161/viru.19013
Riggle, B. A., Manglani, M., Maric, D., Johnson, K. R., Lee, M. H., Neto, O. L. A., et al. (2020). CD8+ T cells target cerebrovasculature in children with cerebral malaria. J. Clin. Invest. 130 (3), 1128–1138. doi: 10.1172/JCI133474
Sahu, P. K., Duffy, F. J., Dankwa, S., Vishnyakova, M., Majhi, M., Pirpamer, L., et al. (2021). Determinants of brain swelling in pediatric and adult cerebral malaria. JCI Insight 6(18). doi: 10.1172/jci.insight.145823
Santos, R. A. S., Sampaio, W. O., Alzamora, A. C., Motta-Santos, D., Alenina, N., Bader, M., et al. (2018). The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1-7). Physiol. Rev. 98 (1), 505–553. doi: 10.1152/physrev.00023.2016
Saraiva, V. B., de Souza Silva, L., Ferreira-DaSilva, C. T., da Silva-Filho, J. L., Teixeira-Ferreira, A., Perales, J., et al. (2011). Impairment of the Plasmodium falciparum erythrocytic cycle induced by angiotensin peptides. PLoS One 6 (2), e17174. doi: 10.1371/journal.pone.0017174
Schmaier, A. H. (2016). The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J. Thromb. Haemost. 14 (1), 28–39. doi: 10.1111/jth.13194
Schmaier, A. H. (2019). The hereditary angioedema syndromes. J. Clin. Invest. 129 (1), 66–68. doi: 10.1172/JCI125378
Schmaier, A. H., Bradford, H., Silver, L. D., Farber, A., Scott, C. F., Schutsky, D., et al. (1986). High molecular weight kininogen is an inhibitor of platelet calpain. J. Clin. Invest. 77 (5), 1565–1573. doi: 10.1172/JCI112472
Schmaier, A. H., Kuo, A., Lundberg, D., Murray, S., Cines, D. B. (1988). The expression of high molecular weight kininogen on human umbilical vein endothelial cells. J. Biol. Chem. 263 (31), 16327–16333.
Seydel, K. B., Kampondeni, S. D., Valim, C., Potchen, M. J., Milner, D. A., Muwalo, F. W., et al. (2015). Brain swelling and death in children with cerebral malaria. N Engl. J. Med. 372 (12), 1126–1137. doi: 10.1056/NEJMoa1400116
Silva, A. F., Alves, F. L., Pedron, C. N., Torres, M. D., Silva, L. S., Pinheiro, A. A., et al. (2015). Anti-plasmodial activity of bradykinin and analogs. Bioorg Med. Chem. Lett. 25 (16), 3311–3313. doi: 10.1016/j.bmcl.2015.05.071
Silva, L. S., Peruchetti, D. B., Silva, C. T. F., Ferreira-DaSilva, A. T., Perales, J., Caruso-Neves, C., et al. (2016). Interaction between bradykinin B2 and Ang-(1-7) Mas receptors regulates erythrocyte invasion by Plasmodium falciparum. Biochim. Biophys. Acta 1860 (11 Pt A), 2438–2444. doi: 10.1016/j.bbagen.2016.07.011
Silva, L. S., Pinheiro, A. S., Teixeira, D. E., Silva-Aguiar, R. P., Peruchetti, D. B., Scharfstein, J., et al. (2019). Kinins Released by Erythrocytic Stages of Plasmodium falciparum Enhance Adhesion of Infected Erythrocytes to Endothelial Cells and Increase Blood Brain Barrier Permeability via Activation of Bradykinin Receptors. Front. Med. (Lausanne) 6. doi: 10.3389/fmed.2019.00075
Silva-Filho, J. L., Caruso-Neves, C., Pinheiro, A. A. (2017). Targeting angiotensin II type-1 receptor (AT(1)R) inhibits the harmful phenotype of plasmodium-specific CD8(+) T cells during blood-stage malaria. Front. Cell Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00042
Silva-Filho, J. L., Souza, M. C., Ferreira-Dasilva, C. T., Silva, L. S., Costa, M. F., Padua, T. A., et al. (2013). Angiotensin II is a new component involved in splenic T lymphocyte responses during Plasmodium berghei ANKA infection. PLoS One 8 (4), e62999. doi: 10.1371/journal.pone.0062999
Skirrow, M. B., Chongsuphajaisiddhi, T., Maegraith, B. G. (1964). The circulation in malaria. Ii. Portal angiography in monkeys (Macaca mulatta) infected with plasmodium knowlesi and in shock following manipulation of the gut. Ann. Trop. Med. Parasitol. 58, 502–510.
Tella, A., Maegraith, B. G. (1966). Studies on bradykinin and bradykininogen in malaria. Ann. Trop. Med. Parasitol. 60 (3), 304–317. doi: 10.1080/00034983.1966.11686421
van der Heyde, H. C., Nolan, J., Combes, V., Gramaglia, I., Grau, G. E. (2006). A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22 (11), 503–508. doi: 10.1016/j.pt.2006.09.002
Verhave, J. P. (2013). Experimental, therapeutic and natural transmission of Plasmodium vivax tertian malaria: scientific and anecdotal data on the history of Dutch malaria studies. Parasit Vectors 6 (1), 19. doi: 10.1186/1756-3305-6-19
Wagner-Jauregg, J., Bruetsch, W. L. (1946). The history of the malaria treatment of general paralysis. Am. J. Psychiatry 102, 577–582. doi: 10.1176/ajp.102.5.577
Wassmer, S. C., Grau, G. E. (2017). Severe malaria: what's new on the pathogenesis front? Int. J. Parasitol. 47 (2-3), 145–152. doi: 10.1016/j.ijpara.2016.08.002
W. H. O (2012). “Management of severe malaria: a practical handbook,” in La prise en charge du paludisme grave: guide pratique– 3ème ed(Geneva).
World Health Organization (2022). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. World Malaria Report 2022. World Health Organization 2022. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022
Wilkerson, R. G., Moellman, J. J. (2023). Hereditary angioedema. Immunol. Allergy Clin. North Am. 43 (3), 533–552. doi: 10.1016/j.iac.2022.10.012
Keywords: cerebral malaria, bradykinin, factor XII, prekallikrein, high molecule weight kininogen, bradykinin B2 and B1 receptors
Citation: Pinheiro AdS, Kazura JW, Pinheiro AA and Schmaier AH (2023) Is there a role for bradykinin in cerebral malaria pathogenesis? Front. Cell. Infect. Microbiol. 13:1184896. doi: 10.3389/fcimb.2023.1184896
Received: 13 March 2023; Accepted: 24 July 2023;
Published: 10 August 2023.
Edited by:
Luiz Juliano, Universidade Federal de São Paulo, BrazilReviewed by:
Alister Craig, Liverpool School of Tropical Medicine, United KingdomCopyright © 2023 Pinheiro, Kazura, Pinheiro and Schmaier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alvin H. Schmaier, U2NobWFpZXJAY2FzZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.