 Morgan Similuk
Morgan Similuk Taco Kuijpers
Taco Kuijpers- 1Centralized Sequencing Program, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
- 2Department of Pediatric Immunology, Rheumatology and Infectious Diseases, Emma Children’s Hospital, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
The overall disease burden of pediatric infection is high, with widely varying clinical outcomes including death. Among the most vulnerable children, those with inborn errors of immunity, reduced penetrance and variable expressivity are common but poorly understood. There are several genetic mechanisms that influence phenotypic variation in inborn errors of immunity, as well as a body of knowledge on environmental influences and specific pathogen triggers. Critically, recent advances are illuminating novel nuances for fundamental concepts on disease penetrance, as well as raising new areas of inquiry. The last few decades have seen the identification of almost 500 causes of inborn errors of immunity, as well as major advancements in our ability to characterize somatic events, the microbiome, and genotypes across large populations. The progress has not been linear, and yet, these developments have accumulated into an enhanced ability to diagnose and treat inborn errors of immunity, in some cases with precision therapy. Nonetheless, many questions remain regarding the genetic and environmental contributions to phenotypic variation both within and among families. The purpose of this review is to provide an updated summary of key concepts in genetic and environmental contributions to phenotypic variation within inborn errors of immunity, conceptualized as including dynamic, reciprocal interplay among factors unfolding across the key dimension of time. The associated findings, potential gaps, and implications for research are discussed in turn for each major influencing factor. The substantial challenge ahead will be to organize and integrate information in such a way that accommodates the heterogeneity within inborn errors of immunity to arrive at a more comprehensive and accurate understanding of how the immune system operates in health and disease. And, crucially, to translate this understanding into improved patient care for the millions at risk for serious infection and other immune-related morbidity.
1 Introduction
Clinical heterogeneity is common among thousands of described Mendelian disorders (Chong et al., 2015; Posey et al., 2019). Likewise, for inborn errors of immunity (IEIs) reduced penetrance and variable expressivity are the rule, not the exception (Gruber and Bogunovic, 2020; Aksentijevich and Schnappauf, 2021). Even the severe combined immune deficiencies, the most paradigmatic of examples, are not uniformly apparent in infancy (Dvorak et al., 2023). Moreover, clinical presentations of IEIs are particularly reliant on environmental triggers, namely, exposure to pathogens, to begin to manifest most symptoms (Casanova and Abel, 2021). The implications that this variability and uncertainty has for families – medically, emotionally, and socially – can be vexing both for the affected individuals and their care teams.
The issue of penetrance is not only a pervasive phenomenon among IEI and other Mendelian diseases, but it is also among the most fundamental questions in human genetics: what is the probability of phenotypic expression given the genotype? Historically, efforts to dissect genotype-phenotype correlation have been largely dichotomized between rare variants of large effect and common variants of small effect on risk for disease, although this has almost certainly obscured the true nature of the underlying biology with disease risk variants being distributed across a continuum of frequencies and effect sizes (Antonarakis et al., 2010; Claussnitzer et al., 2020). Critically, recent advances are illuminating additional nuance for fundamental concepts in understanding penetrance, as well as raising new areas of inquiry. For example, genome-wide analyses in large, population-based biobanks have repeatedly confirmed the suspected inflation of Mendelian disease penetrance estimates when such estimates are clinically ascertained through proband analysis or family studies and have enabled a more accurate, more comprehensive understanding of the relationship between genotype and phenotype for select diseases (Ranola et al., 2019; Tuke et al., 2019; Wright et al., 2019). Concurrently, advances in sequencing have enabled the detection of somatic variants, microbiome composition, and other relevant factors, to an extent not previously possible. If the prior generations of immunologists grappled with the bottlenecks of variant discovery and disease association, the current era will be one defined by efforts to understand the mechanisms by which the full spectrum of genetic variation influences biology and to translate that understanding into clinical care (Lappalainen and MacArthur, 2021). It is a formidable challenge, yet demonstrably tractable, with evidence of progress year after year.
As such, the purpose of this review is to provide an updated summary of key concepts in genetic and environmental contributions to phenotypic variation within IEI, conceptualized as including dynamic, reciprocal interplay among factors (Figure 1). The associated controversies, potential gaps, and implications for research are discussed throughout, as are the connections to patient care.
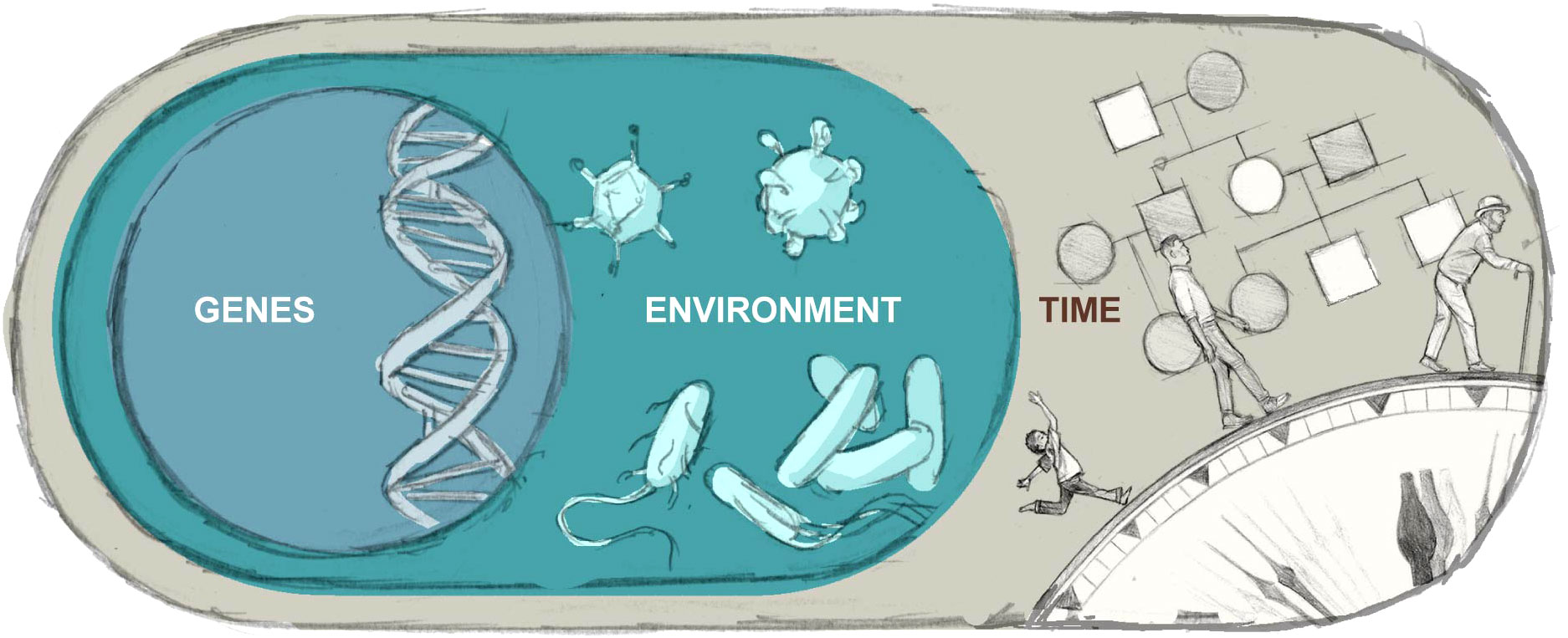
Figure 1 Illustrates the complex interplay among an individual’s genetic and environmental contributions to health and disease over time.
2 Body
2.1 Genetics
We review IEI-specific examples of differential variant impact on phenotype, a foundational concept in genetics, followed by contemporary discussions of mosaicism, allelic bias, and multi-locus genetic variation.
2.1.1 Differential variant impact
Discerning the molecular consequence of a variant is a critical clue in anticipating its potential cellular and ultimate phenotypic impact. There are many related terms used to describe aspects of variant effects, including the effect on the protein sequence (e.g., missense, frameshift, stop gained, synonymous, etc.), the genetic mechanism of disease (e.g., haploinsufficiency, dominant negative), and protein function (e.g., gain-of-function or GoF; loss-of-function or LoF) (Nussbaum et al., 2015; Zschocke et al., 2023). GoF typically refers to the enhancement of the affected protein’s activity, and occasionally refers to a novel or neomorphic function, as is the case in a recently described CEBPE-related autoinflammatory disorder in which the defect lead to enhanced chromatic occupancy and ultimately dysregulated signaling (Göös et al., 2019). See supplemental table for description of GOF IEI. The term LoF is sometimes replaced with more specific descriptors such as null, meaning eliminated activity, or hypomorphic, meaning diminished activity or presence of the encoded protein (Ott et al., 2023). See Figure 2A for illustration.
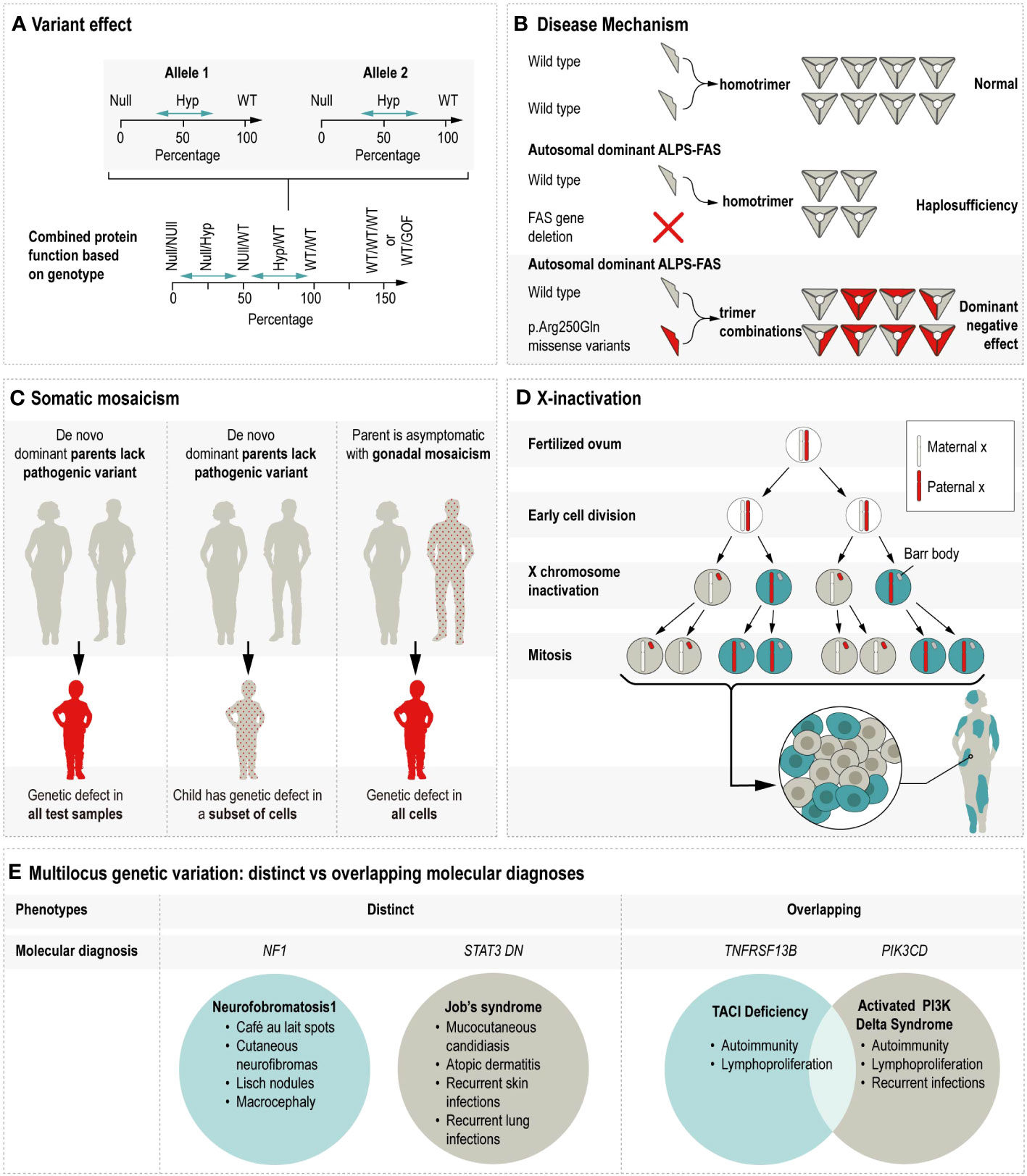
Figure 2 Illustrates the multiple mechanisms by which genetic variation contributes to phenotypic variability. Hyp = Hypomorphic, WT = Wildtype, GOF = Gain of function, ALPS-FAS = Autoimmune lymphoproliferative syndrome due to a defect in FAS, DN = Dominant negative. (A) Variant effect on protein function is shown as the combined impact of allele 1 and allele 2. For example, the combined protein function resulting from two null alleles would be less than the protein function resulting from hypomorphic alleles with some residual function. In some cases, triplosensitivity (WT/WT/WT) or alleles with GOF effects can produce an increased level of protein function. (Adapted from Zschocke et al., 2023.) (B) The disease mechanisms of halploinsufficiency and dominant negative are contrasted using the example of monoallelic ALPS-FAS. Ordinarily, the two wildtype proteins homotrimerize to create the transmembrane protein which initiates caspase cascade upon activation. In cases where one allele is affected by a FAS gene deletion, the single wildtype allele produces half the normal amount of homotrimers, leading to ALPS via haploinsufficiency. In cases where one allele is affected by a pathogenic missense variant, the mutant protein is incorporated in 7 of 8 possible configurations, thus leading to ALPS via dominant negative action. (C) Somatic mosaicism’s impact on autosomal dominant inheritance varies by the timing of the mutational event. In the left panel, the parents lack the pathogenic variant in all their cells, the pathogenic variant arises early in embryonic development, and is present in all tested samples obtained from the affected child. The middle panel is similar, except that in this example the pathogenic variant arises later in development, such that the child is still symptomatic but that the genetic defect is present in some but not all of the tested cells types. In the right panel, the pathogenic variant is present at a low frequency in an unaffected parent and subsequently passed on to the affected child who has the pathogenic variant in all of their cells. (D) X-inactivation creates cellular uniallelic expression in XX females through the inactivation of one X chromosomes early in embryonic development. As a result, XX females mosaically express X chromosomes throughout their body. (E) Patients with multiple molecular diagnoses generally either have molecular diagnoses reflecting distinct or overlapping disorders. For example, the disorders caused by defects in NF1 and dominant negative STAT3 are distinct such that the respective symptoms can be reasonably attributed to either the NF1 or STAT3 defect. In contrast, the symptoms associated with defects in TNFRSF13B and PIK3CD are largely overlapping and not possible to distinguish.
Identifying the diminishment or gain of protein function is a necessary first step in understanding disease mechanisms and is dependent on the variant’s impact on the biological pathway(s) associated with the protein(s) encoded by the gene. The discovery of new mechanisms of disease among known IEI-associated genes accounted for multiple additions to the most recent International Union of Immunological Sciences (IUIS) categorizations of IEI (e.g., STAT2 LoF vs GoF; IKZF1 LoF vs GoF) (Tangye et al., 2021; Tangye et al., 2022). For the discussion at hand, it should be underscored that such differences in protein effect do not contribute to intrafamilial variation in the context of a familial variant.
Indeed, we are only beginning to understand the patterns associated with gain or loss of function at the level of the variant, gene, and protein. A recent comprehensive analysis found that GoF variants are enriched in essential genes, for autosomal dominant inheritance, and localized in protein binding and protein interacting domains; whereas LoF variants are enriched for protein-truncating variants in protein core regions where variants may disrupt the stability and folding of the protein (Sevim Bayrak et al., 2021). Such insights extend our knowledge of the properties associated with LoF or GoF and contribute to a growing ability to accurately predict variant impact.
This progress notwithstanding, GoF vs LoF effects can be very difficult to discern even with direct functional data, particularly when the underlying biology is complex, as highlighted in the case of a CARD11 variant with pathogenic features of both GoF and LoF effects (Desjardins et al., 2018). As such, understanding phenotypic variation in IEI requires further development of in silico tools to prioritize candidate variants and complement the strong history of experimental work in clinical immunology.
Mendelian susceptibility to mycobacterial disease (MSMD) provides an illuminating clinical example of differential variant impact on loss of immune function and subsequent phenotype (Bustamante et al., 2014). MSMD is characterized by predisposition to disease from poorly virulent mycobacteria (e.g., Bacillus Calmette–Guérin (BCG) vaccines and environmental mycobacterial) in otherwise healthy individuals and can be caused by defects in multiple genes (Bustamante et al., 2014; Bustamante, 2020). As one example, multiple defects affecting IFNGR1 cause MSMD. IFNGR1 encodes the IFN-γ receptor-ligand binding chain and thus, when mutated interferon and related signaling are dysregulated. Within IFNGR1-related MSMD, biallelic, null variants cause the most severe phenotype of disseminated, life-threatening infections with nearly complete penetrance in childhood. In these cases, the nonsense or frameshift variants create a premature stop codon and prevent the expression of IFNGR1 at the cell surface (Holland et al., 1998; Filipe-Santos et al., 2006). In contrast, both biallelic, hypomorphic variants and heterozygous monoallelic, autosomal dominant (partial) IFNGR1 deficiency have a milder clinical phenotype and reduced, age-related penetrance (Jouanguy et al., 1997; Jouanguy et al., 1999). Management of the IFNGR1-related MSMD clinical spectrum varies by severity, with poor prognosis in the former case and infrequent need for prophylactic antibiotics or hematopoietic stem cell transplantation (HSCT) in the latter cases (Holland et al., 1998; Filipe-Santos et al., 2006). Thus, differential variant impact is highly consequential for MSMD.
Relatedly, the clinical spectrum arising from RAG (“recombination activating genes”) deficiency provides another example of the effect of variant type on immune function and one that contains both marked inter- and intra-familial variation in clinical manifestations (Bosticardo et al., 2021).
As an additional consideration for variant impact on phenotypic variation, protein function is not equally disrupted by all pathogenic variation along the length of a gene. Understanding the function of a protein’s domains can provide insight into a given variant’s potential consequences. The different molecular and clinical consequences of FAS defects provide one example. The gene FAS encodes a cell surface receptor involved in apoptosis and is critical in maintaining cellular homeostasis (Fisher et al., 1995). Defects in FAS cause autoimmune lymphoproliferative syndrome (ALPS), which is characterized by autoimmunity, particularly against components of the blood; lymphoproliferation including lymphadenopathy; and an increased risk for lymphoma (Price et al., 2014). Heterozygous pathogenic variants in the FAS “death domain” interfere with the formation of the homotrimeric receptor. In the presence of a heterozygous mutation, seven out of eight homotrimers will contain at least one defective copy of the FAS protein, thus, diminishing effective signaling through negative interference (Kuehn et al., 2011).
For variants outside of this domain or FAS deletions, the mechanism is haploinsufficiency, whereby the total amount of FAS protein generated is below the threshold for apoptosis induction (Hsu et al., 2012; Jevtich et al., 2022). Critically, these different mechanisms may influence penetrance, meaning the likelihood that a genetic defect will cause symptoms. The penetrance of ALPS due to negative interferences was reported as up to 60% (Price et al., 2014), whereas haploinsufficiency for FAS is associated with a penetrance estimate of 40% (Kuehn et al., 2011). This is a prime example of distinguishing dominant negative biological effects, which can be distinguished from that of haploinsufficiency. See Figure 2B for illustration. Lastly, apart from the residual surface expression, biallelic inheritance of FAS defects results in a more severe, more penetrant ‘FAS-null’ phenotype (Rieux-Laucat et al., 1995).
AIRE-related autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) also exhibits phenotypic variation based on the location of the pathogenic variant and subsequent genetic mechanism. AIRE (autoimmune regulator) is a gene expressed in the thymus and involved in establishing immune tolerance. Defects results in the peripheral escape of self-reactive T lymphocytes and autoantibodies. APECED is classically characterized by chronic mucocutaneous candidiasis, hypothyroidism, and adrenal insufficiency (Ferré et al., 2021). While biallelic LoF variants are the most common cause of APECED, dominant-negative variants within the SAND and PHD1 domains of the AIRE gene have been reported as a cause of APECED. The dominant-negative variants are associated with reduced penetrance, milder symptoms, and fewer autoimmune manifestations compared to biallelic LoF variants (Ferré et al., 2021), similar as observed with the aforementioned STAT1 biallelic or dominant-negative variants. Genotype and phenotype correlation, particularly by distinct disease mechanisms is a very active area of research and there are many additional and recent examples including disorders caused by defects in WAS, RAC2, IKZF1, and PSTPIP1, among others, with a distinct phenotype when comparing the LoF and GoF variants (Holzinger et al., 2015; Masters et al., 2016; Kuehn et al., 2021; Hoshino et al., 2022; Hsu, 2023).
Lastly, future work will likely continue to provide illustrative examples of differential variant impact for rare IEI, as well as identify additional disease-associated allelic series across the spectrum of frequency and effect size. A current example of allelic series in IEI involves the gene TYK2 (tyrosine kinase 2), which mediates immune and inflammatory signaling (Casanova and Abel, 2022). Rare biallelic variants in TYK2 predispose to infection by intracellular bacteria or viruses, while at the same time, common TYK2 variants are associated with inflammatory and autoimmune disease (Li et al., 2020; Casanova and Abel, 2022). Similarly, rare variants causing haploinsufficiency of CTLA4 (cytotoxic t-lymphocyte associated protein 4) impair Treg function, leading to a syndrome of hypogammaglobulinemia, lymphoproliferation, and autoimmune cytopenia with respiratory, gastrointestinal, and/or neurological features (Schwab et al., 2018). In contrast, select common variants in CTLA4 have been reported in association with complex autoimmune disorders such as rheumatoid arthritis and hypothyroidism (Schwab et al., 2018; Buniello et al., 2019). The same applies to LRBA as CTLA4, being in the same pathway of Treg function and other autoinflammatory conditions such as inflammatory bowel disease, with a potential for therapeutic approaches to improve their regulatory cell function (Gettler et al., 2021). Only by better understanding the full spectrum of how genetic variation influences biology will we arrive at a comprehensive view of genotype-phenotype correlations with translational importance for patient diagnosis, prognosis, and treatment.
2.1.2 Mosaicism
Genetic mosaicism is emerging as an important contributor to many disorders, including IEI molecular diagnostics as well as more complex disease processes. Somatic mosaicism refers to post-zygotic changes to the inherited, germline DNA sequence, which are present at variable allele fractions and distributions among cells throughout the body. Germline mosaicism refers to the heterogenous variant status of the diploid germ cell precursors in one’s gonads: some cells have the variant and some do not (Biesecker and Spinner, 2013). By one estimate, approximately 4% of apparently de novo pathogenic variants are present at a frequency >1% in parental blood, with important implications for recurrence risk counseling (Rahbari et al., 2016). See Figure 2C for illustration.
At one end of the spectrum, high-impact somatic events occurring early in post-zygotic development can lead to IEIs that are phenotypically and genotypically indistinguishable from disorders caused by germline variants, as shown in two cases of STAT5B-related allergic disease and autoimmunity caused by the same somatic variant arising in a hematopoietic progenitor (Ma et al., 2017). The observed frequency of somatic variants leading to ‘monogenic’ IEI is dependent on cohort selection and technologies used, as low allele fraction somatic variants are beyond the limits of detection for common next-generation sequencing approaches.
Somatic mosaicism has been reported as an etiology of multiple IEIs, starting in the mid-2000s (Holzelova et al., 2004; Saito et al., 2005). Autoinflammatory and lymphoproliferative disorders are well represented among IEIs with reported mosaicism and offer key insights. The first example involves NLRP3 (NOD-like receptor proteins 3), which encodes a protein involved in inflammasomes and, ultimately, the inflammatory response. Defects in this gene cause aberrant inflammation and a syndrome called NOMID (neonatal-onset multisystem inflammatory disease), which is characterized by persistent inflammation and tissue damage primarily affecting the nervous system, skin, and joints (Tanaka et al., 2011; Kuemmerle-Deschner et al., 2017; Nishikomori et al., 2019). Compared to individuals with germline NLRP3 pathogenic variants, individuals who have somatic NLRP3 variants restricted to the lymphoid and/or myeloid lineages are reported to have a notable lessening of the neurological impairment classically seen in NOMID. As an extreme example of this concept, a patient was reported with a recurrent urticaria-like rash, fever, conjunctivitis, and oligoarthritis in the 6th decade of life. Genetic studies identified a novel pathogenic NLRP3 variant with a low allele fraction, restricted to the myeloid lineage (Mensa-Vilaro et al., 2016). This case has provocative implications for the potential genetic contributions to other late-onset suspected IEIs as a future area of research and builds on related complex autoinflammatory disease findings (Hoffman and Broderick, 2017).
Other IEIs with somatic mosaicism as a reported etiology include but are not limited to FAS-related autoimmune lymphoproliferative syndrome (Dowdell et al., 2010); TLR8-related immunodeficiency with bone marrow failure (Aluri et al., 2021); and UBA1-related severe and often fatal, adult-onset autoinflammatory disease (Beck et al., 2020). For some of these disorders, the constitutional presence of the pathogenic variant is yet to be reported and may be incompatible with life (e.g., UBA1), while for others somatic mosaicism is a contributing etiology alongside inherited variants (e.g., FAS). Timing of the somatic event is paramount in determining the variant allele fraction and distribution of the variant among cells (Van Horebeek et al., 2019). We anticipate more somatic variants causing IEIs at older ages will be identified in the coming years.
Apart from somatic mutations directly causing disease, acquired, somatic variants may also occur as part of the progressive development of an important underlying disease entity, as is seen in GATA2 deficiency-related myeloid transformation (West et al., 2014; McReynolds et al., 2019; West et al., 2022). GATA2 (GATA binding protein 2) encodes a transcription factor involved in regulating the transcription of genes involved in the development and proliferation of hematopoietic cell lineages (Spinner et al., 2014). Defects in this gene cause a multi-system syndrome encompassing immunodeficiency, myelodysplastic syndrome/acute myeloid leukemia, pulmonary disease, and vascular/lymphatic dysfunction (Spinner et al., 2014; Donadieu et al., 2018). Phenotypic variability observed in these families is striking and can pose clinical challenges regarding the ideal timing of HSCT (Donadieu et al., 2018). By better understanding the cumulative somatic events that foretell myeloid transformation, HSCT timing can be more effectively tailored which can be monitored by NGS-based monitoring with MDS/AML panels, similar to current follow-up of ELANE related severe congenital neutropenia or Shwachman Diamond syndrome with a cumulative risk of clonal myeloid transformation (Beck et al., 2020). Similarly, for families with heterozygous pathogenic variants in FAS, incomplete penetrance of lymphoproliferation and autoimmunity can be partially explained by somatic changes in the second FAS allele in the lymphoid compartment (Magerus-Chatinet et al., 2011), consistent with models of multistep pathogenesis for malignant transformation (Goodnow, 2007).
As a remarkable point of contrast to these relatively precise somatic changes, chromothripsis, the catastrophic cellular event in which chromosomes undergo massive deletion and rearrangement, has been reported as a spontaneous cure for WHIM syndrome (McDermott et al., 2015). WHIM syndrome is caused by defects in the gene CXCR4 and is characterized by neutropenia, B cell lymphopenia, myelokathexis, hypogammaglobulinemia, recurrent infections, and marked susceptibility to human papillomavirus infection with resultant warts (Kawai and Malech, 2009). In the unique case report, the somatic, chromothriptic event deleted the pathogenic CXCR4 allele and other genes in a hematopoietic stem cell. This led to the spontaneous and durable clinical remission of the patient’s long-standing warts, neutropenia, and myelokathexis in her 4th decade of life (McDermott et al., 2015).
Complementing the insights from the compelling clinical course for this patient with WHIM syndrome, somatic reversion has long been described in the setting of IEI and refers to the spontaneous repair of a germline defect, resulting in improvement of the mutated molecular phenotype in progeny cells (Pillay et al., 2021). Over a dozen diseases including but not limited to those caused by defects in DOCK8, WAS, ADA, RAG1, and others have reports of somatic reversion (Miyazawa and Wada, 2021). Such reversion mutations can offer a significant proliferative or survival advantage to affected cells, thus altering the disease course (Humblet-Baron et al., 2007).
The fundamental importance of somatic mosaicisms in explaining phenotype variation within IEIs is underscored by these contrasting examples of somatic events accelerating and ameliorating disease. As a field, we are pivoting our understanding of the genetic contribution to disease from that of a limited, static inheritance to a more sophisticated and accurate model of each individual as a dynamic, complex mosaic of genetically distinct cells that change over time.
2.1.3 Allelic bias
Allelic bias in gene expression is another mechanism that holds key lessons for understanding biology, and potentially, phenotypic variation. Monoallelic expression is an extreme form of allelic bias. Monoallelically expressed genes generally belong to one of three groups: (1.) parent-of-origin imprinted genes, where all cells have the same active allele that is epigenetically determined solely by the allele’s parent-of-origin; many of these genes govern early growth and development and will not be discussed here; (2.) random X-inactivation, and (3.) random allelic bias among autosomal genes (Chess, 2016).
X-inactivation occurring early in female embryonic development is a well-established dosage compensation mechanism ensuring that X-chromosome genes are expressed at comparable levels in males and females. Stochastic deviation from a 50/50 distribution of X inactivation is common (Shvetsova et al., 2019). Inactivation is hypothesized to be further skewed in the presence of negative selection. For example, chromosomal aberrations on the other allele are known to promote skewed inactivation, as is monozygotic twinning. When this X-inactivation is highly skewed toward the expression of an X-chromosome containing a pathogenic variant, females can develop symptoms typically observed in males with the X-linked disease; however, skewed X-inactivation does not seem to explain all cases of manifesting carriers (Migeon, 2020). See Figure 2D for illustration.
Within IEI, this phenomenon is best described in CYBB-related chronic granulomatous disease (CGD). CYBB (gp91phox) encodes a major protein of the NADPH oxidase complex, which plays a critical role in the destruction of pathogens by phagocytes. CYBB-related CGD is characterized by recurrent life-threatening bacterial and fungal infections. In a study of 93 CGD carriers, skewed X-inactivation was associated with infection risk. Interestingly, the carrier state itself, regardless of the balance of X-inactivation, was associated with autoimmunity (Marciano et al., 2018). Smaller cohort studies and case reports replicate this finding (Rupec et al., 2000; Cale et al., 2007; López-Hernández et al., 2019). It is worth noting that female carriers of CYBB-related CGD can exceptionally develop manifestations of CGD, as described in a recent case report of successful HSCT for a female carrier with severe life-threatening, late-onset granulomatous colitis and recurrent lung infections (Trevisan et al., 2022).
Concurrent with the discovery of the mechanisms governing random X-inactivation in the 1960s, random monoallelic expression was described in the autosomal genes encoding antigen receptors in T and B lymphocytes (Jack and Du Pasquier, 2019). For each lymphocyte, only one of the two alleles of the antigen receptor is expressed. To accomplish this, somatic V(D)J rearrangement is completed first on one of the two alleles present. Additional rearrangement is inhibited when a functional receptor is expressed on the surface of the cell (Rada and Ferguson-Smith, 2002; Jack and Du Pasquier, 2019). In subsequent decades, other autosomal genes were found to be subject to random monoallelic expression including, interleukin genes (i.e., IL2, IL3, IL4, IL5, IL10, IL13), and NK cell receptor genes (i.e., KIR genes) (Kelly and Locksley, 2000; Chan et al., 2003; Calado et al., 2006). Further work identified widespread random monoallelic expression affecting more than 5% of assessed genes (Gimelbrant et al., 2007; Chess, 2016). This random monoallelic expression increases the potential for cellular diversity within tissues because it creates up to three distinct cellular states (i.e., cells expressing a gene biallelically or from either of the two alleles) (Chess, 2016).
There are limited examples linking autosomal monoallelic expression to clinical disease in immunology. A recent case report describes a patient with early-onset multi-organ immune dysregulation resulting from a somatic pathogenic variant in JAK1. JAK1 (Janus kinase 1) is a critical player in immune signal transduction. In the reported case, the authors demonstrated that JAK1 was monoallelically expressed (i.e., only the mutated allele was expressed) across multiple immune cell subsets and not in other tissue cells as control (Gruber et al., 2020). When the cellular diversity created by allelic bias is considered in combination with the critical developmental windows in immune maturation, it is possible that such monoallelic expression of immune genes in hematopoietic or non-hematopoietic cells contributes to phenotypic variation in autosomal dominant diseases and may prove to be an important point of focus for future studies of penetrance (Gui et al., 2017).
2.1.4 Multi-locus genetic variation
A final genetic contribution for intra- and interfamilial phenotypic variation for IEI is multi-locus genetic variation. In its simplest form, phenotypic outliers can occur when an individual has multiple genetic disorders. Studies of “dual diagnosis” show that it is more common for co-occurring genetic disorders to affect distinct rather than overlapping organ systems (Posey et al., 2017; Similuk et al., 2022). The co-occurrence of two or more IEIs in one individual is a statistically rare event, although several such cases have been reported (Chinn et al., 2017; Rae et al., 2018). Moderate penetrance alleles maintained at slightly higher population frequencies, such as pathogenic TNFRSF13B variants, have been reported in multiple families (ThaventhIran et al., 2020; Hargreaves et al., 2022; Similuk et al., 2022). Such cases are suggested to be more likely to manifest evidence of autoimmunity, lymphoproliferation, and/or common variable immunodeficiency over time than would otherwise be seen (Salzer and Grimbacher, 2021). See Figure 2E for illustration.
In addition, increasing evidence of mitigated phenotypes in carriers of recessive Mendelian disorders requires us to reconsider what we understood as settled science. A few early studies provided clues to this phenomenon, despite challenges with statistical power and ascertainment bias (Chillón et al., 1995; Girodon et al., 1997). Subsequent SNP-array-based genotyping studies were limited in their sensitivity for detecting rare disease variants for investigation. Only with the current generation of population biobank cohorts can this phenomenon begin to be clarified among a broader set of phenotypes and a wider range of recessive disorders (Barton et al., 2022). Although not an IEI, CFTR-related cystic fibrosis provides a phenotypically-relevant example where CFTR carriers have been shown in multiple studies to be at increased risk of cystic fibrosis-associated phenotypes including asthma, aspergillosis, and bronchiectasis (Çolak et al., 2020; Miller et al., 2020; Barton et al., 2022). For other phenotypically relevant examples: heterozygous carriers of a disease variant in ABCA3 (associated with autosomal recessive interstitial lung disease) show evidence of decreased lung function and heterozygous carriers of a missense variant in TG (associated with autosomal recessive thyroid dyshormonogenesis) show an increased hypothyroidism risk (Barton et al., 2022). Thus, carrier status, long relegated to only being relevant for reproduction, may be relevant in certain circumstances to both complex disorders and individual susceptibility over time. It is yet to be determined the extent to which such information will be integrated into clinical practice, along with the most effective ways to communicate complex, multi-locus risk information. Nonetheless, it is likely that the coming years will further illuminate the spectrum of variant effects in immune defects as demonstrated for TNFRSF13B (TACI) or MBL2 encoding mannose-binding lectin as disease-modifying variants of relevance, including mitigated phenotypes among carriers of recessive gene variants, along with the relevant mechanisms of action.
Digenic inheritance is often described as the simplest form of complex inheritance, although it classically requires the two implicated genes to have interacting gene products, leading to few examples. Within IEI, the best-supported example involves 6 autoinflammatory patients from 4 families with heterozygous pathogenic missense variants in an inducible proteasome subunit (PSMB8 or PSMB9) and a heterozygous pathogenic variant in a constitutive proteasome subunit (PSMB4 or PSMA3). The proteins encoded by these genes form a proteasome complex, which helps maintain protein homeostasis and control inflammation (Torrelo, 2017). For these reported patients, functional evaluation demonstrated proteasome dysfunction, increased type I interferon production, and, subsequently, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (Brehm et al., 2015).
Considerably more complex and without current clinical applications in immunology, polygenic risk modeling is another rapidly evolving technology that may explain aspects of inter- and intra-familial phenotypic variation within IEI. Notably, atopy, autoimmunity, and autoinflammation are highly heritable conditions with partially overlapping susceptibility loci, despite potentially counteracting immune mechanisms (Kreiner et al., 2017). For example, in a cohort of 1540 patients with systemic lupus erythematosus higher polygenic risk scores significantly correlated with earlier age of diagnosis (Dominguez et al., 2021). Additionally, in a large, multi-ethnic cohort analyses identified population-specific effects of common and rare variants associated with inflammatory bowel disease (Gettler et al., 2021). In the future, it may be possible to stratify IEI penetrance via common variant contributions, often in genes known to also explain IEI. Additional controversy exists regarding the extent to which polygenic risk models will be able to capture a meaningful proportion of variation in risk or translate clinically from the cohort to an individual patient (Wand et al., 2021). The integration of variant effects across the frequency spectrum, and in a manner that does not assume identical genetic architecture across ancestral populations, is a rapidly evolving area of investigation.
2.2 Environment
Our discussion of environmental influences on phenotypic variation in IEI will begin with a review of the hygiene hypothesis, a classic body of work in disease epidemiology. This will be followed by contemporary discussions of microbial dysbiosis, hormonal influences, and specific pathogens as triggers.
2.2.1 Early life infectious burden
Much of our understanding of the environmental factors shaping risk for immune-mediated disease can be broadly summarized in the hygiene hypothesis, which postulates that the overall decreased infectious burden over the last several decades has contributed directly to a rise in atopic and autoimmune disease (Garn et al., 2021). It should be emphasized that despite its misnomer, socioeconomic development is more relevant to these trends than one’s hygiene habits. This pattern was first described in the context of atopy more than 30 years ago, has been well supported by epidemiological and experimental data, and has been remarkably adaptable to emerging science (Bach, 2018). While it seems clear that early life infectious burden has a protective effect against immune-mediated diseases across the lifespan, the exact infections, disease-specific developmental windows, and underlying mechanisms are multiple, complex, and not well understood (Bach, 2002; Bach, 2018; Bach, 2020; Garn et al., 2021). The extent to which these patterns influence individuals or families with IEI is unknown.
Other environmental exposures have also been linked to the rise in atopic and autoimmune disorders in the industrialized world, such as smoking, chemical exposures, and air pollution, which likely affect risk via multiple mechanisms including inflammation (Costenbader and Karlson, 2006; Kantor and Silverberg, 2017; Zhao et al., 2019; Luschkova et al., 2021; Woo et al., 2022).
2.2.2 Microbial dysbiosis
The microbiome’s dynamic, bidirectional role in influencing immune system development and function and wide-ranging disease pathogenesis has also garnered great interest. The genomics advances of the 21st century have made it possible to characterize microbiome diversity and composition to an extent not previously possible. Westernized diets (high in fat and simple sugars), broad-spectrum antibiotic use, and other early-life exposures (largely encompassed by the hygiene hypothesis) are all implicated in microbial dysbiosis (Dethlefsen and Relman, 2011; Maurice et al., 2013; Modi et al., 2014; Sonnenburg et al., 2016; Gilbert et al., 2018; Hills et al., 2019). Microbiome composition is uniquely adapted for local body sites and is best understood in the gut. Gut dysbiosis is typically characterized by a decrease in microbiota diversity, with a decrease in specific commensals and an increase in harmful bacteria. While dysbiosis is associated with numerous immune disorders including inflammatory bowel disease, type 1 diabetes, multiple sclerosis, lupus, atopic dermatitis, and allergy, the extent to which dysbiosis plays a role in triggering disease, disease progression, and/or is, in fact, a consequence of the disease itself is unclear to date (Kostic et al., 2015; Bach, 2018; Liu et al., 2022; Xiang et al., 2022). While we are only beginning to understand the complexities involved, current research seems to be pivoting from descriptive and early experimental studies to a more mechanistic and translational emphasis (Gilbert et al., 2018). There is substantial enthusiasm for the therapeutic potential of modulating the microbiome. As a first-in-kind therapy, the FDA approved microbiome transfer for recurrent and refractory Clostridium difficile infection. Many other interventions are under investigation (Hendricks et al., 2019; Waller et al., 2022).
Multiple studies have sought to characterize the microbiome in individuals with IEI and related animal models, as either the cause and/or result of immune dysregulation. A unifying finding has been the depletion of intestinal commensals (Pellicciotta et al., 2019). Despite tremendous challenges, investigators have been eager to translate findings from common, complex disease into the setting of IEI, as well as contribute lessons from the in-depth study of rare disease to common, complex diseases (Pellicciotta et al., 2019; Al-Nesf et al., 2021).
2.2.3 Internal hormonal milieu
Another fundamental vector shaping immunity and disease risk is sex. Differences in male and female immunity are well documented, but not well understood. Generally, females have a more robust immune response as evidenced in response to vaccination, sepsis, and certain other infections, including COVID-19 (Flanagan et al., 2017; Patin et al., 2018; Gupta et al., 2020). Inversely, female sex is a risk factor for multiple autoimmune and inflammatory disorders (Ngo et al., 2014; Billi et al., 2019; Natri et al., 2019; Ortona et al., 2019). Analogous to other environmental factors discussed in this review, sex and gender undoubtedly affect immunity and disease risk through multiple mechanisms. Some mechanisms are likely to be hormonally driven, as has been supported by comparisons in innate immunity amongst healthy controls, subjects with Klinefelter syndrome (XXY) (Holzelova et al., 2004), and pre-pubescent children (Gupta et al., 2020); other mechanisms may be directly linked to genetic factors differentiating males and females, such as those related to X-inactivation; and finally, other effects may be entangled in social lifestyle differences across genders (Billi et al., 2019).
It is not clear if or how the sex- or gender-related trends described above manifest across IEIs or within specific IEIs (Sheikhbahaei et al., 2016; Abolhassani et al., 2020), although the question is complex and the possible mechanisms of effect would likely differentially impact specific IEIs. More work in this area is needed.
2.2.4 Pathogens as triggers
A final and more concrete discussion point amongst environmental factors shaping phenotype variation in IE is the role of pathogens as disease triggers. As a foundational concept, specific viral exposures, most notably cytomegalovirus (CMV), are well appreciated for their broad influence on immune variation as compellingly demonstrated by studies of healthy controls and monozygotic twins (Kuijpers et al., 2003; Brodin et al., 2015; Patin et al., 2018). However, in the setting of an IEI, a viral infection such as CMV, which is typically latent, can profoundly catalyze disease processes with fatal consequences. CMV may be encountered early in life being a frequent viral infection transmitted shortly after birth by breastfeeding (Ruffner et al., 2017; Drutman et al., 2020; Chong-Neto et al., 2022). Also, Epstein-Barr virus (EBV) provides a similar example. Like CMV, EBV is a herpesvirus that latently infects much of the global population and infects predominantly B cells. Multiple IEIs exhibit a striking and specific susceptibility to EBV-related diseases such as mononucleosis, lymphoproliferative disease, hemophagocytic lymphohistiocytosis, and/or EBV+ lymphoma. Notably, individuals may be completely asymptomatic or have a mild clinical course before EBV infection triggers a dramatic progression of disease; examples include disorders related to the genes SH2D1A, XIAP, ITK, MAGT1, CD27, CD70, among others (Tangye et al., 2017; Tangye and Latour, 2020). Other examples of specific viral susceptibility include autosomal recessive OX40-deficiency and human herpes virus 8, leading to exceedingly rare childhood-onset Kaposi sarcoma, as well as, possibly, the recently described TLR7-deficiency leading to aberrant recognition of ssRNA viruses, decreased interferon response to SARS-CoV-2 infection, and increased risk of life-threatening COVID pneumonia (Byun et al., 2013; Asano et al., 2021).
Even for IEIs which are not defined by a severe susceptibility to viral disease, CMV or EBV infection may significantly change the disease course by inducing lymphoproliferation or other complications, as has been observed in RAG-deficiencies, CTLA4-haploinsufficiency, activating PIK3∂ syndrome, and other IEIs (Schwab et al., 2018; Edwards et al., 2019; Bosticardo et al., 2021; Thouenon et al., 2021). Additionally, it was relatively recently discovered that the persistence of vaccine-derived rubella virus appears to be a crucial, triggering factor for many cutaneous and visceral granulomas in individuals with IEI, a phenotype whose only effective management to date is HSCT (Perelygina et al., 2020; Perelygina et al., 2021; Notarangelo, 2022).
There are numerous other examples of pathogens as dramatic triggers of IEI phenotypes, including in otherwise healthy-appearing individuals. BCG vaccination of individuals with MSMD offers a particularly discrete example involving a bacterial pathogen (Fekrvand et al., 2020). Live attenuated virus vaccinations have also been known to reveal previously undiagnosed IEI, with particularly severe consequences in individuals with SCID, as well as individuals with innate immune defects (e.g., STAT2, IRF9) (Gothe et al., 2021). Likewise, CARD9 deficiency underlies invasive fungal infections, primarily reported in individuals who were apparently healthy before the precipitation of their severe, and in some cases, fatal infections (Drewniak et al., 2013; Rieber et al., 2016; Corvilain et al., 2018; Imanaka et al., 2021). Within the broader epidemiological trends increasing the prevalence of common, complex immune disorders across industrialized populations, exposure to specific pathogens remains a serious risk factor for triggering disease in individuals with IEI.
2.3 Time
In the frequently referenced paradigm of gene-environment interplay, time is an often missed “third factor,” which we now highlight (Boyce et al., 2020). As a straightforward interpretation of time’s role in phenotypic variation, one can consider the longitudinal unfolding of a disease’s natural history for a given individual over time. While many IEIs have a fundamentally progressive disease course, exceptions do exist: MYD88 and IRAK4 deficiencies cause early-onset life-threatening infection with a small number of bacterial species, the severity and frequency of which lessen over time, even with decreased reliance on prophylactic antibiotics (von Bernuth et al., 2012). A closely related consideration is time’s role across an individual’s lifecycle: implicating the dynamic, programmed modulation of immune function as an individual progresses from the neonatal period to adulthood, including possible pregnancy, and into later life (Simon et al., 2015; Patin et al., 2018; Natri et al., 2019). Critical developmental windows exist for immune system maturation, particularly in the neonatal period, the disruption of which may have lifelong effects (Al Nabhani and Eberl, 2020). From an alternative and similarly concrete perspective, timing plays a central role in some of the mechanisms discussed above, such as somatic variation where the timing of the somatic event(s) largely determines key characteristics such as somatic variant allele fraction and distribution of the somatic variant across the soma, or even acceleration or amelioration of disease.
The timing of exposure to pathogens and subsequent intervention are deeply consequential for a given individual, as described in the case report of an asymptomatic, EBV-negative young adult male with XIAP deficiency (identified by family history) who required extensive treatment for depression in adjusting to living at-risk of a life-threatening disorder with a ubiquitous viral trigger (Durkee-Shock et al., 2021). These issues of timing are not routinely studied systematically in clinical immunology, suggesting avenues for future research, as well as relevant clinical counseling issues (Brodin, 2020).
3 Discussion
In this review, we have provided an updated summary of key concepts in genetic and environmental contributions to phenotypic variation within IEI. For genetics, this has included differential variant impact, somatic events, allelic bias, and multi-locus genetic contributions. From the environmental perspective, we’ve covered the epidemiological impact of decreased infectious burden over recent generations, microbial dysbiosis, hormonal influences, and pathogens as critical triggers for individuals with IEI. Our review may be limited by the inadvertent omission of relevant literature or missed connections. Although the above factors have been reviewed separately, their impact on phenotype is intrinsically one of reciprocal interplay and is dynamic over time. Reductionist approaches have successfully generated detailed insight into many of the building blocks of biological systems and can fail to capture this dynamic, complex nature.
Across science, we tend to overestimate innovation in the short term but underestimate its impact in the long term. The recent history of genetics is exemplary in this regard: the decade following the completion of the human genome project saw minimal translation into clinical care and certainly not a transformation of medicine. And yet, the early genetic research of this century gave rise to innovations in methods, new applications, and fundamental learning in genome biology. This same period has seen the identification of almost 500 causes of IEI, as well as major advances in our ability to characterize somatic events, the microbiome, and genotypes across large populations. Although the progress has not been linear, these developments have accumulated into an enhanced ability to diagnose and treat IEI, in some cases with effective precision therapy or definitive interventions like HSCT. And yet, anyone who has sat with a family affected by IEI as they grapple with the uncertain prognosis of their disease knows the limits of this progress. The substantial challenge looking forward is to organize information in such a way that accommodates the genotypic and phenotypic heterogeneity within IEI and integrates this information to arrive at a more comprehensive and accurate understanding of how the immune system operates in health and disease (Germain et al., 2011; Germain, 2018).
Author contributions
MS and TK provided joint contributions to the conception and design of the work, drafting the work and revising it critically for important intellectual content. Both authors also provide approval for publication of the content and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported with funds from the NIAID Division of Intramural Research.
Acknowledgments
The authors would like to acknowledge Steven Holland for his involvement and support, as well as Jia Yan for her critical review of early versions of this text.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1183142/full#supplementary-material
References
Abolhassani, H., Azizi, G., Sharifi, L., Yazdani, R., Mohsenzadegan, M., Delavari, S., et al. (2020). Global systematic review of primary immunodeficiency registries. Expert Rev. Clin. Immunol. 16 (7), 717–732. doi: 10.1080/1744666X.2020.1801422
Aksentijevich, I., Schnappauf, O. (2021). Molecular mechanisms of phenotypic variability in monogenic autoinflammatory diseases. Nat. Rev. Rheumatol. 17 (7), 405–425. doi: 10.1038/s41584-021-00614-1
Al Nabhani, Z., Eberl, G. (2020). Imprinting of the immune system by the microbiota early in life. Mucosal Immunol. 13 (2), 183–189. doi: 10.1038/s41385-020-0257-y
Al-Nesf, M. A., Morgan, D., Mohamed-Ali, V. (2021). Primary immunodeficiency and the microbiome. Curr. Opin. Pediatr. 33 (6), 633–638. doi: 10.1097/MOP.0000000000001067
Aluri, J., Bach, A., Kaviany, S., Chiquetto Paracatu, L., Kitcharoensakkul, M., Walkiewicz, M. A., et al. (2021). Immunodeficiency and bone marrow failure with mosaic and germline TLR8 gain of function. Blood 137 (18), 2450–2462. doi: 10.1182/blood.2020009620
Antonarakis, S. E., Chakravarti, A., Cohen, J. C., Hardy, J. (2010). Mendelian disorders and multifactorial traits: the big divide or one for all? Nat. Rev. Genet. 11 (5), 380–384. doi: 10.1038/nrg2793
Asano, T., Boisson, B., Onodi, F., Matuozzo, D., Moncada-Velez, M., Maglorius Renkilaraj, M. R. L., et al. (2021). X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6 (62), eabl4348. doi: 10.1126/sciimmunol.abl4348
Bach, J. F. (2002). The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347 (12), 911–920. doi: 10.1056/NEJMra020100
Bach, J. F. (2018). The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat. Rev. Immunol. 18 (2), 105–120. doi: 10.1038/nri.2017.111
Bach, J. F. (2020). Revisiting the hygiene hypothesis in the context of autoimmunity. Front. Immunol. 11, 615192. doi: 10.3389/fimmu.2020.615192
Barton, A. R., Hujoel, M. L. A., Mukamel, R. E., Sherman, M. A., Loh, P. R. (2022). A spectrum of recessiveness among Mendelian disease variants in UK Biobank. Am. J. Hum. Genet 109 (7), 1298–1307. doi: 10.1016/j.ajhg.2022.05.008
Beck, D. B., Ferrada, M. A., Sikora, K. A., Ombrello, A. K., Collins, J. C., Pei, W., et al. (2020). Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 383 (27), 2628–2638. doi: 10.1056/NEJMoa2026834
Biesecker, L. G., Spinner, N. B. (2013). A genomic view of mosaicism and human disease. Nat. Rev. Genet. 14 (5), 307–320. doi: 10.1038/nrg3424
Billi, A. C., Kahlenberg, J. M., Gudjonsson, J. E. (2019). Sex bias in autoimmunity. Curr. Opin. Rheumatol. 31 (1), 53–61. doi: 10.1097/BOR.0000000000000564
Bosticardo, M., Pala, F., Notarangelo, L. D. (2021). RAG deficiencies: Recent advances in disease pathogenesis and novel therapeutic approaches. Eur. J. Immunol. 51 (5), 1028–1038. doi: 10.1002/eji.202048880
Boyce, W. T., Sokolowski, M. B., Robinson, G. E. (2020). Genes and environments, development and time. Proc. Natl. Acad. Sci. U. S. A. 117 (38), 23235–23241. doi: 10.1073/pnas.2016710117
Brehm, A., Liu, Y., Sheikh, A., Marrero, B., Omoyinmi, E., Zhou, Q., et al. (2015). Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest. 125 (11), 4196–4211. doi: 10.1172/JCI81260
Brodin, P. (2020). New approaches to the study of immune responses in humans. Hum. Genet. 139 (6–7), 795–799. doi: 10.1007/s00439-020-02129-3
Brodin, P., Jojic, V., Gao, T., Bhattacharya, S., Angel, C. J. L., Furman, D., et al. (2015). Variation in the human immune system is largely driven by non-heritable influences. Cell 160 (1–2), 37–47. doi: 10.1016/j.cell.2014.12.020
Buniello, A., MacArthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47 (D1), D1005–D1012. doi: 10.1093/nar/gky1120
Bustamante, J. (2020). Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum. Genet. 139 (6–7), 993–1000. doi: 10.1007/s00439-020-02120-y
Bustamante, J., Boisson-Dupuis, S., Abel, L., Casanova, J. L. (2014). Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 26 (6), 454–470. doi: 10.1016/j.smim.2014.09.008
Byun, M., Ma, C. S., Akçay, A., Pedergnana, V., Palendira, U., Myoung, J., et al. (2013). Inherited human OX40 deficiency underlying classic Kaposi sarcoma of childhood. J. Exp. Med. 210 (9), 1743–1759. doi: 10.1084/jem.20130592
Calado, D. P., Paixão, T., Holmberg, D., Haury, M. (2006). Stochastic monoallelic expression of IL-10 in T cells. J. Immunol. 177 (8), 5358–5364. doi: 10.4049/jimmunol.177.8.5358
Cale, C. M., Morton, L., Goldblatt, D. (2007). Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin. Exp. Immunol. 148 (1), 79–84. doi: 10.1111/j.1365-2249.2007.03321.x
Casanova, J. L., Abel, L. (2021). Lethal infectious diseases as inborn errors of immunity: toward a synthesis of the germ and genetic theories. Annu. Rev. Pathol. 16, 23–50. doi: 10.1146/annurev-pathol-031920-101429
Casanova, J. L., Abel, L. (2022). From rare disorders of immunity to common determinants of infection: Following the mechanistic thread. Cell 185 (17), 3086–3103. doi: 10.1016/j.cell.2022.07.004
Chan, H. W., Kurago, Z. B., Stewart, C. A., Wilson, M. J., Martin, M. P., Mace, B. E., et al. (2003). DNA methylation maintains allele-specific KIR gene expression in human natural killer cells. J. Exp. Med. 197 (2), 245–255. doi: 10.1084/jem.20021127
Chess, A. (2016). Monoallelic gene expression in mammals. Annu. Rev. Genet. 50, 317–327. doi: 10.1146/annurev-genet-120215-035120
Chillón, M., Casals, T., Mercier, B., Bassas, L., Lissens, W., Silber, S., et al. (1995). Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N. Engl. J. Med. 332 (22), 1475–1480. doi: 10.1056/NEJM199506013322204
Chinn, I. K., Sanders, R. P., Stray-Pedersen, A., Coban-Akdemir, Z. H., Kim, V. H. D., Dadi, H., et al. (2017). Novel combined immune deficiency and radiation sensitivity blended phenotype in an adult with biallelic variations in ZAP70 and RNF168. Front. Immunol. 8, 576. doi: 10.3389/fimmu.2017.00576
Chong, J. X., Buckingham, K. J., Jhangiani, S. N., Boehm, C., Sobreira, N., Smith, J. D., et al. (2015). The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 97 (2), 199–215. doi: 10.1016/j.ajhg.2015.06.009
Chong-Neto, H. J., Segundo, G. R. S., Rosário, N. A., IEI-CHC Team (2022). Fatal and unresponsive cytomegalovirus infection in a new homozygous FOXN1 gene variation causing nude SCID. J. Clin. Immunol. 42 (4), 859–861. doi: 10.1007/s10875-021-01192-3
Claussnitzer, M., Cho, J. H., Collins, R., Cox, N. J., Dermitzakis, E. T., Hurles, M. E., et al. (2020). A brief history of human disease genetics. Nature 577 (7789), 179–189. doi: 10.1038/s41586-019-1879-7
Çolak, Y., Nordestgaard, B. G., Afzal, S. (2020). Morbidity and mortality in carriers of the cystic fibrosis mutation CFTR Phe508del in the general population. Eur. Respir. J. 56 (3), 2000558. doi: 10.1183/13993003.00558-2020
Corvilain, E., Casanova, J. L., Puel, A. (2018). Inherited CARD9 deficiency: invasive disease caused by ascomycete fungi in previously healthy children and adults. J. Clin. Immunol. 38 (6), 656–693. doi: 10.1007/s10875-018-0539-2
Costenbader, K. H., Karlson, E. W. (2006). Cigarette smoking and autoimmune disease: what can we learn from epidemiology? Lupus 15 (11), 737–745. doi: 10.1177/0961203306069344
Desjardins, M., Arjunaraja, S., Stinson, J. R., Dorjbal, B., Sundaresan, J., Niemela, J., et al. (2018). A unique heterozygous CARD11 mutation combines pathogenic features of both gain- and loss-of-function patients in a four-generation family. Front. Immunol. 9, 2944. doi: 10.3389/fimmu.2018.02944
Dethlefsen, L., Relman, D. A. (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. U. S. A. 108 Suppl 1, 4554–4561. doi: 10.1073/pnas.1000087107
Dominguez, D., Kamphuis, S., Beyene, J., Wither, J., Harley, J. B., Blanco, I., et al. (2021). Relationship between genetic risk and age of diagnosis in systemic lupus erythematosus. J. Rheumatol. 48 (6), 852–858. doi: 10.3899/jrheum.200002
Donadieu, J., Lamant, M., Fieschi, C., de Fontbrune, F. S., Caye, A., Ouachee, M., et al. (2018). Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 103 (8), 1278–1287. doi: 10.3324/haematol.2017.181909
Dowdell, K. C., Niemela, J. E., Price, S., Davis, J., Hornung, R. L., Oliveira, J. B., et al. (2010). Somatic FAS mutations are common in patients with genetically Undefined autoimmune lymphoproliferative syndrome. Blood 115 (25), 5164–5169. doi: 10.1182/blood-2010-01-263145
Drewniak, A., Gazendam, R. P., Tool, A. T. J., van Houdt, M., Jansen, M. H., van Hamme, J. L., et al. (2013). Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121 (13), 2385–2392. doi: 10.1182/blood-2012-08-450551
Drutman, S. B., Mansouri, D., Mahdaviani, S. A., Neehus, A. L., Hum, D., Bryk, R., et al. (2020). Fatal cytomegalovirus infection in an adult with inherited NOS2 deficiency. N. Engl. J. Med. 382 (5), 437–445. doi: 10.1056/NEJMoa1910640
Durkee-Shock, J. R., Kuehn, H. S., Stoddard, J., Niemela, J. E., Sun, G., Keller, M. D., et al. (2021). Recurrent lymphadenitis in a female XIAP/BIRC4 mutation carrier with normal lyonization. J. Allergy Clin. Immunol. Pract. 9 (2), 1002–1005.e2. doi: 10.1016/j.jaip.2020.10.062
Dvorak, C. C., Haddad, E., Heimall, J., Dunn, E., Buckley, R. H., Kohn, D. B., et al. (2023). The diagnosis of severe combined immunodeficiency (SCID): The Primary Immune Deficiency Treatment Consortium (PIDTC) 2022 Definitions. J. Allergy Clin. Immunol. 151 (2), 539–546. doi: 10.1016/j.jaci.2022.10.022
Edwards, E. S. J., Bier, J., Cole, T. S., Wong, M., Hsu, P., Berglund, L. J., et al. (2019). Activating PIK3CD mutations impair human cytotoxic lymphocyte differentiation and function and EBV immunity. J. Allergy Clin. Immunol. 143 (1), 276–291.e6. doi: 10.1016/j.jaci.2018.04.030
Fekrvand, S., Yazdani, R., Olbrich, P., Gennery, A., Rosenzweig, S. D., Condino-Neto, A., et al. (2020). Primary immunodeficiency diseases and Bacillus Calmette-Guérin (BCG)-vaccine-derived complications: A systematic review. J. Allergy Clin. Immunol. Pract. 8 (4), 1371–1386. doi: 10.1016/j.jaip.2020.01.038
Ferré, E. M. N., Schmitt, M. M., Lionakis, M. S. (2021). Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Front. Pediatr. 9, 723532. doi: 10.3389/fped.2021.723532
Filipe-Santos, O., Bustamante, J., Chapgier, A., Vogt, G., de Beaucoudrey, L., Feinberg, J., et al. (2006). Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin. Immunol. 18 (6), 347–361. doi: 10.1016/j.smim.2006.07.010
Fisher, G. H., Rosenberg, F. J., Straus, S. E., Dale, J. K., Middleton, L. A., Lin, A. Y., et al. (1995). Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81 (6), 935–946. doi: 10.1016/0092-8674(95)90013-6
Flanagan, K. L., Fink, A. L., Plebanski, M., Klein, S. L. (2017). Sex and gender differences in the outcomes of vaccination over the life course. Annu. Rev. Cell Dev. Biol. 33, 577–599. doi: 10.1146/annurev-cellbio-100616-060718
Garn, H., Potaczek, D. P., Pfefferle, P. I. (2021). The hygiene hypothesis and new perspectives-current challenges meeting an old postulate. Front. Immunol. 12, 637087. doi: 10.3389/fimmu.2021.637087
Germain, R. N. (2018). Will systems biology deliver its promise and contribute to the development of new or improved vaccines? What really constitutes the study of “Systems Biology” and how might such an approach facilitate vaccine design. Cold Spring Harb. Perspect. Biol. 10 (8), a033308. doi: 10.1101/cshperspect.a033308
Germain, R. N., Meier-Schellersheim, M., Nita-Lazar, A., Fraser, I. D. C. (2011). Systems biology in immunology: a computational modeling perspective. Annu. Rev. Immunol. 29, 527–585. doi: 10.1146/annurev-immunol-030409-101317
Gettler, K., Levantovsky, R., Moscati, A., Giri, M., Wu, Y., Hsu, N. Y., et al. (2021). Common and rare variant prediction and penetrance of IBD in a large, multi-ethnic, health system-based biobank cohort. Gastroenterology 160 (5), 1546–1557. doi: 10.1053/j.gastro.2020.12.034
Gilbert, J. A., Blaser, M. J., Caporaso, J. G., Jansson, J. K., Lynch, S. V., Knight, R. (2018). Current understanding of the human microbiome. Nat. Med. 24 (4), 392–400. doi: 10.1038/nm.4517
Gimelbrant, A., Hutchinson, J. N., Thompson, B. R., Chess, A. (2007). Widespread monoallelic expression on human autosomes. Science 318 (5853), 1136–1140. doi: 10.1126/science.1148910
Girodon, E., Cazeneuve, C., Lebargy, F., Chinet, T., Costes, B., Ghanem, N., et al. (1997). CFTR gene mutations in adults with disseminated bronchiectasis. Eur. J. Hum. Genet. 5 (3), 149–155. doi: 10.1159/000484750
Goodnow, C. C. (2007). Multistep pathogenesis of autoimmune disease. Cell 130 (1), 25–35. doi: 10.1016/j.cell.2007.06.033
Göös, H., Fogarty, C. L., Sahu, B., Plagnol, V., Rajamäki, K., Nurmi, K., et al. (2019). Gain-of-function CEBPE mutation causes noncanonical autoinflammatory inflammasomopathy. J. Allergy Clin. Immunol. 144 (5), 1364–1376. doi: 10.1016/j.jaci.2019.06.003
Gothe, F., Howarth, S., Duncan, C. J., Hambleton, S. (2021). Monogenic susceptibility to live viral vaccines. Curr. Opin. Immunol. 72, 167–175. doi: 10.1016/j.coi.2021.05.006
Gruber, C., Bogunovic, D. (2020). Incomplete penetrance in primary immunodeficiency: a skeleton in the closet. Hum. Genet. 139 (6–7), 745–757. doi: 10.1007/s00439-020-02131-9
Gruber, C. N., Calis, J. J. A., Buta, S., Evrony, G., Martin, J. C., Uhl, S. A., et al. (2020). Complex autoinflammatory syndrome unveils fundamental principles of JAK1 kinase transcriptional and biochemical function. Immunity 53 (3), 672–684.e11. doi: 10.1016/j.immuni.2020.07.006
Gui, B., Slone, J., Huang, T. (2017). Perspective: is random monoallelic expression a contributor to phenotypic variability of autosomal dominant disorders? Front. Genet. 8, 191. doi: 10.3389/fgene.2017.00191
Gupta, S., Nakabo, S., Blanco, L. P., O’Neil, L. J., Wigerblad, G., Goel, R. R., et al. (2020). Sex differences in neutrophil biology modulate response to type I interferons and immunometabolism. Proc. Natl. Acad. Sci. U. S. A. 117 (28), 16481–16491. doi: 10.1073/pnas.2003603117
Hargreaves, C. E., Dhalla, F., Patel, A. M., de Oteyza, A. C. G., Bateman, E., Miller, J., et al. (2022). Resolving the polygenic aetiology of a late onset combined immune deficiency caused by NFKB1 haploinsufficiency and modified by PIK3R1 and TNFRSF13B variants. Clin. Immunol. 234, 108910. doi: 10.1016/j.clim.2021.108910
Hendricks, A. J., Mills, B. W., Shi, V. Y. (2019). Skin bacterial transplant in atopic dermatitis: Knowns, unknowns and emerging trends. J. Dermatol. Sci. 95 (2), 56–61. doi: 10.1016/j.jdermsci.2019.07.001
Hills, R. D., Pontefract, B. A., Mishcon, H. R., Black, C. A., Sutton, S. C., Theberge, C. R. (2019). Gut microbiome: profound implications for diet and disease. Nutrients 11 (7), E1613. doi: 10.3390/nu11071613
Hoffman, H. M., Broderick, L. (2017). Editorial: it just takes one: somatic mosaicism in autoinflammatory disease. Arthritis Rheumatol. 69 (2), 253–256. doi: 10.1002/art.39961
Holland, S. M., Dorman, S. E., Kwon, A., Pitha-Rowe, I. F., Frucht, D. M., Gerstberger, S. M., et al. (1998). Abnormal regulation of interferon-gamma, interleukin-12, and tumor necrosis factor-alpha in human interferon-gamma receptor 1 deficiency. J. Infect. Dis. 178 (4), 1095–1104. doi: 10.1086/515670
Holzelova, E., Vonarbourg, C., Stolzenberg, M. C., Arkwright, P. D., Selz, F., Prieur, A. M., et al. (2004). Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N. Engl. J. Med. 351 (14), 1409–1418. doi: 10.1056/NEJMoa040036
Holzinger, D., Fassl, S. K., de Jager, W., Lohse, P., Röhrig, U. F., Gattorno, M., et al. (2015). Single amino acid charge switch defines clinically distinct proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1)-associated inflammatory diseases. J. Allergy Clin. Immunol. 136 (5), 1337–1345. doi: 10.1016/j.jaci.2015.04.016
Hoshino, A., Boutboul, D., Zhang, Y., Kuehn, H. S., Hadjadj, J., Özdemir, N., et al. (2022). Gain-of-function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci. Immunol. 7 (69), eabi7160. doi: 10.1126/sciimmunol.abi7160
Hsu, A. P. (2023). Not too little, not too much: The impact of mutation types in Wiskott-Aldrich Syndrome and RAC2 patients. Clin. Exp. Immunol. 212 (2), 137–146. doi: 10.1093/cei/uxad001
Hsu, A. P., Dowdell, K. C., Davis, J., Niemela, J. E., Anderson, S. M., Shaw, P. A., et al. (2012). Autoimmune lymphoproliferative syndrome due to FAS mutations outside the signal-transducing death domain: molecular mechanisms and clinical penetrance. Genet. Med. 14 (1), 81–89. doi: 10.1038/gim.0b013e3182310b7d
Humblet-Baron, S., Sather, B., Anover, S., Becker-Herman, S., Kasprowicz, D. J., Khim, S., et al. (2007). Wiskott-Aldrich syndrome protein is required for regulatory T cell homeostasis. J. Clin. Invest. 117 (2), 407–418. doi: 10.1172/JCI29539
Imanaka, Y., Taniguchi, M., Doi, T., Tsumura, M., Nagaoka, R., Shimomura, M., et al. (2021). Inherited CARD9 Deficiency in a Child with Invasive Disease Due to Exophiala dermatitidis and Two Older but Asymptomatic Siblings. J. Clin. Immunol. 41 (5), 975–986. doi: 10.1007/s10875-021-00988-7
Jack, R., Du Pasquier, L. (2019). Evolutionary Concepts in Immunology (Cham, Switzerland: Springer).
Jevtich, K. C., Price, S., Similuk, M. N., Kulm, E. M., Yan, J., Setzer, M. R., et al. (2022). The contribution of rare copy number variants in FAS towards pathogenesis of autoimmune lymphoproliferative syndrome. Blood Adv 6 (13), 3974–3978. doi: 10.1182/bloodadvances.2021005835
Jouanguy, E., Lamhamedi-Cherradi, S., Altare, F., Fondanèche, M. C., Tuerlinckx, D., Blanche, S., et al. (1997). Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guérin infection and a sibling with clinical tuberculosis. J. Clin. Invest. 100 (11), 2658–2664. doi: 10.1172/JCI119810
Jouanguy, E., Lamhamedi-Cherradi, S., Lammas, D., Dorman, S. E., Fondanèche, M. C., Dupuis, S., et al. (1999). A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat. Genet. 21 (4), 370–378. doi: 10.1038/7701
Kantor, R., Silverberg, J. I. (2017). Environmental risk factors and their role in the management of atopic dermatitis. Expert Rev. Clin. Immunol. 13 (1), 15–26. doi: 10.1080/1744666X.2016.1212660
Kawai, T., Malech, H. L. (2009). WHIM syndrome: congenital immune deficiency disease. Curr. Opin. Hematol. 16 (1), 20–26. doi: 10.1097/MOH.0b013e32831ac557
Kelly, B. L., Locksley, R. M. (2000). Coordinate regulation of the IL-4, IL-13, and IL-5 cytokine cluster in Th2 clones revealed by allelic expression patterns. J. Immunol. 165 (6), 2982–2986. doi: 10.4049/jimmunol.165.6.2982
Kostic, A. D., Gevers, D., Siljander, H., Vatanen, T., Hyötyläinen, T., Hämäläinen, A. M., et al. (2015). The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 17 (2), 260–273. doi: 10.1016/j.chom.2015.01.001
Kreiner, E., Waage, J., Standl, M., Brix, S., Pers, T. H., Couto Alves, A., et al. (2017). Shared genetic variants suggest common pathways in allergy and autoimmune diseases. J. Allergy Clin. Immunol. 140 (3), 771–781. doi: 10.1016/j.jaci.2016.10.055
Kuehn, H. S., Caminha, I., Niemela, J. E., Rao, V. K., Davis, J., Fleisher, T. A., et al. (2011). FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J. Immunol. 186 (10), 6035–6043. doi: 10.4049/jimmunol.1100021
Kuehn, H. S., Gloude, N. J., Dimmock, D., Tokita, M., Wright, M., Rosenzweig, S. D., et al. (2021). Abnormal SCID newborn screening and spontaneous recovery associated with a novel haploinsufficiency IKZF1 mutation. J. Clin. Immunol. 41 (6), 1241–1249. doi: 10.1007/s10875-021-01035-1
Kuemmerle-Deschner, J. B., Ozen, S., Tyrrell, P. N., Kone-Paut, I., Goldbach-Mansky, R., Lachmann, H., et al. (2017). Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann. Rheum. Dis. 76 (6), 942–947. doi: 10.1136/annrheumdis-2016-209686
Kuijpers, T. W., Vossen, M. T., Gent, M. R., Davin, J. C., Roos, M. T., Wertheim-van Dillen, P. M., et al. (2003). Frequencies of circulating cytolytic, CD45RA+CD27-, CD8+ T lymphocytes depend on infection with CMV. J. Immunol. 170 (8), 4342–4348. doi: 10.4049/jimmunol.170.8.4342
Lappalainen, T., MacArthur, D. G. (2021). From variant to function in human disease genetics. Science 373 (6562), 1464–1468. doi: 10.1126/science.abi8207
Li, Z., Rotival, M., Patin, E., Michel, F., Pellegrini, S. (2020). Two common disease-associated TYK2 variants impact exon splicing and TYK2 dosage. PloS One 15 (1), e0225289. doi: 10.1371/journal.pone.0225289
Liu, Y., Du, X., Zhai, S., Tang, X., Liu, C., Li, W. (2022). Gut microbiota and atopic dermatitis in children: a scoping review. BMC Pediatr. 22 (1), 323. doi: 10.1186/s12887-022-03390-3
López-Hernández, I., Deswarte, C., Alcantara-Ortigoza, M. Á., Saez-de-Ocariz, M. D. M., Yamazaki-Nakashimada, M. A., Espinosa-Padilla, S. E., et al. (2019). Skewed X-inactivation in a female carrier with X-linked chronic granulomatous disease. Iran J. Allergy Asthma Immunol. 18 (4), 447–451. doi: 10.18502/ijaai.v18i4.1425
Luschkova, D., Zeiser, K., Ludwig, A., Traidl-Hoffmann, C. (2021). Atopic eczema is an environmental disease. Allergol. Select 5, 244–250. doi: 10.5414/ALX02258E
Ma, C. A., Xi, L., Cauff, B., DeZure, A., Freeman, A. F., Hambleton, S., et al. (2017). Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood 129 (5), 650–653. doi: 10.1182/blood-2016-09-737817
Magerus-Chatinet, A., Neven, B., Stolzenberg, M. C., Daussy, C., Arkwright, P. D., Lanzarotti, N., et al. (2011). Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J. Clin. Invest. 121 (1), 106–112. doi: 10.1172/JCI43752
Marciano, B. E., Zerbe, C. S., Falcone, E. L., Ding, L., DeRavin, S. S., Daub, J., et al. (2018). X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 141 (1), 365–371. doi: 10.1016/j.jaci.2017.04.035
Masters, S. L., Lagou, V., Jéru, I., Baker, P. J., Van Eyck, L., Parry, D. A., et al. (2016). Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 8 (332), 332ra45. doi: 10.1126/scitranslmed.aaf1471
Maurice, C. F., Haiser, H. J., Turnbaugh, P. J. (2013). Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152 (1–2), 39–50. doi: 10.1016/j.cell.2012.10.052
McDermott, D. H., Gao, J. L., Liu, Q., Siwicki, M., Martens, C., Jacobs, P., et al. (2015). Chromothriptic cure of WHIM syndrome. Cell 160 (4), 686–699. doi: 10.1016/j.cell.2015.01.014
McReynolds, L. J., Yang, Y., Yuen Wong, H., Tang, J., Zhang, Y., Mulé, M. P., et al. (2019). MDS-associated mutations in germline GATA2 mutated patients with hematologic manifestations. Leuk. Res. 76, 70–75. doi: 10.1016/j.leukres.2018.11.013
Mensa-Vilaro, A., Teresa Bosque, M., Magri, G., Honda, Y., Martínez-Banaclocha, H., Casorran-Berges, M., et al. (2016). Brief report: late-onset cryopyrin-associated periodic syndrome due to myeloid-restricted somatic NLRP3 mosaicism. Arthritis Rheumatol. 68 (12), 3035–3041. doi: 10.1002/art.39770
Migeon, B. R. (2020). X-linked diseases: susceptible females. Genet. Med. 22 (7), 1156–1174. doi: 10.1038/s41436-020-0779-4
Miller, A. C., Comellas, A. P., Hornick, D. B., Stoltz, D. A., Cavanaugh, J. E., Gerke, A. K., et al. (2020). Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. U. S. A. 117 (3), 1621–1627. doi: 10.1073/pnas.1914912117
Miyazawa, H., Wada, T. (2021). Reversion mosaicism in primary immunodeficiency diseases. Front. Immunol. 12, 783022. doi: 10.3389/fimmu.2021.783022
Modi, S. R., Collins, J. J., Relman, D. A. (2014). Antibiotics and the gut microbiota. J. Clin. Invest. 124 (10), 4212–4218. doi: 10.1172/JCI72333
Natri, H., Garcia, A. R., Buetow, K. H., Trumble, B. C., Wilson, M. A. (2019). The pregnancy pickle: evolved immune compensation due to pregnancy underlies sex differences in human diseases. Trends Genet. 35 (7), 478–488. doi: 10.1016/j.tig.2019.04.008
Ngo, S. T., Steyn, F. J., McCombe, P. A. (2014). Gender differences in autoimmune disease. Front. Neuroendocrinol. 35 (3), 347–369. doi: 10.1016/j.yfrne.2014.04.004
Nishikomori, R., Izawa, K., Kambe, N., Ohara, O., Yasumi, T. (2019). Low-frequency mosaicism in cryopyrin-associated periodic fever syndrome: mosaicism in systemic autoinflammatory diseases. Int. Immunol. 31 (10), 649–655. doi: 10.1093/intimm/dxz047
Notarangelo, L. D. (2022). Rubella virus-associated granulomas in immunocompetent adults-possible implications. JAMA Dermatol 158 (6), 611–613. doi: 10.1001/jamadermatol.2022.0055
Nussbaum, R., McInnes, R., Willard, H. (2015). Thompson & Thompson Genetics in Medicine, 8th. (Philadelphia, PA: Elsevier).
Ortona, E., Pierdominici, M., Rider, V. (2019). Editorial: sex hormones and gender differences in immune responses. Front. Immunol. 10, 1076. doi: 10.3389/fimmu.2019.01076
Ott, N., Faletti, L., Heeg, M., Andreani, V., Grimbacher, B. (2023). JAKs and STATs from a clinical perspective: loss-of-function mutations, gain-of-function mutations, and their multidimensional consequences. J. Clin. Immunol 43, 1326–1359. doi: 10.1007/s10875-023-01483-x
Patin, E., Hasan, M., Bergstedt, J., Rouilly, V., Libri, V., Urrutia, A., et al. (2018). Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat. Immunol. 19 (3), 302–314. doi: 10.1038/s41590-018-0049-7
Pellicciotta, M., Rigoni, R., Falcone, E. L., Holland, S. M., Villa, A., Cassani, B. (2019). The microbiome and immunodeficiencies: Lessons from rare diseases. J. Autoimmun. 98, 132–148. doi: 10.1016/j.jaut.2019.01.008
Perelygina, L., Faisthalab, R., Abernathy, E., Chen, M. H., Hao, L., Bercovitch, L., et al. (2021). Rubella virus infected macrophages and neutrophils define patterns of granulomatous inflammation in inborn and acquired errors of immunity. Front. Immunol. 12, 796065. doi: 10.3389/fimmu.2021.796065
Perelygina, L., Icenogle, J., Sullivan, K. E. (2020). Rubella virus-associated chronic inflammation in primary immunodeficiency diseases. Curr. Opin. Allergy Clin. Immunol. 20 (6), 574–581. doi: 10.1097/ACI.0000000000000694
Pillay, B. A., Fusaro, M., Gray, P. E., Statham, A. L., Burnett, L., Bezrodnik, L., et al. (2021). Somatic reversion of pathogenic DOCK8 variants alters lymphocyte differentiation and function to effectively cure DOCK8 deficiency. J. Clin. Invest. 131 (3), e142434, 142434. doi: 10.1172/JCI142434
Posey, J. E., Harel, T., Liu, P., Rosenfeld, J. A., James, R. A., Coban Akdemir, Z. H., et al. (2017). Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 376 (1), 21–31. doi: 10.1056/NEJMoa1516767
Posey, J. E., O’Donnell-Luria, A. H., Chong, J. X., Harel, T., Jhangiani, S. N., Coban Akdemir, Z. H., et al. (2019). Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 21 (4), 798–812. doi: 10.1038/s41436-018-0408-7
Price, S., Shaw, P. A., Seitz, A., Joshi, G., Davis, J., Niemela, J. E., et al. (2014). Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood 123 (13), 1989–1999. doi: 10.1182/blood-2013-10-535393
Rada, C., Ferguson-Smith, A. C. (2002). Epigenetics: monoallelic expression in the immune system. Curr. Biol. 12 (3), R108–R110. doi: 10.1016/S0960-9822(02)00674-7
Rae, W., Ward, D., Mattocks, C., Pengelly, R. J., Eren, E., Patel, S. V., et al. (2018). Clinical efficacy of a next-generation sequencing gene panel for primary immunodeficiency diagnostics. Clin. Genet. 93 (3), 647–655. doi: 10.1111/cge.13163
Rahbari, R., Wuster, A., Lindsay, S. J., Hardwick, R. J., Alexandrov, L. B., Turki, S. A., et al. (2016). Timing, rates and spectra of human germline mutation. Nat. Genet. 48 (2), 126–133. doi: 10.1038/ng.3469
Ranola, J. M. O., Tsai, G. J., Shirts, B. H. (2019). Exploring the effect of ascertainment bias on genetic studies that use clinical pedigrees. Eur. J. Hum. Genet. 27 (12), 1800–1807. doi: 10.1038/s41431-019-0467-5
Rieber, N., Gazendam, R. P., Freeman, A. F., Hsu, A. P., Collar, A. L., Sugui, J. A., et al. (2016). Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight 1 (17), e89890. doi: 10.1172/jci.insight.89890
Rieux-Laucat, F., Le Deist, F., Hivroz, C., Roberts, I. A., Debatin, K. M., Fischer, A., et al. (1995). Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268 (5215), 1347–1349. doi: 10.1126/science.7539157
Ruffner, M. A., Sullivan, K. E., Henrickson, S. E. (2017). Recurrent and sustained viral infections in primary immunodeficiencies. Front. Immunol. 8, 665. doi: 10.3389/fimmu.2017.00665
Rupec, R. A., Petropoulou, T., Belohradsky, B. H., Walchner, M., Liese, J. G., Plewing, G., et al. (2000). Lupus erythematosus tumidus and chronic discoid lupus erythematosus in carriers of X-linked chronic granulomatous disease. Eur. J. Dermatol. 10 (3), 184–189.
Saito, M., Fujisawa, A., Nishikomori, R., Kambe, N., Nakata-Hizume, M., Yoshimoto, M., et al. (2005). Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheumatol. 52 (11), 3579–3585. doi: 10.1002/art.21404
Salzer, U., Grimbacher, B. (2021). TACI deficiency - a complex system out of balance. Curr. Opin. Immunol. 71, 81–88. doi: 10.1016/j.coi.2021.06.004
Schwab, C., Gabrysch, A., Olbrich, P., Patiño, V., Warnatz, K., Wolff, D., et al. (2018). Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J. Allergy Clin. Immunol. 142 (6), 1932–1946. doi: 10.1016/j.jaci.2018.02.055
Sevim Bayrak, C., Stein, D., Jain, A., Chaudhary, K., Nadkarni, G. N., Van Vleck, T. T., et al. (2021). Identification of discriminative gene-level and protein-level features associated with pathogenic gain-of-function and loss-of-function variants. Am. J. Hum. Genet. 108 (12), 2301–2318. doi: 10.1016/j.ajhg.2021.10.007
Sheikhbahaei, S., Sherkat, R., Roos, D., Yaran, M., Najafi, S., Emami, A. (2016). Gene mutations responsible for primary immunodeficiency disorders: A report from the first primary immunodeficiency biobank in Iran. Allergy Asthma Clin. Immunol. 12, 62. doi: 10.1186/s13223-016-0166-5
Shvetsova, E., Sofronova, A., Monajemi, R., Gagalova, K., Draisma, H. H. M., White, S. J., et al. (2019). Skewed X-inactivation is common in the general female population. Eur. J. Hum. Genet. 27 (3), 455–465. doi: 10.1038/s41431-018-0291-3
Similuk, M. N., Yan, J., Ghosh, R., Oler, A. J., Franco, L. M., Setzer, M., et al. (2022). Clinical Exome Sequencing of 1000 Families with Complex Immune Phenotypes: Towards comprehensive genomic evaluations. J. Allergy Clin. Immunol 150 (4), 947–954. doi: 10.1016/j.jaci.2022.06.009
Simon, A. K., Hollander, G. A., McMichael, A. (2015). Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 282 (1821), 20143085. doi: 10.1098/rspb.2014.3085
Sonnenburg, E. D., Smits, S. A., Tikhonov, M., Higginbottom, S. K., Wingreen, N. S., Sonnenburg, J. L. (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529 (7585), 212–215. doi: 10.1038/nature16504
Spinner, M. A., Sanchez, L. A., Hsu, A. P., Shaw, P. A., Zerbe, C. S., Calvo, K. R., et al. (2014). GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123 (6), 809–821. doi: 10.1182/blood-2013-07-515528
Tanaka, N., Izawa, K., Saito, M. K., Sakuma, M., Oshima, K., Ohara, O., et al. (2011). High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheumatol. 63 (11), 3625–3632. doi: 10.1002/art.30512
Tangye, S. G., Al-Herz, W., Bousfiha, A., Cunningham-Rundles, C., Franco, J. L., Holland, S. M., et al. (2021). The ever-increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J. Clin. Immunol. 41 (3), 666–679. doi: 10.1007/s10875-021-00980-1
Tangye, S. G., Al-Herz, W., Bousfiha, A., Cunningham-Rundles, C., Franco, J. L., Holland, S. M., et al. (2022). Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J. Clin. Immunol 42, 1473–1507. doi: 10.1007/s10875-022-01289-3
Tangye, S. G., Latour, S. (2020). Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood 135 (9), 644–655. doi: 10.1182/blood.2019000928
Tangye, S. G., Palendira, U., Edwards, E. S. J. (2017). Human immunity against EBV-lessons from the clinic. J. Exp. Med. 214 (2), 269–283. doi: 10.1084/jem.20161846
ThaventhIran, J. E. D., Lango Allen, H., Burren, O. S., Rae, W., Greene, D., Staples, E., et al. (2020). Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 583 (7814), 90–95. doi: 10.1038/s41586-020-2265-1
Thouenon, R., Moreno-Corona, N., Poggi, L., Durandy, A., Kracker, S. (2021). Activated PI3Kinase delta syndrome-A multifaceted disease. Front. Pediatr. 9, 652405. doi: 10.3389/fped.2021.652405
Torrelo, A. (2017). CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front. Immunol. 8, 927. doi: 10.3389/fimmu.2017.00927
Trevisan, M., Kang, E. M., CGD Consortium, Tommasini, A., Confalonieri, M. (2022). Hematopoietic stem cell transplantation in late-onset X-linked chronic granulomatous disease in a female carrier. J. Clin. Immunol 42, 1396–1399. doi: 10.1007/s10875-022-01310-9
Tuke, M. A., Ruth, K. S., Wood, A. R., Beaumont, R. N., Tyrrell, J., Jones, S. E., et al. (2019). Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet. Med. 21 (4), 877–886. doi: 10.1038/s41436-018-0271-6
Van Horebeek, L., Dubois, B., Goris, A. (2019). Somatic variants: new kids on the block in human immunogenetics. Trends Genet. 35 (12), 935–947. doi: 10.1016/j.tig.2019.09.005
von Bernuth, H., Picard, C., Puel, A., Casanova, J. L. (2012). Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol. 42 (12), 3126–3135. doi: 10.1002/eji.201242683
Waller, K. M. J., Leong, R. W., Paramsothy, S. (2022). An update on fecal microbiota transplantation for the treatment of gastrointestinal diseases. J. Gastroenterol. Hepatol. 37 (2), 246–255. doi: 10.1111/jgh.15731
Wand, H., Lambert, S. A., Tamburro, C., Iacocca, M. A., O’Sullivan, J. W., Sillari, C., et al. (2021). Improving reporting standards for polygenic scores in risk prediction studies. Nature 591 (7849), 211–219. doi: 10.1038/s41586-021-03243-6
West, R. R., Calvo, K. R., Embree, L. J., Wang, W., Tuschong, L. M., Bauer, T. R., et al. (2022). ASXL1 and STAG2 are common mutations in GATA2 deficiency patients with bone marrow disease and myelodysplastic syndrome. Blood Adv. 6 (3), 793–807. doi: 10.1182/bloodadvances.2021005065
West, R. R., Hsu, A. P., Holland, S. M., Cuellar-Rodriguez, J., Hickstein, D. D. (2014). Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica 99 (2), 276–281. doi: 10.3324/haematol.2013.090217
Woo, J. M. P., Parks, C. G., Jacobsen, S., Costenbader, K. H., Bernatsky, S. (2022). The role of environmental exposures and gene-environment interactions in the etiology of systemic lupus erythematous. J. Intern. Med. 291 (6), 755–778. doi: 10.1111/joim.13448
Wright, C. F., West, B., Tuke, M., Jones, S. E., Patel, K., Laver, T. W., et al. (2019). Assessing the pathogenicity, penetrance, and expressivity of putative disease-causing variants in a population setting. Am. J. Hum. Genet. 104 (2), 275–286. doi: 10.1016/j.ajhg.2018.12.015
Xiang, S., Qu, Y., Qian, S., Wang, R., Wang, Y., Jin, Y., et al. (2022). Association between systemic lupus erythematosus and disruption of gut microbiota: a meta-analysis. Lupus Sci. Med. 9 (1), e000599. doi: 10.1136/lupus-2021-000599
Zhao, C. N., Xu, Z., Wu, G. C., Mao, Y. M., Liu, L. N., Qian-Wu, N., et al. (2019). Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 18 (6), 607–614. doi: 10.1016/j.autrev.2018.12.010
Keywords: inborn error of immunity, genetics, environment, time, penetrance, phenotypic variation
Citation: Similuk M and Kuijpers T (2023) Nature and nurture: understanding phenotypic variation in inborn errors of immunity. Front. Cell. Infect. Microbiol. 13:1183142. doi: 10.3389/fcimb.2023.1183142
Received: 09 March 2023; Accepted: 17 August 2023;
Published: 14 September 2023.
Edited by:
Mingbang Wang, South China Hospital of Shenzhen University, ChinaReviewed by:
Robert Cody Sharp, University of Florida, United StatesRamsay Fuleihan, Columbia University, United States
Copyright © 2023 Similuk and Kuijpers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Morgan Similuk, bW9yZ2FuLnNpbWlsdWtAbmloLmdvdg==