 Bárbara Carvalho Santos Dos Reis1,2,3†
Bárbara Carvalho Santos Dos Reis1,2,3† Roberta Soares Faccion1,2†
Roberta Soares Faccion1,2† Flavia Amendola Anisio de Carvalho1,3
Flavia Amendola Anisio de Carvalho1,3 Daniella Campelo Batalha Cox Moore4,5
Daniella Campelo Batalha Cox Moore4,5 Maria Celia Chaves Zuma1,2
Maria Celia Chaves Zuma1,2 Desirée Rodrigues Plaça6,7Igor Salerno Filgueiras8Dennyson Leandro Mathias Fonseca9
Desirée Rodrigues Plaça6,7Igor Salerno Filgueiras8Dennyson Leandro Mathias Fonseca9 Otavio Cabral-Marques6,8,9,10,11,12Adriana Cesar Bonomo13,14,15,16
Otavio Cabral-Marques6,8,9,10,11,12Adriana Cesar Bonomo13,14,15,16 Wilson Savino13,14,15,16Flávia Cristina de Paula Freitas17
Wilson Savino13,14,15,16Flávia Cristina de Paula Freitas17 Helisson Faoro17
Helisson Faoro17 Fabio Passetti17
Fabio Passetti17 Jaqueline Rodrigues Robaina18
Jaqueline Rodrigues Robaina18 Felipe Rezende Caino de Oliveira19Ana Paula Novaes Bellinat20Raquel de Seixas Zeitel21
Felipe Rezende Caino de Oliveira19Ana Paula Novaes Bellinat20Raquel de Seixas Zeitel21 Margarida dos Santos Salú1,2,20
Margarida dos Santos Salú1,2,20 Mariana Barros Genuíno de Oliveira18
Mariana Barros Genuíno de Oliveira18 Gustavo Rodrigues-Santos18
Gustavo Rodrigues-Santos18 Arnaldo Prata-Barbosa18,22
Arnaldo Prata-Barbosa18,22 Zilton Farias Meira de Vasconcelos2*
Zilton Farias Meira de Vasconcelos2*- 1Programa de Pós Graduação em Pesquisa Aplicada à Saúde da Criança e da Mulher, Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF), Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, RJ, Brazil
- 2Laboratório de Alta Complexidade (LACIFF), Unidade de Pesquisa Clínica, IFF, FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 3Departamento de Imunologia, IFF, FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 4Unidade de Pacientes Graves, Departamento de Pediatria, IFF, FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 5Faculdade de Medicina, Universidade Federal Fluminense, Niterói, Rio de Janeiro, RJ, Brazil
- 6Departamento de Análises Clínicas e Toxicológicas, Faculdade de Ciências Farmacêuticas (FCF), Universidade de São Paulo (USP), São Paulo, SP, Brazil
- 7Programa de Pós-Graduação em Farmácia (Fisiopatologia e Toxicologia), FCF, USP, São Paulo, SP, Brazil
- 8Departamento de Imunologia, Instituto de Ciências Biomédicas (ICB), USP, São Paulo, SP, Brazil
- 9Programa Interunidades de Pós-graduação em Bioinformática, Instituto de Matemática e Estatística (IME), USP, São Paulo, SP, Brazil
- 10Network of Immunity in Infection, Malignancy, and Autoimmunity (NIIMA), Universal Scientific Education and Research Network (USERN), São Paulo, Brazil
- 11Department of Medicine, Division of Molecular Medicine, University of São Paulo School of Medicine, São Paulo, SP, Brazil
- 12Laboratory of Medical Investigation 29, University of São Paulo School of Medicine, São Paulo, Brazil
- 13Laboratoírio de Pesquisas Sobre o Timo, Instituto Oswaldo Cruz (IOC), FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 14Instituto National de Ciencia e Tecnologia em Neuroimunomodulação (INCT/NIM), IOC, FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 15Rede FAPERJ de Pesquisa em Neuroinflamação, IOC, FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 16Rede INOVA-IOC em Neuroimunomodulação, IOC, FIOCRUZ, Rio de Janeiro, RJ, Brazil
- 17Laboratório de Regulação da Expressão Gênica, Instituto Carlos Chagas (ICC), FIOCRUZ, Curitiba, PR, Brazil
- 18Instituto D’Or de Pesquisa e Ensino (IDOR), Rio de Janeiro, Brazil
- 19Pediatric Intensive Care Unit, Hospital Alvorada Moema, São Paulo, SP, Brazil
- 20Pediatric Intensive Care Unit, Hospital Martagão Gesteira, Salvador, BA, Brazil
- 21Pediatric Intensive Care Unit, Hospital Universitário Pedro Ernesto (HUPE), Universidade do Estado do Rio de Janeiro (UERJ), Rio de Janeiro, Brazil
- 22Pediatric Intensive Care Unit, Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG), Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil
Introduction: Despite the existing data on the Multisystem Inflammatory Syndrome in Children (MIS-C), the factors that determine these patients evolution remain elusive. Answers may lie, at least in part, in genetics. It is currently under investigation that MIS-C patients may have an underlying innate error of immunity (IEI), whether of monogenic, digenic, or even oligogenic origin.
Methods: To further investigate this hypothesis, 30 patients with MIS-C were submitted to whole exome sequencing.
Results: Analyses of genes associated with MIS-C, MIS-A, severe covid-19, and Kawasaki disease identified twenty-nine patients with rare potentially damaging variants (50 variants were identified in 38 different genes), including those previously described in IFNA21 and IFIH1 genes, new variants in genes previously described in MIS-C patients (KMT2D, CFB, and PRF1), and variants in genes newly associated to MIS-C such as APOL1, TNFRSF13B, and G6PD. In addition, gene ontology enrichment pointed to the involvement of thirteen major pathways, including complement system, hematopoiesis, immune system development, and type II interferon signaling, that were not yet reported in MIS-C.
Discussion: These data strongly indicate that different gene families may favor MIS- C development. Larger cohort studies with healthy controls and other omics approaches, such as proteomics and RNAseq, will be precious to better understanding the disease dynamics.
1 Introduction
At the end of 2019, the first reports of a new respiratory infectious disease appeared, especially severe in older adults and adults with comorbidities, which developed into a pandemic and became known worldwide as COVID-19. Children appeared to be spared, developing an asymptomatic or oligosymptomatic clinical presentation. Rapidly it was identified that the disease was caused by a coronavirus (SARS-CoV-2). Soon after the onset of the pandemic, children with fever, gastrointestinal symptoms, Kawasaki-like manifestations, acute cardiac involvement, and a hyperinflammatory state started to be reported worldwide (Dufort et al., 2020; Feldstein et al., 2020; Riphagen et al., 2020; Verdoni et al., 2020; Lima-Setta et al., 2021). These manifestations were not caused directly by the SARS-CoV-2, as the peak of reported cases occurred 2-6 weeks after the peak of COVID-19 cases (Dufort et al., 2020; Feldstein et al., 2020; Belay et al., 2021; Lima-Setta et al., 2021). Early reports drew attention to the resemblance between the new disorder and other known conditions such as Kawasaki disease (KD), toxic shock syndrome (TSS), and hemophagocytic lymphohistiocytosis (HLH). The disease, now known as multisystem inflammatory syndrome in children (MIS-C), is characterized as a post-viral complication of unknown pathophysiology associated with SARS-CoV-2 infection. The disease is rare and has also been described in newborns and adults, receiving different designations: MIS-N and MIS-A, respectively (Patel et al., 2021; Shaiba et al., 2022).
The clinical manifestations of COVID-19 vary enormously, from silent infection to lethal disease. Thus, genetic factors have been suspected to be involved in the severity of the disease, especially in young people. Studies on autoantibodies and gene loci with rare genetic variants related to type I interferons (IFNs) were reported in adult cases (Bastard and Rosen, 2020; Zhang and Bastard, 2020). Those were followed by the identification of single nucleotide polymorphisms (SNPs) that increase susceptibility to severe COVID-19 in genes such as TLR7 (van der Made et al., 2020; Asano et al., 2021; Solanich et al., 2021).
Being a rare and heterogeneous disease, it is currently under investigation if genetic factors may also be determinants in the evolution of MIS-C. It is possible that lower penetrance or hypomorphic mutations represent an underlying factor, at least in part of these cases, triggering the phenotype of a pro-inflammatory insult, although not capable of doing so per se (Schulert and Cron, 2020).
Here we describe rare genetic variants identified in a multicenter Brazilian cohort of 30 MIS-C patients and signaling pathways associated with these genes. The data reported herein strongly indicate that different gene families may favor MIS-C development.
2 Materials and methods
2.1 Design, settings, and participants
This cross-sectional analysis was conducted with 30 pediatric patients from eight institutions – six private and two public – from three states in Brazil.
All subjects who met the following CDC case definition for MIS-C (00432, H.A, 2021) were included: 1) fever >38.0 °C for ≥24h (objective or subjective); 2) with multisystem (>2) organ involvement (cardiac, renal, respiratory, hematologic, gastrointestinal, dermatologic or neurological); 3) laboratory evidence of inflammation, including, but not limited to, one or more of the following: high values of C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), fibrinogen, procalcitonin, d-dimer, ferritin, lactic acid dehydrogenase (LDH), or interleukin 6 (IL-6); elevated neutrophils, reduced lymphocytes, and low albumin; 4) no plausible alternative diagnosis; 5) current or recent SARS-CoV-2 infection diagnosed by a positive reverse transcription followed by quantitative polymerase chain reaction (RT-qPCR) or positive serological tests (IgM, IgG or IgA), or exposure to a suspected or confirmed COVID-19 case within the four weeks prior to the onset of symptoms.
The analyses included clinical and laboratory data collected from patients with disease onset from May 2020 to January 2022. Patients were coded chronologically according to their entrance into the study. All recruited subjects underwent whole exome sequencing, and there were no losses due to poor sample quality or refusal to participate.
2.2 Data collection
In cases where the patient had already been discharged from the hospital, the patient and his/her guardian were contacted and invited to participate in the research. When the patient was still hospitalized at the moment of recruitment, during hospitalization, the patient and his/her guardian were invited to participate in the research. In both cases, an informed consent form was presented and signed, followed by blood sample collection that was sent for DNA extraction and whole exome sequencing (WES). For each subject, a medical record review from the intensive care unit was also performed to fill out a specific RedCap electronic form (Supplementary Table 1).
2.3 Whole exome sequencing and analysis of genetic variants
2.3.1 Genomic DNA preparation
Peripheral blood was collected from all patients in heparin tubes, and genomic DNA was extracted from patients via peripheral blood leukocytes using the PureLink® Genomic DNA mini kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The DNA concentration in the samples was assessed by fluorimetry using the Invitrogen Qubit® 4 fluorimeter (Thermo Fisher Scientific). Samples were stored in a biorepository at -80°C until submitted to WES.
2.3.2 Library preparation and sequencing
The DNA libraries were prepared with 50 ng of DNA. Initially an enzymatic fragmentation of the material was performed in portions with an average of 150 bp, subsequently analyzed through the Bioanalyzer software (Agilent). DNA libraries were prepared using Illumina Exome Panel (45Mb) and sequenced on the NextSeq2000 platform to generate 100 bp paired-end reads (2 x 100 bp) at the sequencing facility Centro de Genomica Funcional (ESALQ/USP, Piracicaba, Brazil).
2.3.3 Variant calling and analysis
Reads were processed using the nf-core pipeline sarek aligned to the human reference genome GRCh37 (Garcia et al., 2020). Variants were filtered using panels with genes related to different diseases built from literature data. Since the pathophysiology of MIS-C is still elusive, panels with genes related to severe COVID-19 (van der Made et al., 2020; Zhang and Bastard, 2020; Asano et al., 2021; Solanich et al., 2021), MIS-C (Lee et al., 2020; Chou et al., 2021; Abolhassani et al., 2022b; Abuhammour et al., 2022; Vagrecha et al., 2022), MIS-A (Ronit et al., 2021), and KD (Burgner et al., 2009; Tsai et al., 2011; Lee et al., 2012; Onouchi et al., 2012) were applied, in addition to genes related to inborn errors of immunity (IEI). For this, we used the gene list from the 2021 update of the International Union of Immunological Societies Expert Committee (IUIS) for Human Inborn Errors of Immunity (Bousfiha et al., 2020; Tangye et al., 2021). Gene panels are depicted in Supplementary Table 2.
During analysis of candidate variants (CV), variants with allelic frequency ≤ 5% were selected using the Exome Aggregation Consortium database (ExAC), 1000 Genomes Project database, gnomAD (aggregated), and ABraOM, a Brazilian online mutation archive (http://abraom.ib.usp.br/). To evaluate the pathogenicity of the variants, thirteen predictors were considered, according to the following bioinformatics tools: CADD, BayesDel_addAF, DANN, DEOGEN2, EIGEN, FATHMM-MKL, LIST-S2, M-CAP, MutationAssessor, MutationTaster, SIFT, Polyphen-2, and PrimateAI. Respective websites are seen in Supplementary Table 3. Rare variants classified as damaging/deleterious in at least one of the in silico predictors were selected. The clinical relevance of the variants was evaluated using ClinVar, Polymorphism database (dbSNP), and Human Gene Mutation Database (HGMD). The results are presented in the tables below.
2.3.4 Functional enrichment, pathway, and protein-protein interaction analyses
To identify the functional relationships between genes with CV and the biological processes (BP) that they are involved in, gene ontology (GO) pathway enrichment and protein-protein interaction (PPI) network analysis were performed in String (Szklarczyk et al., 2021). First, network nodes were plotted with the identified genes. Then, a rough estimation of possibly disrupted signaling pathways was obtained by applying an interaction score of medium confidence (0.4). Following the unsupervised analysis, supervised clustering of relevant pathways was performed, and Circos graphs were generated in the circlize R package in R version 4.0.5 (Gu et al., 2014).
2.3.5 Ethical aspects
The study was submitted to the Internal Review Board (IRB) of Instituto D’Or de Pesquisa e Ensino - IDOR (Proponent Institution) under CAAE n° 30272920.0.1001.5249 and was approved. Each participating center evaluated and approved the project, through its own IRB or the associated IRB.
3 Results
3.1 Demographic, clinical, and laboratory data
Thirty patients were included in the study. Ages ranged from 0 to 17 years. The median age of patients was 6 (Q1-Q3 = 3-8.75) years; 63.3% of the patients were female; 58.3% were white; and only 10.0% had comorbidities – asthma was the most common (6.6%) (Table 1).
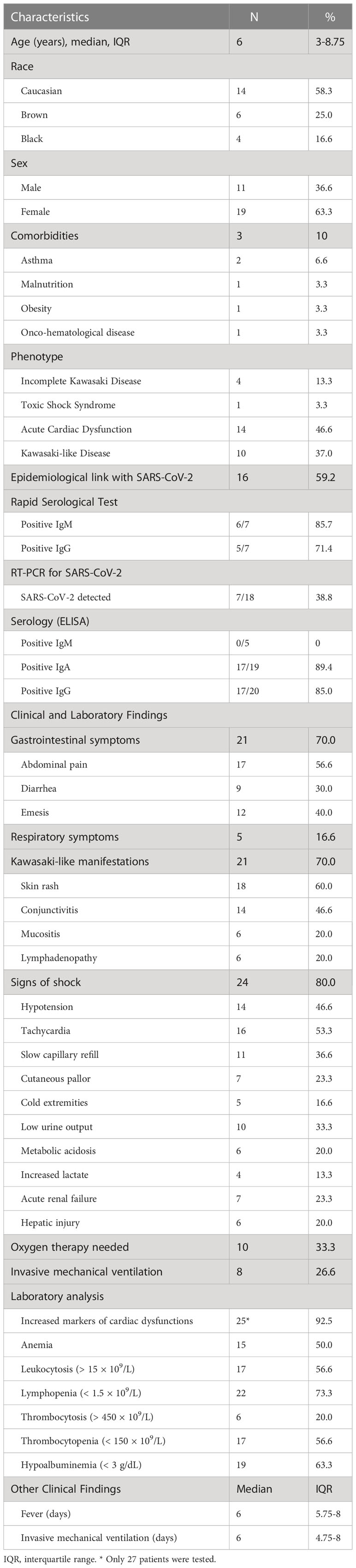
Table 1 Demographic, clinical and laboratory data of the MIS-C patients.
The median duration of fever was six days (Q1-Q3 = 5.75-8), and the most frequent symptoms were skin rash (60.0%), followed by abdominal pain (56.6%), tachycardia (53.3%), conjunctivitis and hypotension (both present in 46.6%). Signs of shock were described in 80.0% of patients, gastrointestinal symptoms in 70.0%, and Kawasaki-like symptoms in almost 70.0%. Most patients had acute cardiac dysfunction (46.6%), followed by the Kawasaki-like phenotype (37.0%) (Table 1). None of the patients reported prior history that might suggest an IEI, autoimmune, or autoinflammatory disease.
A history of contact with a suspected case of COVID-19 was identified in 59.2% of cases. Most individuals with available serologic testing results had positive IgA and IgG titers for SARS-CoV-2 (89.4% and 85.0% of patients, respectively). In addition, a n RT-PCR-detectable SARS-CoV-2 viral load was identified in 38.8% of the patients (Table 1). The most common SARS-CoV-2 variant of concern (VOC) at the time of diagnosis of each case is depicted in Supplementary Table 4.
3.2 WES analysis
A total of 50 CVs in 38 genes were identified in 29 patients (96.6%). Thirty-three genes were comprised in the IEI panel, three in the KD panel, one in the COVID-19 panel, and seven were part of the MIS-C panel. Six genes (15.7%) identified were in more than one gene panel.
The most frequently altered gene was HLA-A. Nine of the 30 patients had multiple HLA-A variants, corroborating the hypothesis that MIS-C encompasses an autoimmune basis. The same G6PD and two different PFR1 CV were detected in five children. The same APOL1 variant, two different CFB variants, and the same FCN3 CV were detected in three children. Two different CFTR, a single JAK3, a single KMT2D, a single PMS2, a single TNFRSF13B, and a single TP53, were detected in two patients. The other CV were detected in only one patient.
One patient harbored CV in six of the evaluated genes (P20), four patients in 4 genes (P11, P19, P21, and P26), and five patients in 3 genes (P1, P5, P12, P13, and P24). In the other patients, only one or two CVs were detected, except in P30, in whom no CV was detected. These results are summarized in Table 2 and Supplementary Table 5.
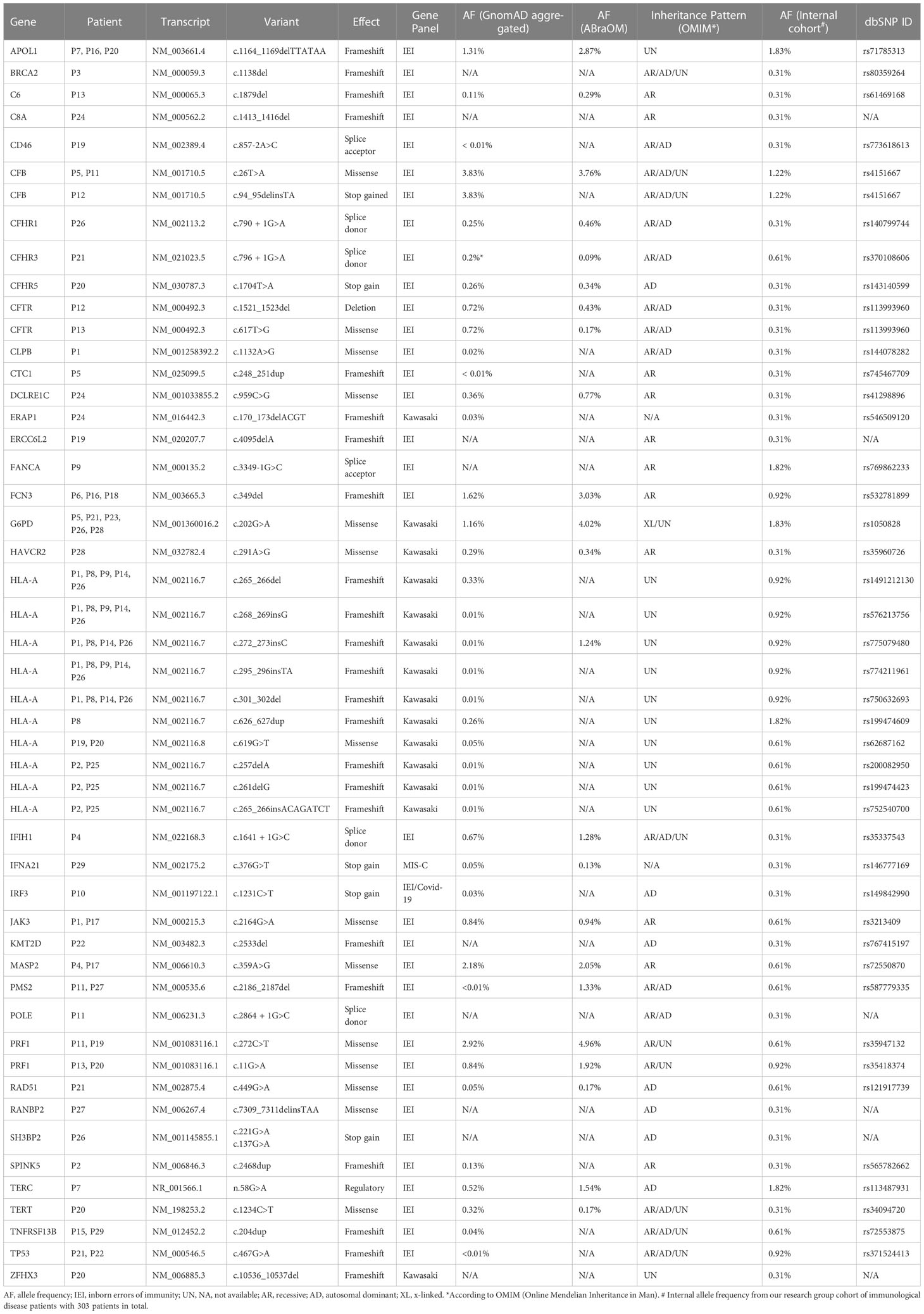
Table 2 Genetic pathogenic and likely pathogenic variants identified in immune-related genes.
Regarding possible haplotypes, P11 and P12 both had variants in CFB and PRF1. P12 and P13 both had variants in CTRF and PRF1. All five patients with PFR1 variants also harbored variants in at least one complement system gene associated with an IEI. We were unable to identify any association between CV profile and most frequent VOC at the time of MISC diagnosis and between patient clinical phenotype (Supplementary Tables 4, 5).
3.3 Gene ontology and biological process analyses
To identify relationships between genes that could indicate possible signaling pathways disruptions in MIS-C, we performed a gene network analysis of CV-bearing genes in String (Szklarczyk et al., 2021). Protein-Protein Interaction (PPI) Networks based on experimental evidence and expert-curated databases are displayed in Figure 1A. As observed in the left panel, our analyses identified CV in 38 genes, with 37 nodes representing proteins and 73 edges representing confidence level (expected number of edges = 19), with a PPI enrichment p-value <1.0−16. These genes can be roughly split into three functional categories as genes that encode complement system proteins (red), genes that encode proteins in “response to virus” pathways (green), including genes related to the interferon pathway (yellow), and genes that regulate DNA processes (blue). Importantly, these DNA processes are mostly linked to hematopoiesis, and immune system activation, mainly lymphocytes.
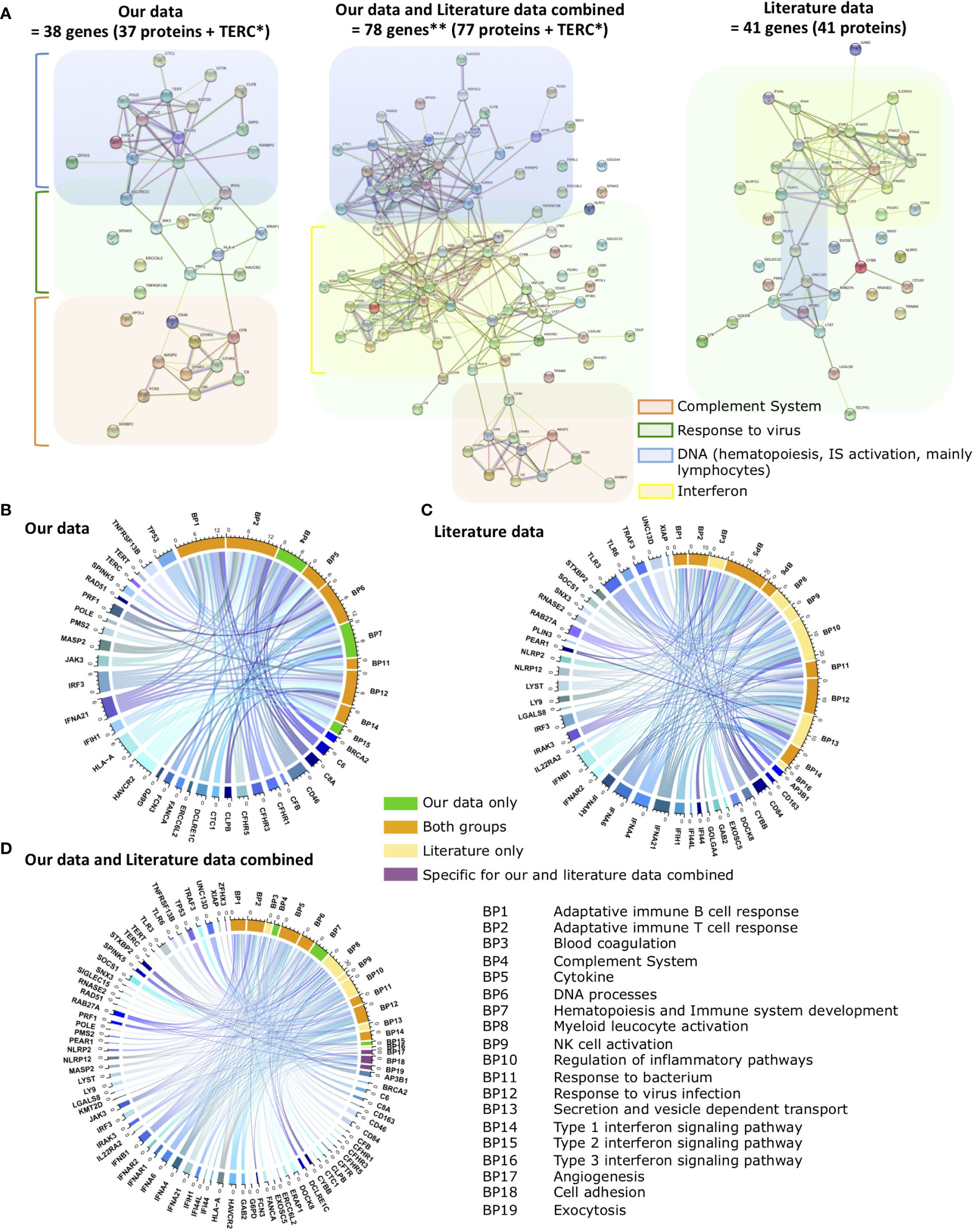
Figure 1 Functional relationships between genes with CV (candidate variants) in MISC patients. (A) Protein-Protein Interaction (PPI) Networks based on experimental evidence and expert-curated databases. Left panel: genes with CV identified in our study. Middle panel: genes with CV identified in our study plus genes with CV previously reported in literature. Right panel: genes with CV previously reported in literature. Genes that encode Complement System proteins are in orange, proteins in Response to virus pathways in green, including interferon pathway genes (yellow), and genes that regulate DNA processes in blue. IS: Immune system. Gene network analyses performed in String. Panels (B–D) depict Chord diagrams illustrating the functional relationships between the genes of (B) our study, (C) literature, and (D) our data and literature data combined and the biological processes (BP) that enrich them according to GO functional enrichment analysis. BPs are indicated by numbers. BPs enriched only by our study genes are in green, BPs enriched only by the literature genes in yellow, BPs that are enriched for both gene sets in orange, and BPs that only appear enriched with the combination of our genes with literature genes in purple. The size of the rectangles is proportional to the involvement of genes in the multiple pathways. Genes from each data set that does not enrich any BP does not appear in the images. The scale outside the circles indicates the number of relationships for each rectangle. Images created using circlize R package in R version 4.0.5. Colors for pathways, genes, and BPs are arbitrary. *Because TERC is not translated into a protein, String Nodes analysis does not include this gene. **We identified 38 genes with CV in MISC patients and other groups had previously described 41 genes. Because we identified CV in 3 genes that had already been described, accounting for those, the current total of genes with CV regarding MISC is 78 genes.
Other research groups previously identified 41 genes with CV in MIS-C patients (right panel) (Lee et al., 2020; Chou et al., 2021; Ronit et al., 2021; Abolhassani et al., 2022b; Abuhammour et al., 2022; Vagrecha et al., 2022). PPI Network analysis of these genes yields 41 nodes representing proteins and 93 edges representing confidence level (expected number of edges = 9), with a PPI enrichment p-value <1.0−16. Following the PPI Network analysis, it is observed that these literature-reported genes are largely related to response to virus pathways, several are interferon pathway- related genes, and a few are linked to DNA processes. When our data and literature data are combined (middle panel), there are 78 genes, 77 nodes representing proteins, and 192 edges representing confidence level (expected number of edges = 50), with a PPI enrichment p-value <1.0−16. All categories remained evident, complement system, DNA processes, and interferon gravitating around the main response to the virus response group. This is coherent with the importance of these genes in ensuring an appropriate immune response to COVID-19 infection, consistent with impairment of adequate infection resolution in MIS-C patients, and depicts a complementarity between genes described in previous studies and genes firstly associated with MIS-C in the present study.
To estimate more precisely which signaling pathways the CV we identified converge to impact on, hence would be more disrupted by pathological mutations in the described genes, we performed a Biological Processes (BP) Enrichment in String. The non-supervised analysis identified 86 BP with statistical significance (Confidence Interval of 95%), 35 of which can be further subcategorized into ten different functional groups (Figure 1B, Table 3, and Supplementary Table 6). To compare with previous findings, we performed the same analysis with the 41 previously described genes and identified 101 BP with statistical significance, 48 of which can be further subcategorized into 13 different functional groups (Figure 1C, Table 3, and Supplementary Table 6).
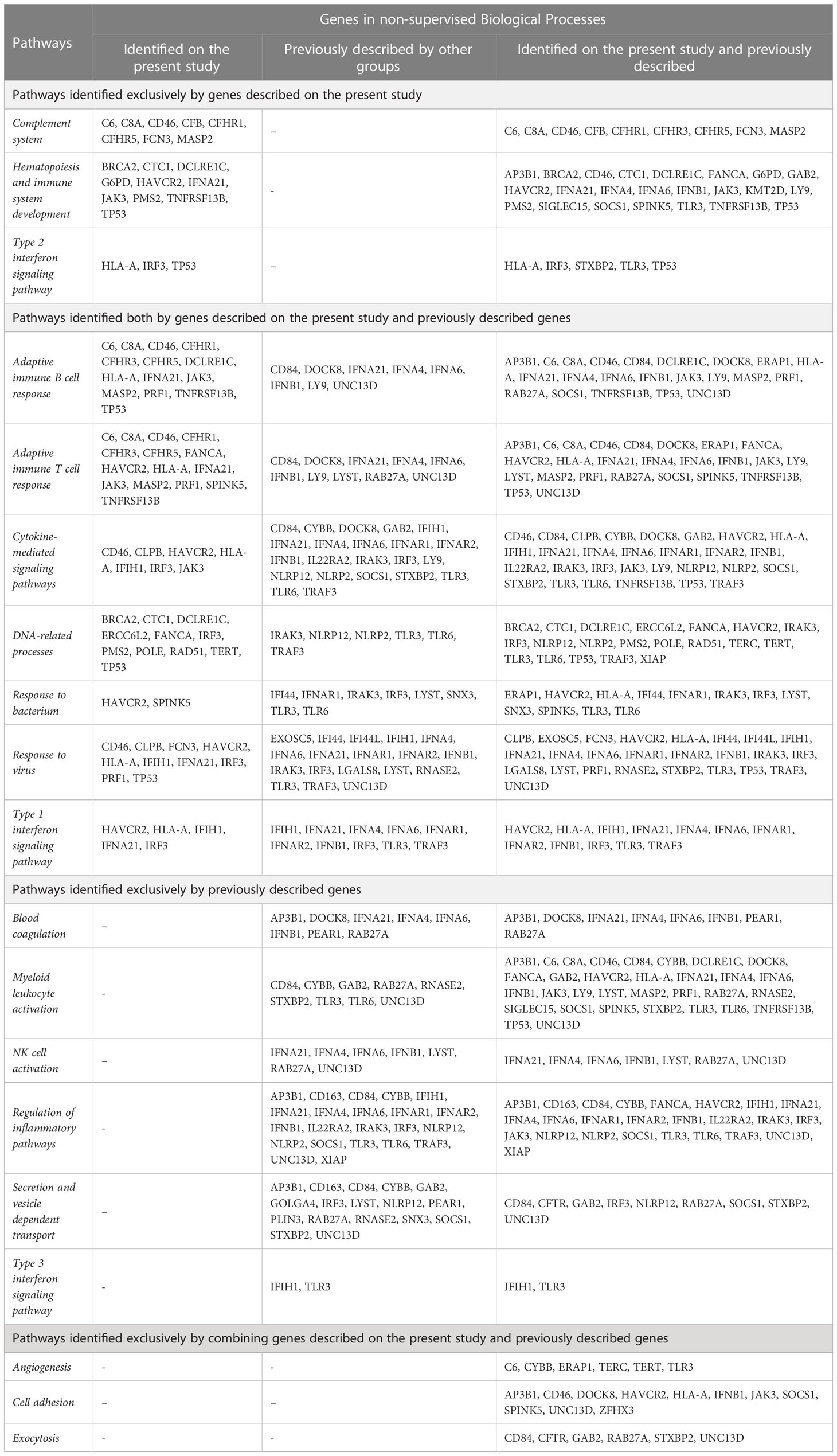
Table 3 String Enrichment non-supervised analysis of Biological Processes involving the identified genes, followed by supervised further categorization displaying genes in each group of pathways.
Notably, the 38 genes with CV described in our study are involved in seven major pathways that previously identified genes are also involved in, namely (1) Adaptive immune T cell response, (2) Adaptive immune B cell response, (3) DNA related processes, (4) Response to virus, (5) Response to bacteria, (6) Cytokine-mediated signaling pathways, and (7) Type 1 interferon-signaling pathway. Additionally, three pathways – (1) Complement system, (2) Hematopoiesis and Immune system development, and (3) Type 2 interferon signaling pathway – were first identified in the present study. Our data suggest that in addition to these previously identified pathways, complement system, hematopoiesis and immune system development, and Type 2 interferon signaling pathway impairment may also play a role in MIS-C pathogenesis.
Following this analysis, we combined the genes with CV identified in our analysis with previously described genes and ran a following unsupervised enrichment analysis of BP to search for possible still unidentified pathways. When taken together, the 78 genes clustered into 180 Biological Processes with statistical significance, from which 44 were not detected when considering only the set of genes we identified or the set of previously described genes. Upon supervised analysis of relevant pathways, six of these BPs could be further clustered into three major pathways, namely (1) Angiogenesis, (2) Cell adhesion, and (3) Exocytosis (Figure 1D, Table 3 and Supplementary Table 6).
4 Discussion
Although the pathophysiology of MIS-C remains unclear, several hypotheses have emerged, including immune dysregulation, autoantibody involvement, antibody-dependent enhancement (ADE), and superantigen-mediated immune activation (Cheng and Zhang, 2020; Consiglio et al., 2020; Kouo and Chaisawangwong, 2021; Ramaswamy et al., 2021; Morita et al., 2022). Also, there are indications that genetic factors may play a role in the pathogenesis of MIS-C, at least in a fraction of the patients, such as the report of siblings with clinical manifestations of MIS-C occurring almost simultaneously (Lim et al., 2021), the similarities with DK and LHH, in which the participation of genetic factors is already established, and the fact that it is a rare complication of SARS-CoV-2 infection (Fujita et al., 1989; Uehara et al., 2003; Sancho-Shimizu et al., 2021).
The present study identified pathogenic or likely pathogenic variants in 29 out of 30 MIS-C patients (96.6%), with a total of 50 CV across 38 different genes (Table 2). Notably, several genes, including HLA-A, PRF1, G6PD, had CVs detected in more than one patient. These findings support the hypothesis that genetic variants may be involved in the pathophysiology of MIS-C. To our knowledge, this is the largest MIS-C cohort that underwent genetic evaluation in Latin-American patients. The data reported here are similar to the largest cohort of MIS-C cases reported so far (Belay et al., 2021), with 1733 US patients in terms of number of days with fever, prevalence of signs, symptoms, and laboratory findings, as well as the frequency of serologic test and RT-PCR positivity for SARS-CoV-2. Concerning male-female rate, there is a slight predominance of males in the literature. Interestingly, our cohort has a peculiar female predominance. This difference likely does not reflect selection bias, as all patients identified at participating centers who met the MIS-C definition criteria were included in the study without any secondary selection.
A notable finding in our cohort is that only 59.2% of the cases had a traceable history of known contact with a suspected or confirmed case of COVID-19. It is possible that the signs and symptoms of COVID-19 were not appreciated by the contact persons of these children, or even that the individuals who transmitted the virus to the patients were asymptomatic and therefore did not realize they were infected, or did not inform these families about the diagnosis of the disease.
4.1 Gene sequencing and MIS-C
In the search for genetic risk factors in patients with MIS-C, one of the strategies has been WES or the use of gene panels to identify deleterious variants in genes related to immune processes. In this sense, Chou et al. (Chou et al., 2021) were pioneers in describing the presence of rare variants in XIAP, CYBB, and SOCS1 genes (Lee et al., 2020) in 3 patients diagnosed with MIS-C. Along with the same line, a homozygous variant in the IFNAR1 gene was reported in a patient diagnosed with MIS-C who had consanguineous parents (Abolhassani et al., 2022b). The authors also described other variants identified in genes related to IEI, including MASP1, POLE2, and KMT2D. Interestingly, we identified CV in MASP2, POLE1, and KMT2D (a different variant from the one identified in the cited study). In 2022, the same research group published a study where 31 patients previously suspected of heaving IEI (but lacking molecular diagnosis) were evaluated after presenting COVID-19 complications, including MIS-C. The cohort had four patients with MIS-C in addition to the one previously described. Two patients had CFH variants. A variant in TBX1 and one in UNG were identified in the two remaining patients (Abolhassani et al., 2022a). Of note, we observed CV in CFHR1, CFHR3, and CFHR5, which as CFH, are also part of the Human Factor H Gene Cluster.
A case report (Vagrecha et al., 2021), described a patient bearing HLH due to a homozygous frameshift mutation of the gene encoding syntaxin 11 (STX11). The subject achieved remission with anakinra monotherapy but subsequently developed an acute SARS-CoV-2 infection followed by a possible MIS-C, both responsive to increased anakinra dosing. A following study involving MIS-C and HLH-related genes described variants in IEI and LHH-related genes in 74% of the patients (29/45) with MIS-C (Vagrecha et al., 2022). One of the genes in which a rare CV (rs779584225) was identified in that study (PRF1) also appears among the genes of interest in our cohort. In addition, they also described variants in another four genes we identified CV in our patients (DCLRE1C, IFIH1, TNFRSF13B, SH3BP2) (Vagrecha et al., 2022).
A further study involving 45 Middle Eastern children with MIS-C and 25 controls identified rare and probably deleterious variants in 42% of the patients, in 16 genes related to the immune system. Interestingly, in the same study, two CVs that were also identified in our cohort were described (Abuhammour et al., 2022): one in the IFIH1 gene (rs35337543) and one in the IFNA21 gene (rs146777169).
Complement-related genes were investigated in 71 pediatric patients from Greece with either MIS-C (7 patients) or COVID-19 (64 patients). Notably, nine patients harbored the same FCN3 variant identified in three of our patients (rs532781899), four in the non-admitted COVID-19 group and 5 in the admitted group, but none in the MIS-C group (Gavriilaki and Tsiftsoglou, 2022). Also, five patients harbored the same MASP2 variant identified in two of our patients (rs72550870). However, once more, none of the patients of the MIS-C group harbored the variant. Despite that, the authors themselves recognized that the size of the MIS-C group was relatively small for conclusions about the absence of these variants. It is also worth noting that 22 patients harbored the rs12614 CFB variant identified in P12, including four of the seven MIS-C patients (Gavriilaki and Tsiftsoglou, 2022).
More recently, next-generation sequencing (NGS) was performed in samples from 37 MIS-C children at hospital admission and 24 healthy controls from Italy for a panel of 386 genes related to autoimmune diseases, autoinflammation, and primary immunodeficiencies (Gelzo et al., 2022). Similarly, to the present study, the authors identified variants in 34 genes, and 83.3% of patients had at least one variant. Most genes were related to autoimmune diseases like ATM, NCF1, MCM4, FCN3, and DOCK8 or autoinflammatory diseases associated with the release of IFN-gamma, such as PRF1, NOD2, and MEF (Gelzo et al., 2022). Interestingly, three variants identified by the authors were also present in our cohort: PRF1 rs35947132, FCN3 rs532781899, and IFIH1 rs35337543. Abuhammour et al. also identified the latter, as seen above (Abuhammour et al., 2022). It is worth noting that 2/30 patients in our cohort and 5/30 patients reported by Gelzo et al. (Gelzo et al., 2022) had the same uncommon PRF1 variant (c.272C>T). Although the pathogenicity of this variant is unclear as it is relatively prevalent (AF of 2,8% on GnomAD aggregated), it leads to reduced stability and abnormal traffic of the protein, leading to impaired NK cell cytotoxic function (Voskoboinik et al., 2007; House et al., 2015; Cabrera-Marante et al., 2020).
In another recent study that enrolled 558 MIS-C patients from different ethnicities, 16 different variants were identified in 12 unrelated children who harbor homozygous or compound heterozygous variants in the genes OAS1, OAS2, OAS3 or RNASE, five of which had rare homozygous variants predicted as loss-of-function. However, none of our patients have any of the variants reported (Lee and Le Pen, 2023).
To our knowledge, apart from the present study, the single study that performed WES in Brazilian children with MISC investigated 16 patients (Santos-Rebouças et al., 2022). They screened for very rare variants (global minor allele frequency less than 0.01) previously associated with any known disease, including those unrelated to MISC or SARS-CoV-2 infection. They identified ten very rare variants in eight genes (FREM1, MPO, POLG, C6, C9, ABCA4, ABCC6, and BSCL2) as promising candidates that could be related to a higher risk of MISC development. From those, seven are classified as pathogenic, and three variants of uncertain significance (VUS) of interest. Interestingly, two of our patients harbor CVs predicted to be pathogenic in their cohort’s most frequently altered gene, ABCA4 (3 patients bearing three different CVs). The CV found in our p4 (ABCA4 rs148460146) is in the nucleotide-binding domain (NBD), as the pathogenic variant found in their EXOC13. The second pathogenic ABCA4 CV found in our cohort (in p25) is in the extracellular domain two (ECD2), whose function is currently not fully understood (ABCA4 rs61750145) (Xie et al., 2021). Another patient in our cohort also harbors a CV in another gene with CV described by them, ABCC6. Although it is not the same variant (our patient harbors the ABCC6 rs72653706, and theirs harbors the ABCC6 rs63750759), both are classified as pathogenic and associated with Pseudoxanthoma Elasticum (Bergen et al., 2000). The variants in ABCA4 are associated with retina, macula, and cone-rod dystrophy, which results in vision loss (Lin et al., 2018). However, these CVs are not among our main findings because neither is associated with MISC, KD, severe COVID-19, or any IEI, and their association with MISC is still unclear. Another similarity between both studies is that, as we, they also detected a CV in the C6 gene. In their study, one patient harbored the CV C6 rs375762365; in ours, one patient harbored the CV C6 rs61469168. Although it is not the same variant, both are frameshift mutations, which usually yield a loss of function effect.
Taken together, these findings reinforce the hypothesis that genes previously related to immune processes and, more specifically, IEI may underlie the clinical manifestations of MIS-C.
4.2 Gene sequencing and MIS-A
The investigation of possible genetic factors in the pathophysiology of MIS-A through WES has also been described in the scarce literature currently available. In one study, 12 rare and potentially disease-causing VUS were found in 5 patients diagnosed with MIS-A (Ronit et al., 2021), including an ERAP1 variant, a gene that also had a CV identified in our cohort.
4.3 HLA and MIS-C
Another strategy used in the investigation of children and adolescents with MIS-C was evaluating the association of the disease with specific HLA alleles in 2 cohorts of Italian patients. In those studies, it was observed that the combination of the alleles HLA-A*02, B*35, and C*04 was more frequent in patients with severe MIS-C, reinforcing the existence of genetic susceptibility related to the disease (Porritt et al., 2021; Sacco and Castagnoli, 2022). Interestingly, in our cohort, one female patient (P4) was identified with this combination of alleles, and is of Italian descent (Supplementary Table 7). Also, 9/30 patients harbor a CV in HLA-A.
4.4 Functional enrichment analysis
Using the gene ontology (GO) enrichment analysis, the functional relationships between genes with CV and the enriched biological processes (BP) were analyzed. With this information combined with supervised annotation of relevant pathways, Circos graphs were generated (Figure 1B). Most of the identified pathways were consistent with a potential over-persistent state of inflammation, possibly due to COVID-19 infection, which could lead to the onset of MIS-C. Our study identified 13 potentially relevant pathways in the context of MIS-C, seven in common with the analysis of genes previously reported in the literature and six not previously described by the WES approach (Figure 2). Three of these are uniquely identified when genes previously reported and genes in our study are analyzed together, and three others emerged exclusively from the analysis of the genes with CV in our patients (Supplementary Table 6). Some of these pathways have also been highlighted in the literature and may shed light on the mechanisms that lead to this disease, as discussed below.
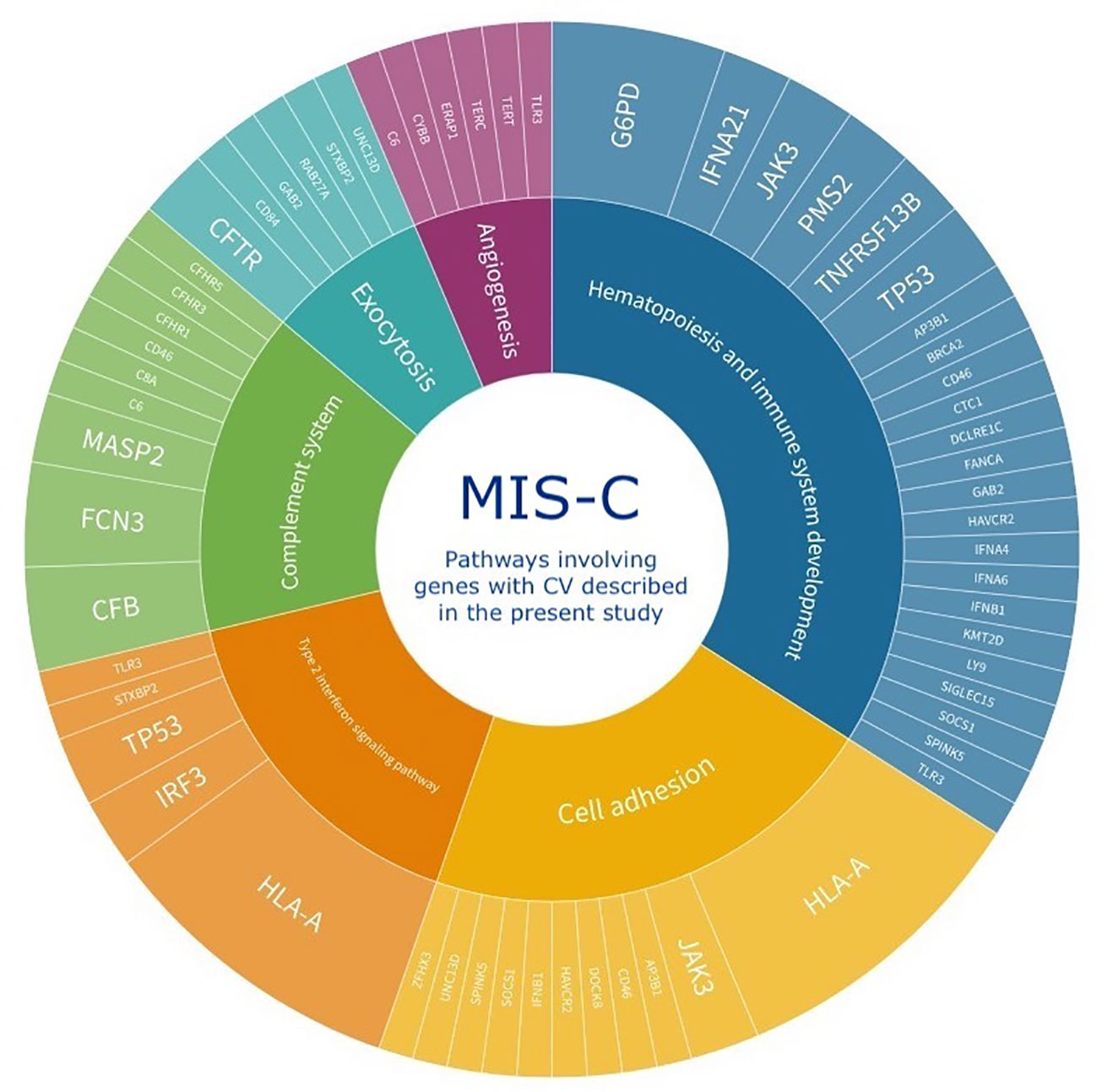
Figure 2 Schematic representation of the six major pathways identified by functional relationships between MISC patients’ genes with CV described by the present study. Three of these are uniquely identified when genes previously reported and genes in our study are analyzed together (Angiogenesis, Cell adhesion, and Exocytosis) and three others emerged exclusively from the analysis of the genes with CV in our patients (Complement system, Hematopoiesis and Immune system development, and Type 2 interferon signaling pathway). The inner circle depicts the pathways and the outer circle the genes with CV involved in each pathway. The size of each pathway in the inner circle is proportional to the number of patients with a respective gene with CV.
The complement system is one of the most activated intracellular pathways during the COVID-19 antiviral response, and deregulation of this pathway has been described in MIS-C (Ali et al., 2021; Defendi et al., 2021; Syrimi et al., 2021; Afzali and Noris, 2022; Kumar et al., 2022). Interestingly, 14/30 of our patients had a CV in complement system genes.
Type I interferons (IFN) have a crucial role in antiviral response and severe COVID-19 (Bastard and Rosen, 2020; Zhang and Bastard, 2020; Chou et al., 2021; Tovo and Garazzino, 2021; Abolhassani et al., 2022b). Patients with MIS-C or severe COVID-19 harbor reduced expression of type I IFN-stimulated genes (ISGs), whereas children with mild COVID-19 exhibit an increased expression pattern (Tovo and Garazzino, 2021). In line with these findings, studies in adults have linked this reduction to a persistent viral load and consequent hyperinflammatory state (Trouillet-Assant et al., 2020). In our cohort, 18/30 patients harbor a CV in Response to Virus Pathways related genes.
In addition to type I IFN involvement in MIS-C, type II IFN-dependent and NF-κB-dependent signatures, matrisome activation, increased cell adhesion biomarkers, and increased levels of circulating spike protein have also been detected in MIS-C (Lee et al., 2020; Chou et al., 2021; Sacco and Castagnoli, 2022). Notably, one of the studies that detected increased type II IFN-dependent signaling in MIS-C also described increased cell adhesion biomarkers, one of the major pathways we detected in our analysis when combining previously described genes with genes detected in our present study (Sacco and Castagnoli, 2022). A total of 12/30 patients in our cohort harbor CV in genes associated with the type II IFN pathway.
Many hematopoietic findings are frequently observed in MIS-C patients, such as leukocytosis, lymphopenia, and thrombocytosis/thrombocytopenia (Belay et al., 2021). Also, very small embryonic-like stem cells (VSEL) and hematopoietic stem cells (HSC) express ACE2, SARS-CoV-2 entry receptor. Hence, VSELs residing in developed tissues could be damaged by SARS-CoV-2, with remote effects on tissue/organ regeneration, which is a feature of MIS-C (Ratajczak et al., 2021). Therefore, variants in genes that disrupt hematopoiesis and immune system development might impair response to SARS-CoV-2, favoring the development of MIS-C. In our study, 14/30 patients harbor CV in hematopoiesis and immune system development related genes.
Finally, our results also support a role in MIS-C of variants in genes involved in multiple pathways or haplotypes. For example, all 5/30 patients with PFR1 variants detected in our study also harbored variants in at least one complement system gene, and co-occurrence of CV in PFR1 with CV in other genes was also observed in the other studies that reported CV in PRF1 (Gelzo et al., 2022; Vagrecha et al., 2022).
4.5 Involvement of genetic factors in MIS-C
The clinical phenotype of MIS-C is variable, and several categorizations of these patients into subgroups have been proposed (Godfred-Cato et al., 2020; Whittaker et al., 2020; Harwood et al., 2021). Also, because of this variability, it is speculated that at least a portion of patients with MIS-C may have an underlying IEI, either of monogenic, digenic, or even oligogenic origin (Sancho-Shimizu et al., 2021). Thus, genetic studies such as whole genome sequencing or WES are important to better understand this disease pathophysiology. Furthermore, to further tackle which are the determinant factors in the development of MIS-C, other strategies should be employed, such as an in-depth genetic investigation through HLA analysis, evaluation of the expression of genes of interest using proteomics, and search for pathways that may be hyper- or hypo-activated, with the use of transcriptomics using bulk and single-cell analysis.
The limitations of the present study include the absence of a control group, the non-consideration of VUS in the analysis, and the absence of functional tests, which are necessary to determine the actual damage caused by the variants described. By searching for variants previously identified as pathogenic or likely pathogenic, we partially addressed this limitation, but further studies are needed to better understand our findings so that they can be translated into clinical advice. It is also important to note that several of the CVs described were identified in only one subject, and more than one CV was found in most subjects. Based on the limitations mentioned above, it is currently not possible to conclude that the cited variants are directly involved in the pathophysiology of MIS-C. As a rare disease, a worldwide multicentric effort would be ideal for strengthening the findings from this cohort and other studies.
What appears to be shared by all WES and other genetic studies carried out on MIS-C so far is that children with MIS-C may take benefit from an approach such as the search for disease-causing mutations based on an IEI-associated gene panel. In addition, early detection before the onset of the disease may help prevent severe clinical scenarios for these children and enable better clinical management.
5 Conclusions
In our cohort, 29/30 MIS-C patients harbored uncommon and rare variants in genes related to immune system disorders. Our results support the hypothesis that MIS-C has an immune-genetic basis, as summarized in Figure 2. Importantly, however, other unidentified genes or factors are also likely to be involved.
To our knowledge, this is the largest MIS-C cohort that underwent genetic evaluation in Latin American patients. In a second vein, our study strengthens the need for larger genomic studies in children with MIS-C to identify other possible variants involved in the pathogenesis of the disease, to better understand the role of genetics in MIS-C, and eventually to provide assistance in the clinical management of patients in a higher risk of poor outcomes.
Many IEIs are silent during the first years of life, and several phenotypes appear after exposure to particular infectious agents. Our data support the use of WES as a diagnostic tool for children with this rare COVID-19 complication. This strategy may help to pinpoint the genetic impairments contributing to the development of the MIS-C phenotype in children, identify children at a higher risk of worse outcomes, and provide guidance to narrow treatment decisions.
Data availability statement
The data presented in the study are deposited in the ARCA Dados Fiocruz repository, accession doi: https://doi.org/10.35078/RDZHNL.
Ethics statement
The studies involving human participants were reviewed and approved by Internal Review Board (IRB) of Instituto D’Or de Pesquisa e Ensino - IDOR (Proponent Institution) under CAAE n° 30272920.0.1001.5249. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceptualization, BR, AP-B, and ZV. Methodology, BR, and ZV. Formal analysis, BR, RSF, FF, HF, DP, ISF, DLMF, and OC-M. Investigation, BR, FC, MZ, DM, ACB, WS, JR, FO, ANB, RZ, MS, MO, and GR-S. Resources, FF, DP, MZ, OC-M, MS. Data curation, BR, FF, and RSF. Writing—original draft preparation, BR, and RSF. Writing—review and editing, FC, DM, ACB, WS, AP-B, and ZV. Visualization, BR, RSF, DP, ISF, and DLMF. Supervision, AP-B and ZV. Project administration, ZV. Funding acquisition, FP, OC-M, AP-B, and ZV. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the following Brazilian research promotion agencies: Inova Fiocruz/Fundação Oswaldo Cruz and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant 308697/2019-7 and 310299/2022-5) to FP. Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (National Council for Scientific and Technological Development – CNPq), grant n° 309482/2022-4 to O-CM and 401597/2020-2, Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (Carlos Chagas Filho Foundation for Research Support of the State of Rio de Janeiro – FAPERJ), Process n° E-26/010.000160/2020, grant n° 2020/0996, Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (São Paulo Research Foundation – FAPESP) grants 2018/18886-9, 2020/01688-0, and 2020/07069-0 to O-CM, 2020/16246-2 to DLMF, and 2020/11710-2 to DP, Post-Doctoral Fellowship from FAPERJ (E-26/203.910/2021) to RSF, The APC was funded by CAPES (Finance Code 001) through our institutional Post-Graduate Program “Pesquisa Aplicada à Sauíde da Criança e da Mulher”.
Acknowledgments
We wish to thank the patients and families for participating in this study. We also thank the healthcare workers who cared for the patients for their guidance and support in consolidating the data and making it available for the study.
Conflict of interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1182257/full#supplementary-material
References
00432, H.A (2021). Available at: https://emergency.cdc.gov/han/2020/han00432.asp (Accessed 23/12/2022).
Abolhassani, H., Delavari, S., Landegren, N., Shokri, S., Bastard, P., Du, L., et al. (2022a). Genetic and immunologic evaluation of children with inborn errors of immunity and severe or critical COVID-19. J. Allergy Clin. Immunol. 150 (5), 1059–1073. doi: 10.1016/j.jaci.2022.09.005
Abolhassani, H., Landegren, N., Bastard, P., Materna, M., Modaresi, M., Du, L., et al. (2022b). Inherited IFNAR1 deficiency in a child with both critical COVID-19 pneumonia and multisystem inflammatory syndrome. Journal of Clinical Immunology 42, 3, 471–483. doi: 10.1007/s10875-022-01215-7
Abuhammour, W., Yavuz, L., Jain, R., Abu Hammour, K., Al-Hammouri, G. F., El Naofal, M., et al. (2022). Genetic and clinical characteristics of patients in the middle East with multisystem inflammatory syndrome in children. JAMA Netw. Open 5 (5), e2214985. doi: 10.1001/jamanetworkopen.2022.14985
Afzali, B., Noris, M. (2022). The state of complement in COVID-19. Nature Reviews Immunology 22, 2, 77–84. doi: 10.1038/s41577-021-00665-1
Ali, Y. M., Ferrari, M., Lynch, N. J., Yaseen, S., Dudler, T., Gragerov, S., et al. (2021). Lectin pathway mediates complement activation by SARS-CoV-2 proteins. Front. Immunol. 12. doi: 10.3389/fimmu.2021.714511
Asano, T., Boisson, B., Onodi, F., Matuozzo, D., Moncada-Velez, M., Maglorius Renkilaraj, M. R. L., et al. (2021). X-Linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6 (62). doi: 10.1126/sciimmunol.abl4348
Bastard, P., Rosen, L. B. (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, 6515. doi: 10.1126/science.abd4585
Belay, E. D., Abrams, J., Oster, M. E., Giovanni, J., Pierce, T., Meng, L., et al. (2021). Trends in geographic and temporal distribution of US children with multisystem inflammatory syndrome during the COVID-19 pandemic. JAMA Pediatr. 175 (8), 837–845. doi: 10.1001/jamapediatrics.2021.0630
Bergen, A. A., Plomp, A. S., Schuurman, E. J., Terry, S., Breuning, M., Dauwerse, H., et al. (2000). Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 25 (2), 228–231. doi: 10.1038/76109
Bousfiha, A., Jeddane, L., Picard, C., Al-Herz, W., Ailal, F., Chatila, T., et al. (2020). Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J. Clin. Immunol. 40 (1), 66–81. doi: 10.1007/s10875-020-00758-x
Burgner, D., Davila, S., Breunis, W. B., Ng, S. B., Li, Y., Bonnard, C., et al. (2009). A genome-wide association study identifies novel and functionally related susceptibility loci for Kawasaki disease. PloS Genet. 5 (1), e1000319. doi: 10.1371/journal.pgen.1000319
Cabrera-Marante, O., Rodríguez de Frías, E., Pleguezuelo, D. E., Allende, L. M., Serrano, A., Laguna-Goya, R., et al. (2020). Perforin gene variant A91V in young patients with severe COVID-19. Haematologica 105 (12), 2844–2846. doi: 10.3324/haematol.2020.260307
Cheng, M. H., Zhang, S. (2020). Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proceedings of the National Academy of Sciences 117, 41, 25254–25262. doi: 10.1073/pnas.2010722117
Chou, J., Platt, C. D., Habiballah, S., Nguyen, A. A., Elkins, M., Weeks, S., et al. (2021). Mechanisms underlying genetic susceptibility to multisystem inflammatory syndrome in children (MIS-c). J. Allergy Clin. Immunol. 148 (3), 732–738.e731. doi: 10.1016/j.jaci.2021.06.024
Consiglio, C. R., Cotugno, N., Sardh, F., Pou, C., Amodio, D., Rodriguez, L., et al. (2020). The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell 183 (4), 968–981.e967. doi: 10.1016/j.cell.2020.09.016
Defendi, F., Leroy, C., Epaulard, O., Clavarino, G., Vilotitch, A., Le Marechal, M., et al. (2021). Complement alternative and mannose-binding lectin pathway activation is associated with COVID-19 mortality. Front. Immunol. 12. doi: 10.3389/fimmu.2021.742446
Dufort, E. M., Koumans, E. H., Chow, E. J. (2020). Multisystem inflammatory syndrome in children in new York state. The New England Journal of Medicine 383, 4, 347–358. doi: 10.1056/NEJMoa2021756
Feldstein, L. R., Rose, E. B., Horwitz, S. M., Collins, J. P., Newhams, M. M., Son, M. B. F., et al. (2020). Multisystem inflammatory syndrome in U.S. Children Adolescents 383 (4), 334–346. doi: 10.1056/NEJMoa2021680
Fujita, Y., Nakamura, Y., Sakata, K., Hara, N., Kobayashi, M., Nagai, M., et al. (1989). Kawasaki Disease in families. Pediatrics 84 (4), 666–669. doi: 10.1542/peds.84.4.666
Garcia, M., Juhos, S., Larsson, M., Olason, P. I., Martin, M. (2020). Sarek: a portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000 Research 9, 63. doi: 10.12688/f1000research.16665.2
Gavriilaki, E., Tsiftsoglou, S. A. (2022). Targeted genotyping of MIS-c patients reveals a potential alternative pathway mediated complement dysregulation during COVID-19 infection. Current Issues in Molecular Biology 44, 7, 2811–2824. doi: 10.3390/cimb44070193
Gelzo, M., Castaldo, A., Giannattasio, A., Scalia, G., Raia, M., Esposito, M. V., et al. (2022). MIS-c: a COVID-19-as sociated condition between hypoimmunity and hyperimmunity. Front. Immunol. 13. doi: 10.3389/fimmu.2022.985433
Godfred-Cato, S., Bryant, B., Leung, J., Oster, M. E., Conklin, L., Abrams, J., et al. (2020). COVID-19-Associated multisystem inflammatory syndrome in children - united states, March-July 2020. MMWR Morb Mortal Wkly Rep. 69 (32), 1074–1080. doi: 10.15585/mmwr.mm6932e2
Gu, Z., Gu, L., Eils, R., Schlesner, M., Brors, B. (2014). Circlize implements and enhances circular visualization in r. Bioinformatics 30 (19), 2811–2812. doi: 10.1093/bioinformatics/btu393
Harwood, R., Allin, B., Jones, C. E., Whittaker, E., Ramnarayan, P., Ramanan, A. V., et al. (2021). A national consensus management pathway for paediatric inflammatory multisystem syndrome temporally associated with COVID-19 (PIMS-TS): results of a national Delphi process. Lancet Child Adolesc. Health 5 (2), 133–141. doi: 10.1016/s2352-4642(20)30304-7
House, I. G., Thia, K., Brennan, A. J., Tothill, R., Dobrovic, A., Yeh, W. Z., et al. (2015). Heterozygosity for the common perforin mutation, p.A91V, impairs the cytotoxicity of primary natural killer cells from healthy individuals. Immunol. Cell Biol. 93 (6), 575–580. doi: 10.1038/icb.2015.1
Kouo, T., Chaisawangwong, W. (2021). SARS-CoV-2 as a superantigen in multisystem inflammatory syndrome in children. J. Clin. Invest. 131 (10). doi: 10.1172/jci149327
Kumar, D., Rostad, C. A., Jaggi, P., Villacis Nunez, D. S., Prince, C., Lu, A., et al. (2022). Distinguishing immune activation and inflammatory signatures of multisystem inflammatory syndrome in children (MIS-c) versus hemophagocytic lymphohistiocytosis (HLH). J. Allergy Clin. Immunol. 149 (5), 1592–1606.e1516. doi: 10.1016/j.jaci.2022.02.028
Lee, Y. C., Kuo, H. C., Chang, J. S., Chang, L. Y., Huang, L. M., Chen, M. R., et al. (2012). Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat. Genet. 44 (5), 522–525. doi: 10.1038/ng.2227
Lee, D., Le Pen, J. (2023). Inborn errors of OAS-RNase l in SARS-CoV-2-related multisystem inflammatory syndrome in children. Nature Genetics 379, 6632. doi: 10.1126/science.abo3627
Lee, P. Y., Platt, C. D., Weeks, S., Grace, R. F., Maher, G., Gauthier, K., et al. (2020). Immune dysregulation and multisystem inflammatory syndrome in children (MIS-c) in individuals with haploinsufficiency of SOCS1. J. Allergy Clin. Immunol. 146 (5), 1194–1200.e1191. doi: 10.1016/j.jaci.2020.07.033
Lim, L., Lim, S. J., Loy, J. S., Ng, D. C. (2021). Multisystem inflammatory syndrome in children (MIS-c) occurring in temporal proximity between siblings. BMJ case reports 14, 9. doi: 10.1136/bcr-2021-246066
Lima-Setta, F., Magalhães-Barbosa, M. C., Rodrigues-Santos, G., Figueiredo, E., Jacques, M. L., Zeitel, R. S., et al. (2021). Multisystem inflammatory syndrome in children (MIS-c) during SARS-CoV-2 pandemic in Brazil: a multicenter, prospective cohort study. J. Pediatr. (Rio J) 97 (3), 354–361. doi: 10.1016/j.jped.2020.10.008
Lin, Y., Gao, H., Chen, C., Zhu, Y., Li, T., Liu, B., et al. (2018). Clinical and next-generation sequencing findings in a Chinese family exhibiting severe familial exudative vitreoretinopathy. Int. J. Mol. Med. 41 (2), 773–782. doi: 10.3892/ijmm.2017.3308
Morita, A., Hosaka, S., Imagawa, K., Ishiodori, T., Nozaki, Y., Murakami, T., et al. (2022). Time course of peripheral immunophenotypes of multisystem inflammatory syndrome in children. Clin. Immunol. 236, 108955. doi: 10.1016/j.clim.2022.108955
Onouchi, Y., Ozaki, K., Burns, J. C., Shimizu, C., Terai, M., Hamada, H., et al. (2012). A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat. Genet. 44 (5), 517–521. doi: 10.1038/ng.2220
Patel, P., DeCuir, J., Abrams, J., Campbell, A. P., Godfred-Cato, S., Belay, E. D. (2021). Clinical characteristics of multisystem inflammatory syndrome in adults: a systematic review. JAMA Netw. Open 4 (9), e2126456. doi: 10.1001/jamanetworkopen.2021.26456
Porritt, R. A., Paschold, L., Rivas, M. N., Cheng, M. H., Yonker, L. M., Chandnani, H., et al. (2021). HLA class I-associated expansion of TRBV11-2 T cells in multisystem inflammatory syndrome in children. J. Clin. Invest. 131 (10). doi: 10.1172/jci146614
Ramaswamy, A., Brodsky, N. N., Sumida, T. S., Comi, M., Asashima, H., Hoehn, K. B., et al. (2021). Immune dysregulation and autoreactivity correlate with disease severity in SARS-CoV-2-associated multisystem inflammatory syndrome in children. Immunity 54 (5), 1083–1095.e1087. doi: 10.1016/j.immuni.2021.04.003
Ratajczak, M. Z., Bujko, K., Ciechanowicz, A., Sielatycka, K., Cymer, M., Marlicz, W., et al. (2021). SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45(-) precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev. Rep. 17 (1), 266–277. doi: 10.1007/s12015-020-10010-z
Riphagen, S., Gomez, X., Gonzalez-Martinez, C., Wilkinson, N., Theocharis, P. (2020). Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 395 (10237), 1607–1608. doi: 10.1016/s0140-6736(20)31094-1
Ronit, A., Jørgensen, S. E., Roed, C., Eriksson, R., Iepsen, U. W., Plovsing, R. R., et al. (2021). Host genetics and antiviral immune responses in adult patients with multisystem inflammatory syndrome. Front. Immunol. 12. doi: 10.3389/fimmu.2021.718744
Sacco, K., Castagnoli, R. (2022). Immunopathological signatures in multisystem inflammatory syndrome in children and pediatric COVID-19. Nature Medicine 28, 5, 1050–1062. doi: 10.1038/s41591-022-01724-3
Sancho-Shimizu, V., Brodin, P., Cobat, A., Biggs, C. M., Toubiana, J., Lucas, C. L., et al. (2021). SARS-CoV-2-related MIS-c: a key to the viral and genetic causes of Kawasaki disease? J. Exp. Med. 218 (6). doi: 10.1084/jem.20210446
Santos-Rebouças, C. B., Piergiorge, R. M., Dos Santos Ferreira, C., Seixas Zeitel, R., Gerber, A. L., Rodrigues, M. C. F., et al. (2022). Host genetic susceptibility underlying SARS-CoV-2-associated multisystem inflammatory syndrome in Brazilian children. Molecular Medicine (Cambridge, Mass) 28, 1, 153. doi: 10.1186/s10020-022-00583-5
Schulert, G. S., Cron, R. Q. (2020). The genetics of macrophage activation syndrome. Genes Immun. 21 (3), 169–181. doi: 10.1038/s41435-020-0098-4
Shaiba, L. A., More, K., Hadid, A., Almaghrabi, R., Al Marri, M., Alnamnakani, M., et al. (2022). Multisystemic inflammatory syndrome in neonates: a systematic review. Neonatology 119 (4), 405–417. doi: 10.1159/000524202
Solanich, X., Vargas-Parra, G., van der Made, C. I., Simons, A., Schuurs-Hoeijmakers, J., Antolí, A., et al. (2021). Genetic screening for TLR7 variants in young and previously healthy men with severe COVID-19. Front. Immunol. 12. doi: 10.3389/fimmu.2021.719115
Syrimi, E., Fennell, E., Richter, A., Vrljicak, P., Stark, R., Ott, S., et al. (2021). The immune landscape of SARS-CoV-2-associated multisystem inflammatory syndrome in children (MIS-c) from acute disease to recovery. iScience 24 (11), 103215. doi: 10.1016/j.isci.2021.103215
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49 (D1), D605–d612. doi: 10.1093/nar/gkaa1074
Tangye, S. G., Al-Herz, W., Bousfiha, A., Cunningham-Rundles, C., Franco, J. L., Holland, S. M., et al. (2021). The ever-increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J. Clin. Immunol. 41 (3), 666–679. doi: 10.1007/s10875-021-00980-1
Tovo, P. A., Garazzino, S. (2021). COVID-19 in children: expressions of type I/II/III interferons, TRIM28, SETDB1, and endogenous retroviruses in mild and severe cases. International Journal of Molecular Sciences 22, 14. doi: 10.3390/ijms22147481
Trouillet-Assant, S., Viel, S., Gaymard, A., Pons, S., Richard, J. C., Perret, M., et al. (2020). Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 146 (1), 206–208.e202. doi: 10.1016/j.jaci.2020.04.029
Tsai, F. J., Lee, Y. C., Chang, J. S., Huang, L. M., Huang, F. Y., Chiu, N. C., et al. (2011). Identification of novel susceptibility loci for kawasaki disease in a han chinese population by a genome-wide association study. PloS One 6 (2), e16853. doi: 10.1371/journal.pone.0016853
Uehara, R., Yashiro, M., Nakamura, Y., Yanagawa, H. (2003). Kawasaki Disease in parents and children. Acta Paediatr. 92 (6), 694–697. doi: 10.1080/08035320310002768
Vagrecha, A., Patel, H. B., Mamdouhi, T., Acharya, S., Appiah-Kubi, A., Aygun, B. (2021). Effect of COVID-19 on anakinra-induced remission in homozygous STX11 hemophagocytosis lymphohistiocytosis. Pediatric Blood and Cancer 68, 6. doi: 10.1002/pbc.28897
Vagrecha, A., Zhang, M., Acharya, S., Lozinsky, S., Singer, A., Levine, C., et al. (2022). Hemophagocytic lymphohistiocytosis gene variants in multisystem inflammatory syndrome in children. Biology (Basel) 11, 3. doi: 10.3390/biology11030417
van der Made, C. I., Simons, A., Schuurs-Hoeijmakers, J., van den Heuvel, G., Mantere, T., Kersten, S., et al. (2020). Presence of genetic variants among young men with severe COVID-19. Jama 324 (7), 663–673. doi: 10.1001/jama.2020.13719
Verdoni, L., Mazza, A., Gervasoni, A., Martelli, L., Ruggeri, M., Ciuffreda, M., et al. (2020). An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 395 (10239), 1771–1778. doi: 10.1016/s0140-6736(20)31103-x
Voskoboinik, I., Sutton, V. R., Ciccone, A., House, C. M., Chia, J., Darcy, P. K., et al. (2007). Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood 110 (4), 1184–1190. doi: 10.1182/blood-2007-02-072850
Whittaker, E., Bamford, A., Kenny, J., Kaforou, M., Jones, C. E., Shah, P., et al. (2020). Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. Jama 324 (3), 259–269. doi: 10.1001/jama.2020.10369
Xie, T., Zhang, Z., Fang, Q., Du, B., Gong, X. (2021). Structural basis of substrate recognition and translocation by human ABCA4. Nature Communications 12, 1, 3853. doi: 10.1038/s41467-021-24194-6
Keywords: multisystem inflammatory syndrome in children, pediatric inflammatory multisystem syndrome, coronavirus infection, mucocutaneous lymph node syndrome, Kawasaki disease, whole exome sequencing, whole exome sequencing
Citation: Reis BCSD, Soares Faccion R, de Carvalho FAA, Moore DCBC, Zuma MCC, Plaça DR, Salerno Filgueiras I, Leandro Mathias Fonseca D, Cabral-Marques O, Bonomo AC, Savino W, Freitas FCdP, Faoro H, Passetti F, Robaina JR, de Oliveira FRC, Novaes Bellinat AP, Zeitel RdS, Salú MdS, de Oliveira MBG, Rodrigues-Santos G, Prata-Barbosa A and Vasconcelos ZFMd (2023) Rare genetic variants involved in multisystem inflammatory syndrome in children: a multicenter Brazilian cohort study. Front. Cell. Infect. Microbiol. 13:1182257. doi: 10.3389/fcimb.2023.1182257
Received: 08 March 2023; Accepted: 23 June 2023;
Published: 31 July 2023.
Edited by:
Alessandra Pierangeli, Sapienza University of Rome, ItalyReviewed by:
Jennifer T. Grier, University of South Carolina, United StatesRandy Q. Cron, University of Alabama at Birmingham, United States
Copyright © 2023 Reis, Soares Faccion, de Carvalho, Moore, Zuma, Plaça, Salerno Filgueiras, Leandro Mathias Fonseca, Cabral-Marques, Bonomo, Savino, Freitas, Faoro, Passetti, Robaina, de Oliveira, Novaes Bellinat, Zeitel, Salú, de Oliveira, Rodrigues-Santos, Prata-Barbosa and Vasconcelos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zilton Farias Meira de Vasconcelos, emlsdG9uLnZhc2NvbmNlbG9zQGZpb2NydXouYnI=
†These authors have contributed equally to this work