
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 05 July 2023
Sec. Clinical Microbiology
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1180297
This article is part of the Research Topic Genomic variation and immune escape of viral pathogens View all 5 articles
 Qin Zhang1Runjie Qiao1Jiaojiao Niu1Xia Xiong1
Qin Zhang1Runjie Qiao1Jiaojiao Niu1Xia Xiong1 Nan Wang1Ruixian Zhang1Sha Luo1Yuwan Guo2Zhonghua Liu2Li Peng2Shaoduo Zhang2Guolei Tan1Keyu Song1Mei Sun1Lulu Xu1
Nan Wang1Ruixian Zhang1Sha Luo1Yuwan Guo2Zhonghua Liu2Li Peng2Shaoduo Zhang2Guolei Tan1Keyu Song1Mei Sun1Lulu Xu1 Rong Zhang2*
Rong Zhang2* Xuping Wu1*†
Xuping Wu1*†The Delta variant of SARS-CoV-2 dominated the COVID-19 pandemic due to its high viral replication capacity and immune evasion, causing massive outbreaks of cases, hospitalizations, and deaths. Currently, variant identification is performed mainly by sequencing. However, the high requirements for equipment and operators as well as its high cost have limited its application in underdeveloped regions. To achieve an economical and rapid method of variant identification suitable for undeveloped areas, we applied an amplification-refractory mutation system (ARMS) based on PCR for the detection of novel coronavirus variants. The results showed that this method could be finished in 90 min and detect as few as 500 copies/mL and not react with SARS-Coronavirus, influenza A H1N1(2009), and other cross-pathogens or be influenced by fresh human blood, α- interferon, and other interfering substances. In a set of double-blind trials, tests of 262 samples obtained from patients confirmed with Delta variant infection revealed that our method was able to accurately identify the Delta variant with high sensitivity and specificity. In conclusion, the ARMS-PCR method applied in Delta variant identification is rapid, sensitive, specific, economical, and suitable for undeveloped areas. In our future study, ARMS-PCR will be further applied in the identification of other variants, such as Omicron.
The emergence of novel coronavirus variants has been continuously reported since late 2019, including Alpha, Beta, Delta, Gamma, etc (Cherian et al., 2021; Grabowski et al., 2021; Singh et al., 2021; Tegally et al., 2021; Vaidyanathan, 2021; da Silva et al., 2022). Alpha, with a key mutation-N501Y in the receptor binding domain (RBD), was first detected in the UK in late 2020 and became the dominant variant in early 2021 (Davies et al., 2021). The Delta variant was first found in India in late 2020 and displaced Alpha as the dominant variant in mid-2021, leading to a resurgence of COVID-19 cases in many countries (Centers for Disease Control and Prevention, 2021; Bolze et al., 2022). A study from the US showed that the Delta variant was more transmissible than previous variants in part because of the higher viral load it caused during acute infection (Earnest et al., 2022). Another earlier study also reported that the Delta variant had a higher replication rate and immune escaping ability (Mlcochova et al., 2021). The Delta variant contains RBD mutation L452R and the proximal furin cleavage site mutation P681R (Tao et al., 2021). In addition to L452R and P681R, Kappa (B.1.617.1), which shares a common ancestor with Delta (B.1.617.2), also has an additional RBD E484Q mutation (Tao et al., 2021). L452R and E484Q mutations increased the interaction between the spike protein and ACE2, possibly leading to increased viral infectivity and immune escape (Cherian et al., 2021; Deng et al., 2021; Augusto et al., 2022). P681R mutation increased the cutting rate between S1/S2, increasing virus replication and ultimately resulting in higher transmissibility (Hoffmann et al., 2020; Cherian et al., 2021; Liu et al., 2022). Moreover, the Delta variant posed the great risk to countries with limited access to vaccines, particularly in Africa, where the vaccination coverage in most countries was less than 5% of their population (Callaway, 2021). Reduced sensitivity of the Delta variant to antibody neutralization and the lower effectiveness of vaccines after the receipt of the first dose against the Delta variant were reported (Liu et al., 2021; Lopez Bernal et al., 2021; Planas et al., 2021).
Due to the increased prevalence and potential dangers posed by the Delta variant, there is a critical need for a more effective method of detection. Real-time reverse transcription PCR, the most common method used to detect SARS-CoV-2, and other methods which have been developed, such as RT-LAMP and CRISPR, could determine only whether samples contain SARS-CoV-2 but do not identify the type of variants (Broughton et al., 2020; Corman et al., 2020; Dao Thi et al., 2020). Currently, the identification of virus variants relies mainly on whole-genome sequencing, in which the nucleotide sequences of unknown samples are obtained and compared with known pathogen sequences (Charre et al., 2020; Lu et al., 2020). However, whole-genome sequencing requires the availability of highly professional instruments, high testing costs, long testing time, and complex results analysis. These difficulties and deficiencies have limited its application in undeveloped areas, which cannot currently meet the requirements of rapid virus variant identification. In this regard, Aoki et al. developed a genotyping platform for SARS-CoV-2 variant identification using high-resolution melting analysis, but it has not been practically tested on clinical samples of COVID-19 patients (Aoki et al., 2021). Doubtless, the development of multiple platforms and methods that can detect the type of SARS-CoV-2 variants is highly necessary.
Amplification-refractory mutation system (ARMS) is a single, rapid, and reliable method for detecting any mutation involving single-base changes or small deletions (Newton et al., 1989; Little, 2001). It is commonly used to perform tests for point mutations in a variety of genetic diseases and cancers, such as phenylketonuria, thalassemia, and BRAF V600E mutation (Huang et al., 2013; Shaykholeslam Esfahani et al., 2018; Gholami et al., 2021). In this study, ARMS was established for detecting the Delta variant, which overcomes the shortcomings of sequencing. The assay was successfully tested on 262 clinical samples from patients infected with the Delta variant.
The combination mutations of L452R and P681R (nucleotide positions T22917G and C23604G) could differentiate B.1.617 lineage from other variants. The E484Q (nucleotide position G23012C) mutation could differentiate other variants from Delta among B.1.617 lineage. We designed three pairs of mutant primers for these three mutant sites. Primers and probes used in this assay were synthesized from Shuoying Biotechnology (Shanghai, China). Negative control consisted of a plasmid containing fragments of internal standard (RNaseP). Both positive control and negative control were synthesized by GeneoDx Biotech Co., LTD (Shanghai, China). The virus-like particles (VLPs) used to determine diagnostic cut-off values were ordered by Shuoying Biotechnology (Shanghai, China). The sequences of the positive and negative control are displayed in Table S1. The sequences of the primers and probes of L452R, E484Q, P681R mutations, ORF1ab gene, and RNaseP used in this experiment are presented in Table 1.
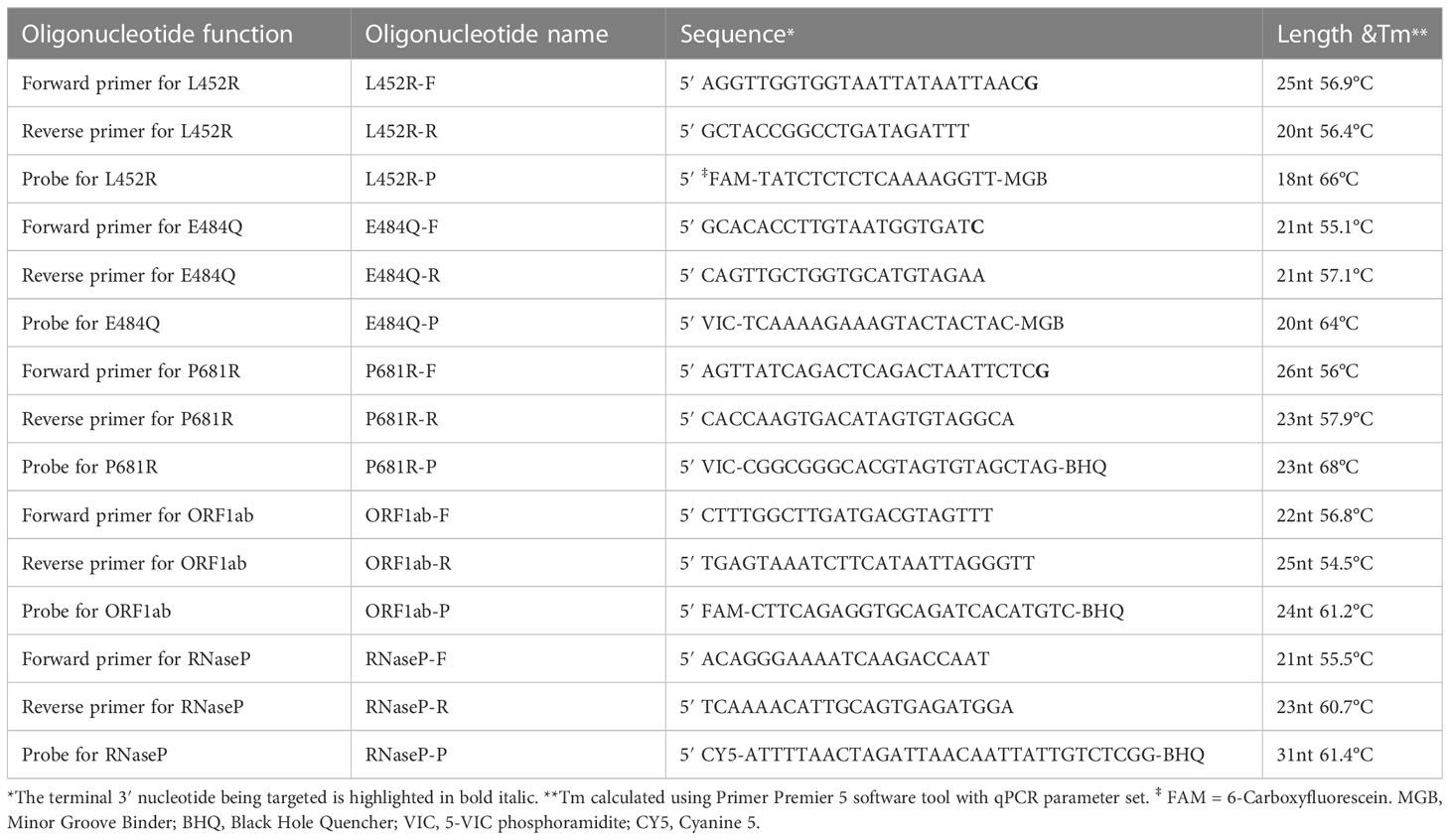
Table 1 Details of probes and primers sequences used in ARMS-PCR assay.
The ARMS-PCR assay, based on an amplification-refractory mutation system and real-time fluorescence PCR technology, was developed for the identification of Delta variant infection (Figure 1). In this experiment, S-gene was used as the target sequence to design mutant primers of L452R, E484Q, and P681R; ORF1ab gene was used as the target sequence to design internal control primer. The probes were labeled with different fluorescence to detect multiple targets in one reaction tube (Table 1). The mutant primers were efficient only for the mutation sequence amplification, while wild-type sequence greatly reduced the amplification efficiency, and even no amplification because of the mismatch between primers and bases. The fluorescence quantitative PCR instrument could automatically draw a real-time amplification curve based on the detected fluorescence signal thus achieving the mutation detection of L452R, E484Q, and P681R mutation sites of coronavirus S-gene according to the difference of Ct value of mutation and ORF1ab gene reaction (ΔCt). In this study, each sample was distributed in two reaction tubes, tube A and tube B. Reaction A mixture contained primers and probes of RNaseP and mutations L452R and P681R on the S-gene. Reaction B mixture contained primers and probes of the ORF1ab gene and mutation E484Q on the S-gene. ARMS-PCR was performed in a final mixture with a volume of 25 uL containing 5 uL of the sample, 7.5 uL of detection solution, 7.5 uL of nucleic acid amplification reaction solution, and 5 uL of enzyme mixture. The specific concentrations of the primers and probes are presented in Table S2. The high concentration of the primer and probe at the E484Q site was to obtain sufficient fluorescence values during the reaction. All amplification reactions were performed on an ABI 7500 fluorescence quantitative PCR instrument (Thermo Fisher Scientific, USA). The PCR reaction was carried out in three stages: at 50°C for 10 min, 97°C for 1 min, 45 cycles of 97°C for 5 sec, and 58°C for 30 sec (fluorescence collection).
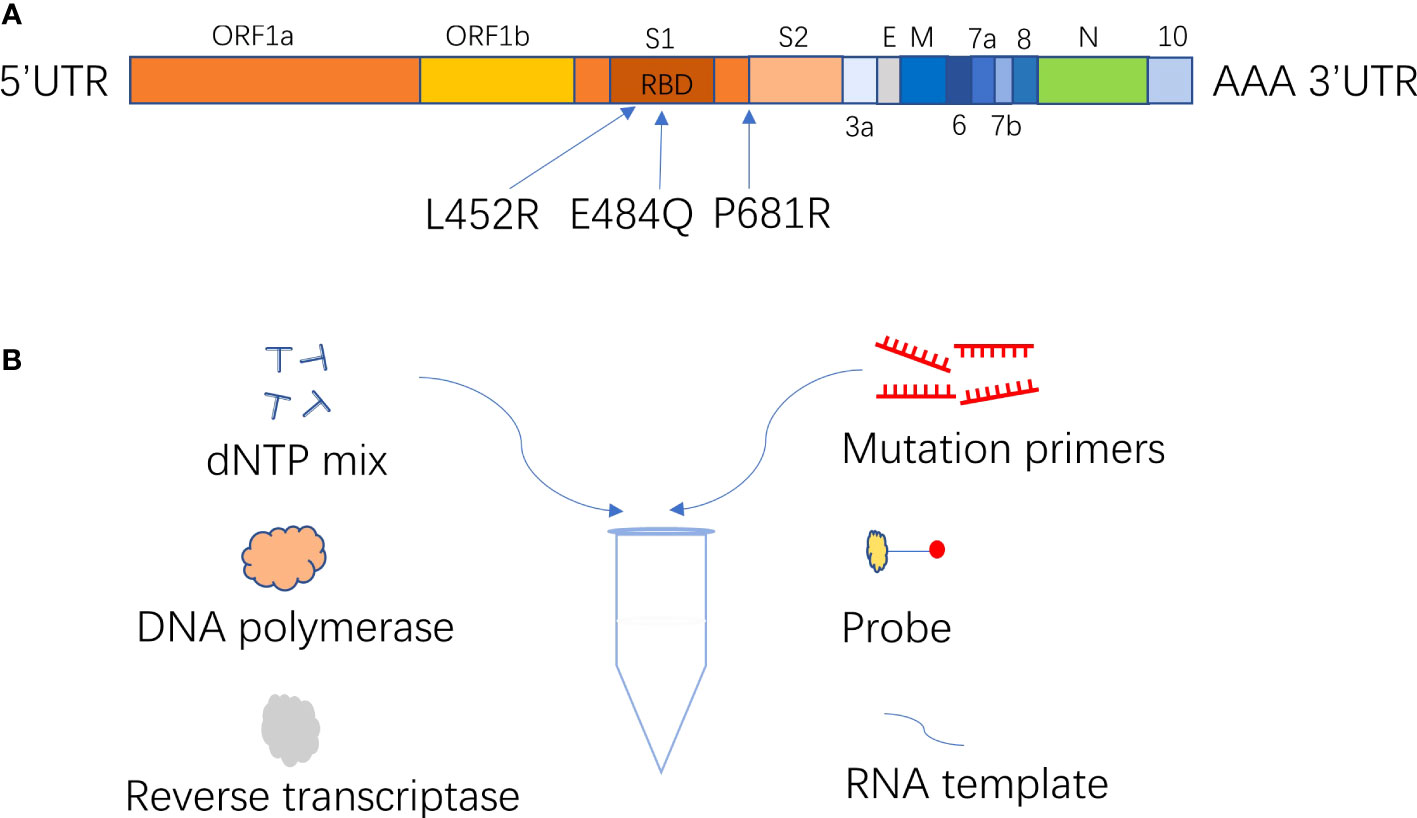
Figure 1 (A) Location of major mutation site of B.1.617; (B) ARMS-PCR diagrammatic diagram.
This method was designed mainly for variant identification rather than quantification. Therefore, calibration curves were not required. Calculate ΔCt of L452R, P681R, and E484Q mutation sites. ΔCt of the mutation X of a sample equals the Ct value of the mutation X (Mx) minus the Ct value of the ORF1ab: ΔCt(x) = Ct(Mx) – Ct(ORF1ab); ΔCt can be negative. If neither reaction produced Ct values in less than 40 cycles, it was interpreted as “no SARS-COV-2 detected.” The combination of mutations L452R and P681R were preliminarily classified into B.1.617 lineage. If the sample had additional mutation E484Q, it would be identified as B.1.617.1/3. Conversely, if the sample excluded the E484Q mutation, it would be identified as Delta variant (B.1.617.2). The test results of non-B.1.617 mutant samples and B.1.617.2 mutant samples can be seen in Figure 2.
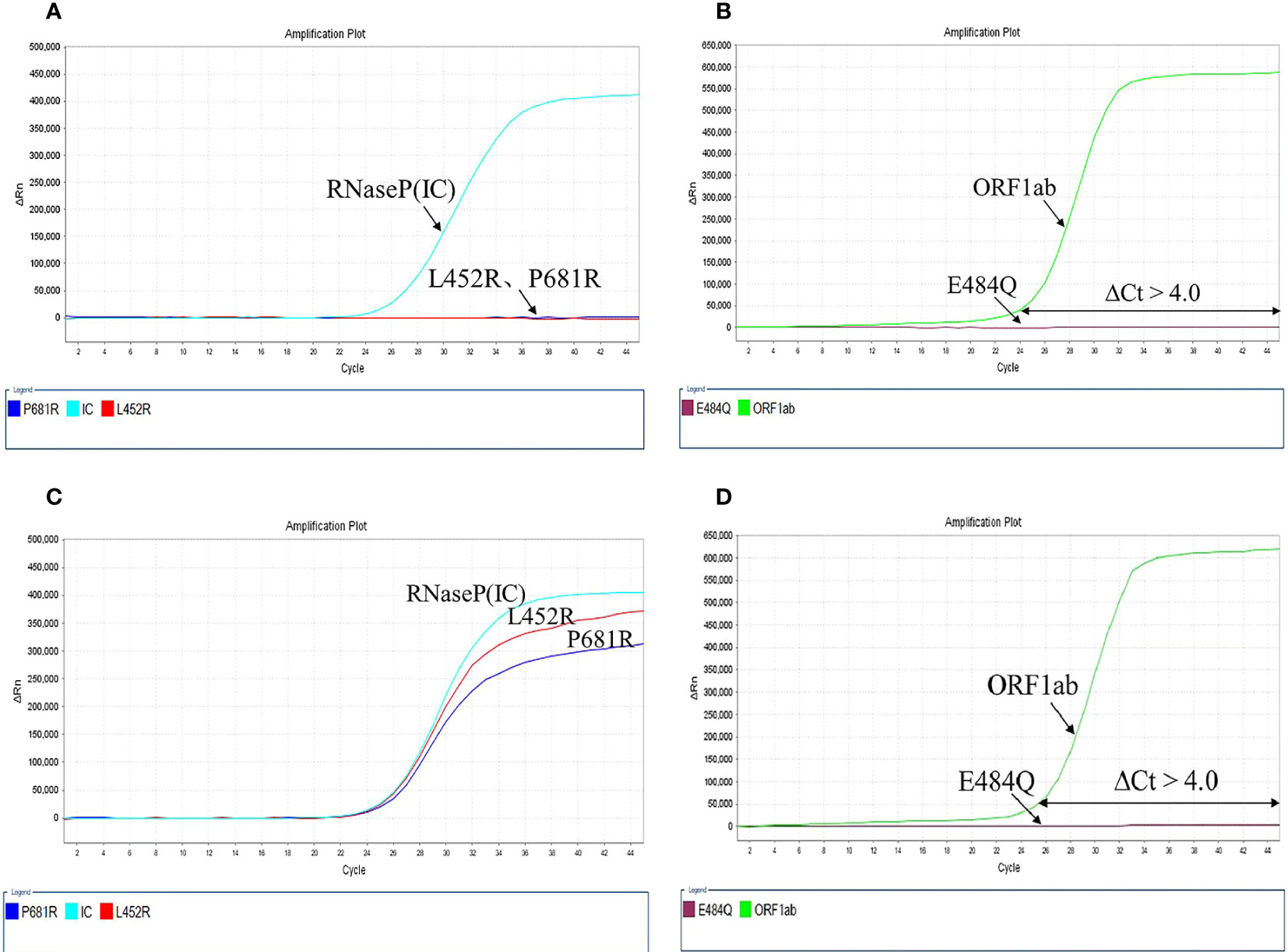
Figure 2 The results of the mutation test. (A, B) Non-B.1.617 mutant, L452R, P681R, and E484Q showed no mutation; (C, D) B1.617.2 mutant, L452R, P681R mutations with E484Q wild-type.
The diagnostic cut-off values of the L452R, E484Q, and P681R mutations and the ORF1ab gene on the S-gene of SARS-CoV-2 in our assay were evaluated based on the Ct values of 200 simulated samples including 100 cases of VLPs-simulated samples and 100 cases of negative samples using Receiver Operating Characteristic (ROC) curve in SPSS19.0 software. ROC curve is an effective method to evaluate the performance of diagnostic tests, which is defined as a plot of test sensitivity as the y coordinate versus its 1-specificity or false positive rate as the x coordinate (Park et al., 2004). The determination of the diagnostic cut-off ΔCt values was based on the ΔCt values results of 100 VLPs-simulated samples and 100 cases of negative samples using ROC curves in SPSS19.0 software.
Limit of detection (LOD) – The lowest amount of analyte in a sample that can be detected with (stated) probability, although perhaps not quantified as an exact value (revised from WHO-BS/95.1793). In this study, the lowest viral concentration level with a positive detection rate of 95% or greater was defined as the LOD. To determine the LOD of this method, we collected three types of samples, including nasopharyngeal swabs, throat swabs, and sputum samples. First, two nasopharyngeal swabs, two throat swabs, and two sputum samples were 10-fold serially diluted in five concentrations: at 5 × 105 copies/mL, 5 × 104 copies/mL, 5 × 103 copies/mL, 5 × 102 copies/mL, and 5 × 101 copies/mL, and each dilution was tested by one lot of reagents for 20 repeats. Then, we determined the concentration in the nasopharyngeal swabs, throat swabs, and sputum samples detected with a positive rate higher than 95% as the LOD. Directly count the number of positive results corresponding to each dilution and calculated the positive detection rate (Number of positive results (n)/Number of total repeated tests of each concentration level (N)). Next, two positive nasopharyngeal swabs, two positive throat swabs, and two positive sputum samples from different patients were diluted to the LOD concentration, and each dilution was tested with three lots of reagents for 20 repeats. The LOD was verified by a positive rate of at least 19/20.
To determine the specificity of the assay, we performed interference tests involving the cross-reactivity of pathogens which produce similar symptoms as SARS-CoV-2 and also involving potential endogenous and exogenous interfering substances. Samples in the cross-reactivity assay were prepared by adding cultured isolates or nucleic acid of the cross-pathogens (as shown in Table S3) into 37 low-concentration VLPs-simulated samples (3 × LOD) and 37 negative samples, respectively. Samples in interference testing were prepared by adding 4 potential endogenous and 6 exogenous interfering substances (as shown in Table S4) into 10 low-concentration VLPs-simulated samples (3 × LOD) and 10 negative samples, respectively. Each cross-pathogen or interfering substance was tested three times in low-concentration and negative samples. The experiments were performed on a QuantStudio™ 5 real-time PCR (Thermo Fisher Scientific, USA) instrument with one lot of reagents.
The clinical samples collected in this study were all obtained from one epidemic (Nanjing Lukou Airport epidemic). Based on the epidemiological investigation and sequencing data, the local CDC (Nanjing CDC) announced that this outbreak was caused by the spread of the Delta variant. On admission, all the patients were identified to have Delta infection. All samples analyzed in this study were from patients infected with the Delta variant. A total number of 262 clinical samples (nasopharyngeal swabs, n=249; throat swabs, n=9; sputum, n=4) were collected from patients diagnosed with Delta variant infection at the Second Hospital of Nanjing, a designated hospital for COVID-19 treatment in Nanjing, China. This study was approved by the Medical Ethics Committee of the Second Hospital of Nanjing (2021-LS-ky037). The nucleic acid extraction was performed on an automatic nucleic acid extraction instrument (Bioperfectus diagnostics, Jiangsu, China) using a nucleic acid isolation kit (magnetic beads) (Bioperfectus diagnostics, Jiangsu, China) following the manufacturer’s instructions. After extraction, RNA was eluted in 60 µL of RNase-free water and stored at -80°C for later use. The interference samples used in this study were provided by Taizhou Center for Disease Control and Prevention (Taizhou, Jiangsu, China).
The assay for detecting SARS-CoV-2 was performed with a commercial RT-PCR fluorescence diagnostic kit (Sansure Biotech, China), which was approved by the Chinese National Medical Products Administration (CNMPA), Food and Drug Administration (FDA), and Conformité Européenne (CE) Certification. This commercial kit has been widely used to detect SARS-CoV-2 in hospitals, CDCs, and third-party detection institutions in China. A Slan-96s real-time PCR machine (Hongshi, China) was used to detect and a Ct value below 40 was considered positive. SARS-CoV-2 RNA was mixed with amplification mixture and put into Slan-96s for reaction at 50°C for 30 min, 95°C for 1 min, and 45 cycles of 95°C for 15 sec, 60°C for 30 sec, and 25°C for 10 sec.
The 262 patients (158 females and 104 males) were clinically classified as 51 mild, 185 moderate, 19 severe, and 7 critical, as shown in Table 2. There was no significant difference in gender distribution, however, the age distribution showed an obvious difference. The average age was 48.61 ± 18.72. Patients older than 50 were more likely to develop into moderate, severe, and critical cases, while the ages of critical cases were all above 50. Besides, patients with basic diseases were more likely to develop into severe and critical cases. Hypertension and diabetes were the main basic diseases.
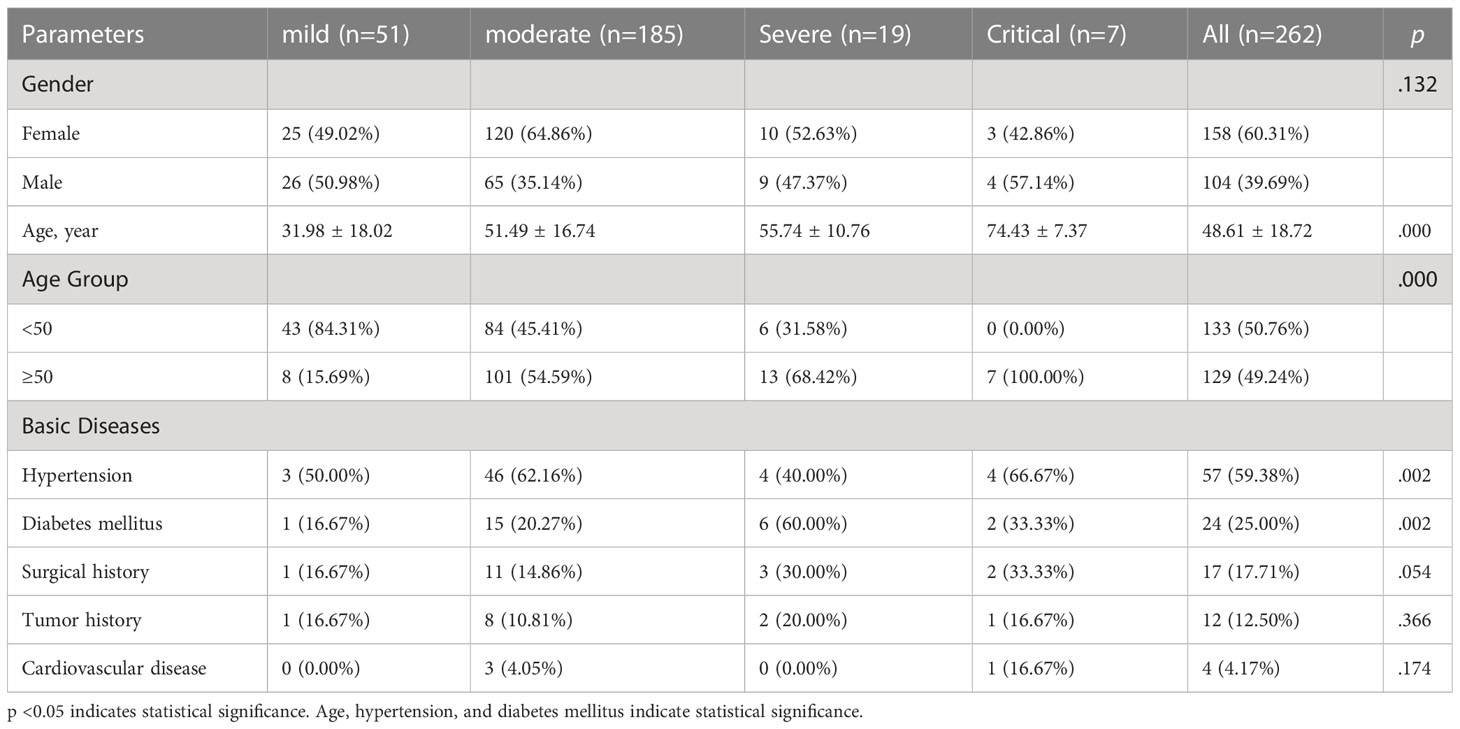
Table 2 Demographic characteristics in 262 patients with Delta variant infection.
The diagnostic cut-off values of L452R, E484Q, and P681R mutations on the S-gene and ORF1ab gene of SARS-CoV-2 were all equal to 40.0 to ensure high sensitivity based on ROC curves, as can be seen in Figures 3A–D and Table S5 (displaying the Youden’s index). Similarly, the diagnostic cut-off ΔCt values of L452R, E484Q, and P681R mutations on the S-gene of SARS-CoV-2 were determined as 10.0, 4.0, and 10.0, respectively, (Figures 3E–G; Table S6).
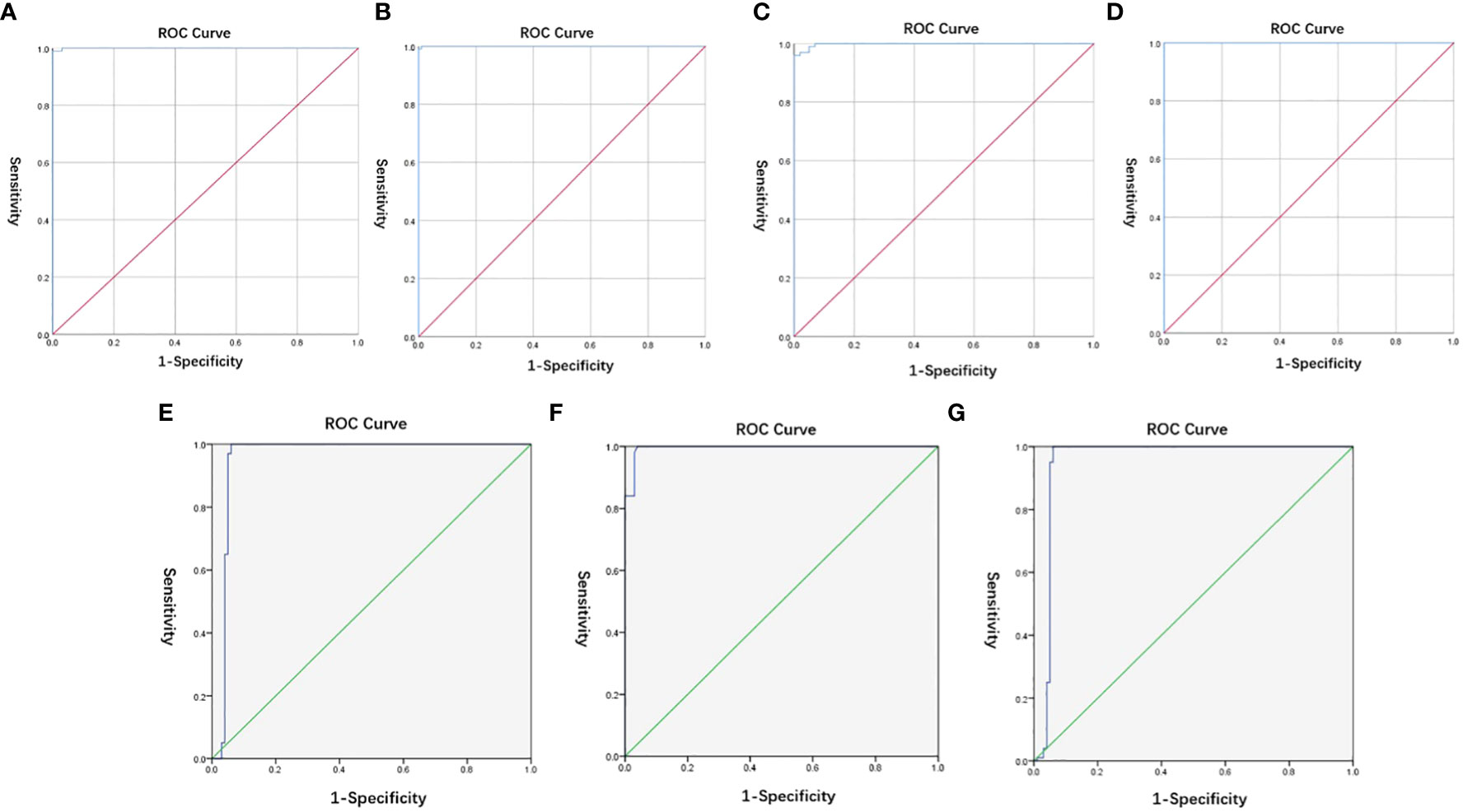
Figure 3 (A–D) ROC of cut-off values of L452R, E484Q, and P681R mutations on the S-gene and ORF1ab gene of SARS-CoV-2; (E-G) ROC of cut-off ΔCt values of L452R, E484Q, and P681R mutations on the S-gene of SARS-CoV-2.
Through 20 replicates performed on the five series of concentrations diluted samples, we found that a concentration of 5 × 102 copies/mL and above could be detected with a positive rate higher than 95% (Table S7). Thus, we determined 5 × 102 copies/mL as the LOD of this assay. Then, we further verified the LOD by testing two nasopharyngeal swabs, two throat swabs, and two sputum samples at a concentration of 5 × 102 copies/mL by three lots of reagents for 20 replicates. As visible in Table 3, three different types of samples were subjected to analysis with a positive detection rate higher than 95%. Therefore, the suggested here assay can detect as few as 5 × 102 copies/mL; hence, 5 × 102 copies/mL was determined and verified as the LOD in this study.

Table 3 Verification of LOD by nasopharyngeal swabs, throat swabs, and sputum.
As can be seen in Table S8, there was no cross-reaction with the cross-pathogens in negative samples and VLPs-simulated samples (3 × LOD) were also undisturbed by cross-pathogens at the test concentration. The potential endogenous and exogenous interfering substances in negative samples and VLPs-simulated samples (3 × LOD) at the test concentration did not interfere with the detection (Table 4). Therefore, the proposed approach has very good specificity which is not influenced by cross-pathogens and interfering substances.
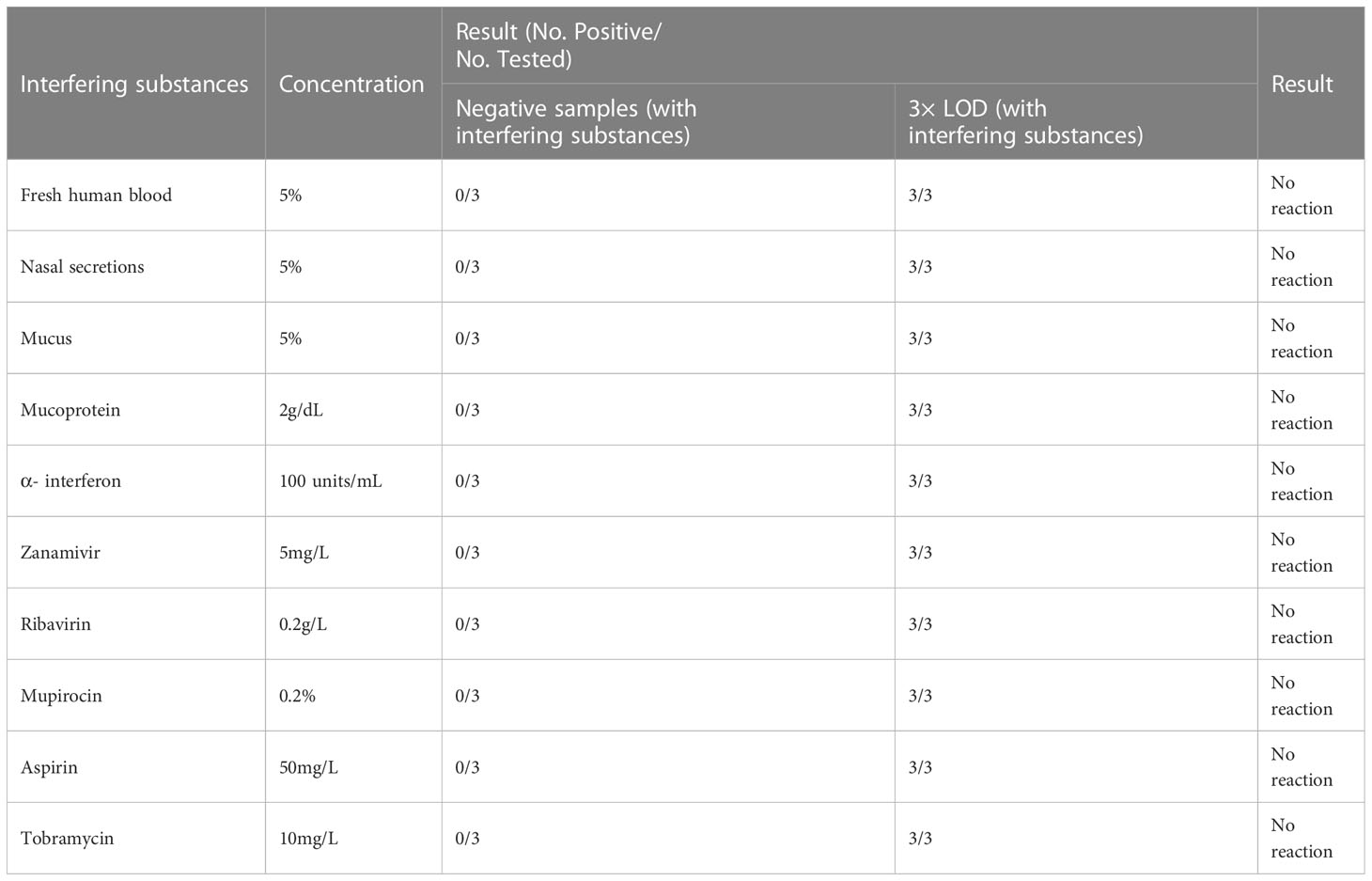
Table 4 Results of testing with potential endogenous and exogenous interfering substances.
After determining the sensitivity and specificity of ARMS-PCR, we further verified its performance in clinical samples. In a set of double-blind trials, we tested 262 clinical samples using both the RT-PCR kit (Sansure Biotech, China) and the ARMS-PCR assay to analyze and compare the differences between the two methods. As visible in Table 5, 245 positive samples and 17 negative samples were detected in the test results of the gold standard RT-PCR. The ARMS-PCR assay tested 235 positives and 27 negatives, of which 10 showed inconsistent results. Among the 17 samples with negative RT-PCR detection, ARMS-PCR detection also tested negative, showing good consistency. To analyze the difference in the test results between the two methods, we further evaluated and compared the performance of ARMS-PCR and RT-PCR in clinical positive samples (Table 6). To intuitively analyze the corresponding results of different Ct values, we divided these positive samples into three groups: a high group with Ct < 20, a middle group with 20≤Ct < 30, and a low group with Ct ≥ 30. Further analysis revealed that the Ct values of the samples with no results detected by ARMS-PCR were concentrated above the value of 35 (mainly distributed around 39), which meant that these samples had a low viral load (Table S9).
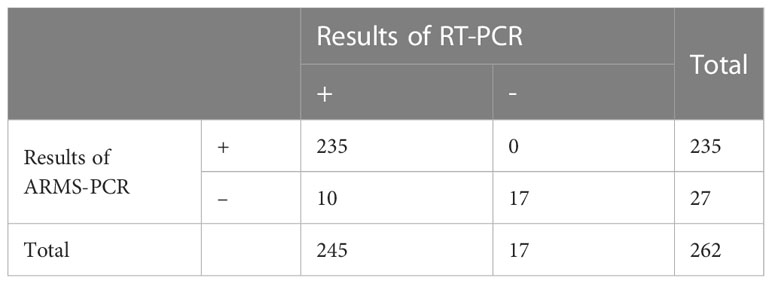
Table 5 The results of ARMS-PCR and RT-PCR in 262 clinical samples, of which 10 showed inconsistent results.
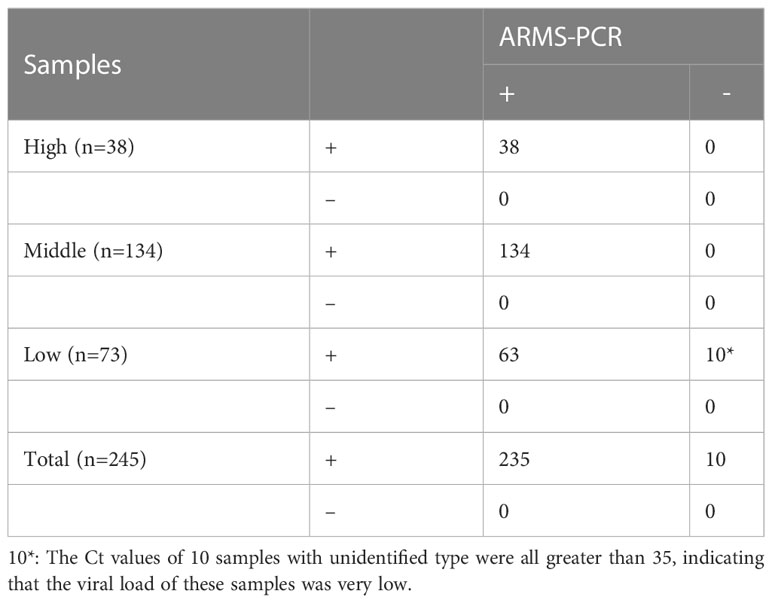
Table 6 The performance between ARMS-PCR and RT-PCR for detection of Delta variant in clinical positive samples.
Based on the data communicated at the press conference held by the local CDC, the transmission variant of this outbreak was identified as the Delta variant by sequencing. The samples used in our experiment were all obtained from COVID-19 patients with Delta variant infection that had been admitted to designated hospitals during the epidemic. As can be observed in Table 7, both L452R and P681R mutations were detected in all 235 clinical positive samples, but E484Q mutations were not detected in any of the 235 samples. Therefore, according to the explanation of this method, all 235 samples tested were identified as Delta variant (B.1.617.2), which was consistent with the epidemic findings reported.

Table 7 The results of type identification of ARMS-PCR in 235 clinical positive samples.
The Delta variant has higher transmissibility and pathogenicity than other variants, resulting in higher rates of hospitalization and mortality. The resurgence of the epidemic put a serious negative impact on the economy and social health welfare. The impact of the outbreak may be minimized by slowing the spread of variants in the bud. At this point, rapid tests play an important role in controlling variant spread. However, commercial rapid detection of SARS-CoV-2 antigens and antibodies has some shortcomings and cannot meet the need for accurate and rapid detection. Antigen tests are often less sensitive than nucleic acid tests due to the lack of amplification (Lai and Lam, 2021). Cross-reactivity with other pathogens causing false positives is a problem of antigen-antibody testing, but nucleic acid testing with specific primers and probes does not present this concern (Lustig et al., 2021). The lack of identification of variants is the critical problem of antigen and antibody tests. Therefore, it is necessary to develop multiple methods based on nucleic acid testing for variant identification.
In this experiment, specific mutant primers were designed for mutation sites L452R, P681R, and E484Q. The base mismatch between the primer and the template could effectively inhibit PCR reaction, thereby achieving the purpose of template differentiation. Because a single-base mutation could not cause a significant difference, an additional mismatch in some loci was introduced to ensure that chain extension could be effectively prevented. It is worth mentioning that we used internal standards (RNaseP) to monitor the collection, transportation, and extraction process of test samples during the experiment to avoid false negative results.
Several ARMS-PCR assays have been developed for disease diagnosis. For example, Esfahani et al. introduced a novel compound-primed multiplex ARMS-PCR (CPMAP) for simultaneous detection of common PAH gene mutations (Shaykholeslam Esfahani et al., 2018). Additionally, Huang et al. developed a method using an ARMS-PCR to detect the BRAF V600E mutation in formalin-fixed, paraffin-embedded (FFPE) tissue (Huang et al., 2013). The results of these studies indicate that ARMS-PCR has great potential in the diagnosis of diseases. In the present study, the ARMS-PCR assay could detect as few as 500 copies/mL of SARS-CoV-2 RNA and showed no cross-reaction with other cross pathogens, such as SARS-coronavirus, MERS-coronavirus, Influenza A H1N1(2009). The assay effectiveness was also not affected by endogenous and exogenous interfering substances, such as fresh human blood, mucus, aspirin, and tobramycin. A total number of 262 clinical samples were detected, whose results showed good agreement with RT-PCR and epidemiological investigation results. Since the detection failed in a small number of samples, we further analyzed the results and found that the Ct values of these samples detected by RT-PCR were basically above 35, mainly concentrated around 39, which was below the LOD of ARMS-PCR. More specifically, the LOD of RT-PCR (Sansure Biotech) used in our study was 200 copies/mL, whereas the LOD of the ARMS-PCR method we proposed was 500 copies/mL.
Nowadays, nucleotide sequencing was used to identify SARS-CoV-2 variants. Our developed ARMS assay has several advantages over conventional sequencing. First, it can be completed in less than 90 min and can be synchronized for high-throughput detection. Second, the whole experiment operation is equivalent to RT-PCR, which is simple and easy to implement in a routine laboratory. Third, for the interpretation of results, only computation of the cut-off values is necessary, whereas no complicated analysis comparing sequences is required, which saves manpower and material resources. Last, the ARMS-PCR method can directly use the existing conventional RT-PCR instruments in the laboratory, unlike sequencing, which needs to buy an expensive sequencer or send it to a third party for testing. Therefore, compared with the sequencing method, our method has the advantages of lower cost, faster detection, stronger availability, and more convenience (Simner et al., 2018; John et al., 2021). In fact, due to resource constraints and unequal distribution, sequencing can be used only for the detection of a small number of samples, even in countries with high sequencing capacity. Nevertheless, the method proposed here can be used for the detection of a large number of samples, requiring only a conventional PCR-level laboratory. In addition, our method can be used for molecular-level identification of samples with low viral loads that are difficult to meet the requirements of sequencing. In addition, compared with routine RT-PCR, this assay not only could detect SARS-CoV-2 but performed also preliminarily identification of its variant. A number of detection methods have been developed to identify novel coronavirus mutant types. For example, Garson et al. introduced double-mismatch allele-specific real-time reverse transcription PCR for the detection of SARS-COV-2 Delta variants, but only for the L452R and T478K sites of S spike (Garson et al., 2022). Further, Fabiani et al. reported a rapid and low-cost technique to distinguish the Alpha, Beta, Gamma, and Delta SARS-CoV-2 variants by detecting spike gene mutations using RT-PCR (Fabiani et al., 2022). However, all the samples in their study had a cycle threshold (Ct) value less than or equal to 30, implying uncertainty with Ct values above 30. Notably, 73 samples analyzed in our study had Ct values over 30 and only 10 samples that had Ct values over 35 were not successfully detected.
Nonetheless, our study still has a few shortcomings. Due to sample collection, most of the clinical samples used in this study were swabs, and the comparison with other sample types was lacking. Another concern of this approach is that due to the excessive mutation sites of S-gene, the relatively conservative ORF1ab gene was used as a substitute for internal control. One important future direction is to design S-gene as the internal control to improve the sensitivity and specificity of this method. Another significant future direction is to explore the use of the assay in different types of clinical samples, such as sputum. The experimental results in this study will facilitate the designing of mutant primers and developing ARMS to detect the latest variants, such as Omicron.
In conclusion, the proposed ARMS method can be used for rapid, sensitive, and specific identification of the B.1.617 lineage, especially the Delta variant. This novel method is simple, economical, and can be implemented in a conventional PCR laboratory, saving time and personnel costs.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of the Second Hospital of Nanjing (2021-LS-ky037). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. All samples used in this study were from the patient’s leftover swabs after clinical inspection and did not involve the patient’s privacy. Patient information collected in the case system did not contain name, address or other personal information, so the patient’s written informed consent was exempt. The study was conducted in accordance with Declaration of Helsinki.
Conceptualization: XW, RoZ. Methodology: XW, RoZ. Funding acquisition: XW. Project administration: XW, QZ, RoZ. Writing – original draft: QZ, XW. Writing – review and editing: XW, RoZ. Sample collection: MS, GT, KS, LX. Nucleic acid extraction: QZ, GT, KS, NW. Experimental operation: QZ, RuZ, YG, ZL. Material preparation: QZ, SL, LP, SZ. Manuscript Revision: XW, QZ, RQ, JN, XX. Data collection and analysis: RQ, JN, XX. All authors contributed to the article and approved the submitted version.
This work was supported by Social Development Project of Jiangsu Provincial Science and Technology Department (BE2022669).
Authors YG, ZL, LP, SZ, and RoZ were employed by the company Jiangsu Bioperfectus Technologies Company Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1180297/full#supplementary-material
Aoki, A., Mori, Y., Okamoto, Y., Jinno, H. (2021). Development of a genotyping platform for SARS-CoV-2 variants using high-resolution melting analysis. J. Infect. Chemother. 27 (9), 1336–1341. doi: 10.1016/j.jiac.2021.06.007
Augusto, G., Mohsen, M. O., Zinkhan, S., Liu, X., Vogel, M., Bachmann, M. F. (2022). In vitro data suggest that Indian delta variant B.1.617 of SARS-CoV-2 escapes neutralization by both receptor affinity and immune evasion. Allergy 77 (1), 111–117. doi: 10.1111/all.15065
Bolze, A., Luo, S., White, S., Cirulli, E. T., Wyman, D., Dei Rossi, A., et al. (2022). SARS-CoV-2 variant delta rapidly displaced variant alpha in the united states and led to higher viral loads. Cell Rep. Med. 3 (3), 100564. doi: 10.1016/j.xcrm.2022.100564
Broughton, J. P., Deng, X., Yu, G., Fasching, C. L., Servellita, V., Singh, J., et al. (2020). CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38 (7), 870–874. doi: 10.1038/s41587-020-0513-4
Callaway, E. (2021). Delta coronavirus variant: scientists brace for impact. Nature 595 (7865), 17–18. doi: 10.1038/d41586-021-01696-3
Centers for Disease Control and Prevention. (2021). Delta variant: what we know about the science. https://stacks.cdc.gov/view/cdc/108671 [Accessed June 20, 2022].
Charre, C., Ginevra, C., Sabatier, M., Regue, H., Destras, G., Brun, S., et al. (2020). Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 6 (2), veaa075. doi: 10.1093/ve/veaa075
Cherian, S., Potdar, V., Jadhav, S., Yadav, P., Gupta, N., Das, M., et al. (2021). SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in maharashtra, India. Microorganisms 9 (7), 1542. doi: 10.3390/microorganisms9071542
Corman, V. M., Landt, O., Kaiser, M., Molenkamp, R., Meijer, A., Chu, D. K., et al. (2020). Detection of 2019 novel coronavirus, (2019-nCoV) by real-time RT-PCR. Euro Surveill 25 (3), 2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045
Dao Thi, V. L., Herbst, K., Boerner, K., Meurer, M., Kremer, L. P., Kirrmaier, D., et al. (2020). A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 12 (556), eabc7075. doi: 10.1126/scitranslmed.abc7075
da Silva, J. F., Esteves, R. J., Siza, C., Soares, E. P., Ramos, T. C., Campelo, E. C., et al. (2022). Cluster of SARS-CoV-2 gamma variant infections, parintins, Brazil, march 2021. Emerg. Infect. Dis. 28 (1), 262–264. doi: 10.3201/eid2801.211817
Davies, N. G., Abbott, S., Barnard, R. C., Jarvis, C. I., Kucharski, A. J., Munday, J. D., et al. (2021). Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372 (6538), eabg3055. doi: 10.1126/science.abg3055
Deng, X., Garcia-Knight, M. A., Khalid, M. M., Servellita, V., Wang, C., Morris, M. K., et al. (2021). Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 184 (13), 3426–3437.e3428. doi: 10.1016/j.cell.2021.04.025
Earnest, R., Uddin, R., Matluk, N., Renzette, N., Turbett, S. E., Siddle, K. J., et al. (2022). Comparative transmissibility of SARS-CoV-2 variants delta and alpha in new England, USA. Cell Rep. Med. 3 (4), 100583. doi: 10.1016/j.xcrm.2022.100583
Fabiani, M., Margiotti, K., Sabatino, M., Viola, A., Mesoraca, A., Giorlandino, C. (2022). A rapid and consistent method to identify four SARS-CoV-2 variants during the first half of 2021 by RT-PCR. Vaccines (Basel) 10 (3), 483. doi: 10.3390/vaccines10030483
Garson, J. A., Badru, S., Parker, E., Tedder, R. S., McClure, M. O. (2022). Highly sensitive and specific detection of the SARS-CoV-2 delta variant by double-mismatch allele-specific real time reverse transcription PCR. J. Clin. Virol. 146, 105049. doi: 10.1016/j.jcv.2021.105049
Gholami, M. S., Shahidi, M., Tabibian, S., Naderi, M., Dorgalaleh, A. (2021). Genotyping of blood groups in alloimmunized patients with beta-thalassemia major by T-ARMS-PCR and multiplex-aso-pcr. Transfus. Apher. Sci. 60 (1), 102984. doi: 10.1016/j.transci.2020.102984
Grabowski, F., Preibisch, G., Gizinski, S., Kochanczyk, M., Lipniacki, T. (2021). SARS-CoV-2 variant of concern 202012/01 has about twofold replicative advantage and acquires concerning mutations. Viruses 13 (3), 392. doi: 10.3390/v13030392
Hoffmann, M., Kleine-Weber, H., Pohlmann, S. (2020). A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell. 78 (4), 779–784.e775. doi: 10.1016/j.molcel.2020.04.022
Huang, T., Zhuge, J., Zhang, W. W. (2013). Sensitive detection of BRAF V600E mutation by amplification refractory mutation system (ARMS)-PCR. biomark. Res. 1 (1), 3. doi: 10.1186/2050-7771-1-3
John, G., Sahajpal, N. S., Mondal, A. K., Ananth, S., Williams, C., Chaubey, A., et al. (2021). Next-generation sequencing (NGS) in COVID-19: a tool for SARS-CoV-2 diagnosis, monitoring new strains and phylodynamic modeling in molecular epidemiology. Curr. Issues Mol. Biol. 43 (2), 845–867. doi: 10.3390/cimb43020061
Lai, C. K. C., Lam, W. (2021). Laboratory testing for the diagnosis of COVID-19. Biochem. Biophys. Res. Commun. 538, 226–230. doi: 10.1016/j.bbrc.2020.10.069
Little, S. (2001). Amplification-refractory mutation system (ARMS) analysis of point mutations. Curr. Protoc. Hum. Genet. Chapter 9, Unit 9, 8. doi: 10.1002/0471142905.hg0908s07
Liu, C., Ginn, H. M., Dejnirattisai, W., Supasa, P., Wang, B., Tuekprakhon, A., et al. (2021). Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 184 (16), 4220–4236.e4213. doi: 10.1016/j.cell.2021.06.020
Liu, Y., Liu, J., Johnson, B. A., Xia, H., Ku, Z., Schindewolf, C., et al. (2022). Delta spike P681R mutation enhances SARS-CoV-2 fitness over alpha variant. Cell Rep. 39 (7), 110829. doi: 10.1016/j.celrep.2022.110829
Lopez Bernal, J., Andrews, N., Gower, C., Gallagher, E., Simmons, R., Thelwall, S., et al. (2021). Effectiveness of covid-19 vaccines against the B.1.617.2 (Delta) variant. N. Engl. J. Med. 385 (7), 585–594. doi: 10.1056/NEJMoa2108891
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395 (10224), 565–574. doi: 10.1016/S0140-6736(20)30251-8
Lustig, Y., Keler, S., Kolodny, R., Ben-Tal, N., Atias-Varon, D., Shlush, E., et al. (2021). Potential antigenic cross-reactivity between severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and dengue viruses. Clin. Infect. Dis. 73 (7), e2444–e2449. doi: 10.1093/cid/ciaa1207
Mlcochova, P., Kemp, S. A., Dhar, M. S., Papa, G., Meng, B., Ferreira, I., et al. (2021). SARS-CoV-2 B.1.617.2 delta variant replication and immune evasion. Nature 599 (7883), 114–119. doi: 10.1038/s41586-021-03944-y
Newton, C. R., Graham, A., Heptinstall, L. E., Powell, S. J., Summers, C., Kalsheker, N., et al. (1989). Analysis of any point mutation in DNA. the amplification refractory mutation system (ARMS). Nucleic Acids Res. 17 (7), 2503–2516. doi: 10.1093/nar/17.7.2503
Park, S. H., Goo, J. M., Jo, C. H. (2004). Receiver operating characteristic (ROC) curve: practical review for radiologists. Korean J. Radiol. 5 (1), 11–18. doi: 10.3348/kjr.2004.5.1.11
Planas, D., Veyer, D., Baidaliuk, A., Staropoli, I., Guivel-Benhassine, F., Rajah, M. M., et al. (2021). Reduced sensitivity of SARS-CoV-2 variant delta to antibody neutralization. Nature 596 (7871), 276–280. doi: 10.1038/s41586-021-03777-9
Shaykholeslam Esfahani, M., Shaykholeslam Esfahani, E., Vallian, S. (2018). A novel compound-primed multiplex ARMS-PCR (CPMAP) for simultaneous detection of common PAH gene mutations. Metab. Brain Dis. 33 (4), 1165–1173. doi: 10.1007/s11011-018-0210-x
Simner, P. J., Miller, S., Carroll, K. C. (2018). Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases. Clin. Infect. Dis. 66 (5), 778–788. doi: 10.1093/cid/cix881
Singh, J., Rahman, S. A., Ehtesham, N. Z., Hira, S., Hasnain, S. E. (2021). SARS-CoV-2 variants of concern are emerging in India. Nat. Med. 27 (7), 1131–1133. doi: 10.1038/s41591-021-01397-4
Tao, K., Tzou, P. L., Nouhin, J., Gupta, R. K., de Oliveira, T., Kosakovsky Pond, S. L., et al. (2021). The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 22 (12), 757–773. doi: 10.1038/s41576-021-00408-x
Tegally, H., Wilkinson, E., Giovanetti, M., Iranzadeh, A., Fonseca, V., Giandhari, J., et al. (2021). Detection of a SARS-CoV-2 variant of concern in south Africa. Nature 592 (7854), 438–443. doi: 10.1038/s41586-021-03402-9
Keywords: SARS-CoV-2, Delta variant, amplification-refractory mutation system, variant identification, molecular identification
Citation: Zhang Q, Qiao R, Niu J, Xiong X, Wang N, Zhang R, Luo S, Guo Y, Liu Z, Peng L, Zhang S, Tan G, Song K, Sun M, Xu L, Zhang R and Wu X (2023) Evaluation of an identification method for the SARS-CoV-2 Delta variant based on the amplification-refractory mutation system. Front. Cell. Infect. Microbiol. 13:1180297. doi: 10.3389/fcimb.2023.1180297
Received: 06 March 2023; Accepted: 19 June 2023;
Published: 05 July 2023.
Edited by:
Nahed Ismail, University of Illinois Chicago, United StatesReviewed by:
Melisa Intan Barliana, Universitas Padjadjaran, IndonesiaCopyright © 2023 Zhang, Qiao, Niu, Xiong, Wang, Zhang, Luo, Guo, Liu, Peng, Zhang, Tan, Song, Sun, Xu, Zhang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuping Wu, eHVwaW5nLnd1QG5qdWNtLmVkdS5jbg==; Rong Zhang, emhhbmdyb25nMTI5QDE2My5jb20=
†Present address: Xuping Wu, The Second Hospital of Nanjing, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, China
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.