 Laurence Don Wai Luu
Laurence Don Wai Luu Vasilli Kasimov
Vasilli Kasimov Samuel Phillips
Samuel Phillips Garry S. A. Myers
Garry S. A. Myers Martina Jelocnik
Martina Jelocnik- 1School of Life Sciences, University of Technology Sydney, Sydney, NSW, Australia
- 2Centre for Bioinnovation, University of the Sunshine Coast, Sippy Downs, QLD, Australia
- 3School of Science, Technology and Engineering, University of the Sunshine Coast, Sippy Downs, QLD, Australia
- 4Australian Institute for Microbiology and Infection, University of Technology Sydney, Sydney, NSW, Australia
The genus Chlamydia contains important obligate intracellular bacterial pathogens to humans and animals, including C. trachomatis and C. pneumoniae. Since 1998, when the first Chlamydia genome was published, our understanding of how these microbes interact, evolved and adapted to different intracellular host environments has been transformed due to the expansion of chlamydial genomes. This review explores the current state of knowledge in Chlamydia genomics and how whole genome sequencing has revolutionised our understanding of Chlamydia virulence, evolution, and phylogeny over the past two and a half decades. This review will also highlight developments in multi-omics and other approaches that have complemented whole genome sequencing to advance knowledge of Chlamydia pathogenesis and future directions for chlamydial genomics.
1 Introduction
Within the last 15 years, advances in whole genome sequencing (WGS) technologies and expansions in publicly available chlamydial genomes have dramatically increased our knowledge on chlamydial evolution and phylogeny, genome structure, metabolic processes, and potential virulence factors of these organisms. Presently, WGS is considered the gold standard for molecular epidemiology, evolution, and genetic diversity, and has become an integral part of cell biology studies in the genetically recalcitrant Chlamydia. The distinguishing feature of Chlamydia is its biphasic developmental cycle which alternates between extracellular infectious elementary bodies (EBs), which attach to and enter the host cell, and intracellular replicative reticulate bodies (RBs) residing in a membrane-enclosed inclusion. Upon RBs replication and inclusion growth, they dedifferentiate into EBs, to be released out of host cells and continue the infectious process (Elwell et al., 2016; Gitsels et al., 2019; Chiarelli et al., 2020). Under stress, Chlamydia RBs can also dedifferentiate into a non-replicating, persistent form, known as aberrant bodies (ABs) (Panzetta et al., 2018). The chlamydial lifecycle is central to the pathogenesis of these bacteria, as their survival depends on the complex host-pathogen interactions to establish an intracellular niche, subvert host cellular processes, acquire host-derived nutrients and evade the host immune response (Elwell et al., 2016; Panzetta et al., 2018; Gitsels et al., 2019; Chiarelli et al., 2020). Currently, the Chlamydiaceae consists of two genera: 1) Chlamydia (Figure 1), which harbours the following characterised species: C. abortus, C. avium, C. buteonis, C. caviae, C. crocodili, C. felis, C. gallinacea, C. muridarum, C. pecorum, C. pneumoniae, C. poikilothermis, C. psittaci, C. serpentis, C. suis, C. trachomatis, in addition to four Candidatus (Ca.) species: Ca. Chlamydia corallus, Ca. Chlamydia ibidis, Ca. Chlamydia sanzinia and Ca. Chlamydia testudines (Cheong et al., 2019; Zaręba-Marchewka et al., 2021) and, 2) the newly described Chlamydiifrater (Figure 1), harbouring the two recent species, C. phoenicopteri and C. volucris (Vorimore et al., 2021b). The availability of genome sequences for all novel and well-characterised species within the Chlamydiaceae provides an unparalleled opportunity to advance our understanding of the genetic diversity and taxonomy, genome biology, and gene content of these fascinating organisms (Figure 1). This review provides an overview of WGS methods and the most common genomic analyses, followed by a description of the chlamydial genome structure and content. It also provides details on the currently known events of genomic recombination as it relates to the threat of genetic transfer of antibiotic resistance genes, and challenges to the identification of lymphogranuloma venereum (LGV) outbreaks. Finally, we conclude with a discussion of anticipated directions for chlamydial genomics.
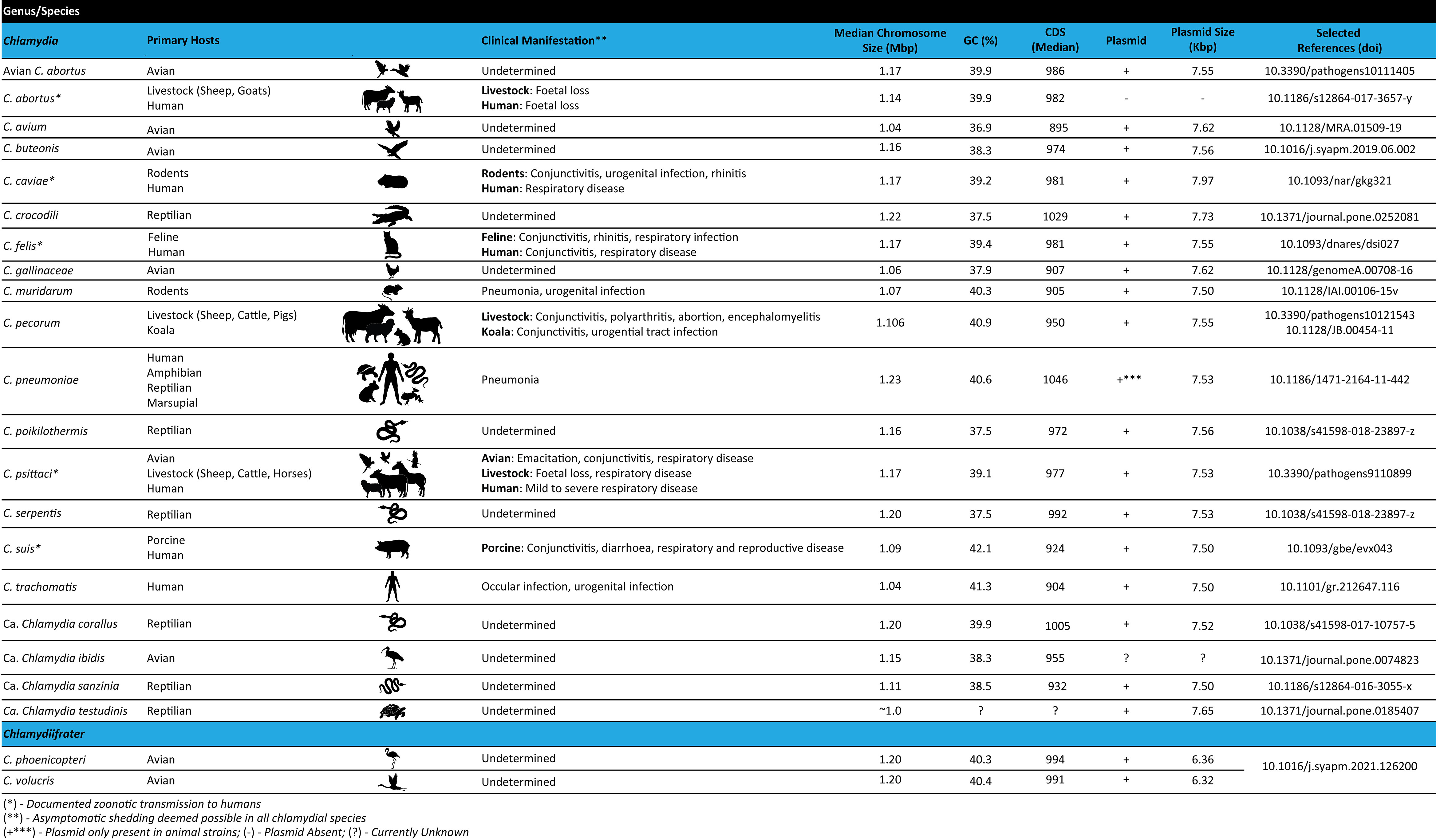
Figure 1 Features of the Chlamydiaceae family including primary host range, clinical manifestations and genome characteristics.
2 Chlamydial genomics: A new gold standard for chlamydial studies
2.1 WGS and genomic analyses
For most bacterial species, obtaining high-yield DNA extracted from cultured organisms remains the gold standard for WGS. Due to the intracellular niche of Chlamydiaceae, culturing and isolation is laborious, generally yielding lower levels of chlamydial genetic material contaminated with host DNA. In the past decade, there have been advances in obtaining chlamydial genomes from contaminated and/or low-yielding DNA samples retrieved from clinical and environmental samples using non-targeted and targeted approaches. These developments have resulted in a substantial increase of WGS data being generated (Bachmann et al., 2014; Taylor-Brown et al., 2018). New targeted WGS approaches are preferred for epidemiological and diagnostic purposes, particularly if the organism has already been sequenced, due to lower costs, higher throughput, and increased specificity (Taylor-Brown et al., 2018). However, for chlamydial discovery (e.g. novel species), non-targeted approaches are mostly utilised. Other methods also include expensive deep sequencing of contaminated samples using bioinformatic filtering to determine chlamydial sequences.
For WGS studies using (high-yield) DNA extracted from cultured isolates, direct next generation sequencing (NGS) is commonly used. Due to decreasing costs and ongoing developments in NGS, hybrid approaches such as nanopore sequencing to complement short-read WGS, are used to improve and/or provide closed complete genomes for Chlamydia (Heijne et al., 2021; Zaręba-Marchewka et al., 2021). In contrast, WGS of Chlamydia-specific and/or as a part of metagenomic sequencing from clinical swabs often yield poor results. However, this limitation was overcome with the development of culture-independent sample preparation methods, further supplemented with targeted capture methods (Joseph et al., 2015; Taylor-Brown et al., 2018; Zaręba-Marchewka et al., 2020). One of these methods is probe-based sequence-specific capture using biotinylated 120mer RNA probes called baits (Christiansen et al., 2014). This approach was successfully performed on WGS of C. trachomatis DNA from clinical samples (Christiansen et al., 2014; Hadfield et al., 2017; Seth-Smith et al., 2021), livestock and koala C. pecorum DNA from mucosal animal samples (Bachmann et al., 2015), DNA from archival C. pneumoniae strains (Roulis et al., 2015a; Roulis et al., 2015b), as well as C. psittaci clinical isolates (White et al., 2022) and DNA samples form a range of hosts (Branley et al., 2016). The recent development of third generation long-read sequencing technologies such as Oxford Nanopore and PacBio has already been used to provide high-quality complete Chlamydia genomes, resolving complex repetitive and recombination regions (Livingstone et al., 2021; Zaręba-Marchewka et al., 2021). These technologies also contain the additional potential for understanding chlamydial biology which is yet to be fully understood. For instance, Nanopore sequencing can directly detect DNA and RNA methylation in bacteria such as 6mA through changes in voltage signals when modified bases pass through the nanopore (Tourancheau et al., 2021), while PacBio Hi-C sequencing can be used to reveal the 3D genome structure and chromatin interactions (Lamy-Besnier et al., 2021). These novel technologies can be used to explore epigenetic modifications and 3D genome structure in different Chlamydia species, strains and/or stages of infection.
With the decreasing costs of deep sequencing, improvements in host DNA depletion techniques and advances in bioinformatic algorithms, shotgun metagenome sequencing which involves direct, untargeted sequencing of all microbial DNA in samples without the need for culture is becoming increasingly common. Shotgun metagenome sequencing provides new opportunities to study both Chlamydia and the microbiome; the microbiome has been shown to impact Chlamydia colonisation (Edwards et al., 2019). The successful recovery of metagenome-assembled genomes (MAGS) from shotgun metagenome data can also be used to discover new genera or species, as illustrated by the discovery of novel chlamydial lineages in anoxic marine sediments (Dharamshi et al., 2020).
Following WGS, analysis of Chlamydia genomes mainly follows a straightforward approach including de novo assembly into contigs and mapping approaches. Assembled sequences are subjected to genome annotation and estimation of genomic variability using single nucleotide polymorphisms (SNPs) and comparative approaches. Finally, a variety of phylogenomic analyses are performed to establish phylogenetic relationships among analysed strains and/or species (Joseph et al., 2015; Wolff et al., 2015; Hadfield et al., 2017; Sigalova et al., 2019; Hölzer et al., 2020; White et al., 2021; Zaręba-Marchewka et al., 2021; White et al., 2022). The reads and/or assembled genomes are deposited in publicly available databases so that they can be re-used across studies, providing a valuable resource for the chlamydial research community.
In chlamydial genomic studies, in silico analyses elucidating virulence factors and/or gene functions to explain phenotypes are also common. However, genes encoding for hypothetical or unknown proteins account for a significant proportion (39 – 45%) of most chlamydial genomes. Such hypothetical protein genes are conserved and considered “chlamydial-specific hypothetical protein genes”, with homologues commonly found among chlamydial species (Sigalova et al., 2019; Hölzer et al., 2020). Despite major advances in the genetic manipulation of chlamydiae, the function of many chlamydial virulence, tissue tropic-, and metabolic factors remain unknown (Bastidas and Valdivia, 2016). WGS and comparative genomics are efficient ways to characterise the genome content for strains of interest, further enabling detailed investigation of specific genotypes and delineating factors for host specificity, pathogenicity and tissue tropisms. One method for determining gene function is querying predicted Open Reading Frame (ORF) or coding DNA sequences (CDS) and assigning gene functions against a reference protein database (e.g. UniProtKB or Pfam-A database). Further gene and protein sequence homology or conserved domain analyses are commonly performed in chlamydial WGS annotation studies utilising publicly available or custom databases (Voigt et al., 2012; Read et al., 2013; Borges and Gomes, 2015; Seth-Smith et al., 2017a; Borges et al., 2019; Sigalova et al., 2019; Hölzer et al., 2020; Heijne et al., 2021; White et al., 2021). Recently, Pillonel and colleagues developed “open-access ChlamDB”, a comparative genomics database containing 277 genomes covering the entire Chlamydiae phylum as well as their closest relatives belonging to the Planctomycetes-Verrucomicrobiae-Chlamydiae (PVC) superphylum. Open-access ChlamDB provides various tools for comparing, analysing and retrieving Chlamydiae-specific genomic data (Pillonel et al., 2020). Such in silico analyses enable predictions of certain phenotypic or pathogenicity traits of chlamydial organisms. However, these require experimental validation, with limitations depending on the computational methods and resources used (Zhao et al., 2020). For genes encoding hypothetical proteins, comparative genomics to reveal conserved or absent genes in different lineages or tropic strains can provide valuable insight into their potential function and help identify genes for further investigation. Finally, using WGS in phylogenetic analyses can provide fine-detailed phylogeny, epidemiological networks and evolutionary pathways of Chlamydia species by estimating the chlamydial molecular clock. Without WGS techniques, this level of resolution would not be achievable, and certain evolutionary important lineages could be missed.
3 Chlamydial genome organisation and content: variability within similarity and synteny
Genomes of Chlamydiaceae are compact, remarkably conserved and syntenic. Similar to most other obligate intracellular bacteria, chlamydial genomes have a significantly reduced, circularised chromosome at approximately 1-1.2 Mbp with 900 - 1500 CDSs (Figures 1, 2) (Collingro et al., 2011; Voigt et al., 2012; Joseph et al., 2015; Pillonel et al., 2018; Sigalova et al., 2019). Almost all Chlamydiaceae species contain a highly conserved chlamydial plasmid of approximately 6.3 - 8 kbp, apart from livestock C. abortus strains, human strains of C. pneumoniae, and rare plasmid-free strains (Zhong, 2017; Szabo et al., 2020). The reduced genome for Chlamydiaceae species is a result of genome streamlining rather than degradation and is thought to be due to their transition to the intracellular lifestyle and co-evolution within their eukaryotic hosts (Moran, 2002; Collingro et al., 2011; Nunes and Gomes, 2014; Dharamshi et al., 2023). Within the same chlamydial species, the genomes are highly syntenic and similar, with a sequence homology of approximately 90% (Figure 2). Most of the differences are attributed to SNPs which have been postulated to play a role in virulence, host, and tissue tropism (Nunes et al., 2013; Read et al., 2013; Bachmann et al., 2014; Bachmann et al., 2015; Wolff et al., 2015).
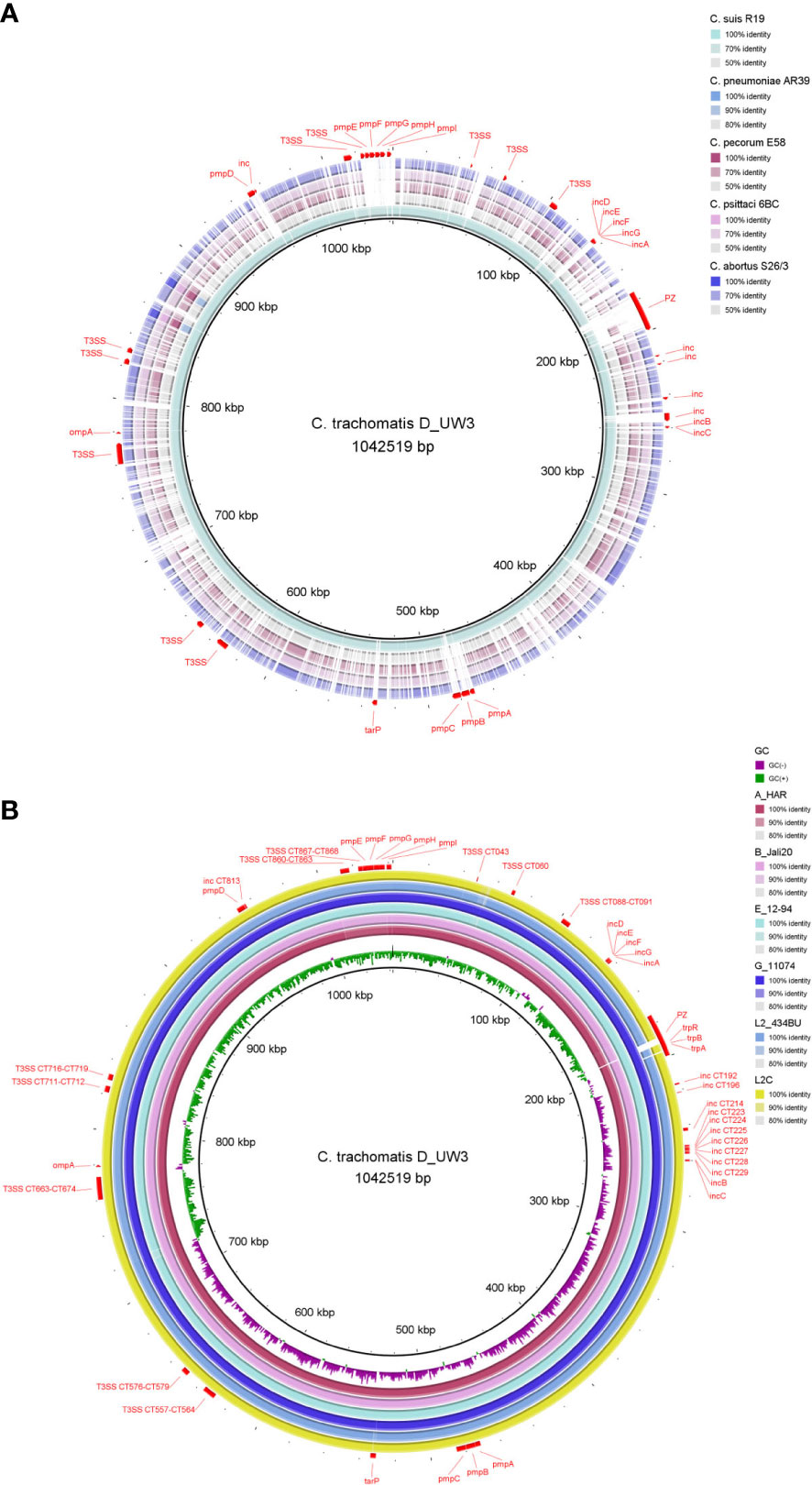
Figure 2 Chromosome comparison of selected genomes from (A) major human and animal Chlamydia pathogens (C. trachomatis, C suis, C pneumoniae, C pecorum, C psittaci and C abortus) and (B) C trachomatis strains from the three main lineages: ocular (A_HAR, B_Jali20), urogenital (D_UW3, E_12-94, G_11074) and LGV (L2_434BU, L2C). The C trachomatis strain D_UW3 was used as the reference. Important genome elements are annotated in red including the plasticity zone (PZ), ompA, pmps, Incs, T3SS, tarP and trp operon. The figure was generated using BRIG (v 0.95).
Almost all Chlamydia spp. genomes possess several common well-characterised metabolic, species-specific, and virulence genes or genomic regions. Recent pan-genomic analyses of 227 Chlamydia genomes demonstrated the degree of chlamydial similarity, with over 81% of genes universally (~700 universal genes) or partially conserved (~967 periphery genes), and only 19% of genes (~380 genes) unique to singular genomes. According to homology analyses, the periphery genes within species accounted for differences in organic molecule transport and metabolism, while unique genes contributed to differences in intracellular trafficking, secretion, and vesicular transport (Sigalova et al., 2019). Due to their host-dependency and reduced genome size, Chlamydiaceae lack genes encoding essential metabolic enzymes, requiring them to siphon amino acids, nucleotides, and cofactors from the infected host cell. This symbiotic relationship results in phenotypic variations involving tissue and host tropisms among different Chlamydiaceae species. The estimated number of pseudogenes in each genome is low possibly due to genome streamlining and a strong selection pressure to remove non-essential genes (Sigalova et al., 2019). The estimated number of pseudogenes ranges from 5 - 8 in C. pecorum, 9 - 12 in C. trachomatis, C. muridarum, and C. suis to 21 in C. pneumoniae and 28 - 29 in C. abortus and C. psittaci, with premature stop codons commonly occurring in genes encoding hypothetical proteins, and virulence-associated genes (pmps, Incs, and cytotoxin) (Voigt et al., 2012; Bachmann et al., 2014; Borges and Gomes, 2015; Sigalova et al., 2019; White et al., 2021).
Despite a high level of genome conservation, Chlamydiaceae possess several distinct polymorphic regions (Figure 2), with the highest variation occurring within the plasticity zone (PZ) (Figure 3). Other highly variable elements include genes encoding polymorphic membrane (pmps) and inclusion membrane proteins (Incs), Type III secretion system (T3SS), and other metabolic pathways, such as tryptophan biosynthesis (Bachmann et al., 2014; Sigalova et al., 2019; Hölzer et al., 2020) (Figure 2). Furthermore, Chlamydiaceae have been shown to possess strong recombination potential, particularly within virulence-associated genes, including the major outer membrane protein (ompA), T3SS effector translocated actin-recruiting protein (tarP), pmp genes and the PZ (Harris et al., 2012; Read et al., 2013; Joseph et al., 2015; Borges et al., 2019; White et al., 2021).

Figure 3 Comparison of the plasticity zone (PZ) from selected major human and animal Chlamydia species (C. suis, C. trachomatis, C. pecorum, C. psittaci, C. pneumoniae and C. abortus). The PZ elements are depicted in different colours, where biotin modification genes (accB, accC) are in purple, hypothetical protein (hyp) genes are in grey, MAC/perforin (MAC/P) in teal, Phospholipase D (PLD) in blue, purine interconversion genes (guaAB, add) in orange, Tryptophan operon genes (trp) in pink, and chlamydial cytotoxin (tox) gene(s) in yellow.
3.1 Tissue-tropic and virulence genes revealed by WGS
3.1.1 The plasticity zone
An evolutionary conserved but highly variable genomic region, the PZ is a genomic region associated with bacterial virulence protein genes and is responsible for distinct niche characteristics specific to different Chlamydia species (Rajaram et al., 2015; Dimond et al., 2021). Among the pathogenically diverse Chlamydia spp., the PZ ranges from ∼5 kb for C. avium (five genes), ~20 – 40 kb for C. abortus, C. psittaci and C. pecorum, (~15 to 20 genes), to ~55 kb and ~81 kb for C. trachomatis and C. muridarum (> 45 genes) (Figure 3) (Hölzer et al., 2020; Dimond et al., 2021; Zaręba-Marchewka et al., 2021). This region commonly includes homologs to biotin modification genes accB and accC, MAC/perforin (MacP), a variable number of phospholipase D-like genes (PLD), cytotoxin (tox) homologs, purine interconversion genes guaA and guaB, and a different subset of species-specific hypothetical proteins. Additionally, a near-full tryptophan (Trp) operon can be found in the PZ regions of C. caviae and C. felis, while in C. trachomatis and C. suis, the PZ regions only contain partial Trp operons (Bachmann et al., 2014; Rajaram et al., 2015; Zaręba-Marchewka et al., 2021).
Of these PZ elements, only accB and accC are common to all Chlamydia and are considered the 5’ boundary of the PZ, while other PZ genes vary between the species. Notably, chlamydial tox genes have significant homology to the large clostridial toxins (LCTs) and are present in almost all Chlamydia spp., however it may vary in copy number. C. muridarum has three tox copies, C. pecorum and C. suis two copies, while C. trachomatis, C. caviae, C. crocodile, C. felis, C. gallinacea, C. psittaci, C. buteonis, and avian C. abortus strains have one tox gene. A cytotoxin gene is absent from the PZs of the human pathogen C. pneumoniae and the ovine pathogen C. abortus (Figure 3) (Bachmann et al., 2014; Heijne et al., 2021; Zaręba-Marchewka et al., 2021). Significantly, tox gene polymorphisms have been useful in defining C. trachomatis disease phenotypes where the tox ORFs can be intact in urogenitotropic strains, truncated in oculotropic strains, or absent altogether in the LGV invasive strains (Figures 2B, 3) (Carlson et al., 2004; Nunes et al., 2013; Bachmann et al., 2014). Some PZ ORFs (e.g. species-specific hypothetical proteins) have no homologs or have been classified into families based on the nucleotide and/or protein sequence homology.
3.1.2 Type 3 secretion system genes
All Chlamydia spp. possess conserved genes encoding for structural, effector and chaperone proteins of the evolutionary conserved T3SS, essential for the translocation of effector proteins directly into host cells. The secreted T3SS effectors are recognised as virulence factors in Chlamydiae (Peters et al., 2007; Bachmann et al., 2014; Mueller et al., 2014; Mojica et al., 2015). While for some species, like C. trachomatis and C. psittaci, T3SS effectors have been experimentally confirmed and/or functionally characterised (Mojica et al., 2015; da Cunha et al., 2017; Marschall et al., 2020). For other less characterised chlamydial species WGS analyses is used to identify and decipher their roles (Hölzer et al., 2020; Heijne et al., 2021; White et al., 2021).
In Chlamydia spp. genomes, T3SS genes consist of 20 – 25 genes, encoding for highly conserved T3SS apparatus scaffolding (e.g. chlamydial CdsC, CdsJ, and CdsD homologs), inner membrane components (e.g. Cds R/S/T/U/V and Cds L/N/Q), extracellular needles (e.g. CdsF, CopN), chaperons (e.g. Ssc1 -3, CdsE and CdsG), and a variable number of genetically diverse effectors (e.g. tarP, SINC), and is usually found in four clusters across the chromosome (Betts-Hampikian and Fields, 2010; Borges and Gomes, 2015; Wolff et al., 2015; Bugalhão and Mota, 2019; Hölzer et al., 2020; Heijne et al., 2021; White et al., 2021).
In Chlamydia spp. genomes, 5 - 8% of the CDS are estimated to be effectors (Valdivia, 2008; Bugalhão and Mota, 2019; Andersen et al., 2021). Of the T3SS effectors, TarP is an extensively studied and functionally characterised T3SS effector, responsible for remodelling the actin cytoskeleton and facilitating the entry of Chlamydia into the host cell (Peters et al., 2007; Bugalhão and Mota, 2019; Andersen et al., 2021). Orthologs of tarP are present in all Chlamydia spp. genomes but show extensive sequence variation. Variations in the tarP gene sequences are hypothesised to contribute to virulence and variations to host tissue tropisms, as shown in C. psittaci and C. trachomatis. This is thought to be due to distinct N-terminal tyrosine-repeat units and C-terminal binding domains in C. trachomatis TarP (Borges and Gomes, 2015; Wolff et al., 2015). Another conserved T3SS secreted protein known as SINC (secreted inner nuclear membrane–associated Chlamydia protein) is a functionally characterised nuclear membrane targeting protein (Mojica et al., 2015; Marschall et al., 2020), which has been identified in C. psittaci, C. abortus, C. pecorum, C. caviae, C. felis, C. gallinacea, and C. avium genomes (Hölzer et al., 2020). The ability of SINC to manipulate infected and non-infected host cell nuclear envelope function has been hypothesised to contribute to the high virulence of C. psittaci (Mojica et al., 2015).
3.1.3 Inc genes
Inc proteins are virulence factors unique to the Chlamydiae phylum (Heinz et al., 2010). They are typified by one or more bilobed hydrophobic domains. These hydrophobic domains consist of two transmembrane helices separated by a hydrophilic loop, allowing Inc proteins to be inserted into an inclusion membrane (Dehoux et al., 2011). Inc proteins are translocated by the T3SS with the N-terminal containing the secretion signal. Inc proteins contribute to the physical structure of the inclusion membrane and perform diverse functions, including subverting vesicular trafficking for nutrient acquisition and avoiding lysosomal degradation (Elwell et al., 2016; Bugalhão and Mota, 2019).
Inc genes constitute a significant portion of the Chlamydia genome, accounting for 6% to 14% of the total coding capacity in the respective species. Comparative genomics revealed a significant variation in the number of predicted inc genes in different Chlamydia species, with 65 - 70 genes in C. trachomatis, C. muridarum and C. suis, 102 - 110 in C. psittaci and C. abortus and 140 - 147 in C. pneumoniae (Sigalova et al., 2019). Only a subset of Incs are conserved between species with ~23 Incs shared between C. trachomatis, C. muridarum, C. felis, C. caviae and C. pneumoniae (Lutter et al., 2012; Dimond et al., 2021). Of the Incs that are shared between species, sequence conservation is low among Chlamydia (Dehoux et al., 2011). Incs conserved across species may point to common protein functions and host interactions important in Chlamydia species, while non-conserved Inc proteins are hypothesised to contribute to host species differences. For instance, Inc genes differences account for approximately one-third of the genetic variations between C. trachomatis and C. pneumoniae. The increased number of Inc genes in C. pneumoniae may allow it to adapt to a broader host range (Dehoux et al., 2011).
In addition to species differences, Inc proteins may be responsible for differences in tissue tropisms and disease severity between C. trachomatis LGV and trachoma biovars. Analysis of C. trachomatis genomes showed that Inc genes were subject to positive selection with four times more non-synonymous SNPs compared to synonymous SNPs (Almeida et al., 2012; Borges and Gomes, 2015). Amino acid changes in Inc proteins were primarily specific to LGV strains and could phylogenetically separate LGV from ocular and urogenital strains (Almeida et al., 2012; Borges et al., 2012; Lutter et al., 2012). These amino acid changes occurred in regions predicted to be exposed to the host cytoplasm and thus may be involved in host-pathogen interactions and/or immune recognition. Also, LGV strains were found to have LGV-specific expression patterns from three Inc genes (CT058, CT192 and CT214). These expression differences correlated with specific LGV-mutations in the CT192 and CT214 promoters and within the CT059-CT058 transcript. These Inc sequence and expression differences may be associated with LGV strain tissue tropism to macrophages, possibly by inhibiting phagolysomal fusion (Almeida et al., 2012).
3.1.4 Pmp genes
Pmps are Chlamydiaceae unique, membrane-bound, type V surface-exposed autotransporters, associated with many chlamydia-host cell functions, including cell adhesion and virulence. All Pmps contain N-terminal Sec-dependent leader sequences (passenger domain) with multiple short repetitive motifs (GGA(I, L, V) and FxxN), a middle region and a C-terminal autotransporter β-domain. This autotransporter β-domain is responsible for translocating the protein to the bacterial surface (Mölleken et al., 2010; Nunes et al., 2015; Vasilevsky et al., 2016). The variable chlamydial pmp genes are grouped into several pmp gene families, where some are more evolutionary conserved in sequence and gene number (such as pmpA, pmpB, pmpD and pmpH families), while others are more varied (such as pmpG and pmpE families). The subtype pmpG is the most expanded in gene copy numbers. Among chlamydial species, the number of pmp genes ranges between seven and nine genes in C. avium, and C. trachomatis, to 18 and 21 genes in C. psittaci and C. pneumoniae, accounting for approximately 14% of the coding capacity of their genomes (Vasilevsky et al., 2016; Sigalova et al., 2019; Hölzer et al., 2020). Furthermore, bioinformatic analyses have shown that the subtype pmpG may undergo phase variation, by exhibiting differences in the lengths of in-frame poly(G) tracts. This may lead to random, high-frequency, on/off reversible switching of gene expression, involved in host adaptation and immune evasion (Nunes et al., 2015; Sigalova et al., 2019).
3.1.5 ompA gene
The ompA gene encodes for the chlamydial major outer membrane protein (MOMP), a ~40kDa surface-exposed porin protein containing four variable domains (VD I to VD IV), contributing to approximately 60% of the chlamydial outer membrane mass (Sun et al., 2007; Confer and Ayalew, 2013; Wen et al., 2016). Due to its antigenic properties, MOMP has been an attractive vaccine candidate against human and veterinary Chlamydia infection (Tifrea et al., 2020; Olsen et al., 2021; Quigley and Timms, 2021). The chlamydial ompA is a highly polymorphic gene specific to the genus Chlamydia, with approximately 30% of the sequence attributed to nucleotide polymorphisms and/or recombination (Nunes et al., 2009; Harris et al., 2012; Hadfield et al., 2017). Despite its highly recombinogenic nature, the chlamydial ompA gene is still a widely utilised marker for clinical, epidemiological, phylogenetic, and public health studies.
3.2 New insights into metabolic capabilities revealed by WGS
3.2.1 Nucleotide scavenging and energy production genes
Comparative genomic studies commonly note differences in metabolic genes and/or lack of pathways to synthesise most amino acids, including biosynthesis of biotin, tryptophan, thiamine, folate, purines, and pyrimidines nucleotides within and among chlamydial species.
Pyrimidine and purine nucleotides are necessary for energy transduction and the biosynthesis of nucleic acids for most bacterial species. However, Chlamydia spp. lack the necessary genes to synthesise pyrimidine and purine nucleotides, instead hijacking host cellular systems for their energy needs (Saka and Valdivia, 2010). Chlamydia genomes contain species-specific differences for purine conversion genes, with only C. caviae, C. felis, C. muridarum, C. psittaci, and C. pecorum possessing a functional guaAB-add operon capable of producing AMP adenosine deaminase, GMP synthase and IMP dehydrogenase. Conversely, species including C. trachomatis, C. gallinaceae, C. suis, C. pneumoniae, and C. abortus, are lacking these conversion genes (Figure 3) (Voigt et al., 2012; Nunes and Gomes, 2014; Hölzer et al., 2020).
Additionally, all Chlamydia genomes contain the pyrimidine interconversion genes pyrH, ndk and pyrG (uridylate kinase, nucleoside diphosphate kinase, and CTP synthase, respectively). These allow for the conversion of uridine monophosphate (UMP) to cytidine triphosphate (CTP). With the exception of C. trachomatis and C. muridarum, chlamydial genomes also contain the pyrE (orotate phosphoribosyltransferase) gene that encodes for uridine monophosphate synthetase (Saka and Valdivia, 2010; Voigt et al., 2012; Nunes and Gomes, 2014; Sigalova et al., 2019). Finally, only C. pneumoniae has a uridine kinase (udk) gene, which converts uridine or cytidine into UMP or CMP, respectively (Voigt et al., 2012; Nunes and Gomes, 2014).
Chlamydia utilises host-cell adenosine triphosphate (ATP), guanosine triphosphate (GTP), thymidine triphosphate (TTP) and uridine triphosphate (UTP) for survival (McClarty and Qin, 1993). Chlamydial species also contain a CTP synthetase gene (Npt1), which converts UTP to CTP and provides an alternative pathway for Chlamydia to obtain ATP (Saka and Valdivia, 2010; Sigalova et al., 2019). However, all other known mechanisms are lacking. Alternatively, WGS analyses have revealed that Chlamydia species contain genes for a complete glycolysis pathway (except for hexokinase, utilising hexose phosphate transporter to uptake host glucose-6-phosphate instead) and sodium-driven oxidative phosphorylation to generate its own ATP for energy or nucleic acid synthesis) and a hexose phosphate transporter, allowing Chlamydia to use glucose-6-phosphate as an energy source (Liang et al., 2018; Sigalova et al., 2019; Ende and Derré, 2020). However, this process would still depend on ADP availability from nucleotide uptake or interconversion.
3.2.2 Tryptophan operon
Trp is an essential amino acid used by bacterial species in protein biosynthesis. This pathway has been identified in chlamydial species, and is of particular interest. Genomic analyses of Chlamydia spp. have identified partial/complete gene loss in the trp operon resulting in the selective abilities of species to synthesise Trp. In human host cells, the pro-inflammatory cytokine, interferon-gamma (IFN-γ) stimulates the indoleamine-2,3-dioxygenase (IDO) enzyme to catabolise host tryptophan to kynurenine, essentially starving the invading organism. Due to this, IFN-γ treatment can be used for infection control (Bachmann et al., 2014; Somboonna et al., 2019).
A (near) complete and functional trp operon, which includes trpR-trpDCFBA and the complementary genes kynU and prsA, has been described in C. caviae, C. felis, C. pecorum. The trpR gene mediates tryptophan-dependent transcriptional repression, while the rest of the operon genes encode for anthranilate phosphoribosyltransferase (trpD), indole-3-glycerol phosphate synthase (trpC), phosphoribosylanthranilate isomerase (trpF), tryptophan synthase beta chain (trpB) and tryptophan synthase alpha chain (trpA). The complementary kynU and prsA encode for kynureninase and phosphoribosylpyrophosphate. However, C. suis and C. trachomatis have only partial trpR-trpBA genes, while the rest of the species lack all trp operon genes (e.g. C. abortus, C. gallinacea, C. pneumoniae and C. psittaci) (Nunes et al., 2013; Bachmann et al., 2014; Hölzer et al., 2020). Interestingly, C. pecorum was reported to contain a trp operon outside the PZ, while C. caviae and C. felis have a near-full trp operon in the PZ. Similarly, C. trachomatis and C. suis have been reported to contain only partial trp operons located in their PZs (Bachmann et al., 2014; Hölzer et al., 2020). Using an example of C. trachomatis, where trp genes are organised as an operon with trpR separated by a 348-base pair (bp) intergenic region from trpBA, a recent study demonstrated that these could be independently transcribed and not necessarily expressed as a monocistronic transcript. Pokorzynski and colleagues showed that iron starvation can switch on the Trp salvage pathway, mediated by the iron-dependent repressor YtgR, which binds to an operator sequence within the trpR-trpBA intergenic region to repress transcription initiation from the alternative promoter for trpBA (Pokorzynski et al., 2019; Pokorzynski et al., 2020).
Furthermore, in ocular strains of C. trachomatis, the trp operon has a loss of function due to a single nucleotide deletion and frameshift in trpA gene encoding a truncated tryptophan synthase alpha (TrpA) subunit. Conversely, the trp operon genes of C. trachomatis urogenital strains (D – K) are intact and functional, having the ability to produce tryptophan from indole, further allowing them to recover from IFN-γ exposure and resume their infectious cycle and this is supported by experimental studies (Fehlner-Gardiner et al., 2002; Ziklo et al., 2016; Somboonna et al., 2019; Bommana et al., 2021). For the chlamydial species, such as C. caviae and C. pecorum, which have the complementary genes, kynU and prsA, it has been shown that these species can synthesise tryptophan when limited in the presence of IFNγ, likely due to the presence of these genes (Wood et al., 2004; Islam et al., 2018). Overall, the biological significance of the identified trp polymorphisms remains uncharacterised. However, for some, one could infer with confidence, such as acquisition of kynU, which mediates anthranilate synthesis from kynurenine, instead of the canonical chorismate-to-anthranilate pathway facilitated by trpE. The intergenic region of the C. trachomatis trp operon has regulatory functions that potentially optimises trp operon expression in specific biological contexts, including iron starvation and mucosal epithelial tropism.
3.2.3 Biotin operon
Biotin is a critical cofactor involved in many central cell metabolism pathways (Fisher et al., 2012). Analysis of chlamydial genomes identified differences across species for biotin acquisition, where C. psittaci, C. abortus, C. felis, C. pecorum and C. pneumoniae have an intact biotin operon (bioBFDA) needed for de novo biotin synthesis. Other species, such as C. trachomatis, C. caviae and C. muridarum lack a functional biotin operon and thus depend on biotin salvaging (Fisher et al., 2012; Voigt et al., 2012; Nunes and Gomes, 2014; Sigalova et al., 2019). Interestingly, some species such as C. psittaci can synthesise biotin de novo and also sequester host biotin (Voigt et al., 2012).
3.3 Chlamydial plasmid
There is increasing evidence that the chlamydial plasmid is a key virulence factor and determinant of pathogenicity. Almost all Chlamydia spp., except C. abortus and human C. pneumoniae strains, contain a small (~7.5 kbp), highly conserved, non-conjugative or integrative chlamydial virulence plasmid (Pawlikowska-Warych et al., 2015; Szabo et al., 2020). Within some of the plasmid-bearing species, naturally occurring plasmid-less strains can be common, as observed in C. pecorum (Jelocnik et al., 2015), or rare, as seen in C. trachomatis (Sigar et al., 2014; Jones et al., 2020).
The chlamydial plasmids contain eight CDSs, named plasmid glycoproteins 1 to 8 (pGP 1-8), and four tandem repeat sequences, with each iteration comprising of 22 base pairs near the origin of replication (Pawlikowska-Warych et al., 2015; Zhong, 2017; Szabo et al., 2020). Briefly, CDSs 1 (pGP7) and 2 (pGP8) encode a putative integrase, involved in plasmid replication, CDS3 (pGP1) is homologous to a helicase, while CDSs 4 (pGP2) is an unknown chlamydia-specific protein. CDS5 (pGP3) encodes a secreted virulence 28 kDa protein essential for establishing persistent infection (Yang et al., 2020) and CDS 6 (pGP4) encodes a chlamydia-specific transcriptional regulator for multiple genes located within both the plasmid and chromosome including pGP3 and glgA, a glycogen synthase gene (Song et al., 2013). A recent study also demonstrated that in C. trachomatis pGP3 or pGP4 are both required for infectivity (Turman et al., 2023). Finally, CDSs7 and 8 (pGP5 and 6) are denoted as partitioning plasmid proteins (Rockey, 2011; Pawlikowska-Warych et al., 2015; Zhong, 2017; Jones et al., 2020).
Sequence and phylogenetic analyses indicate that chlamydial plasmids are evolutionarily conserved and syntenic between species, but within species, chlamydial plasmids contain unique sequence variations, further supporting the notion that chlamydial plasmids have co-evolved with the chromosome (Seth-Smith et al., 2009; Jelocnik et al., 2015; Szabo et al., 2020). In C. trachomatis diagnostics, the chlamydial plasmid CDSs are useful targets for diagnostic testing due to their stability, and high copy number, with up to ten copies per genome (Seth-Smith et al., 2009). However, the emergence of a urogenital mutant strain from Sweden containing a 377 bp deletion in plasmid gene CDS1 led to this plasmid locus being abolished as a diagnostic target due to false negative test results (Seth-Smith et al., 2009). This variant was characterised using WGS, and has since been occasionally reported in other countries (Piñeiro et al., 2014; Dahlberg et al., 2018; Escobedo-Guerra et al., 2019).
3.4 Other genomic elements
3.4.1 Antibiotic resistance genes
There are limited reports of antibiotic-resistant genes (ARGs) in the genomes (including both chromosome and chlamydial plasmid) of Chlamydia spp. However, due to observed treatment failures, WGS studies continue to monitor for chromosomal genes and mutations associated with Chlamydia spp. resistance to antibiotics (Borel et al., 2016; Benamri et al., 2021).
The only ARG identified in Chlamydia spp. is the tetracycline resistance gene tetA(C) found in tetracycline-resistant C. suis strains, the only chlamydial species to have naturally obtained a resistance gene (Borel et al., 2016; Seth-Smith et al., 2017a). The tetA(C) gene encodes a tetracycline efflux pump which has been integrated as a genomic island (Tet-island) into the C. suis invasion-like gene (inv) on the chromosome. This integration most likely occurred during a transposition event directed by the transposase-encoding insertion sequence IScs605 (Joseph et al., 2016; Marti et al., 2022). The Tet-island is currently the only evidence for recent acquisition of foreign DNA from other bacterial species in Chlamydia as it shares high nucleotide identity with a pRAS3-type plasmid from the fish pathogen Aeromonas salmonicida ssp. salmonicida (Marti et al., 2022).
4 Recombination and horizontal gene transfer in chlamydial genomes
Due to the biphasic and intracellular life cycle of Chlamydia species, chlamydiae do not commonly replicate in environments occupied by other microorganisms. Most bacterial species gain foreign DNA, including antibiotic-resistance genes through environmental interactions with other bacterial species. Despite an intracellular niche, chlamydial species have developed an increased ability to undergo genomic recombination (Harris et al., 2012; Read et al., 2013). Although Chlamydia genomic recombination had been identified prior to 2004, Gomes et al. demonstrated that C. trachomatis recombination was frequent. Using phylogenetic analysis of 19 reference strains and 10 clinical isolates, two genomic regions were found to be hotspots for interstrain recombination (Gomes et al., 2007). Genetic recombination hotspots for other Chlamydia species have also been identified in C. suis (Marti et al., 2017; Seth-Smith et al., 2017a), C. psittaci (Read et al., 2013; Branley et al., 2016), C. pecorum (White et al., 2021), and even in small regions in the otherwise monomorphic C. abortus (Seth-Smith et al., 2017b). Genomic regions with evidence of recombination include the genes IncA, pmps, tarp and the PZ (Gomes et al., 2004; Harris et al., 2012; Joseph et al., 2012; Borges and Gomes, 2015; Seth-Smith et al., 2021).
The most convincing evidence indicating the ability of chlamydial species to undergo genetic recombination (horizontal gene transfer (HGT)) was reported in co-culture studies (Marti et al., 2022). Co-culture of C. suis carrying the tetC gene with C. trachomatis, C. muridarum, and C. caviae confirmed genomic integration of the tetC island in both C. trachomatis and C. muridarum (but not C. caviae) (Suchland et al., 2009). Similarly, chlamydial interspecies HGT was analysed using crosses of tetracycline (Tc)-resistant C. trachomatis L2/434 and chloramphenicol (Cam)-resistant C. muridarum VR-123. WGS of the recombinant clones identified examples of duplications, mosaic recombination endpoints, and recombined sequences that were not linked to the selection marker (Suchland et al., 2019). A more recent study co-cultured tetracycline-resistant C. suis with rifamycin group-resistant C. suis resulting in an overall in vitro recombination efficiency of 28% (Marti et al., 2021). However, these observations have not been found in clinical isolates, and tetracyclines still remain part of the first-line defence antibiotics for human chlamydial infections.
5 Whole genome phylogenies resolve fine-detail relationships between Chlamydia species and strains
The use of WGS combined with sample-specific metadata have provided important observations on the contemporary history of current C. trachomatis circulating lineages within global populations (Andersson et al., 2016; Hadfield et al., 2017; Seth-Smith et al., 2021). It is well established that C. trachomatis is comprised of three distinct lineages, where ompA genotypes A, B, Ba and C are associated with trachoma, genotypes D-K associated with non-invasive UGT infections, and genotypes L1-L3 and L2b associated with invasive infections and LGV (Harris et al., 2012; Joseph et al., 2012; Andersson et al., 2016; Hadfield et al., 2017) (Figure 4A). However, phylogenomic studies show that not all strains may fit into this traditional separation, perhaps reflecting C. trachomatis adaptation to their respective niche and the noted recombination events. For example, whole-genome phylogenetic analyses of ocular isolates obtained from Australian Aboriginal people with trachoma placed these isolates into two lineages that fall outside the classical trachoma lineage (A-C). Instead, these genetically distinct strains clustered within lineages that were previously occupied exclusively by UGT isolates (namely genotypes D–K). These trachoma isolates appear to be recombinants with the C. trachomatis UGT genome backbones, where recombination was noted in ompA and pmp loci replacing them with those ompA and pmp genotypes characteristic of ocular isolates (Andersson et al., 2016). Similarly, recent evidence has suggested that clinically relevant recombination events have occurred between two C. trachomatis strains (serovar D and LGV). Recent outbreaks of LGV (L2b strain) within men who have sex with men (MSM) populations have resulted in genetic recombination of the ompA gene from non-LGV strains (Borges et al., 2019; Borges et al., 2021). A previously undetected outbreak of LGV may have gone undiagnosed due to over- reliance on the ompA gene for identification. Further genomic analysis found that the C. trachomatis L2B strain had undergone genetic recombination, acquiring ompA from the non-LGV strain C. trachomatis D-Da (Borges et al., 2019). Furthermore, follow-up studies have indicated that this L2b/D-Da variant is spreading across Europe and, with the continued reliance on ompA for strain identification, the probability of further dissemination across the globe is high (Borges et al., 2021).
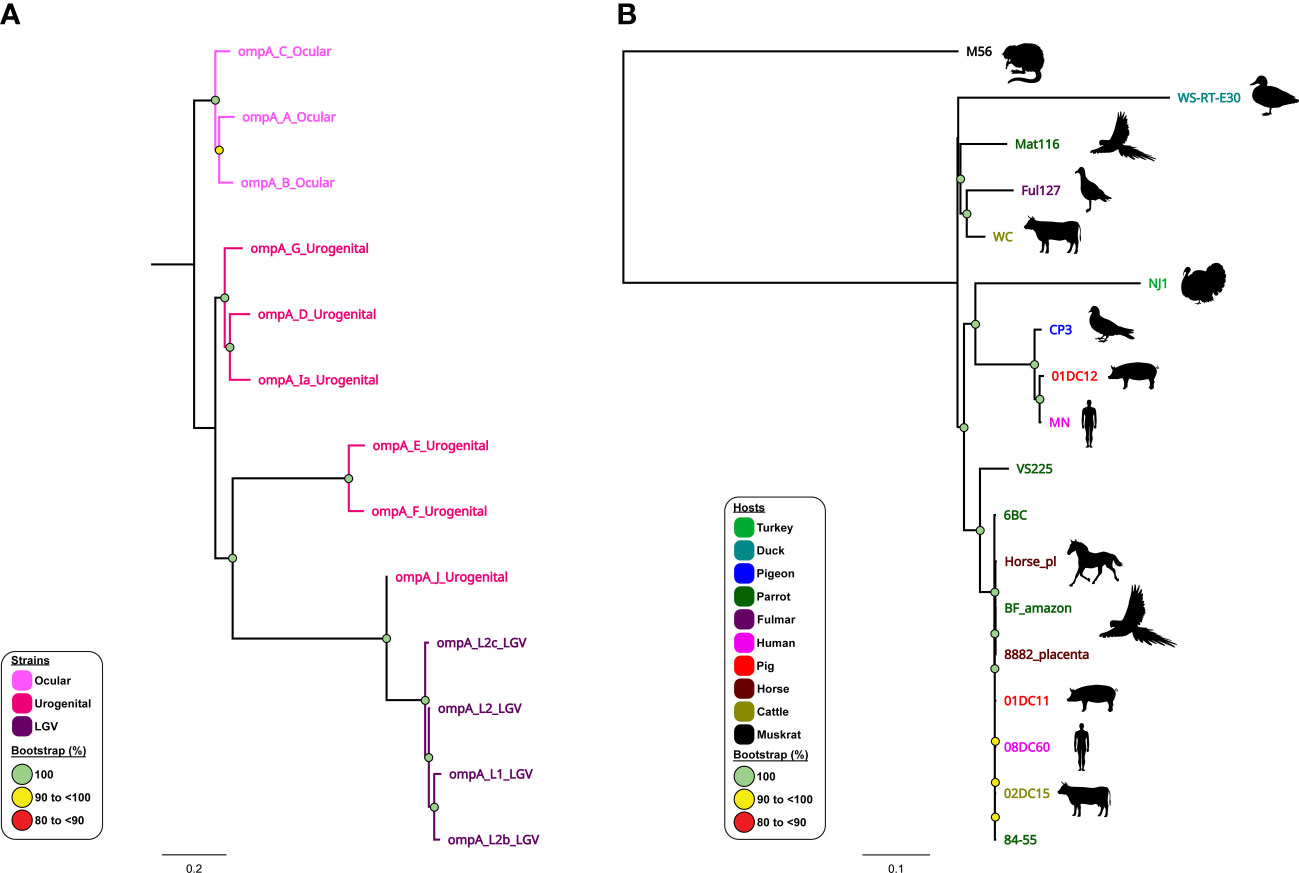
Figure 4 Phylogenomic relationships of C trachomatis and C psittaci reference strains. Midpoint-rooted maximum-likelihood (ML) core-genome phylogenetic trees constructed using Parsnp version 1.2 of (A) 13 aligned publicly available complete C trachomatis genomes; and (B) 18 aligned publicly available complete C psittaci genomes. For C trachomatis, genome sequences are coloured according to tissue tropism/known genotypes (as depicted in the figure legend). For C psittaci, genome sequences are coloured according to their respective host (as depicted in the figure legend). Corresponding host silhouettes are shown adjacent to sequence names. Bootstrap values greater than 0.8 are shown. Branch lengths represent the nucleotide substitutions per site, as indicated by the scale bar.
Similarly, phylogenomic analyses confirmed that modern human C. pneumoniae strains have evolved separately from animal strains (Roulis et al., 2015b), while further demonstrating a distinct Australian indigenous C. pneumoniae clade pre-dating European exploration of the continent (Roulis et al., 2015a). Phylogenomic analyses have demonstrated that whilst the global C. psittaci population is genetically diverse, there are also recently emerged, globally distributed and highly clonal lineages (such as C. psittaci ST24 strains) that infect humans, parrots, and, most recently, horses. Within this highly clonal ST24 lineage, the equine, human and parrot strains differ by less than 200 single nucleotide variants (SNVs), evenly distributed around the highly conserved and syntenic chromosome (Read et al., 2013; Branley et al., 2016; Jenkins et al., 2018; White et al., 2022; White et al., 2023). However, between genetically diverse C. psittaci lineages (such as ST24 and pigeon-associated clade) SNVs and differences in gene content were noted in distinct chromosomal regions, including T3SS, ompA, Inc and pmp genes (Voigt et al., 2012; Wolff et al., 2015; Hölzer et al., 2020; Vorimore et al., 2021a) (Figure 4B). The difference in gene content and SNVs likely plays a role in C. psittaci host tropism, adaptation and species-specific pathogenicity (Wolff et al., 2015; Favaroni et al., 2021). Furthermore, other phylogenomic analyses have genetically separated distinct avian-type C. abortus strains from the traditional ruminant C. abortus and/or avian C. psittaci strains (Longbottom et al., 2021; Zaręba-Marchewka et al., 2021) and confirmed that ruminant C. abortus genomic diversity is low level with no recombination detected, contrasting other related species (Seth-Smith et al., 2017b).
5.1 Complementing WGS studies: Gene-centric molecular epidemiology of chlamydial infections
WGS is considered the norm for evolutionary and epidemiological studies of many pathogens, including the human pathogen C. trachomatis, with the most available WGS data among chlamydial genomes (Harris et al., 2012; Bowden et al., 2021; Seth-Smith et al., 2021). However, numerous molecular epidemiology and/or genetic diversity studies of C. trachomatis and veterinary species still rely on gene-centric typing methods using a single (e.g. ompA) or multiple conserved gene markers (e.g. multi locus sequence typing (MLST)) due to decreased costs. Genotyping using the full-length as well as fragments of the highly variable ompA gene is employed in genetic diversity studies for veterinary and human chlamydial infections, as it provides an initial molecular characterisation of the infecting strains (Liu et al., 2019; Rawre et al., 2019; Robbins et al., 2019).
However, the insufficient resolution and noted recombination in the chlamydial ompA genes, led to use of species-specific higher-resolution genotyping methods, such as MLST (Versteeg et al., 2018a; Jelocnik et al., 2019). The chlamydial MLST is globally adopted as a rapid, universal fine-detailed molecular typing tool for both human C. trachomatis and veterinary chlamydial pathogens (Figure 5). As a technically easier and cheaper alternative to WGS, MLST utilises the Chlamydiales PubMLST database (http://pubMLST.org/chlamydiales) hosted on a platform that provides easy to use sequence and phylogenetic analyses. Furthermore, WGS studies are often supplemented with extended genotyping of samples using MLST due to high congruency with WGS phylogenetic clustering (Guo et al., 2017; Jenkins et al., 2018). Core genome and/or traditional MLST-derived phylogeny is highly congruent with WGS and/or SNP-derived phylogenies, and avoids the need to reconstruct computationally heavy phylogenies (Jolley et al., 2018; Patiño et al., 2018; Versteeg et al., 2018a; Jelocnik et al., 2019; Floridia-Yapur et al., 2021). Chlamydial MLST was effectively used in uncovering the global epidemiology of C. trachomatis strains (Herrmann et al., 2015; Danielewski et al., 2017), identifying clonal psittacine, horse and human strains of C. psittaci during an outbreak (Anstey et al., 2021) as well closely related but genetically diverse avian chlamydial strains (Figure 5A), distinguishing diverse koala C. pecorum strains to aid in translocation of animals (Figure 5B) (Fernandez et al., 2019) and other examples (Batteiger et al., 2014).
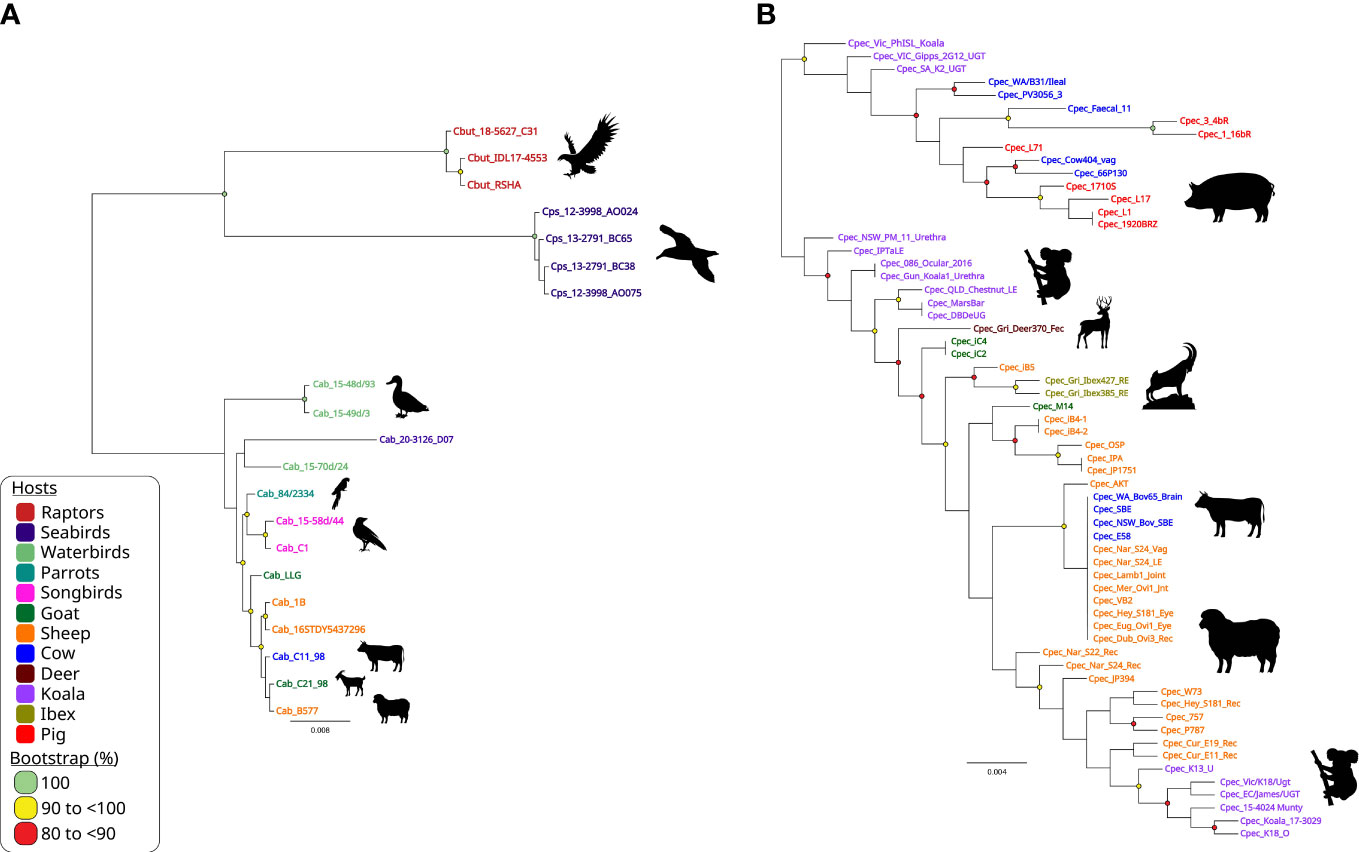
Figure 5 Chlamydia intraspecies genetic diversity. Midpoint-rooted maximum-likelihood (ML) phylogenetic analysis of 3,098 bp concatenated MLST sequences alignment representing (A) 20 avian and livestock STs from closely related C buteonis, C psittaci, traditional livestock and novel avian C abortus; and (B) 62 C. pecorum STs from a range of hosts. Phylogenetic trees were constructed using FastTree version 2.1.11, as implemented in Geneious Prime (available at: https://www.geneious.com/). Corresponding host silhouettes are shown adjacent to sequence names. Bootstrap values greater than 0.8 are shown. Branch lengths represent the nucleotide substitutions per site, as indicated by the scale bar.
6 Complementing WGS studies: Multi-omics integration for a systems biology understanding of chlamydial infections
Systems biology is the large-scale study of how genes, proteins, metabolites and other regulatory elements in an organism interact together using various integrated high-throughput multi-omic techniques, including genomics, transcriptomics, proteomics and metabolomics. Due to their biphasic, intracellular lifecycle, Chlamydia remain difficult to genetically manipulate; multi-omic approaches to understanding Chlamydia biology and pathogenesis are thus essential (Figure 6).
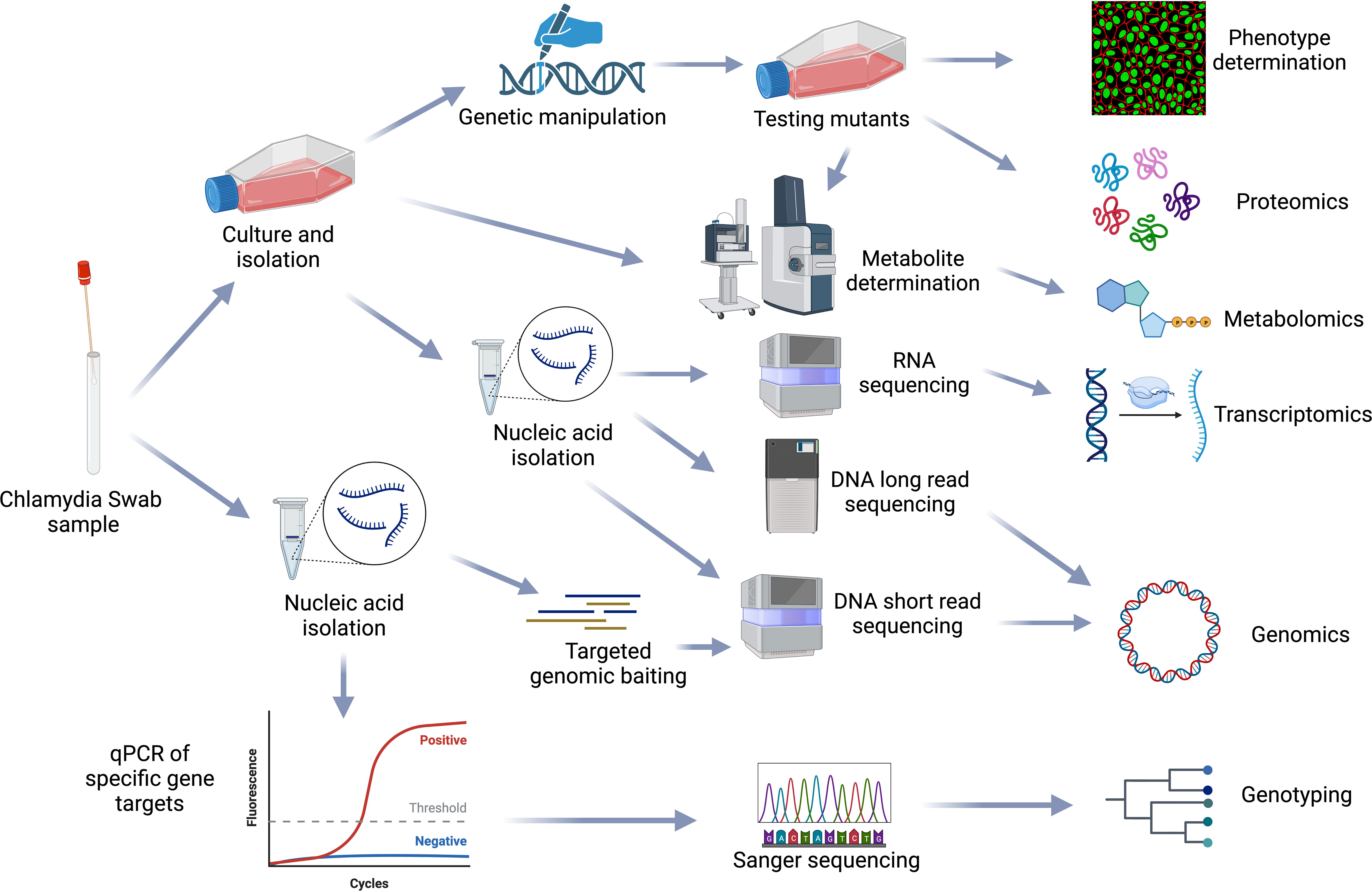
Figure 6 Chlamydial omics. Representation of the workflows from sample processing to multiple omics analysis methods used in the chlamydial field. Figure created using BioRender (available from https://www.biorender.com/).
Genomics forms the basis for systems biology and allows genes/proteins expressed during different conditions to be mapped and identified using complementary transcriptomic and proteomic methods. In Chlamydia, these approaches have provided insights into homologous virulence factor expression differences between C. psittaci and C. abortus which may be related to disease severity in humans (Beder and Saluz, 2018). Transcriptomics/proteomics studies have also revealed the host and pathogen genes expressed during the three phases of chlamydia infection- early, mid and late (Albrecht et al., 2010; Humphrys et al., 2013; Hayward et al., 2019), the chlamydial gene expression changes in response to stress such as iron limitation and exposure to IFN-γ (Belland et al., 2003; Brinkworth et al., 2018) and how C. trachomatis alters host chromatin accessibility by inducing epigenetic modifications (Hayward et al., 2020). These studies also led to the identification of new sRNAs in C. trachomatis (Albrecht et al., 2010), the role of the chlamydial plasmid in infection (Porcella et al., 2015) and insights into chlamydial-induced epithelial to mesenchymal cell transition (Zadora et al., 2019). It has also provided key insights into expression differences between EBs and RBs in C. trachomatis including increased T3SS proteins in EBs (Østergaard et al., 2016; Skipp et al., 2016). Recently, genomics was used to reconstruct the first Chlamydia genome-scale metabolic model (GSMM). GSMMs are mathematical models used to computationally describe the movement of metabolites within an organism. This model was then integrated with proteomic data to elucidate metabolic differences and altered flux between EBs and RBs (Yang et al., 2019).
Finally, multi-omics methods have also been essential for vaccine development and the discovery of new drug targets against Chlamydia. Immunoproteomics have been used to identify new C. trachomatis T-cell antigens for vaccine development and to identify immunogenic proteins in C. abortus outer membrane complexes (Karunakaran et al., 2008; Longbottom et al., 2019). The use of RNA-seq and metabolomics identified essential host metabolites and pathways for chlamydia infection, such as glutamine and guanine nucleotide biosynthesis. Targeting of these host-pathways was shown to inhibit chlamydia infection (Rother et al., 2018; Rajeeve et al., 2020).
Currently, most multi-omics approaches in Chlamydia involve characterising gene expression at the bulk population level from thousands to millions of Chlamydia and/or host cells within a given sample (Hayward et al., 2021; Pokorzynski et al., 2022). However, this approach ignores the important variations between individual cells. Future single-cell and spatial approaches such as single-cell RNA-sequencing (scRNA-seq) in bacteria (Kuchina et al., 2021) and/or host cells (Hayward et al., 2019) for interrogating host-pathogen responses to chlamydia infections may reveal meaningful differences in cell-to-cell responses to and/or from Chlamydia as well as identify the spatial distribution of important cell populations/lineages during infection. These existing and future omics techniques can be applied to Chlamydia infected tissues or advanced organotypic/organoid models, which better represent the host factors encountered (Nogueira et al., 2017; Versteeg et al., 2018b; McQueen et al., 2020; Dolat and Valdivia, 2021; Dolat et al., 2022; Edwards et al., 2022). This may provide insight into the pathogenesis in vivo and shed light on differences in genes or virulence factors required when infecting different host cells.
7 Future directions
The members of family Chlamydiaceae are an important species in the list of intracellular bacteria. They are highly adapted to their environments and depend on host cell machinery to survive and replicate. Their host range is one of the largest known, with predictions of a chlamydial species for every cell type on the planet (Collingro et al., 2020). With respect to C. trachomatis, it is the most common sexually transmitted infection and results in millions of cases of infertility and blindness across the globe. Improvements in WGS techniques have given rise to a deeper understanding of the chlamydial genome and the specific adaptations that have allowed specific species to thrive in different host environments. WGS has also resulted in new understandings of genetic recombination within species (but also interspecies) and has highlighted the issues around single gene sequencing to predict species evolution. In addition to enriching our understanding of Chlamydia, advances in genomics provides opportunities for rapid diagnosis of chlamydial infections and antimicrobial resistance detection using targeted single-gene nanopore sequencing or direct metagenome sequencing (Gu et al., 2020; Zhou et al., 2022). The advantages of using genomics for diagnosis is the ability to reveal the presence or absence of different virulence and antimicrobial-resistant genes, plasmids or important lineage/phylogenetic markers that may guide treatment and/or outbreak management.
The increasing number of chlamydial genomes sequenced and in publicly available databases poses challenges for future genome analysis as phylogenetic trees become more computationally intensive to construct. Thus, the future development of a Chlamydia hierarchical genome typing scheme based on core genome MLST or SNPs, such as those developed for C. trachomatis (Patiño et al., 2018; Versteeg et al., 2018a) will provide a standardised, universal and stable method for rapid chlamydia genotyping and disease surveillance. These hierarchical schemes can rapidly assign sequence types to infer phylogenetic relationships, require less intensive computation, and are made up of multiple SNPs or MLST schemes with increasing resolution to provide both short and long-term epidemiological information for outbreak detection and long-term lineage tracking, respectively. Many aspects of the Chlamydiae are still hidden, but with more and more genome-scale data published every day, our understanding of this complex and widespread bacterial species continues to improve.
Author contributions
Conceptualisation: MJ, LL, GM. Formal analysis: LL, SP, VK, GM and MJ. Data analysis: LL, SP, VK and MJ. Writing, original draft preparation: LL and MJ. Writing, figure preparation,: LL, VK and SP. Writing, review and editing: LL, SP, VK, GM and MJ. All authors have contributed to the article and approved the submitted version.
Funding
LL is funded by a University of Technology Sydney Chancellor’s Postdoctoral Research Fellowship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Albrecht, M., Sharma, C. M., Reinhardt, R., Vogel, J., Rudel, T. (2010). Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38 (3), 868–877. doi: 10.1093/nar/gkp1032
Almeida, F., Borges, V., Ferreira, R., Borrego, M. J., Gomes, J. P., Mota, L. J. (2012). Polymorphisms in inc proteins and differential expression of inc genes among Chlamydia trachomatis strains correlate with invasiveness and tropism of lymphogranuloma venereum isolates. J. Bacteriol. 194 (23), 6574–6585. doi: 10.1128/jb.01428-12
Andersen, S. E., Bulman, L. M., Steiert, B., Faris, R., Weber, M. M. (2021). Got mutants? how advances in chlamydial genetics have furthered the study of effector proteins. Pathog. Dis. 79 (2), ftaa078. doi: 10.1093/femspd/ftaa078
Andersson, P., Harris, S. R., Smith, H. M. B. S., Hadfield, J., O'Neill, C., Cutcliffe, L. T., et al. (2016). Chlamydia trachomatis from Australian aboriginal people with trachoma are polyphyletic composed of multiple distinctive lineages. Nat. Commun. 7, 10688–10688. doi: 10.1038/ncomms10688
Anstey, S. I., Kasimov, V., Jenkins, C., Legione, A., Devlin, J., Amery-Gale, J., et al. (2021). Chlamydia psittaci ST24: clonal strains of one health importance dominate in Australian horse, bird and human infections. Pathogens 10 (8), 1015. doi: 10.3390/pathogens10081015
Bachmann, N. L., Polkinghorne, A., Timms, P. (2014). Chlamydia genomics: providing novel insights into chlamydial biology. Trends Microbiol. 22 (8), 464–472. doi: 10.1016/j.tim.2014.04.013
Bachmann, N. L., Sullivan, M. J., Jelocnik, M., Myers, G. S., Timms, P., Polkinghorne, A. (2015). Culture-independent genome sequencing of clinical samples reveals an unexpected heterogeneity of infections by Chlamydia pecorum. J. Clin. Microbiol. 53 (5), 1573–1581. doi: 10.1128/jcm.03534-14
Bastidas, R. J., Valdivia, R. H. (2016). Emancipating Chlamydia: advances in the genetic manipulation of a recalcitrant intracellular pathogen. Microbiol. Mol. Biol. Rev. 80 (2), 411–427. doi: 10.1128/MMBR.00071-15
Batteiger, B. E., Wan, R., Williams, J. A., He, L., Ma, A., Fortenberry, J. D., et al. (2014). Novel Chlamydia trachomatis strains in heterosexual sex partners, Indianapolis, Indiana, USA. Emerg. Infect. Dis. 20 (11), 1841–1847. doi: 10.3201/2011.140604
Beder, T., Saluz, H. P. (2018). Virulence-related comparative transcriptomics of infectious and non-infectious chlamydial particles. BMC Genom. 19 (1), 575. doi: 10.1186/s12864-018-4961-x
Belland, R. J., Nelson, D. E., Virok, D., Crane, D. D., Hogan, D., Sturdevant, D., et al. (2003). Transcriptome analysis of chlamydial growth during IFN-gamma-mediated persistence and reactivation. Proc. Natl. Acad. Sci. U.S.A. 100 (26), 15971–15976. doi: 10.1073/pnas.2535394100
Benamri, I., Azzouzi, M., Sanak, K., Moussa, A., Radouani, F. (2021). An overview of genes and mutations associated with chlamydiae species' resistance to antibiotics. Ann. Clin. Microbiol. Antimicrob. 20 (1), 59. doi: 10.1186/s12941-021-00465-4
Betts-Hampikian, H., Fields, K. (2010). The chlamydial type III secretion mechanism: revealing cracks in a tough nut. Front. Microbiol. 1. doi: 10.3389/fmicb.2010.00114
Bommana, S., Somboonna, N., Richards, G., Tarazkar, M., Dean, D. (2021). Tryptophan operon diversity reveals evolutionary trends among geographically disparate Chlamydia trachomatis ocular and urogenital strains affecting tryptophan repressor and synthase function. mBio 12 (3), e00605–e00621. doi: 10.1128/mBio.00605-21
Borel, N., Leonard, C., Slade, J., Schoborg, R. V. (2016). Chlamydial antibiotic resistance and treatment failure in veterinary and human medicine. Curr. Clin. Microbiol. Rep. 3, 10–18. doi: 10.1007/s40588-016-0028-4
Borges, V., Cordeiro, D., Salas, A. I., Lodhia, Z., Correia, C., Isidro, J., et al. (2019). Chlamydia trachomatis: when the virulence-associated genome backbone imports a prevalence-associated major antigen signature. Microb. Genom. 5 (11), e000313. doi: 10.1099/mgen.0.000313
Borges, V., Gomes, J. P. (2015). Deep comparative genomics among Chlamydia trachomatis lymphogranuloma venereum isolates highlights genes potentially involved in pathoadaptation. Infect. Genet. Evol. 32, 74–88. doi: 10.1016/j.meegid.2015.02.026
Borges, V., Nunes, A., Ferreira, R., Borrego, M. J., Gomes, J. P. (2012). Directional evolution of Chlamydia trachomatis towards niche-specific adaptation. J. Bacteriol. 194 (22), 6143–6153. doi: 10.1128/jb.01291-12
Borges, V., Isidro, J., Correia, C., Cordeiro, D., Vieira, L., Lodhia, Z., et al (2021). Transcontinental dissemination of the L2b/D-Da recombinant chlamydia trachomatis lymphogranuloma venereum (LGV) strain: Need of broad multi-country molecular surveillance. Clin. Infect. Dis. 73 (4), e1004–e1007. doi: 10.1093/cid/ciab067
Bowden, K. E., Joseph, S. J., Cartee, J. C., Ziklo, N., Danavall, D., Raphael, B. H., et al. (2021). Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from patient clinical vaginal and rectal swabs. mSphere 6 (2), e01302–e01320. doi: 10.1128/mSphere.01302-20
Branley, J., Bachmann, N. L., Jelocnik, M., Myers, G. S. A., Polkinghorne, A. (2016). Australian Human and parrot Chlamydia psittaci strains cluster within the highly virulent 6BC clade of this important zoonotic pathogen. Sci. Rep. 6, 30019–30019. doi: 10.1038/srep30019
Brinkworth, A. J., Wildung, M. R., Carabeo, R. A. (2018). Genomewide transcriptional responses of iron-starved Chlamydia trachomatis reveal prioritization of metabolic precursor synthesis over protein translation. mSystems 3 (1), e00184–17. doi: 10.1128/mSystems.00184-17
Bugalhão, J. N., Mota, L. J. (2019). The multiple functions of the numerous Chlamydia trachomatis secreted proteins: the tip of the iceberg. Microb. Cell 6 (9), 414–449. doi: 10.15698/mic2019.09.691
Carlson, J. H., Hughes, S., Hogan, D., Cieplak, G., Sturdevant, D. E., McClarty, G., et al. (2004). Polymorphisms in the Chlamydia trachomatis cytotoxin locus associated with ocular and genital isolates. Infect. Immun. 72 (12), 7063–7072. doi: 10.1128/iai.72.12.7063-7072.2004
Cheong, H. C., Lee, C. Y. Q., Cheok, Y. Y., Tan, G. M. Y., Looi, C. Y., Wong, W. F. (2019). Chlamydiaceae: diseases in primary hosts and zoonosis. Microorganisms 7 (5), 146. doi: 10.3390/microorganisms7050146
Chiarelli, T. J., Grieshaber, N. A., Omsland, A., Remien, C. H., Grieshaber, S. S. (2020). Single-inclusion kinetics of Chlamydia trachomatis development. mSystems 5 (5), e00689–20. doi: 10.1128/mSystems.00689-20
Christiansen, M. T., Brown, A. C., Kundu, S., Tutill, H. J., Williams, R., Brown, J. R., et al. (2014). Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect. Dis. 14, 591. doi: 10.1186/s12879-014-0591-3
Collingro, A., Köstlbacher, S., Horn, M. (2020). Chlamydiae in the environment. Trends Microbiol. 28 (11), 877–888. doi: 10.1016/j.tim.2020.05.020
Collingro, A., Tischler, P., Weinmaier, T., Penz, T., Heinz, E., Brunham, R. C., et al. (2011). Unity in variety–the pan-genome of the Chlamydiae. Mol. Biol. Evol. 28 (12), 3253–3270. doi: 10.1093/molbev/msr161
Confer, A. W., Ayalew, S. (2013). The OmpA family of proteins: roles in bacterial pathogenesis and immunity. Vet. Microbiol. 163 (3-4), 207–222. doi: 10.1016/j.vetmic.2012.08.019
da Cunha, M., Pais, S. V., Bugalhão, J. N., Mota, L. J. (2017). The Chlamydia trachomatis type III secretion substrates CT142, CT143, and CT144 are secreted into the lumen of the inclusion. PloS One 12 (6), e0178856. doi: 10.1371/journal.pone.0178856
Dahlberg, J., Hadad, R., Elfving, K., Larsson, I., Isaksson, J., Magnuson, A., et al. (2018). Ten years transmission of the new variant of Chlamydia trachomatis in Sweden: prevalence of infections and associated complications. Sex. Transm. Infect. 94 (2), 100–104. doi: 10.1136/sextrans-2016-052992
Danielewski, J. A., Phillips, S., Kong, F. Y. S., Smith, K. S., Hocking, J. S., Guy, R., et al. (2017). A snapshot of Chlamydia trachomatis genetic diversity using multilocus sequence type analysis in an Australian metropolitan setting. Eur. J. Clin. Microbiol. Infect. Dis. 36 (7), 1297–1303. doi: 10.1007/s10096-017-2935-6
Dehoux, P., Flores, R., Dauga, C., Zhong, G., Subtil, A. (2011). Multi-genome identification and characterization of Chlamydiae-specific type III secretion substrates: the inc proteins. BMC Genom. 12, 109. doi: 10.1186/1471-2164-12-109
Dharamshi, J. E., Köstlbacher, S., Schön, M. E., Collingro, A., Ettema, T. J. G., Horn, M. (2023). Gene gain facilitated endosymbiotic evolution of Chlamydiae. Nat. Microbiol. 8 (1), 40–54. doi: 10.1038/s41564-022-01284-9
Dharamshi, J. E., Tamarit, D., Eme, L., Stairs, C. W., Martijn, J., Homa, F., et al. (2020). Marine sediments illuminate Chlamydiae diversity and evolution. Curr. Biol. 30 (6), 1032–1048.e1037. doi: 10.1016/j.cub.2020.02.016
Dimond, Z. E., Suchland, R. J., Baid, S., LaBrie, S. D., Soules, K. R., Stanley, J., et al. (2021). Inter-species lateral gene transfer focused on the Chlamydia plasticity zone identifies loci associated with immediate cytotoxicity and inclusion stability. Mol. Microbiol. 116 (6), 1433–1448. doi: 10.1111/mmi.14832
Dolat, L., Carpenter, V. K., Chen, Y. S., Suzuki, M., Smith, E. P., Kuddar, O., et al. (2022). Chlamydia repurposes the actin-binding protein EPS8 to disassemble epithelial tight junctions and promote infection. Cell Host Microbe 30 (12), 1685–1700.e1610. doi: 10.1016/j.chom.2022.10.013
Dolat, L., Valdivia, R. H. (2021). An endometrial organoid model of interactions between Chlamydia and epithelial and immune cells. J. Cell Sci. 134 (5), 1685–1700. doi: 10.1242/jcs.252403
Edwards, V. L., McComb, E., Gleghorn, J. P., Forney, L., Bavoil, P. M., Ravel, J. (2022). Three-dimensional models of the cervicovaginal epithelia to study host-microbiome interactions and sexually transmitted infections. Pathog. Dis. 80 (1), ftac026. doi: 10.1093/femspd/ftac026
Edwards, V. L., Smith, S. B., McComb, E. J., Tamarelle, J., Ma, B., Humphrys, M. S., et al. (2019). The cervicovaginal microbiota-host interaction modulates Chlamydia trachomatis infection. mBio 10 (4), e01548–19. doi: 10.1128/mBio.01548-19
Elwell, C., Mirrashidi, K., Engel, J. (2016). Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 14 (6), 385–400. doi: 10.1038/nrmicro.2016.30
Ende, R. J., Derré, I. (2020). Host and bacterial glycolysis during Chlamydia trachomatis infection. Infect. Immun. 88 (12), e00545–e00520. doi: 10.1128/IAI.00545-20
Escobedo-Guerra, M. R., Katoku-Herrera, M., Lopez-Hurtado, M., Villagrana-Zesati, J. R., de Haro-Cruz, M. J., Guerra-Infante, F. M. (2019). Identification of a new variant of Chlamydia trachomatis in Mexico. Enferm Infecc Microbiol. Clin. 37 (2), 93–99. doi: 10.1016/j.eimc.2018.02.008
Favaroni, A., Trinks, A., Weber, M., Hegemann, J. H., Schnee, C. (2021). Pmp repertoires influence the different infectious potential of avian and mammalian Chlamydia psittaci strains. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.656209
Fehlner-Gardiner, C., Roshick, C., Carlson, J. H., Hughes, S., Belland, R. J., Caldwell, H. D., et al. (2002). Molecular basis defining human Chlamydia trachomatis tissue tropism. a possible role for tryptophan synthase. J. Biol. Chem. 277 (30), 26893–26903. doi: 10.1074/jbc.M203937200
Fernandez, C. M., Schmertmann, L. J., Higgins, D. P., Casteriano, A., Irinyi, L., Mella, V. S. A., et al. (2019). Genetic differences in Chlamydia pecorum between neighbouring sub-populations of koalas (Phascolarctos cinereus). Vet. Microbiol. 231, 264–270. doi: 10.1016/j.vetmic.2019.02.020
Fisher, D. J., Fernández, R. E., Adams, N. E., Maurelli, A. T. (2012). Uptake of biotin by Chlamydia spp. through the use of a bacterial transporter (BioY) and a host-cell transporter (SMVT). PloS One 7 (9), e46052. doi: 10.1371/journal.pone.0046052
Floridia-Yapur, N., Rusman, F., Diosque, P., Tomasini, N. (2021). Genome data vs MLST for exploring intraspecific evolutionary history in bacteria: much is not always better. Infect. Genet. Evol. 93, 104990. doi: 10.1016/j.meegid.2021.104990
Gitsels, A., Sanders, N., Vanrompay, D. (2019). Chlamydial infection from outside to inside. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02329
Gomes, J. P., Bruno, W. J., Borrego, M. J., Dean, D. (2004). Recombination in the genome of Chlamydia trachomatis involving the polymorphic membrane protein c gene relative to ompA and evidence for horizontal gene transfer. J. Bacteriol 186 (13), 4295–4306. doi: 10.1128/jb.186.13.4295-4306.2004
Gomes, J. P., Bruno, W. J., Nunes, A., Santos, N., Florindo, C., Borrego, M. J., et al. (2007). Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res. 17 (1), 50–60. doi: 10.1101/gr.5674706
Gu, L., Liu, W., Ru, M., Lin, J., Yu, G., Ye, J., et al. (2020). The application of metagenomic next-generation sequencing in diagnosing Chlamydia psittaci pneumonia: a report of five cases. BMC Pulm. Med. 20 (1), 65. doi: 10.1186/s12890-020-1098-x
Guo, W., Jelocnik, M., Li, J., Sachse, K., Polkinghorne, A., Pannekoek, Y., et al. (2017). From genomes to genotypes: molecular epidemiological analysis of Chlamydia gallinacea reveals a high level of genetic diversity for this newly emerging chlamydial pathogen. BMC Genom. 18 (1), 949. doi: 10.1186/s12864-017-4343-9
Hadfield, J., Harris, S. R., Seth-Smith, H. M. B., Parmar, S., Andersson, P., Giffard, P. M., et al. (2017). Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 27 (7), 1220–1229. doi: 10.1101/gr.212647.116
Harris, S. R., Clarke, I. N., Seth-Smith, H. M. B., Solomon, A. W., Cutcliffe, L. T., Marsh, P., et al. (2012). Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 44 (4), 413–419. doi: 10.1038/ng.2214
Hayward, R. J., Humphrys, M. S., Huston, W. M., Myers, G. S. A. (2021). Dual RNA-seq analysis of in vitro infection multiplicity and RNA depletion methods in Chlamydia-infected epithelial cells. Sci. Rep. 11 (1), 10399. doi: 10.1038/s41598-021-89921-x
Hayward, R. J., Marsh, J. W., Humphrys, M. S., Huston, W. M., Myers, G. S. A. (2019). Early transcriptional landscapes of Chlamydia trachomatis-infected epithelial cells at single cell resolution. Front. Cell Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00392
Hayward, R. J., Marsh, J. W., Humphrys, M. S., Huston, W. M., Myers, G. S. A. (2020). Chromatin accessibility dynamics of Chlamydia-infected epithelial cells. Epigenet. Chromatin 13 (1), 45. doi: 10.1186/s13072-020-00368-2
Heijne, M., Jelocnik, M., Umanets, A., Brouwer, M. S. M., Dinkla, A., Harders, F., et al. (2021). Genetic and phenotypic analysis of the pathogenic potential of two novel Chlamydia gallinacea strains compared to Chlamydia psittaci. Sci. Rep. 11 (1), 16516. doi: 10.1038/s41598-021-95966-9
Heinz, E., Rockey, D. D., Montanaro, J., Aistleitner, K., Wagner, M., Horn, M. (2010). Inclusion membrane proteins of Protochlamydia amoebophila UWE25 reveal a conserved mechanism for host cell interaction among the Chlamydiae. J. Bacteriol. 192 (19), 5093–5102. doi: 10.1128/jb.00605-10
Herrmann, B., Isaksson, J., Ryberg, M., Tångrot, J., Saleh, I., Versteeg, B., et al. (2015). Global multilocus sequence type analysis of Chlamydia trachomatis strains from 16 countries. J. Clin. Microbiol. 53 (7), 2172–2179. doi: 10.1128/JCM.00249-15
Hölzer, M., Barf, L.-M., Lamkiewicz, K., Vorimore, F., Lataretu, M., Favaroni, A., et al. (2020). Comparative genome analysis of 33 Chlamydia strains reveals characteristic features of Chlamydia psittaci and closely related species. Pathogens 9 (11), 899. doi: 10.3390/pathogens9110899
Humphrys, M. S., Creasy, T., Sun, Y., Shetty, A. C., Chibucos, M. C., Drabek, E. F., et al. (2013). Simultaneous transcriptional profiling of bacteria and their host cells. PloS One 8 (12), e80597. doi: 10.1371/journal.pone.0080597
Islam, M. M., Jelocnik, M., Huston, W. M., Timms, P., Polkinghorne, A. (2018). Characterization of the in vitro chlamydia pecorum response to gamma interferon. Infect. Immun. 86 (4), e00714–17. doi: 10.1128/iai.00714-17
Jelocnik, M., Bachmann, N. L., Kaltenboeck, B., Waugh, C., Woolford, L., Speight, K. N., et al. (2015). Genetic diversity in the plasticity zone and the presence of the chlamydial plasmid differentiates Chlamydia pecorum strains from pigs, sheep, cattle, and koalas. BMC Genom. 16, 893. doi: 10.1186/s12864-015-2053-8
Jelocnik, M., Polkinghorne, A., Pannekoek, Y. (2019). Multilocus sequence typing (MLST) of Chlamydiales. Methods Mol. Biol. 2042, 69–86. doi: 10.1007/978-1-4939-9694-0_7
Jenkins, C., Jelocnik, M., Micallef, M. L., Galea, F., Taylor-Brown, A., Bogema, D. R., et al. (2018). An epizootic of Chlamydia psittaci equine reproductive loss associated with suspected spillover from native Australian parrots. Emerg. Microbes Infect. 7 (1), 88. doi: 10.1038/s41426-018-0089-y
Jolley, K. A., Bray, J. E., Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Jones, C. A., Hadfield, J., Thomson, N. R., Cleary, D. W., Marsh, P., Clarke, I. N., et al. (2020). The nature and extent of plasmid variation in Chlamydia trachomatis. Microorganisms 8 (3), 373. doi: 10.3390/microorganisms8030373
Joseph, S. J., Didelot, X., Rothschild, J., de Vries, H. J., Morré, S. A., Read, T. D., et al. (2012). Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol. Biol. Evol. 29 (12), 3933–3946. doi: 10.1093/molbev/mss198
Joseph, S. J., Marti, H., Didelot, X., Castillo-Ramirez, S., Read, T. D., Dean, D. (2015). chlamydiaceae genomics reveals interspecies admixture and the recent evolution of Chlamydia abortus infecting lower mammalian species and humans. Genome Biol. Evol. 7 (11), 3070–3084. doi: 10.1093/gbe/evv201
Joseph, S. J., Marti, H., Didelot, X., Read, T. D., Dean, D. (2016). Tetracycline selective pressure and homologous recombination shape the evolution of Chlamydia suis: a recently identified zoonotic pathogen. Genome Biol. Evol. 8 (8), 2613–2623. doi: 10.1093/gbe/evw182
Karunakaran, K. P., Rey-Ladino, J., Stoynov, N., Berg, K., Shen, C., Jiang, X., et al. (2008). Immunoproteomic discovery of novel T cell antigens from the obligate intracellular pathogen Chlamydia. J. Immunol. 180 (4), 2459–2465. doi: 10.4049/jimmunol.180.4.2459
Kuchina, A., Brettner, L. M., Paleologu, L., Roco, C. M., Rosenberg, A. B., Carignano, A., et al. (2021). Microbial single-cell RNA sequencing by split-pool barcoding. Science 371 (6531), eaba5257. doi: 10.1126/science.aba5257
Lamy-Besnier, Q., Koszul, R., Debarbieux, L., Marbouty, M. (2021). Closed and high-quality bacterial genome sequences of the oligo-mouse-microbiota community. Microbiol. Resour. Announc. 10 (17), e01396–20. doi: 10.1128/mra.01396-20
Liang, P., Rosas-Lemus, M., Patel, D., Fang, X., Tuz, K., Juárez, O. (2018). Dynamic energy dependency of Chlamydia trachomatis on host cell metabolism during intracellular growth: role of sodium-based energetics in chlamydial ATP generation. J. Biol. Chem. 293 (2), 510–522. doi: 10.1074/jbc.M117.797209
Liu, S.-Y., Li, K.-P., Hsieh, M.-K., Chang, P.-C., Shien, J.-H., Ou, S.-C. (2019). Prevalence and genotyping of Chlamydia psittaci from domestic waterfowl, companion birds, and wild birds in Taiwan. Vector-Borne Zoonotic Dis. 19 (9), 666–673. doi: 10.1089/vbz.2018.2403
Livingstone, M., Caspe, S. G., Longbottom, D. (2021). Complete genome sequence of Chlamydia abortus MRI-10/19, isolated from a sheep vaccinated with the commercial live C. abortus 1B vaccine strain. Microbiol. Resour. Announc. 10 (18), e00203–21. doi: 10.1128/mra.00203-21
Longbottom, D., Livingstone, M., Aitchison, K. D., Imrie, L., Manson, E., Wheelhouse, N., et al. (2019). Proteomic characterisation of the Chlamydia abortus outer membrane complex (COMC) using combined rapid monolithic column liquid chromatography and fast MS/MS scanning. PloS One 14 (10), e0224070. doi: 10.1371/journal.pone.0224070
Longbottom, D., Livingstone, M., Ribeca, P., Beeckman, D. S. A., van der Ende, A., Pannekoek, Y., et al. (2021). Whole genome de novo sequencing and comparative genomic analyses suggests that Chlamydia psittaci strain 84/2334 should be reclassified as Chlamydia abortus species. BMC Genom. 22 (1), 159. doi: 10.1186/s12864-021-07477-6
Lutter, E. I., Martens, C., Hackstadt, T. (2012). Evolution and conservation of predicted inclusion membrane proteins in Chlamydiae. Comp. Funct. Genomics 2012, 362104. doi: 10.1155/2012/362104
Marschall, M. T., Simnacher, U., Walther, P., Essig, A., Hagemann, J. B. (2020). The putative type iii secreted Chlamydia abortus virulence-associated protein CAB063 targets lamin and induces apoptosis. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01059
Marti, H., Bommana, S., Read, T. D., Pesch, T., Prähauser, B., Dean, D., et al. (2021). Generation of tetracycline and rifamycin resistant Chlamydia suis recombinants. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.630293
Marti, H., Kim, H., Joseph, S. J., Dojiri, S., Read, T. D., Dean, D. (2017). Tet(C) gene transfer between Chlamydia suis strains occurs by homologous recombination after co-infection: implications for spread of tetracycline-resistance among Chlamydiaceae. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00156
Marti, H., Suchland, R. J., Rockey, D. D. (2022). The impact of lateral gene transfer in Chlamydia. Front. Cell Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.861899
McClarty, G., Qin, B. (1993). Pyrimidine metabolism by intracellular Chlamydia psittaci. J. Bacteriol 175 (15), 4652–4661. doi: 10.1128/jb.175.15.4652-4661.1993
McQueen, B. E., Kiatthanapaiboon, A., Fulcher, M. L., Lam, M., Patton, K., Powell, E., et al. (2020). Human fallopian tube epithelial cell culture model to study host responses to Chlamydia trachomatis infection. Infect. Immun. 88 (9), e00105–20. doi: 10.1128/iai.00105-20
Mojica, S. A., Hovis, K. M., Frieman, M. B., Tran, B., Hsia, R.-c., Ravel, J., et al. (2015). SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell 26 (10), 1918–1934. doi: 10.1091/mbc.E14-11-1530
Mölleken, K., Schmidt, E., Hegemann, J. H. (2010). Members of the pmp protein family of Chlamydia pneumoniae mediate adhesion to human cells via short repetitive peptide motifs. Mol. Microbiol. 78 (4), 1004–1017. doi: 10.1111/j.1365-2958.2010.07386.x
Moran, N. A. (2002). Microbial minimalism: genome reduction in bacterial pathogens. Cell 108 (5), 583–586. doi: 10.1016/s0092-8674(02)00665-7
Mueller, K. E., Plano, G. V., Fields, K. A. (2014). New frontiers in type III secretion biology: the Chlamydia perspective. Infect. Immun. 82 (1), 2–9. doi: 10.1128/IAI.00917-13
Nogueira, A. T., Braun, K. M., Carabeo, R. A. (2017). Characterization of the growth of Chlamydia trachomatis in in vitro-generated stratified epithelium. Front. Cell Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00438
Nunes, A., Borrego, M. J., Gomes, J. P. (2013). Genomic features beyond Chlamydia trachomatis phenotypes: what do we think we know? Infect. Genet. Evol. 16, 392–400. doi: 10.1016/j.meegid.2013.03.018
Nunes, A., Borrego, M. J., Nunes, B., Florindo, C., Gomes, J. P. (2009). Evolutionary dynamics of ompA, the gene encoding the Chlamydia trachomatis key antigen. J. Bacteriol 191 (23), 7182–7192. doi: 10.1128/JB.00895-09
Nunes, A., Gomes, J. P. (2014). Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 23, 49–64. doi: 10.1016/j.meegid.2014.01.029
Nunes, A., Gomes, J. P., Karunakaran, K. P., Brunham, R. C. (2015). Bioinformatic analysis of Chlamydia trachomatis polymorphic membrane proteins PmpE, PmpF, PmpG and PmpH as potential vaccine antigens. PloS One 10 (7), e0131695. doi: 10.1371/journal.pone.0131695
Olsen, A. W., Rosenkrands, I., Holland, M. J., Andersen, P., Follmann, F. (2021). A Chlamydia trachomatis VD1-MOMP vaccine elicits cross-neutralizing and protective antibodies against C/C-related complex serovars. NPJ Vaccines 6 (1), 58. doi: 10.1038/s41541-021-00312-9
Østergaard, O., Follmann, F., Olsen, A. W., Heegaard, N. H., Andersen, P., Rosenkrands, I. (2016). Quantitative protein profiling of Chlamydia trachomatis growth forms reveals defense strategies against tryptophan starvation. Mol. Cell Proteom. 15 (12), 3540–3550. doi: 10.1074/mcp.M116.061986
Panzetta, M. E., Valdivia, R. H., Saka, H. A. (2018). Chlamydia persistence: a survival strategy to evade antimicrobial effects in-vitro and in-vivo. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.03101
Patiño, L. H., Camargo, M., Muñoz, M., Ríos-Chaparro, D. I., Patarroyo, M. A., Ramírez, J. D. (2018). Unveiling the multilocus sequence typing (MLST) schemes and core genome phylogenies for genotyping Chlamydia trachomatis. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01854
Pawlikowska-Warych, M., Śliwa-Dominiak, J., Deptuła, W. (2015). Chlamydial plasmids and bacteriophages. Acta Biochim. Pol. 62 (1), 1–6. doi: 10.18388/abp.2014_764
Peters, J., Wilson, D. P., Myers, G., Timms, P., Bavoil, P. M. (2007). Type III secretion à la Chlamydia. Trends Microbiol. 15 (6), 241–251. doi: 10.1016/j.tim.2007.04.005
Pillonel, T., Bertelli, C., Greub, G. (2018). Environmental metagenomic assemblies reveal seven new highly divergent chlamydial lineages and hallmarks of a conserved intracellular lifestyle. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00079
Pillonel, T., Tagini, F., Bertelli, C., Greub, G. (2020). ChlamDB: a comparative genomics database of the phylum Chlamydiae and other members of the Planctomycetes-Verrucomicrobiae-Chlamydiae superphylum. Nucleic Acids Res. 48 (D1), D526–d534. doi: 10.1093/nar/gkz924
Piñeiro, L., Bernal, S., Bordes, A., Palomares, J. C., Gilarranz, R., von Wichmann, M. A., et al. (2014). Minimum spread of the new Swedish variant of Chlamydia trachomatis and distribution of C. trachomatis ompA genotypes in three geographically distant areas of Spain 2011-2012. Infection 42 (5), 905–912. doi: 10.1007/s15010-014-0665-6
Pokorzynski, N. D., Alla, M. R., Carabeo, R. A. (2022). Host cell amplification of nutritional stress contributes to persistence in Chlamydia trachomatis. mBio 13 (6), e0271922. doi: 10.1128/mbio.02719-22
Pokorzynski, N. D., Brinkworth, A. J., Carabeo, R. (2019). A bipartite iron-dependent transcriptional regulation of the tryptophan salvage pathway in Chlamydia trachomatis. Elife 8, e42295. doi: 10.7554/eLife.42295
Pokorzynski, N. D., Hatch, N. D., Ouellette, S. P., Carabeo, R. (2020). The iron-dependent repressor YtgR is a tryptophan-dependent attenuator of the trpRBA operon in Chlamydia trachomatis. Nat. Commun. 11, 6430. doi: 10.1038/s41467-020-20181-5
Porcella, S. F., Carlson, J. H., Sturdevant, D. E., Sturdevant, G. L., Kanakabandi, K., Virtaneva, K., et al. (2015). Transcriptional profiling of human epithelial cells infected with plasmid-bearing and plasmid-deficient Chlamydia trachomatis. Infect. Immun. 83 (2), 534–543. doi: 10.1128/iai.02764-14
Quigley, B. L., Timms, P. (2021). The koala immune response to chlamydial infection and vaccine development-advancing our immunological understanding. Anim. (Basel) 11 (2), 380. doi: 10.3390/ani11020380
Rajaram, K., Giebel, A. M., Toh, E., Hu, S., Newman, J. H., Morrison, S. G., et al. (2015). Mutational analysis of the Chlamydia muridarum plasticity zone. Infect. Immun. 83 (7), 2870–2881. doi: 10.1128/iai.00106-15
Rajeeve, K., Vollmuth, N., Janaki-Raman, S., Wulff, T. F., Baluapuri, A., Dejure, F. R., et al. (2020). Reprogramming of host glutamine metabolism during Chlamydia trachomatis infection and its key role in peptidoglycan synthesis. Nat. Microbiol. 5 (11), 1390–1402. doi: 10.1038/s41564-020-0762-5
Rawre, J., Dhawan, B., Khanna, N., Sreenivas, V., Broor, S., Chaudhry, R. (2019). Distribution of Chlamydia trachomatis ompA genotypes in patients attending a sexually transmitted disease outpatient clinic in new Delhi, India. Indian J. Med. Res. 149 (5), 662–670. doi: 10.4103/ijmr.IJMR_1171_17
Read, T. D., Joseph, S. J., Didelot, X., Liang, B., Patel, L., Dean, D. (2013). Comparative analysis of Chlamydia psittaci genomes reveals the recent emergence of a pathogenic lineage with a broad host range. mBio 4 (2), e00604–e00612. doi: 10.1128/mBio.00604-12
Robbins, A., Hanger, J., Jelocnik, M., Quigley, B. L., Timms, P. (2019). Longitudinal study of wild koalas (Phascolarctos cinereus) reveals chlamydial disease progression in two thirds of infected animals. Sci. Rep. 9 (1), 13194. doi: 10.1038/s41598-019-49382-9
Rockey, D. D. (2011). Unraveling the basic biology and clinical significance of the chlamydial plasmid. J. Exp. Med. 208 (11), 2159–2162. doi: 10.1084/jem.20112088
Rother, M., Gonzalez, E., Teixeira da Costa, A. R., Wask, L., Gravenstein, I., Pardo, M., et al. (2018). Combined human genome-wide rnai and metabolite analyses identify IMPDH as a host-directed target against Chlamydia infection. Cell Host Microbe 23 (5), 661–671.e668. doi: 10.1016/j.chom.2018.04.002
Roulis, E., Bachmann, N., Humphrys, M., Myers, G., Huston, W., Polkinghorne, A., et al. (2015a). Phylogenetic analysis of human Chlamydia pneumoniae strains reveals a distinct Australian indigenous clade that predates European exploration of the continent. BMC Genom. 16, 1094–1094. doi: 10.1186/s12864-015-2281-y
Roulis, E., Bachmann, N. L., Myers, G. S., Huston, W., Summersgill, J., Hudson, A., et al. (2015b). Comparative genomic analysis of human Chlamydia pneumoniae isolates from respiratory, brain and cardiac tissues. Genomics 106 (6), 373–383. doi: 10.1016/j.ygeno.2015.09.008
Saka, H. A., Valdivia, R. H. (2010). Acquisition of nutrients by Chlamydiae: unique challenges of living in an intracellular compartment. Curr. Opin. Microbiol. 13 (1), 4–10. doi: 10.1016/j.mib.2009.11.002
Seth-Smith, H. M. B., Bénard, A., Bruisten, S. M., Versteeg, B., Herrmann, B., Kok, J., et al. (2021). Ongoing evolution of Chlamydia trachomatis lymphogranuloma venereum: exploring the genomic diversity of circulating strains. Microb. Genom 7 (6), 599. doi: 10.1099/mgen.0.000599
Seth-Smith, H. M. B., Busó, L. S., Livingstone, M., Sait, M., Harris, S. R., Aitchison, K. D., et al. (2017b). European Chlamydia abortus livestock isolate genomes reveal unusual stability and limited diversity, reflected in geographical signatures. BMC Genom 18 (1), 344. doi: 10.1186/s12864-017-3657-y
Seth-Smith, H. M. B., Harris, S. R., Persson, K., Marsh, P., Barron, A., Bignell, A., et al. (2009). Co-Evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genom. 10, 239–239. doi: 10.1186/1471-2164-10-239
Seth-Smith, H. M., Wanninger, S., Bachmann, N., Marti, H., Qi, W., Donati, M., et al. (2017a). The Chlamydia suis genome exhibits high levels of diversity, plasticity, and mobile antibiotic resistance: comparative genomics of a recent livestock cohort shows influence of treatment regimes. Genome Biol. Evol. 9 (3), 750–760. doi: 10.1093/gbe/evx043
Sigalova, O. M., Chaplin, A. V., Bochkareva, O. O., Shelyakin, P. V., Filaretov, V. A., Akkuratov, E. E., et al. (2019). Chlamydia pan-genomic analysis reveals balance between host adaptation and selective pressure to genome reduction. BMC Genom 20 (1), 710. doi: 10.1186/s12864-019-6059-5
Sigar, I. M., Schripsema, J. H., Wang, Y., Clarke, I. N., Cutcliffe, L. T., Seth-Smith, H. M., et al. (2014). Plasmid deficiency in urogenital isolates of Chlamydia trachomatis reduces infectivity and virulence in a mouse model. Pathog. Dis. 70 (1), 61–69. doi: 10.1111/2049-632x.12086
Skipp, P. J., Hughes, C., McKenna, T., Edwards, R., Langridge, J., Thomson, N. R., et al. (2016). Quantitative proteomics of the infectious and replicative forms of Chlamydia trachomatis. PloS One 11 (2), e0149011. doi: 10.1371/journal.pone.0149011
Somboonna, N., Ziklo, N., Ferrin, T. E., Hyuk Suh, J., Dean, D. (2019). Clinical persistence of Chlamydia trachomatis sexually transmitted strains involves novel mutations in the functional αββα tetramer of the tryptophan synthase operon. mBio 10 (4), e01464–19. doi: 10.1128/mBio.01464-19
Song, L., Carlson, J. H., Whitmire, W. M., Kari, L., Virtaneva, K., Sturdevant, D. E., et al. (2013). Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect. Immun. 81 (3), 636–644. doi: 10.1128/iai.01305-12
Suchland, R. J., Carrell, S. J., Wang, Y., Hybiske, K., Kim, D. B., Dimond, Z. E., et al. (2019). Chromosomal recombination targets in Chlamydia interspecies lateral gene transfer. J. Bacteriol 201 (23), 4604–4611. doi: 10.1128/jb.00365-19
Suchland, R. J., Sandoz, K. M., Jeffrey, B. M., Stamm, W. E., Rockey, D. D. (2009). Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob. Agents Chemother. 53 (11), 4604–4611. doi: 10.1128/aac.00477-09
Sun, G., Pal, S., Sarcon, A. K., Kim, S., Sugawara, E., Nikaido, H., et al. (2007). Structural and functional analyses of the major outer membrane protein of Chlamydia trachomatis. J. Bacteriol. 189 (17), 6222–6235. doi: 10.1128/JB.00552-07
Szabo, K. V., O’Neill, C. E., Clarke, I. N. (2020). Diversity in chlamydial plasmids. PloS One 15 (5), e0233298. doi: 10.1371/journal.pone.0233298
Taylor-Brown, A., Madden, D., Polkinghorne, A. (2018). Culture-independent approaches to chlamydial genomics. Microb. Genom. 4 (2), e000145. doi: 10.1099/mgen.0.000145
Tifrea, D. F., Pal, S., de la Maza, L. M. (2020). A recombinant Chlamydia trachomatis MOMP vaccine elicits cross-serogroup protection in mice against vaginal shedding and infertility. J. Infect. Dis. 221 (2), 191–200. doi: 10.1093/infdis/jiz438
Tourancheau, A., Mead, E. A., Zhang, X. S., Fang, G. (2021). Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat. Methods 18 (5), 491–498. doi: 10.1038/s41592-021-01109-3
Turman, B. J., Alzhanov, D., Nagarajan, U. M., Darville, T., O'Connell, C. M. (2023). Virulence protein Pgp3 is insufficient to mediate plasmid-dependent infectivity of Chlamydia trachomatis. Infect. Immun. 91 (2), e0039222. doi: 10.1128/iai.00392-22
Valdivia, R. H. (2008). Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr. Opin. Microbiol. 11 (1), 53–59. doi: 10.1016/j.mib.2008.01.003
Vasilevsky, S., Stojanov, M., Greub, G., Baud, D. (2016). Chlamydial polymorphic membrane proteins: regulation, function and potential vaccine candidates. Virulence 7 (1), 11–22. doi: 10.1080/21505594.2015.1111509
Versteeg, B., Bruisten, S. M., Pannekoek, Y., Jolley, K. A., Maiden, M. C. J., van der Ende, A., et al. (2018a). Genomic analyses of the Chlamydia trachomatis core genome show an association between chromosomal genome, plasmid type and disease. BMC Genom 19 (1), 130. doi: 10.1186/s12864-018-4522-3
Versteeg, B., van den Broek, L. J., Bruisten, S. M., Mullender, M., de Vries, H. J. C., Gibbs, S. (2018b). An organotypic reconstructed human urethra to study Chlamydia trachomatis infection. Tissue Eng. Part A 24 (21-22), 1663–1671. doi: 10.1089/ten.TEA.2017.0511
Voigt, A., Schöfl, G., Saluz, H. P. (2012). The Chlamydia psittaci genome: a comparative analysis of intracellular pathogens. PloS One 7 (4), e35097. doi: 10.1371/journal.pone.0035097
Vorimore, F., Aaziz, R., de Barbeyrac, B., Peuchant, O., Szymańska-Czerwińska, M., Herrmann, B., et al. (2021a). A new SNP-based genotyping method for C. psittaci: application to field samples for quick identification. Microorganisms 9 (3), 625. doi: 10.3390/microorganisms9030625
Vorimore, F., Hölzer, M., Liebler-Tenorio, E. M., Barf, L. M., Delannoy, S., Vittecoq, M., et al. (2021b). Evidence for the existence of a new genus Chlamydiifrater gen. nov. inside the family Chlamydiaceae with two new species isolated from flamingo (Phoenicopterus roseus): chlamydiifrater phoenicopteri sp. nov. and Chlamydiifrater volucris sp. nov. Syst. Appl. Microbiol. 44 (4), 126200. doi: 10.1016/j.syapm.2021.126200
Wen, Z., Boddicker, M. A., Kaufhold, R. M., Khandelwal, P., Durr, E., Qiu, P., et al. (2016). Recombinant expression of Chlamydia trachomatis major outer membrane protein in E. coli outer membrane as a substrate for vaccine research. BMC Microbiol. 16 (1), 165. doi: 10.1186/s12866-016-0787-3
White, R. T., Anstey, S. I., Kasimov, V., Jenkins, C., Devlin, J., El-Hage, C., et al. (2022). One clone to rule them all: culture-independent genomics of Chlamydia psittaci from equine and avian hosts in Australia. Microb. Genom. 8 (10), mgen000888. doi: 10.1099/mgen.0.000888
White, R. T., Jelocnik, M., Klukowski, N., Haque, M. H., Sarker, S. (2023). The first genomic insight into Chlamydia psittaci sequence type (ST)24 from a healthy captive psittacine host in Australia demonstrates evolutionary proximity with strains from psittacine, human, and equine hosts. Vet. Microbiol. 280, 109704. doi: 10.1016/j.vetmic.2023.109704
White, R. T., Legione, A. R., Taylor-Brown, A., Fernandez, C. M., Higgins, D. P., Timms, P., et al. (2021). Completing the genome sequence of Chlamydia pecorum strains MC/MarsBar and DBDeUG: new insights into this enigmatic koala (Phascolarctos cinereus) pathogen. Pathogens 10 (12), 1543. doi: 10.3390/pathogens10121543
Wolff, B. J., Morrison, S. S., Pesti, D., Ganakammal, S. R., Srinivasamoorthy, G., Changayil, S., et al. (2015). Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type III secretion system genes. Microbiology 161 (7), 1378–1391. doi: 10.1099/mic.0.000097
Wood, H., Roshick, C., McClarty, G. (2004). Tryptophan recycling is responsible for the interferon-gamma resistance of chlamydia psittaci GPIC in indoleamine dioxygenase-expressing host cells. Mol. Microbiol. 52 (3), 903–916. doi: 10.1111/j.1365-2958.2004.04029.x
Yang, C., Kari, L., Lei, L., Carlson, J. H., Ma, L., Couch, C. E., et al. (2020). Chlamydia trachomatis plasmid gene protein 3 is essential for the establishment of persistent infection and associated immunopathology. mBio 11 (4), e01902–20. doi: 10.1128/mBio.01902-20
Yang, M., Rajeeve, K., Rudel, T., Dandekar, T. (2019). Comprehensive flux modeling of Chlamydia trachomatis proteome and qRT-PCR data indicate biphasic metabolic differences between elementary bodies and reticulate bodies during infection. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02350
Zadora, P. K., Chumduri, C., Imami, K., Berger, H., Mi, Y., Selbach, M., et al. (2019). Integrated phosphoproteome and transcriptome analysis reveals Chlamydia-induced epithelial-to-mesenchymal transition in host cells. Cell Rep. 26 (5), 1286–1302.e1288. doi: 10.1016/j.celrep.2019.01.006
Zaręba-Marchewka, K., Szymańska-Czerwińska, M., Livingstone, M., Longbottom, D., Niemczuk, K. (2021). Whole genome sequencing and comparative genome analyses of Chlamydia abortus strains of avian origin suggests that Chlamydia abortus species should be expanded to include avian and mammalian subgroups. Pathogens 10 (11), 461–467. doi: 10.3390/pathogens10111405
Zaręba-Marchewka, K., Szymańska-Czerwińska, M., Niemczuk, K. (2020). Chlamydiae - what's new? J. Vet. Res. 64 (4), 461–467. doi: 10.2478/jvetres-2020-0077
Zhao, Y., Wang, J., Chen, J., Zhang, X., Guo, M., Yu, G. (2020). A literature review of gene function prediction by modeling gene ontology. Front. Genet. 11. doi: 10.3389/fgene.2020.00400
Zhong, G. (2017). Chlamydial plasmid-dependent pathogenicity. Trends Microbiol. 25 (2), 141–152. doi: 10.1016/j.tim.2016.09.006
Zhou, L., Lopez Rodas, A., Llangarí, L. M., Romero Sandoval, N., Cooper, P., Sadiq, S. T. (2022). Single gene targeted nanopore sequencing enables simultaneous identification and antimicrobial resistance detection of sexually transmitted infections. PloS One 17 (1), e0262242. doi: 10.1371/journal.pone.0262242
Keywords: Chlamydiaceae, Chlamydia trachomatis, genomics, genome content, whole genome sequence, next generation sequencing, tissue tropism, host tropism
Citation: Luu LDW, Kasimov V, Phillips S, Myers GSA and Jelocnik M (2023) Genome organization and genomics in Chlamydia: whole genome sequencing increases understanding of chlamydial virulence, evolution, and phylogeny. Front. Cell. Infect. Microbiol. 13:1178736. doi: 10.3389/fcimb.2023.1178736
Received: 03 March 2023; Accepted: 10 May 2023;
Published: 23 May 2023.
Edited by:
Rey Carabeo, University of Nebraska Medical Center, United StatesReviewed by:
Tian Luo, University of Texas Medical Branch at Galveston, United StatesNick D. Pokorzynski, Yale University, United States
Copyright © 2023 Luu, Kasimov, Phillips, Myers and Jelocnik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martina Jelocnik, bWplbG9jbmlAdXNjLmVkdS5hdQ==