
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 16 May 2023
Sec. Bacteria and Host
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1178248
This article is part of the Research Topic Global Excellence in Bacteriology: Central and South America View all 6 articles
 Maryoris Elisa Soto Lopez1,2*
Maryoris Elisa Soto Lopez1,2* Marco Tulio Pardini Gontijo1,3
Marco Tulio Pardini Gontijo1,3 Rodrigo Rezende Cardoso1
Rodrigo Rezende Cardoso1 Laís Silva Batalha1Monique Renon Eller1Denise Mara Soares Bazzolli4
Laís Silva Batalha1Monique Renon Eller1Denise Mara Soares Bazzolli4 Pedro Marcus Pereira Vidigal5Regina Célia Santos Mendonça1
Pedro Marcus Pereira Vidigal5Regina Célia Santos Mendonça1Introduction: Bacteriophages infecting human pathogens have been considered potential biocontrol agents, and studying their genetic content is essential to their safe use in the food industry. Tequatrovirus ufvareg1 is a bacteriophage named UFV-AREG1, isolated from cowshed wastewater and previously tested for its ability to inhibit Escherichia coli O157:H7.
Methods: T. ufvareg1 was previously isolated using E. coli O157:H7 (ATCC 43895) as a bacterial host. The same strain was used for bacteriophage propagation and the one-step growth curve. The genome of the T. ufvareg1 was sequenced using 305 Illumina HiSeq, and the genome comparison was calculated by VIRIDIC and VIPTree.
Results: Here, we characterize its genome and compare it to other Tequatrovirus. T. ufvareg1 virions have an icosahedral head (114 x 86 nm) and a contracted tail (117 x 23 nm), with a latent period of 25 min, and an average burst size was 18 phage particles per infected E. coli cell. The genome of the bacteriophage T. ufvareg1 contains 268 coding DNA sequences (CDS) and ten tRNA genes distributed in both negative and positive strains. T. ufvareg1 genome also contains 40 promoters on its regulatory regions and two rho-independent terminators. T. ufvareg1 shares an average intergenomic similarity (VIRIDC) of 88.77% and an average genomic similarity score (VipTree) of 88.91% with eight four reference genomes for Tequatrovirus available in the NCBI RefSeq database. The pan-genomic analysis confirmed the high conservation of Tequatrovirus genomes. Among all CDS annotated in the T. ufvareg1 genome, there are 123 core genes, 38 softcore genes, 94 shell genes, and 13 cloud genes. None of 268 CDS was classified as being exclusive of T. ufvareg1.
Conclusion: The results in this paper, combined with other previously published findings, indicate that T. ufvareg1 bacteriophage is a potential candidate for food protection against E. coli O157:H7 in foods.
Enterohemorrhagic Escherichia coli (EHEC), a subgroup of Shiga toxin-producing E. coli (STEC), has emerged as one of the primary foodborne pathogens (Cooley et al., 2013). The infections’ symptoms vary from watery diarrhea to hemorrhagic colitis (HC) and, in more severe cases, hemolytic-uremic syndrome (HUS) (Gambushe et al., 2022). STEC pathogens belong to a wide range of serotypes, and E. coli O157:H7 is the most prevalent outbreak-associated serotype. Human infections are linked to consuming contaminated foods and water (Rani et al., 2021; Gambushe et al., 2022). E. coli O157:H7 has a low infectious dose (10 to 100 CFU.mL-1) due to its stress resistance mechanisms (Rahal et al., 2012), surviving in low-pH environments such as acidic foods. Acid resistance is crucial for the pathogen’s survival in the stomach’s acidic condition before reaching and colonizing the small intestines and/or colon (Leyer et al., 1995; Price et al., 2004). According to the Centers for Disease Control and Prevention (CDC), as of 2022, STEC was responsible for three outbreaks in the US. The number of cases reached 136, accounting for 63 hospitalizations. Similar outbreaks were reported yearly since 2006.
Classical preservation methods, including pasteurization, radiation, food preservatives (Lopez et al., 2018b; Enciso-Martínez et al., 2022), or lactic acid bacteria (Ren et al., 2019; Gontijo et al., 2020), have been used to control pathogenic bacteria in foods, such as E. coli O157:H7. Alternative methods are proposed to control biological contaminants in foods. For example, recent studies have suggested using bacteriophages to control bacteria for food production and processing, showing promising results (Moye et al., 2018; Endersen and Coffey, 2020).
Viruses are the most abundant entities on the planet, present in all living organisms’ ecosystems (Krishnamurthy et al., 2016; Koskella et al., 2022). Bacteriophage application as antibacterial agents in the food industry has some advantages over traditional methods: (i) phages are highly host-specific, infecting closely related species, species or even strains within a species (Peng and Yuan, 2018); (ii) selective toxicity, infection of humans and other eukaryotes by phages has been reported only in rare occasions (Lehti et al., 2017; Naureen et al., 2020); (iii) little or no influence on the gut microbiota (Galtier et al., 2016); (iv) phages do not alter the sensory properties of foods, once their genomes do not code substances that may change the color, flavor or texture of the foods (Moye et al., 2018); (v) phages are self-replicating and self-limiting if their bacterial host is present (Sillankorva et al., 2012); and (vi) the frequency of phage mutation is higher than that of bacteria, which diminishes the chances of bacterial resistance (Sabouri Ghannad and Mohammadi, 2012).
Bacteriophages have been evaluated for clinical use (Morello et al., 2011; Merabishvili et al., 2017; Zaki et al., 2023), agriculture (Czajkowski et al., 2017; Zaczek-Moczydłowska et al., 2020; Liu et al., 2022), and the food industry (Soffer et al., 2017). A few phage-based products have been approved by the Food and Drug Administration (FDA), ListShield (used in salami, sausage, basterami, seafood, food contact surfaces, and environments), EcoShield (applied to red meat parts and trim intended to be grounded) and SalmoFresh (applied to poultry, fish and shellfish, fresh and processed fruits, and vegetables), all produced by Intralytix, Inc. in the USA. LISTEX, produced by Food Safety in the Netherlands for use in ready-to-eat meat, fish, and cheese, and Agriphage, made by Omnilytics in the USA, for use in agriculture on fruits and vegetables (Jamal et al., 2019). However, more studies are being conducted for further approval and regulation. Phage vB-LmoM-SH3-3 was effective in reducing the Listeria spp. count in salmon meat, reducing by approximately 2.67 log CFU.mL-1 after 24 h of phage addition at 4°C and 4.14 log CFU.mL-1 after 48 h. Similar results were found for orange juice (Zhou et al., 2020). Encapsulated phages specific for Salmonella reduced the count of Salmonella Enteritidis and S. Typhimurium in 0.57 and 1.78 log CFU.cm-2 in meat and 0.86 and 1.2 log CFU.g-1, respectively, in sprout (Petsong et al., 2019). Tequatrovirus ufvareg1, in particular, has already been evaluated against E. coli O157:H7 in the biosanitization of cherry tomatoes (Lopez et al., 2018a). The treatment of cherry tomatoes inoculated with E. coli O157:H7 was reduced by 0.95 log CFU.g-1 after adding a cocktail of bacteriophages (T. ufvareg1 included). This value was statistically similar to the decontamination treatment of tomatoes with the sodium dichloroisocyanurate (0.57 log CFU.g-1), hydrogen peroxide (0.98 log CFU.g-1), and peracetic acid (0.9 log CFU.g-1). T. ufvareg1 encapsulated in alginate via microfluidics and applied in a propylene glycol gel showed efficiency in the reduction of E. coli O157:H7 count (reducing from 4 log CFU.g-1 to an undetectable count on the surface) at a similar level to alcohol 70% (Boggione et al., 2017). In addition, the bacteriophage UFV-AREG1 was encapsulated in several food hydrocolloids and remained viable when submitted to pH 2.5. Phage-loaded beads incubated in simulated intestinal fluid (pH 6.8) resulted in a 50% release of the phages in the first 5 min. These results also highlight the potential of phage UFV-AREG1 to control pathogens in livestock (Silva Batalha et al., 2021). Such reductions in foods or food-related environments could substantially decrease the potential risk of foodborne infections caused by E. coli O157:H7.
Phages specific to E. coli O157:H7 have previously been isolated in several niches (Lu and Breidt, 2015; Shahin et al., 2022). To expand our understanding of the E. coli O157:H7-specific Tequatrovirus ufvareg1, we present the analysis of its morphology, growth parameters, host range, and genomic features in detail. In addition, information about T. ufvareg1 will be helpful in the development of phage control of multiple foodborne pathogens.
The strain Escherichia coli O157:H7 (ATCC 43895) was used as the host for bacteriophage propagation and characterization. The strain was cultivated in Brain Heart Infusion Broth (HiMedia, Brazil) growth medium and incubated at 37°C. Bacteriophage UFV-AREG1 from Tequatrovirus ufvareg1 specie was previously isolated and sequenced (Lopez et al., 2016). Bacteriophages were stocked and diluted in SM buffer (gelatin 0,01% [m/v] [Vetec, Brazil], Tris-HCl 50 mM [pH 7,5] [Sigma Aldrich, Brazil], NaCl 100 mM [Vetec, Brazil], MgSO4.7H2O 8 mM [Vetec, Brazil]).
A bacteriophage suspension at 109 PFU.mL-1 was centrifuged at 26,000 ×g for 60 min. The pellet was washed with an ammonium acetate solution (0.1 M) [Sigma Aldrich, Brazil] and centrifuged at 26,000 ×g for 60 min. The pellet was resuspended in distilled water and filtered through a cellulose acetate membrane (0.22 µm pore). A droplet of the suspension was deposited on the surface of an electron microscopy screen coated with formvar resin. Virions were negatively stained with uranyl acetate (2% w/v) [Sigma Aldrich, Brazil], and micrographs were taken under 80 kV transmission electron microscope Zeiss EM 109 at the Núcleo de Microscopia e Microanálise (NMM/UFV).
The one-step growth experiment was performed following the procedure described previously (Lu and Breidt, 2015). The E. coli O157:H7 (ATCC 43895) culture was activated in BHI broth until the exponential growth phase. The cells were centrifuged at 10,000 ×g for 5 min, and a suspension was prepared in saline solution (0.85% w/v NaCl) [Vetec, Brazil] with turbidity equivalent to 0.5 in the McFarland scale (108 CFU.mL-1). The bacterial suspension was 10-fold diluted, and 0.1 mL was added in 0.8 mL of BHI broth preheated to 37°C. A phage suspension was prepared in SM buffer (105 PFU.mL-1), and 0.1 mL was added to the tube with the bacterial culture (MOI: 0.01), followed by incubation at 37°C for 10 min at 100 rpm. After incubation, the suspension was centrifuged at 13,000 ×g for 30 s, and the supernatant was removed to determine the titer of non-adsorbed bacteriophages. The precipitate with absorbed bacteria and bacteriophages was resuspended in 20 mL of BHI broth preheated to 37°C. Subsequently, 1 mL samples were filtered (0.22 μm) and collected every 5 min for 60 min. Phage count was made by the double layer plating technique, and the T. ufvareg1 phage titer was determined on plaque forming units (PFU) plating ten-fold serial dilutions in SM buffer (Green et al., 2012). The latent period was defined as the time interval between the end of the adsorption and the beginning of the lysis, as indicated by the beginning of the period of increase in the bacteriophage titer. Burst size was calculated by dividing the final number of released bacteriophages by the number of infected bacteria. The number of infected bacteria was estimated through the difference between the initial number of bacteriophages and non-adsorbed bacteriophages.
The DNA of the T. ufvareg1 bacteriophage was extracted according to a previously described procedure (Green et al., 2012). A bacteriophage suspension of 1 mL was added to 20 µL of chloroform [Vetec, Brazil] and stirred for 10 min. After stirring, the suspension was centrifuged at 22,000 rpm for 20 min. The aqueous phase was added of DNAse [Thermo Scientific, Brazil] and RNAse [Thermo Scientific, Brazil] to a final concentration of 1 µg.mL-1 for 10 min at room temperature. Subsequently, 25 µL of proteinase K [Thermo Scientific, Brazil] was added to a final concentration of 1 mg.mL-1 and 0.9 µL of SDS [Sigma Aldrich, Brazil] for 10 min at room temperature. A first extraction was made using phenol-chloroform (1:1) [Vetec, Brazil] for 5 min. It proceeded to centrifuge at 12,000 rpm for 10 min, recovering the aqueous phase. In the second extraction, chloroform was added in the proportion (1:1). Afterwards, the samples were centrifuged at 12,000 rpm for 10 min at 4°C. The aqueous phase was recovered. Ammonium acetate was added at a final concentration of 3.5 M for DNA precipitation and isopropanol in the ratio (1:1), storing the samples at -20°C for approximately 4 h. After this procedure, the samples were centrifuged at 12,000 rpm for 30 min, discarded the supernatant, and dried at room temperature. To remove excess salt from the samples, 200 µL of 70% ethanol [Vetec, Brazil] was added by centrifuging at 12,000 rpm for 10 min, discarding the ethanol, and placing the pellet to dry at 37°C for 10 min. The pellet was reconstituted with 40 µL of ice-cold ultrapure water [Sigma Aldrich, Brazil] and homogenizing. Finally, the DNA extraction process was evaluated on 0.8% agarose gel [Vetec, Brazil] with a 50 ng.mL-1 lambda marker. The genome of the T. ufvareg1 was sequenced using Illumina HiSeq by the company Macrogen (Seoul, Korea).
The quality of sequencing data was assessed using FASTQC version 0.11.9 (https://github.com/s-andrews/FastQC). Adapter sequences were detected and removed from sequencing data using the “auto-detection” setting of TrimGalore version 0.6.7 (Krueger et al., 2021). Then, paired reads were trimmed for quality and filtered for length using Trimmomatic version 0.39 (Bolger et al., 2014) by selecting the following parameters: HEADCROP:10, CROP:90, SLIDINGWINDOW:4:20, and MINLEN:50. The de novo assembly of the genome was performed using the method “careful” of SPAdes version 3.15.3 (Bankevich et al., 2012) and testing all odd k-mers between 21 and 89. Then, the paired reads were mapped in the scaffolds using the BWA-MEM algorithm of BWA version 0.7.17 (Li and Durbin, 2009), and the Sequence Alignment Map (SAM) files were converted to ordered Binary Alignment Map (BAM) format using Picard toolkit version 2.26.2 (https://github.com/broadinstitute/picard). The BAM files were processed by SSPACE version 3.0 (Boetzer et al., 2011), which merged the scaffolds into a single circular sequence (Figure S1).
Eighty-four genomes from species of the Tequatrovirus genus (taxonomy ID: 10663) were downloaded from NCBI Reference Sequence (RefSeq) database (https://www.ncbi.nlm.nih.gov/refseq/, accessed on August 29th, 2022) (Table S1) and used as references to predict the genes of the T. ufvareg1 genome. The genes were predicted using Prokka version 1.14.6 (Seemann, 2014) by selecting the following parameters: compliant, kingdom: viruses, gcode: 11, cdsrnaolap, E-value: 1e−10, proteins: Tequatrovirus Refseq GenBank file. The genome was also inspected to identify putative promoters using the PhagePromoter (https://galaxy.bio.di.uminho.pt/) (Sampaio et al., 2019) by selecting the following parameters: threshold: 0.5, phage family: Podoviridae, host bacteria genus: Escherichia coli, and phage type: virulent. The Rho-independent terminators were identified using ARNold (Naville et al., 2011) (http://rssf.i2bc.paris-saclay.fr/toolbox/arnold/) and FinTerm (Solovyev and Salamov, 2011) by considering a free energy threshold value of -11 kcal/mol for stem-loop regions. The phage host range was previously assessed (Lopez et al., 2018a) and expanded using HostPhinder(Villarroel et al., 2016), and the results were compared to previously published data for T. ufvareg1 bacteriophage(Lopez et al., 2018a). Antibiotic resistance genes were predicted using CARD (Alcock et al., 2023).
Endolysins and holins were screened using HmmerWeb version 2.41.2 (Potter et al., 2018) and the protein family database (Pfam), applying default parameters. Possible signal peptides and signal-arrest-release domains in endolysins were characterized previously (Gontijo et al., 2021b; Gontijo et al., 2022). Signal peptides were predicted using SignalP version 5.0 (Petersen et al., 2011) and PrediSi version 1.0 (Hiller et al., 2006) against the Gram-negative database. Transmembrane regions were predicted using SOSUI version 1.1 (Hirokawa et al., 1998), TMHMM version 2.0 (Krogh et al., 2001), Phobius version 1.01 (Käll et al., 2004), and Topcons version 1.0 (Hennerdal and Elofsson, 2011).
The Eighty-four Tequatrovirus genomes, downloaded from NCBI Reference Sequence (RefSeq) database (Table S1), were compared with the genome of T. ufvareg1. All the genomes were opened in the rIIA gene, which is the same point as the Tequatrovirus T4 genome (NCBI accession NC_000866). A pairwise genomic similarity matrix was calculated by the VIRIDIC web server (http://rhea.icbm.uni-oldenburg.de/VIRIDIC/) (Moraru et al., 2020) as previously described (Hungaro et al., 2022), considering a genomic similarity threshold 95% for species assignment and 70% for the genus as that recommended by ICTV for virus taxonomy. In addition, a proteomic tree was calculated by the Viral Proteomic Tree (VIPTree) web server (https://www.genome.jp/viptree/) (Nishimura et al., 2017), with default parameters. The synteny shared among the Tequatrovirus genomes was evaluated by Clinker version 0.0.23 (Gilchrist and Chooi, 2021), with default parameters. The repertoire of genes shared among the Tequatrovirus genomes was predicted by Roary version 3.13.0 (Page et al., 2015), considering a minimum identity threshold of 80% to cluster the protein sequences in the pangenome and setting that the core genes are shared among all genomes.
T. ufvareg1 is a bacteriophage isolated from cowshed wastewater using Escherichia coli ATCC 11229 as the bacterial host (Lopez et al., 2016). A detailed characterization of this bacteriophage is presented in the following topics. Transmission Electron Microscopy analyses showed that T. ufvareg1 virions have an icosahedral head (114 x 86 nm) and a contracted tail (117 x 23 nm) (Figure 1A). Therefore, T. ufvareg1 belongs to Caudoviricetes class and shares a morphological similarity with the Tequatrovirus phages, such as Tequatrovirus hy01(Lee et al., 2016).
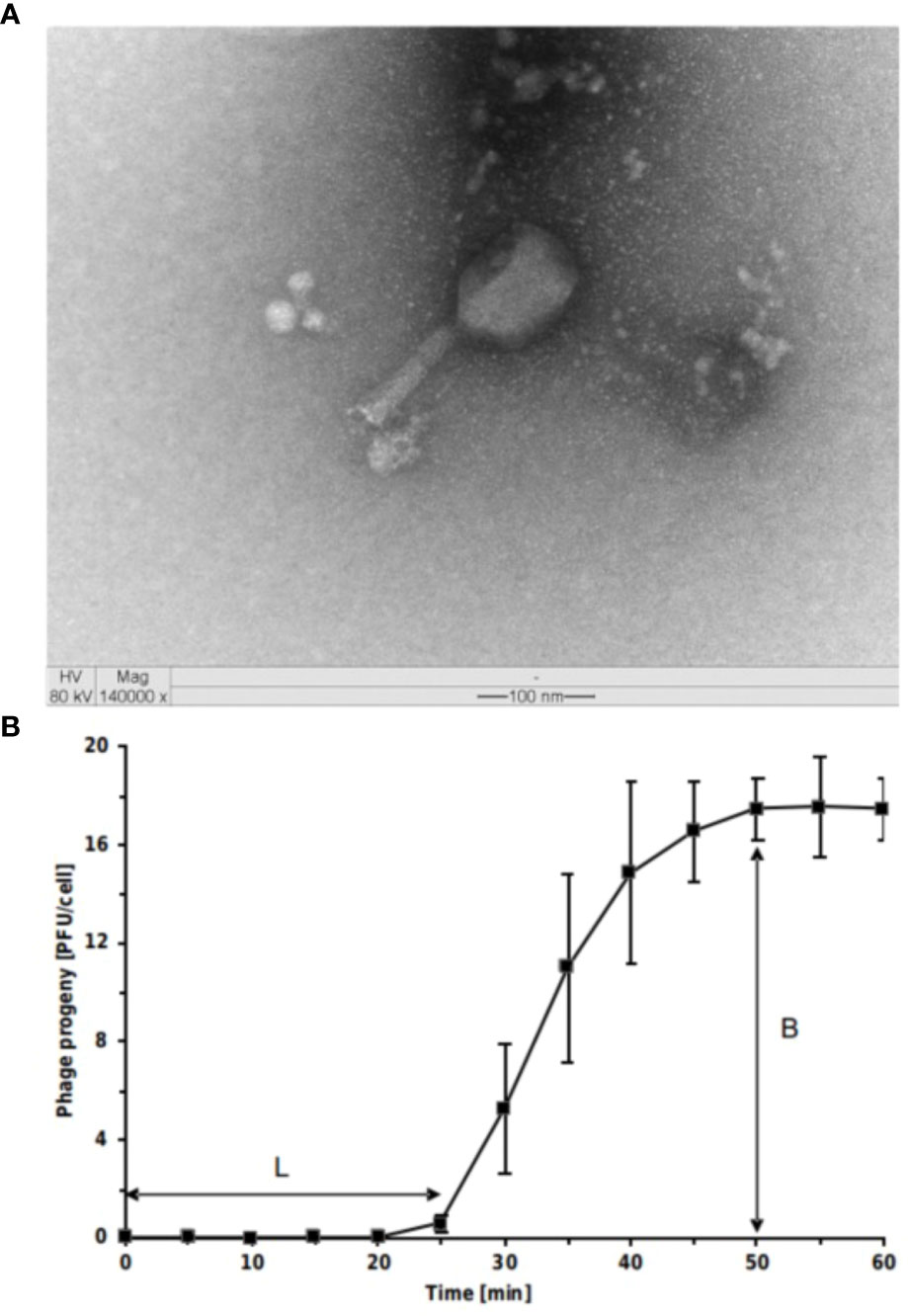
Figure 1 Virions and one-step growth curve of Escherichia coli O157:H7 phage Tequatrovirus ufvareg1. (A) the phage virion was negatively stained with uranyl acetate and observed through transmission electron microscopy (TEM) at × 140,000 magnification, scale bar = 100 nm. (B) The results of one-step growth curves of T. ufvareg1 on E coli O157:H7 (ATCC 43895) are presented as the mean values ± SD from three independent experiments. L, latent period; B, burst size.
To elucidate the ability of T. ufvareg1 bacteriophage to lyse E. coli O157:H7 (ATCC 43895), the latent periods and burst size of the bacteriophage were determined using a one-step growth curve analysis (Figure 1B). The latent period of T. ufvareg1 bacteriophage was 25 minutes, the complete infectious lasted 50 minutes, and the burst size after lysis of the E. coli host was about 18 PFU per infected cell. These results are similar to the one observed for Tequatrovirus hy01 (latent period of 25 minutes and burst size of 25 bacteriophages per infected cell) (Lee et al., 2016). The burst size is an average of bacteriophages released by the bacterial cells in culture, but the capacity of virion production varies from cell to cell (Kannoly et al., 2022). Bacteriophages specific for E. coli O157:H7 have varying latent periods and burst sizes. Bacteriophage vB_Eco4M-7 has a latent period of 10 min, with a burst size of approximately 100 phages per cell (Necel et al., 2020). Bacteriophage Φ241 has a latent period of 15 min and a burst size of 53 phage particles per infected cell (Lu and Breidt, 2015), and bacteriophage SFP10 has a latent period of 25 min and releases about 100 new viral particles after the infectious cycle (Park et al., 2012). The burst size of E. coli O157:H7 bacteriophages varies substantially depending on the bacteriophage. From multiple isolates using the same host strains, it was previously observed a burst size ranging from 91 to 522 virion particles per infected cell (Montso et al., 2019). Generally, a phage with both a short latent period and a large burst size may have a selective advantage over competing phages and might be more effective in phage therapy (Park et al., 2012; Lu and Breidt, 2015; Amarillas et al., 2017). This result might indicate a need for bacteriophage combinations. In the case of T. ufvareg1 bacteriophage, an effective reduction in bacterial count in situ has been observed in a bacteriophage cocktail (Lopez et al., 2018a).
The complete nucleotide sequence of the genome of Tequatrovirus ufvareg1 has been previously assembled using a reference-based approach and deposited in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) under accession number KX009778.4 (Lopez et al., 2016). To understand the genomic organization of the new virus species, we updated this genome using a de novo assembly and deposited it in Genbank under accession KX009778.4. The T. ufvareg1 has a linear dsDNA genome with 167,231 bp and a GC content of 35.35%, similar to what was observed for the Tequatrovirus reference genomes (average values: size = 167,831, GC content = 35.39%) (Supplementary Material, Table S1). The T. ufvareg1 genome has a bidirectional organization with 278 genes corresponding to a gene density of 1.66 genes per 1,000 bp. Information about functional annotation of the coding DNA sequences (CDS) and their respective proteins is detailed in Table S2. Among the 268 predicted CDS, 113 encode hypothetical proteins (40.65%) that share similarities with other Tequatrovirus bacteriophages proteins and have no defined function in the replication and viral infection of T. ufvareg1 (Table S2).
The search for consensus sequences of regulatory elements revealed the presence of 18 phage-specific promoters (score threshold = 0.5) and 325 host promoters (score threshold = 0.9) (Table S3) in the T. ufvareg1 genome. In the positive strand, 13 promoters are found regulating mainly the late genes from the structural gene module. The remaining five promoters are located on the negative strand, including one regulating the gene that encodes the ModB ADP-ribosylase involved in regulating the replication cycle. The host promoters are distributed across the T. ufvareg1 genome, with 108 in the positive strand and 217 in the negative strand. The presence of promoters is a common feature of viral genomes to start and control the gene transcription and consequent protein expression during their infection cycle (Eller et al., 2014; Sampaio et al., 2019). In addition, 20 rho-independent terminator sequences were found by both predictors in the T. ufvareg1 genome, seven located at the positive and 13 at the negative strand (Table S4). As observed for the predicted promoters, the rho-independent terminators are found mainly immediately after the late genes from the structural gene module and after the genes involved in the preparation of the viral genome replication process (Bonocora et al., 2011; Pham et al., 2020). The second terminator is located after the gene encoding DNA polymerase, an essential enzyme for viral replication (Morcinek-Orłowska et al., 2022). The low number of rho-independent terminator sequences (20 terminators), concerning the large number of genes identified in the T. ufvareg1 genome (278 genes), can be explained by the existence of another type of not conserved terminators, the modular structure of the genome of the phage or even the generation of polycistronic mRNAs (Wicke et al., 2021).
Furthermore, ten tRNA genes were found in the genome of the T. ufvareg1 bacteriophage (Tables S1, S5), the same number observed for 24 of 84 Tequatrovirus reference genomes. However, the exact function of the tRNAs encoded by bacteriophages is unclear. These elements of the genome may be involved in adapting to the translation system of the bacterial host, to the demand for codon use pattern by the bacteriophage, or the rapid extinction of the specific translation of the host proteins after phage infection (Miller et al., 2003; Delesalle et al., 2016). A possible function of tRNAs in the phage genome may be to allow greater efficiency in translating specific unessential genes with different codon uses (Yang et al., 2021) or the ability of phages to growth in the host or to infect more hosts (Delesalle et al., 2016).
The 268 CDS predicted in the T. ufvareg1 genome were classified into eight functional categories (“gene expression related proteins,” “metabolism-related proteins,” “cell wall lysis,” “structural proteins,” “direct lysis, host defense, and resistance-genes acquisition proteins,” “hypothetical proteins,” “homing endonucleases,” and “superinfection immunity protein”) (Table S2).
Thirty-four CDS distributed in the positive strand of the UFV-AREG1 bacteriophage genome (CDS numbers 153 to 178, 192 to 197, and 249 to 251) encode “structural proteins” of the virion, such as capsid, tail, and head proteins (Table S2). These CDS can be considered late genes and directly relate to the lysis processes after assembling the virus, DNA maturation, and viral particle release. Other 11 CDS that encode “structural proteins” are in the negative strand of the UFV-AREG1 genome (CDS numbers 27, 35, 39, 146, 148, 150, 152, 182, 183, 190, and 191), and they are probably expressed in parallel to the viral metabolism and replication (Table S2).
Seven CDS encode “direct lysis, host defense, and resistance-genes acquisition proteins,” and four ORFs encode “cell wall lysis” proteins (CDS 103, 120, 154, and 254), including one lysozyme encoded by CDS 120. The lysozyme encoded by CDS 120 is related to the bacteriophage infection process and the degradation of peptidoglycan to allow the entry of viral genetic material into the host. The lysozyme encoded by CDS 154 is an endolysin with the activity that degrades the peptidoglycan and acts together with holin (CDS 254) in the sequential events that lead to the programmed lysis of the host for the release of mature viral particles (Lu et al., 2019). Holin is responsible for permeabilizing the host cell membrane and signaling the exit of lysine to the periplasmic space for peptidoglycan cleavage, facilitating the release of the viral progeny (Wang et al., 2000; Saier and Reddy, 2015). Holin is strongly regulated by antiholin (Guo et al., 2018) encoded by CDS 103, located on the negative strand of the genome of the T. ufvareg1 bacteriophage. The CDS classified as “direct lysis, host defense, and resistance-genes acquisition proteins” are related to the protection of replication processes, in general, to prevent the action by the host from interrupting the viral genome replication process, the assembly of the virus itself and the cell lysis.
In the negative strand of the T. ufvareg1 genome, 102 CDS encode proteins responsible for bacteriophage replication classified as “gene expression-related proteins” or “metabolism-related proteins.” The T. ufvareg1 bacteriophage genome opens with the CDS that encodes the protein rIIA, typical of bacteriophages with lytic activity. The genome of Tequatrovirus bacteriophages has its opening of the genome with the protein rIIA, and the evidence suggests that proteins of type rII are not directly related to the processes of inhibition of cell lysis. When they are absent, an alternative route for the cell lysis process depends on the presence of genes from some possible prophages (Miller et al., 2003; Abedon, 2019). The rIIA protein can be crucial in viral replication if it is found in clusters carrying different enzymes that synthesize deoxyribonucleotides and are associated with both the DNA and membrane of the host, as seen in the representative scheme of the T. ufvareg1 bacteriophage genome. In addition, it might be required to maintain the integrity of the host membrane during infection and replication (Ennis and Kievitt, 1973). Other essential proteins in the DNA replication processes of T. ufvareg1 are the exonuclease (encoded by CDS 13), ModA RNA polymerase ADP-ribosylase (CDS 19), UvsX RecA-like recombination protein (CDS 40), DNA polymerase (CDS 47), UvsW helicase (CDS 184 and 186), DNA ligase (CDS 202) and DNA topoisomerase II (CDS 265). Regarding DNA helicases (CDS 184 and 186), it is interesting to note that they are found on the positive strand of the phage genome. In contrast, the other CDS related to viral replication is in the negative strand.
Three CDS encode proteins related to “superinfection immunity protein” (CDS 37, 44, and 78). CDS 44 encodes an “immunity to a superinfection membrane protein,” which prevents the phage genome from being ejected from the cell cytoplasm (Berngruber et al., 2010; Egido et al., 2022). Likewise, this protein is responsible for preventing infection of the bacterium by other phages of the same family and recognizing the cell receptors on the bacterial surface (Abedon, 2015). In addition, one CDS encodes a “homing endonucleases” in the UFV-AREG1 bacteriophage genome (CDS 244), which might suggest the presence of introns in the genome of this phage, which will be confirmed in the future. The T. ufvareg1 phage contains five CDS classified as “metabolism-related proteins” that encode recombination endonucleases (CDS 55, 58, 77, 229, and 276). These proteins have specific functions in the replication of the viral genome and degradation of the DNA of the bacterial host, and the suppression of the host’s genetic expression by the action of the virus (Murphy, 1998; Sharples et al., 1998). The genome does not contain any conserved antibiotic-resistance genes.
The Holin sequence (CDS 254) belonged to the bacteriophage T holin family (PF11031), and both lytic proteins (CDS 120 and 154) belonged to the glycoside hydrolase family 24 (PF00959). CDS 154 contained one additional Gp5 N-terminal OB domain (PF06714) and three Gp5 C-terminal repeat domains (PF06715), suggesting a cytoplasmatic lysozyme. SignalP and PrediSi did not identify canonical signal peptides within the lytic protein sequences, and transmembrane-predicting software did not identify transmembrane regions at the N-terminal region of putative endolysins. This analysis shows that the lysozymes encoded in the phage genomes must follow the holin-endolysin theory. Lysozyme hydrolyzes the β-(1,4) linkages between N-acetylglucosamine (NAG) and N-acetylmuramic (NAM) acid monomers of peptidoglycan. Holins control lysis timing by forming non-specific pores in the cytoplasmatic membrane. Finally, the pores allow endolysins to reach the peptidoglycan (Gontijo et al., 2021a).
The genomic distance matrix (VIRIDIC) and the viral proteomic tree (ViPTree) confirmed high conservation among 83 of the 85 reference genomes of Tequatovirus and the genome of T. ufvareg1 (Figure 2, Table S1). These genomes showed an average genomic similarity score (VIRIDIC) of 88.77% and a genomic distance score (ViPTree) of 88.91% (Table S1). None of the genomes showed similarity scores above 95%, attributing different isolates to the same species, confirming the isolate UFV-AREG1 as a reference for the Tequatrovirus ufvareg1 specie. Curiously, the genomes of Tequatrovirus efftwo (GenBank accession: NC_054913.1) and Tequatrovirus jaykay (GenBank accession: NC_054940.1) are divergent from other Tequatrovirus. Their genomes showed scores below the 75% recommended by ICTV to cluster species into the same genus. The pangenome analysis confirmed these observations when the number of genes of the core genome of Tequatrovirus increased from 41 to 123 genes after removing the genomes of T. efftwo and T. jaykay from the analysis. Therefore, these two phages represent a distinct genus that is very close evolutionary to the Tequatrovirus genus.

Figure 2 Viral proteomic tree of RefSeq genomes for Tequatrovirus genus. The proteomic tree includes T. ufvareg1 and 84 reference genomes for the Tequatrovirus genus. The genomes were aligned all against all by ViPTree, and the genomic similarity scores (SG) were computed. The genomes were also clustered by VIRIDIC using the genomic similarity (SIM) threshold of 70% for genus assignment (Genus) and 95% for species assignment (Species). The pangenomes were predicted by clustering the protein sequences with an identity threshold of 80%.
The ViPTree included the T. ufvareg1 into a cluster with eight other Tequatrovirus genomes (Figure 2). In this cluster, the most similar genomes to T. ufvareg1 are those from T. hy01 (Escherichia phage HY01) (GenBank accession: NC_027349; VIRIDC score: 91.97%; ViPTree score: 91.87%), T. sh7 (Shigella phage SH7) (GenBank accession: NC_0054942; VIRIDC score: 91.18%; ViPTree score: 91.58%), and Shigella phage psSs-1 (GenBank accession: NC_025829; VIRIDC score: 90.56%; ViPTree score: 90.79%). All the eight Tequatrovirus genomes clustered with T. ufvareg1 are colinear (Figure 3), and the pangenome analysis confirmed the high conservation by identifying 177 core genes, 160 shell genes, and 56 cloud genes. Core genes are found in >95% of the genomes; shell genes are found in 15–95%, while cloud genes are present in less than 15% of genomes (Page et al., 2015). None of the 268 CDS predicted in T. ufvareg1 are exclusive genes in pan-genomic analysis, and 13 CDS are cloud genes showing conservation less or equal to 15% among the genomes (Table 1). The CDS 93 encodes the ModB ADP-rybosilase, involved in the regulation of the replication cycle; the CDS 24 encodes the Mrh transcription modulator, which modulates the host’s heat shock sigma factor (σ32) (Mosig et al., 1998); the CDS 242 encode the Frd.3 hypothetical protein, and the other 9 CDS encode hypothetical proteins with unknown functions.
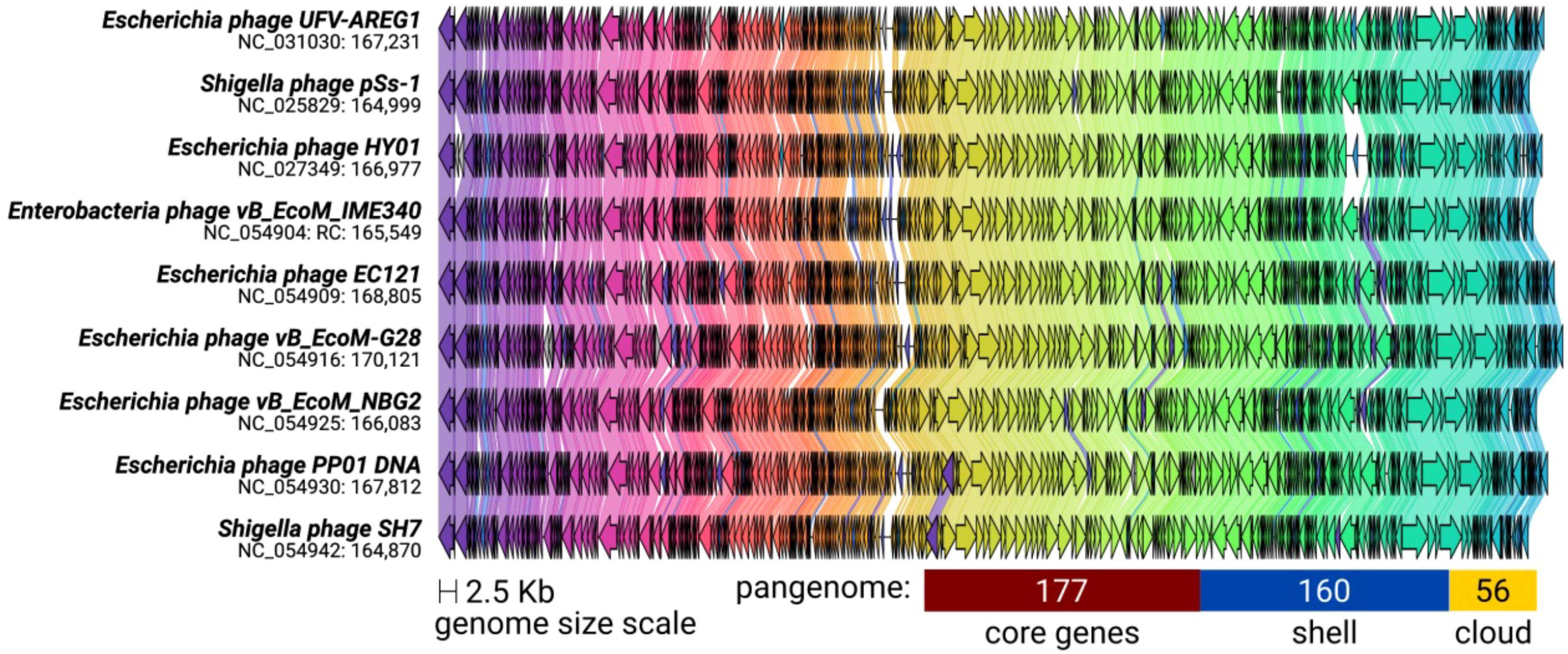
Figure 3 Synteny shared among Tequatrovirus genomes clustered with T. ufvareg1. The genomes have a bidirectional organization with 267 coding DNA sequences on average (represented by arrows). The arrow’s colors represent the gene clusters identified by Clinker, which encode similar proteins. The lines connecting the arrows represent gene-encoding proteins that share a significant sequence identity of more than 40%. The pangenome was predicted by clustering the protein sequences with an identity threshold of 80%.
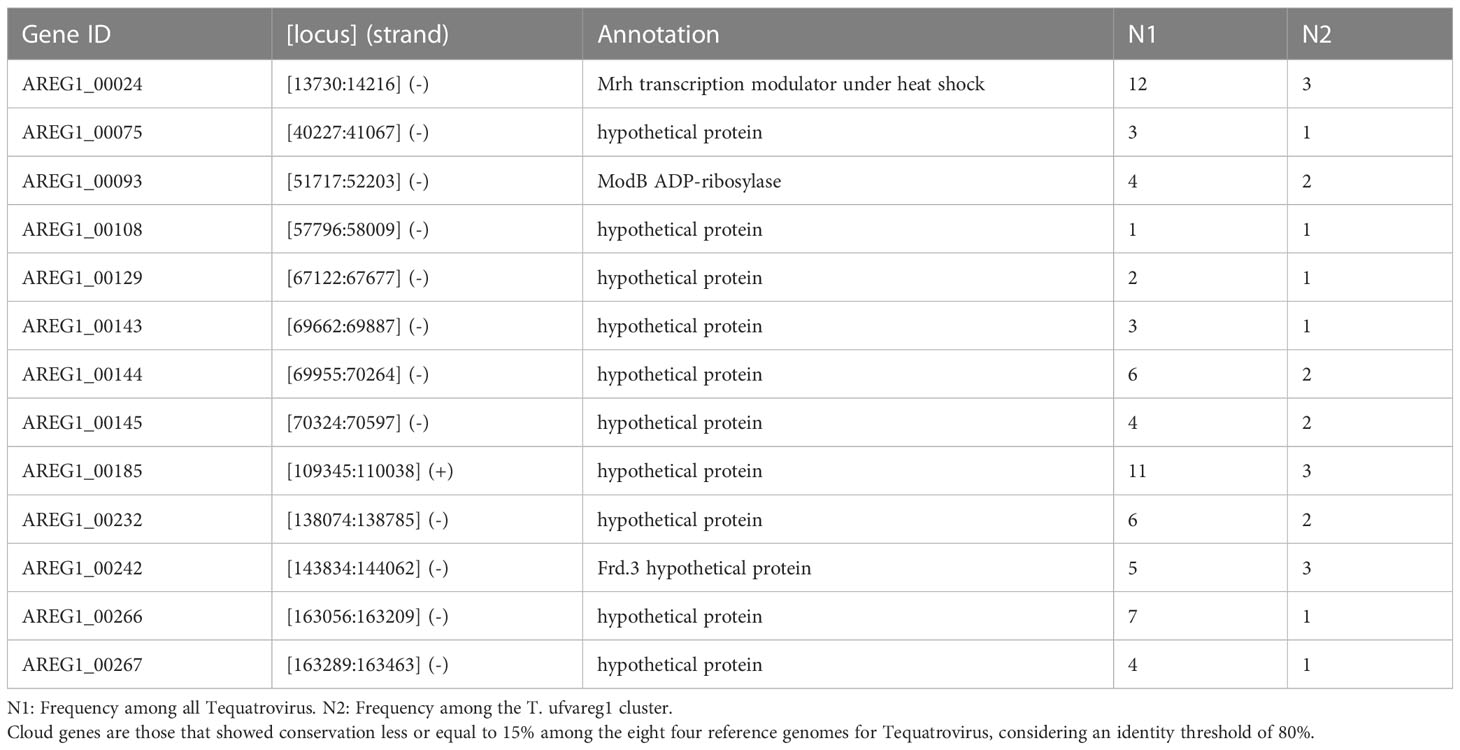
Table 1 Genes of Tequatrovirus ufvareg1 genome classified among cloud genes in pangenome analysis.
The host range of T. ufvareg1 bacteriophage using HostPhinder (Villarroel et al., 2016) revealed that it might be able to infect four bacterial genera: Escherichia (E-value: 5.7e-01), Yersinia (E-value: 5.5e-01), Shigella (E-value: 5.7e-01) and Salmonella (E-value: 1.1e-02), all belonging to the Enterobacteriaceae family. At the species level, T. ufvareg1 is predicted to infect Escherichia coli (E-value: 5.7e-01), Shigella flexneri (E-value: 5.7e-01), Yersinia pestis (E-value: 5.5e-01), Shigella sonnei (E-value: 1.9e-02) and Salmonella enterica (E-value: 1.0e-02). Tequatrovirus ecomufv133 isolated and propagated using Escherichia coli 30 showed similar results after HostPhinder prediction(da Silva Duarte et al., 2018). In vitro determination of host, the range was evaluated against Salmonella Enteritidis, Salmonella Typhi, Salmonella Cholerasius, Escherichia coli, Pseudomonas aeruginosa, Pseudomonas fluorescens, Enterococcus faecium, Enterobacter aerogenes. Lysis was observed only for E. coli strains (Lopez et al., 2018a).
The de novo assembled genome of the Tequatrovirus ufvareg1 bacteriophage isolate has 167,231 bp, a GC content of 35.35%, and 278 predicted genes. The genome annotation is detailed in this study, and 268 CDS encode proteins attributed to eight functional categories. Genomic comparisons confirmed that the UFV-AREG1 isolate only represents the T. ufvareg1 specie. The analysis of Tequatrovirus species revealed high conservation among their genomes and pangenome. The Tequatrovirus ufvareg1 causes rapid cell lysis liberating about 18 viral particles per infected, indicating the bacteriophage has a high potential as an effective biocontrol agent of E. coli O157:H7 in foods.
The datasets generate for this study can be found in figshare: https://figshare.com/s/9ef5934fd016b8cb09e8 and will be published under the following: https://doi.org/10.6084/m9.figshare.21696890.
Conceptualization: ML, ME, DB, and RM; Data curation: ML, MG, and PV; Formal analysis: ML, MG, and PV; Funding acquisition: RM; Investigation: ML, MG, RC, LB, and PV; Methodology: ML, MG, RC, LB, and PV; Project administration: RM; Resources: RM; Software: MG and PV; Supervision: PV, ME, DB, and RM; Validation: MG and PV; Visualization: ML, MG, and PV; Writing – original draft: ML and MG; Writing – review and editing: ME and PV. All authors contributed to the article and approved the submitted version.
The APC was funded by Vicerrectoria de Investigación y Extensión–Universidad de Córdoba, Montería, Colombia. The funders had no role in the study design, data collection, analysis, publication decision, or manuscript preparation.
We acknowledge the Instituto Nacional Controle Qualidade em Saúde (INCQS), a Fundação Oswaldo Cruz (Fiocruz) unit, for kindly providing the bacterial strains to complete this study. We also thank Núcleo de Análise de Biomoléculas (NuBioMol) for supporting data analysis. NuBioMol is supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), CAPES, CNPq, Financiadora de Estudos e Projetos (Finep), and Sistema Nacional de Laboratórios em Nanotecnologias (SisNANO)/Ministério da Ciência, Tecnologia e Informação (MCTI).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1178248/full#supplementary-material
Abedon, S. T. (2015). Bacteriophage secondary infection. Virol. Sin. 30, 3–10. doi: 10.1007/s12250-014-3547-2
Abedon, S. T. (2019). Look who’s talking: T-even phage lysis inhibition, the granddaddy of virus-virus intercellular communication research. Viruses 11, 951. doi: 10.3390/v11100951
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2023). CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51, D690–D699. doi: 10.1093/nar/gkac920
Amarillas, L., Rubí-Rangel, L., Chaidez, C., González-Robles, A., Lightbourn-Rojas, L., León-Félix, J. (2017). Isolation and characterization of phiLLS, a novel phage with potential biocontrol agent against multidrug-resistant escherichia coli. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01355
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berngruber, T. W., Weissing, F. J., Gandon, S. (2010). Inhibition of superinfection and the evolution of viral latency. J. Virol. 84, 10200–10208. doi: 10.1128/JVI.00865-10
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Boggione, D. M. G., Batalha, L. S., Gontijo, M. T. P., Lopez, M. E. S., Teixeira, A. V. N. C., Santos, I. J. B., et al. (2017). Evaluation of microencapsulation of the UFV-AREG1 bacteriophage in alginate-Ca microcapsules using microfluidic devices. Colloids Surfaces B: Biointerfaces 158, 182–189. doi: 10.1016/j.colsurfb.2017.06.045
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonocora, R. P., Zeng, Q., Abel, E. V., Shub, D. A. (2011). A homing endonuclease and the 50-nt ribosomal bypass sequence of phage T4 constitute a mobile DNA cassette. PNAS 108, 16351–16356. doi: 10.1073/pnas.1107633108
Cooley, M. B., Jay-Russell, M., Atwill, E. R., Carychao, D., Nguyen, K., Quiñones, B., et al. (2013). Development of a robust method for isolation of shiga toxin-positive escherichia coli (STEC) from fecal, plant, soil and water samples from a leafy greens production region in California. PloS One 8, e65716. doi: 10.1371/journal.pone.0065716
Czajkowski, R., Smolarska, A., Ozymko, Z. (2017). The viability of lytic bacteriophage ΦD5 in potato-associated environments and its effect on dickeya solani in potato (Solanum tuberosum l.) plants. PloS One 12, e0183200. doi: 10.1371/journal.pone.0183200
da Silva Duarte, V., Dias, R. S., Kropinski, A. M., Campanaro, S., Treu, L., Siqueira, C., et al. (2018). Genomic analysis and immune response in a murine mastitis model of vB_EcoM-UFV13, a potential biocontrol agent for use in dairy cows. Sci. Rep. 8, 1–12. doi: 10.1038/s41598-018-24896-w
Delesalle, V. A., Tanke, N. T., Vill, A. C., Krukonis, G. P. (2016). Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 6, e1219441. doi: 10.1080/21597081.2016.1219441
Egido, J. E., Costa, A. R., Aparicio-Maldonado, C., Haas, P.-J., Brouns, S. J. J. (2022). Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 46, fuab048. doi: 10.1093/femsre/fuab048
Eller, M. R., Vidigal, P. M. P., Salgado, R. L., Alves, M. P., Dias, R. S., da Silva, C. C., et al. (2014). UFV-P2 as a member of the Luz24likevirus genus: a new overview on comparative functional genome analyses of the LUZ24-like phages. BMC Genomics 15, 7. doi: 10.1186/1471-2164-15-7
Enciso-Martínez, Y., González-Aguilar, G. A., Martínez-Téllez, M. A., González-Pérez, C. J., Valencia-Rivera, D. E., Barrios-Villa, E., et al. (2022). Relevance of tracking the diversity of escherichia coli pathotypes to reinforce food safety. Int. J. Food Microbiol. 374, 109736. doi: 10.1016/j.ijfoodmicro.2022.109736
Endersen, L., Coffey, A. (2020). The use of bacteriophages for food safety. Curr. Opin. Food Sci. 36, 1–8. doi: 10.1016/j.cofs.2020.10.006
Ennis, H. L., Kievitt, K. D. (1973). Association of the rIIA protein with the bacterial membrane. PNAS 70, 1468–1472. doi: 10.1073/pnas.70.5.1468
Galtier, M., Sordi, L. D., Maura, D., Arachchi, H., Volant, S., Dillies, M.-A., et al. (2016). Bacteriophages to reduce gut carriage of antibiotic resistant uropathogens with low impact on microbiota composition. Environ. Microbiol. 18, 2237–2245. doi: 10.1111/1462-2920.13284
Gambushe, S. M., Zishiri, O. T., El Zowalaty, M. E. (2022). Review of escherichia coli O157:H7 prevalence, pathogenicity, heavy metal and antimicrobial resistance, African perspective. Infect. Drug Resist. 15, 4645–4673. doi: 10.2147/IDR.S365269
Gilchrist, C. L. M., Chooi, Y.-H. (2021). Clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics 37(16), 2473–2475. doi: 10.1093/bioinformatics/btab007
Gontijo, M. T. P., Jorge, G. P., Brocchi, M. (2021a). Current status of endolysin-based treatments against gram-negative bacteria. Antibiotics 10, 1143. doi: 10.3390/antibiotics10101143
Gontijo, M. T. P., Silva, J., de, S., Vidigal, P. M. P., Martin, J. G. P. (2020). Phylogenetic distribution of the bacteriocin repertoire of lactic acid bacteria species associated with artisanal cheese. Food Res. Int. 128, 108783. doi: 10.1016/j.foodres.2019.108783
Gontijo, M. T. P., Teles, M. P., Vidigal, P. M. P., Brocchi, M. (2022). Expanding the database of signal-Anchor-Release domain endolysins through metagenomics. Probiotics Antimicrob. Proteins 14, 603–612. doi: 10.1007/s12602-022-09948-y
Gontijo, M. T. P., Vidigal, P. M. P., Lopez, M. E. S., Brocchi, M. (2021b). Bacteriophages that infect gram-negative bacteria as source of signal-arrest-release motif lysins. Res. Microbiol. 172, 103794. doi: 10.1016/j.resmic.2020.103794
Green, M. R., Sambrook, J., Sambrook, J. (2012). Molecular cloning: a laboratory manual. 4th ed (Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press).
Guo, T., Xin, Y., Zhang, C., Kong, J. (2018). A cytoplasmic antiholin is embedded in frame with the holin in a lactobacillus fermentum bacteriophage. Appl. Environ. Microbiol. 84, e02518-17. doi: 10.1128/AEM.02518-17
Hennerdal, A., Elofsson, A. (2011). Rapid membrane protein topology prediction. Bioinformatics 27, 1322–1323. doi: 10.1093/bioinformatics/btr119
Hiller, K., Grote, A., Maneck, M., Münch, R., Jahn, D. (2006). JVirGel 2.0: computational prediction of proteomes separated via two-dimensional gel electrophoresis under consideration of membrane and secreted proteins. Bioinformatics 22, 2441–2443. doi: 10.1093/bioinformatics/btl409
Hirokawa, T., Boon-Chieng, S., Mitaku, S. (1998). SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14, 378–379. doi: 10.1093/bioinformatics/14.4.378
Hungaro, H. M., Vidigal, P. M. P., do Nascimento, E. C., Gomes da Costa Oliveira, F., Gontijo, M. T. P., Lopez, M. E. S. (2022). Genomic characterisation of UFJF_PfDIW6: a novel lytic pseudomonas fluorescens-phage with potential for biocontrol in the dairy industry. Viruses 14, 629. doi: 10.3390/v14030629
Jamal, M., Bukhari, S. M. A. U. S., Andleeb, S., Ali, M., Raza, S., Nawaz, M. A., et al. (2019). Bacteriophages: an overview of the control strategies against multiple bacterial infections in different fields. J. Basic Microbiol. 59, 123–133. doi: 10.1002/jobm.201800412
Käll, L., Krogh, A., Sonnhammer, E. L. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Kannoly, S., Oken, G., Shadan, J., Musheyev, D., Singh, K., Singh, A., et al. (2022). Single-cell approach reveals intercellular heterogeneity in phage-producing capacities. Microbiol. Spectr. 11, e02663–e02621. doi: 10.1128/spectrum.02663-21
Koskella, B., Hernandez, C. A., Wheatley, R. M. (2022). Understanding the impacts of bacteriophage viruses: from laboratory evolution to natural ecosystems. Annu. Rev. Virol. 9, 57–78. doi: 10.1146/annurev-virology-091919-075914
Krishnamurthy, S. R., Janowski, A. B., Zhao, G., Barouch, D., Wang, D. (2016). Hyperexpansion of RNA bacteriophage diversity. PloS Biol. 14, e1002409. doi: 10.1371/journal.pbio.1002409
Krogh, A., Larsson, B., von Heijne, G., Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Krueger, F., James, F., Ewels, P., Afyounian, E., Schuster-Boeckler, B. (2021). FelixKrueger/TrimGalore: v0.6.7 - DOI via zenodo. doi: 10.5281/zenodo.5127899
Lee, H., Ku, H.-J., Lee, D.-H., Kim, Y.-T., Shin, H., Ryu, S., et al. (2016). Characterization and genomic study of the novel bacteriophage HY01 infecting both escherichia coli O157:H7 and shigella flexneri: potential as a biocontrol agent in food. PloS One 11, e0168985. doi: 10.1371/journal.pone.0168985
Lehti, T. A., Pajunen, M. I., Skog, M. S., Finne, J. (2017). Internalization of a polysialic acid-binding escherichia coli bacteriophage into eukaryotic neuroblastoma cells. Nat. Commun. 8, 1915. doi: 10.1038/s41467-017-02057-3
Leyer, G. J., Wang, L. L., Johnson, E. A. (1995). Acid adaptation of escherichia coli O157:H7 increases survival in acidic foods. Appl. Environ. Microbiol. 61, 3752–3755. doi: 10.1128/aem.61.10.3752-3755.1995
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Liu, B., Zheng, T., Quan, R., Jiang, X., Tong, G., Wei, X., et al. (2022). Biological characteristics and genomic analysis of a novel vibrio parahaemolyticus phage phiTY18 isolated from the coastal water of xiamen China. Front. Cell. Infection Microbiol. 12. doi: 10.3389/fcimb.2022.1035364
Lopez, M. E. S., Batalha, L. S., Vidigal, P. M. P., Albino, L. A. A., Boggione, D. M. G., Gontijo, M. T. P., et al. (2016). Genome sequence of the enterohemorrhagic escherichia coli bacteriophage UFV-AREG1. Genome Announc. 4, e00412-16. doi: 10.1128/genomeA.00412-16
Lopez, M. E. S., Gontijo, M. T. P., Batalha, L. S., Mendonca, R. C. S. (2018a). Bio-sanitization using specific bacteriophages to control escherichia coli O157:H7 in cherry tomatoes. Advance J. Food Sci. Technol. 16, 92–101. doi: 10.19026/ajfst.16.5942
Lopez, M. E. S., Gontijo, M. T. P., Boggione, D. M. G., Albino, L. A. A., Batalha, L. S., Mendonça, R. C. S. (2018b). “Chapter 3 - microbiological contamination in foods and beverages: consequences and alternatives in the era of microbial resistance,” in Microbial contamination and food degradation handbook of food bioengineering. Eds. Holban, A. M., Grumezescu, A. M. (Academic Press), 49–84. doi: 10.1016/B978-0-12-811515-2.00003-2
Lu, Z., Breidt, F. (2015). Escherichia coli O157:H7 bacteriophage Φ241 isolated from an industrial cucumber fermentation at high acidity and salinity. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00067
Lu, N., Sun, Y., Wang, Q., Qiu, Y., Chen, Z., Wen, Y., et al. (2019). Cloning and characterization of endolysin and holin from streptomyces avermitilis bacteriophage phiSASD1 as potential novel antibiotic candidates. Int. J. Biol. Macromolecules 147, 980–989. doi: 10.1016/j.ijbiomac.2019.10.065
Merabishvili, M., Monserez, R., Belleghem, J., Rose, T., Jennes, S., Vos, D. D., et al. (2017). Stability of bacteriophages in burn wound care products. PloS One 12, e0182121. doi: 10.1371/journal.pone.0182121
Miller, E. S., Kutter, E., Mosig, G., Arisaka, F., Kunisawa, T., Ruger, W. (2003). Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 67, 86–156. doi: 10.1128/MMBR.67.1.86-156.2003
Montso, P. K., Mlambo, V., Ateba, C. N. (2019). Characterization of lytic bacteriophages infecting multidrug-resistant shiga toxigenic atypical escherichia coli O177 strains isolated from cattle feces. Front. Public Health 7. doi: 10.3389/fpubh.2019.00355
Moraru, C., Varsani, A., Kropinski, A. M. (2020). VIRIDIC-a novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 12, E1268. doi: 10.3390/v12111268
Morcinek-Orłowska, J., Zdrojewska, K., Węgrzyn, A. (2022). Bacteriophage-encoded DNA polymerases–beyond the traditional view of polymerase activities. Int. J. Mol. Sci. 23, 1–17. doi: 10.3390/ijms23020635
Morello, E., Saussereau, E., Maura, D., Huerre, M., Touqui, L., Debarbieux, L. (2011). Pulmonary bacteriophage therapy on pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PloS One 6, e16963. doi: 10.1371/journal.pone.0016963
Mosig, G., Colowick, N. E., Pietz, B. C. (1998). Several new bacteriophage T4 genes, mapped by sequencing deletion endpoints between genes 56 (dCTPase) and dda (a DNA-dependent ATPase-helicase) modulate transcription1Published in conjunction with a Wisconsin gathering honoring waclaw szybalski on the occasion of his 75th year and 20years of editorship-in-Chief of gene, 10–11 august 1997, university of Wisconsin, Madison, WI, USA.1. Gene 223, 143–155. doi: 10.1016/S0378-1119(98)00238-8
Moye, Z. D., Woolston, J., Sulakvelidze, A. (2018). Bacteriophage applications for food production and processing. Viruses 10, 1–22. doi: 10.3390/v10040205
Murphy, K. C. (1998). Use of bacteriophage λ recombination functions to promote gene replacement in escherichia coli. J. Bacteriol 180, 2063–2071. doi: 10.1128/JB.180.8.2063-2071.1998
Naureen, Z., Dautaj, A., Anpilogov, K., Camilleri, G., Dhuli, K., Tanzi, B., et al. (2020). Bacteriophages presence in nature and their role in the natural selection of bacterial populations. Acta BioMed. 91, e2020024. doi: 10.23750/abm.v91i13-S.10819
Naville, M., Ghuillot-Gaudeffroy, A., Marchais, A., Gautheret, D. (2011). ARNold: a web tool for the prediction of rho-independent transcription terminators. RNA Biol. 8, 11–13. doi: 10.4161/rna.8.1.13346
Necel, A., Bloch, S., Nejman-Faleńczyk, B., Grabski, M., Topka, G., Dydecka, A., et al. (2020). Characterization of a bacteriophage, vB_Eco4M-7, that effectively infects many escherichia coli O157 strains. Sci. Rep. 10, 3743. doi: 10.1038/s41598-020-60568-4
Nishimura, Y., Yoshida, T., Kuronishi, M., Uehara, H., Ogata, H., Goto, S. (2017). ViPTree: the viral proteomic tree server. Bioinformatics 33, 2379–2380. doi: 10.1093/bioinformatics/btx157
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Park, M., Lee, J.-H., Shin, H., Kim, M., Choi, J., Kang, D.-H., et al. (2012). Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both salmonella enterica and escherichia coli O157:H7. Appl. Environ. Microbiol. 78, 58–69. doi: 10.1128/AEM.06231-11
Peng, Q., Yuan, Y. (2018). Characterization of a newly isolated phage infecting pathogenic escherichia coli and analysis of its mosaic structural genes. Sci. Rep. 8, 1–10. doi: 10.1038/s41598-018-26004-4
Petersen, T. N., Brunak, S., Heijne, G.v., Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Petsong, K., Benjakul, S., Vongkamjan, K. (2019). Evaluation of storage conditions and efficiency of a novel microencapsulated salmonella phage cocktail for controlling s. enteritidis and s. typhimurium in-vitro and in fresh foods. Food Microbiol. 83, 167–174. doi: 10.1016/j.fm.2019.05.008
Pham, J. Y., Ogbunugafor, C. B., Nguyen Ba, A. N., Hartl, D. L. (2020). Experimental evolution for niche breadth in bacteriophage T4 highlights the importance of structural genes. MicrobiologyOpen 9, e968. doi: 10.1002/mbo3.968
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi: 10.1093/nar/gky448
Price, S. B., Wright, J. C., DeGraves, F. J., Castanie-Cornet, M.-P., Foster, J. W. (2004). Acid resistance systems required for survival of escherichia coli O157:H7 in the bovine gastrointestinal tract and in apple cider are different. Appl. Environ. Microbiol. 70, 4792–4799. doi: 10.1128/AEM.70.8.4792-4799.2004
Rahal, E. A., Kazzi, N., Nassar, F. J., Matar, G. M. (2012). Escherichia coli O157:H7–clinical aspects and novel treatment approaches. Front. Cell. Infect. Microbiol. 2. doi: 10.3389/fcimb.2012.00138
Rani, A., Ravindran, V. B., Surapaneni, A., Mantri, N., Ball, A. S. (2021). Review: trends in point-of-care diagnosis for escherichia coli O157:H7 in food and water. Int. J. Food Microbiol. 349, 109233. doi: 10.1016/j.ijfoodmicro.2021.109233
Ren, H., Saliu, E.-M., Zentek, J., Goodarzi Boroojeni, F., Vahjen, W. (2019). Screening of host specific lactic acid bacteria active against escherichia coli from massive sample pools with a combination of in vitro and ex vivo methods. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02705
Sabouri Ghannad, M., Mohammadi, A. (2012). Bacteriophage: time to re-evaluate the potential of phage therapy as a promising agent to control multidrug-resistant bacteria. Iran J. Basic Med. Sci. 15, 693–701.
Saier, M. H., Reddy, B. L. (2015). Holins in bacteria, eukaryotes, and archaea: multifunctional xenologues with potential biotechnological and biomedical applications. J. Bacteriol 197, 7–17. doi: 10.1128/JB.02046-14
Sampaio, M., Rocha, M., Oliveira, H., Dias, O. (2019). Predicting promoters in phage genomes using PhagePromoter. Bioinformatics 35, 5301–5302. doi: 10.1093/bioinformatics/btz580
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shahin, K., Bao, H., Zhu, S., Soleimani-Delfan, A., He, T., Mansoorianfar, M., et al. (2022). Bio-control of O157:H7, and colistin-resistant MCR-1-positive escherichia coli using a new designed broad host range phage cocktail. LWT 154, 112836. doi: 10.1016/j.lwt.2021.112836
Sharples, G. J., Corbett, L. M., Graham, I. R. (1998). λ rap protein is a structure-specific endonuclease involved in phage recombination. Proc. Natl. Acad. Sci. U.S.A. 95, 13507–13512. doi: 10.1073/pnas.95.23.13507
Sillankorva, S. M., Oliveira, H., Azeredo, J. (2012). Bacteriophages and their role in food safety. Int. J. Microbiol. 2012, 863945. doi: 10.1155/2012/863945
Silva Batalha, L., Pardini Gontijo, M. T., Vianna Novaes de Carvalho Teixeira, A., Meireles Gouvêa Boggione, D., Soto Lopez, M. E., Renon Eller, M., et al. (2021). Encapsulation in alginate-polymers improves stability and allows controlled release of the UFV-AREG1 bacteriophage. Food Res. Int. 139, 109947. doi: 10.1016/j.foodres.2020.109947
Soffer, N., Woolston, J., Li, M., Das, C., Sulakvelidze, A. (2017). Bacteriophage preparation lytic for shigella significantly reduces shigella sonnei contamination in various foods. PloS One 12, e0175256. doi: 10.1371/journal.pone.0175256
Solovyev, V., Salamov, A. (2011). “Automatic annotation of microbial genomes and metagenomic sequences,” in Etagenomics and its applications in agriculture, biomedicine and environmental studies. Ed. Li, R. W. (Nova Science Publishers), p.61–p.78.
Villarroel, J., Kleinheinz, K. A., Jurtz, V. I., Zschach, H., Lund, O., Nielsen, M., et al. (2016). HostPhinder: a phage host prediction tool. Viruses 8, 1–22. doi: 10.3390/v8050116
Wang, I. N., Smith, D. L., Young, R. (2000). Holins: the protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 54, 799–825. doi: 10.1146/annurev.micro.54.1.799
Wicke, L., Ponath, F., Coppens, L., Gerovac, M., Lavigne, R., Vogel, J. (2021). Introducing differential RNA-seq mapping to track the early infection phase for pseudomonas phage ΦKZ. RNA Biol. 18, 1099–1110. doi: 10.1080/15476286.2020.1827785
Yang, J. Y., Fang, W., Miranda-Sanchez, F., Brown, J. M., Kauffman, K. M., Acevero, C. M., et al. (2021). Degradation of host translational machinery drives tRNA acquisition in viruses. Cell Syst. 12, 771–779.e5. doi: 10.1016/j.cels.2021.05.019
Zaczek-Moczydłowska, M. A., Young, G. K., Trudgett, J., Plahe, C., Fleming, C. C., Campbell, K., et al. (2020). Phage cocktail containing podoviridae and myoviridae bacteriophages inhibits the growth of pectobacterium spp. under in vitro and in vivo conditions. PloS One 15, e0230842. doi: 10.1371/journal.pone.0230842
Zaki, B. M., Fahmy, N. A., Aziz, R. K., Samir, R., El-Shibiny, A. (2023). Characterization and comprehensive genome analysis of novel bacteriophage, vB_Kpn_ZCKp20p, with lytic and anti-biofilm potential against clinical multidrug-resistant klebsiella pneumoniae. Front. Cell. Infection Microbiol. 13. doi: 10.3389/fcimb.2023.1077995
Keywords: whole genome sequencing (WGS), pan-genome, taxonomy, biocontrol, Enterohemorrhagic Escherichia coli
Citation: Lopez MES, Gontijo MTP, Cardoso RR, Batalha LS, Eller MR, Bazzolli DMS, Vidigal PMP and Mendonça RCS (2023) Complete genome analysis of Tequatrovirus ufvareg1, a Tequatrovirus species inhibiting Escherichia coli O157:H7. Front. Cell. Infect. Microbiol. 13:1178248. doi: 10.3389/fcimb.2023.1178248
Received: 02 March 2023; Accepted: 27 April 2023;
Published: 16 May 2023.
Edited by:
Angel Adrián Cataldi, Instituto Nacional de Tecnología Agropecuaria, ArgentinaReviewed by:
Ariel Fernando Amadio, National Agricultural Technology Institute, ArgentinaCopyright © 2023 Lopez, Gontijo, Cardoso, Batalha, Eller, Bazzolli, Vidigal and Mendonça. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maryoris Elisa Soto Lopez, bWVzb3RvQGNvcnJlby51bmljb3Jkb2JhLmVkdS5jbw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.