 Yasuhiro Suzuki
Yasuhiro Suzuki Jenny Lutshumba
Jenny Lutshumba Kuey Chu Chen
Kuey Chu Chen Mohamed H. Abdelaziz1
Mohamed H. Abdelaziz1- 1Department of Microbiology, Immunology and Molecular Genetics, University of Kentucky College of Medicine, Lexington, KY, United States
- 2Department of Pharmacology and Nutritional Science, University of Kentucky College of Medicine, Lexington, KY, United States
- 3Genomics Core Laboratory, University of Kentucky College of Medicine, Lexington, KY, United States
We previously demonstrated that brain-resident cells produce IFN-γ in response to reactivation of cerebral infection with Toxoplasma gondii. To obtain an overall landscape view of the effects of IFN-γ from brain-resident cells on the cerebral protective immunity, in the present study we employed NanoString nCounter assay and quantified mRNA levels for 734 genes in myeloid immunity in the brains of T and B cell-deficient, bone marrow chimeric mice with and without IFN-γ production by brain-resident cells in response to reactivation of cerebral T. gondii infection. Our study revealed that IFN-γ produced by brain-resident cells amplified mRNA expression for the molecules to activate the protective innate immunity including 1) chemokines for recruitment of microglia and macrophages (CCL8 and CXCL12) and 2) the molecules for activating those phagocytes (IL-18, TLRs, NOD1, and CD40) for killing tachyzoites. Importantly, IFN-γ produced by brain-resident cells also upregulated cerebral expression of molecules for facilitating the protective T cell immunity, which include the molecules for 1) recruiting effector T cells (CXCL9, CXCL10, and CXCL11), 2) antigen processing (PA28αβ, LMP2, and LMP7), transporting the processed peptides (TAP1 and TAP2), assembling the transported peptides to the MHC class I molecules (Tapasin), and the MHC class I (H2-K1 and H2-D1) and Ib molecules (H2-Q1, H-2Q2, and H2-M3) for presenting antigens to activate the recruited CD8+ T cells, 3) MHC class II molecules (H2-Aa, H2-Ab1, H2-Eb1, H2-Ea-ps, H2-DMa, H2-Ob, and CD74) to present antigens for CD4+ T cell activation, 4) co-stimulatory molecules (ICOSL) for T cell activation, and 5) cytokines (IL-12, IL-15, and IL-18) facilitating IFN-γ production by NK and T cells. Notably, the present study also revealed that IFN-γ production by brain-resident cells also upregulates cerebral expressions of mRNA for the downregulatory molecules (IL-10, STAT3, SOCS1, CD274 [PD-L1], IL-27, and CD36), which can prevent overly stimulated IFN-γ-mediated pro-inflammatory responses and tissue damages. Thus, the present study uncovered the previously unrecognized the capability of IFN-γ production by brain-resident cells to upregulate expressions of a wide spectrum of molecules for coordinating both innate and T cell-mediated protective immunity with a fine-tuning regulation system to effectively control cerebral infection with T. gondii.
1 Introduction
Toxoplasma gondii is an obligate intracellular protozoan parasite that can establish a chronic infection in the brain (Montoya and Liesenfeld, 2004; Dubey, 2007). This chronic infection is based on the formation of tissue cysts of the parasite. This infection is prevalent in humans worldwide including developed countries (Montoya and Liesenfeld, 2004). Based on the prevalence of IgG antibodies to T. gondii, which is a standard method to identify the infected individuals, one third of human populations is estimated to be infected with this parasite (Montoya and Liesenfeld, 2004). This chronic infection can reactivate and cause potentially serious toxoplasmic encephalitis (TE), when infected hosts become immunocompromised due to AIDS, neoplasms, or immunosuppressive treatments for organ transplantation (Montoya and Liesenfeld, 2004). The reactivation of chronic T. gondii infection is initiated by ruptures of the tissue cysts, followed by release of bradyzoites from the cysts, conversion of the released bradyzoites to tachyzoites (the acute stage form of the parasite), and proliferation of the tachyzoites.
IFN-γ is required to prevent the cerebral tachyzoite proliferation and the development of TE (Suzuki et al., 1989). CD8+ T cells, and CD4+ T cells at lesser extent, are crucial producers of the IFN-γ to prevent cerebral tachyzoite growth (Gazzinelli et al., 1991; Suzuki et al., 1994; Wang et al., 2005). However, our previous studies identified that the in addition to the T cells, innate immune cells other than NK cells need to produce IFN-γ to prevent reactivation of chronic T. gondii infection (Kang and Suzuki, 2001). Whereas adoptive transfer of immune T cells from infected wild-type (WT) mice to infected athymic nude mice, which lack T cells but have innate immune cells capable of producing IFN-γ, is able to prevent reactivation of cerebral T. gondii infection in the recipients, the transfer of WT immune T cells to infected IFN-γ-knockout mice fails to prevent the reactivation of the infection (Kang and Suzuki, 2001). In addition, a depletion of NK cells from infected nude mice by treatment with anti-asialo GM1 antibody does not ablate the capability of the mice to prevent TE after receiving a transfer of immune T cells (Kang and Suzuki, 2001).
In regard to the non-NK innate immune cells that produce IFN-γ, both CD45+CD11blow microglia and CD45+CD11bhigh blood-derived macrophages purified from the brains of infected, T cell-deficient mice during reactivation of cerebral T. gondii infection are identified to secrete IFN-γ when they are cultured without any additional stimulations in vitro (Wang and Suzuki, 2007). Our recent studies with bone marrow (BM) chimeras revealed that brain-resident cells (non-hematopoietic cells in the brain), but not hematopoietic innate immune cells, need to produce IFN-γ in addition to T cells to prevent TE (Sa et al., 2015). Based on this evidence, in the present study we utilized NanoString myeloid innate immunity panel capable of measuring mRNA levels for 734 immunity-related genes to obtain a comprehensive view of the effects of IFN-γ production by brain-resident cells on the cerebral immune responses during reactivation of cerebral T. gondii infection using the bone marrow chimeric mice with and without IFN-γ production by brain-resident cells. We found that the IFN-γ production by brain-resident cells upregulate mRNA expressions for a wide spectrum of molecules for recruitment and activation of the cells crucial for the innate and T cell-mediated protective immunity to effectively control cerebral tachyzoite proliferation along with increasing an expression of down-regulatory molecules for preventing overly stimulated immune responses that can cause unwanted immunopathology.
2 Materials and methods
2.1 Mice
Female BALB/c, and BALB/c-background RAG1-/- and IFN-γ-/- mice were from the Jackson Laboratories (Bar Harbor, ME). Female Swiss-Webster mice were from Taconic (Germantown, NY). Mice were housed in specific pathogen-free environment and experimental procedures were performed in sterile settings. The studies were performed in accordance with approved protocols from the Institutional Animal Care and Use Committee. Two independent studies were performed and the results from those two studies were combined for the analyses.
2.2 Generation of BM chimeric mice
RAG1-/- and IFN-γ-/- mice received whole body irradiation (950 rads), and 1-2 hrs later, the animals were injected intravenously with 2.4 x 107 BM cells from RAG1-/- mice (namely RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/-, respectively). BM cells were prepared from tibial and femoral bones of the donor mice and suspended in Hanks’ balanced salt solution (HBSS, Hyclone, Logan, UT) with 2% fetal bovine serum (FBS, Sigma, St. Louis, MO). Animals received sulfamethoxazole/trimethoprim (0.8 mg/ml for the former and 0.16mg/ml for the latter) in drinking water beginning at 1 week before the irradiation for 5 weeks.
2.3 Infection with T. gondii
Cysts of the ME49 strain were obtained from brains of chronically infected Swiss-Webster mice (Kang and Suzuki, 2001). BM chimeric mice were infected with 10 cysts orally by gavage at two weeks after discontinuation of sulfamethoxazole/trimethoprim treatment. Animals were then treated with sulfadiazine in the drinking water (400 mg/L) beginning at 4-6 days after infection for 2-3 weeks to control the proliferation of tachyzoites and establish a chronic infection in their brains (Kang and Suzuki, 2001). Their brains were collected on the last day of sulfadiazine treatment (Day 0) and Day 5 after discontinuation of sulfadiazine, which had initiated reactivation of the cerebral infection with T. gondii. The Day 0 is the baseline time point without active tachyzoite proliferation in the brain, and the Day 5 is the early stage of an occurrence of the progressive active tachyzoite proliferation in the brains to elucidate the immune responses activated during the early stage of reactivation of the infection.
2.4 RNA extraction and NanoString gene expression analysis
RNA was isolated from a half brain of each of infected BM chimeric mice using RNA STAT-60 (Tel-test, Friendswood, TX) as previously described (Ochiai et al., 2015) at Day 0 and Day 5 after discontinuation of sulfadiazine treatment. The RNA concentration was measured using Nanodrop ND-2000 (Thermo Fisher Scientific, Waltham MA). Total RNA (100 ng) from each brain sample was quality checked using a Bioanalyzer for RNA Integrity Score (RIN> 9.0) and used to measure mRNA levels for 734 immunity-related genes using NanoString nCounter Mouse Myeloid Innate Immunity Panel (NanoString Technologies, Seattle, WA). The assay was performed by the Genomics Core Laboratory at the University of Kentucky. Data were collected and analyzed using the NanoString SPRINT nCounter and nSolvr 4.0 software (NanoString Technologies), and normalized using positive control and housekeeping genes. When transcript counts of some genes were less than the background value of the assay, which is the average of the negative control transcripts, the data on those genes were excluded from analysis. The major advantage of the use of the NanoString nCounter assay system is that we can apply 12 individual RNA samples to one assay plate for simultaneously quantifying mRNA levels for the 734 immunity-related genes. In this way, we can apply RNA samples from 2 or 3 mice on Day 0 and 3 or 4 mice on Day 5 from each of the RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mouse groups from one experiment to a single assay plate and perform the quantification of the mRNA levels in all of the 12 samples in the identical condition. Two independent studies were performed using the BM chimeric mice, and the mRNA expression levels in the samples from each of these two experiments were measured independently using the same NanoString gene panel. Thereafter, the results from these two studies were combined for the analyses. The total numbers of the samples were five for Day 0 and seven for Day 5 for each of the two groups of BM chimeric mice.
2.5 Statistical analysis
Differences in mRNA expression levels of molecules in NanoString data between experimental groups were determined by ANOVA using PartekFlow software (Partek Inc. MO). Differences that provided P<0.05 were considered significant.
3 Results and discussion
3.1 Expression of mRNA for the molecules involved in the IFN-γ-mediated signaling pathway is upregulated only in the presence of brain-resident cells capable of producing IFN-γ in response to reactivation of cerebral infection with T. gondii
We recently developed an experimental model to examine the roles of IFN-γ production by brain-resident cells in prevention of reactivation of cerebral T. gondii infection (Sa et al., 2015). In this model, RAG1-/- or IFN-γ-/- mice were treated with a whole body irradiation to eradicate hematopoietic cells and thereafter, they received a systemic transfer of BM cells obtained from RAG1-/- mice that lack T and B cells but have innate immune cells such as NK cells and macrophages that can produce IFN-γ (namely RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/-, respectively) (Sa et al., 2015). The brain-resident cells including microglia are irradiation-resistant. Therefore, the only difference between the brains of these two groups of BM chimeric mice was the presence (RAG1-/-→RAG1-/- mice) and the absence (RAG1-/-→IFN-γ-/- mice) of the capability to produce IFN-γ in the brain-resident cells under the presence of the hematopoietic innate immune cells capable of producing this cytokine in the systemic circulation (Sa et al., 2015). Our previous study using this model demonstrated that RAG1-/-→RAG1-/- mice inhibited reactivation of cerebral T. gondii infection approximately 10 times more effectively than did RAG1-/-→IFN-γ-/- mice, and that the efficient control of the reactivation of the infection in the former was associated with approximately 18 times increase in cerebral IFN-γ protein levels, which occurred only in RAG1-/-→RAG1-/- mice, during reactivation of the infection (Sa et al., 2015). In the present study, we utilized this BM chimeric mice model and the NanoString nCounter assay system to obtain an overall landscape view of the effects of IFN-γ production by brain-resident cells on the cerebral immune responses.
RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice were infected with T. gondii and treated with sulfadiazine to control tachyzoite proliferation and establish a chronic infection in their brains. Three to four weeks after the infection, sulfadiazine treatment was discontinued to initiate reactivation of cerebral T. gondii infection. Brains samples were collected from these mice on the last day of sulfadiazine treatment (Day 0) and 5 days after its discontinuation (Day 5). The mRNA levels for the immunity-related genes on Day 0 indicate the baseline expression levels for these genes during the latent chronic T. gondii infection without active tachyzoite proliferation in their brains. The mRNA levels on Day 5 indicate their expression levels in response to an occurrence of reactivation of the infection during the early stage of the progressive active tachyzoite proliferation in the brains of these two groups of mice. The mRNA levels for IFN-γ in response to reactivation of cerebral T. gondii infection (Day 5) were markedly higher in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice as expected (P<0.01), whereas the mRNA levels for this cytokine at Day 0 were similarly low in both of these two groups of animals (Figure 1A). In contrast, mRNA levels for both IFN-α (Ifna1) IFN-β (Ifnb1) did not differ between the brains of these two groups of mice (Figure 1A). Consistently, mRNA levels for the key molecules involved in IFN-γ signaling, signal transducer and activator or transcription 1 (STAT1) and IFN regulatory factor 1 (IRF1), were both more than 5 times grater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.001 for the both) (Figures 1B, C). The mRNA levels for three other IRF molecules that are involved in the IFN-γ signaling (Elser et al., 2002; Kuwata et al., 2002; Farlik et al., 2012) were also significantly upregulated in the former than the latter (1.4-fold increase for IRF2, 3.2-fold increase for IRF7, and 3.8-fold increase for IRF8, P<0.001 for all of these molecules, Figure 1C). In contrast, mRNA levels for the other STAT (STAT 4, 5a, 5b, and 6) and IRF molecules (IRF3, 4, and 5) did not differ between these two groups (Figures 1B, C). Although type I interferon can also upregulate IRF2, IRF7, and IRF8 expression, mRNA levels for both IFN-α and IFN-β were not upregulated in the brains of RAG1-/-→RAG1-/- mice when compare to those of RAG1-/-→IFN-γ-/- mice as described earlier, suggesting that the upregulation of mRNA for these three IRF molecules are due to the increased IFN-γ expression in the former. These results indicate that IFN-γ production by brain-resident cells in response to reactivation of cerebral T. gondii infection is effective to potently activate the IFN-γ signaling pathway to activate the protective immunity to suppress tachyzoite growth and reactivation of the infection.
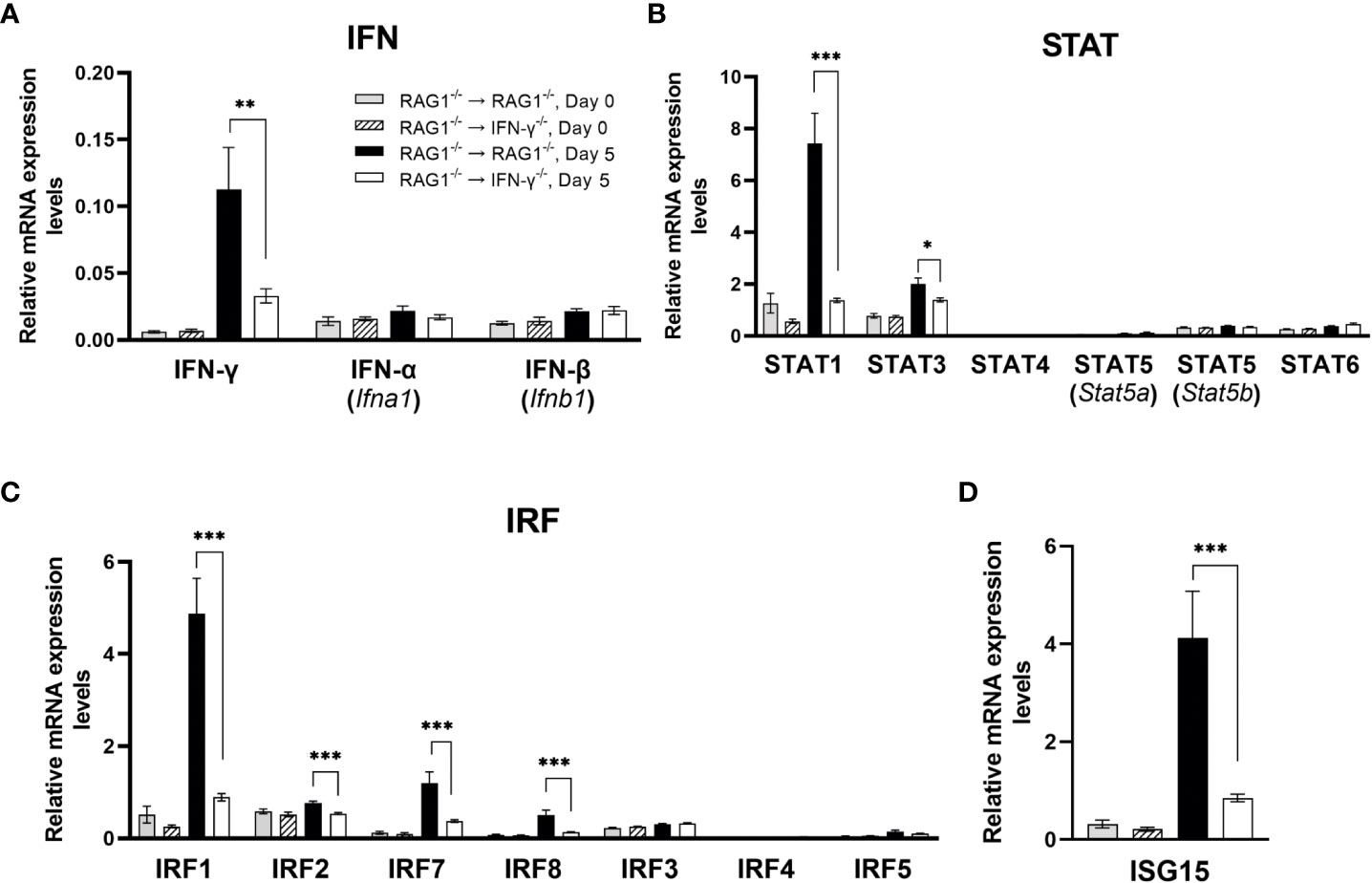
Figure 1 Cerebral mRNA levels for IFN-γ-mediated signaling pathway are upregulated only in the presence of brain-resident cells capable of producing IFN-γ in response to reactivation of cerebral infection with T. gondii. RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice were infected orally with 10 cysts of the ME49 strain of T. gondii and treated with sulfadiazine in the drinking water (400 mg/L) beginning at 4-6 days after infection for 2-3 weeks to control the proliferation of tachyzoites and establish a chronic infection in their brains. Their brains were collected on the last day of sulfadiazine treatment (Day 0) and Day 5 after discontinuation of sulfadiazine (Day 5), which had initiated reactivation of the cerebral infection with T. gondii. The mRNA levels for (A) type I IFN (IFN-α and IFN-β) and type II IFN (IFN-γ), (B) STAT1, 3, 4, 5 and 6, (C) IRF1, 2, 3, 4, 5, 7, and 8, and (D) ISG15 in the brains of both groups of mice at both Day 0 and Day 5 were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
Of interest, mRNA levels for interferon-stimulated gene 15 (ISG15) was also 4.9 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.001) (Figure 1D). A recent study showed that ISG15-/- mice have reduced IFN-γ levels in their sera when compared to WT control mice during the acute stage of T. gondii infection (Napolitano et al., 2018). Therefore, it may be possible that the increased expression of ISG15 contribute to amplifying IFN-γ production by brain-resident cells. ISG15 has also been shown to be involved in killing of tachyzoites within IFN-γ-activated human (HeLa) cells (Bhushan et al., 2020).
3.2 IFN-γ production by brain-resident cells amplifies mRNA expression for the molecules to activate the innate immune functions in response to reactivation of cerebral T. gondii infection
3.2.1 Molecules involved in recruitment of microglia, monocytes, and macrophages
Recent studies by others demonstrated an importance of Ly6C+ inflammatory monocytes in controlling tachyzoite growth in the brains of mice (Biswas et al., 2015). Among 21 CC chemokines that we tested, mRNA expression levels for only CCL8 were 1.7 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.05) (Figure 2A). CCL8 is a chemokine that can recruit various types of immune cell populations including monocytes and macrophages (Charo and Ransohoff, 2006). Although we were unable to find the information on the role of CCL8 in the resistance against T. gondii, it has been reported that CCL8 is associated with control of cytomegalovirus replication in organ transplant recipients (Lisboa et al., 2015).
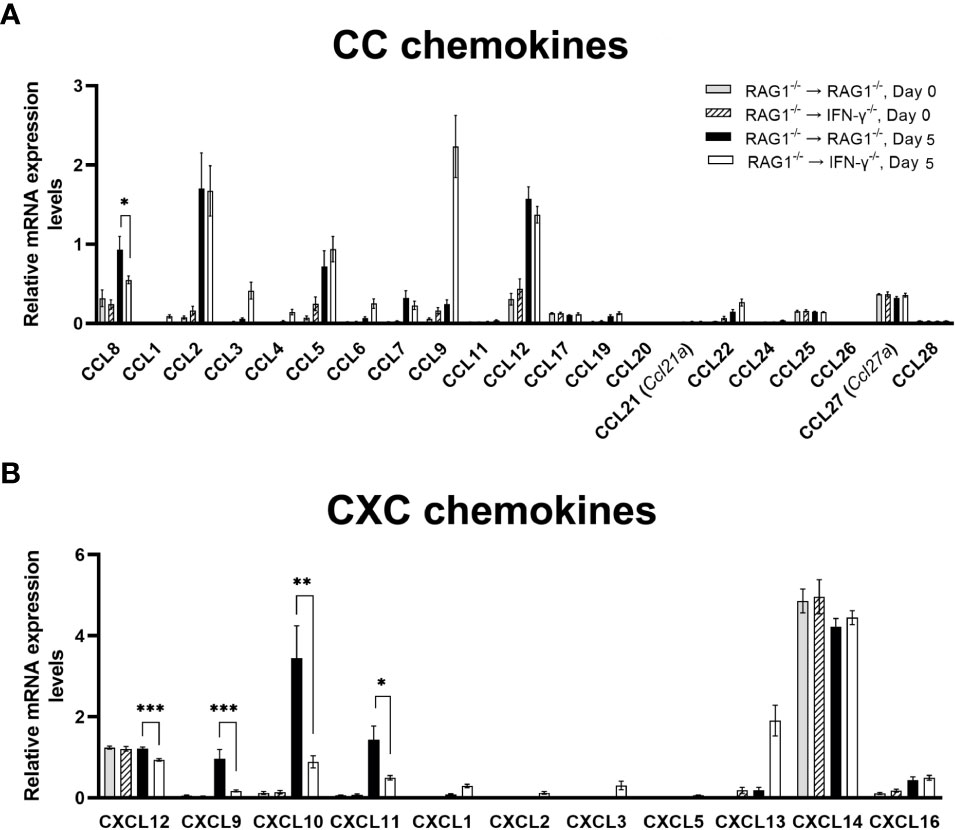
Figure 2 Cerebral mRNA levels for the molecules that recruit microglia, monocytes, macrophages, and T cells are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for (A) 21 CC chemokines and (B) 11 CXC chemokines were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
In regard to CXC chemokines, mRNA levels for CXCL12 along with CXCL9, CXCL10, and CXCL11, were significantly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice at Day 5 after initiation of reactivation of cerebral T. gondii infection (P<0.001) (Figure 2B). CXCL12 is able to stimulate transmigration of monocytes across the blood-brain barrier in vitro (Man et al., 2012). A unique characteristic of this chemokine is that both glial cells and neurons can produce this chemokine (Guyon, 2014). A recent study using a human neuroblastoma cell line demonstrated that transfection of this cell line to express T. gondii rhoptry protein 16 (ROP16) induces increased expression of CXCL12 (Fan et al., 2016). ROP16 is one of the molecules that T. gondii secretes when it invade into host cells. Therefore, it is most likely that neurons, possibly glial cells as well, produce CXCL12 when they become infected with T. gondii tachyzoites. Another study in humans also showed that increased serum levels of CXCL12 are a marker for an occurrence of ocular toxoplasmosis (Marino et al., 2020). Therefore, it is possible that greater levels of CXCL12 mRNA in the brains of RAG1-/-→RAG1-/- mice contribute to increased recruitment of inflammatory monocytes for efficient control of cerebral tachyzoite growth during reactivation of T. gondii infection, when compared to infected RAG1-/-→IFN-γ-/- mice.
CXCR3 is the receptor for each of CXCL9, CXCL10, and CXCL11, and this chemokine receptor is expressed on microglia, resident macrophages in the brain parenchyma, in both normal and disease conditions (Rappert et al., 2004; de Haas et al., 2008; Krauthausen et al., 2014). Microglia activated by IFN-γ are able to kill tachyzoites in vitro (Chao et al., 1993). Whereas the direct evidence on an expression of CXCR3 on microglia in the brain during T. gondii infection is not available yet, our recent studies demonstrated that CXCR3 expression significantly increases during CD8+ T cell-mediated anti-T. gondii cyst immune responses (Lutshumba et al., 2020), in which Iba1+ microglia and Ly6C+ inflammatory macrophages accumulate to the cysts and phagocytose bradyzoites within the cysts for their elimination (Tiwari et al., 2019). Therefore, it is possible that CXCR3+ microglia accumulate into the areas of tachyzoite proliferation and phagocytose them to kill the parasite.
3.2.2 Molecules involved in increasing phagocytosis of microglia and macrophages
IL-18 can enhance phagocytosis of microglia and macrophages (Mori et al., 2001; Henan et al., 2014). Of interest, mRNA expression levels for IL-18 were significantly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection (P<0.05) (Figure 3). Caspase-1 cleaves the precursor of IL-18 and activates this cytokine. In agreement, expression levels of mRNA for caspase-1 were 2.5 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.001) (Figure 3). An importance of IL-18 in resistance against T. gondii was recently demonstrated by using an acute infection model in mice deficient in IL-18 and IL-18 receptor (IL-18R) (Gorfu et al., 2014). They demonstrated that wild-type mice had markedly increased IL-18 levels in sera during the acute stage of infection, and that both IL-18-/- and IL-18R-/- mice showed increased mortality associated with enhanced parasite proliferation during the infection (Gorfu et al., 2014). Consistently, a recent study showed that mice deficient in caspase-1/11 display decreased serum IL-18 levels and increased parasite replication and mortality (Gorfu et al., 2014). Another recent study using macrophages from T. gondii-resistant LEW rats demonstrated that pharmacological inactivation of caspase-1 ablated the capability of their macrophages to kill tachyzoites in vitro (Cavailles et al., 2014), although it is not addressed whether this anti-tachyzoite activity of caspase-1 is mediated by IL-18 or not.
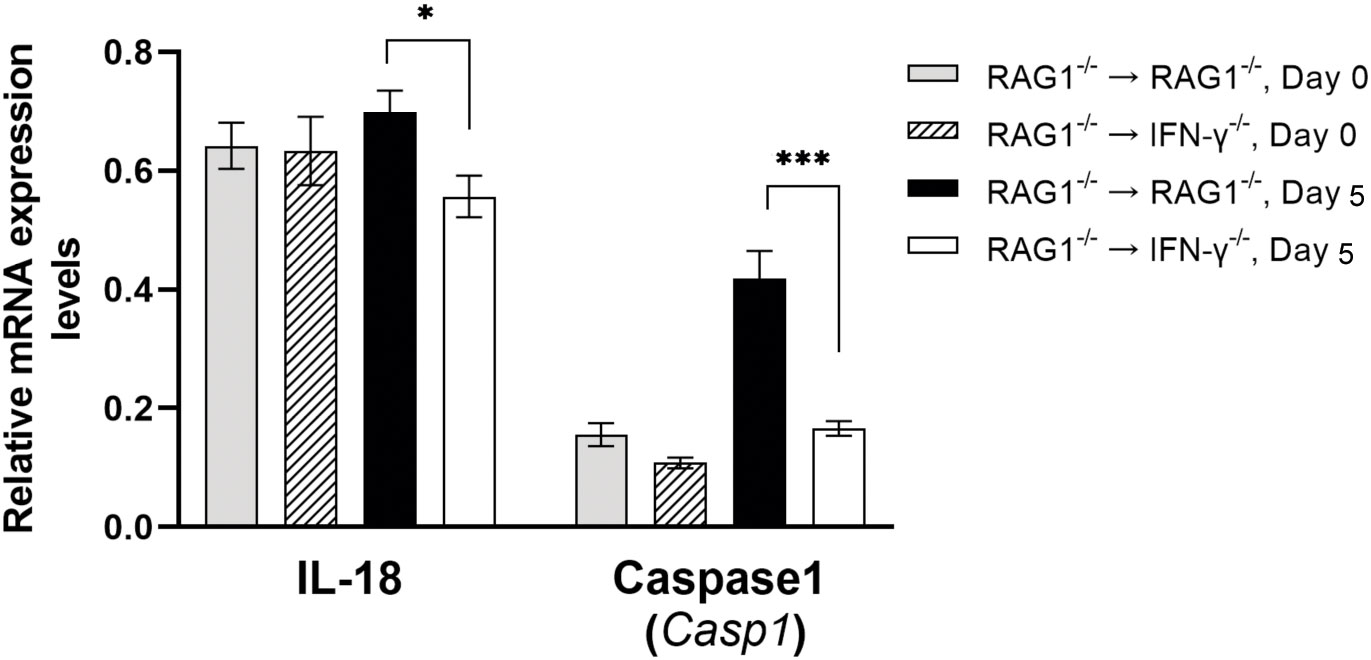
Figure 3 Cerebral mRNA levels for molecules that increase phagocytosis of microglia and macrophages are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for IL-18 and caspase1, which generate active IL-18 molecule from pro-IL-18, were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05 and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
3.2.3 Molecules involved in recognition of the pathogen-associated molecular patterns including Toll-like receptors, and NOD-like receptors
Among 11 TLR tested, mRNA levels for TLR3, TLR9, TLR11, and TLR12 were approximately 2-3 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection (P<0.001 for TLR3, TLR9, and TLR12 and P<0.01 for TLR11) (Figure 4A). TLR11 is the first TLR that was identified to recognize a T. gondii molecule, profilin (Yarovinsky et al., 2005). TLR11 forms heterodimers with TLR12 to recognize T. gondii profilin for activating the IL-12 production (Koblansky et al., 2013; Raetz et al., 2013) and this activity is mediated by IRF8 (Raetz et al., 2013). Notably, IRF8 is one of the molecules whose mRNA levels were markedly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice as described earlier in section 1. TLR9 is known to recognize unmethylated CpG motifs of bacterial DNA. A previous study using WT and TLR9-/- mice demonstrated that TLR9-/- mice have higher parasite burdens than WT mice in the intestine during acute stage of oral infection with T. gondii in association with reduced frequencies of IFN-γ-producing T cells in lamina propria in the former (Minns et al., 2006). TLR3 is known to recognize double stranded RNA. A recent study demonstrated that mice deficient in TLR3/7/9 are more susceptible to T. gondii infection than mice deficient in TLR7/9 (Andrade et al., 2013), suggesting an involvement of TLR3 in resistance against T. gondii infection. Therefore, IFN-γ production by brain-resident cells in RAG1-/-→RAG1-/- mice upregulates mRNA expressions of the TLR molecules that recognize nucleic acids and protein (profilin) of T. gondii to most likely amplify the protective immunity against the parasite.
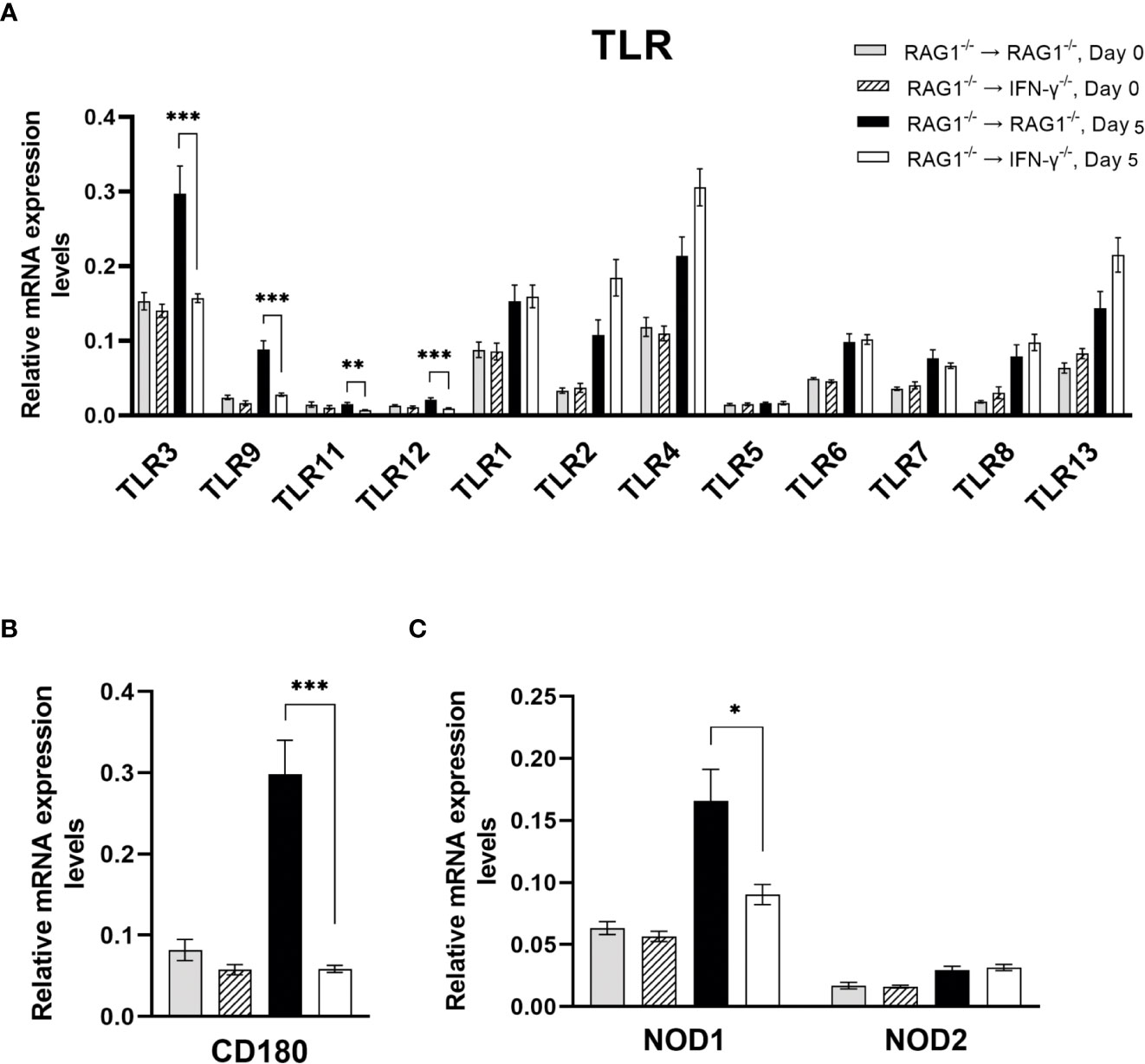
Figure 4 Cerebral mRNA levels for the molecules that recognize the pathogen-associated molecular patterns are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for (A) 12 TLRs, (B) CD180 (a member of TLR family), and (C) NOD1 and NOD2 were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
Expression levels of mRNA for CD180 were 5 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection (P<0.001) (Figure 4B). CD180 is a member of TLR family and known as a member of microglia sensomes (Hickman et al., 2013; Saddala et al., 2021). Although the ligand for this TLR family molecule remains unknown, this sensome is induced by IFN-γ and TNF-α in microglia (Saddala et al., 2021). Therefore, it is possible that CD180 is involved in recognition of T. gondii tachyzoites by microglia to activate innate immunity.
Expression levels of mRNA for NOD-like receptor 1 (NOD1) were also significantly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice following reactivation of T. gondii infection (P<0.05) (Figure 4C) whereas mRNA levels for NOD2 did not differ between these two groups of mice (Figure 4C). Although the protective roles of NOD1 in T. gondii infection remain to be identified, it has been shown that NOD1 plays important roles in resistance against Listeria monocytogenes (Mosa et al., 2009). NOD1-/- mice showed increased dissemination of the bacteria into the brains (Mosa et al., 2009). They also showed that non-hematopoietic cells mediate the NOD1-dependent resistance, and that astrocytes and fibroblasts of NOD1-/- mice displayed enhanced intracellular growth of L. monocytogenes in in vitro (Mosa et al., 2009). Therefore, it may be possible that increased NOD1 mRNA levels in the brains of RAG1-/-→RAG1-/- mice contribute to controlling intracellular proliferation of T. gondii tachyzoites within brain-resident cells during reactivation of the infection.
3.2.4 Molecules involved in CD40
Recent studies demonstrated that an activation of microglia and macrophages through CD40 expressed on their surfaces triggers killing of intracellular T. gondii tachyzoites (Andrade et al., 2006; Portillo et al., 2010), and that CD40-/- mice are susceptible to cerebral toxoplasmosis with impaired killing of the parasite (Portillo et al., 2010). In the present study, mRNA levels for CD40 were approximately 2-fold greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of T. gondii infection (P<0.05) (Figure 5). Astrocytes (Calingasan et al., 2002; Ke et al., 2005) and neurons (McWilliams et al., 2015; Carriba and Davies, 2021) have been shown to express CD40 ligand (CD40L). Therefore, interactions of CD40+ microglia and macrophages with CD40L+ astrocytes and/or neurons will be able to activate them to kill tachyzoites in the brain.
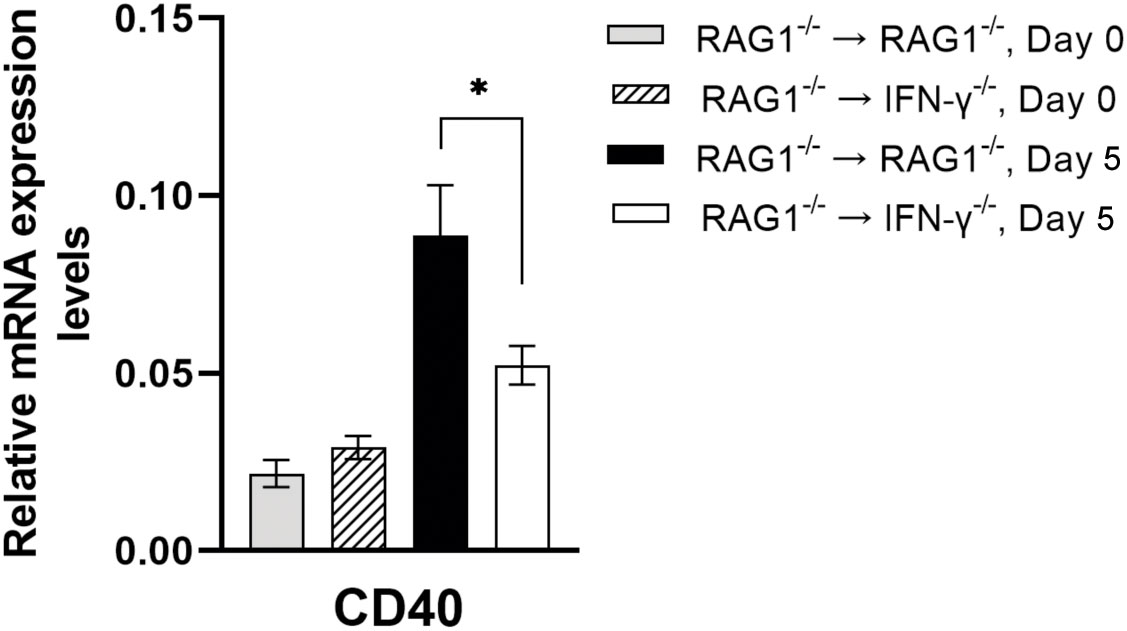
Figure 5 Cerebral mRNA levels for CD40, which activates intracellular killing of tachyzoites by microglia and macrophages, are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for CD40 were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
3.3 IFN-γ production by brain-resident cells amplifies mRNA expression for the molecules to facilitate the protective T cell immunity in response to reactivation of cerebral T. gondii infection
3.3.1 Molecules involved in recruitment of CD4+ and CD8+ effector T cells
Our previous study demonstrated that the majority of both CD4+ and CD8+ T cell populations that have migrated into the brains of infected BALB/c mice are expressing CXCR3 on their cell surfaces (Ochiai et al., 2015). CXCL9, CXCL10, and CXCL11 are the three ligands for CXCR3 as mentioned earlier in section 2.1, and the expression levels for mRNA for each of these three chemokines were much greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (5.9 times for CXCL9 [P<0.001], 4.4 times for CXCL10 [P<0.01], and 2.9 times for CXCL11 [P<0.05]) (Figure 2B). These results on CXCL9 and CXCL10 are consistent with those that we recently reported (Sa et al., 2015). In regard to CXCL11, our previous study showed that mRNA levels for this chemokine were greater in the brains of infected WT control mice than IFN-γ-/- mice, indicating that cerebral expression of this chemokine is mediated by IFN-γ during T. gondii infection (Wen et al., 2010).
In regard to the protective functions of these three chemokines to prevent cerebral tachyzoites growth and reactivation of T. gondii infection, our recent study demonstrated that CXCL9 is critical for recruiting both CD4+ and CD8+ immune T cells not only into the brain but also into the areas of tachyzoite proliferation in the brain to prevent the parasite growth in BALB/c mice that are genetically resistant to this infection (Ochiai et al., 2015). The RAG1-/- and IFN-γ-/- mice that were used to generate the BM chimeric mice in the present study were BALB/c-background. In regard to the roles of CXCL10, it was shown that CXCL10 is important for recruitment of CD8+ T cells into the brain and inhibiting tachyzoite proliferation during the progressive TE in genetically susceptible C57BL/6 mice (Harris et al., 2012). A previous study demonstrated that CXCL9 is mostly expressed in microglia, whereas CXCL10 is mostly expressed in astrocytes in the brains of infected BALB/c mice (Strack et al., 2002). Therefore, it would be possible that IFN-γ production by brain-resident cells in response to cerebral tachyzoite proliferation activates expression of CXCL9 in microglia and CXCL10 in astrocytes in the areas of tachyzoite growth and these chemokines produced by the different glial cell populations both contribute to recruiting immune T cells into those areas to prevent the pathogen growth. Although we were unable to find the information on the cell populations that express CXCL11 or the protective role of this chemokine in controlling T. gondii in the brain, an expression of CXCL11 in neurons has been reported in other disease models (Guo et al., 2017; Yu et al., 2019). Therefore, it would be possible that neurons in the areas of tachyzoite growth produce CXCL11 and contribute to recruiting immune T cells into those areas.
3.3.2 Molecules involved in processing, transporting, and presentation of antigens through the MHC class I molecules for activating CD8+ T cells
CD8+ T cells play a crucial role in controlling cerebral tachyzoite growth to prevent reactivation of T. gondii infection (Gazzinelli et al., 1991; Suzuki et al., 1994; Wang et al., 2005). After CD8+ immune T cells migrate into the areas of tachyzoite proliferation in the brain, they need to be activated by recognizing their target antigens presented by the MHC class I molecules. This antigen-presentation process consists of the following four steps: 1) antigen processing and peptide generation, 2) peptide transport, 3) assembly of peptide-MHC class I complex, 4) presentation of the antigen by the MHC class I molecules on the surface of the cell membrane.
3.3.2.1 Antigen processing and peptide generation
Immunoproteasome brakes ubiquitinated protein antigens into peptides and release them into cytoplasm to initiate the antigen presentation process. NanoString nCounter Mouse Myeloid Innate Immunity Panel contains three genes, Psme2, Psmb8, and Psmb9, for the molecules that compose immunoproteasome, and mRNA levels for each of these three genes were markedly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (2.2 times for Psme2, 5.0 times for Psmb8, and 5.9 times for Psmb9, P<0.001 for each molecule) during reactivation of T. gondii infection (Figure 6A). Immunoproteasome gate is normally closed, and PA28αβ coded by the Pmse2 gene is one of the regulatory particles that open the gate of the immunoproteasome (Whitby et al., 2000). The Psmb8, and Psmb9 genes encode proteasome subunit-β5i (also called low molecular weight protein [LMP] 7) and proteasome subunit-β1i (also called LMP2) of immunoproteasome, respectively. These subunits are critical for generating epitopes of multiple antigens for MHC class I molecules (Sijts et al., 2000; Kincaid et al., 2011). Notably, each of these three subunits is induced by IFN-γ. Therefore, the greater mRNA levels for these three subunits of immunoproteasome in the brains of infected RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice are consistent with the higher IFN-γ levels derived from the production of this cytokine by brain-resident cells in the former.
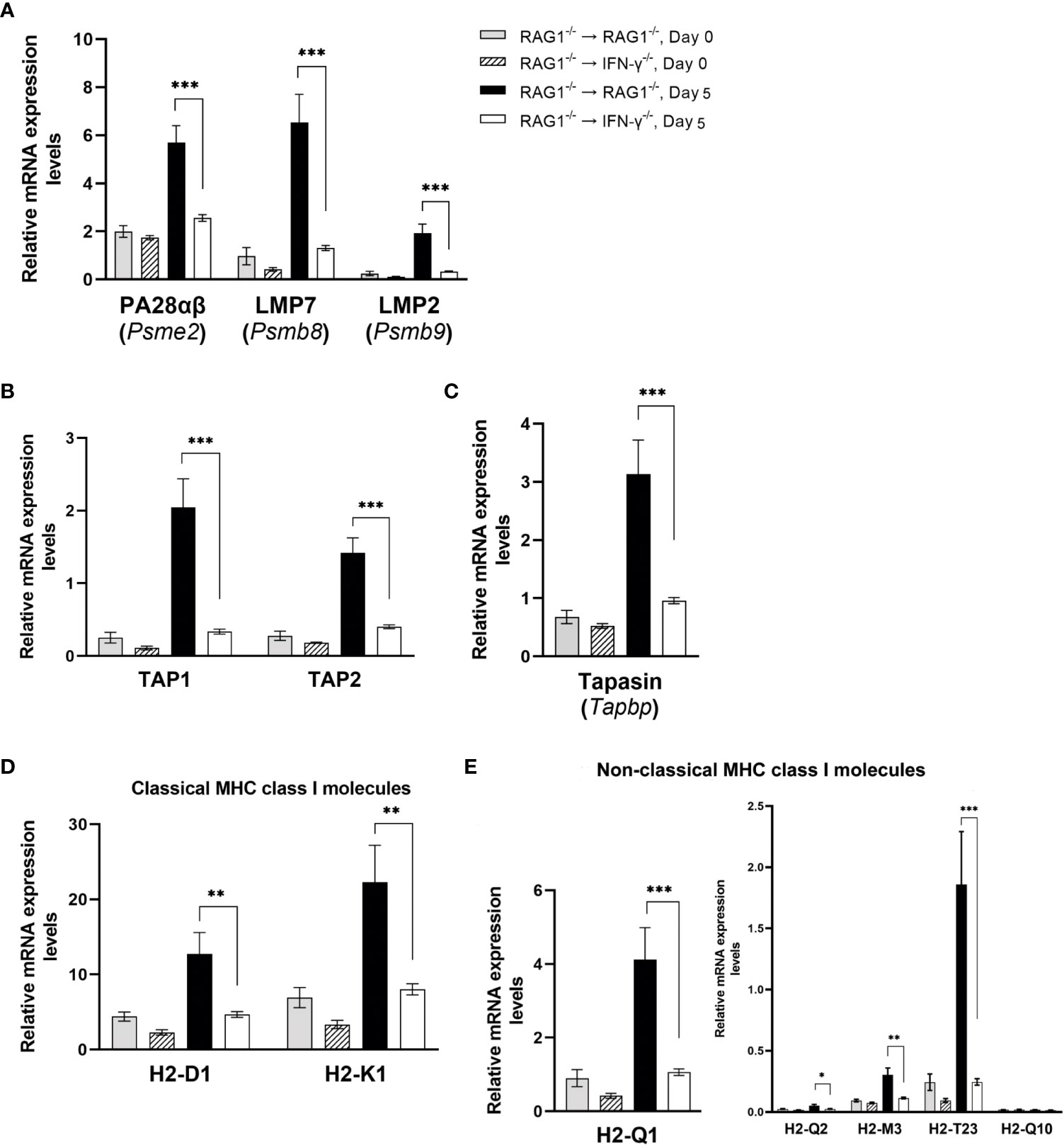
Figure 6 Cerebral mRNA levels for the molecule involved in processing, transporting, and presentation of antigens through the MHC class I molecules for activating CD8+ T cells are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for the molecules for (A) antigen processing and peptide generation, (B) Peptide transport, (C) assembling peptide-MHC class I complex, and (D, E) presenting the peptides by classical MHC class I molecules (D) and non-classical MHC class I molecules (E) to activate CD8+ T cells were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
3.3.2.2 Peptide transport
The peptides generated by the immunoproteasone are actively transported from the cytoplasm to endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP) composed of TAP1 and TAP2, that forms a transmembrame pore in the ER membrane. Expression levels for mRNA for TAP1 and TAP2 were 6.1 and 3.5 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice in response to reactivation of T. gondii infection (P<0.001 for both) (Figure 6B).
3.3.2.3 Assembly of peptide-MHC class I complex
There are four chaperone proteins that facilitate loading of transported peptides onto the MHC class I molecules within the ER. Tapasin is one of the chaperone proteins that bridge the MHC class I molecule to TAP and allow peptides, which are transported into the ER, to gain access to the MHC class I molecules (Sadasivan et al., 1996; Lehner et al., 1998). The mRNA levels for Tapbp, which encodes Tapasin, in the brains of infected RAG1-/-→RAG1-/- mice were 3.3 times greater than those of infected RAG1-/-→IFN-γ-/- mice during reactivation of the infection (P<0.001) (Figure 6C).
3.3.2.4 Antigen presentation
The assembled peptide-MHC class I complex are then placed into vesicles, and the vesicles leave the ER, pass over the Golgi, and transferred to the cell membranes to fuse with it. This process allows the peptides bound to the MHC class I molecules exposed extracellularly for activating CD8+ T cells. Expression levels for mRNA for classical MHC class I molecule genes, H2-K1 and H2-D1, were both approximately three times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of T. gondii infection (P<0.01) (Figure 6D). The mRNA levels for H2-T23, which is the molecule predicted to be a part of the classical MHC class I protein and non-classical MHC class Ib protein complex, were 7.6 times greater in the former than the latter (P<0.001) (Figure 6E). The mRNA levels for three non-classical MHC class Ib complex molecules, H2-Q1, H-2Q2, and H2-M3, were also 2 to 3.9 times greater in the former than the latter (P<0.001 for H2-Q1 P<0.05 for H2-Q2, P<0.01 for H2-M3) (Figure 6E). Whereas the contributions of the non-classical MHC class Ib molecules in activating the protective CD8+ T cells against T. gondii remain to be determined, an importance of this CD8+ T cell activation pathway has been shown in infections with various pathogens including bacteria (D'Orazio et al., 2003) and viruses (Mazzarino et al., 2005; Swanson et al., 2008).
The non-classical MHC class Ib molecules are involved in activation of not only CD8+ T cells but also NK cells and NKT cells (Gomes et al., 2007). NK cells produce IFN-γ following T. gondii infection and contribute to controlling tachyzoite growth (Hunter et al., 1994). Therefore, the increased expression of H2-Q1, H-2Q2, H2-M3, and H2-T23 induced by IFN-γ production by brain-resident cells in RAG1-/-→RAG1-/- mice could contribute to preventing cerebral tachyzoite proliferation by activating not only CD8+ T cells but also NK cells during reactivation of cerebral T. gondii infection. The results shown in Figures 6A–E all together strongly suggest that IFN-γ production by brain-resident cells in the brains of RAG1-/-→RAG1-/- mice is able to upregulate cerebral mRNA expression for the molecules critical for all of the four steps to present T. gondii antigens through the MHC class I molecules to activate CD8+ immune T cells and possibly NK cells recruited into the brain during reactivation of cerebral T. gondii infection.
3.3.3 Molecules involved in antigen presentation for activating CD4+ T cells
CD4+ T cells recognize their target antigens presented by the MHC class II molecules. Among 10 MHC class II molecules tested, mRNA levels for 7 of those molecules were significantly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of T. gondii infection (P<0.001 for H2-Aa, H2-Ab1,H2-DMa, H2-Ea-ps, H2-Eb1, and CD74, and P<0.05 for H2-Ob) (Figure 7). These results indicate that IFN-γ production by brain-resident cells, which occurs in the brains of RAG1-/-→RAG1-/- mice in response to reactivation of T. gondii infection, is able to enhance expression of most of the MHC class II molecules. In addition to CD8+ T cells, CD4+ T cells contribute to the protective immunity (Gazzinelli et al., 1991; Suzuki et al., 1994; Wang et al., 2005). Therefore, the upregulation of cerebral expression of the MHC class II molecules induced by brain-resident cell IFN-γ production most likely contribute to preventing cerebral tachyzoite growth by facilitating activation of CD4+ T cells in response to reactivation of cerebral T. gondii infection.
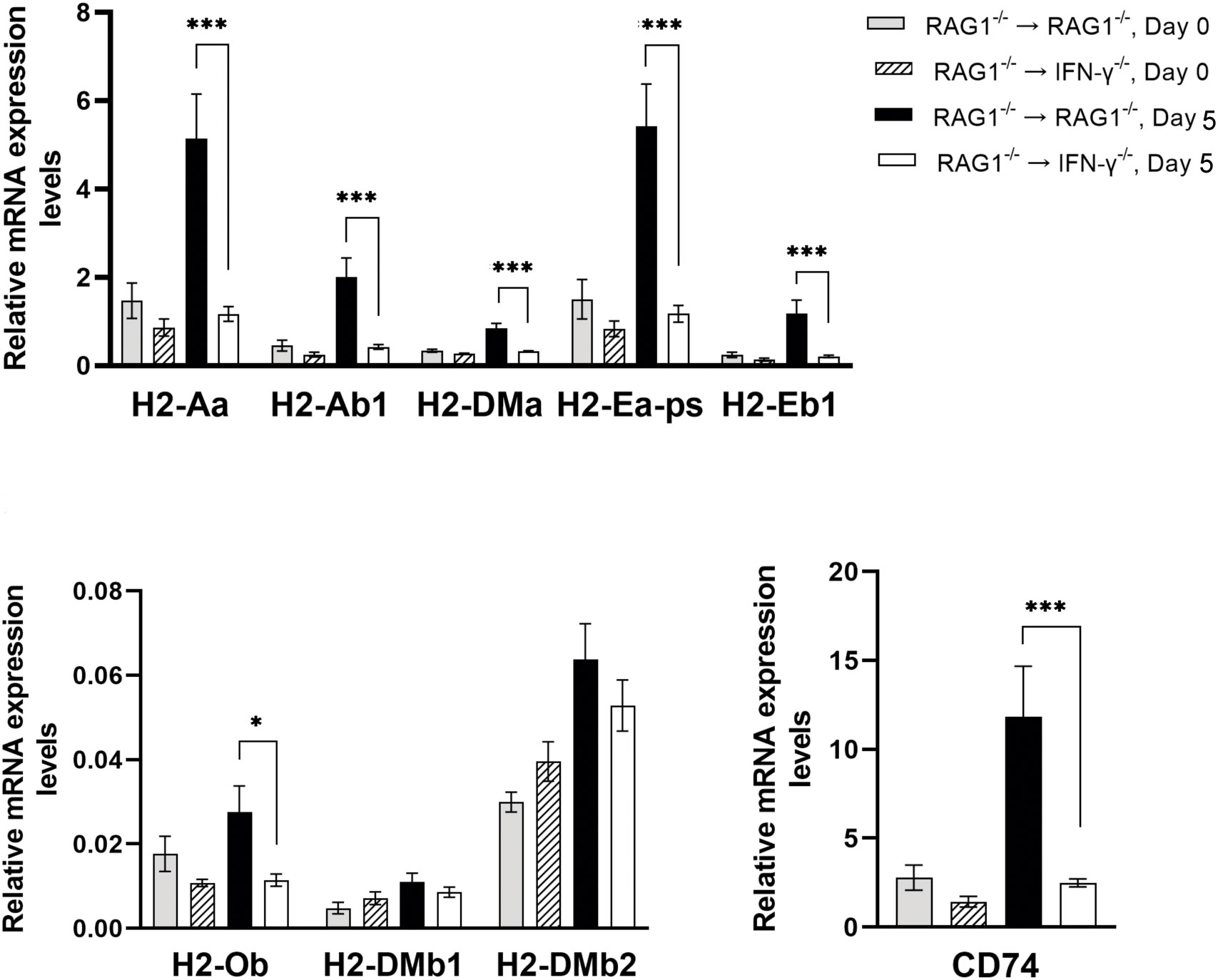
Figure 7 Cerebral mRNA levels for the molecules involved in antigen presentation for activating CD4+ T cells are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for 9 MHC class II molecules were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05 and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
3.3.4 Co-stimulatory molecules for T cell activation
Interactions of CD28 expressed on CD4+ T cells with CD80 and CD86 expressed on antigen-presenting cells provide the co-stimulatory signal required for an activation of the T cells in addition to their recognition of target antigens presented by the MHC class II molecules. An interaction of the inducible co-stimulatory molecule (ICOS) on CD4+ T cells with ICOS ligand (ICOSL) on antigen-presenting cells is another co-stimulatory pathway for CD4+ T cells. Expression levels of mRNA for ICOSL in the brains of RAG1-/-→RAG1-/- mice were approximately twice greater than those in RAG1-/-→IFN-γ-/- mice during reactivation of T. gondii infection (P<0.01) (Figure 8A). A recent study demonstrated that ICOS is required for optimal proliferation of CD4+ T cells following T. gondii infection (Wilson et al., 2006). The absence of ICOS resulted in reduced numbers of CD4+ T cells and reduced production of IFN-γ by mononuclear cells isolated from the brains of infected mice following stimulation with tachyzoite lysate antigens in vitro (Wilson et al., 2006). Therefore, it is possible that the increased expression levels of ICOSL induced by IFN-γ production by brain-resident cells in the brains of RAG1-/-→RAG1-/- mice contribute to enhancing proliferation and IFN-γ production of CD4+ T cells following their migration into their brains for efficient control of reactivation of cerebral T. gondii infection.
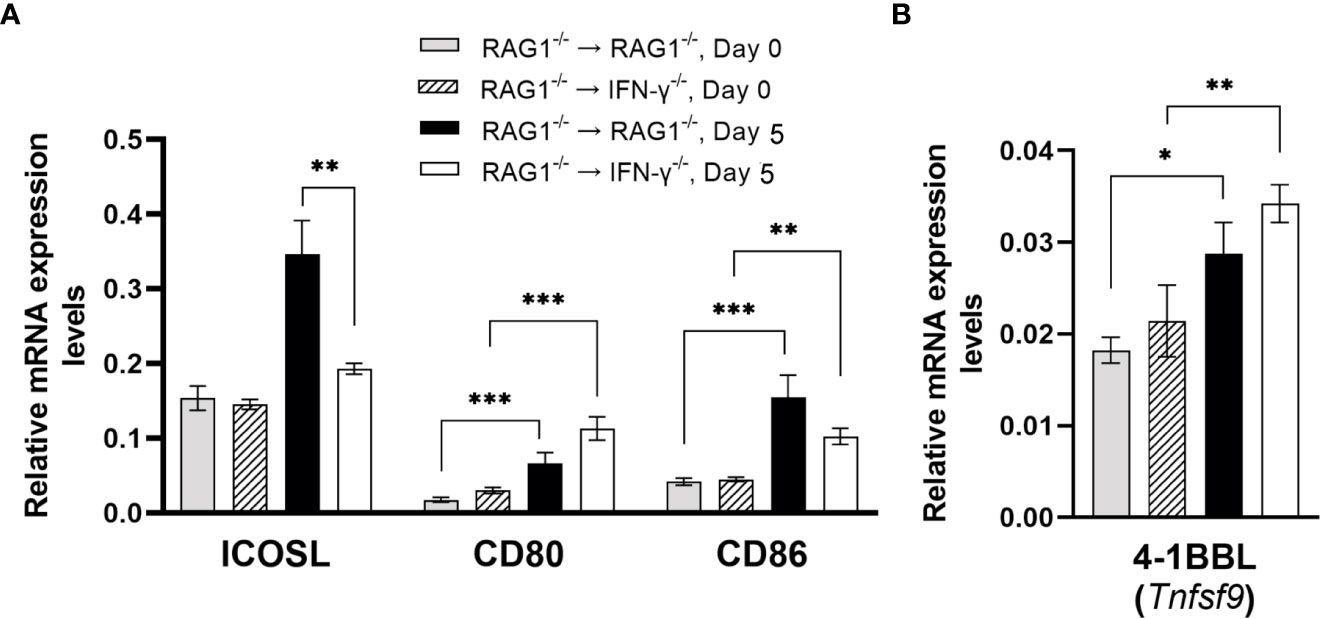
Figure 8 Cerebral mRNA levels for co-stimulatory molecules are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for (A) ICOS, CD80, CD86 and (B) 4-1BBL were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
Although mRNA levels for both CD80 and CD86 did not differ between the brains of infected RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection (Figure 8A), mRNA expression levels for these co-stimulatory molecules were both significantly greater after initiation of reactivation of the infection (Day 5) than their expression levels before reactivation of the infection (Day 0) in either of these two groups of BM chimeric mice (P<0.01or P<0.001) (Figure 8A). Previous studies showed that both CD80 and CD86 are important for proliferation and IFN-γ production of primed T cells from T. gondii-seropositive individuals during their secondary response to the parasite antigens in vitro (Subauste et al., 1998). Another study demonstrated that T. gondii infection triggers upregulation of CD86 in macrophages from BALB/c mice genetically resistant to cerebral T. gondii infection, and that the upregulated CD86 facilitate antigen-specific proliferation of CD4+ T cells (Fischer et al., 1999). Interestingly, the upregulation of CD86 did not occur in infected macrophages of BALB.B mice that are genetically susceptible to the infection (Fischer et al., 1999).
In activation of CD8+ T cells, the interaction between 4-1BB expressed on the T cells and 4-1BBL (Tnfsf9) expressed on the antigen-presenting cells provides the co-stimulation signal for their activation. Although mRNA levels for the Tnfsf9 did not differ between the brains of RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice after reactivation of cerebral T. gondii infection (Figure 8B), their 4-1BBL mRNA levels after reactivation of the infection were significantly greater those of prior to initiation of the reactivation of the infection (P<0.05 or P<0.01) (Figure 8B). In our previous study, CD8+ T cells that migrated into the brains of RAG1-/-→RAG1-/- mice following adoptive T cell transfer become activated and efficiently produce IFN-γ (Sa et al., 2015). Therefore, those upregulated 4-1BBL expression induced independently from IFN-γ produced by brain-resident cells in response to reactivation of T. gondii infection appears to provide the sufficient co-stimulatory signal for the recruited CD8+ T cells. As mentioned earlier, enhanced recruitment of CD8+ T cells and amplified antigen presentation by the MHC class I molecules induced by IFN-γ produced by brain-resident cells in RAG1-/-→RAG1-/- mice in combination with sufficient levels of co-stimulation signal from the 4-1BB/4-1BBL interactions can most likely provide the sufficient protective immunity to control reactivation of cerebral T. gondii infection.
3.3.5 Cytokines for facilitating IFN-γ production by NK cells and T cells
IL-12 plays an important role in activating IFN-γ production by NK cells and T cells during T. gondii infection (Hunter et al., 1994). Expression levels for IL12B mRNA were 3.2 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection (P<0.001) (Figure 9). Therefore, IFN-γ produced by brain-resident cells in response to reactivation of the infection probably induces an environment to further facilitate IFN-γ-production by NK and T cells to prevent cerebral tachyzoite growth.
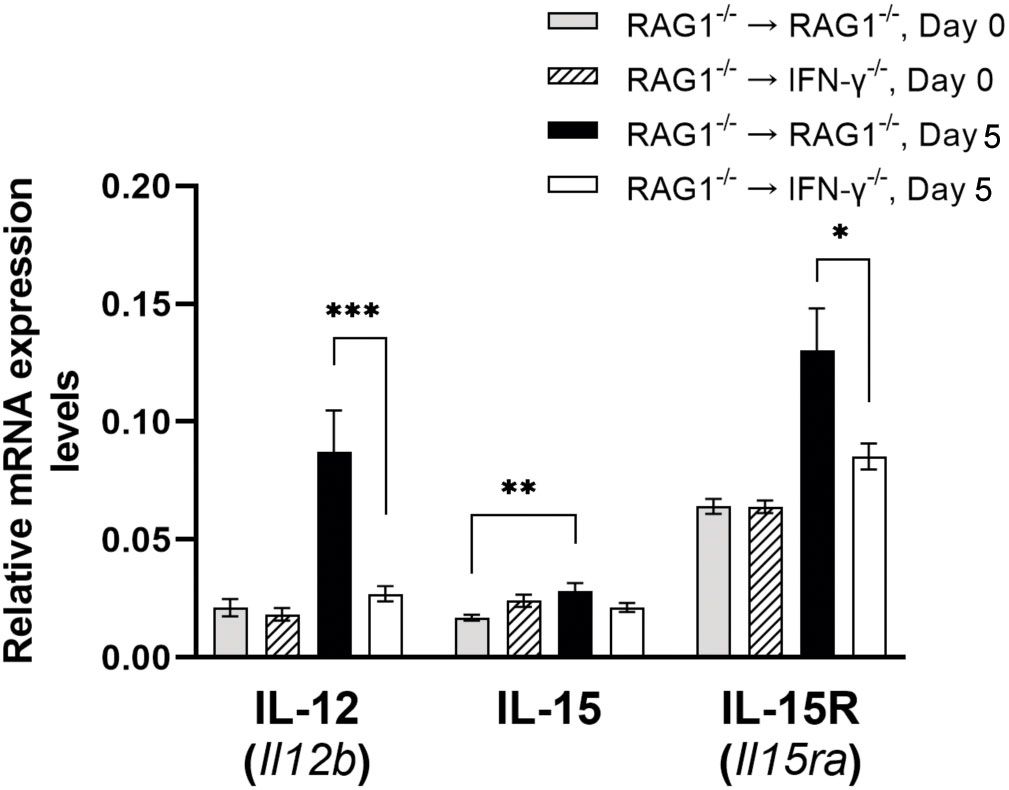
Figure 9 Cerebral mRNA levels for the cytokines that facilitate IFN-γ production by NK and T cells are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for IL-12, IL-15, and IL-15R were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
IL-15 is another cytokine that promotes the protective CD8+ T cell responses including both IFN-γ production and cytotoxic activity against T. gondii (Khan and Kasper, 1996; Bhadra et al., 2010). Whereas IL-15 mRNA levels tended to be slightly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection, the difference did not reach statistical significance (Figure 9). However, when compared between before and after initiation of reactivation of the infection, only RAG1-/-→RAG1-/- mice showed significant increases in IL-15 mRNA levels in response to reactivation of the infection (P<0.05) (Figure 9). In addition, mRNA levels for IL-15 receptor (IL-15RA) were significantly greater in the brains of the RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of T. gondii infection (P<0.05, Figure 9). Since IL-15 is also able to activate IFN-γ production of NK cells, the increased IL-15RA mRNA expression may reflect the activation of NK cells.
In addition to IL-12 and IL-15, mRNA expression levels for IL-18 were significantly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during reactivation of cerebral T. gondii infection as mentioned earlier in section 3.2, (P<0.05, Figure 3). IL-18 has the capability to upregulate IFN-γ production by CD4+ T cells (Okamura et al., 1995; Robinson et al., 1997). A recent study using T. gondii-induced intestinal inflammation demonstrated that IL-18-/- mice have decreased IFN-γ levels in the small intestine and serum, which are associated with decreased intestinal necrosis, following oral infection with T. gondii (Vossenkamper et al., 2004). We previously demonstrated that the intestinal necrosis induced by oral infection with this parasite is caused by IFN-γ production by CD4+ T cells (Liesenfeld et al., 1996). IL-18 also acts with IL-12 and induces IFN-γ production by CD8+ T cells (Okamoto et al., 1999). As described earlier in this section, IL-12 expression is markedly greater in brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.001) (Figure 9). Therefore, it is possible that increased expression of IL-18 induced by IFN-γ production by brain-resident cells in infected RAG1-/-→RAG1-/- mice contributes to facilitate IFN-γ production of both CD4+ and CD8+ T cells once they are recruited into the brain in response to reactivation of cerebral T. gondii infection.
3.4 The activations of the protective innate and T cell-mediated protective immune responses induced by IFN-γ production by brain-resident cells are associated with increased mRNA expression for the molecules that down-regulate the activated protective immunity
Since IFN-γ is a potent proinflammatory cytokine, overly stimulated IFN-γ-mediated immune responses can cause serious tissue damages during T. gondii infection (Gazzinelli et al., 1996; Liesenfeld et al., 1996). Therefore, it is critical to have a finely tuned regulation of this powerful immune responses. IL-10 has been shown to play a critical role in downregulating IFN-γ-mediated protective immune responses against T. gondii for maintaining the protective immune responses within the desirable levels (Gazzinelli et al., 1996; Liesenfeld et al., 1996). Importantly, the potent upregulations for mRNA expression for the molecules involved in the IFN-γ-mediated protective immunity in the brains of RAG1-/-→RAG1-/- mice were associated with relatively less (1.8-fold) but statistically significant upregulation of mRNA for IL-10R (Il10ra) (P<0.01) (Figure 10A). In addition, IL-10 mRNA levels significantly increased only in the brains of after the RAG1-/-→RAG1-/- mice after reactivation of cerebral T. gondii infection when compared to those before the reactivation of the infection (P<0.05) (Figure 10A). The combination of increased expression levels for IL-10 and IL-10R most likely contributes to down-regulating the IFN-γ-mediated immune responses for its finely tuned regulation. This possibility is supported by the evidence that mRNA levels for the IL-10 signaling molecule, STAT3, were 1.4 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.05) (Figure 10A).
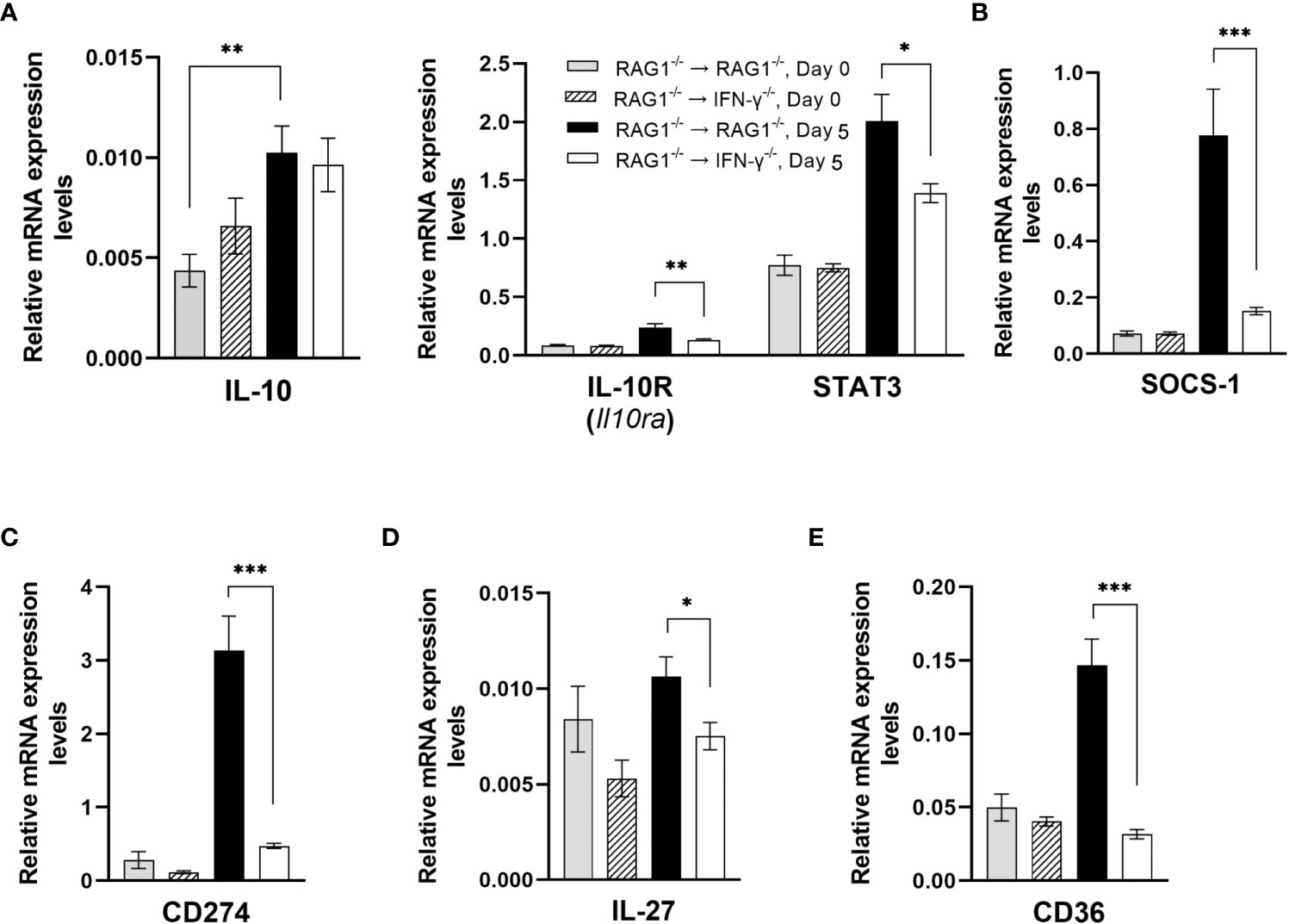
Figure 10 Cerebral mRNA levels for the molecules that down-regulate the IFN-γ-mediated protective immunity to prevent overly stimulated immune responses are upregulated by IFN-γ production by brain-resident cells during reactivation of cerebral T. gondii infection. For the details of experimental methods, see the legend of Figure 1. The mRNA levels for (A) IL-10, IL-10R, STAT3, (B) SOCS-1, (C) CD274, (D) IL-27, and (E) CD36 were measured at both Day 0 and Day 5 in the brains of both groups of mice were measured using NanoString nCounter Mouse Myeloid Innate Immunity Panel. *P<0.05, **P<0.01, and ***P<0.001. The data were combined from two independent experiments (n=5 for Day 0 and n=7 for Day 5 in both RAG1-/-→RAG1-/- and RAG1-/-→IFN-γ-/- mice).
In addition to IL-10, IL-10R and STAT3, mRNA for suppressor of cytokine signaling-1 (SOCS1) was more than 5 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice during the reactivation of cerebral T. gondii infection (P<0.001) (Figure 10B). SOCS1 can inhibit STAT1-mediaed signaling for IFN-γ by directly inhibiting JAK1/2 (Liau et al., 2018). Therefore, an activation of IFN-γ production by brain-resident cells in RAG1-/-→RAG1-/- mice in response to reactivation of the infection occurs in association with upregulations of multiple downregulatory mechanisms to secure an optimum protection against cerebral tachyzoite proliferation without causing unwanted tissue damages caused by overly stimulated IFN-γ-mediated inflammatory responses.
An interaction between CD274 (PD-L1) and CD279 (PD-1) is a key component of the immune checkpoint inhibitors that prevent unwanted overly stimulated or overly prolonged immune responses. Expression levels of mRNA for CD274 (PD-L1) were also more than 6 times greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice (P<0.001) (Figure 10C).
IL-27 is also an important downregulator of the Th1 CD4+ T cell responses. Expression levels of IL-27 mRNA were significantly greater in the brains of RAG1-/-→RAG1-/- than RAG1-/-→IFN-γ-/- mice in response to reactivation of T. gondii infection (P<0.05) (Figure 10D). It has been shown that IL-27 limits IL-2 production by the Th1 cells to prevent their overly stimulated IL-2 production that can cause a lethal inflammatory immunopathology associated with highly upregualted Th1 immune responses during acute T. gondii infection (Villarino et al., 2006). IL-27 also downregulates the development of Th17 cells during T. gondii infection, and IL-27R-/- mice developed severe inflammatory histopathology associated a prominent IL-17 response in the brain during the later stage of the infection and showed increased mortality (Stumhofer et al., 2006).
Expression levels of mRNA for CD36 (scavenger receptor) markedly increased in the brains of RAG1-/-→RAG1-/- mice when compared to RAG1-/-→IFN-γ-/- mice during reactivation of T. gondii infection (P<0.001) (Figure 10E). A recent study demonstrated that CD36-/- mice displayed increased weight losses and mortality associated with heightened IFN-γ response by NK cells as well as increased serum levels for biomarkers for tissue damages, GDF-15 and FGF-12 (Zhao et al., 2021). Therefore, the increased CD36 expression induced by IFN-γ production by brain-resident cells in the brains of RAG1-/-→RAG1-/- mice could be involved in the finely tuned regulation of the IFN-γ-mediated protective immunity in the brain during reactivation of cerebral T. gondii infection to prevent tissue damages.
The only difference in the brains between RAG1-/-→RAG1-/-and RAG1-/-→IFN-γ-/- mice is the presence (the former) and the absence (the latter) of the capability to produce IFN-γ in the brain-resident cells in the presence of the hematopoietic innate immune cells capable of producing this cytokine in the systemic circulation (Sa et al., 2015) as mentioned earlier. The present study revealed that the IFN-γ production by brain-resident cells in T. gondii-infected RAG1-/-→RAG1-/- mice significantly upregulates cerebral expressions of mRNA for a wide spectrum of molecules critical for both innate and T cell-mediated protective immunity to control tachyzoite growth during reactivation of chronic infection. These molecules include the molecules involved in IFN-γ signaling pathway, confirming that IFN-γ production by brain-resident cells is indeed able to activate the signaling pathway of this cytokine against cerebral tachyzoite proliferation. In the protective innate immunity, brain-resident cell IFN-γ production enhanced the expression of molecules for chemokines for recruitment of microglia and macrophages (CXCL9, CXCL10, and CXCL11) and an activation of those recruited phagocytes (IL-18, TLR family molecules [TLR3, TLR9, TLR11, TLR12, and CD180], NOD1, and CD40) for killing tachyzoites during reactivation of cerebral infection with T. gondii.
For activation of T cell-mediated protective immunity, the present study revealed that the IFN-γ production by brain-resident cells in T. gondii-infected RAG1-/-→RAG1-/- mice is able to upregulate cerebral expressions of mRNA for chemokines for recruiting CD4+ and CD8+ effector T cells into the areas of tachyzoite proliferation (CXCL9, CXCL10, and CXCL11) and the molecules for presenting target antigens through the MHC class I and II molecules to activate the recruited CD4+ and CD8+ T cells. The upregulated molecules for activating CD8+ T cells includes those for the processing of antigens (PA28αβ, LMP2, and LMP7), transporting the processed peptides (TAP1 and TAP2), assembling the transported peptides to the MHC class I molecules (Tapasin), and the MHC class I (H2-K1 and H2-D1) and MHC class Ib molecules (H2-Q1, H-2Q2, H2-M3, and H2-T23). For CD4+ T cell activation, the present study showed upregulations of mRNA for eight MHC class II molecules (H2-Aa, H2-Ab1, H2-DMa, H2-Ea-ps, H2-Eb1, H2-Ob, and CD74) through IFN-γ production by brain-resident cells. In addition, our study revealed that brain-resident cell-derived IFN-γ is able to upregulate cerebral expression of co-stimulatory molecules (ICOSL), and cytokines (IL-12, IL-15, and IL-18) facilitating IFN-γ production by NK cells and CD4+ and CD8+ T cells.
Since IFN-γ-mediated protective immunity is a powerful proinflammatory process, an important aspect from the host defense is to maintain this protective immunity within desirable levels through finely tuned regulatory mechanisms to avoid overly stimulated or overly prolonged IFN-γ-mediated pro-inflammatory responses that cause serious tissue damages. Notably, the present study revealed that IFN-γ production by brain-resident cells in T. gondii-infected RAG1-/-→RAG1-/- mice also upregulates cerebral expressions of mRNA for those down-regulatory molecules (IL-10, IL-10R, STAT3, SOCS1, CD274 [PD-L1], IL-27, and CD36) in response to reactivation of the infection to maintain the protective immune responses within a desirable range for an effective host defense. Thus, the present study uncovered the previously unrecognized but highly effective capability of IFN-γ production by brain-resident cells to orchestrate both the innate and T cell-mediated protective immunity with a fine-tuning regulation system to control cerebral infection with T. gondii. IFN-γ plays crucial roles in resistance against infections with various intracellular microorganisms in the brain (Chesler and Reiss, 2002; van den Broek et al., 1995). Therefore, the finely regulated activation of the innate and T cell-mediated protective immunity organized by IFN-γ production by brain-resident cells presented in the present study using T. gondii infection could function as a key first line defense system in the brain to effectively control cerebral infections with the other various microorganisms as well. Performing further functional analyses will be able to elucidate the mechanisms by which the IFN-γ production by brain-resident cells effectively orchestrate cerebral protective immune responses against the infection.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by IACUC committee at the University of Kentucky.
Author contributions
Designing research studies: YS. Funding acquisition: YS. Conducting experiments and acquiring the data: YS, JL, QS, and EO. Analyzing the data: KC, MA, and YS. Writing manuscript: YS, Reviewing the manuscript: YS, JL, KC, MA, QS, and EO. All authors contributed to the article and approved the submitted version.
Funding
The study was supported in part by the funding from the National Institutes of Health (AI152597, AI095032, and AI136821).
Acknowledgments
The authors appreciate the service and assistance in flow cytometry and NanoString assay provided by Markey Shared Resource Facility at University of Kentucky supported by P30CA177558.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andrade, W. A., Souza Mdo, C., Ramos-Martinez, E., Nagpal, K., Dutra, M. S., Melo, M. B., et al. (2013). Combined action of nucleic acid-sensing toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 13, 42–53. doi: 10.1016/j.chom.2012.12.003
Andrade, R. M., Wessendarp, M., Gubbels, M. J., Striepen, B., Subauste, C. S. (2006). CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 116, 2366–2377. doi: 10.1172/JCI28796
Bhadra, R., Guan, H., Khan, I. A. (2010). Absence of both IL-7 and IL-15 severely impairs the development of CD8 T cell response against Toxoplasma gondii. PloS One 5, e10842. doi: 10.1371/journal.pone.0010842
Bhushan, J., Radke, J. B., Perng, Y. C., McAllaster, M., Lenschow, D. J., Virgin, H. W., et al. (2020). ISG15 connects autophagy and IFN-gamma-Dependent control of toxoplasma gondii infection in human cells. mBio 11, e00852–20. doi: 10.1128/mBio.00852-20
Biswas, A., Bruder, D., Wolf, S. A., Jeron, A., Mack, M., Heimesaat, M. M., et al. (2015). Ly6C(high) monocytes control cerebral toxoplasmosis. J. Immunol. 194, 3223–3235. doi: 10.4049/jimmunol.1402037
Calingasan, N. Y., Erdely, H. A., Altar, C. A. (2002). Identification of CD40 ligand in alzheimer's disease and in animal models of alzheimer's disease and brain injury. Neurobiol. Aging 23, 31–39. doi: 10.1016/S0197-4580(01)00246-9
Carriba, P., Davies, A. M. (2021). CD40L/CD40 bidirectional signaling is a major regulator of neuronal morphology in the developing nervous system. Neural Regener. Res. 16, 1539–1541. doi: 10.4103/1673-5374.303025
Cavailles, P., Flori, P., Papapietro, O., Bisanz, C., Lagrange, D., Pilloux, L., et al. (2014). A highly conserved Toxo1 haplotype directs resistance to toxoplasmosis and its associated caspase-1 dependent killing of parasite and host macrophage. PloS Pathog. 10, e1004005. doi: 10.1371/journal.ppat.1004005
Chao, C. C., Anderson, W. R., Hu, S., Gekker, G., Martella, A., Peterson, P. K. (1993). Activated microglia inhibit multiplication of Toxoplasma gondii via a nitric oxide mechanism. Clin. Immunol. Immunopathol. 67, 178–183. doi: 10.1006/clin.1993.1062
Charo, I. F., Ransohoff, R. M. (2006). The many roles of chemokines and chemokine receptors in inflammation. N Engl. J. Med. 354, 610–621. doi: 10.1056/NEJMra052723
Chesler, D. A., Reiss, C. S. (2002). The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 13, 441–454. doi: 10.1016/s1359-6101(02)00044-8
D'Orazio, S. E., Halme, D. G., Ploegh, H. L., Starnbach, M. N. (2003). Class ia MHC-deficient BALB/c mice generate CD8+ T cell-mediated protective immunity against Listeria monocytogenes infection. J. Immunol. 171, 291–298. doi: 10.4049/jimmunol.171.1.291
de Haas, A. H., Boddeke, H. W., Biber, K. (2008). Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 56, 888–894. doi: 10.1002/glia.20663
Dubey, J. P. (2007). “The history and life cycle of toxoplasma gondii,” in Toxoplasma gondii: The model apicomplexan parasite: Perspectives and methods. Ed. Weiss LMaK, K. (London, United Kingdom: Elsevier), 1–18.
Elser, B., Lohoff, M., Kock, S., Giaisi, M., Kirchhoff, S., Krammer, P. H., et al. (2002). IFN-gamma represses IL-4 expression via IRF-1 and IRF-2. Immunity 17, 703–712. doi: 10.1016/s1074-7613(02)00471-5
Fan, W., Chang, S., Shan, X., Gao, D., Zhang, S. Q., Zhang, J., et al. (2016). Transcriptional profile of SH-SY5Y human neuroblastoma cells transfected by toxoplasma rhoptry protein 16. Mol. Med. Rep. 14, 4099–4108. doi: 10.3892/mmr.2016.5758
Farlik, M., Rapp, B., Marie, I., Levy, D. E., Jamieson, A. M., Decker, T. (2012). Contribution of a TANK-binding kinase 1-interferon (IFN) regulatory factor 7 pathway to IFN-gamma-induced gene expression. Mol. Cell Biol. 32, 1032–1043. doi: 10.1128/MCB.06021-11
Fischer, H. G., Dorfler, R., Schade, B., Hadding, U. (1999). Differential CD86/B7-2 expression and cytokine secretion induced by Toxoplasma gondii in macrophages from resistant or susceptible BALB h-2 congenic mice. Int. Immunol. 11, 341–349. doi: 10.1093/intimm/11.3.341
Gazzinelli, R. T., Hakim, F. T., Hieny, S., Shearer, G. M., Sher, A. (1991). Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-gamma production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J. Immunol. 146, 286–292. doi: 10.4049/jimmunol.146.1.286
Gazzinelli, R. T., Wysocka, M., Hieny, S., Scharton-Kersten, T., Cheever, A., Kuhn, R., et al. (1996). In the absence of endogenous IL-10, mice acutely infected with toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157, 798–805. doi: 10.4049/jimmunol.157.2.798
Gomes, A. Q., Correia, D. V., Silva-Santos, B. (2007). Non-classical major histocompatibility complex proteins as determinants of tumour immunosurveillance. EMBO Rep. 8, 1024–1030. doi: 10.1038/sj.embor.7401090
Gorfu, G., Cirelli, K. M., Melo, M. B., Mayer-Barber, K., Crown, D., Koller, B. H., et al. (2014). Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 5, e01117–13. doi: 10.1128/mBio.01117-13
Guo, G., Peng, Y., Xiong, B., Liu, D., Bu, H., Tian, X., et al. (2017). Involvement of chemokine CXCL11 in the development of morphine tolerance in rats with cancer-induced bone pain. J. Neurochem. 141, 553–564. doi: 10.1111/jnc.13919
Guyon, A. (2014). CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front. Cell Neurosci. 8, 65. doi: 10.3389/fncel.2014.00065
Harris, T. H., Banigan, E. J., Christian, D. A., Konradt, C., Tait Wojno, E. D., Norose, K., et al. (2012). Generalized levy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486, 545–548. doi: 10.1038/nature11098
Henan, X., Toyota, N., Yanjiang, X., Fujita, Y., Zhijun, H., Touma, M., et al. (2014). Enhancement of phagocytosis and cytotoxicity in macrophages by tumor-derived IL-18 stimulation. BMB Rep. 47, 286–291. doi: 10.5483/BMBRep.2014.47.5.152
Hickman, S. E., Kingery, N. D., Ohsumi, T. K., Borowsky, M. L., Wang, L. C., Means, T. K., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Hunter, C. A., Subauste, C. S., Van Cleave, V. H., Remington, J. S. (1994). Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect. Immun. 62, 2818–2824. doi: 10.1128/iai.62.7.2818-2824.1994
Kang, H., Suzuki, Y. (2001). Requirement of non-T cells that produce gamma interferon for prevention of reactivation of Toxoplasma gondii infection in the brain. Infect. Immun. 69, 2920–2927. doi: 10.1128/IAI.69.5.2920-2927.2001
Ke, Z. J., Calingasan, N. Y., DeGiorgio, L. A., Volpe, B. T., Gibson, G. E. (2005). CD40-CD40L interactions promote neuronal death in a model of neurodegeneration due to mild impairment of oxidative metabolism. Neurochem. Int. 47, 204–215. doi: 10.1016/j.neuint.2005.03.002
Khan, I. A., Kasper, L. H. (1996). IL-15 augments CD8+ T cell-mediated immunity against Toxoplasma gondii infection in mice. J. Immunol. 157, 2103–2108. doi: 10.4049/jimmunol.157.5.2103
Kincaid, E. Z., Che, J. W., York, I., Escobar, H., Reyes-Vargas, E., Delgado, J. C., et al. (2011). Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 13, 129–135. doi: 10.1038/ni.2203
Koblansky, A. A., Jankovic, D., Oh, H., Hieny, S., Sungnak, W., Mathur, R., et al. (2013). Recognition of profilin by toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38, 119–130. doi: 10.1016/j.immuni.2012.09.016
Krauthausen, M., Saxe, S., Zimmermann, J., Emrich, M., Heneka, M. T., Muller, M. (2014). CXCR3 modulates glial accumulation and activation in cuprizone-induced demyelination of the central nervous system. J. Neuroinflamm. 11, 109. doi: 10.1186/1742-2094-11-109
Kuwata, T., Gongora, C., Kanno, Y., Sakaguchi, K., Tamura, T., Kanno, T., et al. (2002). Gamma interferon triggers interaction between ICSBP (IRF-8) and TEL, recruiting the histone deacetylase HDAC3 to the interferon-responsive element. Mol. Cell Biol. 22, 7439–7448. doi: 10.1128/MCB.22.21.7439-7448.2002
Lehner, P. J., Surman, M. J., Cresswell, P. (1998). Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line .220. Immunity 8, 221–231. doi: 10.1016/S1074-7613(00)80474-4
Liau, N. P. D., Laktyushin, A., Lucet, I. S., Murphy, J. M., Yao, S., Whitlock, E., et al. (2018). The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 9, 1558. doi: 10.1038/s41467-018-04013-1
Liesenfeld, O., Kosek, J., Remington, J. S., Suzuki, Y. (1996). Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J. Exp. Med. 184, 597–607. doi: 10.1084/jem.184.2.597
Lisboa, L. F., Egli, A., Fairbanks, J., O'Shea, D., Manuel, O., Husain, S., et al. (2015). CCL8 and the immune control of cytomegalovirus in organ transplant recipients. Am. J. Transplant. 15, 1882–1892. doi: 10.1111/ajt.13207
Lutshumba, J., Ochiai, E., Sa, Q., Anand, N., Suzuki, Y. (2020). Selective upregulation of transcripts for six molecules related to T cell costimulation and phagocyte recruitment and activation among 734 immunity-related genes in the brain during perforin-dependent, CD8(+) T cell-mediated elimination of Toxoplasma gondii cysts. mSystems 5, e00189–20. doi: 10.1128/mSystems.00189-20
Man, S., Tucky, B., Cotleur, A., Drazba, J., Takeshita, Y., Ransohoff, R. M. (2012). CXCL12-induced monocyte-endothelial interactions promote lymphocyte transmigration across an in vitro blood-brain barrier. Sci. Transl. Med. 4, 119ra114. doi: 10.1126/scitranslmed.3003197
Marino, A. P., Dos Santos, L. I., Henriques, P. M., Roffe, E., Vasconcelos-Santos, D. V., Sher, A., et al. (2020). Circulating inflammatory mediators as biomarkers of ocular toxoplasmosis in acute and in chronic infection. J. Leukoc. Biol. 108, 1253–1264. doi: 10.1002/JLB.4MA0420-702R
Mazzarino, P., Pietra, G., Vacca, P., Falco, M., Colau, D., Coulie, P., et al. (2005). Identification of effector-memory CMV-specific T lymphocytes that kill CMV-infected target cells in an HLA-e-restricted fashion. Eur. J. Immunol. 35, 3240–3247. doi: 10.1002/eji.200535343
McWilliams, T. G., Howard, L., Wyatt, S., Davies, A. M. (2015). Regulation of autocrine signaling in subsets of sympathetic neurons has regional effects on tissue innervation. Cell Rep. 10, 1443–1449. doi: 10.1016/j.celrep.2015.02.016
Minns, L. A., Menard, L. C., Foureau, D. M., Darche, S., Ronet, C., Mielcarz, D. W., et al. (2006). TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J. Immunol. 176, 7589–7597. doi: 10.4049/jimmunol.176.12.7589
Montoya, J. G., Liesenfeld, O. (2004). Toxoplasmosis. Lancet 363, 1965–1976. doi: 10.1016/S0140-6736(04)16412-X
Mori, I., Hossain, M. J., Takeda, K., Okamura, H., Imai, Y., Kohsaka, S., et al. (2001). Impaired microglial activation in the brain of IL-18-gene-disrupted mice after neurovirulent influenza a virus infection. Virology 287, 163–170. doi: 10.1006/viro.2001.1029
Mosa, A., Trumstedt, C., Eriksson, E., Soehnlein, O., Heuts, F., Janik, K., et al. (2009). Nonhematopoietic cells control the outcome of infection with Listeria monocytogenes in a nucleotide oligomerization domain 1-dependent manner. Infect. Immun. 77, 2908–2918. doi: 10.1128/IAI.01068-08
Napolitano, A., van der Veen, A. G., Bunyan, M., Borg, A., Frith, D., Howell, S., et al. (2018). Cysteine-reactive free ISG15 generates IL-1beta-Producing CD8alpha(+) dendritic cells at the site of infection. J. Immunol. 201, 604–614. doi: 10.4049/jimmunol.1701322
Ochiai, E., Sa, Q., Brogli, M., Kudo, T., Wang, X., Dubey, J. P., et al. (2015). CXCL9 is important for recruiting immune T cells into the brain and inducing an accumulation of the T cells to the areas of tachyzoite proliferation to prevent reactivation of chronic cerebral infection with Toxoplasma gondii. Am. J. Pathol. 185, 314–324. doi: 10.1016/j.ajpath.2014.10.003
Okamoto, I., Kohno, K., Tanimoto, T., Ikegami, H., Kurimoto, M. (1999). Development of CD8+ effector T cells is differentially regulated by IL-18 and IL-12. J. Immunol. 162, 3202–3211. doi: 10.4049/jimmunol.162.6.3202
Okamura, H., Tsutsi, H., Komatsu, T., Yutsudo, M., Hakura, A., Tanimoto, T., et al. (1995). Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 378, 88–91. doi: 10.1038/378088a0
Portillo, J. A., Okenka, G., Reed, E., Subauste, A., Van Grol, J., Gentil, K., et al. (2010). The CD40-autophagy pathway is needed for host protection despite IFN-gamma-dependent immunity and CD40 induces autophagy via control of P21 levels. PloS One 5, e14472. doi: 10.1371/journal.pone.0014472
Raetz, M., Kibardin, A., Sturge, C. R., Pifer, R., Li, H., Burstein, E., et al. (2013). Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to toxoplasma gondii profilin. J. Immunol. 191, 4818–4827. doi: 10.4049/jimmunol.1301301
Rappert, A., Bechmann, I., Pivneva, T., Mahlo, J., Biber, K., Nolte, C., et al. (2004). CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J. Neurosci. 24, 8500–8509. doi: 10.1523/JNEUROSCI.2451-04.2004
Robinson, D., Shibuya, K., Mui, A., Zonin, F., Murphy, E., Sana, T., et al. (1997). IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity 7, 571–581. doi: 10.1016/S1074-7613(00)80378-7
Sadasivan, B., Lehner, P. J., Ortmann, B., Spies, T., Cresswell, P. (1996). Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity 5, 103–114. doi: 10.1016/S1074-7613(00)80487-2
Saddala, M. S., Yang, X., Tang, S., Huang, H. (2021). Transcriptome-wide analysis reveals core sets of transcriptional regulators of sensome and inflammation genes in retinal microglia. Genomics 113, 3058–3071. doi: 10.1016/j.ygeno.2021.07.001
Sa, Q., Ochiai, E., Tiwari, A., Perkins, S., Mullins, J., Gehman, M., et al. (2015). Cutting edge: IFN-gamma produced by brain-resident cells is crucial to control cerebral infection with Toxoplasma gondii. J. Immunol. 195, 796–800. doi: 10.4049/jimmunol.1500814
Sijts, A. J., Ruppert, T., Rehermann, B., Schmidt, M., Koszinowski, U., Kloetzel, P. M. (2000). Efficient generation of a hepatitis b virus cytotoxic T lymphocyte epitope requires the structural features of immunoproteasomes. J. Exp. Med. 191, 503–514. doi: 10.1084/jem.191.3.503
Strack, A., Asensio, V. C., Campbell, I. L., Schluter, D., Deckert, M. (2002). Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol. (Berl.) 103, 458–468. doi: 10.1007/s00401-001-0491-7
Stumhofer, J. S., Laurence, A., Wilson, E. H., Huang, E., Tato, C. M., Johnson, L. M., et al. (2006). Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat. Immunol. 7, 937–945. doi: 10.1038/ni1376
Subauste, C. S., de Waal Malefyt, R., Fuh, F. (1998). Role of CD80 (B7.1) and CD86 (B7.2) in the immune response to an intracellular pathogen. J. Immunol. 160, 1831–1840. doi: 10.4049/jimmunol.160.4.1831
Suzuki, Y., Conley, F. K., Remington, J. S. (1989). Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J. Immunol. 143, 2045–2050. doi: 10.4049/jimmunol.143.6.2045
Suzuki, Y., Joh, K., Kwon, O. C., Yang, Q., Conley, F. K., Remington, J. S. (1994). MHC class I gene(s) in the D/L region but not the TNF-alpha gene determines development of toxoplasmic encephalitis in mice. J. Immunol. 153, 4649–4654. doi: 10.4049/jimmunol.153.10.4649
Swanson, P. A., 2nd, Pack, C. D., Hadley, A., Wang, C. R., Stroynowski, I., Jensen, P. E., et al. (2008). An MHC class ib-restricted CD8 T cell response confers antiviral immunity. J. Exp. Med. 205, 1647–1657. doi: 10.1084/jem.20080570
Tiwari, A., Hannah, R., Lutshumba, J., Ochiai, E., Weiss, L. M., Suzuki, Y. (2019). Penetration of CD8(+) cytotoxic T cells into Large target, tissue cysts of Toxoplasma gondii, leads to its elimination. Am. J. Pathol 189, 1594–1607. doi: 10.1016/j.ajpath.2019.04.018
van den Broek, M. F., Muller, U., Huang, S., Zinkernagel, R. M., Aguet, M. (1995). Immune defence in mice lacking type I and/or type II interferon receptors. Immunol. Rev. 148, 5–18. doi: 10.1111/j.1600-065X.1995.tb00090.x
Villarino, A. V., Stumhofer, J. S., Saris, C. J., Kastelein, R. A., de Sauvage, F. J., Hunter, C. A. (2006). IL-27 limits IL-2 production during Th1 differentiation. J. Immunol. 176, 237–247. doi: 10.4049/jimmunol.176.1.237
Vossenkamper, A., Struck, D., Alvarado-Esquivel, C., Went, T., Takeda, K., Akira, S., et al. (2004). Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur. J. Immunol. 34, 3197–3207. doi: 10.1002/eji.200424993
Wang, X., Claflin, J., Kang, H., Suzuki, Y. (2005). Importance of CD8(+)Vbeta8(+) T cells in IFN-gamma-mediated prevention of toxoplasmic encephalitis in genetically resistant BALB/c mice. J. Interferon Cytokine Res. 25, 338–344. doi: 10.1089/jir.2005.25.338
Wang, X., Suzuki, Y. (2007). Microglia produce IFN-gamma independently from T cells during acute toxoplasmosis in the brain. J. Interferon Cytokine Res. 27, 599–605. doi: 10.1089/jir.2006.0157
Wen, X., Kudo, T., Payne, L., Wang, X., Rodgers, L., Suzuki, Y. (2010). Predominant interferon-gamma-mediated expression of CXCL9, CXCL10, and CCL5 proteins in the brain during chronic infection with Toxoplasma gondii in BALB/c mice resistant to development of toxoplasmic encephalitis. J. Interferon Cytokine Res. 30, 653–660. doi: 10.1089/jir.2009.0119
Whitby, F. G., Masters, E. I., Kramer, L., Knowlton, J. R., Yao, Y., Wang, C. C., et al. (2000). Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 408, 115–120. doi: 10.1038/35040607
Wilson, E. H., Zaph, C., Mohrs, M., Welcher, A., Siu, J., Artis, D., et al. (2006). B7RP-1-ICOS interactions are required for optimal infection-induced expansion of CD4+ Th1 and Th2 responses. J. Immunol. 177, 2365–2372. doi: 10.4049/jimmunol.177.4.2365
Yarovinsky, F., Zhang, D., Andersen, J. F., Bannenberg, G. L., Serhan, C. N., Hayden, M. S., et al. (2005). TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629. doi: 10.1126/science.1109893
Yu, S. P., Ong, K. C., Perera, D., Wong, K. T. (2019). Neuronal transcriptomic responses to Japanese encephalitis virus infection with a special focus on chemokine CXCL11 and pattern recognition receptors RIG-1 and MDA5. Virology 527, 107–115. doi: 10.1016/j.virol.2018.10.015
Keywords: IFN-γ, protective immunity, Toxoplasma gondii, brain-resident cells, chemokine, cytokine, antigen presentation, regulatory molecule
Citation: Suzuki Y, Lutshumba J, Chen KC, Abdelaziz MH, Sa Q and Ochiai E (2023) IFN-γ production by brain-resident cells activates cerebral mRNA expression of a wide spectrum of molecules critical for both innate and T cell-mediated protective immunity to control reactivation of chronic infection with Toxoplasma gondii. Front. Cell. Infect. Microbiol. 13:1110508. doi: 10.3389/fcimb.2023.1110508
Received: 28 November 2022; Accepted: 18 January 2023;
Published: 15 February 2023.
Edited by:
Emma Harriet Wilson, University of California, Riverside, United StatesReviewed by:
Renato Augusto DaMatta, State University of the North Fluminense Darcy Ribeiro, BrazilMaria M. Costa, Instituto de Investigaciones Marinas, Spain
Copyright © 2023 Suzuki, Lutshumba, Chen, Abdelaziz, Sa and Ochiai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasuhiro Suzuki, eWFzdS5zdXp1a2lAdWt5LmVkdQ==