 Jennifer A. Black
Jennifer A. Black João Luís Reis-Cunha3
João Luís Reis-Cunha3 Angela. K. Cruz
Angela. K. Cruz Luiz. R.O. Tosi
Luiz. R.O. Tosi- 1Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil
- 2The Wellcome Centre for Integrative Parasitology, School of Infection, Immunity, and Inflammation, University of Glasgow, Glasgow, United Kingdom
- 3Biomedical research institute, University of York, York, United Kingdom
Leishmania are kinetoplastid pathogens that cause leishmaniasis, a debilitating and potentially life-threatening infection if untreated. Unusually, Leishmania regulate their gene expression largely post-transcriptionally due to the arrangement of their coding genes into polycistronic transcription units that may contain 100s of functionally unrelated genes. Yet, Leishmania are capable of rapid and responsive changes in gene expression to challenging environments, often instead correlating with dynamic changes in their genome composition, ranging from chromosome and gene copy number variations to the generation of extrachromosomal DNA and the accumulation of point mutations. Typically, such events indicate genome instability in other eukaryotes, coinciding with genetic abnormalities, but for Leishmania, exploiting these products of genome instability can provide selectable substrates to catalyse necessary gene expression changes by modifying gene copy number. Unorthodox DNA replication, DNA repair, replication stress factors and DNA repeats are recognised in Leishmania as contributors to this intrinsic instability, but how Leishmania regulate genome plasticity to enhance fitness whilst limiting toxic under- or over-expression of co-amplified and co-transcribed genes is unclear. Herein, we focus on fresh, and detailed insights that improve our understanding of genome plasticity in Leishmania. Furthermore, we discuss emerging models and factors that potentially circumvent regulatory issues arising from polycistronic transcription. Lastly, we highlight key gaps in our understanding of Leishmania genome plasticity and discuss future studies to define, in higher resolution, these complex regulatory interactions.
1 Introduction
Pathogenic organisms can rapidly adapt to challenging environments by altering their genome composition. Mutagenesis, genetic exchanges, abnormal chromosome number (aneuploidy and chromosome instability; CIN), DNA insertions and deletions (indels), single nucleotide polymorphisms (SNPs), gene copy number variations (CNVs), and other DNA rearrangements can give rise to genome heterogeneity and selectable fitness enhancing traits (Merlo et al., 2006; Żmieńko et al., 2014; Lee et al., 2016; Bolhaqueiro et al., 2019; Todd et al., 2019; López et al., 2020; Watkins et al., 2020). For a host, a genetically flexible pathogen has important clinical consequences, including the selection and emergence of drug resistance, ultimately limiting treatment options (Yang et al., 2019; Sah et al., 2021; Kukurudz et al., 2022). Nevertheless, a flexible genome requires limits to prevent the accumulation of deleterious mutations and catastrophic genome collapse. By improving our understanding of how genome plasticity is harnessed in pathogens, we may uncover key targetable dependencies in these processes, ultimately improving the clinical management of numerous medically important infections.
Over the last decade, single cell sequencing (SCS) technologies (Imamura et al., 2020; Louradour et al., 2020; Bussotti et al., 2021; Negreira et al., 2022), novel screening strategies (Baker et al., 2021), improved genetic engineering using CRISPR/Cas9 (Zhang and Matlashewski, 2015; Espada et al., 2021; Beneke et al., 2022) and inducible gene deletion (Duncan et al., 2016; Damasceno et al., 2020b; Yagoubat et al., 2020) have seen the Kinetoplastid parasite Leishmania, a single-celled eukaryote, emerge as a strong model of adaptive genome plasticity due to its surprising tolerance for extensive genomic alterations (Rogers et al., 2011; Sterkers et al., 2011; Lachaud et al., 2014; Ubeda et al., 2014). Over 20 species of Leishmania cause the vector-borne, neglected tropical disease (NTD) leishmaniasis in humans and animals. Leishmaniasis primarily affects poverty-stricken regions in the tropics and sub-tropics of the world (Torres-Guerrero et al., 2017; Burza et al., 2018), with the symptoms and disease outcomes partially determined by the infecting species. Broadly, the disease manifests as one of two main forms: tegumentary and visceral leishmaniasis. Tegumentary leishmaniasis includes Cutaneous Leishmaniasis (CL), Mucocutaneous Leishmaniasis (MCL) and Diffuse Cutaneous Leishmaniasis (DCL) which typically range from self-healing but potentially disfiguring skin lesions (i.e. CL) or disseminated skin nodules (i.e. DCL), to severe damage to the nose and mouth mucosa (i.e. MCL). Visceral Leishmaniasis (VL) is a systemic disease and often lethal if untreated (Burza et al., 2018). Currently, these infections are managed clinically by chemotherapy, however drug toxicity and emerging resistance to front line treatments highlight a need for novel treatment options (Ponte-Sucre et al., 2017; Capela et al., 2019).
Hallmarks of genome instability (i.e. aneuploidy, CNVs and SNPs) are widespread in Leishmania, reported in natural isolates and laboratory populations (Reis-Cunha et al., 2018; Dumetz et al., 2018; Patino et al., 2019; Cupolillo et al., 2020). Like fungi and cancer cells (Sheltzer et al., 2011; Pfau and Amon, 2012; Lukow et al., 2021; Sah et al., 2021), some of these genomic rearrangements coincide with drug resistance and environmental adaptations (Dumetz et al., 2017; Dumetz et al., 2018; Patino et al., 2019), yet how Leishmania balance potentially beneficial instability whilst retaining genome fidelity is unknown. Furthermore, whether (or how) this plasticity directly contributes to the spectrum and severity of disease is unclear. Collectively, in vitro evidence points to DNA repair (Laffitte et al., 2014; Laffitte et al., 2016a), DNA repeats (Ubeda et al., 2008; Ubeda et al., 2014), unusual DNA replication dynamics and enhanced DNA replication stress as plasticity drivers (Damasceno et al., 2018; Damasceno et al., 2020a; Damasceno et al., 2020b) implying this phenomenon is multifactorial and intimately linked with specific features of the Leishmania genome and wider biological processes.
2 Repeated DNA sequences can catalyse Leishmania genome plasticity
In eukaryotes, repeated sequences of DNA can drive gene expression changes and genome diversification (reviewed by Biscotti et al., 2015; Kratochwil and Meyer, 2019; Brown and Freudenreich, 2021). In Leishmania, ~10% of the genome is populated with repetitive DNA, which is considerably less than predicated for the related pathogens Trypanosoma brucei (~20%) and Trypanosoma cruzi (~50%) (Pita et al., 2019). However, recent analyses implicate a wide variety of Leishmania DNA repeats catalyse their extreme genome plasticity (Ubeda et al., 2014; Bussotti et al., 2021).
Approximately 2000 low complexity Direct Repeats (DRs) and Inverted Repeats (IRs), named in relation to their genomic orientations, are present in the Leishmania genome. From these DNA repeats, ~3000-4000 unique and selectable extrachromosomal circular or linear amplicons are estimated to arise (Ubeda et al., 2014), originating from the genome and carrying potential fitness enhancing traits. Amplification is proposed to occur stochastically with subsequent changes to the abundance of beneficial amplicons leading to alterations in RNA levels under stressful environments, for instance following drug exposure (Ubeda et al., 2008; Leprohon et al., 2009; Ubeda et al., 2014; Laffitte et al., 2014; Bussotti et al., 2021). Broadly, the locations of DRs and IRs are syntenic across different Leishmania species (Dias et al., 2007; Ubeda et al., 2008; Ubeda et al., 2014) most (~68%) belonging to a family of extinct transposable elements (TEs), known as Short Interspersed DEgenerate Retroposons (SIDERs), that became expanded in Leishmania. Two subfamilies of SIDER elements have been described in these parasites: SIDER1 and SIDER2. Experimentally, SIDER elements can destabilise messengerRNA (mRNA) and may perform broader functions relating to the regulation of three prime untranslated regions (3’UTRs) (Bringaud et al., 2007; Smith et al., 2009; Müller et al., 2010; Requena et al., 2017), although further study is required to understand these roles. Nonetheless, no evidence suggests DRs or IRs perform functions outside of their described roles in extrachromosomal genome amplification.
Current data supports two distinct pathways orchestrate Leishmania extrachromosomal amplification: one for linear amplification and one for circular amplification (summarised in Figure 1) however to date, neither pathway has been completely described. Extrachromosomal DNA circles, and tandem duplications in Leishmania exploit the activity of the recombinase RAD51 that facilitates a recombination reaction between DRs, subsequently leading to the formation of a circular amplicon or a duplication event (Figure 1). RAD51 is a key orchestrator of the homologous recombination (HR) pathway (Wright et al., 2018; Elbakry and Löbrich, 2021), required for double strand break (DSB) repair, thus the involvement of RAD51 is suggestive of unstable DNA or DNA injuries as catalysts. Additionally, RAD51 paralogues are also known regulators of RAD51 activity (Sullivan and Bernstein, 2018) and in Leishmania, RAD51-4, one of three Leishmania RAD51 paralogues, acts during circular amplification (Genois et al., 2015). Whether this role relates to the regulation of RAD51 activity remains untested. Direct interactions between Leishmania RAD51 and the mediator protein BRCA2 have also been experimentally confirmed but outside of the context of circular amplification (Genois et al., 2012). Thus far, we still lack key insights into three important events: 1) what triggers circular amplification, 2) what factors initiate amplification and, 3) what processes regulate amplicon abundance and consequently, their expression. Recent studies now shed light on some of these events (discussed below); nonetheless, wider identification and examination of circular amplification pathway members are still required.
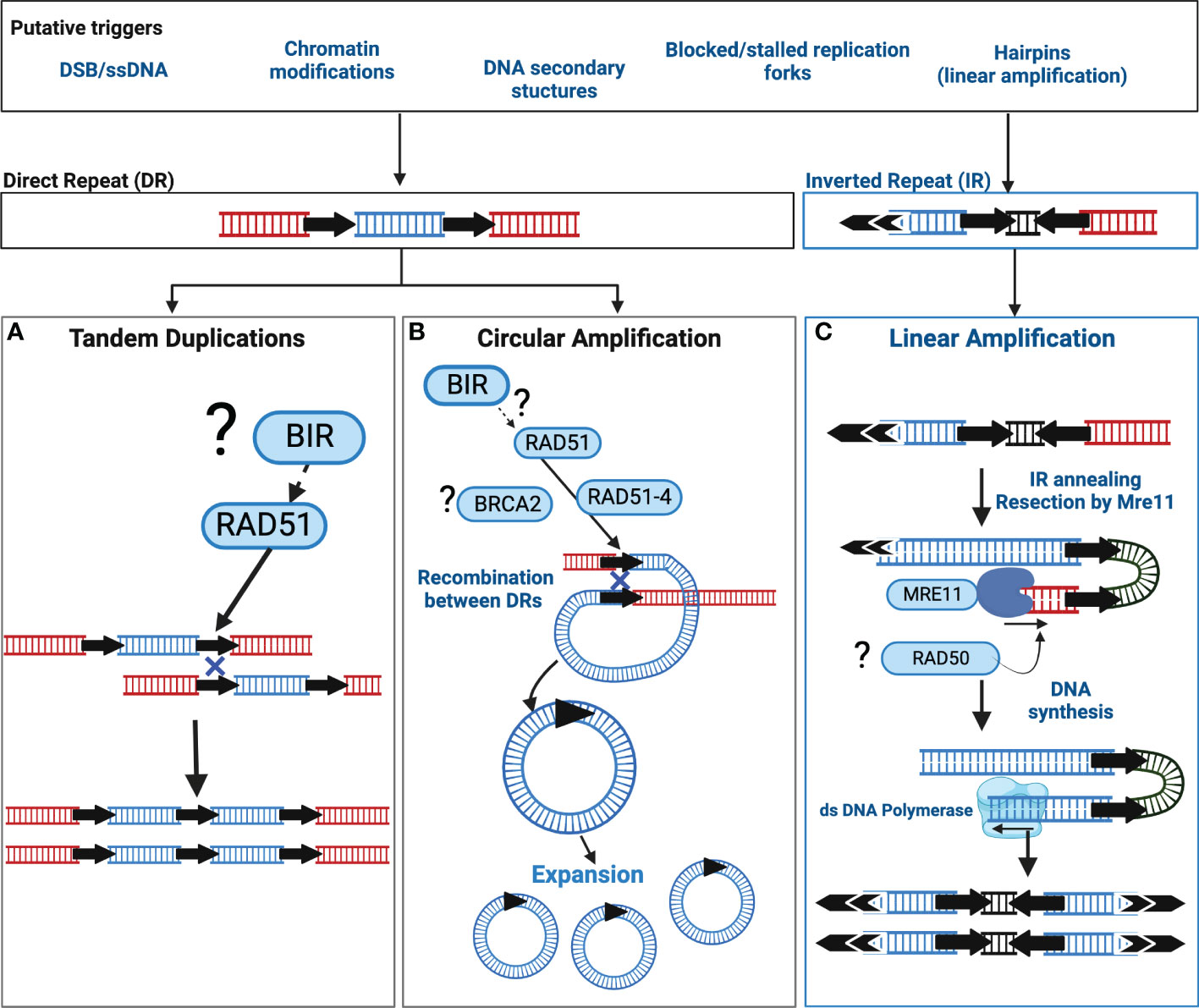
Figure 1 Putative models of extrachromosomal amplification in Leishmania driven by DNA Repeats. In Leishmania, extrachromosomal DNA amplification can be catalysed by either Direct Repeats (DRs) or Inverted Repeats (IR). Though the precise trigger(s) are unknown, putative sources of DNA instability are listed in the corresponding box above that may contribute to DNA amplification in Leishmania. Recombination reactions associated with DRs can result in tandem duplications and circular amplicons. The recombinase RAD51 facilitates a homology driven recombination between DRs that may result in (A) a tandem duplication, or (B) extrachromosomal circular amplicons. The mechanism driving tandem duplication events is unclear and may be the result of Break Induced Replication (BIR) or form an uneven exchange of genetic information between sister chromatids. Black arrows = DRs, (?) = the involvement of this factor or pathway requires experimental confirmation, Blue Cross indicates homologous recombination. (C) Linear amplification is driven through an annealing reaction between IRs. Here, the exonuclease activity of Mre11 may process a DNA lesion, for example a single strand break, or a hairpin structure formed due to DNA replication, after which the IRs anneal, and the DNA is replicated. Double arrows = telomeric sequences. Black arrows = IRs. Diagram adapted from (Laffitte et al., 2016b).
The events initiating linear amplification are also largely elusive, though DSBs, single strand breaks (SSBs) and DNA hairpin structures are proposed triggers (see Figure 1 for more details). In contrast, linear amplification does not rely on RAD51 or RAD51-4. Instead, the DNA repair enzyme MRE11 (Meiotic REcombination 11), a component of the Mre11-Rad50-Nbs1 (MRN) complex, plays a key role in facilitating annealing reactions between IRs (Figure 1); disrupting Mre11 activity impairs linear but not circular amplification (Laffitte et al., 2014).
Common to both is the co-option of DNA repair enzymes (and potentially wider pathways) supporting intrinsic DNA instability as a putative trigger. In agreement, the activities of RAD51 or Mre11 are not solely restricted to extrachromosomal amplification: Mre11 inactivation alone or in combination with RAD50 disruption is associated with chromosome translocations and broader instability (Laffitte et al., 2016a) whereas the loss of RAD51, via rapamycin induced LoxP excision, disrupts core chromosome duplication (Damasceno et al., 2020b). Indeed, the study by Damasceno and colleagues highlights DNA replication as potential contributor to this instability, with replication stress, a phenomenon that describes abnormal replication machinery progression, experimentally enhancing Leishmania genome diversity (Louradour et al., 2020) and driving subtelomeric duplication (Damasceno et al., 2020a). Whether the DNA repeats themselves are the source of instability (i.e prone to DNA breaks or secondary structures) requires testing. One other feature of these DNA repeats yet to be investigated is the relevance of their genomic positioning; DRs are dispersed more evenly across the chromosome, whereas IRs are concentrated at chromosome ends (subtelomere and telomere proximal regions) (Ubeda et al., 2014). Whether these sites impact upon the type of DNA amplicon is not known.
If, and how, linear amplicons are transmitted is undetermined, however circular DNA amplicons experimentally transmit via two distinct routes: 1) trans-generationally during cell division, 2) as part of the contents of extracellular vesicles (EV’s). During cell division, Leishmania daughter cells can inherit circular amplicons, but the processes that govern circular amplicon inheritance are undefined. In cancer cells, extrachromosomal circular DNA transmission appears to be random during cellular division (Lange et al., 2022), therefore it is possible the inheritance of circular amplicons in Leishmania is also random (Lange et al., 2022). Moreover, circular amplicons are typically lost once stressors are removed (Beverley et al., 1984; Ubeda et al., 2008; Leprohon et al., 2009), thus likely they pose a fitness cost in less restrictive circumstances. A second route of transmission emerged more recently, in which circular amplicons containing drug resistance genes were found within EVs, correlating with the emergence of drug resistant parasites in response when exposed to a specific compound (Douanne et al., 2022).
To date, all these experiments were performed using promastigotes, and currently it is unknown if these drug resistance genes re-integrate into the genome and/or are maintained after amastigote differentiation. Nevertheless, such findings could have significant impacts on our understanding of Leishmania- host and -vector interactions. Whether Leishmania utilise these amplification products to directly modulate their immediate extracellular environment and potentially alter disease progression, requires testing. Thus far, exposing immune cells to Leishmania EVs correlates with a Th2 directed anti-inflammatory response (da Silva Lira Filho et al., 2022) suggesting Leishmania excreted products can influence the host immune response. Indeed, in some human cancers, extracellular extrachromosomal circular DNAs have been reported in connection with altered disease outcome, acting as putative biomarkers of tumour severity (as reviewed by Li et al., 2022; Noer et al., 2022).
Additionally, the transmission of DNA amplicons could have consequences for species evolution. It is exciting to consider that mixed species infections of Leishmania provide opportunities for inter-species DNA transmission, and indeed such hybrids have been detected (Romano et al., 2014; Louradour et al., 2020). Currently, it is unknown whether circular or linear amplicons contribute. One final striking gap in our understanding, as alluded to previously, is how circular (and linear) DNA amplicons are copied. Whether similar processes duplicate the chromosomes and extrachromosomal DNA is unclear, or at which cell cycle stage these processes occur. The ability of Leishmania to duplicate exogenous sources of DNAs (i.e plasmids or cosmids of bacterial origins), suggests the replication pathway for extrachromosomal DNA is unlikely to rely on Leishmania specific sequences or factors (Papadopoulou et al., 1994).
Low complexity repeats, LDPR1, TATE and LINE elements are also found in the genome of Leishmania (Pita et al., 2019; Bussotti et al., 2021), yet their functions are understudied. Furthermore, 8 additional repetitive elements were linked to CNVs (Bussotti et al., 2021), mapping proximal to known CNV sites (~1 kb outside the variable region to ~ 150 bp within). Future studies will be key in deciphering their contributions to Leishmania genome variability.
3 Mosaic aneuploidy in the Leishmania genome
3.1 The origins of Leishmania aneuploidy
Aneuploidy and CIN describe imbalances in chromosome numbers. Typically, CIN describes an inability to retain the same number of chromosomes from one division to the next, whereas aneuploidy explains a state of abnormal chromosome number. Though an aneuploid cell does not always experience CIN, often both coexist, particularly in cancers (Potapova et al., 2013). In humans, aneuploidy commonly correlates with early miscarriage (Ben-David and Amon, 2020), and developmental syndromes including Down Syndrome (Trisomy 21) (Antonarakis et al., 2020). Yet, in unicellular eukaryotes like yeast (Hose et al., 2020) and Leishmania, aneuploidy and CIN may enhance genome diversity. Disomy (i.e. two chromosome copies) likely predominates in Leishmania, however mosaic aneuploidy (variable aneuploidy states) is common in vitro and within natural populations, suggesting it is a constitutive feature of their genome (Rogers et al., 2011; Sterkers et al., 2011; Negreira et al., 2022). Why aneuploidy is frequent in Leishmania is unclear but like extrachromosomal amplification, varying chromosome number may provide an additional method of mRNA regulation by increasing DNA copies. In fact, correlations exist between chromosomal copies and gene expression for all chromosomes, except for chromosome 31 (Dumetz et al., 2017; Prieto Barja et al., 2017). Conversely, CNVs arising from aneuploidy do not always mirror protein abundance (Cuypers et al., 2022) suggesting additional layers of regulation operate, perhaps to mitigate wider effects due to haploinsufficiency or toxic overexpression of co-amplified genes. Besides providing populational variability, Leishmania could also use chromosomal duplication and loss to exclude whole chromosome variants leading to loss of heterozygosity (LOH), a process termed haplotype selection (Prieto Barja et al., 2017). During this process Leishmania cells may duplicate a disomic chromosome (chromosomes AB), becoming trisomic (chromosomes AAB), and lose the unwanted copy (chromosomes AA), reducing its heterozygosity. However, the relevance of this process to Leishmania evolution is poorly understood. Thus, the origins of aneuploidy in Leishmania are likely multifactorial, arising from lax chromosome segregation (i.e CIN), hybridisation via cell-cell fusions and from the unusual replication dynamics of the parasite, or a combination of these events (as summarised in Figure 2).
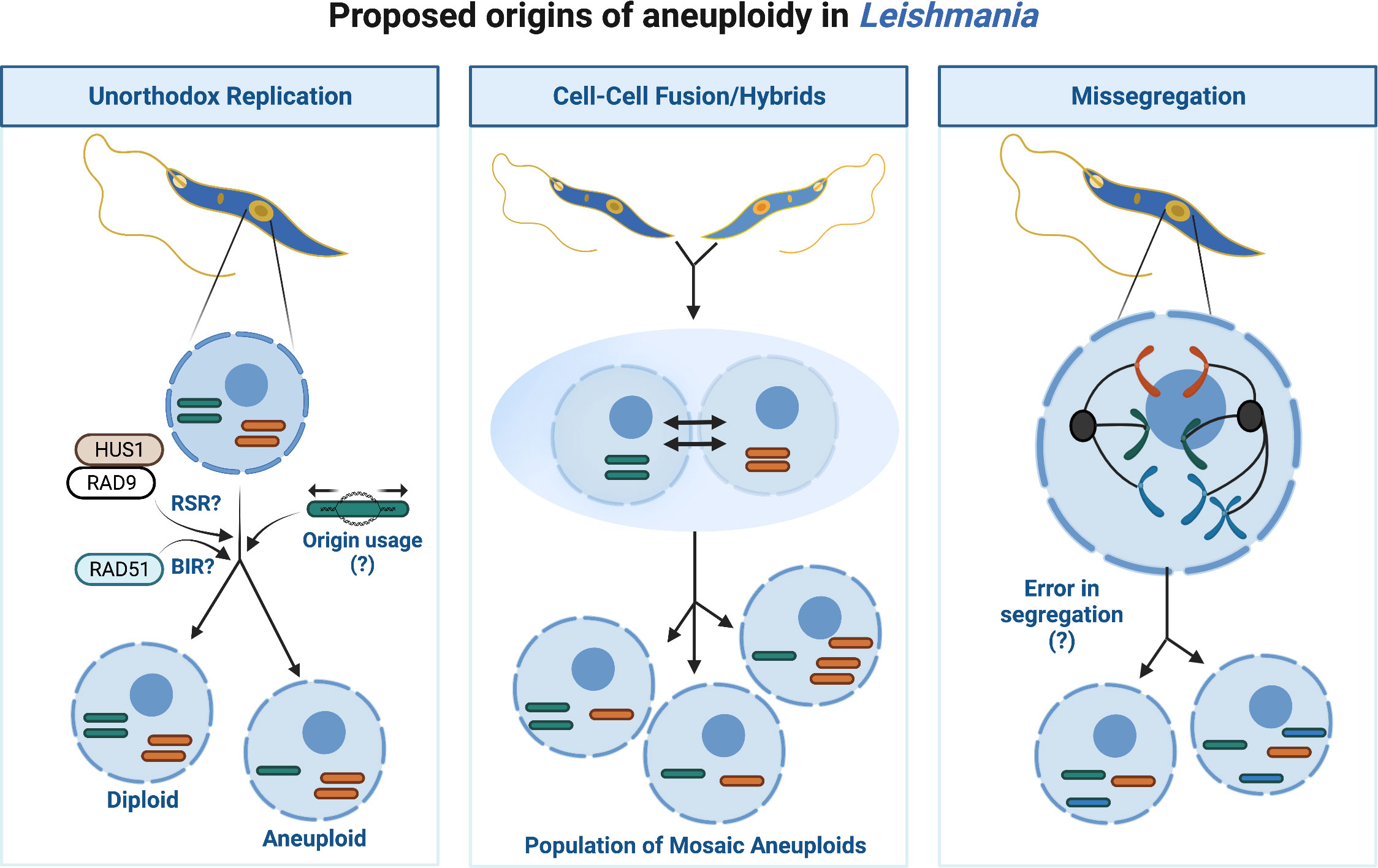
Figure 2 Proposed origins of aneuploidy in Leishmania. Aneuploidy in Leishmania may arise through several different processes or from a combination of events as illustrated. Unorthodox DNA replication may enhance opportunities for replication associated stress, for instance the potential usage of replication stress response (RSR) machinery for subtelomeric duplication and/or BIR to duplicate the chromosome cores may lead to under- or -over replication of chromosomes and thus aneuploid daughters. Cell-Cell fusions/hybrids may arise within the population leading to a fusion of cells each containing different chromosome numbers or putative hybrid events between different Leishmania species. Missegregation of chromosomes from mother cell to daughters may perpetuate aneuploidy, though the underlying biological processes permitting abnormal segregation are unclear.
DNA replication in Leishmania (reviewed by da Silva et al., 2017; Damasceno et al., 2021) could contribute to aneuploidy in several ways. A unanimous model for Leishmania DNA replication has yet to be reached (Lombraña et al., 2016; Stanojcic et al., 2016; Damasceno et al., 2020a; da Silva et al., 2020), though current data supports replication preferentially initiates from a single site (an ‘origin’) per chromosome during early S-phase. Generally, the origin site is positioned at a transcription unit boundary (or strand-switch region: SSR), however why replication initiates here is still unknown. No defined sequence motifs have been identified at such sites; instead, the co-localisation of transcription associated epigenetic marks Base J (a modified thymidine) and acetylated Histone H3 (AchH3), in addition to the presence of the kinetochore protein KKT1 designate replication initiation permissive SSRs (Damasceno et al., 2020a). This unusual replication program may pose problems for chromosome duplication. For smaller chromosomes, firing a singular origin could accommodate complete duplication however, larger chromosomes may fail to duplicate prior to S-phase completion. Alternative ‘dormant’ origins could exist, as detected in T. cruzi (Calderano et al., 2015), however inducible deletion of RAD51 revealed a potential ‘origin-independent’ process may operate. Break Induced Replication (BIR), a mutagenic HR-like pathway which tackles single ended DSBs (reviewed by Kramara et al., 2018), has been proposed to complete Leishmania core chromosome duplication (Damasceno et al., 2020b). For subtelomeric sites, separate replicative processes appear to act, relying on replication stress response (RSR) machinery post-S phase (Damasceno et al., 2020a). Thus, the temporal organisation of Leishmania DNA replication may enhance opportunities for chromosomes to become over- or under-replicated. In support, cells in varying ‘somy’ states exist during mitosis, coinciding with the emergence of aneuploid daughters (Sterkers et al., 2011; Sterkers et al., 2014). Furthermore, DNA duplication involving DNA repair pathways (i.e. BIR) and post-S-phase synthesis correlates with enhanced mutagenesis in other eukaryotes (Ivanova et al., 2020 and reviewed by Saxena and Zou, 2022). Evidence of BIR or a BIR-like pathway during Leishmania DNA synthesis requires further testing however, together, these unusual replication dynamics could support frequent chromosome losses or gains and increased mutagenesis, particularly at subtelomeric sites, which are common instability ‘hotspots’. Repeated DNA and expanded gene families typically populate eukaryotic subtelomeres and, consequently, can undergo rapid evolution due these elevated levels of mutagenesis and recombination (Freitas-Junior et al., 2000; Linardopoulou et al., 2005; Rudd et al., 2007; Chen et al., 2018). The subtelomeres of T. brucei and T. cruzi harbour variable gene families that play key roles during host immune evasion (Ramirez, 2020; Sima et al., 2022), and thus are vital to parasite survival. Perplexingly, Leishmania subtelomeres, unlike T. brucei and T. cruzi, are devoid of variable gene families, thus why diversification would be required is unclear.
Aneuploidy could arise from cell-cell fusions (i.e hybridisation) in Leishmania. Cellular fusion with temporary tetraploidy (4 chromosome copies), followed by genome erosion with chromosomal loss was recently shown to occur in hybrids from the Leishmania close-related parasite, T. cruzi (Matos et al., 2022). Heterozygosity is rarer in natural isolates, relative to experimental strains; nonetheless, inter-, and intra-species hybrids exist (Volf et al., 2007; Romano et al., 2014; Louradour et al., 2020). It is possible interspecies hybridisation events, in addition or as an alternative to, extrachromosomal DNA transmission could explain the origin of certain Leishmania species. For instance, two chromosome fusion events in Leishmania mexicana may indicate genetic streamlining from the original parents. Moreover, a meiotic-like cycle may exist in Leishmania (Lanotte and Rioux, 1990; Weedall and Hall, 2015; Inbar et al., 2017; Inbar et al., 2019), and the generation of viable experimental hybrids can be enhanced following parasite exposure to genotoxins, indicating DNA repair plays a role in this process (Louradour et al., 2020; Ferreira and Sacks, 2022). However, exposure to genotoxic agents results in polyploid hybrids, which are different to the typical disomic hybrids observed in natural non-genotoxic exposed sandfly infections (Inbar et al., 2019). On the other hand, a mix of diploid, triploid and tetraploid hybrids were observed following L. tropica hybridisation in vitro, suggesting that polyploidy could arise even in the absence of genotoxic agents (Louradour et al., 2020; Ferreira and Sacks, 2022). Moreover, a recent study by Ferreira et al. (Ferreira et al., 2022), demonstrated the ability of Leishmania to generate self-hybrids in the insect vector. Indeed, the use of self-hybridisation could potentially limit the accumulation of deleterious mutations that could arise from rounds of asexual reproduction (Muller, 1964). That said, as certain meiotic cycle regulators appear absent from the genome, and no haploid intermediate stages have been identified in Leishmania, this cycle could be atypical as proposed previously (i.e. parasexual) (Sterkers et al., 2014). Irrespective, a meiotic-like process could explain chromosome shuffling and limited recombination events between parental chromosomes leading to altered chromosome dynamics in the offspring. Such a process may have important implications for mixed species infections, particularly if they occur in the mammalian host. It is still unclear whether genetic exchange occurs at this stage given the rarity of aneuploidy events in amastigote stages (Domagalska et al., 2019).
Lastly, imperfect chromosome segregation may generate CNVs in Leishmania. Less is known about the cell cycle checkpoints of Leishmania and the apparent absence of some Spindle Assembly Checkpoint (SAC) factors in the genome (Wheeler et al., 2019; Kops et al., 2020) could suggest lax or absent spindle checkpoint controls thereby supporting lenient spindle attachments, asymmetrical allotments, and potentially partial chromosome deletions. An alternative checkpoint exists during metaphase in procyclic (insect) forms of T. brucei which becomes instead activated in response to damaged DNA (Zhou et al., 2019) though in Leishmania, such a checkpoint remains undescribed.
Together, Leishmania aneuploidy and CNVs could arise from several sources, perhaps enhanced by this parasite’s unusual biology.
3.2 Leishmania aneuploidy is stochastic
Studying CNV regulation and its biological relevance in Leishmania is challenging. Foremost, we lack functional data for ~ 50% of the coding content of the genome, with less known about non-coding elements. Such gaps impair our ability to evaluate the consequences of aneuploidy events without subsequent targeted phenotyping. Secondly, the extreme malleability of the Leishmania genome often hinders basic reverse genetics approaches for phenotyping. Thirdly, the polycistronic transcription of functionally unrelated genes complicates how parasites balance beneficial dose alterations whilst mitigating toxic effects. Lastly, CNVs are often studied in the context of a phenotype, thus we likely lose resolution on the events initially promoting amplification or deletion prior to phenotype emergence. For instance, the detection and expansion of drug resistant phenotypes already threatens the clinical management of the disease. However, recent works by Negreira et al. (Negreira et al., 2022) and Bussotti et al. (Bussotti et al., 2021) are now refining our view on these processes by uncovering patterns of CNV that lead to parasite population heterogeneity associated with changes in gene expression and parasite evolution.
SCS used to study aneuploidy in Leishmania promastigotes by Negreira et al. produced several key findings. By comparing two independent clonal lines, one predominantly euploid (BPK081, clone 8) and one with variable somies (BPK282, clone 4), in in vitro cultured L. donovani promastigotes, a diversity of complex karyotypes was found co-existing within the population at any given time indicative of a genome under stochastic flux. Such diversity surfacing from both predominantly euploid and aneuploid founder populations reinforces this aneuploidy as stochastic. Their data supports an initial expansion of karyotype complexity that refines over time, leading to the emergence of more dominant (‘common’) karyotypes. This suggests that Leishmania steadily accumulate chromosome expansions in culture, which is a permissive environment. Subsequent alterations may further shape beneficial genomic changes. A small proportion of cells carrying rarer karyotypes persist in the population, perhaps because of the rich culture medium environment. Nonetheless, rare karyotypes could act as additional diversity reservoirs for overcoming subsequent bottlenecks including differentiation across lifecycle stages and vector or host entry.
One puzzling aspect of aneuploidy in Leishmania is the seeming preference for certain chromosomes to readily increase or decrease in copy, whilst others remain disomic or monosomic, at least in these two evaluated clones and their derived populations. Thus, restrictions presumably operate to limit supernumerary chromosomes. However, it could be possible to explain this effect as experimental limitations. Their data supports a model in which all chromosomes may possess the potential for amplification and additional selective pressures likely define which subset are frequently polysomic. Therefore, despite chromosome CNV itself being constrained, some underlying flexibility is retained if required (Negreira et al., 2022). It will be interesting to evaluate if the chromosomes consistently observed as disomic or polysomic by Negreira et al. will also maintain this pattern when other Leishmania populations or species are evaluated. Given ‘somy’ alterations often reverse if disadvantageous, aneuploidy likely imposes fitness costs for the parasite despite its frequency and seeming significance to Leishmania gene regulation. One surprising finding was the discovery of some parasites in vitro lacking entire chromosomes (i.e. nullisomic). Chromosome loss correlates with reduced genetic diversity within populations, and therefore counterintuitive for population diversification. Nullisomy is common in several plant species (i.e. wheat), often coinciding with the amplification of other homologous chromosomes to mitigate consequences of entire chromosome content depletion (Zhang et al., 2017). Whether true nullisomy naturally occurs in Leishmania is unclear but if supported, this strategy could serve as a ‘last-resort’ to remove survival-limiting genes under highly restricted environments. On the other hand, these nullisomic cells may arise from unbalanced cell division, and may lack long term viability.
Taken together, stochastic aneuploidy in Leishmania could represent a unique opportunity for genomic pre-adaption in the absence of stochastic alterations to transcription levels. These events may occur more freely in permissive conditions such as during in vitro culture and potentially within the sandfly environment.
4 Do epistatic pathways direct chromosome and gene copy number?
A routinely cited example of chromosome polyploidy in Leishmania is chromosome 31 of L. major and all other evaluated species to date (Rogers et al., 2011). Why chromosome 31 is apparently always supernumeric in copy number is unknown. However, recent data suggests this polyploidy may correlate with increased chromosome 15 amplification (Negreira et al., 2022), suggestive of unknown physical and/or functional inter- and intra-chromosome interactions. Indeed, in T. brucei chromosome interactions regulate transcription and splicing of the variant surface glycoprotein (VSG) required for host immune evasion (Faria et al., 2021) highlighting the importance of these events in host evasion. Yet, we currently lack evidence linking chromosome interactions to gene expression changes in Leishmania as detailed maps of such interactions are still to surface.
One emerging explanation to describe these correlative ploidy changes between chromosomes pertains to the non-coding RNAs (ncRNAs) content (Bussotti et al., 2021; Negreira et al., 2022). Although ncRNAs do not encode proteins, they are key regulators of cellular metabolism (Cech and Steitz, 2014). In Leishmania, the ‘RNAome’ may contain upwards of 12,000 ncRNAs per species but limited studies have functionally characterised their activities (Ruy et al., 2019; Fort et al., 2022). Now, links between ncRNAs and parasite development suggest these elements do directly regulate key parasite processes, for example the recent description of a long ncRNA required for differentiation to the quiescent, transmissible form (the ‘stumpy’ form) of T. brucei (Guegan et al., 2022) or the variable expression of ncRNAs across the Leishmania lifecycle (Ruy et al., 2019). Whether ncRNAs play roles in Leishmania genome plasticity is unknown, though thus far, small nucleolar RNA (snoRNAs), transfer RNA (tRNAs) and ribosomal RNA (rRNAs) appear to associate with chromosome polyploidy and gene CNVs (Bussotti et al., 2021; Negreira et al., 2022), though the natures of these relationships require further clarification. Nevertheless, differential snoRNA expression in Leishmania correlates with rRNA changes and the production of modified ribosomes, in turn altering mRNA turnover and translation (Piel et al., 2022). Together, these data could explain the lack of a defined relationship between the coding content of co-amplified chromosomes.
One study exploring these effects in culture adapted L. infantum promastigotes uncovered evidence of putative relationships between co-amplified genes and those of similar functionalities (Bussotti et al., 2021), attributing their findings to an underlying and functional ‘epistatic’ network. Epistasis is a phenomenon that broadly describes the outcome of a mutation or mutations as functions of the genetic background they appear in. For example, a mutation of a gene which enhances gene expression in one genetic background, may instead have differing effects in another (Domingo et al., 2019). In Leishmania, the spontaneous deletion of an 11kb region containing an essential NIMA-related kinase led to viable in vitro promastigotes suggesting an unknown compensatory method(s) operates, independently, to limit potentially fatal genomic alterations. Therein, the authors reported an increased abundance of 350 transcripts including ncRNA elements and metabolic enzymes in their deletion mutants (Bussotti et al., 2021). Similarly, it was demonstrated in another recent study that non-targeted deletions can be induced as compensatory mechanisms in Leishmania when targeting an essential gene (Alpizar-Sosa et al., 2022). Thus far, definitive evidence of epistatic interactions in Leishmania is still required. These data are frequently challenging to interpret and the wider implications of such interaction networks in the context of an infection must be investigated.
In summary, during early adaptions, flexible gene dosage variation, that may include non-coding elements, could alter translation and RNA stability regulation thereby regulating expression rapidly. Later (and likely more stable) adaptions appear to require more extensive alterations to genomic content.
5 Concluding remarks and future directions
Possession of a plastic genome presents Leishmania with benefits and challenges. Likely arising from multiple sources (summarised in Figure 3), the ability of Leishmania to maximise and harness stochastic instability, generated by core biological processes, may favour the frequent discovery of beneficial traits in harsh and changing environments. Adjustments to the abundance of favourable genes, followed by putative regulatory interactions by ncRNAs, DNA modifications and chromatin alterations could allow Leishmania to fine-tune gene expression further by adapting translation efficiency. Moreover, the recent discovery of extrachromosomal DNAs within Leishmania EVs provides opportunity for the population-wide dissemination of fitness enhancing traits, offering naïve individuals a means of survival, and putatively maximising the persistence of the infection. Similarly, DNA exchanges in mixed species infections, for instance in the insect vector, may contribute to species diversification through the exchange and incorporation of amplified DNA from others.
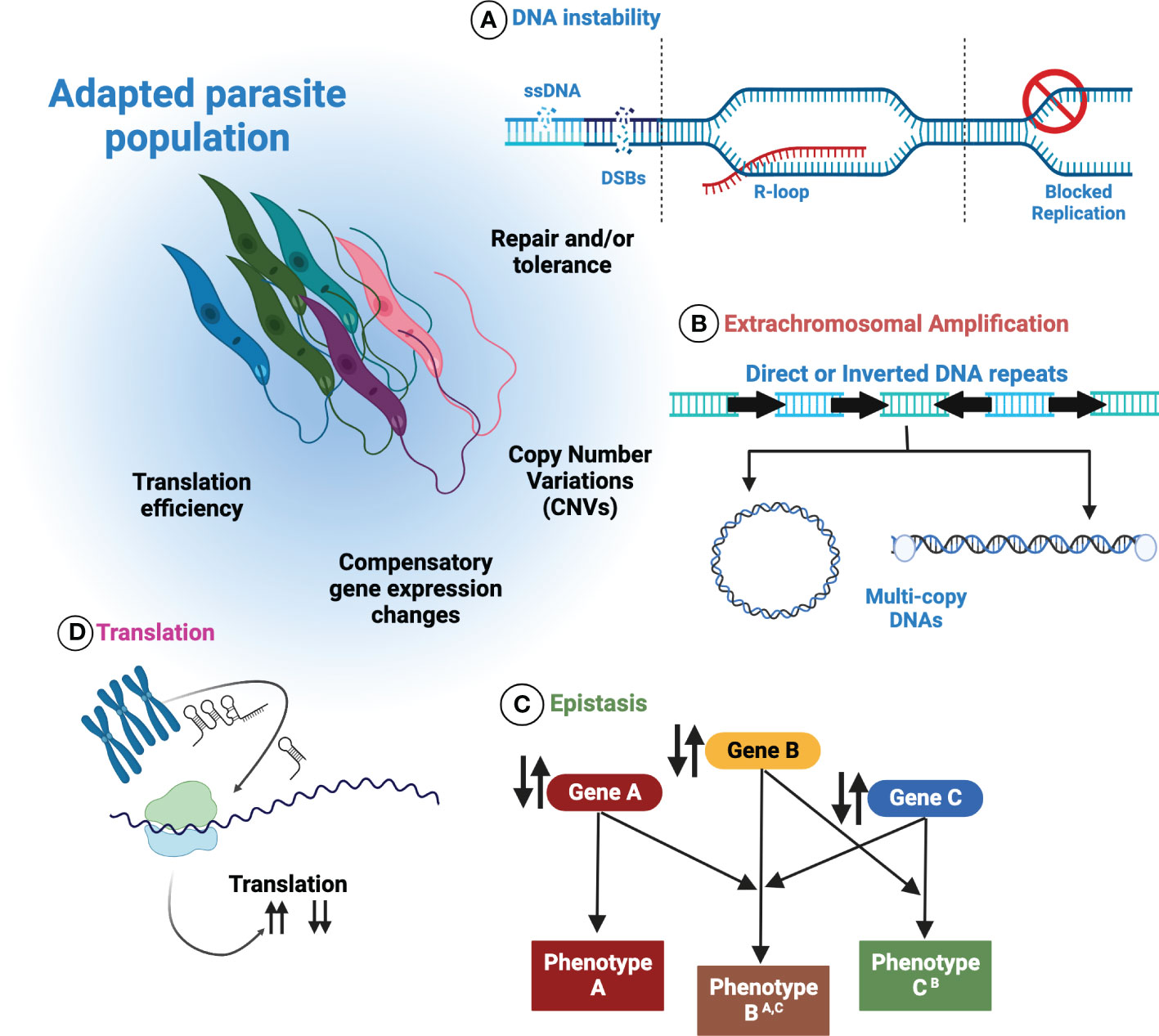
Figure 3 Control over genome plasticity in Leishmania is multifactorial. Numerous processes are thought to contribute to Leishmania genome plasticity. (A) Inherent DNA instability may be a key source of genome plasticity events in Leishmania. How these events are repaired and/or tolerated may offer deeper insights into how they are exploited by Leishmania. (B) By increasing or decreasing the abundance of extrachromosomal amplicons, Leishmania can modulate the corresponding mRNA levels in response to changing environments. (C) Epistatic interactions may operate in Leishmania, allowing compensatory mutations or genomic rearrangements to arise. (D) The putative regulation of translation by non-coding RNAs may permit the modulation of translation efficiency.
Whilst the related pathogens T. brucei and T. cruzi can utilise variable antigen gene families to evade host defences, no such strategy appears to operate in Leishmania. Thus, a genetically diverse population of parasites with flexible, and rapidly evolving genomes may offer an alternative strategy for overcoming host defences. Whether such extensive diversity arises in the context of a clinical infection requires further experimentation given aneuploidy appears rarer in the mammalian stage parasites (amastigotes), and to some extent, in naturally isolated promastigotes. Technical limitations often thwart direct investigations in amastigotes from clinical isolates, namely poor sample size leading to an inevitable passage through mice or into in vitro culture. That said, it is possible that the exclusively intracellular lifecycle of amastigotes may impose fewer extreme demands for genetic plasticity.
In contrast, a genome under constant, stochastic flux is problematic. Too many random alterations could impede survival under restrictive environments, for instance within neutrophils or macrophages. Toxic rearrangements, persistent damage, and the irreversible loss of genetic information are serious consequences of unregulated genome instability and may compromise the parasite population in the face of further stressors. Thus, a deeper understanding of how Leishmania regulate their genome composition is crucial as currently Leishmania genome plasticity is a key barrier to the development of novel compounds for the treatment of leishmaniasis. Finally, even less known about the impact of these genomic changes on the host and subsequent future infections.
Author contributions
All authors contributed equally to the writing and preparation of the manuscript and to the figure design.
Funding
JB is supported by a FAPESP post-doctoral fellowship (20/01883-7). LT and AC are supported by FAPESP, grant number 18/14398-0. JR-C is funded by an MRC New Investigator Research Grant, grant number (MR/T016019/1).
Acknowledgments
We thank our funders for their support. All figures were produced using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Żmieńko, A., Samelak, A., Kozłowski, P., Figlerowicz, M. (2014). Copy number polymorphism in plant genomes. Theor. Appl. Genet. 127, 1–18. doi: 10.1007/s00122-013-2177-7
Alpizar-Sosa, E. A., Kumordzi, Y., Wei, W., Whitfield, P. D., Barrett, M. P., Denny, P. W. (2022). Genome deletions to overcome the directed loss of gene function in leishmania. Front. Cell Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.988688
Antonarakis, S. E., Skotko, B. G., Rafii, M. S., Strydom, A., Pape, S. E., Bianchi, D. W., et al. (2020). Down syndrome. Nat. Rev. Dis. Primers 6, 9. doi: 10.1038/s41572-019-0143-7
Baker, N., Catta-Preta, C. M. C., Neish, R., Sadlova, J., Powell, B., Alves-Ferreira, E. V. C., et al. (2021). Systematic functional analysis of leishmania protein kinases identifies regulators of differentiation or survival. Nat. Commun. 12 (1), 1244. doi: 10.1038/s41467-021-21360-8
Ben-David, U., Amon, A. (2020). Context is everything: aneuploidy in cancer. Nat. Rev. Genet. 21, 44–62. doi: 10.1038/s41576-019-0171-x
Beneke, T., Madden, R., Makin, L., Valli, J., Sunter, J., Gluenz, E. (2022). A CRISPR Cas9 high-throughput genome editing toolkit for kinetoplastids. R Soc. Open Sci. 4, 170095. doi: 10.1098/rsos.170095
Beverley, S. M., Coderre, J. A., Santi, D. V, Schimke, R. T. (1984). Unstable DNA amplifications in methotrexate resistant leishmania consist of extrachromosomal circles which relocalize during stabilization. Cell 38, 431–439. doi: 10.1016/0092-8674(84)90498-7
Biscotti, M. A., Olmo, E., Heslop-Harrison, J.S.(. (2015). Repetitive DNA in eukaryotic genomes. Chromosome Res. 23, 415–420. doi: 10.1007/s10577-015-9499-z
Bolhaqueiro, A. C. F., Ponsioen, B., Bakker, B., Klaasen, S. J., Kucukkose, E., van Jaarsveld, R. H., et al. (2019). Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 51, 824–834. doi: 10.1038/s41588-019-0399-6
Bringaud, F., Müller, M., Cerqueira, G. C., Smith, M., Rochette, A., El-Sayed, N. M. A., et al. (2007). Members of a Large retroposon family are determinants of post-transcriptional gene expression in leishmania. PloS Pathog. 3, e136-. doi: 10.1371/journal.ppat.0030136
Brown, R. E., Freudenreich, C. H. (2021). Structure-forming repeats and their impact on genome stability. Curr. Opin. Genet. Dev. 67, 41–51. doi: 10.1016/j.gde.2020.10.006
Burza, S., Croft, S. L., Boelaert, M. (2018). Leishmaniasis. Lancet 392, 951–970. doi: 10.1016/S0140-6736(18)31204-2
Bussotti, G., Piel, L., Pescher, P., Domagalska, M. A., Rajan, K. S., Cohen-Chalamish, S., et al. (2021). Genome instability drives epistatic adaptation in the human pathogen leishmania. Proc. Natl. Acad. Sci. 118, e2113744118. doi: 10.1073/pnas.2113744118
Calderano, S. G., Drosopoulos, W. C., Quaresma, M. M., Marques, C. A., Kosiyatrakul, S., McCulloch, R., et al. (2015). Single molecule analysis of trypanosoma brucei DNA replication dynamics. Nucleic Acids Res. 43, 2655–2665. doi: 10.1093/nar/gku1389
Capela, R., Moreira, R., Lopes, F. (2019). An overview of drug resistance in protozoal diseases. Int. J. Mol. Sci. 20, 5748. doi: 10.3390/ijms20225748
Cech, T. R., Steitz, J. A. (2014). The noncoding RNA revolution–trashing old rules to forge new ones. Cell 157, 77–94. doi: 10.1016/j.cell.2014.03.008
Chen, N. W. G., Thareau, V., Ribeiro, T., Magdelenat, G., Ashfield, T., Innes, R. W., et al. (2018). Common bean subtelomeres are hot spots of recombination and favor resistance gene evolution. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01185
Cupolillo, E., Cavalcanti, A. S., Ferreira, G. E. M., Boité, M. C., Morgado, F. N., Porrozzi, R. (2020). Occurrence of multiple genotype infection caused by leishmania infantum in naturally infected dogs. PloS Negl. Trop. Dis. 14 (7). doi: 10.1371/journal.pntd.0007986
Cuypers, B., Meysman, P., Erb, I., Bittremieux, W., Valkenborg, D., Baggerman, G., et al. (2022). Four layer multi-omics reveals molecular responses to aneuploidy in leishmania. PloS Pathog. 18, e1010848-. doi: 10.1371/journal.ppat.1010848
Damasceno, J. D., Marques, C. A., Beraldi, D., Crouch, K., Lapsley, C., Obonaga, R., et al. (2020a). Genome duplication in leishmania major relies on persistent subtelomeric DNA replication. Elife 9, e58030. doi: 10.7554/eLife.58030
Damasceno, J. D., Marques, C. A., Black, J., Briggs, E., McCulloch, R. (2021). Read, write, adapt: Challenges and opportunities during kinetoplastid genome replication. Trends Genet. 37, 21–34. doi: 10.1016/j.tig.2020.09.002
Damasceno, J. D., Obonaga, R., Silva, G. L. A., Reis-Cunha, J. L., Duncan, S. M., Bartholomeu, D. C., et al. (2018). Conditional genome engineering reveals canonical and divergent roles for the Hus1 component of the 9–1–1 complex in the maintenance of the plastic genome of leishmania. Nucleic Acids Res. 46, 11835–11846. doi: 10.1093/nar/gky1017
Damasceno, J. D., Reis-Cunha, J., Crouch, K., Beraldi, D., Lapsley, C., Tosi, L. R. O., et al. (2020b). Conditional knockout of RAD51-related genes in leishmania major reveals a critical role for homologous recombination during genome replication. PloS Genet. 16, e1008828-. doi: 10.1371/journal.pgen.1008828
da Silva Lira Filho, A., Fajardo, E. F., Chang, K. P., Clément, P., Olivier, M. (2022). Leishmania Exosomes/Extracellular vesicles containing GP63 are essential for enhance cutaneous leishmaniasis development upon Co-inoculation of leishmania amazonensis and its exosomes. Front. Cell Infect. Microbiol. 11 (5), 523. doi: 10.3389/fcimb.2021.709258
da Silva, M. S., Pavani, R. S., Damasceno, J. D., Marques, C. A., McCulloch, R., Tosi, L. R. O., et al. (2017). Nuclear DNA replication in trypanosomatids: There are no easy methods for solving difficult problems. Trends Parasitol. 33, 858–874. doi: 10.1016/j.pt.2017.08.002
da Silva, M. S. O., Vitarelli, M. F., Souza, B., Elias, M. C. (2020). Comparative analysis of the minimum number of replication origins in trypanosomatids and yeasts. Genes (Basel) 11 (5), 523. doi: 10.3390/genes11050523
Dias, F. C., Ruiz, J. C., Lopes, W. C. Z., Squina, F. M., Renzi, A., Cruz, A. K., et al. (2007). Organization of h locus conserved repeats in leishmania (Viannia) braziliensis correlates with lack of gene amplification and drug resistance. Parasitol. Res. 101, 667–676. doi: 10.1007/s00436-007-0528-5
Domagalska, M. A., Imamura, H., Sanders, M., van den Broeck, F., Bhattarai, N. R., Vanaerschot, M., et al. (2019). Genomes of leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent. PloS Negl. Trop. Dis. 13, e0007900-. doi: 10.1371/journal.pntd.0007900
Domingo, J., Baeza-Centurion, P., Lehner, B. (2019). The causes and consequences of genetic interactions (Epistasis). Annu. Rev. Genomics Hum. Genet. 20, 433–460. doi: 10.1146/annurev-genom-083118-014857
Douanne, N., Dong, G., Amin, A., Bernardo, L., Blanchette, M., Langlais, D., et al. (2022). Leishmania parasites exchange drug-resistance genes through extracellular vesicles. Cell Rep. 40, 111121. doi: 10.1016/j.celrep.2022.111121
Dumetz, F., Cuypers, B., Imamura, H., Zander, D., D’Haenens, E., Maes, I., et al. (2018). Molecular preadaptation to antimony resistance in leishmania donovani on the Indian subcontinent. mSphere 3, e00548–e00517. doi: 10.1128/mSphere.00548-17
Dumetz, F., Imamura, H., Sanders, M., Seblova, V., Myskova, J., Pescher, P., et al. (2017). Modulation of aneuploidy in leishmania donovani during adaptation to different In vitro and In vivo environments and its impact on gene expression. mBio 8, e00599–e00517. doi: 10.1128/mBio.00599-17
Duncan, S. M., Myburgh, E., Philipon, C., Brown, E., Meissner, M., Brewer, J., et al. (2016). Conditional gene deletion with DiCre demonstrates an essential role for CRK3 in leishmania mexicana cell cycle regulation. Mol. Microbiol. 100, 931–944. doi: 10.1111/mmi.13375
Elbakry, A., Löbrich, M. (2021). Homologous recombination subpathways: A tangle to resolve. Front. Genet. 12. doi: 10.3389/fgene.2021.723847
Espada, C. R., Quilles, J. C., Albuquerque-Wendt, A., Cruz, M. C., Beneke, T., Lorenzon, L. B., et al. (2021). Effective genome editing in leishmania (Viannia) braziliensis stably expressing Cas9 and T7 RNA polymerase. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.772311
Faria, J., Luzak, V., Müller, L. S. M., Brink, B. G., Hutchinson, S., Glover, L., et al. (2021). Spatial integration of transcription and splicing in a dedicated compartment sustains monogenic antigen expression in African trypanosomes. Nat. Microbiol. 6, 289–300. doi: 10.1038/s41564-020-00833-4
Ferreira, T. R., Inbar, E., Shaik, J., Jeffrey, B. M., Ghosh, K., Dobson, D. E., et al. (2022). Self-hybridization in leishmania major. mBio 13 (6). doi: 10.1128/mbio.02858-22
Ferreira, T. R., Sacks, D. L. (2022). Experimental hybridization in leishmania: Tools for the study of genetic exchange. Pathogens 11, 580. doi: 10.3390/pathogens11050580
Fort, R. S., Chavez, S., Trinidad Barnech, J. M., Oliveira-Rizzo, C., Smircich, P., Sotelo-Silveira, J. R., et al. (2022). Current status of regulatory non-coding RNAs research in the tritryp. Noncoding RNA 8 (4), 54. doi: 10.3390/ncrna8040054
Freitas-Junior, L. H., Bottius, E., Pirrit, L. A., Deitsch, K. W., Scheidig, C., Guinet, F., et al. (2000). Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of p. falciparum. Nature 407, 1018–1022. doi: 10.1038/35039531
Genois, M.-M., Mukherjee, A., Ubeda, J.-M., Buisson, R., Paquet, E., Roy, G., et al. (2012). Interactions between BRCA2 and RAD51 for promoting homologous recombination in leishmania infantum. Nucleic Acids Res. 40, 6570–6584. doi: 10.1093/nar/gks306
Genois, M.-M., Plourde, M., Éthier, C., Roy, G., Poirier, G. G., Ouellette, M., et al. (2015). Roles of Rad51 paralogs for promoting homologous recombination in leishmania infantum. Nucleic Acids Res. 43, 2701–2715. doi: 10.1093/nar/gkv118
Guegan, F., Rajan, K. S., Bento, F., Pinto-Neves, D., Sequeira, M., Gumińska, N., et al. (2022). A long noncoding RNA promotes parasite differentiation in African trypanosomes. Sci. Adv. 8, eabn2706. doi: 10.1126/sciadv.abn2706
Hose, J., Escalante, L. E., Clowers, K. J., Dutcher, H. A., Robinson, D., Bouriakov, V., et al. (2020). The genetic basis of aneuploidy tolerance in wild yeast. Elife 9, e52063. doi: 10.7554/eLife.52063
Imamura, H., Monsieurs, P., Jara, M., Sanders, M., Maes, I., Vanaerschot, M., et al. (2020). Evaluation of whole genome amplification and bioinformatic methods for the characterization of leishmania genomes at a single cell level. Sci. Rep. 10, 15043. doi: 10.1038/s41598-020-71882-2
Inbar, E., Hughitt, V. K., Dillon, L. A., Ghosh, K., El-Sayed, N. M., Sacks, D. L. (2017). The transcriptome of Leishmania major developmental stages in their natural sand fly vector. MBio 8 (2).
Inbar, E., Shaik, J., Iantorno, S. A., Romano, A., Nzelu, C. O., Owens, K., et al. (2019). Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in leishmania. PloS Genet. 15 (5), e1008042. doi: 10.1371/journal.pgen.1008042
Ivanova, T., Maier, M., Missarova, A., Ziegler-Birling, C., Dam, M., Gomar-Alba, M., et al. (2020). Budding yeast complete DNA synthesis after chromosome segregation begins. Nat. Commun. 11, 2267. doi: 10.1038/s41467-020-16100-3
Kops, G. J. P. L., Snel, B., Tromer, E. C. (2020). Evolutionary dynamics of the spindle assembly checkpoint in eukaryotes. Curr. Biol. 30 (10), R589–R602. doi: 10.1016/j.cub.2020.02.021
Kramara, J., Osia, B., Malkova, A. (2018). Break-induced replication: The where, the why, and the how. Trends Genet. 34, 518–531. doi: 10.1016/j.tig.2018.04.002
Kratochwil, C. F., Meyer, A. (2019). Fragile DNA contributes to repeated evolution. Genome Biol. 20, 39. doi: 10.1186/s13059-019-1655-x
Kukurudz, R. J., Chapel, M., Wonitowy, Q., Adamu Bukari, A.-R., Sidney, B., Sierhuis, R., et al. (2022). Acquisition of cross-azole tolerance and aneuploidy in candida albicans strains evolved to posaconazole. G3 Genes|Genomes|Genetics 12, jkac156. doi: 10.1093/g3journal/jkac156
Lachaud, L., Bourgeois, N., Kuk, N., Morelle, C., Crobu, L., Merlin, G., et al. (2014). Constitutive mosaic aneuploidy is a unique genetic feature widespread in the leishmania genus. Microbes Infect. 16, 61–66. doi: 10.1016/j.micinf.2013.09.005
Laffitte, M.-C. N., Genois, M.-M., Mukherjee, A., Légaré, D., Masson, J.-Y., Ouellette, M. (2014). Formation of linear amplicons with inverted duplications in leishmania requires the MRE11 nuclease. PloS Genet. 10, e1004805-. doi: 10.1371/journal.pgen.1004805
Laffitte, M.-C. N., Leprohon, P., Hainse, M., Légaré, D., Masson, J.-Y., Ouellette, M. (2016a). Chromosomal translocations in the parasite leishmania by a MRE11/RAD50-independent microhomology-mediated end joining mechanism. PloS Genet. 12, e1006117-. doi: 10.1371/journal.pgen.1006117
Laffitte, M.-C. N., Leprohon, P., Papadopoulou, B., Ouellette, M. (2016b). Plasticity of the leishmania genome leading to gene copy number variations and drug resistance. F1000Res 5, 2350. doi: 10.12688/f1000research.9218.1
Lange, J. T., Rose, J. C., Chen, C. Y., Pichugin, Y., Xie, L., Tang, J., et al. (2022). The evolutionary dynamics of extrachromosomal DNA in human cancers. Nat. Genet. 54, 1527–1533. doi: 10.1038/s41588-022-01177-x
Lanotte, G., Rioux, J. A. (1990). [Cell fusion in Leishmania (Kinetoplastida, Trypanosomatidae)]. C R Acad Sci III 310 (7), 285–288.
Lee, J.-K., Choi, Y.-L., Kwon, M., Park, P. J. (2016). Mechanisms and consequences of cancer genome instability: Lessons from genome sequencing studies. Annu. Rev. Pathology: Mech. Dis. 11, 283–312. doi: 10.1146/annurev-pathol-012615-044446
Leprohon, P., Légaré, D., Raymond, F., Madore, É., Hardiman, G., Corbeil, J., et al. (2009). Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant leishmania infantum. Nucleic Acids Res. 37, 1387–1399. doi: 10.1093/nar/gkn1069
Linardopoulou, E., Williams, E. M., Fan, Y., Friedman, C., Young, J. M., Trask, B. J. (2005). Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature 437, 94–100. doi: 10.1038/nature04029
Li, R., Wang, Y., Li, J., Zhou, X. (2022). Extrachromosomal circular DNA (eccDNA): an emerging star in cancer. biomark. Res. 10, 53. doi: 10.1186/s40364-022-00399-9
Lombraña, R., Álvarez, A., Fernández-Justel, J. M., Almeida, R., Poza-Carrión, C., Gomes, F., et al. (2016). Transcriptionally driven DNA replication program of the human parasite leishmania major. Cell Rep. 16, 1774–1786. doi: 10.1016/j.celrep.2016.07.007
López, S., Lim, E. L., Horswell, S., Haase, K., Huebner, A., Dietzen, M., et al. (2020). Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 52, 283–293. doi: 10.1038/s41588-020-0584-7
Louradour, I., Ferreira, T. R., Ghosh, K., Shaik, J., Sacks, D. (2020). In vitro generation of leishmania hybrids. Cell Rep. 31, 107507. doi: 10.1016/j.celrep.2020.03.071
Lukow, D. A., Sausville, E. L., Suri, P., Chunduri, N. K., Wieland, A., Leu, J., et al. (2021). Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 56, 2427–2439.e4. doi: 10.1016/j.devcel.2021.07.009
Matos, G. M., Lewis, M. D., Talavera-López, C., Yeo, M., Grisard, E. C., Messenger, L. A., et al. (2022). Microevolution of trypanosoma cruzi reveals hybridization and clonal mechanisms driving rapid genome diversification. Elife 11, e75237. doi: 10.7554/eLife.75237
Merlo, L. M. F., Pepper, J. W., Reid, B. J., Maley, C. C. (2006). Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 6, 924–935. doi: 10.1038/nrc2013
Muller, H. J. (1964). The relation of recombination to mutational advance. Mutat. Research/Fundamental Mol. Mech. Mutagenesis 1, 2–9. doi: 10.1016/0027-5107(64)90047-8
Müller, M., Padmanabhan, P. K., Papadopoulou, B. (2010). Selective inactivation of SIDER2 retroposon-mediated mRNA decay contributes to stage- and species-specific gene expression in Leishmania. Molecular Microbiology 77, 471–491. doi: 10.1111/j.1365-2958.2010.07226.x
Negreira, G. H., Monsieurs, P., Imamura, H., Maes, I., Kuk, N., Yagoubat, A., et al. (2022). High throughput single-cell genome sequencing gives insights into the generation and evolution of mosaic aneuploidy in leishmania donovani. Nucleic Acids Res. 50, 293–305. doi: 10.1093/nar/gkab1203
Noer, J. B., Hørsdal, O. K., Xiang, X., Luo, Y., Regenberg, B. (2022). Extrachromosomal circular DNA in cancer: history, current knowledge, and methods. Trends Genet. 38, 766–781. doi: 10.1016/j.tig.2022.02.007
Papadopoulou, B., Roy, G., Ouellette, M. (1994). Autonomous replication of bacterial DNA plasmid oligomers in leishmania. Mol. Biochem. Parasitol. 65, 39–49. doi: 10.1016/0166-6851(94)90113-9
Patino, L. H., Imamura, H., Cruz-Saavedra, L., Pavia, P., Muskus, C., Méndez, C., et al. (2019). Major changes in chromosomal somy, gene expression and gene dosage driven by SbIII in leishmania braziliensis and leishmania panamensis. Sci. Rep. 9, 9485. doi: 10.1038/s41598-019-45538-9
Pfau, S. J., Amon, A. (2012). Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 13, 515–527. doi: 10.1038/embor.2012.65
Piel, L., Rajan, K. S., Bussotti, G., Varet, H., Legendre, R., Proux, C., et al. (2022). Experimental evolution links post-transcriptional regulation to leishmania fitness gain. PloS Pathog. 18, e1010375-. doi: 10.1371/journal.ppat.1010375
Pita, S., Díaz-Viraqué, F., Iraola, G., Robello, C. (2019). The tritryps comparative repeatome: Insights on repetitive element evolution in trypanosomatid pathogens. Genome Biol. Evol. 11, 546–551. doi: 10.1093/gbe/evz017
Ponte-Sucre, A., Gamarro, F., Dujardin, J.-C., Barrett, M. P., López-Vélez, R., García-Hernández, R., et al. (2017). Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PloS Negl. Trop. Dis. 11, e0006052-. doi: 10.1371/journal.pntd.0006052
Potapova, T. A., Zhu, J., Li, R. (2013). Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 32, 377–389. doi: 10.1007/s10555-013-9436-6
Prieto Barja, P., Pescher, P., Bussotti, G., Dumetz, F., Imamura, H., Kedra, D., et al. (2017). Haplotype selection as an adaptive mechanism in the protozoan pathogen leishmania donovani. Nat. Ecol. Evol. 1, 1961–1969. doi: 10.1038/s41559-017-0361-x
Ramirez, J. L. (2020). An evolutionary view of trypanosoma cruzi telomeres. Front. Cell Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00439
Reis-Cunha, J. L., Valdivia, H. O., Bartholomeu, D. C. (2018). Gene and chromosomal copy number variations as an adaptive mechanism towards a parasitic lifestyle in trypanosomatids. Curr. Genomics 19. doi: 10.2174/1389202918666170911161311
Requena, J. M., Rastrojo, A., Garde, E., López, M. C., Thomas, M. C., Aguado, B. (2017). Genomic cartography and proposal of nomenclature for the repeated, interspersed elements of the leishmania major SIDER2 family and identification of SIDER2-containing transcripts. Mol. Biochem. Parasitol. 212, 9–15. doi: 10.1016/j.molbiopara.2016.12.009
Rogers, M. B., Hilley, J. D., Dickens, N. J., Wilkes, J., Bates, P. A., Depledge, D. P., et al. (2011). Chromosome and gene copy number variation allow major structural change between species and strains of leishmania. Genome Res. 21, 2129–2142. doi: 10.1101/gr.122945.111
Romano, A., Inbar, E., Debrabant, A., Charmoy, M., Lawyer, P., Ribeiro-Gomes, F., et al. (2014). Cross-species genetic exchange between visceral and cutaneous strains of leishmania in the sand fly vector. Proc. Natl. Acad. Sci. 111, 16808–16813. doi: 10.1073/pnas.1415109111
Rudd, M. K., Friedman, C., Parghi, S. S., Linardopoulou, E. V, Hsu, L., Trask, B. J. (2007). Elevated rates of sister chromatid exchange at chromosome ends. PloS Genet. 3, e32-. doi: 10.1371/journal.pgen.0030032
Ruy, P. D. C., Monteiro-Teles, N. M., Miserani Magalhães, R. D., Freitas-Castro, F., Dias, L., Aquino Defina, T. P., et al. (2019). Comparative transcriptomics in leishmania braziliensis: disclosing differential gene expression of coding and putative noncoding RNAs across developmental stages. RNA Biol. 16, 639–660. doi: 10.1080/15476286.2019.1574161
Sah, S. K., Hayes, J. J., Rustchenko, E. (2021). The role of aneuploidy in the emergence of echinocandin resistance in human fungal pathogen candida albicans. PloS Pathog. 17, e1009564-. doi: 10.1371/journal.ppat.1009564
Saxena, S., Zou, L. (2022). Hallmarks of DNA replication stress. Mol. Cell 82, 2298–2314. doi: 10.1016/j.molcel.2022.05.004
Sheltzer, J. M., Blank, H. M., Pfau, S. J., Tange, Y., George, B. M., Humpton, T. J., et al. (2011). Aneuploidy drives genomic instability in yeast. Sci. (1979) 333, 1026–1030. doi: 10.1126/science.1206412
Sima, N., McLaughlin, E. J., Hutchinson, S., Glover, L. (2022). Escaping the immune system by DNA repair and recombination in African trypanosomes. Open Biol. 9, 190182. doi: 10.1098/rsob.190182
Smith, M., Bringaud, F., Papadopoulou, B. (2009). Organization and evolution of two SIDER retroposon subfamilies and their impact on the leishmania genome. BMC Genomics 10, 240. doi: 10.1186/1471-2164-10-240
Stanojcic, S., Sollelis, L., Kuk, N., Crobu, L., Balard, Y., Schwob, E., et al. (2016). Single-molecule analysis of DNA replication reveals novel features in the divergent eukaryotes leishmania and trypanosoma brucei versus mammalian cells. Sci. Rep. 6, 23142. doi: 10.1038/srep23142
Sterkers, Y., Crobu, L., Lachaud, L., Pagès, M., Bastien, P. (2014). Parasexuality and mosaic aneuploidy in leishmania: alternative genetics. Trends Parasitol. 30, 429–435. doi: 10.1016/j.pt.2014.07.002
Sterkers, Y., Lachaud, L., Crobu, L., Bastien, P., Pagès, M. (2011). FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in leishmania major. Cell Microbiol. 13, 274–283. doi: 10.1111/j.1462-5822.2010.01534.x
Sullivan, M. R., Bernstein, K. A. (2018). RAD-ical new insights into RAD51 regulation. Genes (Basel) 9, 629. doi: 10.3390/genes9120629
Todd, R. T., Wikoff, T. D., Forche, A., Selmecki, A. (2019). Genome plasticity in candida albicans is driven by long repeat sequences. Elife 8, e45954. doi: 10.7554/eLife.45954
Torres-Guerrero, E., Quintanilla-Cedillo, M. R., Ruiz-Esmenjaud, J., Arenas, R. (2017). Leishmaniasis: a review. F1000Res 6, 750. doi: 10.12688/f1000research.11120.1
Ubeda, J.-M., Légaré, D., Raymond, F., Ouameur, A. A., Boisvert, S., Rigault, P., et al. (2008). Modulation of gene expression in drug resistant leishmania is associated with gene amplification, gene deletion and chromosome aneuploidy. Genome Biol. 9, R115. doi: 10.1186/gb-2008-9-7-r115
Ubeda, J.-M., Raymond, F., Mukherjee, A., Plourde, M., Gingras, H., Roy, G., et al. (2014). Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite leishmania. PloS Biol. 12, e1001868-. doi: 10.1371/journal.pbio.1001868
Volf, P., et al. (2007). Increased transmission potential of Leishmania major/Leishmania infantum hybridsLeishmania. Int. J. Parasitol. 37, 589–593. doi: 10.1016/j.ijpara.2007.02.002
Watkins, T. B. K., Lim, E. L., Petkovic, M., Elizalde, S., Birkbak, N. J., Wilson, G. A., et al. (2020). Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 587, 126–132. doi: 10.1038/s41586-020-2698-6
Weedall, G. D., Hall, N. (2015). Sexual reproduction and genetic exchange in parasitic protists. Parasitology 142 (Suppl 1), S120–7.
Wheeler, R. J., Gull, K., Sunter, J. D. (2019). Coordination of the cell cycle in trypanosomes. Annu. Rev. Microbiol. 73, 133–154. doi: 10.1146/annurev-micro-020518-115617
Wright, W. D., Shah, S. S., Heyer, W.-D. (2018). Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535. doi: 10.1074/jbc.TM118.000372
Yagoubat, A., Crobu, L., Berry, L., Kuk, N., Lefebvre, M., Sarrazin, A., et al. (2020). Universal highly efficient conditional knockout system in leishmania, with a focus on untranscribed region preservation. Cell Microbiol. 22, e13159. doi: 10.1111/cmi.13159
Yang, F., Teoh, F., Tan, A. S. M., Cao, Y., Pavelka, N., Berman, J. (2019). Aneuploidy enables cross-adaptation to unrelated drugs. Mol. Biol. Evol. 36, 1768–1782. doi: 10.1093/molbev/msz104
Zhang, A., Li, N., Gong, L., Gou, X., Wang, B., Deng, X., et al. (2017). Global analysis of gene expression in response to whole-chromosome aneuploidy in hexaploid wheat. Plant Physiol. 175, 828–847. doi: 10.1104/pp.17.00819
Zhang, W., Matlashewski, G. (2015). CRISPR-Cas9-Mediated genome editing in leishmania donovani. mBio 6, e00861–e00815. doi: 10.1128/mBio.00861-15
Keywords: Leishmania, genome plasticity, replication, adaption, aneuploidy, DNA instability
Citation: Black JA, Reis-Cunha JL, Cruz AK and Tosi LRO (2023) Life in plastic, it’s fantastic! How Leishmania exploit genome instability to shape gene expression. Front. Cell. Infect. Microbiol. 13:1102462. doi: 10.3389/fcimb.2023.1102462
Received: 18 November 2022; Accepted: 05 January 2023;
Published: 26 January 2023.
Edited by:
Igor Cestari, McGill University, CanadaReviewed by:
Tamara Sternlieb, McGill University, CanadaNilmar Silvio Moretti, Federal University of São Paulo, Brazil
Copyright © 2023 Black, Reis-Cunha, Cruz and Tosi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luiz. R.O. Tosi, bHVpenRvc2lAZm1ycC51c3AuYnI=; Jennifer A. Black, SmVubmlmZXIuU3RvcnR6QGdsYXNnb3cuYWMudWs=