 Trisha Parbhoo
Trisha Parbhoo Jacoba M. Mouton
Jacoba M. Mouton Samantha L. Sampson
Samantha L. Sampson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 27 September 2022
Sec. Bacteria and Host
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.956607
This article is part of the Research Topic Mycobacterial Dormancy, Culturability, and Resuscitation: State-of-the-art, Challenges, and Future Prospects View all 10 articles
Mycobacterium tuberculosis exhibits a remarkable ability to interfere with the host antimicrobial response. The pathogen exploits elaborate strategies to cope with diverse host-induced stressors by modulating its metabolism and physiological state to prolong survival and promote persistence in host tissues. Elucidating the adaptive strategies that M. tuberculosis employs during infection to enhance persistence is crucial to understanding how varying physiological states may differentially drive disease progression for effective management of these populations. To improve our understanding of the phenotypic adaptation of M. tuberculosis, we review the adaptive strategies employed by M. tuberculosis to sense and coordinate a physiological response following exposure to various host-associated stressors. We further highlight the use of animal models that can be exploited to replicate and investigate different aspects of the human response to infection, to elucidate the impact of the host environment and bacterial adaptive strategies contributing to the recalcitrance of infection.
The interaction between a pathogen and its host is extremely complex. During infection with Mycobacterium tuberculosis, bacilli reside in various host microenvironments, which influence their physiological and metabolic state (Liu et al., 2016; Vijay et al., 2017a). As an adaptive mechanism in response to host-associated stressors, heterogeneous bacterial responses at a single-cell and population-wide level may drive the formation of phenotypically diverse subpopulations that enable survival in the host for prolonged periods. The majority of antibiotic-susceptible bacteria will succumb to host immune pressures, whilst a subpopulation of M. tuberculosis may transition into a persister state (Jain et al., 2016; Mouton et al., 2016). Persister bacteria are defined here as viable, but non- or slowly replicating, reversibly drug-tolerant subpopulations, with the potential to later resuscitate and cause active tuberculosis (TB) (Balaban et al., 2019; Moldoveanu et al., 2021). Persisters are able to survive exposure to high concentrations of antibiotics and/or host-associated stressors without known genetic mutations (Balaban et al., 2019). This is distinct from genetic resistance, which describes the ability of bacteria to grow at high concentrations of antibiotics through acquisition of resistance-conferring mutations. It is important to make the fine distinction between persister bacteria, and persistence of the infection. While the two are not mutually exclusive, in this review, the term “persistence” refers to failure to clear the infection.
M. tuberculosis has evolved to cope with diverse stressors in the host by manipulating specific host processes, in addition to modulating its metabolism to promote persister formation, enhance virulence, and ultimately prolong survival (persistence) within the host (Zimmermann et al., 2017; Huang et al., 2018; Russell et al., 2019). Improved knowledge regarding the strategies employed by M. tuberculosis to establish a persistent infection in the host, more specifically to sense and coordinate a physiological response, and trigger persister formation, is essential. The use of animal models (rabbits, guinea pigs, various mice and non-human primates) that recapitulate conditions experienced in the host, in combination with improved imaging technology is shedding light on the multifactorial mechanisms that contribute to M. tuberculosis persistence (Via et al., 2008; Lin et al., 2013; Gregg et al., 2018). Such animals display similarities in the pathophysiology and progression of infection compared to humans, however each model presents drawbacks, as highlighted in Table 1. Dependent on the animal model used, response to infection could differ, as observed in C3HeB/FeJ (Kramnik) versus BALB/c mice (Driver et al., 2012). The former model more accurately recapitulates certain aspects of human pathophysiology of infection progression where granuloma formation is observed. Further, C3HeB/FeJ mice display an increased susceptibility to M. tuberculosis, whilst the BALB/c model does not develop granuloma formation nor necrotic lesions (Table 1). However, the mycobacterial physiological state and adaptive mechanisms that promote a persister state during different stages of infection are largely unknown, and may be characterized by exploiting animal models that replicate different aspects of the human response to M. tuberculosis infection.
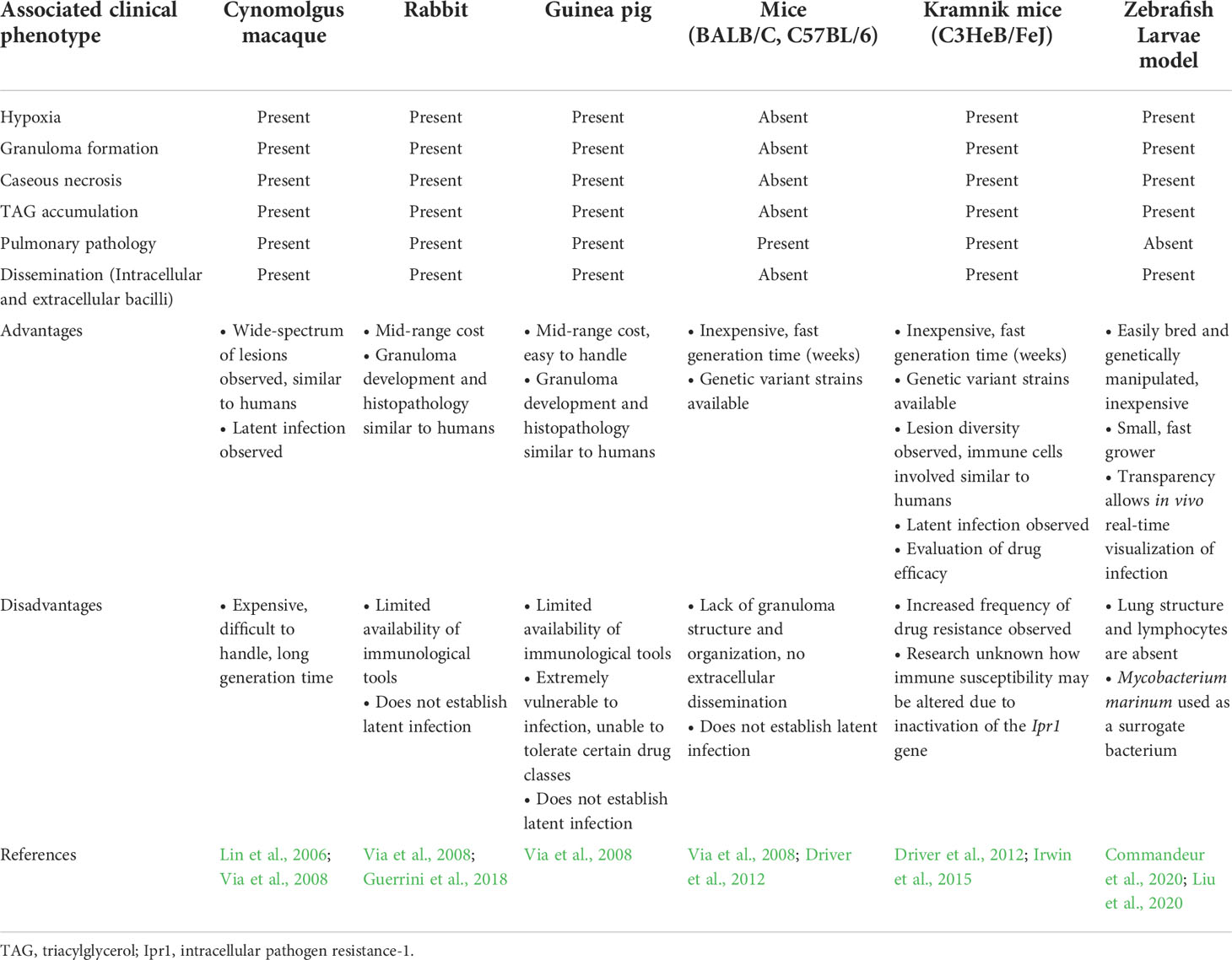
Table 1 M. tuberculosis progression of infection in animal models.
Elucidating the impact of the host environment and bacterial adaptive strategies during infection may guide future research into the recalcitrance of infection to treatment. To improve our understanding of the physiological state of M. tuberculosis persisters, this review will focus on the phenotypic adaptation of M. tuberculosis in response to various host-associated stressors and the strategies exploited by M. tuberculosis to not only survive, but to enter a persister state and ultimately drive disease progression.
Upon inhalation, M. tuberculosis bacilli are transported to lung alveoli where alveolar macrophages engulf bacilli into phagosomal compartments. Dependent on the polarization state of alveolar macrophages, bacterial replication may occur in these compartments (Refai et al., 2018; Huang et al., 2019). Alveolar macrophages respond rapidly to M. tuberculosis through the secretion of cytokines [Interleukin (IL)-12, IL-23, IL-18] to induce interferon-gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) production by natural killer and T-cells (de Martino et al., 2019). Following activation by IFN-γ and TNF-α, macrophages undergo a considerable phenotypic transformation, leading to the increased production of chemokines, cytokines and pro-angiogenic factors to assist with mycobacterial killing (Flynn and Chan, 2001). This exposes M. tuberculosis to a nutrient-limited macrophage environment, where the bacilli experience acid stress, hypoxia, and exposure to reactive oxygen and nitrogen intermediates (Manina et al., 2015). The unique metabolism and immune profile of macrophages is critical for promoting either a restrictive (M1 polarized) or permissive (M2 polarized) environment for M. tuberculosis (Marino et al., 2015; Huang et al., 2018; Pisu et al., 2020). These host environments have shown to alter M. tuberculosis growth and induce persister formation in M. tuberculosis, and will be discussed in the following sections.
A hallmark feature of M. tuberculosis infection is the formation of granulomas, organized immunological structures rich in immune cells that are recruited to the site of inflammation. Granulomas are composed of lymphocytes, neutrophils and macrophages; M. tuberculosis primarily inhabits the latter. Growth restriction and extracellular dissemination of M. tuberculosis is contained by granulomas, which acts in synergy with IFN-γ and TNF-α for structural maintenance of granulomas during early and late stages of infection (Mezouar et al., 2019). Mature macrophages may additionally undergo phenotypic alterations by fusing into multinucleated giant cells and differentiating into lipid-loaded foam cells (Sia and Rengarajan, 2019). Whilst M. tuberculosis is largely contained within the granuloma, the phagosome becomes highly vulnerable to rupture during later stages of infection, leading to cytosolic escape of M. tuberculosis (Simeone et al., 2015).
Changes in oxygen tension within the granuloma result in a shift to anaerobic metabolism whereby M. tuberculosis adapts to exploit alternative pathways and enzymes [such as regulation of the stringent response (Dutta et al., 2019), cytochrome bd oxidase, cytochrome bcc/aa3 (Mascolo and Bald, 2019), fumarate reductase, nitrate reductase, isocitrate lyase, and succinate] (Supplementary Table 1), which are functional at lower metabolic rates (Salina et al., 2014). These pathways are however regarded as energetically demanding, implying that alternative metabolites are being utilized for the maintenance of the proton motive force (PMF), adenosine triphosphate (ATP) and cofactor recycling (Prosser et al., 2017).
M. tuberculosis shifts its metabolism to the glyoxylate shunt when β-oxidation of fatty acids provides the main carbon and energy source (Hoff et al., 2011). Additionally, the presence of the non-proton-translocating electron donor, NADH dehydrogenase 2, along with nitrate reduction (NAD+ recycling) (Eoh and Rhee, 2013; Prosser et al., 2017) plays a key role in maintaining an energized membrane, ATP production and carbon precursors during hypoxia and nutrient limitation. These cofactors may additionally serve as a reserve of metabolic energy that can facilitate the extracellular spread of M. tuberculosis (Tan et al., 2010). The thick cell wall of M. tuberculosis additionally offers protection from reactive oxygen species (ROS) by scavenging oxygen radicals (Ehrt and Schnappinger, 2009). Remodeling of bacterial respiration additionally mitigates intracellular redox stress, as excessive accumulation of NADH/NADPH enhances ROS via the Fenton reaction, which can induce intracellular damage to bacterial proteins and DNA (Vilchèze et al., 2013; Vilchèze et al., 2017).
Further research to identify host factors manipulated by M. tuberculosis will enhance our understanding of how M. tuberculosis modulates the antimicrobial host response. The host-pathogen interaction may be crucial for determining the fate of infection, and whether specific pathways are manipulated for eradication of bacilli, containment and persistence, or bacterial dissemination.
Phagocytosis of M. tuberculosis by macrophages plays a crucial role in pathogen detection, engulfment and immune regulation. Depending on the host cell receptors engaged (e.g. antigen- and pattern recognition receptors), specific microbial ligand sequences will be recognized, which could alter the initial interaction between host and pathogen, and subsequent intracellular processing (Kumar et al., 2019). Following internalization into the phagosome, M. tuberculosis exhibits a remarkable ability to interfere with the host antimicrobial response and inhibit phagosome-lysosome fusion (Figure 1, Supplementary Table 1).
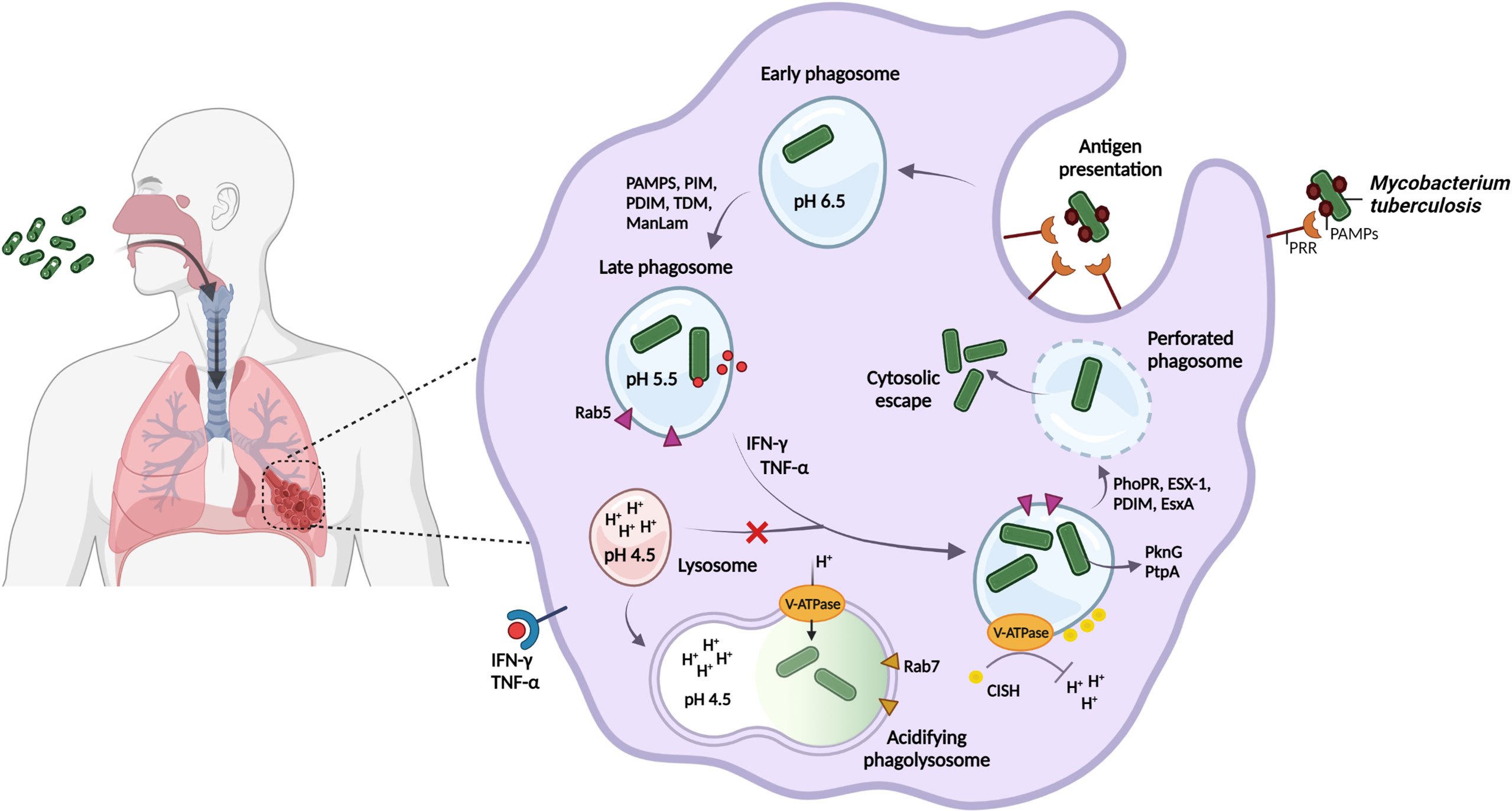
Figure 1 M. tuberculosis regulates phagosomal pH and blocks phagolysosome formation to avoid degradation by host acidic stressors and promote persister formation. M. tuberculosis is spread through the inhalation of aerosolized droplets that are released into the air when an infected individual coughs or sneezes. Host recognition of M. tuberculosis-specific PAMPs results in the internalization into the phagosome of alveolar macrophages for bacterial containment. M. tuberculosis may however interfere with host cellular processing and the host immune response by retaining its interaction with the host early phagosomal marker, Rab5, and by selectively engaging host receptors, as a strategy to enhance intracellular survival in the phagosome. Following host immune activation by IFN-ƴ and TNF-α, phagosomal acidification to pH 4.5 is proposed. M. tuberculosis inhibits phagolysosomal fusion and regulates the phagosomal pH by the V-ATPase proton pump to resist degradation by the acidic environment. M. tuberculosis recruits the host CISH protein to degrade V-ATPase for neutralization of the vacuole. Finally, ESX-1 and the mycobacterial cell envelope lipid PDIM, act in synergy to perforate the phagosome, which promotes cytosolic displacement of bacteria, and is a crucial step for intracellular persistence. Created with BioRender.com.
M. tuberculosis possesses a diverse array of cell-surface ligands and attempts to manipulate host cell recognition and internalization through pathogen-associated molecular patterns (PAMPs, Figure 1), including trehalose-6,6-dimycolate (TDM, cord factor) (Patin et al., 2017; Lerner et al., 2020), phosphatidylinositol mannoside (PIM) and mannose-capped lipoarabinomannan (ManLam) (Torrelles and Schlesinger, 2010; Marakalala and Ndlovu, 2017; Queval et al., 2017a). Phagocytosis of beads coated with ManLam (Fratti et al., 2003; Kang et al., 2005; Shui et al., 2011) and TDM (Geisel et al., 2005; Axelrod et al., 2008) has been shown to interfere with phagosome maturation and promote granuloma formation, respectively.
TDM is a significant contributor to intracellular cording of M. tuberculosis, whereby large lipid pellicles aggregate on the cell surface, preventing cytosolic recognition and phagocytosis into space-constrained vacuoles (Lerner et al., 2020). In contrast, dead bacilli are rapidly transported to lysosomes for degradation (Malik et al., 2000; Kyei et al., 2006). Since host immune cells express varying combinations of receptors, their interaction with PAMPS may differ (Liu et al., 2017; Dubé et al., 2021). M. tuberculosis may preferentially target specific host cell receptors to manipulate the host immune response, however, the mechanism of how manipulation by PAMPs influences macrophage metabolism, the delay of phagosome-lysosome fusion and subsequent intracellular processing, remains to be explored.
Phagosome acidification represents a dynamic antimicrobial defense strategy used by the host, and despite M. tuberculosis being sensitive to high acidity (low pH), the pathogen appears to resist being killed in macrophages by maintaining its intra-bacterial pH (Vandal et al., 2008; Levitte et al., 2016). Several mechanisms that assist in neutralizing phagosomal pH have been proposed (Figure 1), including membrane proteins, proton pumps, ammonia production, amino acid decarboxylation and cell wall modifications (Vandal et al., 2008; Singh et al., 2019).
During phagosomal maturation, highly conserved vacuolar-type ATPase (V-ATPase) proton pumps are rapidly recruited to the phagosomal membrane to promote acidification. M. tuberculosis has however developed the ability to target and trigger degradation of the V-ATPase complex by inducing the expression of the host cytokine-inducible SH2-containing protein (CISH) (Queval et al., 2017b). This allows the phagosomal pH to remain above that required for the activity of lysosomal digestive enzymes and ROS (Pauwels et al., 2017; Baker et al., 2019).
The ability of M. tuberculosis to rapidly sense and respond to the external pH is crucial for pathogenesis, and involves strong induction of the PhoPR two-component regulatory system in vitro and in macrophages (Abramovitch et al., 2011). PhoPR is further required for intracellular pathogenesis and redox homeostasis, such as induction of the type VII secretion system ESX-1, regulation of heat shock-responsive genes (Sevalkar et al., 2019), and lipid biosynthesis regulated by the cytoplasmic redox sensor, WhiB3 (Johnson et al., 2015; Mehta et al., 2016; Feng et al., 2018) (Figures 1, 2).
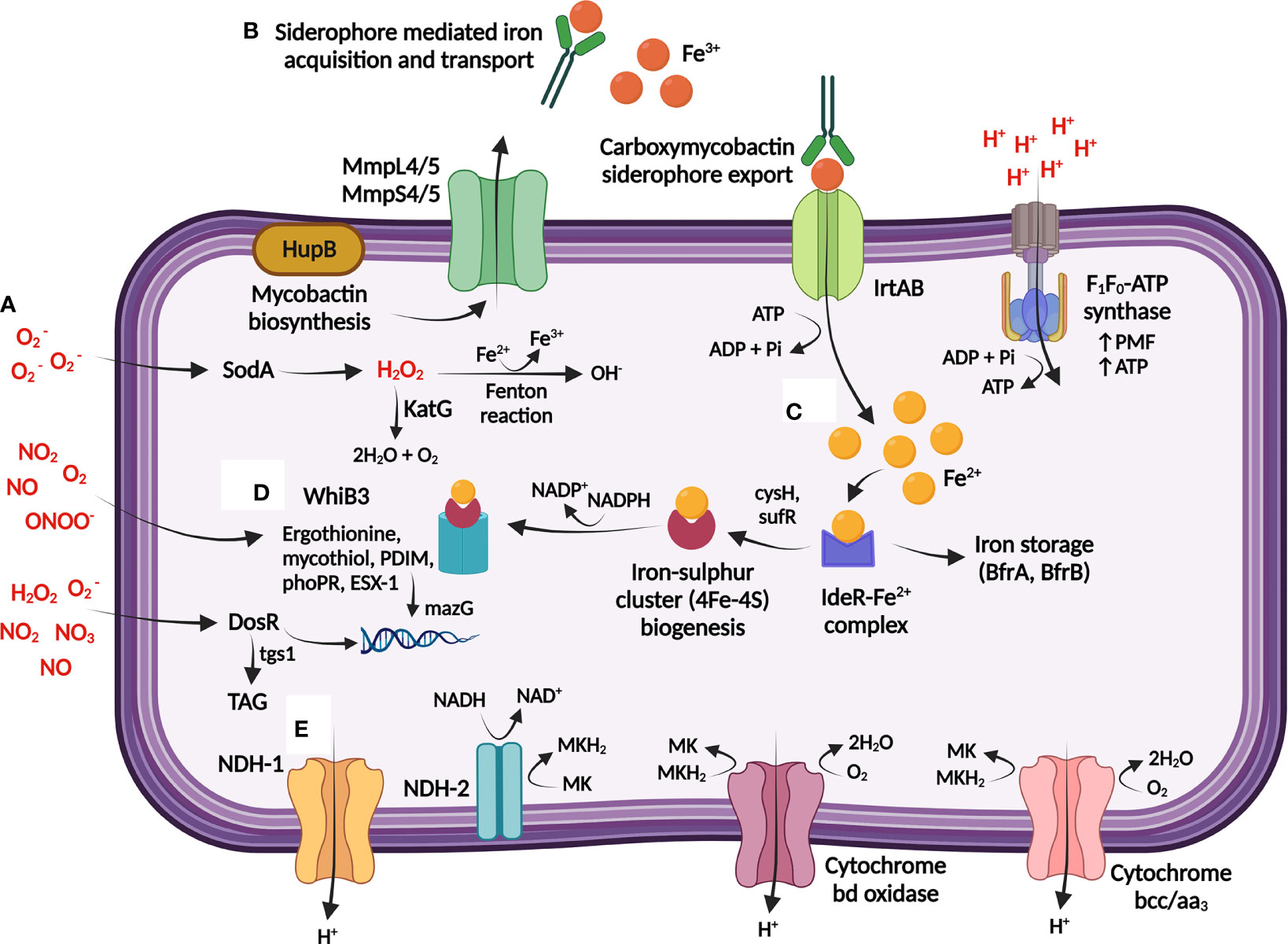
Figure 2 M. tuberculosis maintains bioenergetic metabolism to ensure redox homeostasis during persistence. (A) The host generates toxic reactive oxygen species (ROS) and reactive nitrogen species (RNS) [shown in red] through single-electron reactions, which can result in damage to M. tuberculosis DNA and proteins. (B) Redox homeostasis is maintained through the sensing and regulation of iron, obtained from the host via Fe3+-specific iron scavenging siderophores that is imported into M. tuberculosis by the ABC exporter, IrtAB. (C) Iron is either stored or utilized directly for generation of iron sulphur clusters (4Fe-4S), and further encoded to 4Fe-4S redox sensor proteins by WhiB3. (D) WhiB3 produces detoxification buffers (ergothionine and mycothiol) and in combination with DosR directs the reducing equivalents produced during β-oxidation of fatty acids to produce cell wall lipids and protect DNA from oxidation. DosR further regulates lipid storage within M. tuberculosis through the regulation of TAG, providing a lipid rich nutrient source. (E) Proton balance during redox homeostasis is maintained through regulation of internal protons which can be utilised for the production of menaquinol (MKH2) [via NDH-1/NDH-2], which is subsequently transferred to the respiratory complexes (cytochrome bd and cytochrome bcc/aa3) to maintain the PMF and ATP production via F1F0-ATP synthase. TAG: triacylglycerol; MK: Menaquinone; NDH: NADH dehydrogenase; Fe3+: ferric ion; Fe2+: ferrous ion; : superoxide; NO: nitric oxide; NO2: nitrogen dioxide; ONOO-: peroxynitrite; H2O2: hydrogen peroxide; MmpL: mycobacterial membrane protein large; MmpS: mycobacterial membrane protein small. Created with BioRender.com.
Transport of the ESX-1-dependent 6-kDa early secretory antigenic target (ESAT-6/EsxA) to the host cytosol (Simeone et al., 2015; Conrad et al., 2017) acts synergistically with the mycobacterial cell envelope lipid phthiocerol dimycocerosate (PDIM) in perforating the phagosome (Augenstreich et al., 2017; Barczak et al., 2017; Quigley et al., 2017; Lerner et al., 2020) and preventing recruitment of V-ATPase, thereby neutralizing the vacuole (Astarie-Dequeker et al., 2009; Passemar et al., 2014). Further membrane damage by continued perforation or complete rupture of the phagosome will allow translocation of the bacilli to the host cytosol (Jamwal et al., 2016; Schnettger et al., 2017) (Figure 1). This is crucial for intracellular persistence, as PDIM or esxA mutants failed to perforate the phagosomal membrane, possessed altered PAMPs, and were attenuated in human monocyte-derived macrophages (hMDMs) (Augenstreich et al., 2017). This suggests that remodeling of the cell envelope and central metabolism pathways may improve intracellular survival of persisters during acid stress by controlling the flux of lipid precursors and reducing equivalents (Baker and Abramovitch, 2018; Singh et al., 2019).
Host immune activation of macrophages stimulates multiple antimicrobial defense pathways, exposing M. tuberculosis to an environment where hydrolases, reactive nitrogen species (RNS), and ROS effectively function (Ehrt et al., 2001; Jamaati et al., 2017). M. tuberculosis effectively resists degradation by these reactive molecules by activating detoxification and redox buffering mechanisms to maintain bioenergetic homeostasis (Saini et al., 2016) (Figure 2; Supplementary Table 1).
Exposure to toxic nitric oxides slows bacterial growth by competing for oxygen binding, and in the process, reversibly inhibits cytochrome c oxidase, and thus aerobic respiration (Voskuil et al., 2003). M. tuberculosis metabolically adapts to acquire nitrate via oxidation of nitric oxide, thereby restoring survival due to maintenance of the PMF and ATP (Sohaskey, 2008; Tan et al., 2010). Induction of cytochrome bd by M. tuberculosis additionally plays a respiratory protective role against ROS/RNS by scavenging oxygen radicals upon breakdown of oxidative species (Small et al., 2013; Boot et al., 2017).
Host maintenance of the oxidative balance in the lung is controlled via antioxidant mechanisms, and M. tuberculosis utilizes the ROS scavenging enzymes, superoxide dismutase (SOD) and catalase-peroxidase (KatG) to degrade superoxide to water and molecular oxygen to neutralize the free radicals generated in the host (Kumar et al., 2011) (Figure 2). Subsequent downregulation of ATP synthase genes may further slow bacterial growth by restricting proton translocation into the cytoplasm, thereby replenishing pools of oxidized cofactors to maintain redox and pH homeostasis (Baker et al., 2014; Baker et al., 2019). These metabolic adaptations prevent an excess of RNS/ROS, which may trigger damage to proteins, lipids and nucleic acids.
The pyrimidine-specific housekeeping enzyme, MazG, prevents DNA mutagenesis by specifically degrading and preventing the incorporation of oxidized deoxynucleotides into genomic DNA (Lyu et al., 2013; Shi et al., 2019) (Figure 2). Deletion of mazG decreases the NADH/NAD+ redox balance towards an oxidizing state, resulting in DNA instability, disruption to pyrimidine metabolism, and hindrance to iron and carbon uptake in vitro (Shi et al., 2019) and in mice (Lyu et al., 2013).
Free intracellular iron within M. tuberculosis catalyses ROS formation, thus homeostasis of iron is maintained by M. tuberculosis in a tightly regulated manner via the iron-dependent regulator (IdeR) (Rodriguez et al., 2002; Pandey and Rodriguez, 2014). Mycobactin and cysteine biosynthesis is maintained by M. tuberculosis to replace iron (Rodriguez et al., 2002) and sulphur (Song and Niederweis, 2012), respectively, for reconstruction of the damaged iron-sulphur clusters (Figure 2). This is essential for persister adaptation as iron-sulphur clusters are highly susceptible to oxidative and nitrosative stress as they undergo various oxidation-reduction reactions (Pandey et al., 2018). Paradoxically, iron uptake and cysteine synthesis promote DNA damage by forming reactive and damaging hydroxyl radicals via the Fenton reaction (Figure 2). However, the requirement of these proteins to repair damage appears to outweigh the negative effects of intermediate hydrogen peroxide and nitric oxide levels (Ford et al., 2011; Voskuil et al., 2011; Lyu et al., 2013).
Protection against ROS (Bhaskar et al., 2014) and nutrient starvation (Richard-Greenblatt et al., 2015; Saini et al., 2016) respectively involves WhiB3-dependent upregulation of the M. tuberculosis redox buffers, mycothiol and ergothioneine (Figure 2; Supplementary Table 1). These low molecular weight buffers maintain redox and bioenergetic homeostasis, including consumption of excess NADH/NADPH (Singh et al., 2009; Saini et al., 2016; Mehta and Singh, 2019). Lipid anabolism may therefore counteract reductive stress by oxidizing NADH/NADPH, as phoP mutants displayed a structurally altered cell envelope consisting of limited methyl branched fatty acids and diminished acid-fast staining (Walters et al., 2006).
As granulomas mature, gradual accumulation of necrotic cellular debris from lysed or damaged host and bacterial cells forms a caseous core (Hoff et al., 2011). Enlargement of the caseum during advanced stages of active TB disease compresses the adjacent lung tissue, damaging the vasculature in the process, and distributing extracellular M. tuberculosis to the caseous center (Blanc et al., 2018; Dartois, 2014). The reduced vascularization additionally decreases oxygen availability (Datta et al., 2016), and in response, M. tuberculosis undergoes rapid and substantial metabolic and phenotypic adaptations (Qualls and Murray, 2016; Sershen et al., 2016) (Figure 3; Supplementary Table 1).
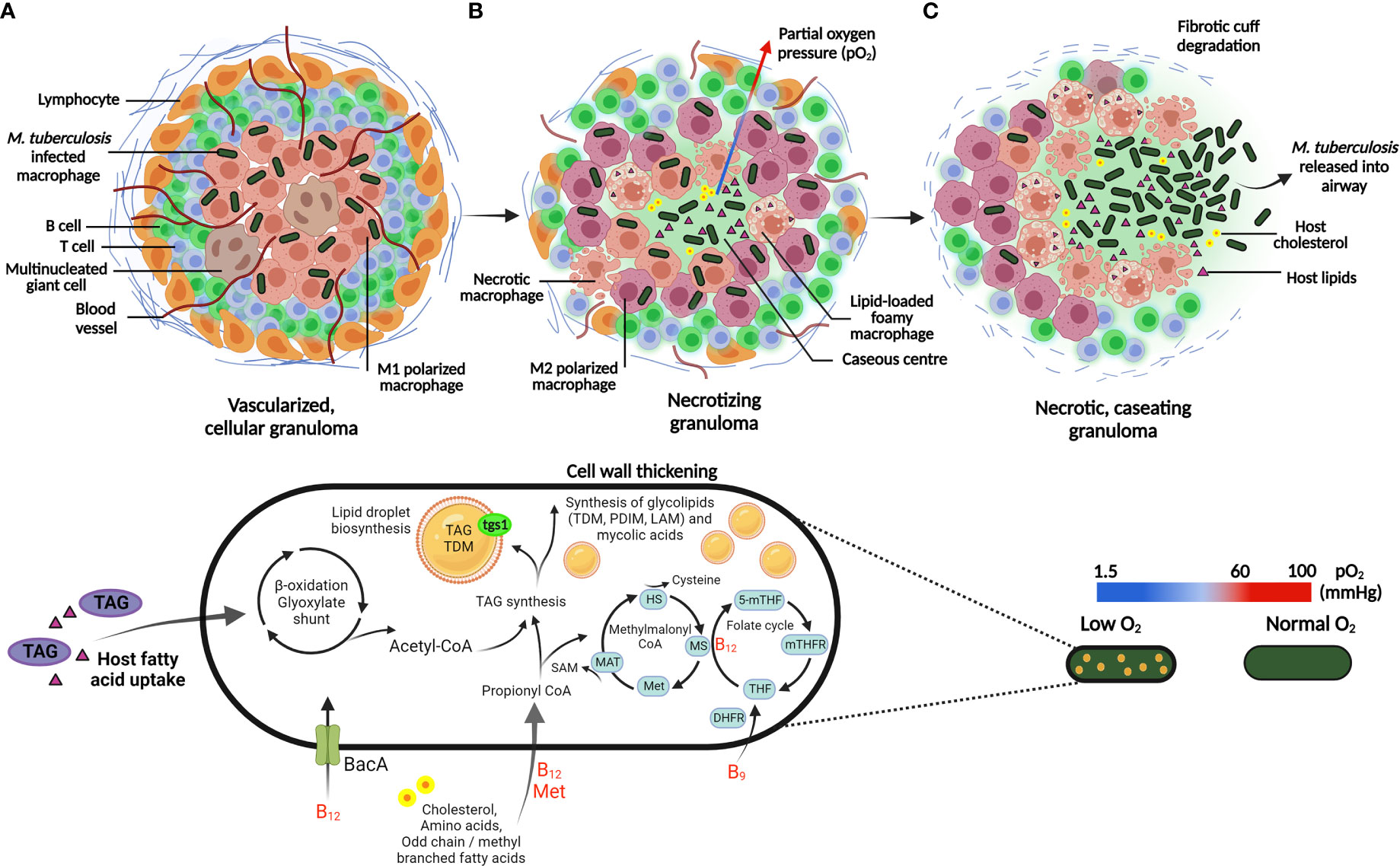
Figure 3 Adaptation of M. tuberculosis to the host granuloma environment and associated stressors. (A) M. tuberculosis is phagocytosed by alveolar macrophages and contained within the center of the granuloma, which is an organized aggregate of immune cells recruited to the site of inflammation. The pro-inflammatory cytokines, IFN-y and TNF-α activate macrophages (M1 polarized; pink macrophages), which is essential for the control of infection. Following host immune activation, recruitment of lymphocytes, largely comprising of T and B cells, surround the periphery of the granuloma. (B) Mature granulomas develop heterogeneous vasculature, displaying a gradient of pro-inflammatory host cells (M1 and M2 polarized; pink and purple macrophages, respectively) and a decreasing oxygen gradient toward the caseum. In response to hypoxia, M. tuberculosis exploit lipid-loaded foamy macrophages to store host TAG during persistence. Host fatty acids and cholesterol are metabolized by the β-oxidation and methylmalonyl CoA pathway respectively, which generates a nutritional carbon source that can be stored in lipid droplets or utilized to generate mycobacterial cell wall components. Essential vitamins utilized in the respective metabolic pathways (shown in red) are acquired by M. tuberculosis from the host environment via the ABC-transporter, BacA. (C) Necrotic macrophages contributes to an accumulation of lipid debris, resulting in central caseation in mature granulomas. Eventual rupture of the granuloma leads to extracellular dissemination of M. tuberculosis. HS: homocysteine; MS: methionine synthase; Met: methionine; MAT: methionine adenosyltransferase; 5-mTHF: 5-methyltetrahydrofolate; mTHFR: methylenetetrahydrofolate reductase; THF: tetrahydrofolate; DHFR: dihydrofolate reductase. Created with BioRender.com.
Foamy macrophages in necrotic regions accumulate triacylglycerol (TAG) within lipid droplets, a process exploited by M. tuberculosis to store host TAG intracellularly, providing a lipid-rich microenvironment for the persistence of M. tuberculosis during hypoxia (Guerrini et al., 2018; Del Portillo et al., 2019). Additionally, M. tuberculosis expression of triacylglycerol synthase (tgs1) induces lipid body formation in persisters, whereby lipid body-positive M. tuberculosis has been detected in sputum prior to the onset of treatment (Figure 3). The respiratory state of these bacilli displayed a shift from oxygen electron transfer to anaerobic respiration, thus challenging the belief that all bacilli in sputum respire aerobically (Garton et al., 2008).
ATP production in aerobic and anaerobic conditions is responsible for generating the membrane potential and proton concentration gradient that drives the PMF (Rao et al., 2008; Reichlen et al., 2017). Inhibitors of the membrane potential and proton concentration gradient are cidal towards persisters and lead to a loss in viability, suggesting that the cytoplasmic membrane of non-replicating hypoxic M. tuberculosis, induced in the Wayne model, is energized (Rao et al., 2008). The mechanisms involved in the maintenance of ATP suggest this process as essential for the survival of non-replicating M. tuberculosis under hypoxic conditions.
Emerging evidence suggests that the active depletion of nutrients by the host immune system may create a nutrient-scarce environment within the phagosome (Appelberg, 2006; Eisenreich et al., 2013). Restricted access to transition metals (Kehl-Fie and Skaar, 2010; Hood and Skaar, 2012), carbon (Pandey and Sassetti, 2008; MacMicking, 2014), and amino acids (Zhang et al., 2013; MacMicking, 2014) drives the downregulation of key metabolism and replication machinery, possibly leading to auxotrophy in M. tuberculosis.
Auxotrophy is described as the inability of an organism to synthesize a specific metabolite required for its growth. M. tuberculosis auxotrophs for methionine (Berney et al., 2015), threonine (Hasenoehrl et al., 2019), lysine (Pavelka et al., 2003), leucine (Hondalus et al., 2000), and arginine (Tiwari et al., 2018) are severely attenuated in vivo due to their inability to scavenge these metabolites from the host. The M. tuberculosis stringent response pathway RelMtb, initiates production of the alarmones (p)ppGpp during amino acid deprivation, hypoxia and oxidative stress (Dutta et al., 2019). (p)ppGpp assists in enhancing survival during nutrient starvation in mice (Dahl et al., 2003) and guinea pigs (Klinkenberg et al., 2010; Thayil et al., 2011; Dutta et al., 2019) by inhibiting RNA synthesis for conservation of energy. Therefore, M. tuberculosis is incapable of entering persistence upon disruption to its stringent response (Dutta et al., 2019).
Certain metabolic processes are energetically expensive; M. tuberculosis has however developed elaborate strategies to synthesize or sequester essential host nutrients to satisfy its bioenergetic and biosynthetic requirements (Eisenreich et al., 2013). M. tuberculosis additionally exploits specific nutrients or metabolic processes that facilitate adaptation to persistence (Supplementary Table 1, Supplementary Table 2).
Amino acids represent a critical source of nutrients used to fuel central metabolic pathways, and many pathogens are required to scavenge them in the host. The host compartments either restrict metabolite availability, or M. tuberculosis attempts to evade host detection by remaining metabolically independent from the host (Berney et al., 2015). M. tuberculosis has evolved to synthesize all 20 proteinogenic amino acids (Cole et al., 1998), of which certain amino acids are rapidly metabolized as carbon, sulphur, nitrogen or energy sources, whilst others are stored. Amino acids may additionally function as both carbon and nitrogen sources, indicating that the pathways required for their degradation and assimilation could differ (Lofthouse et al., 2013). M. tuberculosis furthermore exploits amino acids for their ability to provide cofactors for generating intermediate molecules or proteins, which are integral components for metabolic adaptation (Supplementary Table 2).
Amino acids acquired from the macrophage, such as alanine, glutamate and asparagine/aspartate contribute to the intracellular nutrition of M. tuberculosis (Beste et al., 2013). Alanine is an essential structural component of peptidoglycan (Beste et al., 2013); cell wall homeostasis is maintained by M. tuberculosis during persistence by conversion of L-alanine to D-alanine, whilst an impaired conversion severely restricts intracellular growth in bone marrow-derived macrophages (BMDMs) and mice (Awasthy et al., 2012). During persistence, regulation of the aspartate pathway is involved in essential cell wall processes, such as biosynthesis of the cell wall cofactor, S-adenosylmethionine (SAM), generation of the essential amino acids methionine, isoleucine, threonine and lysine, and the precursor metabolite homoserine (Hasenoehrl et al., 2019). The majority of the methionine pool is converted to SAM via the aspartate pathway (Berney et al., 2015; Hasenoehrl et al., 2019), therefore it is suggested that metabolic regulation of the aspartate pathway is essential for M. tuberculosis persistence.
M. tuberculosis preferentially utilizes carbon-based metabolism during persistence. C-flux metabolism analysis indicates the adaptation of intracellular M. tuberculosis to simultaneously co-catabolize multiple carbon sources, including amino acids, carbon dioxide, vitamins and fatty acids; the latter derived from host lipids and cholesterol (de Carvalho et al., 2010; Beste et al., 2013; Noy et al., 2016; Borah et al., 2021). M. tuberculosis possesses the unusual ability to metabolize host fatty acids as substrates for β-oxidation. This yields substrates required for production of mycolic acids, or enables assimilation directly into TAG and phospholipids for maintenance of the cytoplasmic membrane integrity (Pandey and Sassetti, 2008) (Figure 2).
Lipid storage becomes the primary energy-conserving metabolic process in M. tuberculosis persisters, whereby intracellular accumulation of TAG has been observed (Russell et al., 2009; Daniel et al., 2011; Maurya et al., 2018). M. tuberculosis persisters may rapidly accumulate host lipids to form lipid bodies in resting macrophages, whilst access to lipid bodies is limited once macrophages are IFN-ƴ activated due to production of host protective eicosanoids (Knight et al., 2018). Instead, M. tuberculosis accumulates lipids from host macrophage lipid bodies stimulated by extracellular free fatty acids or by hypoxia (Daniel et al., 2011; Knight et al., 2018). Observed within necrotic lesions, lipid-loaded foamy macrophages further represent a lipid-rich niche whereby host cholesterol serves as a carbon source for persisters (Pandey and Sassetti, 2008; Russell et al., 2009; Singh et al., 2012) (Figure 3).
β-oxidation of fatty acids accumulated by M. tuberculosis, generates a pool of coenzyme-A carriers and carbon units; even-chain fatty acids are degraded to acetyl coenzyme A (acetyl-CoA) via oxidation by the glyoxylate shunt to replenish central metabolites, whilst odd-chain fatty acids are degraded to propionyl-CoA (Warner et al., 2007; Savvi et al., 2008). Detoxification of the latter via the methylmalonyl-CoA and methyl citrate cycle is a vitamin B12-dependent and B12-independent mechanism, respectively (Figure 3). Additionally, MetH and MetE involved in methionine biosynthesis encode vitamin B12-dependent and B12-independent methionine synthase, respectively (Warner et al., 2007; Savvi et al., 2008). This indicates the essentiality of methionine biosynthesis, as M. tuberculosis may need to rapidly detect vitamin B12 bioavailability for prevention of toxic accumulation of propionyl-CoA (Figure 3). It is proposed that M. tuberculosis scavenges vitamin B12 from the host via the ATP-binding cassette (ABC) transporter, BacA (Gopinath et al., 2013). Deletion of bacA is speculated to contribute to the accumulation of cholesterol-rich lipid bodies (Gopinath et al., 2013), and since cholesterol provides a source of propionyl-CoA, efficient assimilation of propionyl-CoA is required for effective cholesterol metabolism (Supplementary Table 2).
An imbalance in vitamin B12 and methionine synthase is often associated with disruption to folate (Vitamin B9) metabolism, as methionine synthase utilizes 5-methyltetrahydrofolate (5-MTHF) as a one-carbon (methyl group) donor to catalyze the methylation of homocysteine to methionine (Nixon et al., 2014). This is essential for production of purines, thymidine, methionine, glycine, serine, homocysteine and SAM. Inhibitors of folate synthesis exhibit distinct metabolic disruptions to methionine and SAM in vitro, with an upregulation of genes associated with DNA repair (Nixon et al., 2014). Disruption to SAM may be attributed to imbalances in DNA methylation, thereby introducing single nucleotide polymorphisms (SNPs) and DNA damage. This subsequently starved cells of crucial reduced folate precursors critical for the synthesis of DNA, RNA and protein (Chakraborty et al., 2013; Zheng et al., 2013; Minato et al., 2015). It has been suggested that methionine-mediated antagonism of anti-folate drugs may enhance methylation by increasing SAM abundance (Howe et al., 2018). This could be an adaptive process; however, the antagonistic mechanism has yet to be characterized. This further confirms that metabolic remodeling may offer an advantageous adaptation mechanism for conserving carbon and energy sources for prolonged persistence and possibly resuscitation.
Persistence of M. tuberculosis during iron starvation is controlled by strict regulation of iron utilization and storage via the iron-dependent regulator IdeR; following acquisition, iron not immediately utilized is stored in the host as protein-bound iron in the form of ferritin to prevent the generation of toxic free radicals via the Fenton reaction (Pandey and Rodriguez, 2014) (Figure 2). Key cellular processes such as electron transfer, DNA replication and repair, and regulation of gene expression in response to ROS/RNS are dependent on iron and iron-sulphur clusters as a cofactor (Rodriguez and Smith, 2003). Iron is however not only restricted within granulomas, but the abundance of free iron is scarce due to its low solubility (Kurthkoti et al., 2017).
M. tuberculosis strongly promotes sequestration of iron following macrophage uptake (Abreu et al., 2018), and in necrotic and cavitary granulomas, where M. tuberculosis synthesizes and secretes high-affinity Fe3+-specific siderophores (mycobactins) to actively compete for host iron (Kurthkoti et al., 2017) (Figure 2, Supplementary Table 2). This process is further dependent on the ESX-3 secretion system, as M. tuberculosis lacking the ESX-3 secretion system are defective in acquisition of bound iron from siderophores, and display severely impaired growth in THP-1 macrophages (Serafini et al., 2013), murine macrophages (Siegrist et al., 2009), and human granulomas (Kurthkoti et al., 2017).
Upon iron starvation, the M. tuberculosis heme enzyme KatG is downregulated, therefore reduced bioactivation of isoniazid (INH) likely mediates enhanced tolerance, contributing to antibiotic-tolerant persister formation (Kurthkoti et al., 2017). Metabolic reprogramming of iron-starved bacilli displayed repression in the majority of enzyme-encoding genes from the citric acid cycle (TCA) and oxidative phosphorylation, since these pathways require iron-sulphur clusters or heme proteins (Kurthkoti et al., 2017). DNA repair genes, including iron-sulphur cluster repair proteins were upregulated, poising bacteria to respond to oxidative damage (Tyagi et al., 2015). Additionally, biosynthesis of most amino acids decreased, with exception to lysine (Kurthkoti et al., 2017), which is the backbone of siderophore biosynthesis (McMahon et al., 2012).
Sulphur assimilation pathways in M. tuberculosis produce reduced sulphur-containing metabolites, such as cysteine and methionine, and have been shown to play a critical role in facilitating bacterial adaptation and protection during persistence (Rhee et al., 2005; Senaratne et al., 2006), and upon exposure to oxidative stress and hypoxia (Pinto et al., 2004; Voskuil et al., 2011). Controlled by cys genes, sulphur is required to maintain redox reactions through iron-sulphur clusters, translation initiation, methylation of DNA and RNA, biotin and menaquinone synthesis, and mycolic acid modification by SAM-dependent methyltransferases (Hasenoehrl et al., 2019).
Disruption in CysH renders M. tuberculosis unable to utilize inorganic sulphate for the synthesis of the cytosolic reducing buffer, mycothiol (Senaratne et al., 2006), which serves in detoxification of bactericidal agents and host oxidative damage (Buchmeier et al., 2003; Senaratne et al., 2006; Mishra et al., 2019). Perturbation in mycothiol redox homeostasis elevated endogenous ROS, inducing a long-lasting and irreversible oxidative shift in M. tuberculosis (Tyagi et al., 2015). Adaptation may have favored the release of secretory antioxidants in the phagosome to neutralize exogenous oxidative stress as a defense mechanism, playing a role in facilitating persistence (Tyagi et al., 2015).
To further neutralize host-mediated oxidative stress, M. tuberculosis overexpresses cysteine desulfurase to repair oxidatively damaged iron-sulphur clusters (Ayala-Castro et al., 2008; Tyagi et al., 2015). Accordingly, sufR enabled persistence in the host by maintaining iron homeostasis, and downregulating genes responsible for iron-sulphur cluster biogenesis (Pandey et al., 2018).
M. tuberculosis appears to not require tight control of its nitrogen sources, as it has evolved the ability to co-metabolize a variety of alternate amino acids as nitrogen sources during infection (Agapova et al., 2019; Borah et al., 2019). It has been established that M. tuberculosis utilizes asparagine and glutamate as sole nitrogen sources during persistence (Song et al., 2011; Song and Niederweis, 2012). Originally described as a porin-forming protein, OmpATb (Song et al., 2011), and the asparaginase transporter AnsA (Gouzy et al., 2014) rapidly mediates ammonia secretion from these amino acids, thus neutralizing the acidic phagosomal pH. This may be an adaptive mechanism for pH homeostasis and indicates that asparaginase has evolved two independent key functions, to conduct nitrogen metabolism and counteract the host defense acidification (Figure 2, Supplementary Table 2).
To circumvent nitric oxide toxicity in macrophages, M. tuberculosis acquires nitrate by oxidation of nitric oxide. Upon persister formation, an increase in nitrate reductase occurs, indicating that M. tuberculosis utilizes nitrate as an alternative electron acceptor during anaerobic respiration since reduction in electron flow associated with decreasing oxygen levels results in an increased concentration of reduced cofactors, such as NADH (Sohaskey, 2008; Williams et al., 2015). Furthermore, PhoP and DosR regulates expression of nitrite and nitrate reductases for persistence during hypoxia (Singh et al., 2020).
Potassium (K+) is a crucial cation required for maintenance of an electrochemical gradient and PMF, and regulation of intracellular pH and osmotic pressure. During phagosome maturation, a change in host ions and K+ uptake occurs, thus M. tuberculosis tightly controls regulation of its ionic signals via the Trk and Kdp K+ uptake systems. Disruption of K+ homeostasis results in the inability of M. tuberculosis to respond to low pH and high chloride levels in BMDMs (MacGilvary et al., 2019), and has been shown to negatively affect M. tuberculosis growth rate, as determined by uracil incorporation as a measure of transcriptional activity (Salina et al., 2014).
Interestingly, exposure to low environmental K+ repressed iron uptake in M. tuberculosis, subsequently exposing bacilli to oxidative stress (MacGilvary et al., 2019). Following K+ deficiency, M. tuberculosis persisters maintained a stable, yet low abundance of transcripts coding for biosynthetic enzymes or proteins involved in adaptation, repair, and management of transcription initiation. Once reintroduced into K+ supplemented media, these mRNA transcripts likely assisted in rapid translation following resuscitation (Ignatov et al., 2015).
Phosphorous is essential for the synthesis of nucleotides, DNA, RNA and phospholipids, and is acquired from the host via the phosphate-specific transporter (Pst) to form phosphate compounds through interaction with the two-component regulatory system SenX3-RegX3 (Rifat et al., 2009; Tischler et al., 2013; Namugenyi et al., 2017). SenX3-RegX3 likely plays a role in virulence during persistence as deletion of pstA1 constitutively activates RegX3, regardless of phosphate availability, and subsequently triggers the ESX-5 secretion system, resulting in hypersecretion of ESX-5 substrates in Irgm1-deficient mice (Elliott et al., 2019). While the ESX-5 system is required for virulence, tight control in secretion of ESX-5 substrates may limit recognition by the host immune system or prevent secretion of harmful cytokines (Elliott et al., 2019). This may represent a defense mechanism to evade the host adaptive immune response, however it is unknown whether ESX-5 substrates directly assist in phosphate scavenging or whether constitutive secretion of specific ESX-5 antigens may explain the impaired intracellular survival of M. tuberculosis.
During nutrient starvation, polyphosphate (polyP) accumulation provides a reservoir of energy and a phosphate donor, and may be the preferential energy store since decreased ATP levels were observed during most stress conditions in guinea pigs (Singh et al., 2013). This was associated with down-regulation of reductive TCA cycle intermediates (succinate, fumarate, malate), and up-regulation of oxidative TCA intermediates, as indicated using 13C isotope labelling (Dutta et al., 2019). Additionally, regulation of polyP is dependent on (p)ppGpp levels, as disruption to the stringent response pathway contributed to enhanced antibiotic susceptibility and defective bacterial growth in guinea pig lungs (Singh et al., 2013; Chuang et al., 2015). This suggests that tight regulation of RelMtb and polyP homeostasis is critical for M. tuberculosis persister formation following exposure to host-induced stressors and/or antibiotics.
Granulomas develop a spectrum of lesion types, driven by variable cytokine profiles and lesion histopathology (Lin et al., 2013; Martin et al., 2017). Importantly, previous work has shown that granuloma lesions harbor physiologically distinct bacterial populations with varying rates of replication and different metabolic states (Lenaerts et al., 2007; Hoff et al., 2011). Since heterogeneity within lung lesion types influences the bacterial phenotype, this could provide essential information about the microenvironment and how this impacts the bacterial physiology to favor and induce persistence (Dartois, 2014; Irwin et al., 2015; Gregg et al., 2018).
M. tuberculosis may exploit the host response by tweaking the inflammatory balance within granulomas. Detailed immunohistochemical analysis of granulomatous lesions from M. tuberculosis-infected cynomolgus macaques demonstrated a pro-inflammatory phenotype to be localized to the center of the granuloma (Mattila et al., 2013), which has been shown to contribute to persistence and extracellular survival of M. tuberculosis (Tan and Russell, 2015) (Figure 3). Contrarily, the tissue within the granuloma surrounding the caseum displays a gradient of anti-inflammatory phenotypes and gradually increasing oxygen tension (Mattila et al., 2013). This suggests that the pathways involved in macrophage metabolism influence the inflammatory signals and effector functions generated in response to infection.
Macrophage heterogeneity attributed to ontogeny and polarization states can further influence macrophage function, and the subsequent progression of varying granuloma types that coexist in individual patients. Using experimental data from nonhuman primates, the development of a computational model was used to investigate the temporal dynamics and spatial organization of M1 and M2 polarized macrophages to determine how cytokine signaling impacts the outcome of infection (Marino et al., 2015). Independent of adaptive immunity, alveolar macrophages in the mouse lung exhibit an M2 phenotype and favor fatty acid oxidation, therefore providing a metabolic and nutritional advantage for intracellular M. tuberculosis compared to bacilli residing in interstitial macrophages (Huang et al., 2018). Whilst this mechanism could protect the host against excessive inflammation, it is unclear how M. tuberculosis alters the metabolic state of macrophages towards M2 polarization to enhance intracellular persistence and sequestration of host nutrients.
M. tuberculosis increases TAG accumulation in the caseum, which contributes to antibiotic tolerance (Daniel et al., 2011; Sarathy et al., 2018), and correlates with the presence of lipid-body positive M. tuberculosis in sputum (Sloan et al., 2015). It is thought that foamy macrophages surround the caseum, as their lipid composition resembles the environment encountered in the caseum (Peyron et al., 2008; Kim et al., 2010). Additionally, necrotic death of foamy macrophages would release lipid droplets and cellular debris at the caseum interface (Peyron et al., 2008). Since foamy macrophages have lost their phagocytic ability (Peyron et al., 2008), it is suggested that their formation is exploited by M. tuberculosis to enhance cavitation of the granuloma.
Histopathological analysis of lesions indicates that antibiotic treatment induces changes in the granuloma structure, whereby the majority of caseous granulomas evolve to fibrotic or necrotizing tissue that lack proper structure. Hypoxic conditions and poor antibiotic diffusion into the devascularized caseous center lead to heterogeneous susceptibility of bacteria, whereby residual bacterial growth remains in the caseum or acellular rim of necrotic granulomas following treatment in guinea pigs (Lenaerts et al., 2007), rabbits (Sarathy et al., 2018) and macaques (Lin et al., 2014). These bacilli were found to grow as multicellular pellicles, characteristic of biofilms (Ojha et al., 2008; Islam et al., 2012; Wright et al., 2017). It is hypothesized that this cording phenotype contributes to mycobacterial persistence in the host as the abundance of extracellular free mycolic acids during pellicle maturation creates a waxy-layered lipid-rich matrix that harbors and protects drug-tolerant bacilli (Ojha et al., 2008; Islam et al., 2012; Wright et al., 2017).
The host tissue environment and complex granuloma composition likely drives heterogeneous bacterial susceptibility and antibiotic-tolerant persister formation, as some lesions are sterilized prior to antibiotic treatment in macaques (Lin et al., 2014), whilst other lesions may endure following completion of treatment in humans (Malherbe et al., 2016). Differential drug partitioning between cellular and necrotic lesions (Irwin et al., 2016; Sarathy et al., 2016; Zimmerman et al., 2017; Blanc et al., 2018) result in sub-inhibitory antibiotic concentrations, thereby impacting the rate of sterilization and treatment success. The prodrugs isoniazid and pyrazinamide additionally require bioactivation by M. tuberculosis KatG and PncA, respectively, to be functional. Whilst decreased expression of KatG is associated with isoniazid-treated persisters (Wakamoto et al., 2013; Kurthkoti et al., 2017), genes other than pncA, such as sigE and panD are implicated in pyrazinamide resistance (Thiede et al., 2022). It remains to be determined whether the latter genes mediate tolerance in M. tuberculosis. Inhibition of F1F0-ATP synthase following treatment with bedaquiline involves rapid metabolic adaptation by M. tuberculosis, whereby multiple metabolic pathways associated with the dosR dormancy regulon, lipid homeostasis and cell wall remodeling contribute to antibiotic tolerance (Peterson et al., 2016).
Persister formation and associated antibiotic tolerance may be triggered by host exposure, independent of antibiotic exposure. Increased antibiotic tolerance may thus be attributed to a combination of alternative metabolic pathways being utilized that may not be bioactivated, and inconsistent drug distribution attributed to lesional heterogeneity. In previous work from our group, macrophage uptake resulted in enrichment for non- or slowly replicating M. tuberculosis. Treatment with D-cycloserine revealed that this population is highly enriched for persisters, based on its antibiotic tolerant phenotype (Mouton et al., 2016). This is in line with work from Jain et al. (2016) who observed the presence of persisters in sputum from TB patients prior to antibiotic treatment. Vilchèze et al. (2017) additionally demonstrated that compounds that enhanced respiration and induced ROS in M. tuberculosis persisters prevented M. tuberculosis from entering a low metabolic state and rendered mycobacteria susceptible to bactericidal antibiotics (Vilchèze et al., 2017). This suggests that host pressures drive formation of antibiotic-tolerant persisters, and may be exploited by targeting processes required during maintenance of persistence in the host.
To mimic intracellular confinement and spatially monitor the impact of phagosomes on M. tuberculosis antibiotic-tolerant persister formation, space-confined cell culture chambers (Luthuli et al., 2015) and zebrafish larval models (Adams et al., 2011) have been exploited. This revealed differential progression of lesions in response to antibiotic treatment. Following macrophage uptake, drug tolerance arose within a few days, whereby antibiotic-tolerant persisters residing in macrophages exploited granulomas for their expansion or migrated to disseminate infection (Adams et al., 2011; Luthuli et al., 2015). Recent approaches to tracking the fate of individual lesions provides insight into the heterogeneous granuloma formation and size, and lesion-specific dissemination (Martin et al., 2017; Gregg et al., 2018; Cicchese et al., 2020), which may guide our understanding of how specific tissue environments influence responses to antibiotics.
The varying microenvironments that M. tuberculosis colonizes greatly enhance phenotypic adaptations that enable host evasion and promotes intracellular survival and persistence. A major adaptation is an altered growth rate. A slower growth rate has been associated with modifications to the mycobacterial cell wall architecture, whereby M. tuberculosis reconstructs its surface lipid and mycolic acid structure by undergoing mycolic acid biosynthesis and cell wall thickening (Hampshire et al., 2004; Rohde et al., 2012; Vilchèze and Kremer, 2017; Zimmermann et al., 2017; Baker and Abramovitch, 2018). An altered colony morphology (Salina et al., 2014) and cell length distribution observed in sputum (Vijay et al., 2017b) have been further shown to influence heterogeneity in metabolism, antibiotic sensitivity and response to stressors.
Prolonged periods under multiple stress conditions drastically increases the number of intracellular lipid bodies in vitro (Deb et al., 2009) and in sputum (Garton et al., 2008; Vijay et al., 2018), whereby a correlation between TAG accumulation and loss of acid fastness is observed (Deb et al., 2009; Daniel et al., 2011). Interestingly, transcriptomic analyses of these bacilli revealed downregulation of kasB, one of two M. tuberculosis genes encoding distinct ß-ketoacyl-ACP synthases, during persistence (Deb et al., 2009; Vilchèze et al., 2014). Deletion of kasB led to alterations in mycolic acid structure by producing mycolic acids that were two to four carbons shorter, and resulted in the loss of cording and acid-fastness (Gao et al., 2003; Bhatt et al., 2007). This may negatively affect cell wall permeability since these mycolic acids were not synthesized in dense bundles (Yamada et al., 2012). The precise mechanism resulting in loss of acid fastness is however not well understood, and it is uncertain whether retention of the acid-fast stain is hindered or whether reorganization of the cell wall components prevents access of the stain.
The current acid-fast staining approach is dependent on mycolic acid chain length and structure, which has been shown to substantially differ in the cell wall of M. tuberculosis (Barry et al., 1998; Beken et al., 2011). Persisters residing in the caseum frequently go undetected and appear acid-fast negative in guinea pigs (Hoff et al., 2011), highlighting the inconsistencies of acid-fast staining. Histological representation of various lung lesions from patients non-compliant to treatment display heterogeneous morphology and distribution of acid-fast bacilli. Acid-fast bacilli were largely located at the granuloma surface, with connection to airways, whereas acid-fast bacilli in necrotizing granulomas were scarcely detected (Kaplan et al., 2003; Lenaerts et al., 2007). Interestingly, following resuscitation of M. tuberculosis persisters generated in vitro, upregulation of genes responsible for fatty acid and mycolic acid biosynthesis was observed (Du et al., 2016; Salina et al., 2019), whilst pathways involved in lipid uptake and catabolism were down-regulated (Du et al., 2016). This characteristic may reflect differences in the cell wall components and lipid profiles prior to resuscitation; it is thus intriguing whether these bacilli regained their acid-fastness.
Nutrient availability further influences variations in morphology, whereby phosphate- (Rifat et al., 2014) or potassium-starved bacilli (Salina et al., 2014) display a more elongated, or spherical and shorter morphology, respectively. This may alter the ultrastructure and could explain the loss in acid-fastness of M. tuberculosis persisters. The enhancement in surface-to-volume ratio for elongated cells may be advantageous in sequestering additional nutrients from its environment.
Asymmetric cellular division is commonly observed in mycobacteria during cell elongation and division (Aldridge et al., 2012). As bacterial cells divide, the progeny inherits an ‘old pole’ generated from previous cell divisions, and a ‘new pole’ from the most recent cell division. Cell length heterogeneity introduced by asymmetrical cell division has been associated with differential susceptibility to host- and antibiotic-induced stressors (Aldridge et al., 2012; Vijay et al., 2017a). Asymmetry-promoting proteins LamA and Wag31 regulate polar growth and cell elongation at each pole (Rego et al., 2017). Cells inheriting the old pole generate longer cells with faster elongation rates than cells that inherit the new pole (Aldridge et al., 2012; Richardson et al., 2016; Nair et al., 2019; Vijay et al., 2017a). Unequal partitioning of cells, with a stronger bias towards daughter cells inheriting the old pole, display higher tolerance to host and antibiotic stress (Richardson et al., 2016; Nair et al., 2019). Similarly, multidrug-resistant (MDR) strains were observed to increase their cell length distribution in macrophages (Vijay et al., 2017b), whilst shorter cells with slower elongation rates were more antibiotic susceptible (Aldridge et al., 2012; Nair et al., 2019). Computational tools that cytologically profile M. tuberculosis response to antibiotic treatment (Smith et al., 2020) may further be adapted to predict whether specific cell morphologies prior to treatment influence treatment outcome.
Synthesis of cell envelope components such as peptidoglycan and arabinogalactan varies in polar localization (Botella et al., 2017), thus asymmetrical division may impact on cell envelope assembly, resulting in distinct localization of cellular or metabolic machinery near poles that will allow for efficient growth. Repeated inheritance of the old poles at cellular division has been linked to limited growth rates, increased antibiotic susceptibility and cell death (Dhar and McKinney, 2007; Aldridge et al., 2012). This may be attributed to partitioning of damaged cell components, which would describe replicative aging and cell death. However, whether pole age influences survival or entry into persistence is unknown.
Alternative carbon sources (Priestman et al., 2017) or nutrient starvation (Hayashi et al., 2018) may further reflect variations in growth rate and cell length. Oxidative stress and iron deficiency have been shown to increase cell length heterogeneity in sensitive and MDR strains (Vijay et al., 2017b). A higher level of NADH oxidase in shorter cells generates significantly higher hydroxyl radical levels via the Fenton reaction. Since this occurs to a lesser extent in normal/long-length cells, this inherent predisposition could offer a survival advantage attributed to differential antibiotic susceptibility, and may provide a mechanism underlying tolerance (Vijay et al., 2017b). Interestingly, shorter cells possessed a lower buoyant density fraction in Percoll gradient, indicative of a high lipid content. The presence of lipid-rich membrane vesicles on the bacilli cell surface of shorter cells may indicate molecular differences to normal/long-length cells (Vijay et al., 2017a). Regardless of the growth media, the short and normal/long-length cells comprised ~ 10% and ~ 90% of the population, respectively, in vitro and in clinical isolates (Vijay et al., 2017a). Stressed M. tuberculosis additionally distributes irreversibly oxidized proteins asymmetrically (Vaubourgeix et al., 2015). This may represent an inherent mechanism for regulation of mycobacterial cell growth, further enhancing population heterogeneity, and requires further investigation.
Bacterial phenotypic heterogeneity signifies a survival strategy to allow rapid adaptation and persister formation in response to altering conditions (Ryan et al., 2010). M. tuberculosis is simultaneously exposed to multiple host-related stressors, contributing to the spectrum of adaptive responses that induce phenotypically heterogeneous subpopulations. Research highlights the induction of overlapping adaptive responses that may assist M. tuberculosis persisters to flourish in various microenvironments within the host, and further contributes to antibiotic tolerance (Kurthkoti et al., 2017; Huang et al., 2018; Pisu et al., 2020). M. tuberculosis appears to exploit macrophages and granuloma formation for its expansion. Therefore, utilizing animal models that reflect the diversity within lesion phenotypes as observed in humans can divulge how host interactions drive bacterial phenotypic heterogeneity, and subsequent disease progression (Driver et al., 2012; Martin et al., 2017). Many technical challenges are however associated with investigation of M. tuberculosis persisters, thus approaches involving flow cytometry and high-content imaging-based techniques in combination with M. tuberculosis-specific probes could improve the characterization of persisters (Parbhoo et al., 2020). This could highlight specific mechanisms utilized by M. tuberculosis for adaptation to the host environment; understanding and exploiting such pathways may highlight key mechanisms utilized by M. tuberculosis to maintain persistence, possibly providing a target for therapeutic intervention.
TP researched data, constructed figures, wrote and edited the manuscript. JM and SS critically reviewed the manuscript and provided insightful discussions and ideas. All authors contributed to the article and approved the submitted version.
This work was supported by funding from the South African Medical Research Council (SA MRC), and the South African National Research Foundation (NRF). SS is funded by the South African Research Chairs Initiative of the Department of Science and Technology and NRF of South Africa, award number UID 86539. TP was funded by the Deutscher Akademischer Austauschdienst and NRF of South Africa, award number UID 111868. This work was supported by the GCRF Networks in Vaccines Research and Development VALIDATE Network which was co-funded by the MRC and BBSRC (ref MR/R005850/1). This UK funded award is part of the EDCTP2 programme supported by the European Union.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.956607/full#supplementary-material
Abramovitch, R. B., Rohde, K. H., Hsu, F.-F., Russell, D. G. (2011). aprABC: A mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol. Microbiol. 80, 678–694. doi: 10.1111/j.1365-2958.2011.07601.x
Abreu, R., Essler, L., Loy, A., Quinn, F., Giri, P. (2018). Heparin inhibits intracellular mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci. Rep. 8, 7296. doi: 10.1038/s41598-018-25480-y
Adams, K. N., Takaki, K., Connolly, L. E., Wiedenhoft, H., Winglee, K., Humbert, O., et al. (2011). Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53. doi: 10.1016/j.cell.2011.02.022
Agapova, A., Serafini, A., Petridis, M., Hunt, D. M., Garza-Garcia, A., Sohaskey, C. D., et al. (2019). Flexible nitrogen utilisation by the metabolic generalist pathogen mycobacterium tuberculosis. eLife 8, e41129. doi: 10.7554/eLife.41129
Aldridge, B. B., Fernandez-Suarez, M., Heller, D., Ambravaneswaran, V., Irimia, D., Toner, M., et al. (2012). Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335, 100–104. doi: 10.1126/science.1216166
Appelberg, R. (2006). Macrophage nutriprive antimicrobial mechanisms. J. Leukoc. Biol. 79, 1117–1128. doi: 10.1189/jlb.0206079
Astarie-Dequeker, C., Guyader, L. L., Malaga, W., Seaphanh, F.-K., Chalut, C., Lopez, A., et al. (2009). Phthiocerol dimycocerosates of m. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PloS Pathog. 5, e1000289. doi: 10.1371/journal.ppat.1000289
Augenstreich, J., Arbues, A., Simeone, R., Haanappel, E., Wegener, A., Sayes, F., et al. (2017). ESX-1 and phthiocerol dimycocerosates of mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell. Microbiol. 19, e12726. doi: 10.1111/cmi.12726
Awasthy, D., Bharath, S., Subbulakshmi, V., Sharma, U. (2012). Alanine racemase mutants of mycobacterium tuberculosis require d-alanine for growth and are defective for survival in macrophages and mice. Microbiol. Read. Engl. 158, 319–327. doi: 10.1099/mic.0.054064-0
Axelrod, S., Oschkinat, H., Enders, J., Schlegel, B., Brinkmann, V., Kaufmann, S. H. E., et al. (2008). Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell. Microbiol. 10, 1530–1545. doi: 10.1111/j.1462-5822.2008.01147.x
Ayala-Castro, C., Saini, A., Outten, F. W. (2008). Fe-s cluster assembly pathways in bacteria. Microbiol. Mol. Biol. Rev. MMBR 72, 110–125. doi: 10.1128/MMBR.00034-07
Baker, J. J., Abramovitch, R. B. (2018). Genetic and metabolic regulation of mycobacterium tuberculosis acid growth arrest. Sci. Rep. 8, 1–16. doi: 10.1038/s41598-018-22343-4
Baker, J. J., Dechow, S. J., Abramovitch, R. B. (2019). Acid fasting: Modulation of mycobacterium tuberculosis metabolism at acidic pH. Trends Microbiol. 27, 942–953. doi: 10.1016/j.tim.2019.06.005
Baker, J. J., Johnson, B. K., Abramovitch, R. B. (2014). Slow growth of mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol. Microbiol. 94, 56–69. doi: 10.1111/mmi.12688
Balaban, N. Q., Helaine, S., Lewis, K., Ackermann, M., Aldridge, B., Andersson, D. I., et al. (2019). Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448. doi: 10.1038/s41579-019-0196-3
Barczak, A. K., Avraham, R., Singh, S., Luo, S. S., Zhang, W. R., Bray, M.-A., et al. (2017). Systematic, multiparametric analysis of mycobacterium tuberculosis intracellular infection offers insight into coordinated virulence. PloS Pathog. 13, e1006363. doi: 10.1371/journal.ppat.1006363
Barry, C. E., Lee, R. E., Mdluli, K., Sampson, A. E., Schroeder, B. G., Slayden, R. A., et al. (1998). Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37, 143–179. doi: 10.1016/s0163-7827(98)00008-3
Beken, S. V., Dulayymi, J. R. A., Naessens, T., Koza, G., Maza-Iglesias, M., Rowles, R. R., et al. (2011). Molecular structure of the mycobacterium tuberculosis virulence factor, mycolic acid, determines the elicited inflammatory pattern. Eur. J. Immunol. 41, 450–460. doi: 10.1002/eji.201040719
Berney, M., Berney-Meyer, L., Wong, K.-W., Chen, B., Chen, M., Kim, J., et al. (2015). Essential roles of methionine and s-adenosylmethionine in the autarkic lifestyle of mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 112, 10008–10013. doi: 10.1073/pnas.1513033112
Beste, D. J. V., Nöh, K., Niedenführ, S., Mendum, T. A., Hawkins, N. D., Ward, J. L., et al. (2013). 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular mycobacterium tuberculosis. chem. Biol. 20, 1012–1021. doi: 10.1016/j.chembiol.2013.06.012
Bhaskar, A., Chawla, M., Mehta, M., Parikh, P., Chandra, P., Bhave, D., et al. (2014). Reengineering redox sensitive GFP to measure mycothiol redox potential of mycobacterium tuberculosis during infection. PloS Pathog. 10, e1003902. doi: 10.1371/journal.ppat.1003902
Bhatt, A., Fujiwara, N., Bhatt, K., Gurcha, S. S., Kremer, L., Chen, B., et al. (2007). Deletion of kasB in mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc. Natl. Acad. Sci. U. S. A. 104, 5157–5162. doi: 10.1073/pnas.0608654104
Blanc, L., Daudelin, I. B., Podell, B. K., Chen, P.-Y., Zimmerman, M., Martinot, A. J., et al. (2018). High-resolution mapping of fluoroquinolones in TB rabbit lesions reveals specific distribution in immune cell types. eLife 7, e41115. doi: 10.7554/eLife.41115
Boot, M., Jim, K. K., Liu, T., Commandeur, S., Lu, P., Verboom, T., et al. (2017). A fluorescence-based reporter for monitoring expression of mycobacterial cytochrome bd in response to antibacterials and during infection. Sci. Rep. 7, 1–10. doi: 10.1038/s41598-017-10944-4
Borah, K., Beyß, M., Theorell, A., Wu, H., Basu, P., Mendum, T. A., et al. (2019). Intracellular mycobacterium tuberculosis exploits multiple host nitrogen sources during growth in human macrophages. Cell Rep. 29, 3580–3591.e4. doi: 10.1016/j.celrep.2019.11.037
Borah, K., Mendum, T. A., Hawkins, N. D., Ward, J. L., Beale, M. H., Larrouy-Maumus, G., et al. (2021). Metabolic fluxes for nutritional flexibility of mycobacterium tuberculosis. Mol. Syst. Biol. 17, e10280. doi: 10.15252/msb.202110280
Botella, H., Yang, G., Ouerfelli, O., Ehrt, S., Nathan, C. F., Vaubourgeix, J. (2017). Distinct spatiotemporal dynamics of peptidoglycan synthesis between mycobacterium smegmatis and mycobacterium tuberculosis. mBio 8, e01183–17. doi: 10.1128/mBio.01183-17
Buchmeier, N. A., Newton, G. L., Koledin, T., Fahey, R. C. (2003). Association of mycothiol with protection of mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol. Microbiol. 47, 1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x
Chakraborty, S., Gruber, T., Barry, C. E., Boshoff, H. I., Rhee, K. Y. (2013). Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in mycobacterium tuberculosis. Science 339, 88–91. doi: 10.1126/science.1228980
Chuang, Y.-M., Bandyopadhyay, N., Rifat, D., Rubin, H., Bader, J. S., Karakousis, P. C. (2015). Deficiency of the novel exopolyphosphatase Rv1026/PPX2 leads to metabolic downshift and altered cell wall permeability in mycobacterium tuberculosis. mBio 6, e02428–e02414. doi: 10.1128/mBio.02428-14
Cicchese, J. M., Dartois, V., Kirschner, D. E., Linderman, J. J. (2020). Both pharmacokinetic variability and granuloma heterogeneity impact the ability of the first-line antibiotics to sterilize tuberculosis granulomas. Front. Pharmacol. 11. doi: 10.3389/fphar.2020.00333
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537. doi: 10.1038/31159
Commandeur, S., Iakobachvili, N., Sparrius, M., Nur, M. M., Mukamolova, G. V., Bitter, W. (2020). Zebrafish embryo model for assessment of drug efficacy on mycobacterial persisters. Antimicrob. Agents Chemother. 64, e00801–20. doi: 10.1128/AAC.00801-20
Conrad, W. H., Osman, M. M., Shanahan, J. K., Chu, F., Takaki, K. K., Cameron, J., et al. (2017). Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc. Natl. Acad. Sci. U. S. A. 114, 1371–1376. doi: 10.1073/pnas.1620133114
Dahl, J. L., Kraus, C. N., Boshoff, H. I. M., Doan, B., Foley, K., Avarbock, D., et al. (2003). The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. U. S. A. 100, 10026–10031. doi: 10.1073/pnas.1631248100
Daniel, J., Maamar, H., Deb, C., Sirakova, T. D., Kolattukudy, P. E. (2011). Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PloS Pathog. 7, e1002093. doi: 10.1371/journal.ppat.1002093
Dartois, V. (2014). The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 12, 159–167. doi: 10.1038/nrmicro3200
Datta, M., Via, L. E., Chen, W., Baish, J. W., Xu, L., Barry, C. E., et al. (2016). Mathematical model of oxygen transport in tuberculosis granulomas. Ann. Biomed. Eng. 44, 863–872. doi: 10.1007/s10439-015-1415-3
Deb, C., Lee, C.-M., Dubey, V. S., Daniel, J., Abomoelak, B., Sirakova, T. D., et al. (2009). A novel in vitro multiple-stress dormancy model for mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PloS One 4, e6077. doi: 10.1371/journal.pone.0006077
de Carvalho, L. P. S., Fischer, S. M., Marrero, J., Nathan, C., Ehrt, S., Rhee, K. Y. (2010). Metabolomics of mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17, 1122–1131. doi: 10.1016/j.chembiol.2010.08.009
Del Portillo, P., García-Morales, L., Menéndez, M. C., Anzola, J. M., Rodríguez, J. G., Helguera-Repetto, A. C., et al. (2019). Hypoxia is not a main stress when mycobacterium tuberculosis is in a dormancy-like long-chain fatty acid environment. Front. Cell. Infect. Microbiol. 8. doi: 10.3389/fcimb.2018.00449
de Martino, M., Lodi, L., Galli, L., Chiappini, E. (2019). Immune response to mycobacterium tuberculosis: A narrative review. Front. Pediatr. 7. doi: 10.3389/fped.2019.00350
Dhar, N., McKinney, J. D. (2007). Microbial phenotypic heterogeneity and antibiotic tolerance. Curr. Opin. Microbiol. 10, 30–38. doi: 10.1016/j.mib.2006.12.007
Driver, E. R., Ryan, G. J., Hoff, D. R., Irwin, S. M., Basaraba, R. J., Kramnik, I., et al. (2012). Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against mycobacterium tuberculosis. antimicrob. Agents Chemother. 56, 3181–3195. doi: 10.1128/AAC.00217-12
Dubé, J.-Y., Fava, V. M., Schurr, E., Behr, M. A. (2021). Underwhelming or misunderstood? genetic variability of pattern recognition receptors in immune responses and resistance to mycobacterium tuberculosis. Front. Immunol. 12. doi: 10.3389/fimmu.2021.714808
Du, P., Sohaskey, C. D., Shi, L. (2016). Transcriptional and physiological changes during mycobacterium tuberculosis reactivation from non-replicating persistence. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01346
Dutta, N. K., Klinkenberg, L. G., Vazquez, M.-J., Segura-Carro, D., Colmenarejo, G., Ramon, F., et al. (2019). Inhibiting the stringent response blocks mycobacterium tuberculosis entry into quiescence and reduces persistence. Sci. Adv. 5, eaav2104. doi: 10.1126/sciadv.aav2104
Ehrt, S., Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: Defense against host stresses. Cell. Microbiol. 11, 1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x
Ehrt, S., Schnappinger, D., Bekiranov, S., Drenkow, J., Shi, S., Gingeras, T. R., et al. (2001). Reprogramming of the macrophage transcriptome in response to interferon-γ and mycobacterium tuberculosis. J. Exp. Med. 194, 1123–1140. doi: 10.1084/jem.194.8.1123
Eisenreich, W., Heesemann, J., Rudel, T., Goebel, W. (2013). Metabolic host responses to infection by intracellular bacterial pathogens. Front. Cell. Infect. Microbiol. 3. doi: 10.3389/fcimb.2013.00024
Elliott, S. R., White, D. W., Tischler, A. D. (2019). Mycobacterium tuberculosis requires regulation of ESX-5 secretion for virulence in Irgm1-deficient mice. Infect. Immun. 87, e00660–18. doi: 10.1128/IAI.00660-18
Eoh, H., Rhee, K. Y. (2013). Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 110, 6554–6559. doi: 10.1073/pnas.1219375110
Feng, L., Chen, S., Hu, Y. (2018). PhoPR positively regulates whiB3 expression in response to low pH in pathogenic mycobacteria. J. Bacteriol. 200, e00766–17. doi: 10.1128/JB.00766-17
Flynn, J. L., Chan, J. (2001). Immunology of tuberculosis. Annu. Rev. Immunol. 19, 93–129. doi: 10.1146/annurev.immunol.19.1.93
Ford, C. B., Lin, P. L., Chase, M., Shah, R. R., Iartchouk, O., Galagan, J., et al. (2011). Use of whole genome sequencing to estimate the mutation rate of mycobacterium tuberculosis during latent infection. Nat. Genet. 43, 482–486. doi: 10.1038/ng.811
Fratti, R. A., Chua, J., Vergne, I., Deretic, V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. 100, 5437–5442. doi: 10.1073/pnas.0737613100
Gao, L.-Y., Laval, F., Lawson, E. H., Groger, R. K., Woodruff, A., Morisaki, J. H., et al. (2003). Requirement for kasB in mycobacterium mycolic acid biosynthesis, cell wall impermeability and intracellular survival: implications for therapy. Mol. Microbiol. 49, 1547–1563. doi: 10.1046/j.1365-2958.2003.03667.x
Garton, N. J., Waddell, S. J., Sherratt, A. L., Lee, S.-M., Smith, R. J., Senner, C., et al. (2008). Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PloS Med. 5, e75. doi: 10.1371/journal.pmed.0050075
Geisel, R. E., Sakamoto, K., Russell, D. G., Rhoades, E. R. (2005). In vivo activity of released cell wall lipids of mycobacterium bovis bacillus calmette-guérin is due principally to trehalose mycolates. J. Immunol. 174, 5007–5015. doi: 10.4049/jimmunol.174.8.5007
Gopinath, K., Venclovas, Č., Ioerger, T. R., Sacchettini, J. C., McKinney, J. D., Mizrahi, V., et al. (2013). A vitamin B12 transporter in mycobacterium tuberculosis. Open Biol. 3, 120175. doi: 10.1098/rsob.120175
Gouzy, A., Larrouy-Maumus, G., Bottai, D., Levillain, F., Dumas, A., Wallach, J. B., et al. (2014). Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PloS Pathog. 10, e1003928. doi: 10.1371/journal.ppat.1003928
Gregg, R. W., Maiello, P., Borish, H. J., Coleman, M. T., Reed, D. S., White, A. G., et al. (2018). Spatial and temporal evolution of lung granulomas in a cynomolgus macaque model of mycobacterium tuberculosis infection. Radiol. Infect. Dis. 5, 110–117. doi: 10.1016/j.jrid.2018.08.001
Guerrini, V., Prideaux, B., Blanc, L., Bruiners, N., Arrigucci, R., Singh, S., et al. (2018). Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PloS Pathog. 14, e1007223. doi: 10.1371/journal.ppat.1007223
Hampshire, T., Soneji, S., Bacon, J., James, B. W., Hinds, J., Laing, K., et al. (2004). Stationary phase gene expression of mycobacterium tuberculosis following a progressive nutrient depletion: a model for persistent organisms? Tuberc. Edinb. Scotl. 84, 228–238. doi: 10.1016/j.tube.2003.12.010
Hasenoehrl, E. J., Sajorda, D. R., Berney-Meyer, L., Johnson, S., Tufariello, J. M., Fuhrer, T., et al. (2019). Derailing the aspartate pathway of mycobacterium tuberculosis to eradicate persistent infection. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-12224-3
Hayashi, J. M., Richardson, K., Melzer, E. S., Sandler, S. J., Aldridge, B. B., Siegrist, M. S., et al. (2018). Stress-induced reorganization of the mycobacterial membrane domain. mBio 9, e01823–17. doi: 10.1128/mBio.01823-17
Hoff, D. R., Ryan, G. J., Driver, E. R., Ssemakulu, C. C., De Groote, M. A., Basaraba, R. J., et al. (2011). Location of intra- and extracellular m. tuberculosis populations in lungs of mice and guinea pigs during disease progression and after drug treatment. PloS One 6, e17550. doi: 10.1371/journal.pone.0017550
Hondalus, M. K., Bardarov, S., Russell, R., Chan, J., Jacobs, W. R., Bloom, B. R. (2000). Attenuation of and protection induced by a leucine auxotroph of mycobacterium tuberculosis. infect. Immun. 68, 2888–2898. doi: 10.1128/iai.68.5.2888-2898.2000
Hood, M. I., Skaar, E. P. (2012). Nutritional immunity: Transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 10, 525–537. doi: 10.1038/nrmicro2836
Howe, M. D., Kordus, S. L., Cole, M. S., Bauman, A. A., Aldrich, C. C., Baughn, A. D., et al. (2018). Methionine antagonizes para-aminosalicylic acid activity via affecting folate precursor biosynthesis in mycobacterium tuberculosis. front. Cell. Infect. Microbiol. 8, 399. doi: 10.3389/fcimb.2018.00399
Huang, L., Nazarova, E. V., Russell, D. G. (2019). Mycobacterium tuberculosis: Bacterial fitness within the host macrophage. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.BAI-0001-2019
Huang, L., Nazarova, E. V., Tan, S., Liu, Y., Russell, D. G. (2018). Growth of mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med. 215, 1135–1152. doi: 10.1084/jem.20172020
Ignatov, D. V., Salina, E. G., Fursov, M. V., Skvortsov, T. A., Azhikina, T. L., Kaprelyants, A. S. (2015). Dormant non-culturable mycobacterium tuberculosis retains stable low-abundant mRNA. BMC Genomics 16, 954. doi: 10.1186/s12864-015-2197-6
Irwin, S. M., Driver, E., Lyon, E., Schrupp, C., Ryan, G., Gonzalez-Juarrero, M., et al. (2015). Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with mycobacterium tuberculosis. dis. Model. Mech. 8, 591–602. doi: 10.1242/dmm.019570
Irwin, S. M., Prideaux, B., Lyon, E. R., Zimmerman, M. D., Brooks, E. J., Schrupp, C. A., et al. (2016). Bedaquiline and pyrazinamide treatment responses are affected by pulmonary lesion heterogeneity in mycobacterium tuberculosis infected C3HeB/FeJ mice. ACS Infect. Dis. 2, 251–267. doi: 10.1021/acsinfecdis.5b00127
Islam, M. S., Richards, J. P., Ojha, A. K. (2012). Targeting drug tolerance in mycobacteria: a perspective from mycobacterial biofilms. Expert Rev. Anti Infect. Ther. 10, 1055–1066. doi: 10.1586/eri.12.88
Jain, P., Weinrick, B. C., Kalivoda, E. J., Yang, H., Munsamy, V., Vilcheze, C., et al. (2016). Dual-reporter mycobacteriophages (Φ2DRMs) reveal preexisting mycobacterium tuberculosis persistent cells in human sputum. mBio 7, e01023-16. doi: 10.1128/mBio.01023-16
Jamaati, H., Mortaz, E., Pajouhi, Z., Folkerts, G., Movassaghi, M., Moloudizargari, M., et al. (2017). Nitric oxide in the pathogenesis and treatment of tuberculosis. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02008
Jamwal, S. V., Mehrotra, P., Singh, A., Siddiqui, Z., Basu, A., Rao, K. V. S. (2016). Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci. Rep. 6, 23089. doi: 10.1038/srep23089
Johnson, B. K., Colvin, C. J., Needle, D. B., Mba Medie, F., Champion, P. A. D., Abramovitch, R. B. (2015). The carbonic anhydrase inhibitor ethoxzolamide inhibits the mycobacterium tuberculosis PhoPR regulon and esx-1 secretion and attenuates virulence. Antimicrob. Agents Chemother. 59, 4436–4445. doi: 10.1128/AAC.00719-15
Kang, P. B., Azad, A. K., Torrelles, J. B., Kaufman, T. M., Beharka, A., Tibesar, E., et al. (2005). The human macrophage mannose receptor directs mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202, 987–999. doi: 10.1084/jem.20051239
Kaplan, G., Post, F. A., Moreira, A. L., Wainwright, H., Kreiswirth, B. N., Tanverdi, M., et al. (2003). Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 71, 7099–7108. doi: 10.1128/IAI.71.12.7099-7108.2003
Kehl-Fie, T. E., Skaar, E. P. (2010). Nutritional immunity beyond iron: a role for manganese and zinc. Curr. Opin. Chem. Biol. 14, 218–224. doi: 10.1016/j.cbpa.2009.11.008
Kim, M.-J., Wainwright, H. C., Locketz, M., Bekker, L.-G., Walther, G. B., Dittrich, C., et al. (2010). Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2, 258–274. doi: 10.1002/emmm.201000079
Klinkenberg, L. G., Lee, J.-H., Bishai, W. R., Karakousis, P. C. (2010). The stringent response is required for full virulence of mycobacterium tuberculosis in guinea pigs. J. Infect. Dis. 202, 1397–1404. doi: 10.1086/656524
Knight, M., Braverman, J., Asfaha, K., Gronert, K., Stanley, S. (2018). Lipid droplet formation in mycobacterium tuberculosis infected macrophages requires IFN-γ/HIF-1α signaling and supports host defense. PloS Pathog. 14, e1006874. doi: 10.1371/journal.ppat.1006874
Kumar, A., Farhana, A., Guidry, L., Saini, V., Hondalus, M., Steyn, A. J. C. (2011). Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev. Mol. Med. 13, e39. doi: 10.1017/S1462399411002079
Kumar, R., Singh, P., Kolloli, A., Shi, L., Bushkin, Y., Tyagi, S., et al (2019). Immunometabolism of phagocytes during Mycobacterium tuberculosis infection. Front. Mol. Biosci. 6, 105. doi: 10.3389/fmolb.2019.00105
Kurthkoti, K., Amin, H., Marakalala, M. J., Ghanny, S., Subbian, S., Sakatos, A., et al. (2017). The capacity of mycobacterium tuberculosis to survive iron starvation might enable it to persist in iron-deprived microenvironments of human granulomas. mBio 8, e01092-17. doi: 10.1128/mBio.01092-17
Kyei, G. B., Vergne, I., Chua, J., Roberts, E., Harris, J., Junutula, J. R., et al. (2006). Rab14 is critical for maintenance of mycobacterium tuberculosis phagosome maturation arrest. EMBO J. 25, 5250–5259. doi: 10.1038/sj.emboj.7601407
Lenaerts, A. J., Hoff, D., Aly, S., Ehlers, S., Andries, K., Cantarero, L., et al. (2007). Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob. Agents Chemother. 51, 3338–3345. doi: 10.1128/AAC.00276-07
Lerner, T. R., Queval, C. J., Lai, R. P., Russell, M. R. G., Fearns, A., Greenwood, D. J., et al. (2020). Mycobacterium tuberculosis cords within lymphatic endothelial cells to evade host immunity. JCI Insight 5, 136937. doi: 10.1172/jci.insight.136937
Levitte, S., Adams, K. N., Berg, R. D., Cosma, C. L., Urdahl, K. B., Ramakrishnan, L. (2016). Mycobacterial acid tolerance enables phagolysosomal survival and establishment of tuberculous infection in vivo. Cell Host Microbe 20, 250–258. doi: 10.1016/j.chom.2016.07.007
Lin, P. L., Coleman, T., Carney, J. P. J., Lopresti, B. J., Tomko, J., Fillmore, D., et al. (2013). Radiologic responses in cynomolgus macaques for assessing tuberculosis chemotherapy regimens. Antimicrob. Agents Chemother. 57, 4237–4244. doi: 10.1128/AAC.00277-13
Lin, P. L., Ford, C. B., Coleman, M. T., Myers, A. J., Gawande, R., Ioerger, T., et al. (2014). Sterilization of granulomas is common in both active and latent tuberculosis despite extensive within-host variability in bacterial killing. Nat. Med. 20, 75–79. doi: 10.1038/nm.3412
Lin, P. L., Pawar, S., Myers, A., Pegu, A., Fuhrman, C., Reinhart, T. A., et al. (2006). Early events in mycobacterium tuberculosis infection in cynomolgus macaques. Infect. Immun. 74, 3790–3803. doi: 10.1128/IAI.00064-06
Liu, C. H., Liu, H., Ge, B. (2017). Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell. Mol. Immunol. 14, 963–975. doi: 10.1038/cmi.2017.88
Liu, Y., Tan, S., Huang, L., Abramovitch, R. B., Rohde, K. H., Zimmerman, M. D., et al. (2016). Immune activation of the host cell induces drug tolerance in mycobacterium tuberculosis both in vitro and in vivo. J. Exp. Med. 213, 809–825. doi: 10.1084/jem.20151248
Liu, D., Zhang, J., Pan, Z., Mai, J., Mei, H., Dai, Y., et al. (2020). Over-expression of Tgs1 in mycobacterium marinum enhances virulence in adult zebrafish. Int. J. Med. Microbiol. 310, 151378. doi: 10.1016/j.ijmm.2019.151378
Lofthouse, E. K., Wheeler, P. R., Beste, D. J. V., Khatri, B. L., Wu, H., Mendum, T. A., et al. (2013). Systems-based approaches to probing metabolic variation within the mycobacterium tuberculosis complex. PloS One 8, e75913. doi: 10.1371/journal.pone.0075913
Luthuli, B. B., Purdy, G. E., Balagaddé, F. K. (2015). Confinement-induced drug-tolerance in mycobacteria mediated by an efflux mechanism. PloS One 10, e0136231. doi: 10.1371/journal.pone.0136231
Lyu, L.-D., Tang, B.-K., Fan, X.-Y., Ma, H., Zhao, G.-P. (2013). Mycobacterial MazG safeguards genetic stability via housecleaning of 5-OH-dCTP. PloS Pathog. 9, e1003814. doi: 10.1371/journal.ppat.1003814
MacGilvary, N. J., Kevorkian, Y. L., Tan, S. (2019). Potassium response and homeostasis in mycobacterium tuberculosis modulates environmental adaptation and is important for host colonization. PloS Pathog. 15, e1007591. doi: 10.1371/journal.ppat.1007591
MacMicking, J. D. (2014). Cell-autonomous effector mechanisms against mycobacterium tuberculosis. Cold Spring Harb. Perspect. Med. 4, a018507. doi: 10.1101/cshperspect.a018507
Malherbe, S. T., Shenai, S., Ronacher, K., Loxton, A. G., Dolganov, G., Kriel, M., et al. (2016). Persisting positron emission tomography lesion activity and mycobacterium tuberculosis mRNA after tuberculosis cure. Nat. Med. 22, 1094–1100. doi: 10.1038/nm.4177
Malik, Z. A., Denning, G. M., Kusner, D. J. (2000). Inhibition of Ca2+ signaling by mycobacterium tuberculosisIs associated with reduced phagosome–lysosome fusion and increased survival within human macrophages. J. Exp. Med. 191, 287–302. doi: 10.1084/jem.191.2.287
Manina, G., Dhar, N., McKinney, J. D. (2015). Stress and host immunity amplify mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46. doi: 10.1016/j.chom.2014.11.016
Marakalala, M. J., Ndlovu, H. (2017). Signaling c-type lectin receptors in antimycobacterial immunity. PloS Pathog. 13, e1006333. doi: 10.1371/journal.ppat.1006333
Marino, S., Cilfone, N. A., Mattila, J. T., Linderman, J. J., Flynn, J. L., Kirschner, D. E. (2015). Macrophage polarization drives granuloma outcome during mycobacterium tuberculosis infection. Infect. Immun. 83, 324–338. doi: 10.1128/IAI.02494-14
Martin, C. J., Cadena, A. M., Leung, V. W., Lin, P. L., Maiello, P., Hicks, N., et al. (2017). Digitally barcoding mycobacterium tuberculosis reveals in vivo infection dynamics in the macaque model of tuberculosis. mBio 8, e00312–17. doi: 10.1128/mBio.00312-17
Mascolo, L., Bald, D. (2019). Cytochrome bd in mycobacterium tuberculosis: A respiratory chain protein involved in the defense against antibacterials. Prog. Biophys. Mol. Biol 152, 55–63. doi: 10.1016/j.pbiomolbio.2019.11.002
Mattila, J. T., Ojo, O. O., Kepka-Lenhart, D., Marino, S., Kim, J. H., Eum, S. Y., et al. (2013). Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J. Immunol. 191, 773–784. doi: 10.4049/jimmunol.1300113
Maurya, R. K., Bharti, S., Krishnan, M. Y. (2018). Triacylglycerols: Fuelling the hibernating mycobacterium tuberculosis. front. Cell. Infect. Microbiol. 8. doi: 10.3389/fcimb.2018.00450
McMahon, M. D., Rush, J. S., Thomas, M. G. (2012). Analyses of MbtB, MbtE, and MbtF suggest revisions to the mycobactin biosynthesis pathway in mycobacterium tuberculosis. J. Bacteriol. 194, 2809–2818. doi: 10.1128/JB.00088-12
Mehta, M., Rajmani, R. S., Singh, A. (2016). Mycobacterium tuberculosis WhiB3 responds to vacuolar pH-induced changes in mycothiol redox potential to modulate phagosomal maturation and virulence. J. Biol. Chem. 291, 2888–2903. doi: 10.1074/jbc.M115.684597
Mehta, M., Singh, A. (2019). Mycobacterium tuberculosis WhiB3 maintains redox homeostasis and survival in response to reactive oxygen and nitrogen species. Free Radic. Biol. Med. 131, 50–58. doi: 10.1016/j.freeradbiomed.2018.11.032
Mezouar, S., Diarra, I., Roudier, J., Desnues, B., Mege, J.-L. (2019). Tumor necrosis factor-alpha antagonist interferes with the formation of granulomatous multinucleated giant cells: New insights into mycobacterium tuberculosis infection. Front. Immunol. 10, 1947. doi: 10.3389/fimmu.2019.01947
Minato, Y., Thiede, J. M., Kordus, S. L., McKlveen, E. J., Turman, B. J., Baughn, A. D. (2015). Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob. Agents Chemother. 59, 5097–5106. doi: 10.1128/AAC.00647-15
Mishra, R., Kohli, S., Malhotra, N., Bandyopadhyay, P., Mehta, M., Munshi, M., et al. (2019). Targeting redox heterogeneity to counteract drug tolerance in replicating mycobacterium tuberculosis. Sci. Transl. Med. 11, eaaw6635. doi: 10.1126/scitranslmed.aaw6635
Moldoveanu, A. L., Rycroft, J. A., Helaine, S. (2021). Impact of bacterial persisters on their host. Curr. Opin. Microbiol. 59, 65–71. doi: 10.1016/j.mib.2020.07.006
Mouton, J. M., Helaine, S., Holden, D. W., Sampson, S. L. (2016). Elucidating population-wide mycobacterial replication dynamics at the single-cell level. Microbiol. Read. Engl 162, 966–78. doi: 10.1099/mic.0.000288
Nair, R. R., Sharan, D., Ajitkumar, P. (2019). A minor subpopulation of mycobacteria inherently produces high levels of reactive oxygen species that generate antibiotic resisters at high frequency from itself and enhance resister generation from its major kin subpopulation. Front. Microbiol 10, 1842. doi: 10.3389/fmicb.2019.01842
Namugenyi, S. B., Aagesen, A. M., Elliott, S. R., Tischler, A. D. (2017). Mycobacterium tuberculosis PhoY proteins promote persister formation by mediating Pst/SenX3-RegX3 phosphate sensing. mBio 8, e00494-17. doi: 10.1128/mBio.00494-17
Nixon, M. R., Saionz, K. W., Koo, M.-S., Szymonifka, M. J., Jung, H., Roberts, J. P., et al. (2014). Folate pathway disruption leads to critical disruption of methionine derivatives in mycobacterium tuberculosis. chem. Biol. 21, 819–830. doi: 10.1016/j.chembiol.2014.04.009
Noy, T., Vergnolle, O., Hartman, T. E., Rhee, K. Y., Jacobs, W. R., Berney, M., et al. (2016). Central role of pyruvate kinase in carbon Co-catabolism of mycobacterium tuberculosis. J. Biol. Chem. 291, 7060–7069. doi: 10.1074/jbc.M115.707430
Ojha, A. K., Baughn, A. D., Sambandan, D., Hsu, T., Trivelli, X., Guerardel, Y., et al. (2008). Growth of mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69, 164–174. doi: 10.1111/j.1365-2958.2008.06274.x
Pandey, R., Rodriguez, G. M. (2014). IdeR is required for iron homeostasis and virulence in mycobacterium tuberculosis. Mol. Microbiol. 91, 98–109. doi: 10.1111/mmi.12441
Pandey, A. K., Sassetti, C. M. (2008). Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. 105, 4376–4380. doi: 10.1073/pnas.0711159105
Pandey, M., Talwar, S., Bose, S., Pandey, A. K. (2018). Iron homeostasis in mycobacterium tuberculosis is essential for persistence. Sci. Rep. 8, 17359. doi: 10.1038/s41598-018-35012-3
Parbhoo, T., Sampson, S. L., Mouton, J. M. (2020). Recent developments in the application of flow cytometry to advance our understanding of mycobacterium tuberculosis physiology and pathogenesis. Cytometry A 97, 683–693. doi: 10.1002/cyto.a.24030
Passemar, C., Arbués, A., Malaga, W., Mercier, I., Moreau, F., Lepourry, L., et al. (2014). Multiple deletions in the polyketide synthase gene repertoire of mycobacterium tuberculosis reveal functional overlap of cell envelope lipids in host–pathogen interactions. Cell. Microbiol. 16, 195–213. doi: 10.1111/cmi.12214
Patin, E. C., Geffken, A. C., Willcocks, S., Leschczyk, C., Haas, A., Nimmerjahn, F., et al. (2017). Trehalose dimycolate interferes with FcγR-mediated phagosome maturation through mincle, SHP-1 and FcγRIIB signalling. PLo One 12, e0174973. doi: 10.1371/journal.pone.0174973
Pauwels, A.-M., Trost, M., Beyaert, R., Hoffmann, E. (2017). Patterns, receptors, and signals: Regulation of phagosome maturation. Trends Immunol. 38, 407–422. doi: 10.1016/j.it.2017.03.006
Pavelka, M. S., Chen, B., Kelley, C. L., Collins, F. M., Jacobs, W. R. (2003). Vaccine efficacy of a lysine auxotroph of mycobacterium tuberculosis. infect. Immun. 71, 4190–4192. doi: 10.1128/IAI.71.7.4190-4192.2003
Peterson, E. J. R., Ma, S., Sherman, D. R., Baliga, N. S. (2016). Network analysis identifies Rv0324 and Rv0880 as regulators of bedaquiline tolerance in mycobacterium tuberculosis. nat. Microbiol. 1, 16078. doi: 10.1038/nmicrobiol.2016.78
Peyron, P., Vaubourgeix, J., Poquet, Y., Levillain, F., Botanch, C., Bardou, F., et al. (2008). Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for m. tuberculosis persistence. PloS Pathog. 4, e1000204. doi: 10.1371/journal.ppat.1000204
Pinto, R., Tang, Q. X., Britton, W. J., Leyh, T. S., Triccas, J. A. (2004). The mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex. Microbiol. Read. Engl. 150, 1681–1686. doi: 10.1099/mic.0.26894-0
Pisu, D., Huang, L., Grenier, J. K., Russell, D. G. (2020). Dual RNA-seq of mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 30, 335–350.e4. doi: 10.1016/j.celrep.2019.12.033
Priestman, M., Thomas, P., Robertson, B. D., Shahrezaei, V. (2017). Mycobacteria modify their cell size control under Sub-optimal carbon sources. Front. Cell Dev. Biol. 5. doi: 10.3389/fcell.2017.00064
Prosser, G., Brandenburg, J., Reiling, N., Barry, C. E., Wilkinson, R. J., Wilkinson, K. A. (2017). The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect. 19, 177–192. doi: 10.1016/j.micinf.2016.10.001
Qualls, J. E., Murray, P. J. (2016). Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin. Immunopathol. 38, 139–152. doi: 10.1007/s00281-015-0534-0
Queval, C. J., Brosch, R., Simeone, R. (2017a). The macrophage: A disputed fortress in the battle against mycobacterium tuberculosis. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02284
Queval, C. J., Song, O.-R., Carralot, J.-P., Saliou, J.-M., Bongiovanni, A., Deloison, G., et al. (2017b). Mycobacterium tuberculosis controls phagosomal acidification by targeting CISH-mediated signaling. Cell Rep. 20, 3188–3198. doi: 10.1016/j.celrep.2017.08.101
Quigley, J., Hughitt, V. K., Velikovsky, C. A., Mariuzza, R. A., El-Sayed, N. M., Briken, V. (2017). The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of mycobacterium tuberculosis. mBio 8, e00148-17. doi: 10.1128/mBio.00148-17
Rao, S. P. S., Alonso, S., Rand, L., Dick, T., Pethe, K. (2008). The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 105, 11945–11950. doi: 10.1073/pnas.0711697105
Refai, A., Gritli, S., Barbouche, M.-R., Essafi, M. (2018). Mycobacterium tuberculosis virulent factor ESAT-6 drives macrophage differentiation toward the pro-inflammatory M1 phenotype and subsequently switches it to the anti-inflammatory M2 phenotype. Front. Cell. Infect. Microbiol 8, 327. doi: 10.3389/fcimb.2018.00327
Rego, E. H., Audette, R. E., Rubin, E. J. (2017). Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157. doi: 10.1038/nature22361
Reichlen, M. J., Leistikow, R. L., Scobey, M. S., Born, S. E. M., Voskuil, M. I. (2017). Anaerobic mycobacterium tuberculosis cell death stems from intracellular acidification mitigated by the DosR regulon. J. Bacteriol. 199, e00320-17. doi: 10.1128/JB.00320-17
Rhee, K. Y., Erdjument-Bromage, H., Tempst, P., Nathan, C. F. (2005). S-nitroso proteome of mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc. Natl. Acad. Sci. U. S. A. 102, 467–472. doi: 10.1073/pnas.0406133102
Richard-Greenblatt, M., Bach, H., Adamson, J., Peña-Diaz, S., Li, W., Steyn, A. J. C., et al. (2015). Regulation of ergothioneine biosynthesis and its effect on mycobacterium tuberculosis growth and infectivity. J. Biol. Chem. 290, 23064–23076. doi: 10.1074/jbc.M115.648642
Richardson, K., Bennion, O. T., Tan, S., Hoang, A. N., Cokol, M., Aldridge, B. B. (2016). Temporal and intrinsic factors of rifampicin tolerance in mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 113, 8302–8307. doi: 10.1073/pnas.1600372113
Rifat, D., Belchis, D. A., Karakousis, P. C. (2014). senX3-independent contribution of regX3 to mycobacterium tuberculosis virulence. BMC Microbiol. 14, 265. doi: 10.1186/s12866-014-0265-8
Rifat, D., Bishai, W. R., Karakousis, P. C. (2009). Phosphate depletion: A novel trigger for mycobacterium tuberculosis persistence. J. Infect. Dis. 200, 1126–1135. doi: 10.1086/605700
Rodriguez, G. M., Smith, I. (2003). Mechanisms of iron regulation in mycobacteria: role in physiology and virulence. Mol. Microbiol. 47, 1485–1494. doi: 10.1046/j.1365-2958.2003.03384.x
Rodriguez, G. M., Voskuil, M. I., Gold, B., Schoolnik, G. K., Smith, I. (2002). ideR, an essential gene in mycobacterium tuberculosis: Role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect. Immun. 70, 3371–3381. doi: 10.1128/IAI.70.7.3371-3381.2002
Rohde, K. H., Veiga, D. F. T., Caldwell, S., Balázsi, G., Russell, D. G. (2012). Linking the transcriptional profiles and the physiological states of mycobacterium tuberculosis during an extended intracellular infection. PloS Pathog. 8, e1002769. doi: 10.1371/journal.ppat.1002769
Russell, D. G., Cardona, P.-J., Kim, M.-J., Allain, S., Altare, F. (2009). Foamy macrophages and the progression of the human TB granuloma. Nat. Immunol. 10, 943–948. doi: 10.1038/ni.1781
Russell, D. G., Huang, L., VanderVen, B. C. (2019). Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 19, 291–304. doi: 10.1038/s41577-019-0124-9
Ryan, G. J., Hoff, D.R., Driver, E.R., Voskuil, M.I., Gonzalez-Juarrero, M., Basaraba, R.J., et al (2010). Multiple M. tuberculosis Phenotypes in mouse and guinea pig lung tissue revealed by a dual-staining approach. PLOS ONE 5, e11108. doi: 10.1371/journal.pone.0011108
Saini, V., Cumming, B. M., Guidry, L., Lamprecht, D. A., Adamson, J. H., Reddy, V. P., et al. (2016). Ergothioneine maintains redox and bioenergetic homeostasis essential for drug susceptibility and virulence of mycobacterium tuberculosis. Cell Rep. 14, 572–585. doi: 10.1016/j.celrep.2015.12.056
Salina, E. G., Grigorov, A. S., Bychenko, O. S., Skvortsova, Y. V., Mamedov, I. Z., Azhikina, T. L., et al. (2019). Resuscitation of dormant “Non-culturable” mycobacterium tuberculosis is characterized by immediate transcriptional burst. Front. Cell. Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00272
Salina, E. G., Waddell, S. J., Hoffmann, N., Rosenkrands, I., Butcher, P. D., Kaprelyants, A. S. (2014). Potassium availability triggers mycobacterium tuberculosis transition to, and resuscitation from, non-culturable (dormant) states. Open Biol. 4, 140106–140106. doi: 10.1098/rsob.140106
Sarathy, J. P., Via, L. E., Weiner, D., Blanc, L., Boshoff, H., Eugenin, E. A., et al. (2018). Extreme drug tolerance of mycobacterium tuberculosis in caseum. Antimicrob. Agents Chemother. 62, e02266–17. doi: 10.1128/AAC.02266-17
Sarathy, J. P., Zuccotto, F., Hsinpin, H., Sandberg, L., Via, L. E., Marriner, G. A., et al. (2016). Prediction of drug penetration in tuberculosis lesions. ACS Infect. Dis. 2, 552–563. doi: 10.1021/acsinfecdis.6b00051
Savvi, S., Warner, D. F., Kana, B. D., McKinney, J. D., Mizrahi, V., Dawes, S. S. (2008). Functional characterization of a vitamin B12-dependent methylmalonyl pathway in mycobacterium tuberculosis: Implications for propionate metabolism during growth on fatty acids. J. Bacteriol. 190, 3886–3895. doi: 10.1128/JB.01767-07
Schnettger, L., Rodgers, A., Repnik, U., Lai, R. P., Pei, G., Verdoes, M., et al. (2017). A Rab20-dependent membrane trafficking pathway controls m. tuberculosis replication by regulating phagosome spaciousness and integrity. Cell Host Microbe 21, 619–628.e5. doi: 10.1016/j.chom.2017.04.004
Senaratne, R. H., Silva, A. D. D., Williams, S. J., Mougous, J. D., Reader, J. R., Zhang, T., et al. (2006). 5′-adenosinephosphosulphate reductase (CysH) protects mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol. Microbiol. 59, 1744–1753. doi: 10.1111/j.1365-2958.2006.05075.x
Serafini, A., Pisu, D., Palù, G., Rodriguez, G. M., Manganelli, R. (2013). The ESX-3 secretion system is necessary for iron and zinc homeostasis in mycobacterium tuberculosis. PloS One 8, e78351. doi: 10.1371/journal.pone.0078351
Sershen, C. L., Plimpton, S. J., May, E. E. (2016). Oxygen modulates the effectiveness of granuloma mediated host response to mycobacterium tuberculosis: A multiscale computational biology approach. Front. Cell. Infect. Microbiol. 6. doi: 10.3389/fcimb.2016.00006
Sevalkar, R. R., Arora, D., Singh, P. R., Singh, R., Nandicoori, V. K., Karthikeyan, S., et al. (2019). Functioning of mycobacterial heat shock repressors requires the master virulence regulator PhoP. J. Bacteriol. 201, e00013–19. doi: 10.1128/JB.00013-19
Shi, K.-X., Wu, Y.-K., Tang, B.-K., Zhao, G.-P., Lyu, L.-D. (2019). Housecleaning of pyrimidine nucleotide pool coordinates metabolic adaptation of nongrowing mycobacterium tuberculosis. emerg. Microbes Infect. 8, 40–44. doi: 10.1080/22221751.2018.1559706
Shui, W., Petzold, C. J., Redding, A., Liu, J., Pitcher, A., Sheu, L., et al. (2011). Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J. Proteome Res. 10, 339–348. doi: 10.1021/pr100688h
Sia, J. K., Rengarajan, J. (2019). Immunology of mycobacterium tuberculosis infections. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.GPP3-0022-2018
Siegrist, M. S., Unnikrishnan, M., McConnell, M. J., Borowsky, M., Cheng, T.-Y., Siddiqi, N., et al. (2009). Mycobacterial esx-3 is required for mycobactin-mediated iron acquisition. Proc. Natl. Acad. Sci. U. S. A. 106, 18792–18797. doi: 10.1073/pnas.0900589106
Simeone, R., Sayes, F., Song, O., Gröschel, M. I., Brodin, P., Brosch, R., et al. (2015). Cytosolic access of mycobacterium tuberculosis: Critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PloS Pathog. 11, e1004650. doi: 10.1371/journal.ppat.1004650
Singh, A., Crossman, D. K., Mai, D., Guidry, L., Voskuil, M. I., Renfrow, M. B., et al. (2009). Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PloS Pathog. 5, e1000545. doi: 10.1371/journal.ppat.1000545
Singh, S., Goswami, N., Tyagi, A. K., Khare, G. (2019). Unraveling the role of the transcriptional regulator VirS in low pH-induced responses of mycobacterium tuberculosis and identification of VirS inhibitors. J. Biol. Chem. 294, 10055–75. doi: 10.1074/jbc.RA118.005312
Singh, V., Jamwal, S., Jain, R., Verma, P., Gokhale, R., Rao, K. V. S. (2012). Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12, 669–681. doi: 10.1016/j.chom.2012.09.012
Singh, R., Singh, M., Arora, G., Kumar, S., Tiwari, P., Kidwai, S. (2013). Polyphosphate deficiency in mycobacterium tuberculosis is associated with enhanced drug susceptibility and impaired growth in Guinea pigs. J. Bacteriol. 195, 2839–2851. doi: 10.1128/JB.00038-13
Singh, P. R., Vijjamarri, A. K., Sarkar, D. (2020). Metabolic switching of mycobacterium tuberculosis during hypoxia is controlled by the virulence regulator PhoP. J. Bacteriol. 202, e00705–19. doi: 10.1128/JB.00705-19
Sloan, D. J., Mwandumba, H. C., Garton, N. J., Khoo, S. H., Butterworth, A. E., Allain, T. J., et al (2015). Pharmacodynamic modeling of bacillary elimination rates and detection of bacterial lipid bodies in sputum to predict and understand outcomes in treatment of pulmonary tuberculosis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 61, 1–8. doi: 10.1093/cid/civ195
Small, J. L., Park, S. W., Kana, B. D., Ioerger, T. R., Sacchettini, J. C., Ehrt, S. (2013). Perturbation of cytochrome c maturation reveals adaptability of the respiratory chain in mycobacterium tuberculosis. mBio 4, e00475–00413. doi: 10.1128/mBio.00475-13
Smith, T. C., Pullen, K. M., Olson, M. C., McNellis, M. E., Richardson, I., Hu, S., et al. (2020). Morphological profiling of tubercle bacilli identifies drug pathways of action. Proc. Natl. Acad. Sci. 117, 18744–18753. doi: 10.1073/pnas.2002738117
Sohaskey, C. D. (2008). Nitrate enhances the survival of mycobacterium tuberculosis during inhibition of respiration. J. Bacteriol. 190, 2981–2986. doi: 10.1128/JB.01857-07
Song, H., Huff, J., Janik, K. E., Walter, K., Keller, C. E. M., Ehlers, S., et al. (2011). Expression of the ompATb operon accelerates ammonia secretion and adaptation of mycobacterium tuberculosis to acidic environments. Mol. Microbiol. 80, 900–918. doi: 10.1111/j.1365-2958.2011.07619.x
Song, H., Niederweis, M. (2012). Uptake of sulfate but not phosphate by mycobacterium tuberculosis is slower than that for mycobacterium smegmatis. J. Bacteriol. 194, 956–964. doi: 10.1128/JB.06132-11
Tan, S., Russell, D. G. (2015). Trans-species communication in the mycobacterium tuberculosis-infected macrophage. Immunol. Rev. 264, 233–248. doi: 10.1111/imr.12254
Tan, M. P., Sequeira, P., Lin, W. W., Phong, W. Y., Cliff, P., Ng, S. H., et al. (2010). Nitrate respiration protects hypoxic mycobacterium tuberculosis against acid- and reactive nitrogen species stresses. PloS One 5, e13356. doi: 10.1371/journal.pone.0013356
Thayil, S. M., Morrison, N., Schechter, N., Rubin, H., Karakousis, P. C. (2011). The role of the novel exopolyphosphatase MT0516 in mycobacterium tuberculosis drug tolerance and persistence. PloS One 6, e28076. doi: 10.1371/journal.pone.0028076
Thiede, J. M., Dillon, N. A., Howe, M. D., Aflakpui, R., Modlin, S. J., Hoffner, S. E., et al. (2022). Pyrazinamide susceptibility is driven by activation of the SigE-dependent cell envelope stress response in mycobacterium tuberculosis. mBio 13, e00439-21. doi: 10.1128/mbio.00439-21
Tischler, A. D., Leistikow, R. L., Kirksey, M. A., Voskuil, M. I., McKinney, J. D. (2013). Mycobacterium tuberculosis requires phosphate-responsive gene regulation to resist host immunity. Infect. Immun. 81, 317–328. doi: 10.1128/IAI.01136-12
Tiwari, S., Tonder, A. J., Vilchèze, C., Mendes, V., Thomas, S. E., Malek, A., et al. (2018). Arginine-deprivation–induced oxidative damage sterilizes mycobacterium tuberculosis. Proc. Natl. Acad. Sci. 115, 9779–9784. doi: 10.1073/pnas.1808874115
Torrelles, J. B., Schlesinger, L. S. (2010). Diversity in mycobacterium tuberculosis mannosylated cell wall determinants impacts adaptation to the host. Tuberc. Edinb. Scotl. 90, 84–93. doi: 10.1016/j.tube.2010.02.003
Tyagi, P., Dharmaraja, A. T., Bhaskar, A., Chakrapani, H., Singh, A. (2015). Mycobacterium tuberculosis has diminished capacity to counteract redox stress induced by elevated levels of endogenous superoxide. Free Radic. Biol. Med. 84, 344–354. doi: 10.1016/j.freeradbiomed.2015.03.008
Vandal, O. H., Pierini, L. M., Schnappinger, D., Nathan, C. F., Ehrt, S. (2008). A membrane protein preserves intrabacterial pH in intraphagosomal mycobacterium tuberculosis. Nat. Med. 14, 849–854. doi: 10.1038/nmXXXX
Vaubourgeix, J., Lin, G., Dhar, N., Chenouard, N., Jiang, X., Botella, H., et al. (2015). Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe 17, 178–190. doi: 10.1016/j.chom.2014.12.008
Via, L. E., Lin, P. L., Ray, S. M., Carrillo, J., Allen, S. S., Eum, S. Y., et al. (2008). Tuberculous granulomas are hypoxic in Guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 76, 2333–2340. doi: 10.1128/IAI.01515-07
Vijay, S., Hai, H. T., Thu, D. D. A., Johnson, E., Pielach, A., Phu, N. H., et al. (2018). Ultrastructural analysis of cell envelope and accumulation of lipid inclusions in clinical mycobacterium tuberculosis isolates from sputum, oxidative stress, and iron deficiency. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02681
Vijay, S., Nair, R. R., Sharan, D., Jakkala, K., Mukkayyan, N., Swaminath, S., et al. (2017a). Mycobacterial cultures contain cell size and density specific Sub-populations of cells with significant differential susceptibility to antibiotics, oxidative and nitrite stress. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00463
Vijay, S., Vinh, D. N., Hai, H. T., Ha, V. T. N., Dung, V. T. M., Dinh, T. D., et al. (2017b). Influence of stress and antibiotic resistance on cell-length distribution in mycobacterium tuberculosis clinical isolates. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02296
Vilchèze, C., Hartman, T., Weinrick, B., Jacobs, W. R. (2013). Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin c-induced fenton reaction. Nat. Commun. 4, 1881. doi: 10.1038/ncomms2898
Vilchèze, C., Hartman, T., Weinrick, B., Jain, P., Weisbrod, T. R., Leung, L. W., et al. (2017). Enhanced respiration prevents drug tolerance and drug resistance in mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 114, 4495–4500. doi: 10.1073/pnas.1704376114
Vilchèze, C., Kremer, L. (2017). Acid-fast positive and acid-fast negative mycobacterium tuberculosis: The Koch paradox. Microbiol. Spectr. 5, 5.2.15. doi: 10.1128/microbiolspec.TBTB2-0003-2015
Vilchèze, C., Molle, V., Carrère-Kremer, S., Leiba, J., Mourey, L., Shenai, S., et al. (2014). Phosphorylation of KasB regulates virulence and acid-fastness in mycobacterium tuberculosis. PloS Pathog. 10, e1004115. doi: 10.1371/journal.ppat.1004115
Voskuil, M. I., Bartek, I. L., Visconti, K., Schoolnik, G. K. (2011). The response of mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2, 105. doi: 10.3389/fmicb.2011.00105
Voskuil, M. I., Schnappinger, D., Visconti, K. C., Harrell, M. I., Dolganov, G. M., Sherman, D. R., et al. (2003). Inhibition of respiration by nitric oxide induces a mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713. doi: 10.1084/jem.20030205
Wakamoto, Y., Dhar, N., Chait, R., Schneider, K., Signorino-Gelo, F., Leibler, S., et al. (2013). Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95. doi: 10.1126/science.1229858
Walters, S. B., Dubnau, E., Kolesnikova, I., Laval, F., Daffe, M., Smith, I. (2006). The mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 60, 312–330. doi: 10.1111/j.1365-2958.2006.05102.x
Warner, D. F., Savvi, S., Mizrahi, V., Dawes, S. S. (2007). A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in mycobacterium tuberculosis: Implications for differential methionine synthase function in strains H37Rv and CDC1551. J. Bacteriol. 189, 3655–3659. doi: 10.1128/JB.00040-07
Williams, M. J., Shanley, C. A., Zilavy, A., Peixoto, B., Manca, C., Kaplan, G., et al. (2015). Bis-molybdopterin guanine dinucleotide is required for persistence of mycobacterium tuberculosis in Guinea pigs. Infect. Immun. 83, 544–550. doi: 10.1128/IAI.02722-14
Wright, C. C., Hsu, F. F., Arnett, E., Dunaj, J. L., Davidson, P. M., Pacheco, S. A., et al. (2017). The mycobacterium tuberculosis MmpL11 cell wall lipid transporter is important for biofilm formation, intracellular growth, and nonreplicating persistence. Infect. Immun. 85, e00131–17. doi: 10.1128/IAI.00131-17
Yamada, H., Bhatt, A., Danev, R., Fujiwara, N., Maeda, S., Mitarai, S., et al. (2012). Non-acid-fastness in mycobacterium tuberculosis ΔkasB mutant correlates with the cell envelope electron density. Tuberculosis 92, 351–357. doi: 10.1016/j.tube.2012.02.006
Zhang, Y. J., Reddy, M. C., Ioerger, T. R., Rothchild, A. C., Dartois, V., Schuster, B. M., et al. (2013). Tryptophan biosynthesis protects mycobacteria from CD4 T cell-mediated killing. Cell 155, 1296–1308. doi: 10.1016/j.cell.2013.10.045
Zheng, J., Rubin, E. J., Bifani, P., Mathys, V., Lim, V., Au, M., et al. (2013). Para-aminosalicylic acid is a prodrug targeting dihydrofolate reductase in mycobacterium tuberculosis. J. Biol. Chem. 288, 23447–23456. doi: 10.1074/jbc.M113.475798
Zimmerman, M., Lestner, J., Prideaux, B., O’Brien, P., Dias-Freedman, I., Chen, C., et al. (2017). Ethambutol partitioning in tuberculous pulmonary lesions explains its clinical efficacy. Antimicrob. Agents Chemother. 61, e00924–17. doi: 10.1128/AAC.00924-17
Keywords: Mycobacterium tuberculosis, persistence, persisters, bacterial heterogeneity, host-pathogen interaction
Citation: Parbhoo T, Mouton JM and Sampson SL (2022) Phenotypic adaptation of Mycobacterium tuberculosis to host-associated stressors that induce persister formation. Front. Cell. Infect. Microbiol. 12:956607. doi: 10.3389/fcimb.2022.956607
Received: 30 May 2022; Accepted: 24 August 2022;
Published: 27 September 2022.
Edited by:
Bavesh Kana, University of the Witwatersrand, South AfricaReviewed by:
Lee-Ann H Allen, University of Missouri, United StatesCopyright © 2022 Parbhoo, Mouton and Sampson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samantha L. Sampson, c3NhbXBzb25Ac3VuLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.