
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell. Infect. Microbiol. , 19 October 2022
Sec. Clinical Microbiology
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.955481
This article is part of the Research Topic Improving the Clinical Effectiveness of Metagenomic Next Generation Sequencing (mNGS) in Infection Disease Diagnosis and Treatment: Linking the NGS Specialists and Clinicians View all 44 articles
 Milena Pitashny1,2*
Milena Pitashny1,2* Balqees Kadry1Raya Shalaginov2Liat Gazit2Yaniv Zohar3Moran Szwarcwort2Yoav Stabholz4Mical Paul4
Balqees Kadry1Raya Shalaginov2Liat Gazit2Yaniv Zohar3Moran Szwarcwort2Yoav Stabholz4Mical Paul4We hypothesized that targeted NGS sequencing might have an advantage over Sanger sequencing, especially in polymicrobial infections. The study included 55 specimens from 51 patients. We compared targeted NGS to Sanger sequencing in clinical samples submitted for Sanger sequencing. The overall concordance rate was 58% (32/55) for NGS vs. Sanger. NGS identified 9 polymicrobial and 2 monomicrobial infections among 19 Sanger-negative samples and 8 polymicrobial infections in 11 samples where a 16S gene was identified by gel electrophoresis, but could not be mapped to an identified pathogen by Sanger. We estimated that NGS could have contributed to patient management in 6/18 evaluated patients and thus has an advantage over Sanger sequencing in certain polymicrobial infections.
Most clinical infections are not microbiologically confirmed. The problem is especially pertinent in deep seated invasive infections, where microbiological diagnosis is critical and the specimen is precious, such as osteomyelitis, deep organ abscesses, brain empyema or others. For these cases, there has been a growing interest and implementation of broad-range polymerase chain reaction (PCR) based Sanger sequencing of the 16S ribosomal RNA (rRNA) bacterial gene (panbacterial PCR), directly from clinical specimens (Rampini et al., 2011). Sanger sequencing significantly improved the diagnostic yield in clinical culture isolates as well as mono-microbial infections (Shachor-Meyouhas et al., 2013; Khoury et al., 2019). However, when more than one species are present in the specimen, Sanger sequencing template reads are superimposed and are generally uninterpretable (Salipante et al., 2013). Results from such specimens are reported as negative or as a mixture of bacteria, without further identification.
As in other fields of medicine, next generation sequencing (NGS) technologies have expanded diagnostic capabilities in clinical microbiology laboratories. Recent studies have highlighted the ability of 16S rRNA NGS to accurately reach speciation and quantify bacterial abundances in complex polymicrobial infections (Salipante et al., 2013; Abayasekara et al., 2017; Culbreath et al., 2019).
We hypothesized that for difficult-to-diagnose infections, especially when polymicrobial, targeted NGS of the 16S rRNA gene has better diagnostic performance than panbacterial Sanger sequencing.
We used residual nucleic acids from clinical specimens that were submitted to our reference molecular laboratory at RHCC for panbacterial, panfungal or mycobacterial PCR (Sanger sequencing). Samples were collected between 2020-2021 at Rambam Health Care Campus (RHCC) or other hospitals. DNA stored at -20oC was retrieved and tested by NGS retrospectively. Clinical information was available only for samples sent from within RHCC.
Broad-range 16S rRNA gene Sanger sequencing was performed using in-house protocols at the molecular bacteriology laboratory (Shachor-Meyouhas et al., 2013).
NGS library preparation and analyses were performed blinded to clinical information, culture or Sanger sequencing results. DNA extraction of bacterial DNA from each specimen was carried out using the QIAAmp® DNA Mini kit (QiagenGroup) according to manufacturer’s instructions. Each batch of specimens were extracted with negative controls (extraction control). PCR amplification of the hypervariable region V4 of the 16s rRNA gene was conducted using PCRBIO HS Taq Mix Red according to the Earth Microbiome Project primer pairs (Apprill et al., 2015; Parada et al., 2016), and in selected cases, the addition of V1-2 segments (F27-R338) of the same gene to better identify certain bacteria and improve speciation (such as for Staphylococcus and Enterobacterales) (Walker et al., 2015). PCR products were run on a 1.5% agarose gel. Final library products were purified using Qiaquick PCR Purification Kit (Qiagen Groups) according to manufacturer’s instructions and quantified using Qubit™ dsDNA HS and BR Assay Kits (Invitrogen).
We sequenced the amplified V1-2 or V4 regions of 16s rRNA gene with Ion S5™ System (Thermo Fisher Scientific). Data were analyzed using the Ion Reporter bioinformatics Software pipeline (Thermo Fisher Scientific), using a threshold of 1000 mapped reads for designating significant pathogens. BAM files uploaded to the Ion Reporter were mapped to the Silva 138 SSU database. Typical contaminants found in negative controls, such as Acinetobacter lwofii, Acinetobacter schendleri, or Xanthomonadaceae which are water tolerant bacteria, were subtracted from overall reads obtained on the clinical samples.
Concordance between Sanger sequencing and NGS results was evaluated. For the RHCC samples, we estimated the potential clinical added value of NGS, had it been available in real time. Two physicians (infectious diseases and clinical microbiologist) evaluated each case and assigned the potential contribution of NGS to patient management independently (diagnosis and treatment). Disagreements were resolved by consensus.
The study was approved by the local ethics committee with a waiver of informed consent. NGS results were not conveyed to clinicians.
The study included 55 specimens from 51 patients; 22 specimens from RHCC and all others from other hospitals. Of the 55 specimens evaluated, 25 specimens were Sanger positive with one organism reported. Of those, 24 were concordant by NGS (24/25) that identified the exact Sanger pathogen alone (N=12) or with additional pathogen/s (N=12). The only discordant result was a Propionibacterium spp identified by Sanger, that was missed by NGS. Sanger was negative in 19 samples, of which 8 were negative by NGS, 9 polymicrobial and 2 monomicrobial by NGS. In 11 samples, broad-range 16S gene was identified by gel electrophoresis, but could not be mapped to an identified pathogen within available databases when sequenced by Sanger technology (possible polymicrobial infection); among these, 3/11 were negative on NGS and all others were positive with polymicrobial identification (Figure 1 and Table 1).
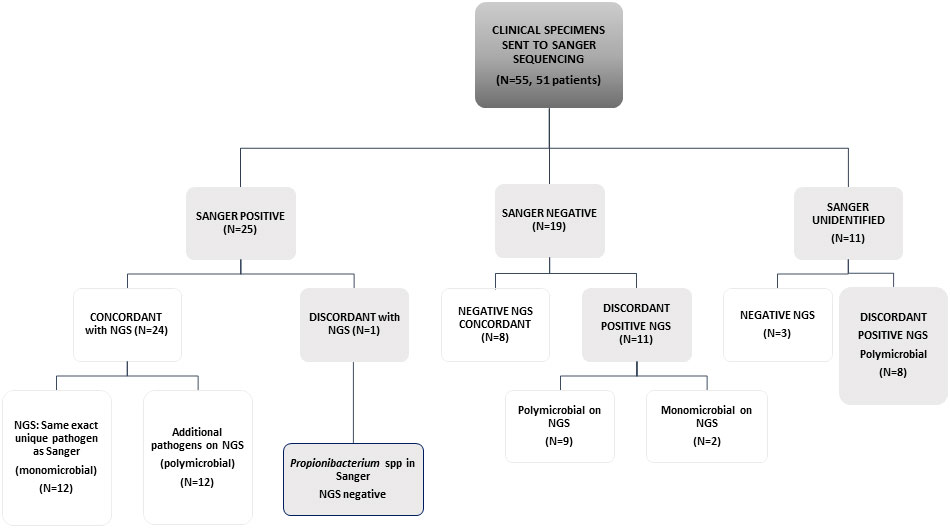
Figure 1 Concordance between Sanger sequencing and NGS in a scheme. Sanger negative are those samples that did not give any signal on PCR gels. "Sanger unidentified" are those samples that presented a band in agarose gels, but on Sanger sequencing it was not possible to define a unique organism against public databases.
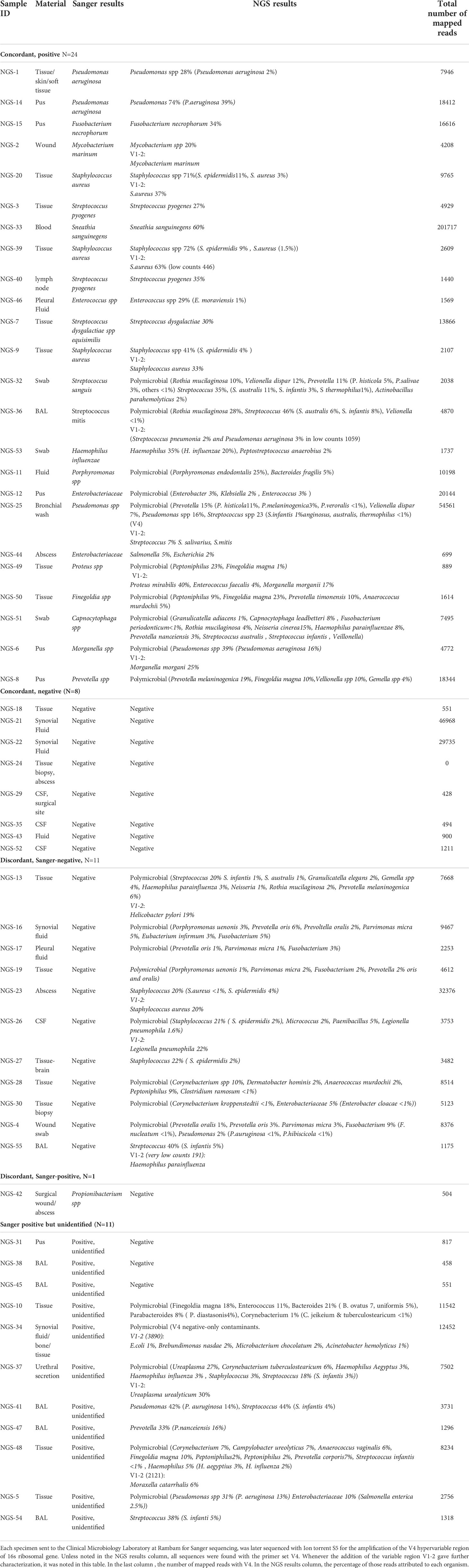
Table 1 Comparison between Sanger and NGS results.
Among the 22 RHCC specimens for which clinical information was available (18 patients), eight could have benefited from NGS for diagnosis (Figure 2 and Table 2): 4 polymicrobial (NGS-26, 30, 32, 34), 3 monomicrobial findings (NGS-23, 27, 54) and 1 genus identification of E.coli/Salmonella spp. in specimen NGS-44. We estimated that 6/18 patients would have benefited from antibiotic adjustment following NGS results and they all belong to the discordant group (Figure 2 and Table 2), while the other 12 patients would have been treated properly without any change in empiric treatment.
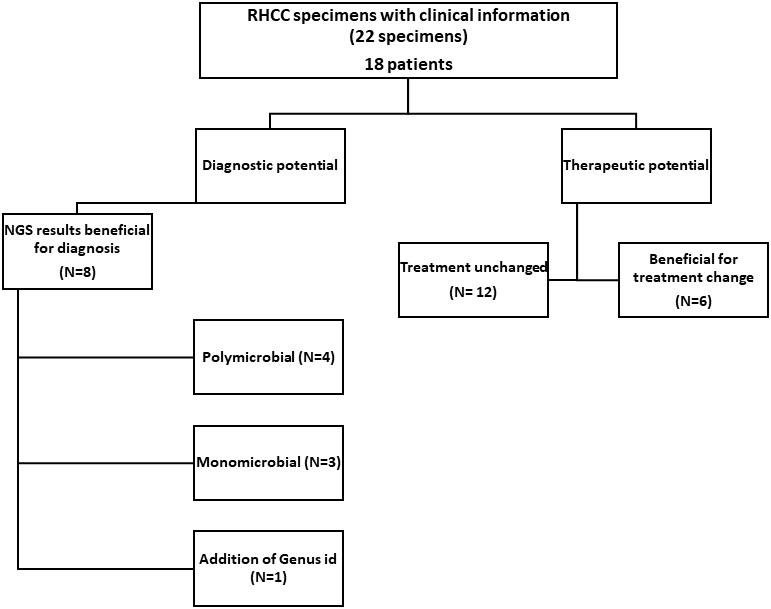
Figure 2 Effect of NGS findings on patient management. In 8/22 samples the result obtained with NGS contributed to diagnosis (also congruent when there were repeated samples from the same patient). In 6/18 patients, treatment could have been changed to a more appropriate one had NGS results been available at the time of diagnosis.
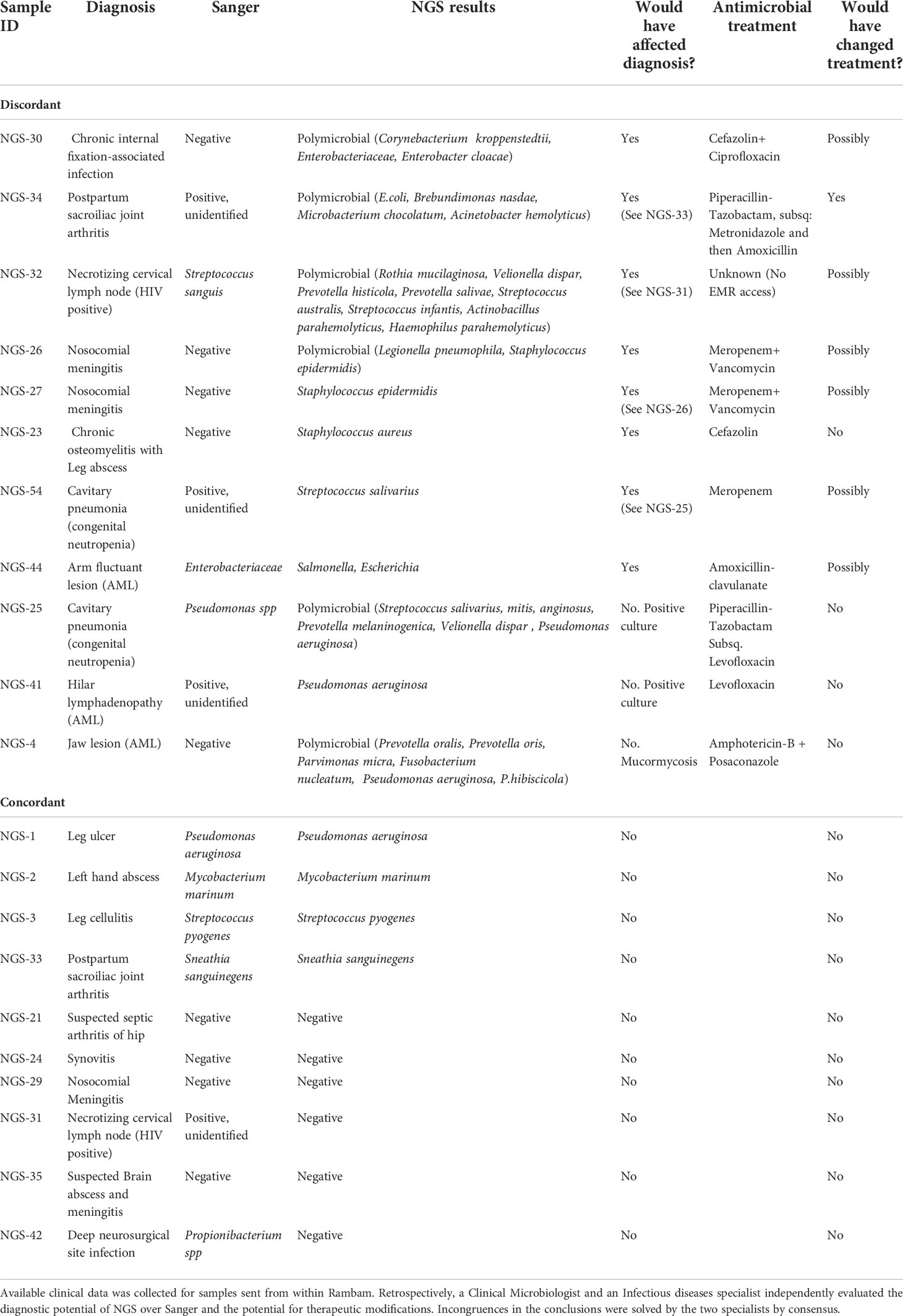
Table 2 Potential added value of NGS over Sanger for patient management.
In this study we describe the comparison between broad range Sanger panbacterial sequencing and targeted deep sequencing (NGS) on clinical samples submitted for panbacterial PCR. Overall, the concordance rate was 58% (32/55) for NGS vs. Sanger. Concordance was more frequent in Sanger-positive samples 24/25 (96%) than for Sanger-negative 8/19 (42%). Among five discordant Sanger-negative results with clinical information (Table 2), the positive NGS result would have been considered clinically-significant and might have improved diagnosis and/or management. In addition, NGS was able to identify possible pathogens in 8/11 Sanger-positive but pathogen-unidentified specimens. These results in a diagnostic advantage to targeted NGS. Moreover, polymicrobial communities identified by NGS may point to particular infection processes that may contribute to patients’ evaluation and optimal management. This is consistent with previous studies that validated the use of NGS for pathogen detection (Culbreath et al., 2019) and compared NGS to culture-based diagnosis (Abayasekara et al., 2017).
The targeted NGS in-house assay was performed on the Ion torrent S5 instrument, used for microbiome purposes, with a predefined threshold of > 1000 mapped reads. The presence of certain pathogens, however, should always be considered as a potential cause of infection, even if the number of reads is below predefined cutoffs, in polymicrobial or monomicrobial results. Such is the case in specimens NGS-44 (699 mapped reads) where Salmonella identified by NGS was considered clinically -significant, and NGS-49 (889 reads) where Proteus mirabilis magna among other intestinal pathogens might have been clinically-significant. Conversely, the presence of common commensals should be interpreted carefully. The clinician and the clinical microbiologist must work together to attribute clinical significance to the NGS results.
NGS technology allows for the parallel coverage of all taxa present in a clinical specimen, resulting in the identification of complex microbial communities. The polymicrobial findings in our study likely represented polymicrobial infections. However, alternative explanations should be considered, such as the possibility of commensal microbiota present in non-sterile or sterile body sites, a non-sterile specimen collection technique, or contamination during laboratory workflows. To overcome the latter, species found in negative controls were considered contaminants in our study and their sequences were subtracted from results. During specimen collection and transportation, polymicrobial communities may change in composition. Sanger identifies the best amplified organism which is not necessarily representative of the dominant or pathogenic one
One central limitation of this study is that only bacterial organisms were targeted (V4 and V1-2 regions of 16S rRNA gene). In addition, samples were selected randomly for this analysis, but not consecutively. The study was non-interventional – NGS results were not used in clinical practice, thus its true effect on patient management remain unknown. One advantage of this study is that NGS was performed blinded to other microbiological and clinical information.
In conclusion, in this validation study we demonstrated superior pathogen identification with targeted 16s NGS compared to Sanger sequencing in clinical samples. We propose to consider NGS upfront in cases where polymicrobial infections are suspected. Further developments of NGS should include the addition of other important targets such as viral targets or Internal Transcribed Space (ITS) for fungi, as well as antimicrobial resistance genes. To better characterize the accuracy of results, comparison with shotgun metagenomics is necessary.
The original contributions presented in the study are publicly available. This data can be found here: [DOI: 10.5281/zenodo.7119981].
The studies involving human participants were reviewed and approved by Helsinki committee at Rambam HCC, Israel. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Authors’ contributions: All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by MPi, BK, and YS. The first draft of the manuscript was written by MPi and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abayasekara, L. M., Perera, J., Chandrasekharan, V., Gnanam, V. S., Udunuwara, N. A., Liyanage, D. S., et al. (2017). Detection of bacterial pathogens from clinical specimens using conventional microbial culture and 16S metagenomics: a comparative study. BMC Infect. Dis. 17 (1), 631.
Apprill, A., Mcnally, S., Parsons, R., Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb. Ecol. 75 (2), 129–137.
Culbreath, K., Melanson, S., Gale, J., Baker, J., Li, F., Saebo, O., et al. (2019). Validation and retrospective clinical evaluation of a quantitative 16S rRNA gene metagenomic sequencing assay for bacterial pathogen detection in body fluids. J. Mol. Diagn. 21 (5), 913–923.
Khoury, N., Amit, S., Geffen, Y., Adler, A. (2019). Clinical utility of pan-microbial PCR assays in the routine diagnosis of infectious diseases. Diagn. Microbiol. Infect. Dis. 93 (3), 232–237.
Parada, A. E., Needham, D. M., Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18 (5), 1403–1414. doi: 10.1111/1462-2920.13023
Rampini, S. K., Bloemberg, G. V., Keller, P. M., Büchler, A. C., Dollenmaier, G., Speck, R. F., et al. (2011). Broad-range 16S rRNA gene polymerase chain reaction for diagnosis of culture-negative bacterial infections. Clin. Infect. Dis. 53 (12), 1245–1251.
Salipante, S. J., Sengupta, D. J., Rosenthal, C., Costa, G., Spangler, J., Sims, E. H., et al. (2013). Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PloS One 8 (5), e65226.
Shachor-Meyouhas, Y., Sprecher, H., Moscoviz, D., Zaidman, I., Haimi, M., Kassis, I. (2013). Molecular-based diagnosis of bacteremia in the setting of fever with or without neutropenia in pediatric hematology-oncology patients. J. Pediatr. Hematol. Oncol. 35 (7), 500–503.
Keywords: NGS, 16s, clinical microbiology, next generation sequencing, polymicrobial infections, polymicrobial
Citation: Pitashny M, Kadry B, Shalaginov R, Gazit L, Zohar Y, Szwarcwort M, Stabholz Y and Paul M (2022) NGS in the clinical microbiology settings. Front. Cell. Infect. Microbiol. 12:955481. doi: 10.3389/fcimb.2022.955481
Received: 28 May 2022; Accepted: 26 August 2022;
Published: 19 October 2022.
Edited by:
Xin Zhou, Stanford University, United StatesReviewed by:
Jiyuan Hu, New York University, United StatesCopyright © 2022 Pitashny, Kadry, Shalaginov, Gazit, Zohar, Szwarcwort, Stabholz and Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milena Pitashny, TV9QSVRBU0hOWUByYW1iYW0uaGVhbHRoLmdvdi5pbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.