
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 16 September 2022
Sec. Clinical Microbiology
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.947486
This article is part of the Research Topic Interactions Between Pathogens and Host Immune System in Patients with Immunodeficiency: Estimation from High-throughput Sequencing View all 14 articles
 Zhongdong Lv1†Yong Chen2,3†Houqing Zhou4†Zhonglin Chen2,3
Zhongdong Lv1†Yong Chen2,3†Houqing Zhou4†Zhonglin Chen2,3 Qianru Yao2,3Jiali Ren2,3Xianglu Liu1Shuang Liu1Xiaomei Deng5Yingchen Pang1Weijun Chen2,3Huiling Yang6*
Qianru Yao2,3Jiali Ren2,3Xianglu Liu1Shuang Liu1Xiaomei Deng5Yingchen Pang1Weijun Chen2,3Huiling Yang6* Ping Xu1*
Ping Xu1*Whipple’s disease is a rare chronic systemic disease that affects almost any organ system of the body caused by the intracellular bacterium Tropheryma whipplei, which is found ubiquitously in the environment. Sequencing of the T. whipplei genome has revealed that it has a reduced genome (0.93 Mbp), a characteristic shared with other intracellular bacteria. Until our research started, 19 T. whipplei strains had been sequenced from cultures originated in France, Canada, and Germany. The genome of T. whipplei bacterium has not been studied in Asia yet. Here, two metagenome-assembled genomes (MAGs) of T. whipplei from China were reconstructed through metagenomic next-generation sequencing (mNGS) and genome binning. We also provided genomic insights into the geographical role and genomic features by analyzing the whole genome. The whole-genome phylogenetic tree was constructed based on single-nucleotide polymorphism (SNP) distance calculations and then grouped by distance similarity. The phylogenetic tree shows inconsistencies with geographic origins, thus suggesting that the variations in geographical origins cannot explain the phylogenetic relationships among the 21 T. whipplei strains. The two Chinese strains were closely related to each other, and also found to be related to strains from Germany (T. whipplei TW08/27) and France (T. whipplei Bcu26 and T. whipplei Neuro1). Furthermore, the Average Nucleotide Identity (ANI) matrix also showed no association between geographic origins and genomic similarities. The pan-genome analysis revealed that T. whipplei has a closed pan-genome composed of big core-genomes and small accessory genomes, like other intracellular bacteria. By examining the genotypes of the sequenced strains, all 21 T. whipplei strains were found to be resistant to fluoroquinolones, due to the genetic mutations in genes gyrA, gyrB, parC, and parE. The 21 T. Whipplei strains shared the same virulence factors, except for the alpC gene, which existed in 7 out of the 21 T. whipplei strains. When comparing 21 entire T. whipplei pan-genomes from various nations, it was discovered that the bacterium also possessed a closed genome, which was a trait shared by intracellular pathogens.
The Gram-positive bacterium Tropheryma whipplei causes a rare multi-systematic infectious disease known as Whipple’s disease, which has clinical manifestations of fever, weight loss, lymphadenopathy, and polyarthritis, as well as cardiac manifestations and central nervous system complications (Ratliff et al., 1984; Durand et al., 1997). The “intestinal lipodystrophy” disease was first reported by George H. Whipple in 1907 and renamed Whipple’s disease by Black-Schaffer in 1949 (Black-Schaffer, 1949). A bacterial infection was long believed to be responsible for Whipple’s disease until in 1961 when the real origin of the disease was discovered by electron microscopy. Researchers detected bacterial inclusions in macrophages and monocytes, which together constituted the predominant infected cell types of this disease (Yardley and Hendrix, 1961). Researchers discovered that T. whipplei is a fastidious bacterium and extremely difficult to culture. The bacterium was first successfully isolated and grown in inactivated human mononuclear phagocytes by Schoedon in 1997, but culture could not be reproduced (Schoedon et al., 1997). In 2000, Raoult isolated the bacterium T. whipplei Twist from the aortic valve of a patient with endocarditis and propagated it in a human fibroblast cell line with the doubling time of 18 days; however, it could not be cultivated in the absence of living eukaryotic cells (Raoult et al., 2000). Subsequently, Raoult found a doubling time of 32 to 34 h for T. whipplei Twist strain, when propagated in the MRC5 cell culture system (Masselot et al., 2003). However, this doubling time was still longer than the slowly growing bacterium M. tuberculosis (14.3 h to 24 h) (James et al., 2000). In 2001, the T. whipplei strain Twist-Marseille was proposed by La Scola as the type strain of a new species of a new genus. The detailed characterization of the bacterium was described and deposited at the Collection Nationale de Culture de Micro-organisms de l’Institut Pasteur, Paris, France (La Scola et al., 2001).
The cultivation of T. whipplei made it possible to reveal the genome. So far, 19 strains of T. whipplei had been successfully cultured, and their genomes were sequenced by Bentley et al. (2003); Raoult et al. (2003), and Wetzstein (2017). Similar to other intracellular bacteria with rudimentary metabolic functions, genomic sequencing revealed that T. whipplei had a reduced genome (T. whipplei Twist, 0.93 Mbp). T. whipplei is ubiquitous in the environment, and it can lead to widespread colonization of the lower respiratory tract of healthy children and adults (Dickson et al., 2015). The bacterium could result in endocarditis, gastrointestinal infection, neurological complications and pulmonary infection, but the incident of Whipple’s disease is very rare. There were some reports of pulmonary infection caused by T. whipplei, which were diagnosed by metagenomic next-generation sequencing (Li et al., 2021; Zhu et al., 2021). T. whipplei is a commensal bacterium that only causes Whipple’s disease in a small number of individuals. Our understanding of the genome of T. whipplei from China is still not clear, due to the harsh cultivation conditions and the lengthy culture period. Metagenomic next-generation sequencing (mNGS) is a high-throughput sequencing technique that sequences all nucleic acids in a sample simultaneously in situ, which includes T. whipplei DNA if an individual had been infected by Whipple’s disease. The possible clinical mNGS applications are tremendous, including diagnosis of infectious diseases, outbreak tracking, infection control surveillance, and new pathogen discovery, among many other purposes. This emerging approach is an unbiased hypothesis-free diagnostic tool. It has been widely applied to guiding infectious disease management and developing treatment strategies. Next-generation sequencing (NGS) makes it possible to analyze genomes precisely and accurately, which aids the detection of single-nucleotide polymorphisms (SNPs) on a large scale. A metagenome-assembled genome T. whipplei shenzhen1 was assembly based on binned metagenome data of bronchoalveolar lavage samples from 26 non-immunodeficient patients, and metagenome-assembled genome T. whipplei shenzhen2 genome was assembled based on metagenome data of a bronchoalveolar lavage sample from an immunodeficient patient. All bronchoalveolar lavage samples from patients were collected by Peking University Shenzhen Hospital in China. The purpose of this study is to answer whether there was any difference between T. whipplei shenzhen1 and T. whipplei shenzhen2 genome, as well as the 19 genomes of T. whipplei that had been deposited in the NCBI database, including the two completed genomes of T. whipplei Twist and T. whipplei TW08/27 (Bentley et al., 2003; Raoult et al., 2003; Wetzstein, 2017).
Bronchoalveolar lavage fluid (BALF) samples were collected from one immunodeficient patient and 26 non-immunodeficient patients who had been admitted to Shenzhen Peking University Shenzhen Hospital with pulmonary infection. We subsequently extracted total genomic DNA from each BALF sample. Briefly, 0.6 ml of BALF and 250 μl of 0.5-mm glass beads in a 1.5-ml microcentrifuge tube were attached to a horizontal platform on a vortex mixer and agitated vigorously at 2,800–3,200 rpm for 30 min. Then, 7.2 μl of lysozyme was added for wall-breaking reaction. A 0.3-ml sample was separated into a new 1.5-ml microcentrifuge tube and DNA was extracted using the TIANamp Micro DNA Kit (DP316, TIANGEN BIOTECH) according to the manufacturer’s recommendation.
The extracted DNA obtained in the previous step was first fragmented to yield 300-bp fragments using enzymatic digestion (RM0434, BGI Wuhan Biotechnology). To construct the DNA library, fragmented DNA was further end-repaired, ligated to adapters, and amplified using PCR with the PMseq high-throughput gene detection kit for infectious pathogens (combined probe anchored polymerization sequencing method, BGI-Shenzhen, China, RM0438), according to the manufacturer’s instruction. Based on the qualified double-strand DNA library, the single-stranded circular DNA library was then generated through DNA denaturation and circularization. Then, DNA nanoballs (DNBs) were formed by rolling circle amplification (RCA) using a universal kit for sequencing reaction (Combinatorial Probe-Anchor Synthesis, BGI-Shenzhen, China, RM0170). DNBs were qualified by the Qubit® ssDNA Assay Kit (Thermo Fisher Scientific) and were further sequenced by the MGISEQ-2000 platform (MGI, China).
A quality control step was conducted on the metagenomic sequencing data by using the fastp tool to filter out low-quality and contaminated reads. By utilizing the Burrows-Wheeler alignment algorithm, the human DNA reads that aligned to human reference genome HG19 were eliminated, and only microbe reads were reserved. The remaining data were mapped and classified by aligning the reads to genomes of bacteria, fungi, viruses, and parasites from the Pathogens Metagenomics Database (PMDB). The classification reference databases were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/). T. whipplei was found in all 27 BALF samples, according to the mNGS diagnostic results. The T. whipplei shenzhen1 genome was assembled by combining metagenomic sequencing data of BALF samples from 26 non-immunodeficient patients with low-quality reads, and host genomes were removed. The T. whipplei shenzhen2 genome was assembled using metagenomic sequencing data of BALF sample from one immunodeficient patient. Briefly, the reads were subjected to de novo metagenomic assembly through metaSPAdes, and contigs shorter than 1,000 nt were discarded from further processing. Reads were mapped to contigs using Bowtie2, and the mapping output was used for contig binning through MetaBAT2. Lastly, putative genomes were subjected to quality control to generate the final set of reconstructed draft genomes. Two metagenome assembled genomes of T. whipplei from China were obtained. Table 1 summarizes the origin and the genome information of T. whipplei strains from China in this study and the 19 T. whipplei genomes reported by other researchers.
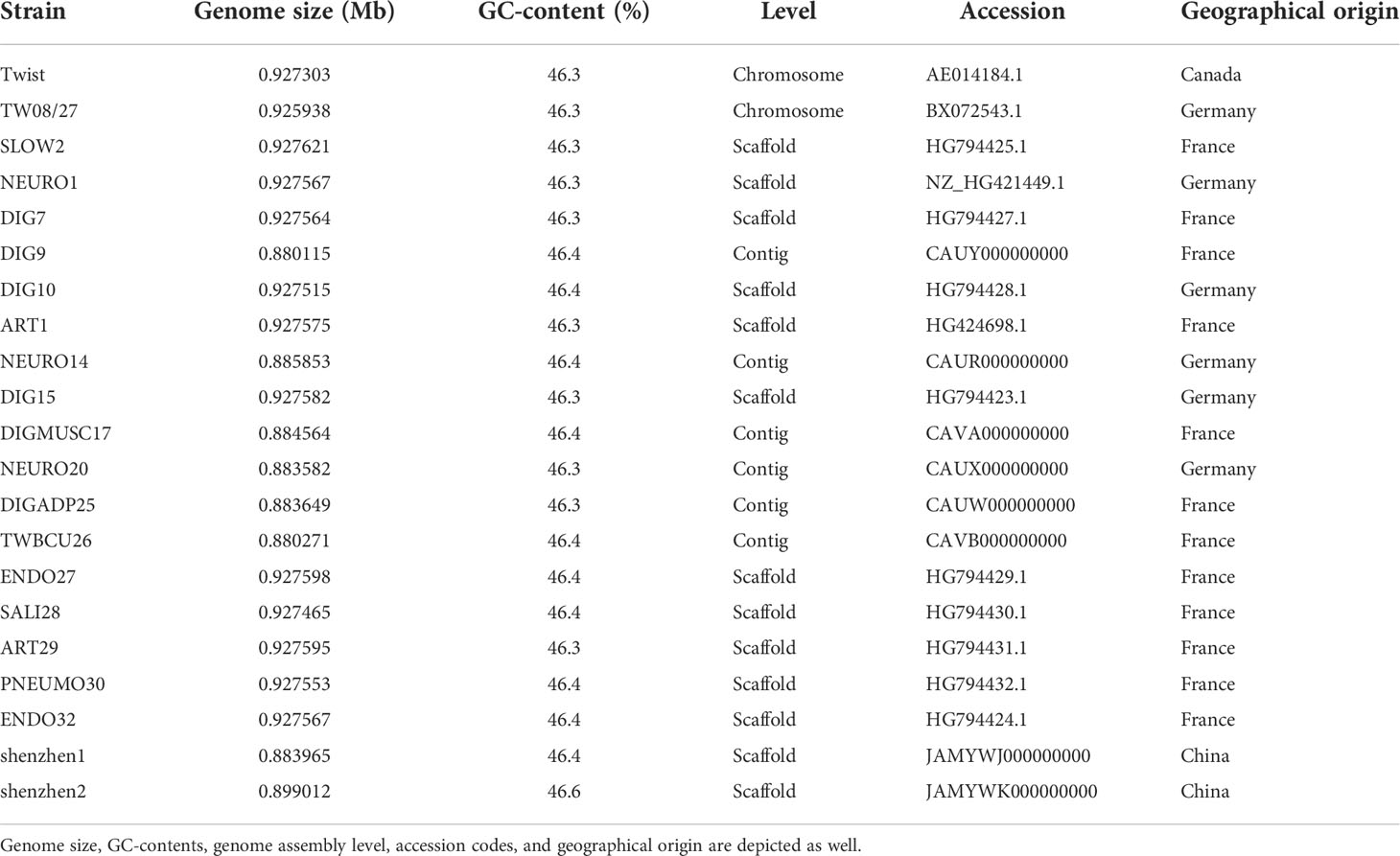
Table 1 Summary of 21 T. whipplei strains studied in this study.
Conducting whole-genome phylogenetic tree for microbial pathogens is a powerful approach that assists scientists to gain a better understanding of how species have evolved while explaining the similarities and differences among species. A wide range of genomic features can be observed across the entire genome derived from mNGS. These characteristics make phylogeny building extremely accurate. To discover the evolution of the 21 T. whipplei strains’ origin from different countries, the phylogenetic trees were constructed based on SNP datasets.
To characterize the core- and pan-genomes of the 21 strains of the T. whipplei genome, the PEPPAN pipeline, which can construct pan-genomes from thousands of genetically diverse bacterial genomes, was applied (Zhou et al., 2020). Genes presented in all 21 T. whipplei genomes were considered to be the core-genome, genes presented in more than 95% but not in all strains were defined as the softcore genes, genes presented in 15%–95% of the genomes were considered the shell genes, while genes presented in lower than 15% of the genomes were defined as the cloud genes. The pan-genome analysis of the 21 T. whipplei was performed by Anvi`o workflow to display the genome, which is an advanced analysis and visualization platform that offers both automated and/or user-specified characterization of metagenomic assembly genomes. (Eren et al., 2015)
Average nucleotide identity analysis is a useful approach to compare genetic relatedness among prokaryotic genomes The whole-genome average nucleotide identity (ANI) values for the 21 T. whipplei strains belonging to diverse geographic origins (Goris et al., 2007; Jain et al., 2018) were calculated to assess the genome similarities by using the FastANI v.0.1.3 software, which produces accurate ANI estimates and is a more efficient approach than alignment (e.g., BLAST)-based approaches.
To comprehend the virulence genes of the T. whipplei pathogens, 21 T. whipplei genomes were annotated with the Prokka annotation pipeline (ProkkaAnnotation v.3.2.1), and we then utilized the BLAST search tool for all known VF-related genes found in the virulence factor database (VFDB) (Chen et al., 2005). At the same time, the virulence factors were compared between T. whipplei strains originating from different countries. Reliable virulence genes were confirmed if the sequence identities were greater than 80% and the query coverages were greater than 80%, in which the values are used as benchmarks for virulence factor detection. The aligned amino acid sequences of GyrA, GyrB, ParC, and ParE for 21 T. whipplei strains were submitted to ESPript 3 to perform the sequence similarities, respectively (Robert and Gouet, 2014).
For the 21 genomes of T. whipplei, antibiotic resistance genes were predicted by aligning the hybrid assembled sequences in the CARD database using RGI v4.2.2 (Resistance Gene Identifier). Subsequently, the genes and the subsequent protein sequence were predicted using Prodigal. During this step, sequences that had an identity of less than 75% or a length coverage of less than 50% with the resistant genes denoted in the database were removed (Jia et al., 2017). The antibiotic resistance genes of T. whipplei were predicted using RAST (Rapid Annotation using Subsystem Technology, https://rast.nmpdr.org/), which is an automated service for annotating bacterial genomes.
The pathogen diagnosis of severe respiratory diseases was carried out using clinical metagenomic next-generation sequencing on 27 BALF samples from one immunodeficient patient and 26 non-immunodeficient patients who were admitted to Shenzhen Peking University Hospital. The metagenome-assembled genome of T. whipplei shenzhen1 was constructed based on the binned metagenomes of 26 BALF samples from non-immunodeficient patients, whereas the metagenome-assembled genome T. whipplei shenzhen2 genome was built based on one metagenome of a BALF sample from an immunodeficient patient.
The whole-genome phylogenetic tree constructed with 21 strains of the T. whipplei genome comprises three major clades, while one clade contains two subclades (Figure 1). The two strains T. whipplei shenzhen1 and T. whipplei shenzhen2 from China, the two strains T. whipplei TW08/27 and T. whipplei Meuro1 from Germany, and the one strain T. whipplei Bcu26 from France are located within the same subclade in the phylogenetic tree. Furthermore, the clades and subclades in the phylogenetic tree are not correlated with the geographical origins of T. whipplei.
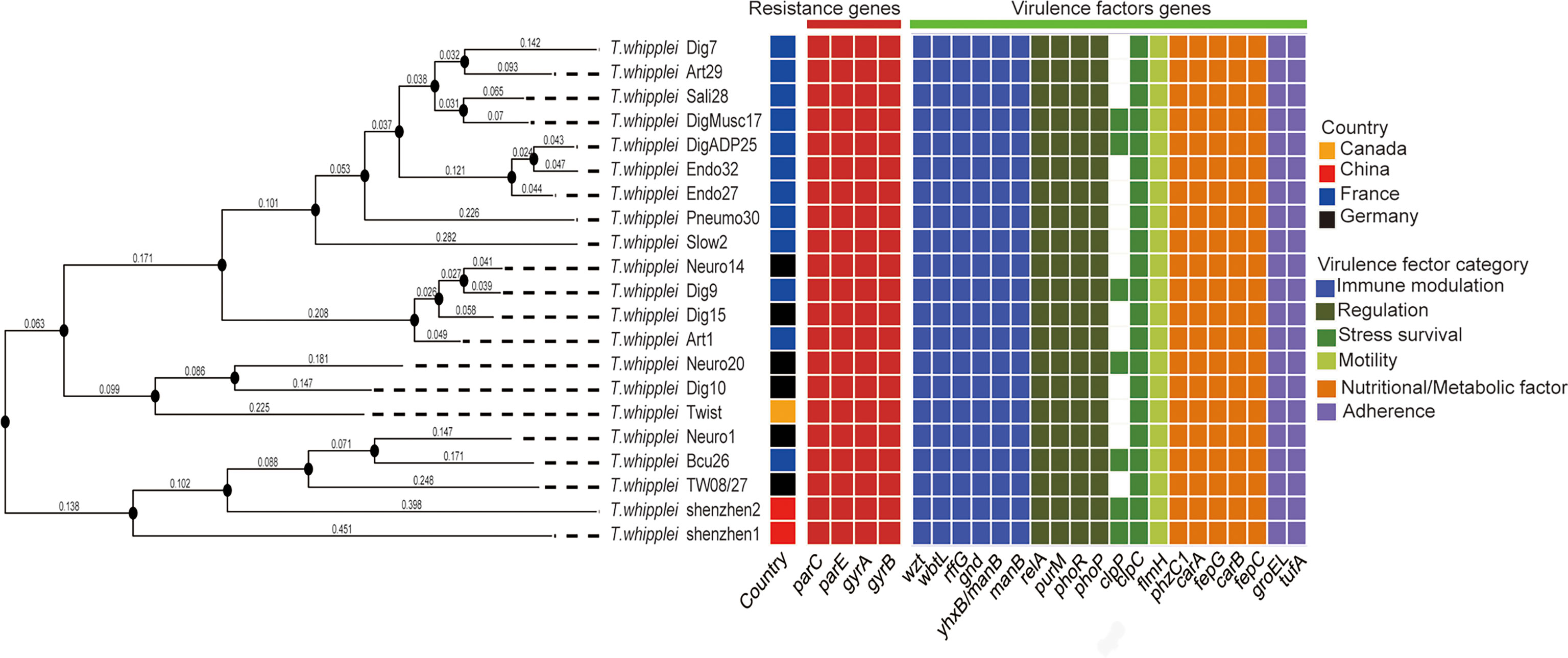
Figure 1 Whole-genome phylogenetic tree of 21 T. whipplei was constructed by single-nucleotide polymorphism (SNP) distance calculation, and the geographical origin of strains was obtained. The heatmaps represent the predicted resistance genes of chromosomal mutations known to confer resistance (gyrase and topoisomerase mutations conferring fluoroquinolone resistance) and the predicted virulence factors of T. whipplei.
To uncover the view of T. whipplei genome contents, the core-genome and pan-genome for 21 T. whipplei assembled genomes were calculated. The pan-genome of T. whipplei contained 977 genes, which were predicted by using the PEPPAN pipeline. The core-genome contained 809 genes that are common to all 21 strains of T. whipplei, and the core-genome accounts for 82.8% of the pan-genome, indicating that T. whipplei has a closed pan-genome. An additional 22 genes belong to the softcore genes, 58 genes form the shell genes, and 88 genes belong to the cloud genes. The pan-genome of 21 T. whipplei genomes was further analyzed by Anvi`o pan-genomic pipeline to visualize the pan-genome (Figure 2). As Figure 2 shows, the single-copy core genes occupy a major part of the pan-genome. Thirteen singleton gene clusters were found from T. whipplei TW08/27, which is the maximum number of singleton gene clusters of T. whipplei. We could thus hypothesize that T. whipplei is a strictly intracellular living organism that has a reduced genome, and no significant horizontal gene transfer has taken place in the past.
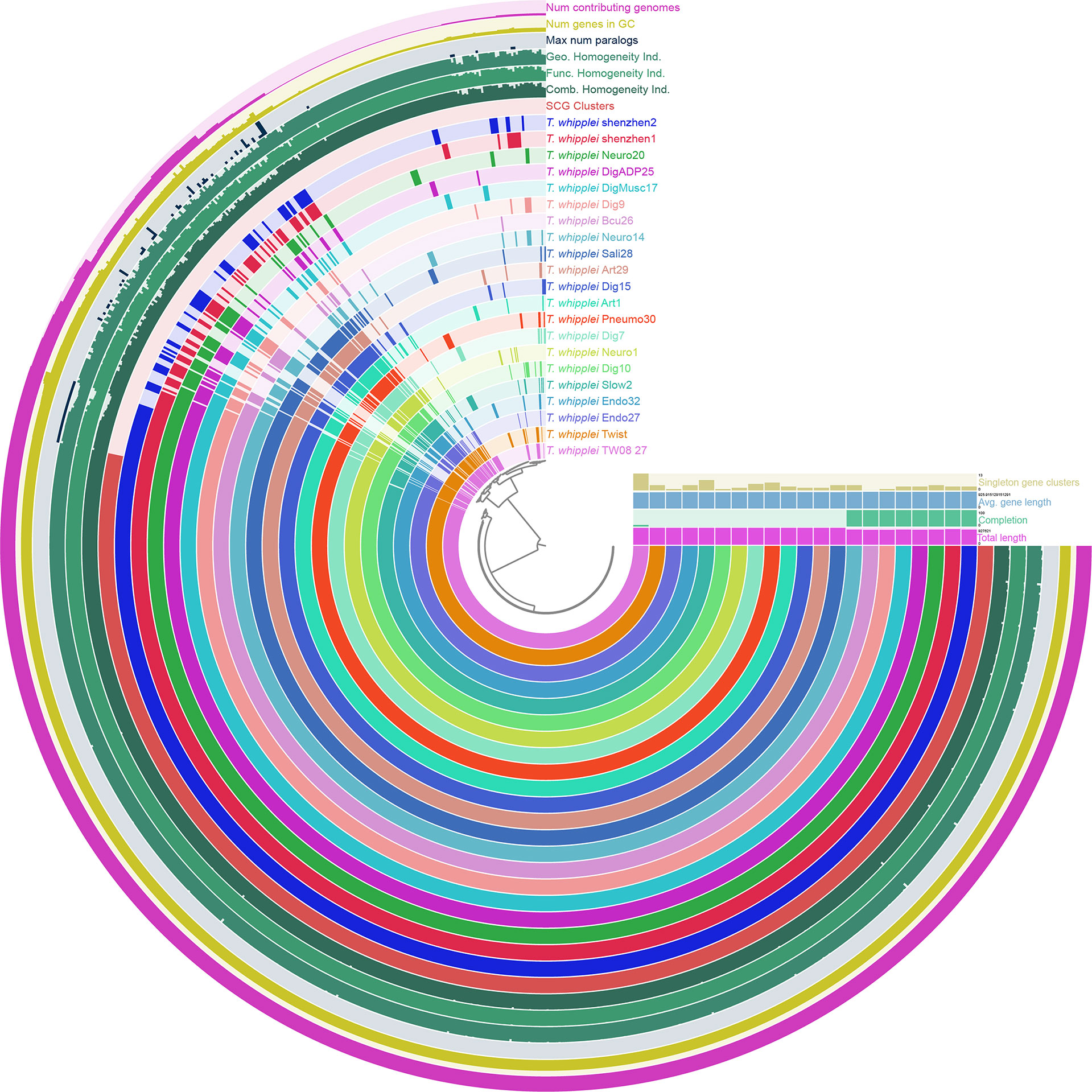
Figure 2 Anvi’o representation of the pan-genome of T. whipplei shenzhen1, T. whippleishenzhen2, and 19 other T. whipplei based on the presence/absence of gene clusters. The inner layers represent individual genomes organized by their phylogenetic relationships as indicated by the dendrogram. The first 21 layers represent each genome. Circle bars represent the occurrence of gene clusters in each genome, and the dark colors indicate the existence of the gene cluster. The subsequent seven layers correspond to various statistics related to the analysis, i.e., single-copy core gene clusters, combine homogeneity index, functional homogeneity index, geometric homogeneity index, maximum number of paralogs per gene cluster, number of genes per gene cluster, and the number of contributing genomes per gene cluster.
The whole-genome ANI lay between 99.11% and 99.98%, whereas the median ANI of all sequenced strains is 99.54% when all 21 T. whipplei genomes were compared with each other. The genomes with maximum and minimum ANI values for T. whipplei shenzhen1 are T. whipplei Bcu26 (99.74%) and T. whipplei strain Endo32 (99.38%) originating from France. The genome with maximum ANI for T. whipplei strain shenzhen2 is T. whipplei Bcu26. At the same time, the ANI between the two T. whipplei strains from China is 99.65%. As Figure 3 illustrates, the average nucleotide identity value calculated between all pairs of strains shows no discernible difference associated with geographical origins.
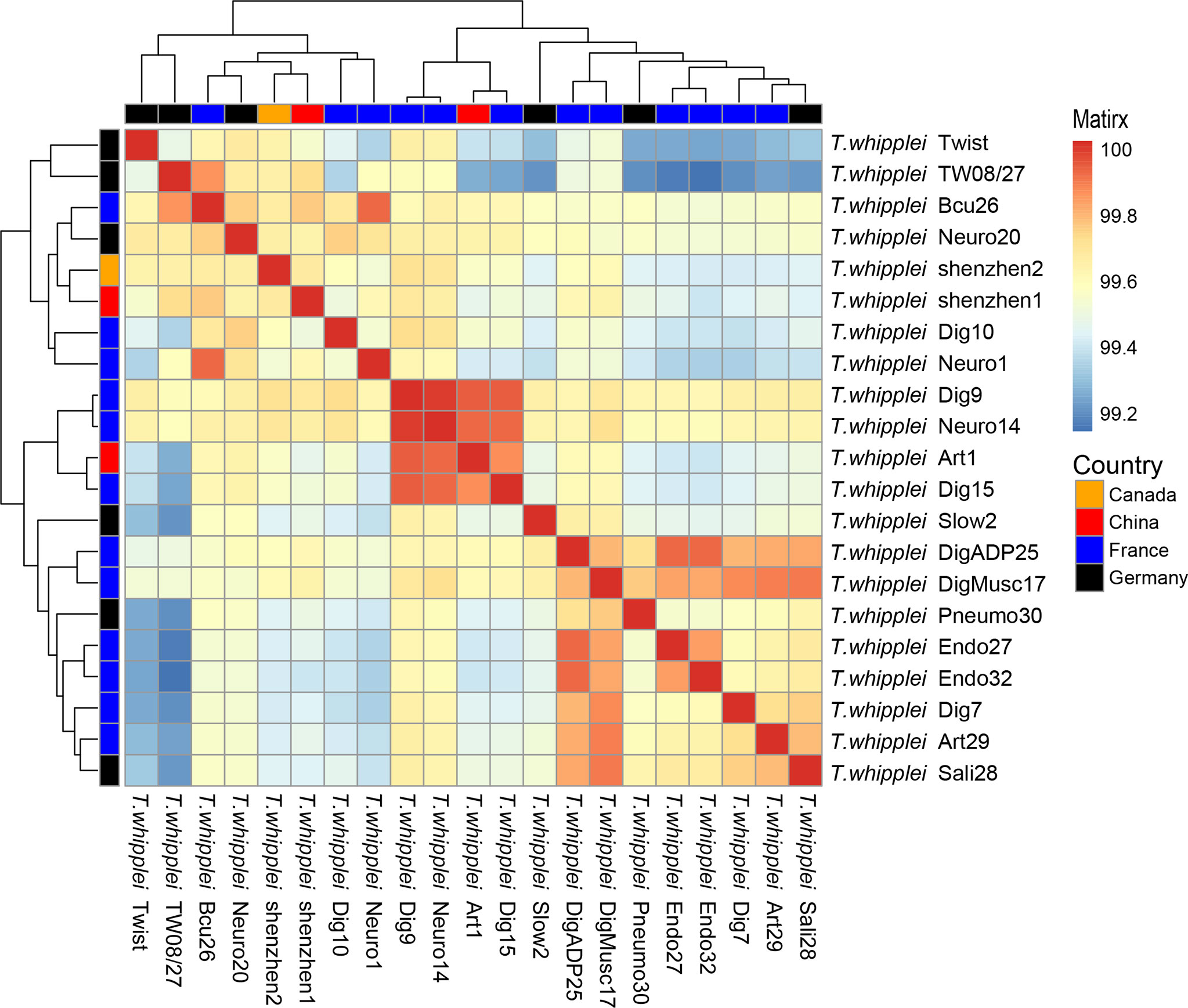
Figure 3 Heatmap of Average Nucleotide Identity (ANI) values for 21 whole genomes of T. whipplei strains from the different geographical origins (orange, Canada; red, China; blue, France; black, Germany).
Submitting the 21 strains of T. whipplei sequenced genomes to the Resistance Gene Identifier failed to detect any antibiotic resistance genes. However, after utilizing RAST-Annotation, the specific mutation in the genes for DNA gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE) leading to the genotypic antibiotic resistance to fluoroquinolones was found in T. whipplei. Some reports proved that gyrA, gyrB, parC, and parE gene mutations induce resistance to fluoroquinolones, due to the altered structures of the target proteins of fluoroquinolones (Gonzalez et al., 1998; Pantel et al., 2012; Johnning et al., 2015; Chien et al., 2016). Alignment of the amino acid sequences of GyrA, GyrB, ParC, and ParE of the 21 T. whipplei strains showed that these genes are highly conserved within the species (Figures S1–S4). The T. whipplei GyrA and ParC quinolone resistance-determining regions (QRDRs) are shown in Figures S1, S3. According to Didier Raoult’s report (Masselot et al., 2003), the T. whipplei GyrA QRDR extends from the alanine at position 65 to the glutamine at position 104, and the ParC QRDR extends from the alanine at position 80 to the histidine a position 119. The amino acid sequences of GyrA and ParC QRDR from the 21 T. whipplei strains and that of Escherichia coli were aligned, and the Ser-to-Ala mutation is indicated in Figure 4. The positions of this mutation are at positions 81 and 96, respectively. Alanines at these positions have previously been associated with increased fluoroquinolone resistance in T. whipplei, E. coli, and Mycobacteria (Cullen et al., 1989; Yagupsky et al., 1990; Masselot et al., 2003). The T. whipplei GyrB and ParE QRDR were identified by aligning with known homologous QRDR sequences of Mycobacterium fortuitum H37Rv (Cole et al., 1998). The GyrB QRDR of T. whipplei extends from the serine at position 474 to the glutamine at position 512. The ParE QRDR of T. whipplei extends from the alanine at position 488 to alanine at position 526. The mutations that likely to cause the fluoroquinolone resistance (e.g., Asp to Asn) were not detected in the GyrB and ParE QRDR regions.

Figure 4 Alignment of amino acid sequences of GyrA and ParC QRDRs from the 21 T. whipplei strains and E. coli K-12. Numbers refer to the amino acid positions in the T. whipplei GyrA and ParC sequence.
The virulence factors of T. whipplei were investigated through the virulence factor database, which harbors information of bacterial virulence factors from various known pathogens. Twenty virulence factors had been predicted to play a role in the pathogenesis of T. whipplei (Figure 1). These include six bacterial VF categories: adherence (groEL and ufa), immune modulation (wzt, wbtL, rffG, gnd, manB, and yhxB/manB), nutritional/metabolic factor (phzC1, carA, fepG, carB, and fepC), regulation (relA, purM, phoR, and phoP), stress survival (clpC and clpP), and motility (flmH), but further studies need to be done to understand the role of these virulence factors. Among the 21 T. whipplei strains, 19 virulence factors are shared among the strains, while the clpC gene is found in 7 out of 21 T. whipplei strains. It encodes a general stress protein belonging to the HSP-100/Clp family, which promotes intracellular bacteria Listeria monocytogenes to escape from the macrophage phagocytosis (Rouquette et al., 1998).
T. whipplei is a fastidious bacterium that is difficult to culture, and not until after 2000 could the researchers successfully culture this bacterium in the human fibroblast cell line in the laboratory (Raoult et al., 2000). There is increasing evidence to suggest that the predominant reservoir of T. whipplei is found in humans, as T. whipplei is known to be viable in the human respiratory tract, fecal, and saliva samples. It also suggests that T. whipplei might be transmitted through both fecal–oral and oral–oral routes (La Scola et al., 2011; Pightling et al., 2018). In previous studies, 19 T. whipplei strains were isolated from various specimens, including the aortic valve, small intestine biopsy, mesenterial lymph node, and bronchoalveolar lavage cutaneous biopsy, in which the pathogens were cultured and the genomes were sequenced (Bentley et al., 2003; Raoult et al., 2003; Wetzstein, 2017). The cultivation of T. whipplei requires living eukaryotic cells, while the bacterium has a very slow replication with a doubling time of 18 days, which severely impedes routine culture-based diagnostics and genomic analysis. On the other hand, obtaining the bacterial whole genome is significant to understanding the properties of this pathogen, including antibiotic resistance, molecular epidemiology, and pathogenic virulence. Utilizing whole-genome sequence analyses could supplement epidemiological studies and trace back evidence for epidemic regulations (Pightling et al., 2018). Moreover, it could help identify sources of pathogens during disease outbreaks. In this study, we report two genome sequences of T. whipplei shenzhen1 and T. whipplei shenzhen2, which were assembled using clinical metagenomic sequencing, without the need to conduct long-term bacterial cultivation.
In comparative genomics, the genetic content of 21 T. whipplei was compared to each other. The antibiotic resistance genes and virulence genes were predicted, and the phylogenetic relationships between the strains were determined. Interestingly, even strains of T. whipplei that originate from different geographical regions have close relationships, including the isolate T. whipplei shenzhen2 from an immunodeficient patient. T. whipplei is an intracellular bacterium that has a closed pan-genome, suggested by investigating the core and pan-genomes of T. whipplei. It has a limited capacity to acquire foreign genes likely due to its limited horizontal gene transfer mechanisms. As shown by the whole-genome phylogenetic tree and the ANI matrix, there is no correlation between the strains and their geographical origins. All T. whipplei strains exhibit genotypic resistance to fluoroquinolones, due to mutations found in the gyrA, gyrB, parC, and parE genes in the RAST annotation. Mutations in the quinolone resistance-determining region (QRDR) of gyrA, gyrB, parC, and parE leading to reduce susceptibility to fluoroquinolone have been reported in many bacterial species (Gonzalez et al., 1998; Pantel et al., 2012; Johnning et al., 2015; Chien et al., 2016). All T. whipplei strains exhibit genotypic resistance to fluoroquinolones, due to the GyrA and ParC QRDR with an alanine residue at positions 81 and 96 (Masselot et al., 2003). The QRDRs of T. whipplei GyrB and ParE were defined as codons 474 to 512 in GyrB, and 488 to 526 in the ParE, but the specific amino acid relative to fluoroquinolone resistance was not discovered. Although the mechanisms of quinolone resistance of GyrB and ParE have not been fully investigated, we still hypothesize that T. whipplei has a natural resistance to fluoroquinolones.
In conclusion, we have obtained two metagenome-assembled genomes of T. whipplei from China using metagenomic next-generation sequencing (mNGS). The 21 T. whipplei strains share highly similar genomic characteristics despite originating from different countries. Aided by the mNGS culture-independent characterization of pathogens, we therefore propose that clinical mNGS could be considered as an approach to obtain and analyze genomic information for difficult or “unculturable” microorganisms.
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: NCBI; PRJNA831609.
The studies involving human participants were reviewed and approved by The Peking University Shenzhen Hospital Ethics Committee. The patients/participants provided their written informed consent to participate in this study.
PX, YH participated in study conception and design. ZL, SL, XLL, XMD enrolled and managed patients, CY, CZ and YQ carried out collection and assembly of data. ZL, CY were involved in data analysis and interpretation. PX, YH, ZL, CY prepared the manuscript and manuscript figures. PX, YH and ZL edited, critically read, and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Key Research and Development Program of China (2019YFC1200500 and 2019YFC1200501), the Shenzhen Science and Technology Innovation Commission Foundation (grant no. JCYJ20190809103203711), the Shenzhen Science and Technology Innovation Commission foundation (grant no. JCYJ20210324105411031), the Fund of “Sanming” Project of Medicine in Shenzhen (no. SZSM201811096), the Shenzhen High-level Hospital Construction Fund (LCYJ2021022), and the Shenzhen High-level Hospital Construction Fund Discipline Construction Project of Guangdong Medical University.
Authors YC, ZC, QY, JR and WC are employed by BGI.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.947486/full#supplementary-material
Bentley, S. D., Maiwald, M., Murphy, L. D., Pallen, M. J., Yeats, C. A., Dover, L. G., et al. (2003). Sequencing and analysis of the genome of the whipple’s disease bacterium tropheryma whipplei. Lancet 361, 637–644. doi: 10.1016/S0140-6736(03)12597-4
Black-Schaffer, B. (1949). The tinctoral demonstration of a glycoprotein in whipple’s disease. Proc. Soc. Exp. Biol. Med. 72, 225–227. doi: 10.3181/00379727-72-17388
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chien, J. Y., Chiu, W. Y., Chien, S. T., Chiang, C. J., Yu, C. J., Hsueh, P. R. (2016). Mutations in gyrA and gyrB among fluoroquinolone- and multidrug-resistant mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 60, 2090–2096. doi: 10.1128/AAC.01049-15
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Cullen, M. E., Wyke, A. W., Kuroda, R., Fisher, L. M. (1989). Cloning and characterization of a DNA gyrase a gene from escherichia coli that confers clinical resistance to 4-quinolones. Antimicrob. Agents Chemother. 33, 886–894. doi: 10.1128/AAC.33.6.886
Dickson, R. P., Erb-Downward, J. R., Freeman, C. M., Mccloskey, L., Beck, J. M., Huffnagle, G. B., et al. (2015). Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann. Am. Thorac. Soc. 12, 821–830. doi: 10.1513/AnnalsATS.201501-029OC
Durand, D. V., Lecomte, C., Cathebras, P., Rousset, H., Godeau, P. (1997). Whipple Disease. clinical review of 52 cases. The SNFMI research group on Whipple disease. societe nationale francaise de medecine interne. Med. (Baltimore) 76, 170–184. doi: 10.1097/00005792-199705000-00003
Eren, A. M., Esen, O. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. doi: 10.7717/peerj.1319
Gonzalez, I., Georgiou, M., Alcaide, F., Balas, D., Linares, J., de la Campa, A. G. (1998). Fluoroquinolone resistance mutations in the parC, parE, and gyrA genes of clinical isolates of viridans group streptococci. Antimicrob. Agents Chemother. 42, 2792–2798. doi: 10.1128/AAC.42.11.2792
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., Tiedje, J. M. (2007). DNA-DNA Hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T., Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
James, B. W., Williams, A., Marsh, P. D. (2000). The physiology and pathogenicity of mycobacterium tuberculosis grown under controlled conditions in a defined medium. J. Appl. Microbiol. 88, 669–677. doi: 10.1046/j.1365-2672.2000.01020.x
Jia, B., Raphenya, A.R., Alcock, B., Waglechner, N., Guo, P., Dave, B.M., et al (2017). CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45, D566–73. doi: 10.1093/nar/gkw1004
Johnning, A., Kristiansson, E., Fick, J., Weijdegard, B., Larsson, D. G. (2015). Resistance mutations in gyrA and parC are common in escherichia communities of both fluoroquinolone-polluted and uncontaminated aquatic environments. Front. Microbiol. 6 1355. doi: 10.3389/fmicb.2015.01355
La Scola, B., Fenollar, F., Fournier, P. E., Altwegg, M., Mallet, M. N., Raoult, D. (2001). Description of tropheryma whipplei gen. nov., sp. nov., the whipple’s disease bacillus. Int. J. Syst. Evol. Microbiol. 51, 1471–1479. doi: 10.1099/00207713-51-4-1471
La Scola, B., Fenollar, F., Perreal, C., Raoult, D. (2011). Epidemiologic implications of the first isolation and cultivation of tropheryma whipplei from a saliva sample. Ann. Intern. Med. 154, 443–444. doi: 10.7326/0003-4819-154-6-201103150-00018
Li, W., Zhang, Q., Xu, Y., Zhang, X., Huang, Q., Su, Z. (2021). Severe pneumonia in adults caused by tropheryma whipplei and candida sp. infection: A 2019 case series. BMC Pulm Med. 21, 29. doi: 10.1186/s12890-020-01384-4
Masselot, F., Boulos, A., Maurin, M., Rolain, J. M., Raoult, D. (2003). Molecular evaluation of antibiotic susceptibility: Tropheryma whipplei paradigm. Antimicrob. Agents Chemother. 47, 1658–1664. doi: 10.1128/AAC.47.5.1658-1664.2003
Pantel, A., Petrella, S., Veziris, N., Brossier, F., Bastian, S., Jarlier, V., et al. (2012). Extending the definition of the GyrB quinolone resistance-determining region in mycobacterium tuberculosis DNA gyrase for assessing fluoroquinolone resistance in m. tuberculosis. Antimicrob. Agents Chemother. 56, 1990–1996. doi: 10.1128/AAC.06272-11
Pightling, A. W., Pettengill, J. B., Luo, Y., Baugher, J. D., Rand, H., Strain, E. (2018). Interpreting whole-genome sequence analyses of foodborne bacteria for regulatory applications and outbreak investigations. Front. Microbiol. 9 1482. doi: 10.3389/fmicb.2018.01482
Raoult, D., Birg, M. L., La Scola, B., Fournier, P. E., Enea, M., Lepidi, H., et al. (2000). Cultivation of the bacillus of whipple’s disease. N Engl. J. Med. 342, 620–625. doi: 10.1056/NEJM200003023420903
Raoult, D., Ogata, H., Audic, S., Robert, C., Suhre, K., Drancourt, M., et al. (2003). Tropheryma whipplei twist: A human pathogenic actinobacteria with a reduced genome. Genome Res. 13, 1800–1809. doi: 10.1101/gr.1474603
Ratliff, N. B., Mcmahon, J. T., Naab, T. J., Cosgrove, D. M. (1984). Whipple’s disease in the porcine leaflets of a carpentier-edwards prosthetic mitral valve. N Engl. J. Med. 311, 902–903. doi: 10.1056/NEJM198410043111407
Robert, X., Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. doi: 10.1093/nar/gku316
Rouquette, C., De Chastellier, C., Nair, S., Berche, P. (1998). The ClpC ATPase of listeria monocytogenes is a general stress protein required for virulence and promoting early bacterial escape from the phagosome of macrophages. Mol. Microbiol. 27, 1235–1245. doi: 10.1046/j.1365-2958.1998.00775.x
Schoedon, G., Goldenberger, D., Forrer, R., Gunz, A., Dutly, F., Hochli, M., et al. (1997). Deactivation of macrophages with interleukin-4 is the key to the isolation of tropheryma whippelii. J. Infect. Dis. 176, 672–677. doi: 10.1086/514089
Wetzstein, N. (2017). Genotyping and genomotyping of tropheryma whipplei – the causative agent of whipple’s disease.
Yagupsky, P. V., Kaminski, D. A., Palmer, K. M., Nolte, F. S. (1990). Cord formation in BACTEC 7H12 medium for rapid, presumptive identification of mycobacterium tuberculosis complex. J. Clin. Microbiol. 28, 1451–1453. doi: 10.1128/jcm.28.6.1451-1453.1990
Yardley, J. H., Hendrix, T. R. (1961). Combined electron and light microscopy in whipple’s disease. demonstration of “bacillary bodies” in the intestine. Bull. Johns Hopkins Hosp 109, 80–98.
Zhou, Z., Charlesworth, J., Achtman, M. (2020). Accurate reconstruction of bacterial pan- and core genomes with PEPPAN. Genome Res. 30, 1667–1679. doi: 10.1101/gr.260828.120
Keywords: tropheryma whipplei, whole-genome analysis, bronchoalveolar lavage (BAL), immunodeficiency – primary, metagenome-assembled genome (MAGs)
Citation: Lv Z, Chen Y, Zhou H, Chen Z, Yao Q, Ren J, Liu X, Liu S, Deng X, Pang Y, Chen W, Yang H and Xu P (2022) Genomic characterization of two metagenome-assembled genomes of Tropheryma whipplei from China. Front. Cell. Infect. Microbiol. 12:947486. doi: 10.3389/fcimb.2022.947486
Received: 18 May 2022; Accepted: 22 August 2022;
Published: 16 September 2022.
Edited by:
Eda Altan, University of Turku, FinlandReviewed by:
Thomas Jové, INSERM U1092 Anti-Infectieux supports moléculaires des résistances et innovations thérapeutiques, FranceCopyright © 2022 Lv, Chen, Zhou, Chen, Yao, Ren, Liu, Liu, Deng, Pang, Chen, Yang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiling Yang, eWFuZ2h1aWxpbmczMDE4QHNpbmEuY29t; Ping Xu, cGluZy14dUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.