
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 06 July 2022
Sec. Microbiome in Health and Disease
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.907323
 Jeanne Couturier1,2*
Jeanne Couturier1,2* Patricia Lepage3Sarah Jolivet4Johanne Delannoy1Victoria Mesa1Pierre-Yves Ancel5,6Jean-Christophe Rozé7Marie-José Butel1
Patricia Lepage3Sarah Jolivet4Johanne Delannoy1Victoria Mesa1Pierre-Yves Ancel5,6Jean-Christophe Rozé7Marie-José Butel1 Frédéric Barbut1,2,4†Julio Aires1†
Frédéric Barbut1,2,4†Julio Aires1†In adults, Clostridioides difficile infections are associated with alterations of the intestinal bacterial populations. Although preterm neonates (PN) are frequently colonized by C. difficile, limited data are available regarding the relationship between C. difficile and the intestinal microbiota of this specific population. Therefore, we studied the intestinal microbiota of PN from two multicenter cohorts using high-throughput sequencing of the bacterial 16S rRNA gene. Our results showed that alpha diversity was significantly higher in children colonized by C. difficile than those without colonization. Beta diversity significantly differed between the groups. In multivariate analysis, C. difficile colonization was significantly associated with the absence of postnatal antibiotherapy and higher gestational age. Taxa belonging to the Lachnospiraceae, Enterobacteriaceae, Oscillospiraceae families and Veillonella sp. were positively associated with C. difficile colonization, whereas Bacteroidales and Bifidobacterium breve were negatively associated with C. difficile colonization. After adjustment for covariables, Clostridioides, Rothia, Bifidobacterium, Veillonella, Eisenbergiella genera and Enterobacterales were more abundant in the gut microbiota of colonized children. There was no significant association between C. difficile colonization and necrotizing enterocolitis in PN. Our results suggest that C. difficile colonization in PN is related to the establishment of physiological microbiota.
Asymptomatic Clostridioides difficile colonization with both toxigenic and non-toxigenic strains has been reported in 17–70% of healthy children less than three years of age (Enoch et al., 2011; Jangi and Lamont, 2010; Rousseau et al., 2011; Stoesser et al., 2017). Among pediatric populations, few studies have targeted C. difficile colonization in preterm neonates (PN); moreover, these studies are either outdated or based on culturing approaches (Cardines et al., 1988; el-Mohandes et al., 1993; Tina et al., 1994; Chang et al., 2012; Ferraris et al., 2012; Pichler et al., 2018). Depending on the study, the colonization rate in PN ranges from 0% (Tina et al., 1994) to 63% (Cardines et al., 1988) and increases during the first month of life. Preterm neonates are more exposed to hospital environments and antibiotics than full-term neonates. These factors influence gut colonization dynamics; for instance, the colonization rate is correlated to the hospital stay length and the resulting spore exposition (Bacon et al., 1988). Antibiotherapy is also associated with more frequent C. difficile colonization in PN (Tina et al., 1994; Pichler et al., 2018). Moreover, gut immaturity disturbs the gut microbiota, i.e., reduced bacterial diversity and richness and a low proportion of genera associated with health benefits (Lactobacillus, Bacteroides, Bifidobacteria) (Gewolb et al., 1999; Drell et al., 2014).
The link between intestinal dysbiosis and C. difficile infection (CDI) or colonization is gaining interest (Chen et al., 2019; Crobach et al., 2020; Han et al., 2020). C. difficile establishment in the adult gut occurs during dysbiosis, and CDI is associated with gut microbiota disruption leading to an increased risk of relapse (Barbut and Couturier, 2019). By sequencing the 16S rRNA gene of fecal samples from infected children aged 28–48 months, Ling et al. observed a decreased bacterial diversity compared to healthy controls (Ling et al., 2014). In addition, pediatric populations can act as reservoirs for adult CDI (Rousseau et al., 2012). Given the rising incidence of CDI in adults and children, understanding the early C. difficile colonization process and its role in gut microbiota establishment is of utmost importance.
The aim of this study was to evaluate C. difficile colonization in PN and its relationship with the intestinal microbiota. Using next-generation sequencing (NGS) of the 16S rRNA gene, we analyzed the intestinal microbiota of 599 PN from two multicenter cohorts, ClosNEC (n=116) and EPIFLORE (n=483), to assess bacterial signatures associated with C. difficile colonization. The impact of perinatal factors on colonization was also determined.
The fecal samples used in this study were obtained from PN recruited during two prospective multicenter studies, ClosNEC (2015–2016) and EPIFLORE (2011), including 20 neonatal intensive care unit facilities (NICUs). The aim of the ClosNEC cohort was to assess the relationship between bacterial colonization and necrotizing enterocolitis (NEC) in 159 recruited PN (<32 weeks of gestation) (Schönherr-Hellec et al., 2018). In this study, 118 fecal samples (35 from PN with NEC at its onset and 83 from matched controls) were analyzed using NGS 16S rRNA gene sequencing. The EPIFLORE cohort is an ancillary study of EPIPAGE 2, which aimed to describe the gut microbiota of PN born between 24–31 weeks of gestation (Rozé et al., 2017). A total of 751 children were recruited, and stool samples were collected in the first week [between day 3 (D3) and D7] (n=673) and 1 month [between D21 and D28 (n=620)]. The gut microbiota was analyzed by NGS for 142 and 486 fecal samples at one week (EPIFLORE_D7 dataset) and one month (EPIFLORE_D28 dataset), respectively. For 116 children whose samples were collected during the first week of life, a second sample was obtained at one month (EPIFLORE_D7_2 dataset). The flowcharts in Figure S1 represent subject recruitment and stool samplings for both cohorts.
For the ClosNEC cohort (Clinical trial no. NCT02444624), approval was obtained from the National Data Protection Authority (Commission Nationale de l’Informatique et des Libertés, approval no. 915094) and the Consultative Committee on the Treatment of Information on Personal Health Data for Research Purposes (approval no. 15.055).
For the EPIFLORE cohort, approval was obtained from the National Data Protection Authority (Commission Nationale de l’Informatique et des Libertés, approval no. 911009), the Consultative Committee on the Treatment of Information on Personal Health Data for Research Purposes (approval no. 10.626), and the Committee for the Protection of People Participating in Biomedical Research (approval no. CPP SC-2873).
Informed consent was obtained from all the children’s parents involved in the study.
Fresh fecal samples were collected from the diapers and stored at −80°C until microbiota analysis. Total DNA was extracted according to the International Human Microbiome Standards standard operating procedure 7 (IHMS_SOP 007) (Doré et al., 2015). The following template-specific primers were used: forward (5′-TACGGR(G/A)AGGCAGCAG-3′) and reverse (5′-TACCAGGGTATCTAAT-3′), targeting the V3-V4 hypervariable region of the 16S rRNA gene (Wilson et al., 1990). Quality control, sequencing library construction, and sequencing (MiSeq Illumina, San Diego, CA, United States) were performed using the Genotoul bioinformatics platform (Toulouse, France). Sequencing data were analyzed using the Galaxy-supported bioinformatics pipeline find, rapidly, OTUs with Galaxy solution (FROGS v3.0, http://bioinfo.genotoul.fr/, accessed 30/04/2020) (Afgan et al., 2018; Escudié et al., 2018). Primers were trimmed using Cutadapt (Martin, 2011). Sequences shorter than 410 bp and longer than 480 bp were excluded. Clustering was performed using the Swarm algorithm (Mahé et al., 2014) following the FROGS guidelines, that is, two successive steps with aggregation parameters d=1 and d=3. Chimeras were detected using the VSEARCH tool with the de novo UCHIME algorithm (Edgar et al., 2011; Rognes et al., 2016) and removed from further analysis. Filters were applied as follows: operational taxonomic units (OTUs) present in at least three samples and representing at least 0.00005% of the sequences were retained. Samples with fewer than 1000 amplified sequences were excluded from the analysis. OTUs were taxonomically classified using the basic local alignment search tool (BLAST) (Altschul et al., 1990) in the SILVA release 128 database (accessed 30/04/2020) (Quast et al., 2013). Taxonomic lineages were combined with a cluster abundance matrix to generate a raw OTU table.
Samples were defined as “C. difficile positive” (CD+) when at least 0.1% of the amplified sequences were assigned to the C. difficile OTU. The taxonomic affiliation of “C. difficile” was verified using BLAST (NCBI standard nucleotide blast https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed 30/04/2020) by comparing the OTU nucleotide sequence with the RefSeq_rna database. Taxonomic affiliation was considered “exact” if the identity percentage was 100%.
We compared the microbiota composition between the CD+ and CD− (“C. difficile negative”) groups using the FROGS Phyloseq v1.19.1 tool (accessed 02/05/2020) (McMurdie and Holmes, 2013). The data were normalized prior to diversity analysis. Results were considered significant at p < 0.05. Analysis of variance (ANOVA) was used to compare Chao 1 species richness estimation and Shannon diversity index between the patient groups. Microbial diversity was visualized using multidimensional scaling (MDS). To investigate associations between microbiota composition and C. difficile colonization, we performed a permutational multivariate analysis of variance (PERMANOVA) using the “Adonis” function with 9999 permutations, and Jaccard and weighted UniFrac (wUnifrac) distances. Biomarkers associated with colonization status were determined by the linear discriminant analysis (LDA) effect size (LefSe) algorithm (Segata et al., 2011).
Through a mixed linear model, the association of the most abundant OTUs transformed logarithmically with the patient individual variables were evaluated using the RStudio’s lme4 and nlme packages (Bates et al., 2015; Pinheiro et al., 2022). For OTUs that showed a significant association (p < 0.05), the model was adjusted to control the effects of confounding variables. In statistical analysis, a p < 0.05 value was considered significant.
Metagenomic functional prediction was carried out using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2 v2.5.0) based on 16S rRNA sequencing data (Langille et al., 2013; Douglas et al., 2020). The obtained prediction of metagenomic functional abundances was combined with descriptions from the KEGG Orthology (KO) database. Differentially present pathways between CD+ and CD− groups were analysed with welch test using STAMP (v2.1.3) (Parks et al., 2014).
For C. difficile isolation, stool samples were processed as previously described (Couturier et al., 2020). Briefly, fecal samples were spread on the C. difficile-selective medium CLO-M (bioMérieux®, Marcy-l’Etoile, France) and incubated for 24 h at 37°C under anaerobic conditions (CO2:H2:N2, 10:10:80, anaerobic chamber). C. difficile isolates were identified using routine laboratory procedures. Toxigenic strains were identified by screening for C. difficile tpi, tcdA, and tcdB genes using multiplex PCR, as previously described (Lemee et al., 2004). Isolates were stored at −80°C in brain-heart infusion liquid medium supplemented with 15% glycerol.
Clinical data, including birth mode, neonatal antibiotherapy, maternal antibiotherapy, and NEC onset, were prospectively collected during hospitalization until discharge for both PN cohorts. PN colonized by C. difficile were compared to those that were non-colonized. All variables were compared using the χ2 test or Fischer’s exact method, as required, or a non-parametric Mann-Whitney test for qualitative variables. Variables with p < 0.10 on univariable analysis were entered into a multivariate logistic regression model. A backward stepwise approach was used to identify independent predictors. Two-sided p < 0.05 defined significance. Statistical analyses were performed using Stata v15.1 software (StataCorp, College Station, Texas, USA).
The characteristics of the different datasets and results of the gut bacterial 16S rRNA gene sequencing analysis are listed in Table 1. Less than 1000 sequences were retrieved for two samples in the ClosNEC dataset and three samples in the EPIFLORE_D28 dataset (Figure S1). Therefore, these samples were excluded from the analysis. Rarefaction curves of the three datasets are available in Supplementary material (Figures S2–S4). Because of the low number of CD+ samples at D7 in the EPIFLORE_D7 dataset (n=2), we considered C. difficile colonization at D28 for the same child (n = 9) for the statistical analysis. Therefore, we used a subset of the EPIFLORE_D7 dataset called EPIFLORE_D7_2, which included children with samples at D7 and D28 (n=116). Groups were defined as C. difficile-positive at D28 (CD+D28) and C. difficile-negative at D28 (CD−D28), indicating the gut microbiota at D7 was compared according to the ulterior colonization status of the child at D28. For both children with a CD+ sample at D7, C. difficile was also detected in the D28 sample.
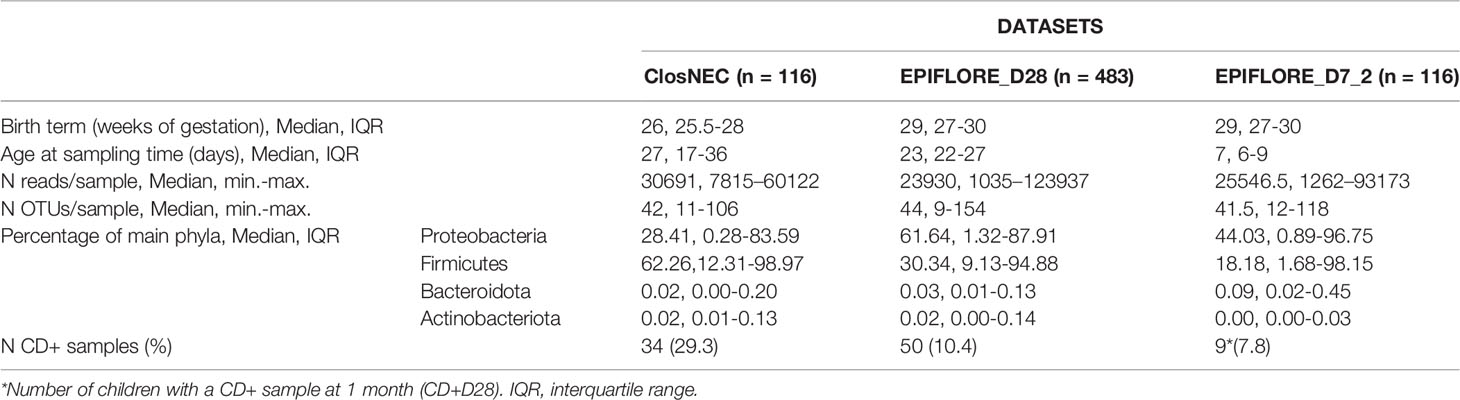
Table 1 Characteristics of datasets and results of the NGS analysis of the intestinal microbiota.
For all datasets, the microbiota was dominated by Firmicutes and Proteobacteria, with important inter-individual variations (Table 1). Among Proteobacteria, the most abundant family was Enterobacteriaceae, with median proportions of 10.63% [interquartile (IQR) 0.11–79.38], 57.33% (IQR 0.53–86.71), and 6.8% (IQR 0.51–95.95) for ClosNEC, EPIFLORE_D28, and EPIFLORE_D7_2, respectively. The most represented families of Firmicutes were Clostridiaceae for ClosNEC, Enterococcaceae for EPIFLORE_D28, and Staphylococcaceae for EPIFLORE_D7_2, with median proportions of 1.11% (IQR 0.03–43.8), 4.00% (IQR 0.28–17.28), and 2.07% (IQR 0.13–42.22), respectively. The median proportions of Lactobacillaceae and Bifidobacteriaceae were < 0.01% in all datasets.
Culture results were compared to C. difficile detection by NGS of the 16S rRNA gene for samples from the ClosNEC cohort (n=116) (Table S1). The overall agreement between the two methods was 86.2%. Among the five culture-positive NGS-negative samples, C. difficile sequences were detected in two samples, but in proportion < 0.1%, and therefore defined as “CD−”. Among the 28 C. difficile strains, 26 (92.9%) were non-toxigenic, and 2 (7.1%) were toxigenic. Owing to the low number of samples analyzed by both culture and NGS (n=27), we could not compare the results from both methods in the EPIFLORE cohort.
We evaluated the differences in alpha diversity between CD+ and CD− samples (ClosNEC and EPIFLORE_D28 datasets) and CD+D28 and CD−D28 samples (EPIFLORE_D7_2 dataset). The median Chao1 and Shannon indices of CD+ samples were significantly higher than that of CD− samples (p < 0.01 and p < 0.01 for ClosNEC, respectively; p < 0.01 and p < 0.001 for EPIFLORE_D28, respectively) (Figure 1). At the first week of life, the median Shannon index in samples of PN colonized by C. difficile at one month (CD+D28 samples) was also higher than that of PN not colonized at one month (CD−D28 samples) (EPIFLORE_D7_2, p < 0.05) (Figure 1).
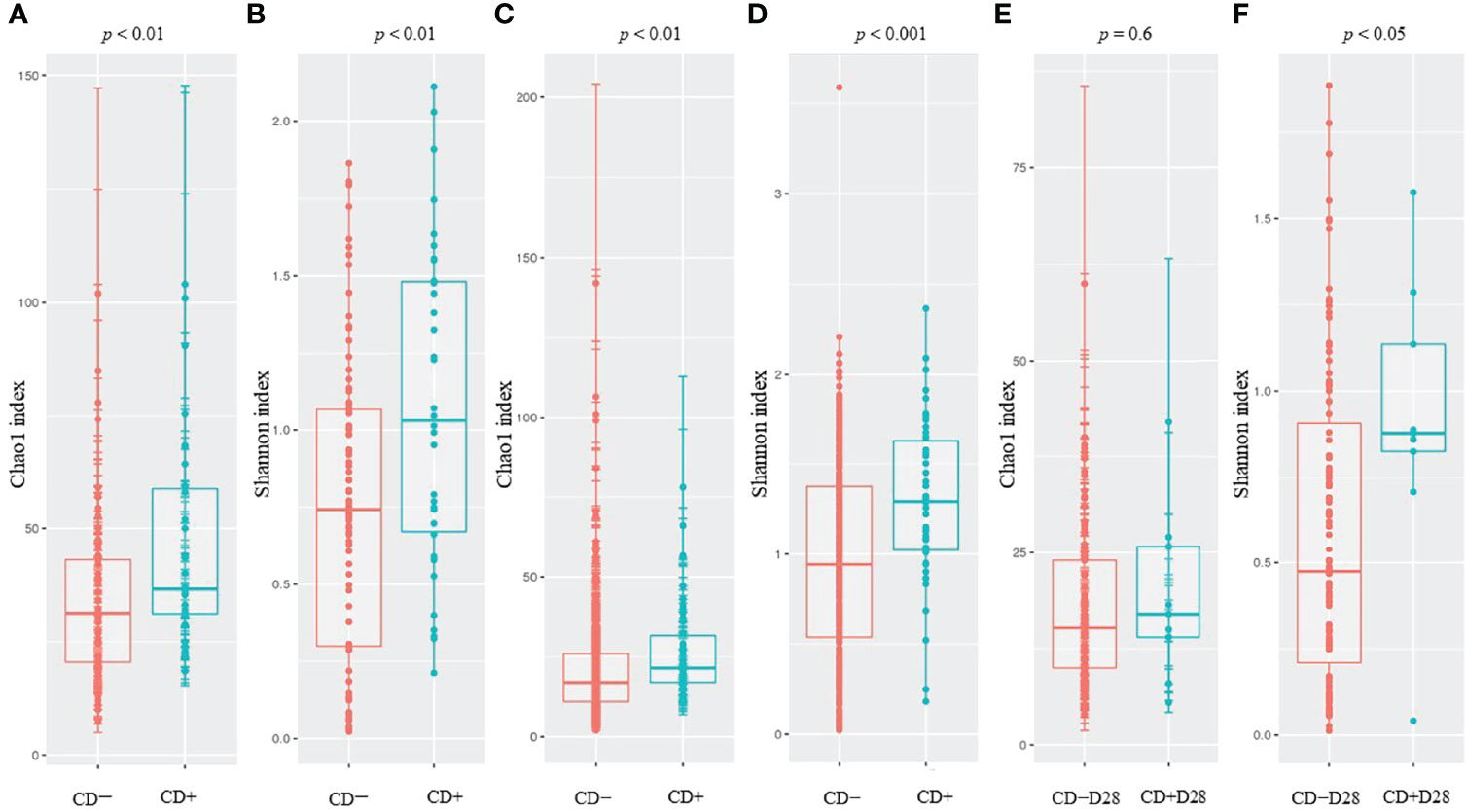
Figure 1 Comparison of the Chao1 and Shannon indices for ClosNEC [(A) and (B)], EPIFLORE_D28 [(C) and (D)] and EPIFLORE_D7_2 [(E) and (F)], respectively. Red, CD− (ClosNEC, EPIFLORE_D28) or CD−D28 (EPIFLORE_D7_2); blue, CD+ (ClosNEC, EPIFLORE_D28) or CD+D28 (EPIFLORE_D7_2). Boxplots represent the median and the 1st and 3rd quartiles.
To evaluate the beta diversity between CD-colonized and non-colonized groups, we performed MDS using Jaccard and wUnifrac matrices for the three datasets (Figure 2). Jaccard matrix analysis showed that the CD+ and CD− groups clustered separately for both the ClosNEC and EPIFLORE_D28 datasets (p < 0.001 and p < 0.001, PERMANOVA), as well as the CD+D28 and CD−D28 groups for the EPIFLORE_D7_2 dataset (p < 0.05, PERMANOVA). In the wUnifrac analysis, groups clustered separately only for the EPIFLORE_D28 dataset (p < 0.001 by PERMANOVA).
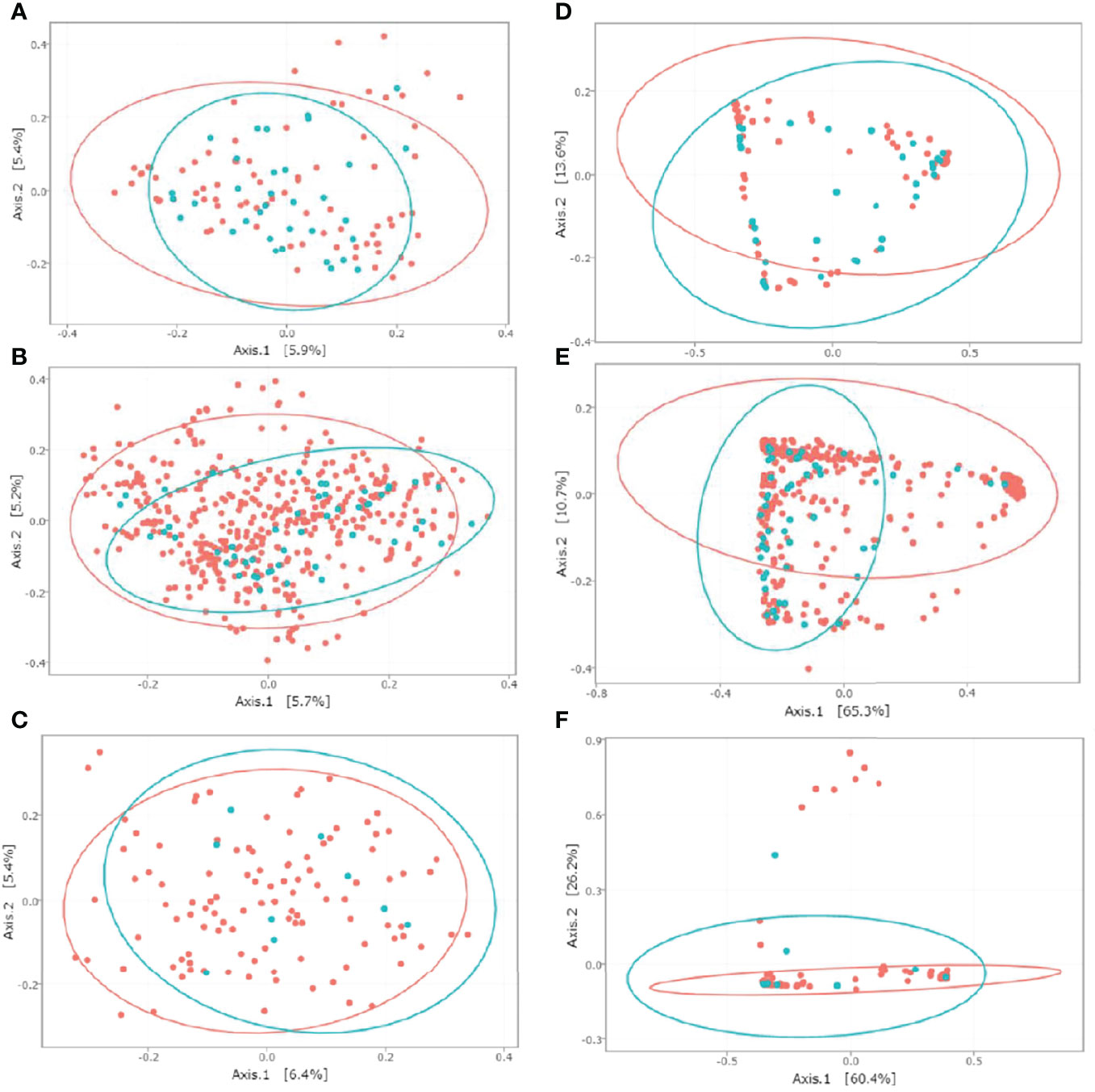
Figure 2 MDS representation of the intestinal microbiota based on Jaccard and wUnifrac indices for ClosNEC [(A) and (D)], EPIFLORE_D28 [(B) and (E)] and EPIFLORE_D7_2 [(C) and (F)], respectively. Each sample is represented by a color according to its category: red, CD− (ClosNEC, EPIFLORE_D28) or CD−D28 (EPIFLORE_D7_2); blue, CD+ (ClosNEC, EPIFLORE_D28) or CD+D28 (EPIFLORE_D7_2). The percentage of variation explained by the two first MDS dimensions is indicated on the respective axes.
Functional predictions identified 22 and 76 differentially present KO pathways in ClosNEC (Figure S5) and EPIFLORE_D28 (Figure S6) datasets, respectively. In the ClosNEC dataset, the largest significant differences were pathways for energy metabolism, starch and sucrose metabolism, peptidases and amino acid metabolism; those pathways had higher proportions in the CD+ group. Energy metabolism, starch and sucrose metabolism, and peptidases had also higher proportions in the CD+ group of the EPIFLORE_D28 dataset. Largest significant differences in this dataset included bacterial motility proteins, flagellar assembly, and sporulation (higher in the CD+ group); DNA repair and recombination proteins, and ribosome (higher in the CD− group).
Using univariate analysis, perinatal factors significantly associated with C. difficile colonization at one month in the EPIFLORE cohort included the absence of postnatal antibiotherapy [odds ratio (OR) 0.35, 95% confidence interval (CI) 0.17–0.72, p = 0.004], enterotype 2 profile as described by Rozé et al. (Rozé et al., 2020) (OR 2.36, 95% CI 1.18–4.72, p = 0.015), and a higher gestational age (OR 1.25, 95% CI 1.05–1.47, p = 0.010) (Table 2). In multivariate analysis, the only significantly associated variable was the absence of postnatal antibiotherapy. The most frequently used antibiotics in the EPIFLORE cohort were vancomycin (34%), aminosides (27%) and third generation cephalosporins (22%). Among PN who received a postnatal antibiotherapy, 93% were treated by at least two different antibiotics.
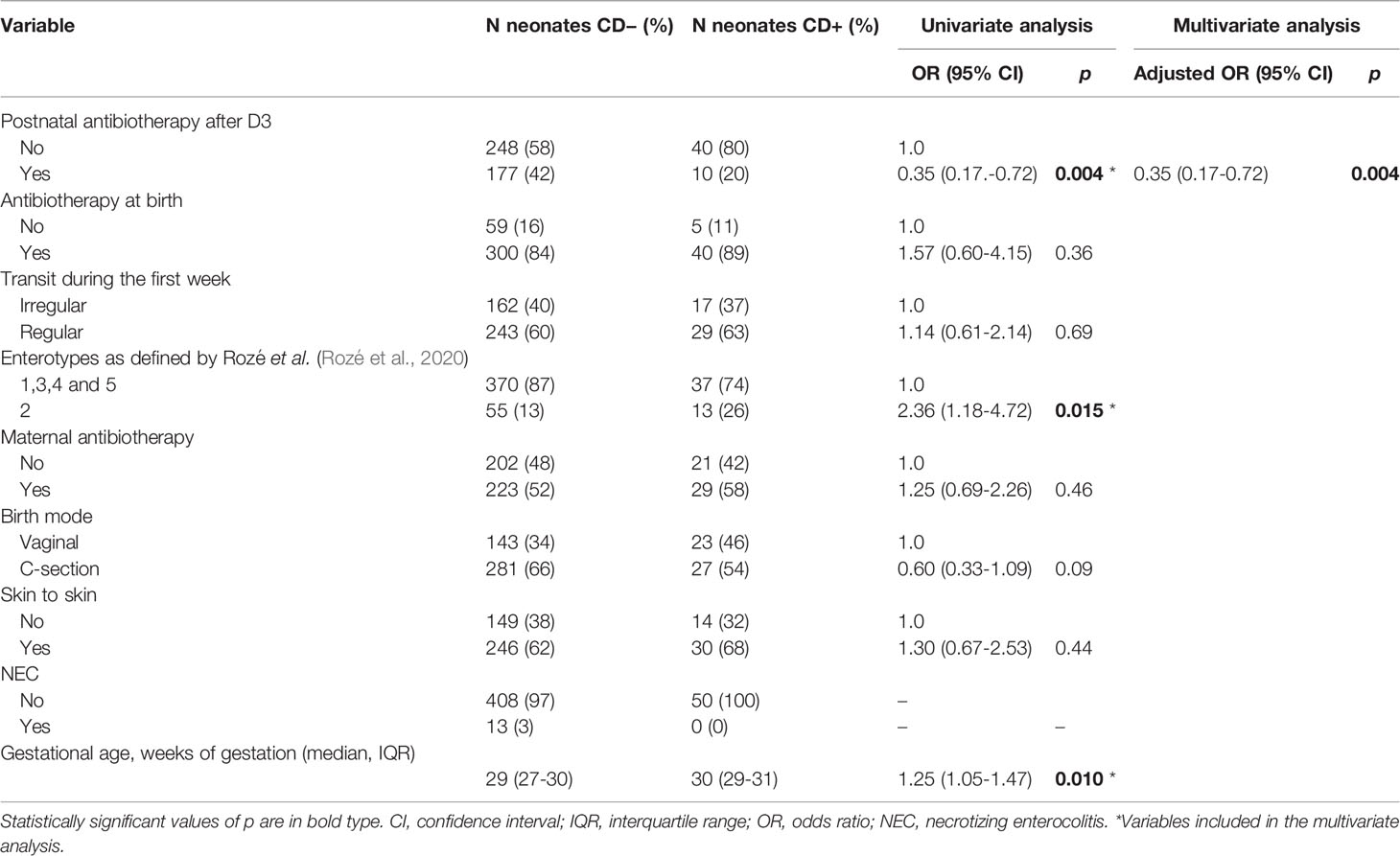
Table 2 Factors associated with C. difficile colonization at 1 month in the EPIFLORE cohort.
In univariate analysis, factors significantly associated with C. difficile colonization in the ClosNEC cohort were transit considered normal at D7 (OR 3.83, 95% CI 1.06–13.91, p = 0.041) and a higher gestational age (OR 1.26, 95% CI 1.03–1.52, p = 0.019) (Table 3). In the multivariate analysis, higher gestational age was the only significantly associated factor. PN of the ClosNEC cohort received third generation cephalosporins (68%), aminosides (59%), vancomycin (59%) or other antibiotics (30%). Among PN who received a postnatal antibiotherapy, 97% were treated by at least two different antibiotics. Maternal antibiotherapy, birth mode, and NEC were not identified as factors associated with C. difficile colonization.
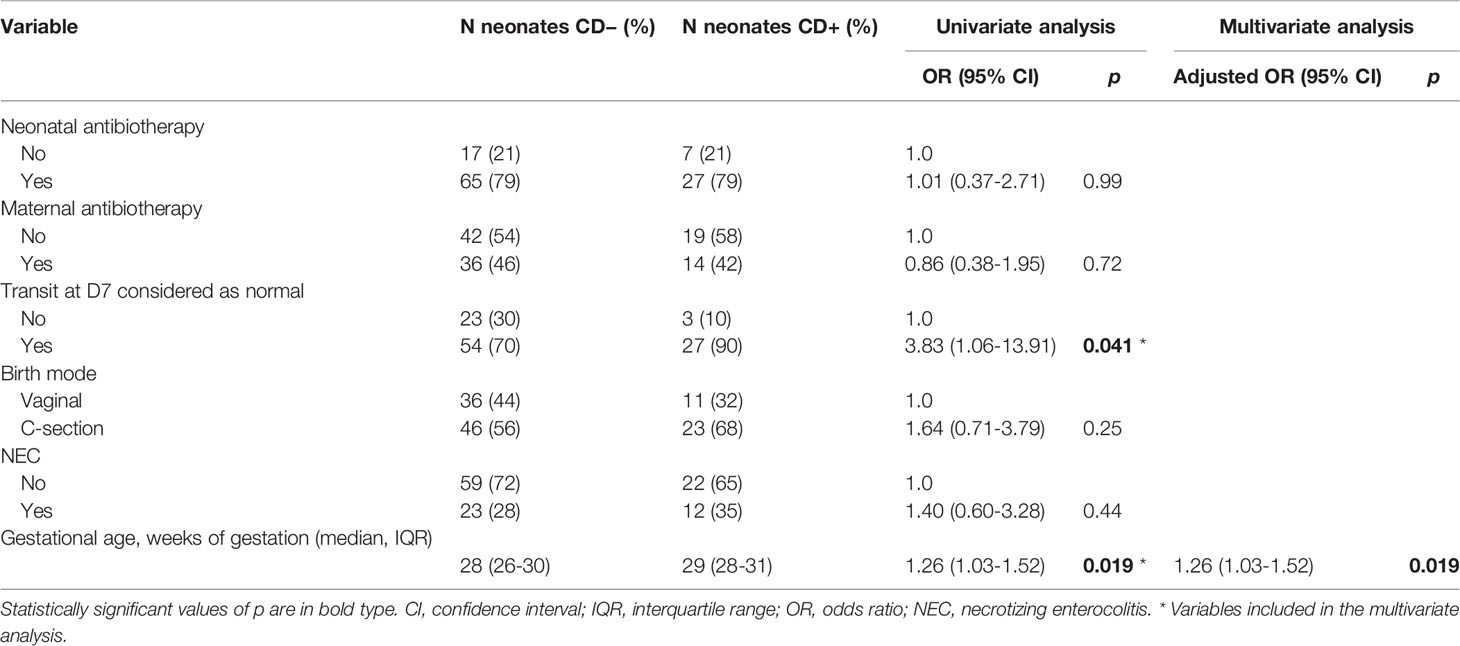
Table 3 Factors associated with C. difficile colonization in the ClosNEC cohort.
Bacterial biomarkers associated with the presence or absence of C. difficile in each dataset (at the sampling time for ClosNEC and EPIFLORE_D28, and 1 month for EPIFLORE_D7_2) are shown in Figure 3. We identified several biomarkers associated with C. difficile colonization, particularly Veillonella sp., and taxa belonging to families such as Lachnospiraceae, Oscillospiraceae, and Enterobacteriaceae (Enterobacter ludwigii for ClosNEC, Escherichia–Shigella for EPIFLORE_D7_2, Klebsiella oxytoca, and Kluyvera for EPIFLORE_D28). Bifidobacterium breve and Bacteroidales were significantly more abundant in samples negative for C. difficile.
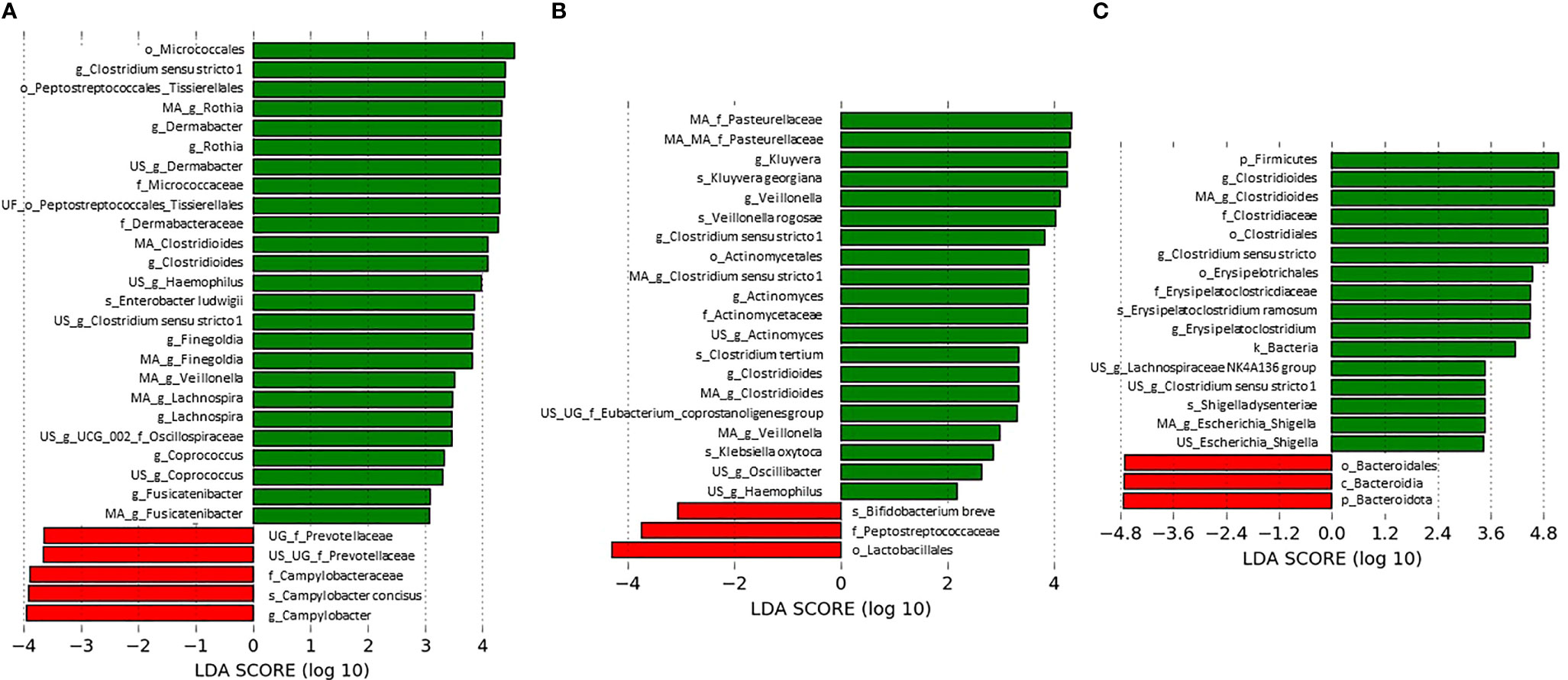
Figure 3 Most differentially abundant taxa associated with C. difficile absence (red) or presence (green) (A) at the sampling time for the ClosNEC dataset, (B) at the sampling time for the EPIFLORE_D28 dataset and (C) at D28 for the EPIFLORE_D7_2 dataset. LDA scores are expressed in log10 values. Only taxa meeting an LDA significant threshold > 2 are shown. MA: multi-affiliation; US: unknown species; UG: unknown genus; UF: unknown family; k: kingdom; p: phylum; c: class; o: order; f: family; g: genus; s: species.
From the mixed linear model analysis, variables identified as significantly associated with the colonization status were integrated with the specifically associated OTUs (Table S2), i.e. for ClosNEC, gestational age and transit at D7 considered as normal, and for EPIFLORE, postnatal antibiotherapy after D3, enterotype profile according to Rozé et al. (Rozé et al., 2020), and gestational age. For OTUs that showed a significant association (p < 0.05), the model was fitted to control the effect of other variables. For ClosNEC, 4 OTUs maintained a positive significant interaction with the CD+ group after fitting the model: Clostridioides, Rothia, Bifidobacterium and Veillonella. For EPIFLORE, Clostridioides, Enterobacterales and Eisenbergiella maintained a positive significant interaction with the CD+ group after fitting the model.
We investigated the intestinal microbiota of PN, focusing on variations in bacterial populations associated with C. difficile colonization. This is an original study, as it included PN from two multicentric cohorts with microbiota analysis based on both culture analysis and 16S rRNA gene sequencing.
Our results showed that the median number of OTUs in each sample ranged from 41.5–44 for the three datasets analyzed, confirming the low richness of the intestinal ecosystem in PN (Drell et al., 2014). The most represented phyla were Proteobacteria and Firmicutes, whereas the abundance of Bifidobacteriaceae and Lactobacillaceae was low (median < 0.01%), in agreement with previous data (Drell et al., 2014; Ho et al., 2018). Among the PN of the EPIFLORE cohort with a first-week sample (n=142), only two (1.3%) were CD+, whereas the prevalence of C. difficile in stools sampled at one month was 10.4% (50/483). C. difficile was detected in 29.3% of the samples (n=116) for the ClosNEC cohort. These colonization rates were lower than those observed in the previous PREMAFLORA cohort (Ferraris et al., 2019) where 20% of PN were colonized at one week and 61% at one month. This difference could be explained by the fact that PREMAFLORA was a single-center study with potential cross-transmission of C. difficile within the ward. Moreover, PN included in PREMAFLORA had a higher gestational age than that in the ClosNEC and EPIFLORE cohorts (median gestational age 32 weeks, range 24–36 weeks). Notably, most strains isolated by culture were non-toxigenic (92.9%).
The reported rates of C. difficile colonization in PN are highly variable and originate primarily from outdated, single-center, culture-based studies (Cardines et al., 1988; Chang et al., 2012; Pichler et al., 2018). The C. difficile colonization rates depend on the detection method used. In our study, the agreement between bacterial culture and NGS of the 16S rRNA gene was 86.2%. The detection threshold for the culture-based method was 3.3 log10 CFU/g stools (Ferraris et al., 2019). The detection limits of NGS-based methods vary from one study to another, particularly because of the use of different primers targeting different regions of the 16S rRNA gene. In samples less complex than feces (urine, water), bacteria can be detected at 101–102 CFU/mL (Sabat et al., 2017; Brandt and Albertsen, 2018). However, it has been suggested that sequence-based techniques can miss bacteria present in the adult gut microbiota at concentrations lower than 105 CFU/mL (Lagier et al., 2012). Detection of the 16S rRNA gene does not indicate the viability of the microorganisms in the sample. Crobach et al. assigned patients to colonized, infected, and control groups according to culture results, and sequences affiliated with the Clostridioides OTU were detected in only 26/41 (63.4%) colonized patients and 38/41 (92.7%) infected patients (Crobach et al., 2020).
We showed that alpha diversity at D28 (for both ClosNEC and EPIFLORE_D28 datasets) increased when C. difficile was detected in stool samples. Alpha diversity at D7 (EPIFLORE_D7_2 dataset) was also higher when C. difficile was detected in samples from the same PN at D28. These findings are unusual compared to available data from 16S rRNA gene NGS analysis suggesting that C. difficile colonization is associated with a decreased alpha diversity of the gut microbiota in adults and children (Chen et al., 2019; Han et al., 2019; Crobach et al., 2020; Han et al., 2020). In a study performed in two Dutch NICUs, Crobach et al. reported that the Shannon index was significantly higher in 43 control adult individuals than in 41 C. difficile-colonized or 41 -infected patients (p < 0.01), whereas no difference was seen between colonized and infected patients (Crobach et al., 2020). Similar results were obtained in a Korean single-center study including 99 adult patients, where the mean Chao1 index of patients who were tcdB-positive was significantly lower than that of the control group (p < 0.001) (Han et al., 2019). In a pediatric population (median age 14 years, range 4–19 years, n=113), alpha diversity was also significantly lower in the C. difficile-colonized group (p = 0.029) (Chen et al., 2019). However, another study showed no difference in alpha diversity (Chao1 index) between C. difficile-colonized (n=93) and control (n=93) adults (p = 0.797) (Han et al., 2020). Unlike older populations, our results suggest that in PN, C. difficile colonization is concomitant with physiological establishment and diversification of the gut microbiota. Moreover, PN included in our study were mostly colonized by non-toxigenic strains, which may have different effects on the gut microbiota.
PERMANOVA analysis of the gut microbiota in the ClosNEC cohort showed that beta diversity estimated by the Jaccard index differed significantly between CD+ and CD− samples, but not according to the wUnifrac method, suggesting that abundant and phylogenetically-related species were shared. In the EPIFLORE cohort, the microbiota composition significantly differed between CD+ and CD− samples at D28 (EPIFLORE_D28 dataset) for both beta diversity matrices. For samples at D7 (EPIFLORE_D7_2 dataset), only beta diversity according to the Jaccard index showed a significantly different composition between PN subsequently colonized at D28 and PN not colonized at D28. These results align with the progressive maturation and diversification of the gut microbiota between one week and one month of life.
The functional pathway prediction analysis showed that several KO pathways differed according to the colonization status. Of note, C. difficile-colonized PN had higher proportions of energy metabolism, starch and sucrose metabolism, and peptidases pathways in both ClosNEC and EPIFLORE cohorts. However, this analysis is only predictive and further studies are needed to confirm these findings.
Using LefSe analysis, we identified several bacterial biomarkers associated with C. difficile colonization of PN, including Veillonella, Lachnospiraceae, Oscillospiraceae, and Enterobacteriaceae. Veillonella are commensal bacteria of the oral cavity that have been previously associated with CDI (Han et al., 2020). Veillonella is associated with low rates of coprostanol, a cholesterol metabolite. Antharam et al. discussed these metabolic abilities as causes of CDI susceptibility, although the molecular mechanisms involved remain unknown (Antharam et al., 2016). Lachnospiraceae family have been previously associated with C. difficile colonization in children aged 4–21 (Chen et al., 2019). Bacteria belonging to the Lachnospiraceae family produce short-chain fatty acids, which are end products of dietary carbohydrates with a beneficial effect on host health (Wong et al., 2006). We hypothesized that the Lachnospiraceae taxa play a role in the permissiveness of the microbiota in C. difficile colonization. Our results on Enterobacteriaceae are consistent with those of previous studies on full-term neonates (Rousseau et al., 2011) and adult patients (Han et al., 2020). A high abundance of facultative anaerobic bacteria, fostering an anaerobic environment by consuming oxygen, has been reported to enable C. difficile colonization (Rousseau et al., 2011). Contradictory data are available for other biomarkers such as Oscillospiraceae. In a Chinese study, Oscillibacter was more abundant in healthy children than in infected children (Ling et al., 2014), whereas Oscillospira was more abundant in C. difficile-colonized than in infected adults (Han et al., 2019). We showed that Bifidobacterium breve and Bacteroidales, previously reported to protect against C. difficile colonization (Rousseau et al., 2011) and infection (Ling et al., 2014), were more abundant in PN without C. difficile colonization. The barrier effect of these taxa could explain our results (Roberfroid et al., 2010; Yang and Yang, 2019). To further identify these potential mechanisms, metabolite measures in stools and competition assays between C. difficile and identified biomarkers are necessary.
Using a mixed linear model, we identified several bacterial groups more abundant in the microbiota of CD+ children after adjustment for covariables, i.e. in the ClosNEC cohort, Clostridioides, Rothia, Bifidobacterium and Veillonella, and in the EPIFLORE cohort, Clostridioides, Enterobacterales and Eisenbergiella. With both LefSe analysis and the mixed linear model, bacterial biomarkers associated with the presence or absence of C. difficile were different between ClosNEC and EPIFLORE cohorts. This difference might be explained by i) the fact that the ClosNEC cohort included more children suffering from NEC than EPIFLORE cohort, ii) the fact that PN in the ClosNEC cohort received more frequently antibiotics, iii) the fact that the range of sampling times was different between the two cohorts (Table 1).
The C. difficile colonization rate increased when PN did not receive postnatal antibiotics (EPIFLORE) and had a higher gestational age (ClosNEC). Our results are unusual compared to those of other studies in adults showing that C. difficile is established in the gut as a result of dysbiosis, especially after antibiotherapy (Vincent et al., 2013). Several hypotheses may explain the relationship between antibiotics, the gut microbiota and C. difficile colonization: antibiotics might protect from colonization either via a direct activity on C. difficile; or via the induced dysbiosis which would be unfavourable for C. difficile implantation. In the EPIFLORE cohort, the fact that predictive differences in the gut microbiota diversity were observed at D7 between PN who would be colonized or not at D28 favours the latter hypothesis. The most frequently received antibiotics by PN of both cohorts were third generation cephalosporins, aminosides, and IV vancomycin, which are inefficient against C. difficile. Our statistical analyses corroborated the higher microbial diversity in colonized PN in accordance with the physiological nature of C. difficile implantation. Notably, we showed that C. difficile colonization did not differ between PN with or without NEC, supporting the fact that C. difficile is not involved in NEC pathogenesis (Lishman et al. 1984; Waligora-Dupriet et al., 2005).
This study has several limitations. When applying filters for OTUs, we lowered the minimum abundance threshold from 0.005% (recommended in (Bokulich et al., 2013)) to 0.00005% to account for the scarcity of PN microbiota (Drell et al., 2014) and to maintain potentially rare OTUs. To avoid artifactual OTUs resulting from sequencing errors, we retained OTUs present in at least three samples. Owing to highly variable sequencing coverage among samples, normalization before diversity analyses leads to a potential loss of rare OTUs in samples with a high number of amplified sequences. The small number of PN included in some groups, particularly children of the EPIFLORE cohort with a sample at D7 and subsequently colonized by C. difficile at D28 (n=9), reduced statistical power. This could partly explain why few diversity changes were observed between the groups in the EPIFLORE_D7_2 dataset. The variable “C. difficile presence or absence”-defining groups for microbiota analysis is based on the detection of sequences corresponding to C. difficile and not culture-based.
The strengths of this study are the high number of PN included (n total = 599), their recruitment from multicentric cohorts, and the extensive clinical data collected. Fecal samples obtained at both D7 and D28 for 116 PN enabled an analysis of the intestinal microbiota predictive of the subsequent colonization status, which is of utmost interest.
This study provides new insights into understanding C. difficile colonization in PN and the associated gut microbiota modifications. Variations in bacterial populations and associated perinatal factors favor physiological C. difficile colonization, occurring in PN with a less disturbed microbiota. However, the timeline of this process and the mechanisms regulating the relationship between C. difficile and the establishment of gut microbiota are yet to be explained.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA819954.
For the ClosNEC cohort (Clinical trial no. NCT02444624), approval was obtained from the National Data Protection Authority (Commission Nationale de l’Informatique et des Libertés, approval no. 915094) and the Consultative Committee on the Treatment of Information on Personal Health Data for Research Purposes (approval no. 15.055). For the EPIFLORE cohort, approval was obtained from the National Data Protection Authority (Commission Nationale de l’Informatique et des Libertés, approval no.911009), the Consultative Committee on the Treatment of Information on Personal Health Data for Research Purposes (approval no.10.626), and the Committee for the Protection of People Participating in Biomedical Research (approval no. CPP SC-2873). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Conceptualization, JA, M-JB, and FB. Methodology, JA, FB, and JC. Software, PL, VM, and JC. Validation, JA, FB, and JC. Formal analysis, JC, PL, JD, VM, and SJ. Investigation, JC and JD. Resources, JA, P-YA, M-JB, PL, and J-CR. Data curation, JC and JD. Writing—original draft preparation, JC. Writing—review and editing, all authors. Visualization, JC. Supervision, JA, and FB. Project administration, JA, FB, and JC. Funding acquisition, JA, P-YA, M-JB, and J-CR. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the Agence Nationale de la Recherche (grants ANR-12-SV, ANR-12-BSV3-0025001/EPIFLORE, and ANR-13-PRTS-0018) and the PremUp Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the INRA MIGALE bioinformatics facility (MIGALE; INRA, 2018. Migale bioinformatics Facility, doi: 10.15454/1.5572390655343293E12) and the Genotoul bioinformatics platform, Toulouse Midi-Pyrenees, and Sigenae group for providing help, computing, and storage resources (Galaxy instance https://vm-galaxy-prod.toulouse.inra.fr). We would like to thank Editage (www.editage.com) for English language editing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.907323/full#supplementary-material
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Cech, M., et al. (2018). The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 46 (W1), W537−44. doi: 10.1093/nar/gky379
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215 (3), 403−10. doi: 10.1016/S0022-2836(05)80360-2
Antharam, V. C., McEwen, D. C., Garrett, T. J., Dossey, A. T., Li, E. C., Kozlov, A. N., et al. (2016). An Integrated Metabolomic and Microbiome Analysis Identified Specific Gut Microbiota Associated With Fecal Cholesterol and Coprostanol in Clostridium Difficile Infection. PLos One 11 (2), e0148824. doi: 10.1371/journal.pone.0148824
Bacon, A. E., Fekety, R., Schaberg, D. R., Faix, R. G. (1988). Epidemiology of Clostridium Difficile Colonization in Newborns: Results Using a Bacteriophage and Bacteriocin Typing System. J. Infect. Dis. 158 (2), 349−54. doi: 10.1093/infdis/158.2.349
Barbut, F., Couturier, J. (2019). [Interactions Between Intestinal Microbiota and Clostridioides Difficile]. Rev. Prat. 69 (7), 784−91.
Bates, D., Mächler, M., Bolker, B., Walker, S. (2015). Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Software 67, 1−48. doi: 10.18637/jss.v067.i01
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-Filtering Vastly Improves Diversity Estimates From Illumina Amplicon Sequencing. Nat. Methods 10 (1), 57−9. doi: 10.1038/nmeth.2276
Brandt, J., Albertsen, M. (2018). Investigation of Detection Limits and the Influence of DNA Extraction and Primer Choice on the Observed Microbial Communities in Drinking Water Samples Using 16s rRNA Gene Amplicon Sequencing. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02140
Cardines, R., Luzzi, I., Menichella, G., Virgili, Q., Mastrantonio, P. (1988). Clostridium Difficile in Preterm Neonates. Microbiologica 11 (3), 259−61.
Chang, J. Y., Shim, J. O., Ko, J. S., Seo, J. K., Lee, J. A., Kim, H. S., et al. (2012). Monitoring of Clostridium Difficile Colonization in Preterm Infants in Neonatal Intensive Care Units. Pediatr. Gastroenterology Hepatol. Nutr. 15 (1), 29−37. doi: 10.5223/pghn.2012.15.1.29
Chen, L. A., Hourigan, S. K., Grigoryan, Z., Gao, Z., Clemente, J. C., Rideout, J. R., et al. (2019). Decreased Fecal Bacterial Diversity and Altered Microbiome in Children Colonized With Clostridium Difficile. J. Pediatr. Gastroenterol. Nutr. 68 (4), 502−8. doi: 10.1097/MPG.0000000000002210
Couturier, J., Franconeri, L., Janoir, C., Ferraris, L., Syed-Zaidi, R., Youssouf, A., et al. (2020). Characterization of Non-Toxigenic Clostridioides Difficile Strains Isolated From Preterm Neonates and In Vivo Study of Their Protective Effect. J. Clin. Med. 9 (11), 1–14. doi: 10.3390/jcm9113650
Crobach, M. J. T., Ducarmon, Q. R., Terveer, E. M., Harmanus, C., Sanders, I. M. J. G., Verduin, K. M., et al. (2020). The Bacterial Gut Microbiota of Adult Patients Infected, Colonized or Noncolonized by Clostridioides Difficile. Microorganisms 8 (5), 1–13. doi: 10.3390/microorganisms8050677
Doré, J., Ehrlich, S. D., Levenez, F., Pelletier, E., Alberti, A., Bertrand, L., et al. (2015). “IHMS_SOP 07 V1: Standard Operating Procedure for Fecal Samples DNA Extraction, Protocol H,” in International Human Microbiome Standards Consortium. Available at: http://www.microbiome-standards.org.
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 38 (6), 685−8. doi: 10.1038/s41587-020-0548-6
Drell, T., Lutsar, I., Stšepetova, J., Parm, U., Metsvaht, T., Ilmoja, M. L., et al. (2014). The Development of Gut Microbiota in Critically Ill Extremely Low Birth Weight Infants Assessed With 16S rRNA Gene Based Sequencing. Gut Microbes 5 (3), 304−12. doi: 10.4161/gmic.28849
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., Knight, R. (2011). UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 27 (16), 2194−200. doi: 10.1093/bioinformatics/btr381
el-Mohandes, A. E., Keiser, J. F., Refat, M., Jackson, B. J. (1993). Prevalence and Toxigenicity of Clostridium Difficile Isolates in Fecal Microflora of Preterm Infants in the Intensive Care Nursery. Biol. Neonate 63 (4), 225−9. doi: 10.1159/000243935
Enoch, D. A., Butler, M. J., Pai, S., Aliyu, S. H., Karas, J. A. (2011). Clostridium Difficile in Children: Colonisation and Disease. J. Infect. 63, 2, 105−113. doi: 10.1016/j.jinf.2011.05.016
Escudié, F., Auer, L., Bernard, M., Mariadassou, M., Cauquil, L., Vidal, K., et al. (2018). FROGS: Find, Rapidly, OTUs With Galaxy Solution. Bioinformatics 34 (8), 1287−94. doi: 10.1093/bioinformatics/btx791
Ferraris, L., Butel, M. J., Campeotto, F., Vodovar, M., Rozé, J. C., Aires, J. (2012). Clostridia in Premature Neonates’ Gut: Incidence, Antibiotic Susceptibility, and Perinatal Determinants Influencing Colonization. PLos One 7 (1), 1–7. doi: 10.1371/journal.pone.0030594
Ferraris, L., Couturier, J., Eckert, C., Delannoy, J., Barbut, F., Butel, M. J., et al. (2019). Carriage and Colonization of C. Difficile in Preterm Neonates: A Longitudinal Prospective Study. PLos One 14 (2), e0212568. doi: 10.1371/journal.pone.0212568
Gewolb, I. H., Schwalbe, R. S., Taciak, V. L., Harrison, T. S., Panigrahi, P. (1999). Stool Microflora in Extremely Low Birthweight Infants. Arch. Dis. Child Fetal Neonatal Ed 80 (3), F167–F173. doi: 10.1136/fn.80.3.F167
Han, S. H., Yi, J., Kim, J. H., Lee, S., Moon, H. W. (2019). Composition of Gut Microbiota in Patients With Toxigenic Clostridioides (Clostridium) Difficile: Comparison Between Subgroups According to Clinical Criteria and Toxin Gene Load. PLos One 14 (2), 1–12. doi: 10.1371/journal.pone.0212626
Han, S. H., Yi, J., Kim, J. H., Moon, H. W. (2020). Investigation of Intestinal Microbiota and Fecal Calprotectin in Non-Toxigenic and Toxigenic Clostridioides Difficile Colonization and Infection. Microorganisms 8 (6), 882. doi: 10.3390/microorganisms8060882
Ho, T. T. B., Groer, M. W., Kane, B., Yee, A. L., Torres, B. A., Gilbert, J. A., et al. (2018). Dichotomous Development of the Gut Microbiome in Preterm Infants. Microbiome 6, 1–13. doi: 10.1186/s40168-018-0547-8
Jangi, S., Lamont, J. T. (2010). Asymptomatic Colonization by Clostridium Difficile in Infants: Implications for Disease in Later Life. J. Pediatr. Gastroenterol. Nutr. 51 (1), 2−7. doi: 10.1097/MPG.0b013e3181d29767
Lagier, J. C., Armougom, F., Million, M., Hugon, P., Pagnier, I., Robert, C., et al. (2012). Microbial Culturomics: Paradigm Shift in the Human Gut Microbiome Study. Clin. Microbiol. Infection 18 (12), 1185−93. doi: 10.1111/1469-0691.12023
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 31 (9), 814−21. doi: 10.1038/nbt.2676
Lemee, L., Dhalluin, A., Testelin, S., Mattrat, M. A., Maillard, K., Lemeland, J. F., et al. (2004). Multiplex PCR Targeting Tpi (Triose Phosphate Isomerase), tcdA (Toxin A), and tcdB (Toxin B) Genes for Toxigenic Culture of Clostridium Difficile. J. Clin. Microbiol. 42 (12), 5710−4. doi: 10.1128/JCM.42.12.5710-5714.2004
Ling, Z., Liu, X., Jia, X., Cheng, Y., Luo, Y., Yuan, L., et al. (2014). Impacts of Infection With Different Toxigenic Clostridium Difficile Strains on Faecal Microbiota in Children. Sci. Rep. 4, 7485. doi: 10.1038/srep07485
Lishman, A. H., Jumaili, I. J. A., Elshibly, E., Hey, E., Record, C. O. (1984). Clostridium Difficile Isolation in Neonates in a Special Care Unit Lack of Correlation with Necrotizing Enterocolitis. Scand. J. Gastroenterol 19 (3), 441–444
Mahé, F., Rognes, T., Quince, C., de Vargas, C., Dunthorn, M. (2014). Swarm: Robust and Fast Clustering Method for Amplicon-Based Studies. PeerJ 2, e593. doi: 10.7717/peerj.593
Martin, M. (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal 17 (1), 10−2. doi: 10.14806/ej.17.1.200
McMurdie, P. J., Holmes, S. (2013). Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLos One 8 (4), e61217. doi: 10.1371/journal.pone.0061217
Parks, D. H., Tyson, G. W., Hugenholtz, P., Beiko, R. G. (2014). STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 30 (21), 3123−4. doi: 10.1093/bioinformatics/btu494
Pichler, K., Bausenhardt, B., Huhulescu, S., Lindtner, C., Indra, A., Allerberger, F., et al. (2018). Impact and Time Course of Clostridium Difficile Colonization in Very Low Birth Weight Infants. Neonatology 114 (2), 149−54. doi: 10.1159/000488025
Pinheiro, J., Bates, D. R., Core Team (2022). “Nlme: Linear and Nonlinear Mixed Effects Models,” in R Package Version 3.1-157.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Roberfroid, M., Gibson, G. R., Hoyles, L., McCartney, A. L., Rastall, R., Rowland, I., et al. (2010). Prebiotic Effects: Metabolic and Health Benefits. Br. J. Nutr. 104 Suppl 2, S1–63. doi: 10.1017/S0007114510003363
Rognes, T., Flouri, T., Nichols, B., Quince, C., Mahé, F. (2016). VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584
Rousseau, C., Lemée, L., Le Monnier, A., Poilane, I., Pons, J. L., Collignon, A. (2011). Prevalence and Diversity of Clostridium Difficile Strains in Infants. J. Med. Microbiol. 60 (8), 1112−8. doi: 10.1099/jmm.0.029736-0
Rousseau, C., Levenez, F., Fouqueray, C., Doré, J., Collignon, A., Lepage, P. (2011). Clostridium Difficile Colonization in Early Infancy Is Accompanied by Changes in Intestinal Microbiota Composition. J. Clin. Microbiol. 49 (3), 858−65. doi: 10.1128/JCM.01507-10
Rousseau, C., Poilane, I., De Pontual, L., Maherault, A. C., Le Monnier, A., Collignon, A. (2012). Clostridium Difficile Carriage in Healthy Infants in the Community: A Potential Reservoir for Pathogenic Strains. Clin. Infect. Dis. 55 (9), 1209−15. doi: 10.1093/cid/cis637
Rozé, J. C., Ancel, P. Y., Lepage, P., Martin-Marchand, L., Al Nabhani, Z., Delannoy, J., et al. (2017). Nutritional Strategies and Gut Microbiota Composition as Risk Factors for Necrotizing Enterocolitis in Very-Preterm Infants. Am. J. Clin. Nutr. 106 (3), 821−30. doi: 10.3945/ajcn.117.152967
Rozé, J. C., Ancel, P. Y., Marchand-Martin, L., Rousseau, C., Montassier, E., Monot, C., et al. (2020). Assessment of Neonatal Intensive Care Unit Practices and Preterm Newborn Gut Microbiota and 2-Year Neurodevelopmental Outcomes. JAMA Netw. Open 3 (9), e2018119−e2018119. doi: 10.1001/jamanetworkopen.2020.18119
Sabat, A. J., van Zanten, E., Akkerboom, V., Wisselink, G., van Slochteren, K., de Boer, R. F., et al. (2017). Targeted Next-Generation Sequencing of the 16S-23S rRNA Region for Culture-Independent Bacterial Identification - Increased Discrimination of Closely Related Species. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-03458-6
Schönherr-Hellec, S., Klein, G. L., Delannoy, J., Ferraris, L., Rozé, J. C., Butel, M. J., et al. (2018). Clostridial Strain-Specific Characteristics Associated With Necrotizing Enterocolitis. Appl. Environ. Microbiol. 84 (7), 1–8. doi: 10.1128/AEM.02428-17
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic Biomarker Discovery and Explanation. Genome Biol. 24 juin 12 (6), R60. doi: 10.1186/gb-2011-12-6-r60
Stoesser, N., Eyre, D. W., Quan, T. P., Godwin, H., Pill, G., Mbuvi, E., et al. (2017). Epidemiology of Clostridium Difficile in Infants in Oxfordshire, UK: Risk Factors for Colonization and Carriage, and Genetic Overlap With Regional C. Difficile Infection Strains. PLos One 12 (8), e0182307. doi: 10.1371/journal.pone.0182307
Tina, L. G., Proto, N., Sciacca, A. (1994). Asymptomatic Intestinal Colonization by Clostridium Difficile in Preterm Neonates. Pediatr. Infect. Dis. J. 13 (12), 1158. doi: 10.1097/00006454-199412000-00023
Vincent, C., Stephens, D. A., Loo, V. G., Edens, T. J., Behr, M. A., Dewar, K., et al. (2013). Reductions in Intestinal Clostridiales Precede the Development of Nosocomial Clostridium Difficile Infection. Microbiome 1, 18. doi: 10.1186/2049-2618-1-18
Waligora-Dupriet, A. J., Dugay, A., Auzeil, N., Huerre, M., Butel, M. J. (2005). Evidence for Clostridial Implication in Necrotizing Enterocolitis Through Bacterial Fermentation in a Gnotobiotic Quail Model. Pediatr. Res. 58 (4), 629−35. doi: 10.1203/01.PDR.0000180538.13142.84
Wilson, K. H., Blitchington, R. B., Greene, R. C. (1990). Amplification of Bacterial 16S Ribosomal DNA With Polymerase Chain Reaction. J. Clin. Microbiol. 28 (9), 1942−6. doi: 10.1128/jcm.28.9.1942-1946.1990
Wong, J. M. W., de Souza, R., Kendall, C. W. C., Emam, A., Jenkins, D. J. A. (2006). Colonic Health: Fermentation and Short Chain Fatty Acids. J. Clin. Gastroenterol. 40 (3), 235−43. doi: 10.1097/00004836-200603000-00015
Keywords: Clostridioides (Clostridium) difficile, colonization, preterm neonates, gut microbiota, microbial diversity, 16S rRNA gene sequencing, necrotizing enterocolitis
Citation: Couturier J, Lepage P, Jolivet S, Delannoy J, Mesa V, Ancel P-Y, Rozé J-C, Butel M-J, Barbut F and Aires J (2022) Gut Microbiota Diversity of Preterm Neonates Is Associated With Clostridioides Difficile Colonization. Front. Cell. Infect. Microbiol. 12:907323. doi: 10.3389/fcimb.2022.907323
Received: 29 March 2022; Accepted: 02 June 2022;
Published: 06 July 2022.
Edited by:
Zhong Peng, Huazhong Agricultural University, ChinaReviewed by:
Qi Wang, Huazhong University of Science and Technology, ChinaCopyright © 2022 Couturier, Lepage, Jolivet, Delannoy, Mesa, Ancel, Rozé, Butel, Barbut and Aires. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeanne Couturier, amVhbm5lLmNvdXR1cmllckBhcGhwLmZy
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.