
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 11 May 2022
Sec. Clinical Microbiology
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.878137
This article is part of the Research TopicCharacterization of Rare and Recently First Described Human Pathogenic BacteriaView all 11 articles
 Stefan Monecke1,2*
Stefan Monecke1,2* Frieder Schaumburg3
Frieder Schaumburg3 Adebayo O. Shittu3,4
Adebayo O. Shittu3,4 Stefan Schwarz5,6
Stefan Schwarz5,6 Kristin Mühldorfer7
Kristin Mühldorfer7 Christian Brandt8
Christian Brandt8 Sascha D. Braun1,2
Sascha D. Braun1,2 Maximilian Collatz1,2
Maximilian Collatz1,2 Celia Diezel1,2
Celia Diezel1,2 Darius Gawlik9Dennis Hanke5,6Helmut Hotzel10‡
Darius Gawlik9Dennis Hanke5,6Helmut Hotzel10‡ Elke Müller1,2Martin Reinicke1,2
Elke Müller1,2Martin Reinicke1,2 Andrea T. Feßler5,6†
Andrea T. Feßler5,6† Ralf Ehricht1,2,11†
Ralf Ehricht1,2,11†The phylogenetic tree of the Staphylococcus aureus complex consists of several distinct clades and the majority of human and veterinary S. aureus isolates form one large clade. In addition, two divergent clades have recently been described as separate species. One was named Staphylococcus argenteus, due to the lack of the “golden” pigment staphyloxanthin. The second one is S. schweitzeri, found in humans and animals from Central and West Africa. In late 2021, two additional species, S. roterodami and S. singaporensis, have been described from clinical samples from Southeast Asia. In the present study, isolates and their genome sequences from wild Straw-coloured fruit bats (Eidolon helvum) and a Diamond firetail (Stagonopleura guttata, an estrildid finch) kept in a German aviary are described. The isolates possessed staphyloxanthin genes and were closer related to S. argenteus and S. schweitzeri than to S. aureus. Phylogenetic analysis revealed that they were nearly identical to both, S. roterodami and S. singaporensis. We propose considering the study isolates, the recently described S. roterodami and S. singaporensis as well as some Chinese strains with MLST profiles stored in the PubMLST database as different clonal complexes within one new species. According to the principle of priority we propose it should be named S. roterodami. This species is more widespread than previously believed, being observed in West Africa, Southeast Asia and Southern China. It has a zoonotic connection to bats and has been shown to be capable of causing skin and soft tissue infections in humans. It is positive for staphyloxanthin, and it could be mis-identified as S. aureus (or S. argenteus) using routine procedures. However, it can be identified based on distinct MLST alleles, and “S. aureus” sequence types ST2470, ST3135, ST3952, ST3960, ST3961, ST3963, ST3965, ST3980, ST4014, ST4075, ST4076, ST4185, ST4326, ST4569, ST6105, ST6106, ST6107, ST6108, ST6109, ST6999 and ST7342 belong to this species.
The phylogenetic tree of bacteria traditionally considered Staphylococcus (S.) aureus consists of several distinct clades. Most human and veterinary S. aureus isolates from all over the world form one large clade. In addition, two divergent clades have recently been elevated to the status of species (Tong et al., 2015; Becker et al., 2019). One has been named S. argenteus, due to its lack of the “golden” carotenoid pigment staphyloxanthin (Holt et al., 2011) regarded as a unique property of S. aureus. Isolates assigned or re-assigned to this new species have been described in many countries including Australia (Ng et al., 2009), Thailand (Indrawattana et al., 2019; Pumipuntu, 2019), Laos (Yeap et al., 2017), Cambodia (Ruimy et al., 2009), Myanmar (Aung et al., 2019), Japan (Ohnishi et al., 2018; Aung et al., 2019; Kitagawa et al., 2020), China and Taiwan (Chen et al., 2018), on Indian Ocean islands such as the Comoros or Mayotte (Dupieux et al., 2015), Gabon (Schuster et al., 2017), Trinidad and Tobago (Monecke et al., 2014) and Brazil (Rossi et al., 2020). Sporadic isolates also have been identified in the United Arab Emirates (Senok et al., 2020), several European countries (Dupieux et al., 2015; Rigaill et al., 2018; Tång Hallbäck et al., 2018; Diot et al., 2020; Kukla et al., 2020; Söderquist et al., 2020) as well as in Canada and the United States of America (Eshaghi et al., 2021). These observations could be associated with travel and migration. Aside from the lack of the carotenoid pigment gene cluster, S. argenteus isolates carry the same genes as S. aureus, albeit they occur as distinct allelic variants (Monecke et al., 2010), and its genes are located in the genome following the same order as in S. aureus. Traditional PCR-based multilocus sequence typing (MLST) can be performed on S. argenteus using slightly modified primers (Ng et al., 2009; Ruimy et al., 2009; Holt et al., 2011) and as in S. aureus, the resulting sequence types (ST) cluster into closely related groups known as clonal complexes (CCs). Moreover, CC affiliation also correlates with the presence or absence of certain genomic islands essentially identical to their counterparts in S. aureus. Thus, ST1223, ST1850 (formerly ST75), ST2198, ST2250, ST2596/2793 (Aung et al., 2019; Hsu et al., 2020), ST2854 and ST4587 can be regarded as founders of homonymous CCs. Some mobile genetic elements from S. aureus have also been identified in S. argenteus. This includes SCCmec IV and V elements that carry the methicillin/beta-lactam resistance gene mecA, phages harbouring the Panton-Valentine leukocidin gene (Dupieux et al., 2015; Aung et al., 2017; Senok et al., 2020) and the pathogenicity-island-borne tst1 [encoding toxic shock syndrome toxin 1 (Aung et al., 2017)]. S. argenteus can asymptomatically be carried in the nares. It also can cause the same types of infections as S. aureus (Becker et al., 2019), i.e., skin and soft tissue infections (Ohnishi et al., 2018), osteomyelitis (Rigaill et al., 2018) or endoprosthesis infections (Diot et al., 2020; Söderquist et al., 2020) and sepsis (Chen et al., 2018; Kitagawa et al., 2020). S. argenteus also has been implicated in food poisoning (Suzuki et al., 2017). Some lineages of S. argenteus have been identified in animals such as rabbits (Indrawattana et al., 2019), dairy cattle (Pumipuntu, 2019; Rossi et al., 2020) and a wild gorilla (Schuster et al., 2017).
The other entity, S. schweitzeri consists of several sequence types (ST1857, ST1872, ST1873, ST1874, ST2022, ST2058, ST2059, ST2067, ST2071, ST2463, ST2464, ST2465, ST2467, ST3952, ST3958, ST3960, ST3961, ST3962, ST3963, ST3980, ST4316, ST4326, ST5117, ST5600 and ST5602). The alleles of core genome genes of S. schweitzeri are distinct from those of S. aureus and S. argenteus. However, published genome sequences of S. schweitzeri and experiments with DNA microarrays (Okuda et al., 2016) indicate that certain genomic island genes (agr alleles, capsule type, egc, cna, seh, carotenoid locus genes) closely resemble their S. aureus counterparts. The presence of these genes in S. schweitzeri is related to their CC affiliation, as also noted in S. aureus. So far, S. schweitzeri has been observed in four different regions. It was first identified in Gabon (Tong et al., 2015) from where it was also reported in other studies (Schaumburg et al., 2012; Schaumburg et al., 2015; Okuda et al., 2016) as well as in Côte d’Ivoire (Schaumburg et al., 2012; Schaumburg et al., 2015), Nigeria (Akobi et al., 2012) and the Democratic Republic of Congo, DRC (Schaumburg et al., 2015). Most of these isolates originated from non-human primates (Schaumburg et al., 2012) or from “bush-meat”, i.e., poached or hunted wildlife sold on local markets (Schaumburg et al., 2015). Some isolates from healthy humans have been identified, suggesting that humans carry this lineage sporadically and asymptomatically (Tong et al., 2015; Schaumburg et al., 2015; Okuda et al., 2016). In Nigeria, S. schweitzeri was recovered from faecal samples of the Straw-coloured fruit bats (Eidolon helvum) on the premises of a university campus (Akobi et al., 2012). This observation caused concerns of zoonotic transmission as these isolates were also detected on fomites in the same university (Shittu et al., 2020). However, a transmission of S. schweitzeri from animals to humans has not yet been observed. A large study from the DRC, Gabon and Côte d’Ivoire investigated rural populations and did not identify S. schweitzeri among humans despite close contact with bushmeat and wildlife (Schaumburg et al., 2015). The pathogenicity of S. schweitzeri remains unclear as humans were found to be colonised rather than infected (Schaumburg et al., 2015; Okuda et al., 2016). However, in vitro experiments suggest that S. schweitzeri is as virulent as S. aureus (Grossmann et al., 2021). Generally, one might assume that it is a zoonotic species that might asymptomatically colonise humans (Becker et al., 2019) and appears to be restricted to Central/West Africa. However, more data are needed to assess the distribution and a possible clinical significance of S. schweitzeri.
In autumn 2021, two new species of “S. aureus-like” staphylococci were described from human samples. These were named S. roterodami and S. singaporensis (Schutte et al., 2021; Chew et al., 2021). A single isolate of S. roterodami was identified from an infected wound of a Dutch traveller returning from Bali, Indonesia, prompting bacteriological investigations and genome sequencing (Schutte et al., 2021). A study (Chew et al., 2021) investigating a possible presence of S. argenteus/schweitzeri in Singapore identified 37 S. argenteus and six “unknowns” assigned to five novel STs and described as a new species, S. singaporensis. Four of these six isolates were associated with skin and soft tissue infections.
We describe a group of animal isolates submitted to the authors´ laboratories for characterisation as suspected S. argenteus or S. schweitzeri. These were characterised and sequenced. We also review their relationship to S. aureus, S. argenteus, S. schweitzeri, S. roterodami and S. singaporensis.
Seven isolates originated from faecal samples of the Straw-coloured fruit bat (Eidolon helvum), collected on a university campus in Ile-Ife, Nigeria, for earlier studies (Akobi et al., 2012; Olatimehin et al., 2018). The eighth isolate was recovered from a pulmonary specimen of a captive Diamond firetail (Stagonopleura guttata), an estrildid finch. The bird was kept in an aviary in a zoological collection in Berlin, Germany. The carcass of the deceased bird was submitted for necropsy. Lung tissue samples revealing disseminated white to yellowish miliary lesions were subsequently forwarded for microbiological investigations with suspected avian mycobacteriosis, and indeed pulmonary smears were positive for acid-fast bacilli.
An overview of isolates and typing data is provided in Table 1. The isolates were characterised by microarray (see below and Supplemental File 1). Three isolates, two from bats and the one from the finch were selected for phenotypic characterisation and whole-genome sequencing (WGS).
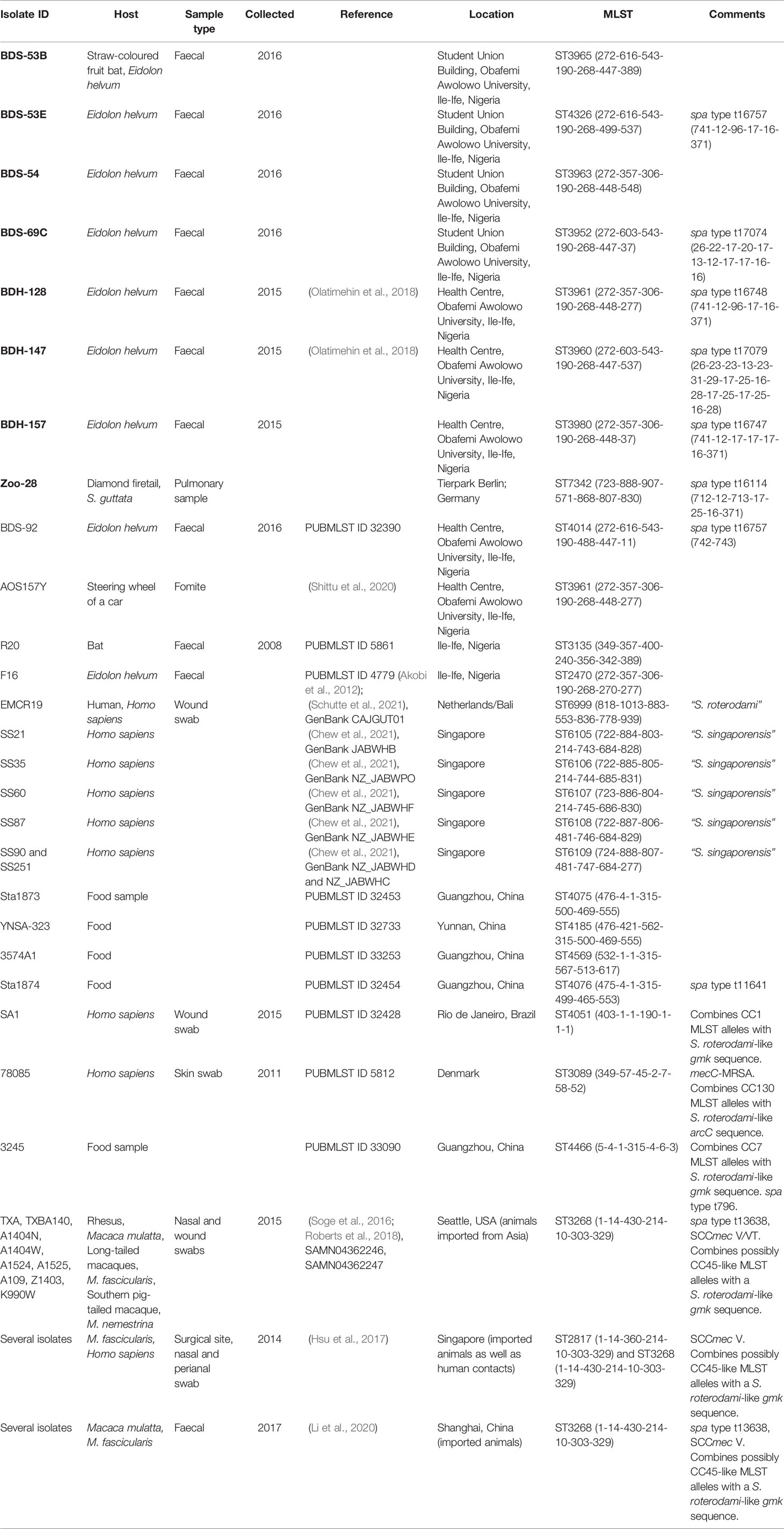
Table 1 Details of animals and isolates described herein (bold font), as well as of related isolates described otherwise or listed in the MLST database.
Antimicrobial susceptibility testing was performed by the Vitek 2 automated system (bioMérieux, Nuertingen, Germany; Supplemental File 2) using the AST-P608 panel (benzylpenicillin, oxacillin, cefoxitin, gentamicin, tobramycin, ciprofloxacin, levofloxacin, moxifloxacin, erythromycin, clindamycin incl. inducible resistance, linezolid, teicoplanin, vancomycin, tetracycline, fosfomycin, nitrofurantoin, fusidic acid, mupirocin, rifampicin) according to manufacturer´s instructions. EUCAST clinical breakpoints for S. aureus (https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_11.0_Breakpoint_Tables.pdf) were applied to classify the isolates as susceptible, intermediate or resistant. S. aureus ATCC® 29213 served as quality control strain. In addition, biochemical tests were performed using the bioMérieux Gram-positive identification card (GP) for the same device.
MLST is based on sequencing seven housekeeping genes, arcC, aroE, glpF, gmk, pta, tpi and yqiL. Sequencing of PCR products was performed as previously described (Enright et al., 2000), or the sequences of the target genes were extracted from assembled whole-genome sequence data. The sequences were assigned to MLST alleles and STs using the S. aureus section (https://pubmlst.org/bigsdb?db=pubmlst_saureus_seqdef&page=sequenceQuery) of the PubMLST website (Jolley et al., 2018).
In addition, spa typing was performed as previously described (Harmsen et al., 2003) using repeat definitions and nomenclature as provided on the Ridom website (http://spa.ridom.de/).
Isolates were characterised using the DNA microarray-based kit (Interarray S. aureus, fzmb GmbH, Research Centre for Medical Technology and Biotechnology, Bad Langensalza, Germany). Primer and probe sequences have been published previously (Monecke et al., 2008; Monecke et al., 2011). The array covers 333 different targets related to approximately 170 different genes and their allelic variants allowing detection of virulence and resistance factors. Isolates were assigned to clonal complexes (CCs) by automated comparison to a reference database. The procedures followed the manufacturer’s instructions as previously described (Monecke et al., 2008; Monecke et al., 2011). Briefly, S. aureus was cultured overnight on Colombia blood agar. DNA extraction was performed after enzymatic lysis. The next step was a multiplexed linear amplification using one specific primer per target. During that non-exponential amplification, biotin-16-dUTP was incorporated into single-stranded amplicons. After incubation and washing, hybridisation was performed to probes immobilised on the array. Hybridisations were detected by adding streptavidin horseradish peroxidase that triggered a localised dye precipitation resulting in a formation of visible spots. Microarrays were then scanned and analysed using an Arraymate (Alere Technologies GmbH (Abbott), Jena, Germany) reading device. A second microarray (Alere, Monecke et al., 2016) was used to detect additional markers (Supplemental File 1), including the staphyloxanthin locus.
Three isolates were subjected to WGS with the Illumina MiSeq platform (Illumina, Inc., San Diego, USA). The whole-cell DNA was extracted using the QIAamp® DNA Mini Kit (QIAGEN, Hilden, Germany) with some adaptations for staphylococci as described previously (Scholtzek et al., 2019). The libraries for WGS were prepared using the Nextera XT DNA Library Preparation Kit (Illumina, Inc., San Diego, USA) according to the manufacturer’s recommendations. The 2×300 bp paired-end sequencing in 40-fold multiplexes was performed on the Illumina MiSeq platform (Illumina, Inc., San Diego, USA).
Oxford Nanopore Technology (ONT) sequencing of the study isolates, i.e., BDS-53E, BDS-54 and Zoo-28, was performed using two different MinION flow cells (IDs: FL1339 and FAO01531; rev: FLO-MIN106D containing an R9.4.1 pore). Library preparations were done using the 1D genomic DNA by ligation kit (SQK-LSK109, ONT), and the native barcoding expansion kit (EXP-NBD104, ONT) following manufacturer’s instructions with minor adaptations. In summary, an AMPure bead (Agencourt AMPure XP, Beckman Coulter, Krefeld, Germany) clean-up step was performed before the library preparation. Potential nicks in DNA and DNA ends were repaired in a combined step using NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End repair/dA-tailing Module (NewEngland Biolabs, Ipswich, USA) by tripling the incubation time. A subsequent second AMPure bead purification was followed by the ligation of sequencing adapters onto prepared ends and a third clean-up step with AMPure beads. An additional barcoding and clean-up step was performed prior to adapter ligation. Sequencing buffer and loading beads were added to the library. At the start of sequencing, an initial quality check of the flow cells showed 1289 (FL1339) and 1388 (FAO01531) active pores. Genomic DNA samples used for loading comprised a total amount of around 25.5 ng per strain (measured by Qubit 4 Fluorometer; ThermoFisher Scientific, Waltham, USA). The sequencing ran for 48 hrs using the MinKNOW software version 20.06.5.
For all nanopore data sets, the guppy basecaller (v4.2.2, Oxford Nanopore Technologies, Oxford, UK) translated and trimmed the MinION raw data (fast5) into quality tagged sequence reads (4,000 reads per fastq-file). Filtlong (v0.2.0) was used for bacterial DNA with a median read quality of 14 and a minimum read length of 1,000 bp to get a smaller and better subset of reads. The median read quality of 15.5 and a N50 read length of approximately 13,000 bp for each sample was highly suitable for assembly. Flye (v2.8.3) was used to assemble the reads to provide high quality contigs. Then, a racon-medaka (4-times racon v1.4.3; 1-time medaka v1.2.0) pipeline was applied for polishing. Moreover, pilon (v1.23) polished the sequences using Illumina sequence data (Supplemental Files 3/4/5a). The NCBI Prokaryotic Genome Annotation Pipeline (PGAP version 2021-01-11.build 5132) was used for annotating all assembled contigs in combination with an in-house database of published staphylococcal gene sequences (Supplemental Files 3/4/5b).
We selected a panel of 154 core genome markers for tree construction using SplitsTree (Monecke et al., 2021). These genes and genome sequences used for visualisation (Figure 1) are listed in Supplemental File 6. Inclusion criteria were the presence of the genes in all CCs of S. aureus/argenteus/schweitzeri clonal complexes and uniform length in all published genomes. Sequences were concatenated and analysed using SplitsTree 4.0 (Huson and Bryant, 2006) using default settings (characters transformation, uncorrected P; distance transformation, Neighbour-Net; and variance, ordinary least squares).
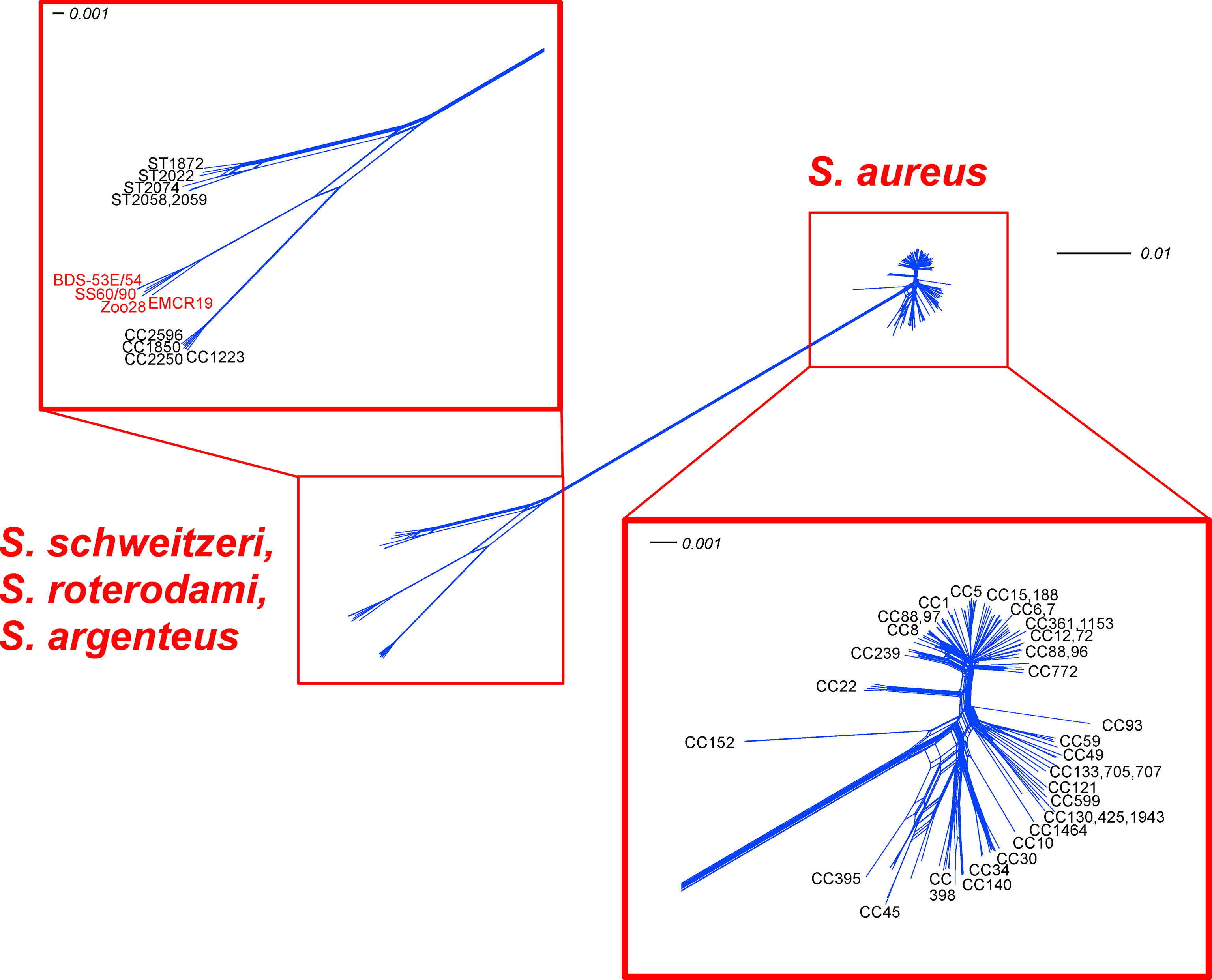
Figure 1 SplitsTree analysis for 154 core genomic markers of the study isolates, “S. roterodami” EMCR19 as well as “S. singaporensis” SS60 and SS90 compared to diverse S. aureus, S. argenteus and S. schweitzeri CCs. Note, for the genes that were found inverted in the Zoo-28 genome, reverse complement sequences were used for analysis.
A comprehensive analysis of existing sequence databases and available literature led to the definition of a set of 2,167 genes (Supplemental File 7A) in the core genome of S. aureus, S. argenteus and S. schweitzeri. These genes are almost universally present although in few cases sequences might be absent due to random mutation or sequencing/assembly artefacts. These genes always appeared in the same order within the genome, regardless of the identity of the isolate. In addition, 125 markers from major genomic islands were considered always present or usually present (with presence or absence being linked to species or CC affiliation). They also appear in the same positions within the genome. Genes in this category are the staphyloxanthin gene cluster, the set/ssl loci and the egc enterotoxin gene cluster. Genomic islands that might occupy variable locations within the genome were not considered. The sequences of these core genome and major genomic island genes were analysed and compared to each other and to reference sequences. A few genomic island genes not present in any of the isolates compared in the present study were excluded. Thus, a total of 2,292 genes were considered representing roughly 2,040,000 nucleotide positions per genome. For each gene, the number of nucleotides different from the comparator isolate was counted and expressed as a percentage of the length of the respective gene (Supplemental File 7B). Gaps in the alignment of different alleles of a given target gene were treated as mismatches. If a gene was present in one isolate but absent in the other isolate, this percentage was set as 100%. In addition, percentages were plotted over the positions in the genome (Figure 2). When comparing two isolates, median values for these percentages of all genes were calculated.

Figure 2 (A) SNP analysis comparing 2292 non-motile genes of BDS-53E with the Zoo-28 (green), “S. roterodami” EMCR19 (blue) as well as with “S. singaporensis” SS60 and SS90 (red and yellow). For each single gene, the number of differences (Supplemental File 7B) to the comparator strains was counted and expressed in percentage. For genes that were present in one, but absent in the other isolate, this percentage was set as 100%. Note, for the genes that were found inverted in the Zoo-28 genome, reverse complement sequences were used for analysis. (B) SNP analysis comparing 2292 non-motile genes of BDS-53E with S. schweitzeri (NCTC13712, LR134304.1, brown), S. argenteus (MSHR1132, FR821777.2, grey), S. aureus CC1 (MW2, BA000033.2, yellow) and S. aureus CC152 (SA17_S6, CP010941.1, red). The genes around pos. 1550 represent a genomic island that is absent in the study isolates (protease genes splF, splE, splD2, splC, splB, splA, leukocidin lukD/E, lantibiotic epidermin gene cluster). The highly conserved genes around pos. 1850 are those encoding ribosomal proteins.
We utilized GTDB-tk, a software toolkit for assigning objective taxonomic classifications to bacterial and archaeal genomes to determine if the genomes of the study isolates were novel species (Parks et al., 2018; Parks et al., 2020; Parks et al., 2022). To taxonomically assess the genomes, GTDB-Tk version 1.6.0 (Chaumeil et al., 2019) and the GTDB database version 202 (ref: https://gtdb.ecogenomic.org/stats/r202) containing 254,090 bacteria and 4,316 archaeal genomes) were utilised. Briefly, the target genomes were classified by the GTDB-Tk’s “classify” workflow (using the default settings) by placing them into the GTDB’s reference tree. We considered the average nucleotide identity (ANI), alignment fraction (AF) and its relative evolutionary divergence (RED) (Scholtzek et al., 2019) to the closely related reference genomes.
Based on the bioMérieux´ Gram-positive identification card (GP) for VITEK-2, the biochemical test results are summarised in Table 2 (see also Supplemental File 2). Profiles were in accordance with S. aureus.
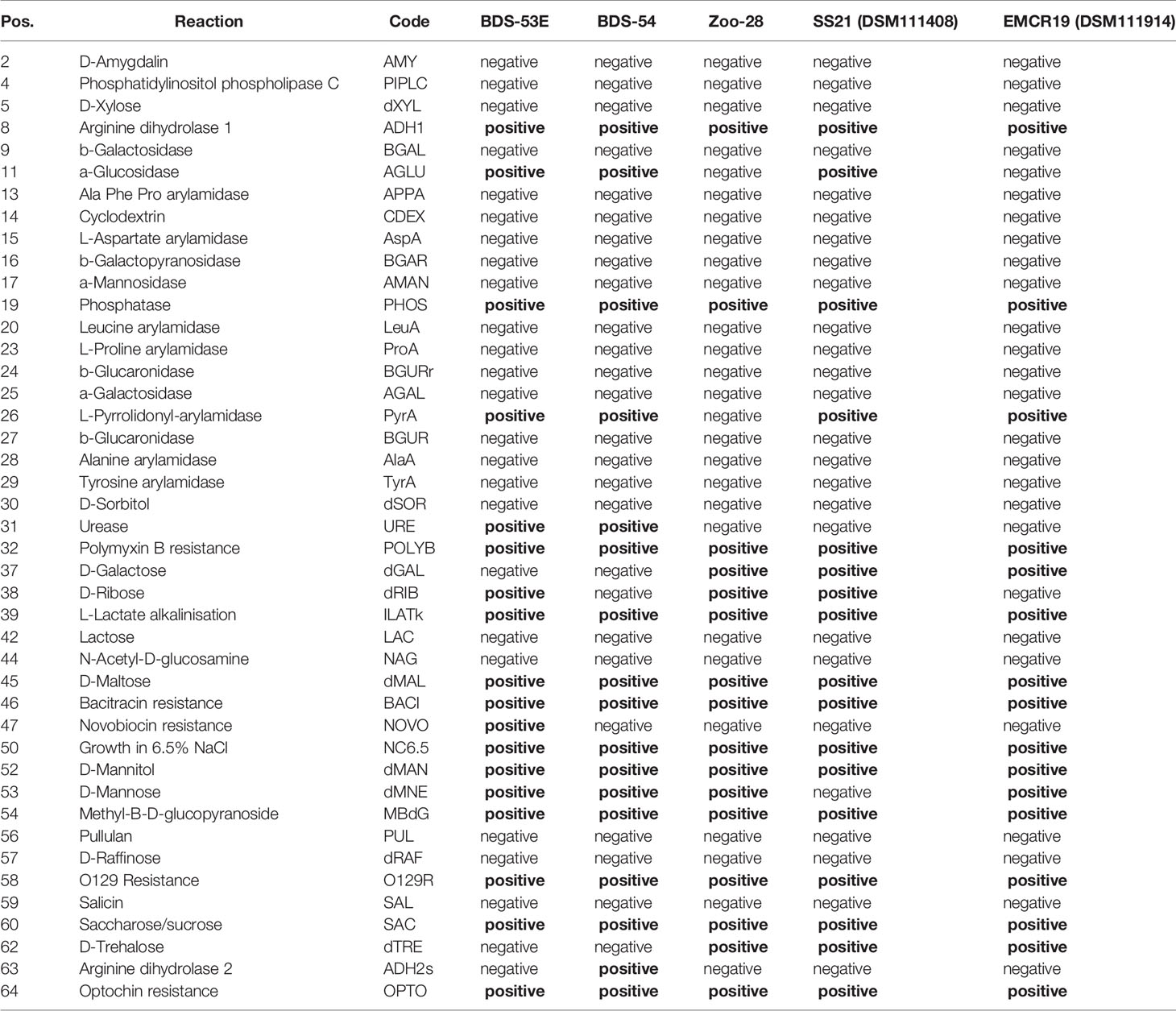
Table 2 Biochemical profiles, based on results for bioMérieux´ Gram-positive identification card (GP) for VITEK-2.
Based on their irregular microarray hybridisation patterns (see Supplemental File 1), an assignment of the isolates to any known lineage of S. aureus was not possible, suggesting affiliation to either S. argenteus or S. schweitzeri. However, isolates yielded positive signals for one crtM probe and weak signals for crtP. Since these probes recognise genes from the staphyloxanthin cluster, which by definition should be absent from S. argenteus, the isolates could not be assigned to this species. On the other hand, all three isolates carried ycjY, a marker on a genomic island identified in S. argenteus and some S. aureus lineages (CC12, CC361 and CC398), but absent from any S. schweitzeri tested or sequenced. The orfX-associated (Holt et al., 2011) cas1 CRISPR-endonuclease 1 (FR821777.2; pos. 62,418...63,323, which can be observed in S. argenteus CC1850 and CC2250) was not detected. The isolates clearly clustered into two distinct putative CCs.
The Nigerian bat isolates were highly similar to each other. They yielded signals with hld, agrC/D-I and with S. argenteus-specific agr probes, indicating a presence of an agr gene cluster albeit an atypical allele. Similarly, icaA was the only ica gene detected by the array (while the others were detected by sequencing; see Supplemental Files 3, 4). Capsule genes were not detected by the array although sequencing showed a presence of specific alleles. The cna gene was absent, while sasG was present. The ssl genes (encoding staphylococcal superantigen-like protein locus 1) were not detected by the array although one gene of this cluster was found by sequencing.
The bat isolates could further be subdivided based on reactivities with either sdrC or sdrD probes. For sequencing, one sdrC- and one sdrD-positive isolate (BDS-53E and BDS-54, respectively) were selected.
The German isolate Zoo-28 shared agr and ica reactivities. It differed in the absence of sasG and in the allelic variants of several adhesion factors. In contrast to the bat isolates, several ssl genes were present (see Supplemental Files 1, 5). It also harboured the leukocidin genes lukD/E, which were absent from the bat isolates (although the latter component was identified only by sequencing).
MLST yielded profiles that are shown in the first half of Table 1. Previously published MLST profiles that appeared related are listed in the second half of the table.
An MLST-like approach based on 154 core genomic markers (Figure 1; Supplemental File 6) led to the clustering of all S. aureus, with three major groups and three separate branches. One group comprised CC1, CC5, CC8 and most of the other S. aureus lineages. A second one included CC59, CC121 and several, mostly animal-associated, lineages such as CC49, CC130 and CC1464 (“S. aureus subsp. anaerobius”). The third group consisted mainly of CC30, CC45, CC398. The separate branches, CC22, and more conspicuously, CC93 and CC152, appeared to be more distant from other S. aureus lineages.
Another very distant branch consisted of S. argenteus lineages (CC1223, CC1850, CC2198, CC2250, CC2596 and CC4587). S. schweitzeri (ST1872, ST2022, ST2058, ST2059, ST(206-303-253-142-196-202-197); accession number CCEO01000001-CCEO01000054) was also clearly separate, but much closer to S. argenteus than S. aureus. The genomes of BDS-53E, BDS-54 and Zoo-28 were closely related to “S. roterodami/singaporensis” (EMCR19, SS60, SS90). The SplitsTree in Figure 1 shows that the aforementioned “S. roterodami/singaporensis” isolates as well as the three sequenced study isolates were located between S. argenteus and S. schweitzeri.
For the Nigerian bat isolates, the order of the predefined core genomic markers and major genomic island markers within the genomes was identical as observed with the published sequences of S. aureus, S. argenteus and S. schweitzeri. In the isolate Zoo-28, a large part of the genome was found inverted (already in the Nanopore sequence before polishing with Illumina) and integrated further downstream, into the map/eap gene, with map/eap fragments identified at pos. 1,991,191...1,992,912 and 2,090,178...2,091,407 of the genome. This inverted part of the chromosome contained approximately 69,000 bp or 60 genes from namA (NADH:flavin oxidoreductase) to yrbD (alanine:cation symporter family protein), including the chromosomal oligopeptide ABC transporter cluster oppA/oppF/oppD/oppC/oppB. It was flanked at both sides by transposase genes and genes of yet unknown function resulting in a total size of the insert of about 97 kb. Another transposase gene was identified at the original position where the inserted genes were supposed to be localised.
The analysis and comparison of the core genomic and major genomic island genes of the three isolates and reference sequences indicated that the differences of BDS-53E compared to the reference sequences are somewhat uniformly scattered all across the genome. Moreover, BDS-53E and Zoo-28 are similar to S. roterodami and S. singaporensis (Figure 2). Using the median values for the differences of all genes to compare sequences of the study isolates, we observed that these sequences and those of S. roterodami and S. singaporensis differed by approximately 1% (Figures 2A, 3). Furthermore, we observed approximately 5% differences compared to both, S. argenteus and S. schweitzeri, and of about 11-12% compared to different S. aureus CCs (Figures 2B, 3).
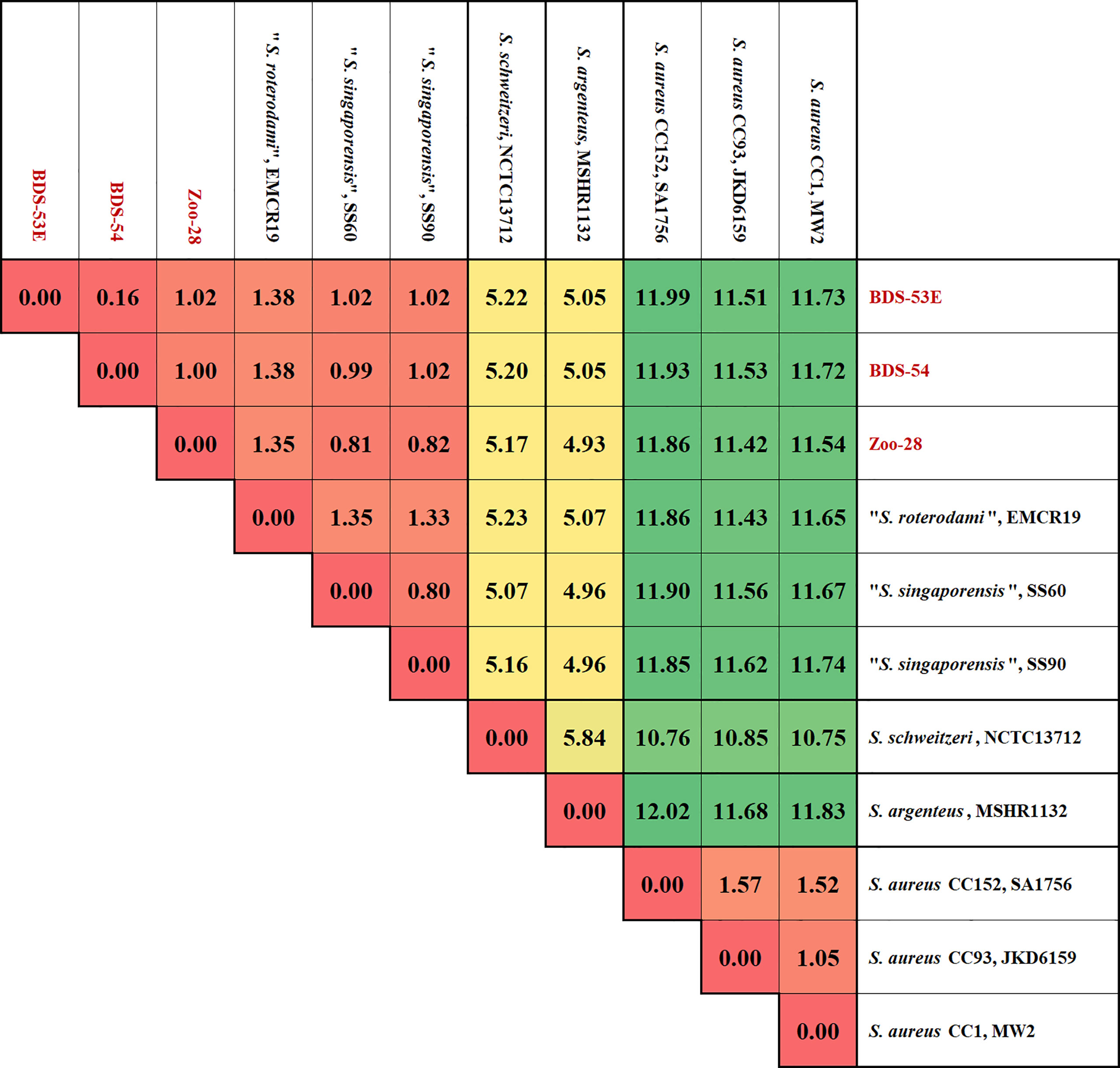
Figure 3 Matrix of differences between BDS-53E, Zoo-28, “S. roterodami” (EMCR19, CAJGUT01), “S. singaporensis” (SS60 and SS90, NZ_JABWHF and NZ_JABWHD), S. schweitzeri, (NCTC13712, LR134304.1), S. argenteus (MSHR1132, FR821777.2) and three S. aureus lineages, CC1 (MW2, BA000033.2), CC93 (JKD6159, CP002114.2) and CC152 (SA17_S6, CP010941.1). The percentages were calculated as explained for Figure 2 and the image shows the median values over all these 2292 genes for each genome sequence compered to all others.
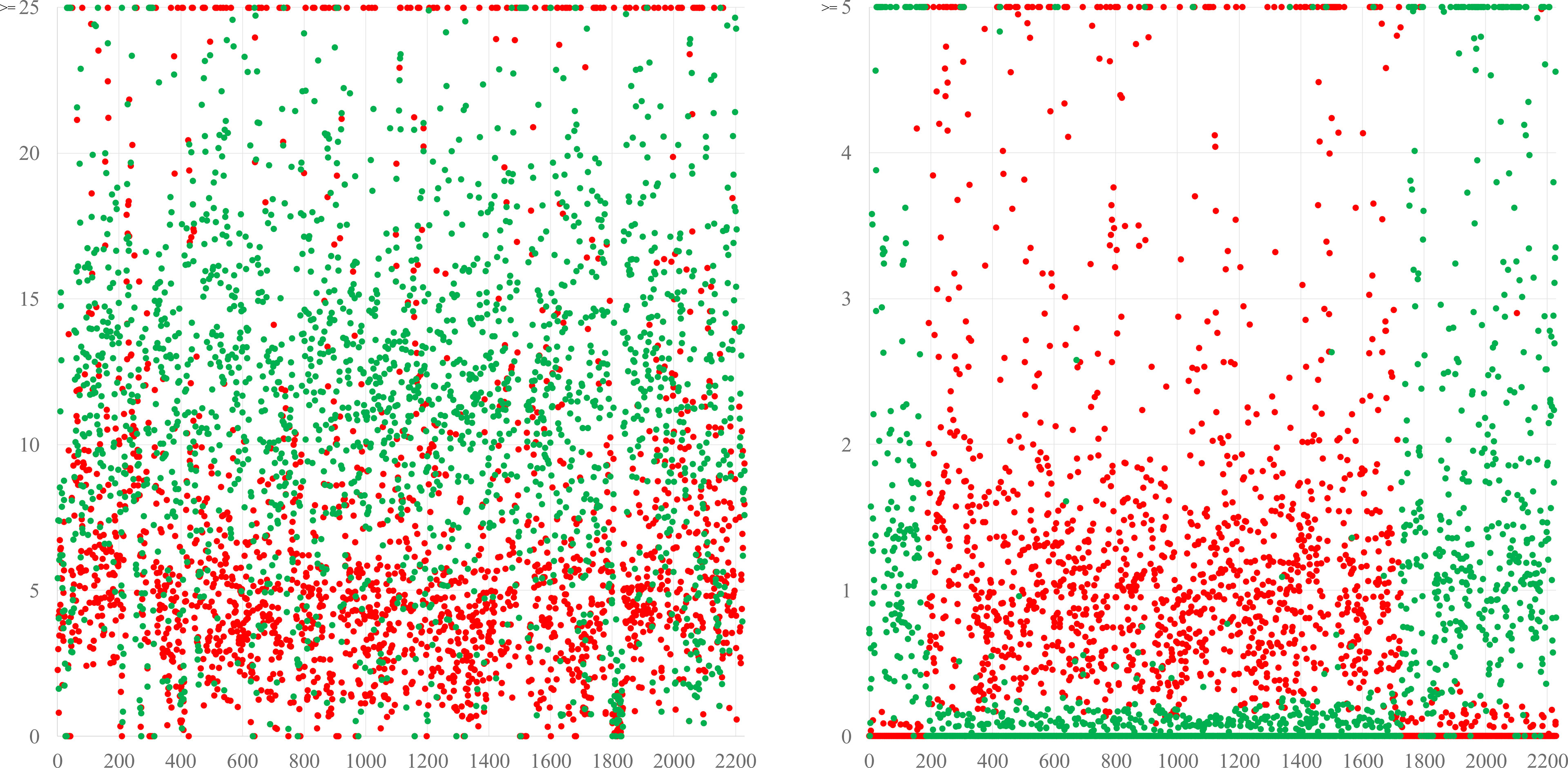
Figure 4 The left diagram shows a simplified version of the blot from Figure 2, comparing BDS-53E to the reference sequences of S. schweitzeri NCTC13712 (red) and the S. aureus CC1 strain MW2 (green). Differences are evenly distributed all across the genome, suggesting a continuous evolution and an accumulation of more mutations compared to S. aureus than to S. schweitzeri corresponding supposedly to a longer time of separate evolution. The right diagram shows the same analysis for a chimeric strain in which a fragment of “red” origin spanning from approximately position 1750 over oriC (pos. 0) to pos. 200 is integrated into a backbone genome of “green” provenance. These are actually S. aureus CC140 and CC8, in a ST6610 strain, see (Burgold-Voigt et al., 2021). For the Y-axes, different scales were used because differences between the different species are larger than those between different CCs of S. aureus.
The genome sequences of the study isolates were taxonomically placed in the genus of Staphylococcus without a species assignment as their closest relative, S. argenteus (GCF_000236925.1), shared only an average nucleotide identity of 93.86% to 94.01% (Table 3). The second most closely related species was S. schweitzeri (average nucleotide identity, ANI, 93.53 to 93.63), followed by S. aureus (ANI, 88.98 to 89.22). S. singaporensis and S. roterodami are not (yet) covered by the database.
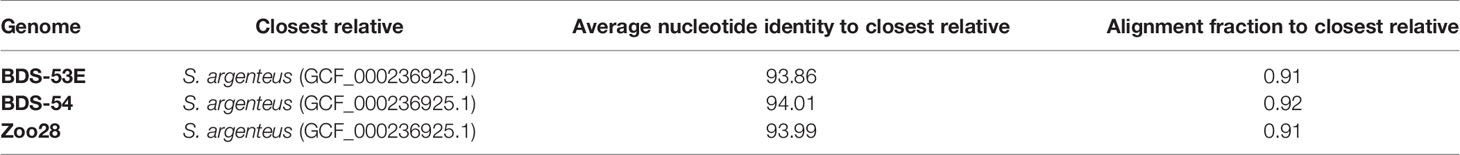
Table 3 GTDB-tk results.
All study isolates were methicillin susceptible and negative by both, array and sequencing, for mecA/mecC genes.
Antimicrobial susceptibility testing using the AST-P608 panel showed that BDS-53E was fully susceptible to all compounds tested. BDS-54 was susceptible to all compounds but intermediate to fluoroquinolones. However, in grlA, some single nucleotide polymorphisms (SNPs) were either observed in BDS-53E and BDS-54 (E310K, K650N, H767Q) or in the three study isolates and in S. argenteus and S. schweitzeri. Similarly, most SNPs in the gyrA gene separated the three study isolates plus S. argenteus and S. schweitzeri from S. aureus (e.g., E248V, V623I). Previously described mutations (Wang et al., 1998) were not identified.
A distinct result for BDS-53E (from the Gram-positive ID panel) was its novobiocin resistance. As mutations in gyrB (CP000253.1; 5,034...6,968) and parE=grlB (CP000253.1; 1,292,206...1,294,197) might be related to novobiocin resistance (Fujimoto-Nakamura et al., 2005), the sequences of these genes were analysed. The BDS-53E sequence of gyrB showed a unique mutation resulting in a substitution of aspartic acid by valine at amino acid position 494, which was not present neither in the other two study isolates nor in some reference sequences (MSHR1132, MW2, SA17_S6, JKD6159 and NCTC13712). The BDS-53E sequence of parE=grlB did not contain unique SNPs although in some positions, SNPs were observed that either all three study isolates shared (I490L), or had in common with S. argenteus and S. schweitzeri.
BDS-53E and BDS-54 carried aadK, a gene encoding an aminoglycoside 6-adenyltransferase gene not covered by the microarrays used. It was nearly identical (in 826 of 846 nt) to CP010526.1, 421,013...421,858. In both isolates, it was localised on putative pathogenicity islands integrated between the gene guaA (glutamine-hydrolysing GMP synthase) and Q8NY63 (putative protein), around position 400,000 in the genome. These pathogenicity islands additionally included several genes for “hypothetical proteins”, a DNA primase, an integrase and a small terminase subunit.
The isolate Zoo-28 was resistant to benzylpenicillin and tetracycline. A blaZ/I/R operon was identified together with yolD and tnpA/B/C genes as part of a transposon that showed 99.58% identity to Tn553, a non-conjugative transposon of the Tn554 family. This transposon has recently been described in porcine S. aureus ST9 from China (Krüger et al., 2021). Like the original Tn553, the Tn553 in Zoo-28 was also integrated into the chromosomal Q5HEJ7/yolD-like gene. Tetracycline resistance was attributable to the tetracycline resistance gene tet(M). It was accompanied by a Tn916-transposase, a Tn916-excisionase, a gene encoding D2N5T7 (a conjugative transposon protein), yddH (encoding a putative peptidase cell wall hydrolase), ardA2 (coding for an antirestriction protein) and a couple of hypothetical proteins. The entire transposon was identical to Tn916 (GenBank, U09422.1), a transposon first described in enterococci but also known from staphylococci such as CC398-MRSA-VT (S0385, GenBank AM990992.1 and 08BA02176, GenBank CP003808.1).
All three sequenced study isolates did not harbour SCCmec elements. BDS-53E and BDS-54 carried clusters of twelve (BDS-54; see Supplemental File 4B) or twenty genes (BDS-53E; see Supplemental File 3B) associated with, and located directly downstream of orfX. These were automatically annotated as “hypothetical proteins”, transposases, helicases, methyltransferases, oxidoreductases and hydrolases. In addition, two of the un-sequenced bat isolates (BDS-69C, BDH-147) were positive in array experiments for B2Y834 (a marker usually associated with SCCmec IV A, IV E and IV c) as well as another one (BDH-128) with ccrA-1.
Zoo-28 lacked these genes, carrying transposase and helicase genes instead, as well as one gene, B6VQU0, which is otherwise known to occur in CC705 (GenBank AJ938182, pos. 34,735...35,634, downstream of orfX), as well as in SCCmec IV h/j elements.
Downstream of the SCC integration site, a large genomic island is situated whose gene content is related to CC affiliation. Typical genes of that genomic island, such as seh and ORF CM14 (characteristic for, e.g., CC1, CC10, CC34, or CC93 or CC772), were absent from the study isolates. BDS-53E and BDS54 harboured in this position mcrB (type IV 5-methylcytosine-specific restriction enzyme subunit B), mcrC (subunit C) and Q6GD44 (putative acetyltransferase, GNAT family) genes. Zoo-28 differed, carrying C1PH96 (putative protein, carboxymuconolactone decarboxylase family), lrpC (HTH-type transcriptional regulator Lrp/AsnC family) and Q6GD44 genes.
BDS-53E and BDS-54 carried prophages that could be assigned to Siphoviridae based on sequence similarity to known S. aureus phages. In both cases, phages were integrated between the genes encoding a putative protein A5ITW8 and tRNA for serine, approximately at position 1,800,000 in the genomes. Phages were similar but not identical (see Supplemental Files 3B, 4B). Both isolates harboured putative pathogenicity islands as described above.
In Zoo-28, no prophage was identified, but it carried as much as three different pathogenicity islands with genes for “hypothetical proteins”, integrases as well as for small terminase subunits. Finally, Zoo-28 carried Tn916 and Tn553-like transposon as discussed above.
There was no evidence for plasmids in the three study isolates.
Evolution is a continuous process that does not occur in discrete steps. Thus, the classification of “evolving live forms” into discrete or distinct species is always problematic regardless of whether they are bacteria or higher organisms, such as herring gulls (Liebers et al., 2004), or cervids (Ludt et al., 2004). This distinction cannot be made without a certain element of arbitrariness. This is caused by an emphasis on specific features of the target organism that are considered sufficient to define a species and by the fact that different observers might prioritise different properties. For instance, traditionally, Shigella is a distinct taxonomic entity from Escherichia (E.) coli because of the “severity of dysentery” and its lethality in humans. However, an investigation of Shigella gene sequences shows only minimal differences compared to E. coli which do not justify its recognition as distinct genus or species (van den Beld and Reubsaet, 2012). For other life forms, taxonomists argue about the fertility of hybrids, or, in the case of the herring gulls mentioned above, mating calls and feet colour (Collinson et al., 2008). A numerical approach to gene analysis might reduce arbitrariness although different “threshold values” must carefully be considered for various clades of life forms.
The numerical approach for assessing nucleotide differences per gene length yielded some interesting results in the present study. First, within S. aureus, the median difference for all 2,292 genes considered was only 1 to 1.5%, even when comparing a CC1 reference sequence to such diverse, or deviant, lineages as CC93 or CC152. This observation gives an impression or benchmark for the largest possible difference within one established species. Median differences between S. aureus and S. argenteus or S. schweitzeri were 10-12%, while it was about 5% between S. argenteus and S. schweitzeri. These data might provide an orientation on the level of distinction of valid species to one another. The study isolates differed from each other by 0.16% to 1.38% but differed from S. argenteus and S. schweitzeri by about 5% and from diverse S. aureus lineages (CC1, CC93, and CC152) by 11-12%. These observations suggest that the study isolates belonged to one distinct species, with the African isolates in one CC and the German zoo isolate in another one. However, a comparison of the genomes of the study isolates to the recently published genomes of the isolates assigned to the new species S. roterodami and S. singaporensis (Schutte et al., 2021; Chew et al., 2021) yielded median differences of around 1%, and the difference between these two species was about the same.
Our observations provide two options. First, one might conclude that a difference of more than 10% was required for recognition as a full species. Hence, S. aureus would be one species, while S. schweitzeri, S. argenteus, S. roterodami, S. singaporensis and the study isolates clustered to another one. Second, one might define a median difference of about 5% as a threshold for a species. This is implemented in GTDB-tk where a query genome is regarded as the same species as the closest reference sequence if it falls within an ANI of at least 95% and an AF of 65%. In this case, S. aureus, S. schweitzeri and S. argenteus would be three species, while a forth one comprised both S. roterodami and S. singaporensis, as well as the study isolates. However, phenotypical tests allow no clear distinction of these isolates from the other members of this “species” raising the question of whether sequence analysis should have priority over biochemical tests or not. Hence, a discussion about the definition of clear criteria for recognition as a discrete species is necessary especially as new technologies and lower costs facilitate sequencing of isolates that could not be sequenced before, including those from faeces of exotic animals, resulting in an unprecedented increase in the number of available genome sequences.
We present evidence for recognising a new species of coagulase- and staphyloxanthin-positive staphylococci positioned between S. argenteus and S. schweitzeri. The previously described species “S. roterodami” and “S. singaporensis” as well as isolates described herein all together should be regarded as a single species, i.e., as the fourth one in the S. aureus complex in addition to S. aureus, S. argenteus and S. schweitzeri. According to the principle of priority, that species should be named S. roterodami as this name was published first, in September 2021 (Schutte et al., 2021).
This species has a much wider geographical range than previously thought, i.e, Nigeria, Southern China, Indonesia, Singapore and possibly Australia.
The majority of isolates described herein and two previously published MLST profiles originate from bat faeces from Nigeria. These two MLST profiles were posted to the MLST database by a Japanese and a British group (ST2470 and ST3135). Sequences are not identical albeit similar, and the sampling location was approximately the same as for the bat isolates described herein. One isolate (Zoo-28) was sampled from an estrildid finch living in a zoo in Germany. While this species is native and endemic to Australia, this individual zoo animal might have been colonised/infected by contact with other animals such as flying foxes kept in close proximity. Unfortunately, we cannot investigate that issue anymore due to alterations to the building and the aviary kept inside. “S. roterodami” and “S. singaporensis” were isolated from humans returning from Bali or living in Singapore, respectively, as discussed above. Further related STs, ST4075, ST4076, ST4569, were observed in isolates from unspecified food from the Guangzhou region, in the southern part of China. Another food isolate, ST4185 originated from Yunnan, a province in Southwestern China. These four STs share a unique gmk allele (gmk-315) suggesting affiliation to yet another CC. They also present ordinary S. aureus-like aroE-alleles and unique, deviant arcC-alleles.
S. roterodami is a polymorphic species, consisting of at least nine distinct CCs with the animal isolates described herein constituting two CCs. The Bali isolate originally described as S. roterodami represents another one. The six “S. singaporensis” isolates could be classified into five different CCs. Differences between the CCs of S. roterodami include carriage, or absence, of gene clusters that also define complexes within S. aureus or S. argenteus. These include agr locus genes, the set/ssl cluster, the enterotoxin gene cluster egc, edinB+etD and sasG. The four sequence types from Southern China might represent at least one additional CC.
In addition to the CCs discussed above, there are several STs in the MLST database that appear to be S. aureus although they contain one S. roterodami MLST allele each in addition to six regular S. aureus MLST markers. Whether this was evidence for cross-species hybridisation or chimerism involving S. aureus and S. roterodami, an accidentally identical accumulation of mutations, or merely technical issues, still needs to be clarified. These STs include ST4051 with gmk-190 while the other markers are in accordance with a CC1 profile, ST3089 that differs from CC130 in arcC-0349 and ST4466 that carries gmk-0315 although it otherwise resembles CC7. Finally, a MRSA lineage associated with imported macaques (Macaca sp.), ST3268/ST2817, was identified in the USA, China, and Singapore (Soge et al., 2016; Hsu et al., 2017; Roberts et al., 2018; Li et al., 2020) in which a S. roterodami-like gmk allele (gmk-214) is present, among other MLST alleles that could be derived from S. aureus CC45.
With regard to chimerism, one might argue that the S. roterodami complex was a group of chimeric S. argenteus isolates that acquired the gene cluster encoding the “golden” carotenoid pigment staphyloxanthin by chromosomal replacement, hybridisation, or chimerism. Figure 1 contains information rendering that concept rather unlikely. The differences compared to the reference isolates affect all parts of the genome essentially and are distributed evenly across the genome (with the notable exception of a few highly conserved genes encoding ribosomal proteins). Previous work (Nimmo et al., 2015; Burgold-Voigt et al., 2021) showed how a part of the genome of a chimeric isolate genome would match the corresponding part from one parent strain, and differ from the same region of the other one, while this would be conversely for the rest of the genome (Figure 4). Therefore, we can assume that the similarities and differences of the S. roterodami complex compared to S. aureus, S. argenteus and S. schweitzeri do not result from a large-scale chromosomal replacement or chimerism. Thus, the isolates cannot be considered S. argenteus that by chance acquired the staphyloxanthin locus from elsewhere. The more likely explanation was a continuous evolution and accumulation of mutations over time. Based on the median differences calculated for core genomic markers (Figure 3), we postulate that the split from S. aureus occurred earlier than the one from S. argenteus and S. schweitzeri.
Identifying S. aureus, S. argenteus, S. schweitzeri and S. roterodami from humans and/or wild animals in Africa suggest that they could have originated from the continent, disseminating to other parts of the world through human migration. Thus, it would be interesting to screen African wildlife for other possible branches of the staphylococcal phylogenetic tree to understand the co-evolution of humans and animals with their coagulase-positive colonisers/pathogens.
S. roterodami has been found in symptomatically ill humans as well as in wild bats and a captive finch, suggesting a relatively broad zoonotic host spectrum as well as a certain virulence in humans. Besides, it can acquire resistance genes known from other staphylococci such as blaZ, tet(M), aadK, aacA-aphD and aadD as demonstrated in the study isolates and in those identified in Singapore (Chew et al., 2021). Thus, its clinical relevance could be comparable to that of S. aureus or S. argenteus.
Further studies should focus on staphylococcal isolates from humans, bats, rodents, birds, and atypical S. aureus isolates from Western and Central Africa, Southern and Southeastern Asia as well as Australia. While phenotypic tests might not be conclusive, unique MLST alleles (see Table 1) should help identifying S. roterodami isolates. However, a non-molecular algorithm for diagnostic procedures to identify the new species from routine diagnostic samples is needed.
The genome sequences of the study isolates were submitted to GenBank. The BioProject accession number is PRJNA810320, BioSamples are SAMN26244312 to 314 and the GenBank accession numbers are CP092781, CP092782 and CP092783. All other data are provided within the manuscript, or as Supplemental Files.
No animal experiments were performed and no animal was sacrificed for this study. No ethical clearance was necessary as no animals were captured and no invasive samples were taken. The strains originated from environmental samples (i.e., bat faeces collected under trees used by wild bats for roosting) and from a post mortem sample from a zoo animal submitted for routine diagnostic procedures.
SM designed the study. SM, FS, AS, and ATF wrote the manuscript. FS, AS, ATF, and KM obtained samples and performed experiments (bacteriological work). HH, CD, MR, and SB performed experiments (sequencing). SM, CB, MC, and DH analysed sequence data. EM and DG performed experiments (bacteriological work and arrays). ATF, SS, and RE supervised the work and revised the manuscript. All authors contributed to the article and approved the submitted version.
The Jena group acknowledges support by the German Federal Ministry of Education and Research, within the framework of the ADA project (13GW0456C) aiming to develop rapid tests for the detection and characterization of resistance genes and virulence factors in zoonotic S. aureus/MRSA. The FU Berlin group acknowledges funding by the German Federal Ministry of Education and Research under project number 01KI2009D a as part of the Research Network Zoonotic Infectious Diseases. AS was an awardee of the Georg Forster Research Fellowship (for Experienced Researchers) of the Alexander von Humboldt Foundation.
DG is employed by a company, Illumina, but he performed experiments for this study before commencing this employment.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Antina Lübke-Becker, Inga Eichhorn (Institute of Microbiology and Epizootics, Freie Universität Berlin), Nadine Jahn, Mirjam Grobbel (Leibniz Institute for Zoo and Wildlife Research, Berlin), Anja Hackbart, Byrgit Hofmann (FLI Jena) and the staff of the Tierpark Berlin for support and assistance as well as Gudrun Wibbelt (Leibniz Institute for Zoo and Wildlife Research, Berlin) for necropsy data. We also acknowledge the German Collection of Microorganisms and Cell Cultures, DSMZ, for providing strains S. singaporensis SS21 (=DSM111408) and S. roterodami EMCR19 (=DSM111914).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.878137/full#supplementary-material
Supplemental File 1 | Hybridisation profiles of study isolates and reference strains (pdf).
Supplemental File 2 | Vitek Data (pdf).
Supplemental File 3 | (A) Genome sequence of BDS-53E (fasta). (B) Genes identified within the genome sequence of BDS-53E (fasta).
Supplemental File 4 | (A) Genome sequence of BDS-54 (fasta). (B) Genes identified within the genome sequence of BDS-54 (fasta).
Supplemental File 5 | (A) Genome sequence of Zoo-28 (fasta). (B) Genes identified within the genome sequence of Zoo-28 (fasta).
Supplemental File 6 | Strains and concatenated Sequences used for the Splitstree analysis.
Supplemental File 7 | (A) Sequences for analyses of strain similarities as shown in and (fasta). (B) Percentages difference as used for Figures 2A, B.
Akobi, B., Aboderin, O., Sasaki, T., Shittu, A. (2012). Characterization of Staphylococcus aureus Isolates From Faecal Samples of the Straw-Coloured Fruit Bat (Eidolon helvum) in Obafemi Awolowo University (OAU), Nigeria. BMC Microbiol. 12, 279. doi: 10.1186/1471-2180-12-279
Aung, M. S., San, T., Aye, M. M., Mya, S., Maw, W. W., Zan, K. N., et al. (2017). Prevalence and Genetic Characteristics of Staphylococcus aureus and Staphylococcus argenteus Isolates Harboring Panton-Valentine Leukocidin, Enterotoxins, and TSST-1 Genes From Food Handlers in Myanmar. Toxins (Basel) 9 (8). doi: 10.3390/toxins9080241
Aung, M. S., San, T., San, N., Oo, W. M., Ko, P. M., Thet, K. T., et al. (2019). Molecular Characterization of Staphylococcus argenteus in Myanmar: Identification of Novel Genotypes/Clusters in Staphylocoagulase, Protein A, Alpha-Haemolysin and Other Virulence Factors. J. Med. Microbiol. 68 (1), 95–104. doi: 10.1099/jmm.0.000869
Aung, M. S., Urushibara, N., Kawaguchiya, M., Sumi, A., Takahashi, S., Ike, M., et al. (2019). Molecular Epidemiological Characterization of Staphylococcus argenteus Clinical Isolates in Japan: Identification of Three Clones (ST1223, ST2198, and ST2550) and a Novel Staphylocoagulase Genotype XV. Microorganisms 7 (10). doi: 10.3390/microorganisms7100389
Becker, K., Schaumburg, F., Kearns, A., Larsen, A. R., Lindsay, J. A., Skov, R. L., et al. (2019). Implications of Identifying the Recently Defined Members of the Staphylococcus aureus Complex S. argenteus and S. Schweitzeri: A Position Paper of Members of the ESCMID Study Group for Staphylococci and Staphylococcal Diseases (ESGS). Clin. Microbiol. Infect. 25 (9), 1064–1070. doi: 10.1016/j.cmi.2019.02.028
Burgold-Voigt, S., Monecke, S., Simbeck, A., Holzmann, T., Kieninger, B., Liebler-Tenorio, E. M., et al. (2021). Characterisation and Molecular Analysis of an Unusual Chimeric Methicillin Resistant Staphylococcus aureus Strain and Its Bacteriophages. Front. Genet. 12 (1823). doi: 10.3389/fgene.2021.723958
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., Parks, D. H. (2019). GTDB-Tk: A Toolkit to Classify Genomes With the Genome Taxonomy Database. Bioinformatics 36 (6), 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen, S. Y., Lee, H., Wang, X. M., Lee, T. F., Liao, C. H., Teng, L. J., et al. (2018). High Mortality Impact of Staphylococcus argenteus on Patients With Community-Onset Staphylococcal Bacteraemia. Int. J. Antimicrob. Agents 52 (6), 747–753. doi: 10.1016/j.ijantimicag.2018.08.017
Chew, K. L., Octavia, S., Lai, D., Lin, R. T. P., Teo, J. W. P. (2021). Staphylococcus singaporensis Sp. Nov., a New Member of the Staphylococcus aureus Complex, Isolated From Human Clinical Specimens. Int. J. Syst. Evol. Microbiol. 71 (10). doi: 10.1099/ijsem.0.005067
Collinson, J., Parkin, D., Knox, A., Sangster, G., Svensson, L. (2008). Species Boundaries in the Herring and Lesser Black-Backed Gull Complex. Brit. Birds 101 (7), 340–363.
Diot, A., Dyon-Tafani, V., Bergot, M., Tasse, J., Martins-Simões, P., Josse, J., et al. (2020). Investigation of a Staphylococcus argenteus Strain Involved in a Chronic Prosthetic-Joint Infection. Int. J. Mol. Sci. 21 (17). doi: 10.3390/ijms21176245
Dupieux, C., Blondé, R., Bouchiat, C., Meugnier, H., Bes, M., Laurent, S., et al. (2015). Community-Acquired Infections Due to Staphylococcus argenteus Lineage Isolates Harbouring the Panton-Valentine Leucocidin, France, 2014. Eurosurveillance 20 (23), 21154. doi: 10.2807/1560-7917.ES2015.20.23.21154
Enright, M. C., Day, N. P., Davies, C. E., Peacock, S. J., Spratt, B. G. (2000). Multilocus Sequence Typing for Characterization of Methicillin-Resistant and Methicillin-Susceptible Clones of Staphylococcus aureus. J. Clin. Microbiol. 38 (3), 1008–1015. doi: 10.1128/JCM.38.3.1008-1015.2000
Eshaghi, A., Bommersbach, C., Zittermann, S., Burnham, C. A., Patel, R., Schuetz, A. N., et al. (2021). Phenotypic and Genomic Profiling of Staphylococcus argenteus in Canada and the United States and Recommendations for Clinical Result Reporting. J. Clin. Microbiol. 59 (6), e02470–20. doi: 10.1128/jcm.02470-20
Fujimoto-Nakamura, M., Ito, H., Oyamada, Y., Nishino, T., Yamagishi, J. (2005). Accumulation of Mutations in Both gyrB and parE Genes Is Associated With High-Level Resistance to Novobiocin in Staphylococcus aureus. Antimicrob. Agents Chemother. 49 (9), 3810–3815. doi: 10.1128/aac.49.9.3810-3815.2005
Grossmann, A., Froböse, N. J., Mellmann, A., Alabi, A. S., Schaumburg, F., Niemann, S. (2021). An In Vitro Study on Staphylococcus schweitzeri Virulence. Sci. Rep. 11 (1), 1157. doi: 10.1038/s41598-021-80961-x
Harmsen, D., Claus, H., Witte, W., Rothgänger, J., Claus, H., Turnwald, D., et al. (2003). Typing of Methicillin-Mesistant Staphylococcus aureus in a University Hospital Setting by Using Novel Software for Spa Repeat Determination and Database Management. J. Clin. Microbiol. 41 (12), 5442–5448. doi: 10.1128/JCM.41.12.5442-5448.2003
Holt, D. C., Holden, M. T. G., Tong, S. Y. C., Castillo-Ramirez, S., Clarke, L., Quail, M. A., et al. (2011). A Very Early-Branching Staphylococcus aureus Lineage Lacking the Carotenoid Pigment Staphyloxanthin. Genome Biol. Evol. 3, 881–895. doi: 10.1093/gbe/evr078
Hsu, L. Y., Holden, M. T. G., Koh, T. H., Pettigrew, K. A., Cao, D., Hon, P. Y., et al. (2017). ST3268: A Geographically Widespread Primate MRSA Clone. J. Antimicrob. Chemother. 72 (8), 2401–2403. doi: 10.1093/jac/dkx120
Hsu, J. C., Wan, T. W., Lee, H., Wang, X. M., Lin, Y. T., Jung, C. J., et al. (2020). Heterogeneity of Molecular Characteristics Among Staphylococcus argenteus Clinical Isolates (ST2250, ST2793, ST1223, and ST2198) in Northern Taiwan. Microorganisms 8 (8). doi: 10.3390/microorganisms8081157
Huson, D. H., Bryant, D. (2006). Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 23 (2), 254–267. doi: 10.1093/molbev/msj030
Indrawattana, N., Pumipuntu, N., Suriyakhun, N., Jangsangthong, A., Kulpeanprasit, S., Chantratita, N., et al. (2019). Staphylococcus argenteus From Rabbits in Thailand. Microbiologyopen 8 (4), e00665. doi: 10.1002/mbo3.665
Jolley, K. A., Bray, J. E., Maiden, M. C. J. (2018). Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.org Website and Their Applications. Wellcome Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Kitagawa, H., Ohge, H., Hisatsune, J., Masuda, K., Aziz, F., Hara, T., et al. (2020). Low Incidence of Staphylococcus argenteus Bacteremia in Hiroshima, Japan. J. Infect. Chemother. 26 (1), 140–143. doi: 10.1016/j.jiac.2019.07.011
Krüger, H., Ji, X., Wang, Y., Feßler, A. T., Wang, Y., Wu, C., et al. (2021). Identification of Tn553, a Novel Tn554-Related Transposon That Carries a Complete blaZ-Blar1-blaI β-Lactamase Operon in Staphylococcus aureus. J. Antimicrob. Chemother. 76 (10), 2733–2735. doi: 10.1093/jac/dkab210
Kukla, R., Neradová, K., Petráš, P., Kekláková, J., Ryšková, L., Žemličková, H. (2020). The First Confirmed Detection of Staphylococcus argenteus in the Czech Republic. Epidemiol. Mikrobiol. Imunol. 69 (1), 48–52.
Liebers, D., de Knijff, P., Helbig, A. J. (2004). The Herring Gull Complex is Not a Ring Species. Proc. Biol. Sci. 271 (1542), 893–901. doi: 10.1098/rspb.2004.2679
Li, Y., Tang, Y., Ren, J., Huang, J., Li, Q., Ingmer, H., et al. (2020). Identification and Molecular Characterization of Staphylococcus aureus and Multi-Drug Resistant MRSA From Monkey Faeces in China. Transbound. Emerg. Dis. 67 (3), 1382–1387. doi: 10.1111/tbed.13450
Ludt, C. J., Schroeder, W., Rottmann, O., Kuehn, R. (2004). Mitochondrial DNA Phylogeography of Red Deer (Cervus elaphus). Mol. Phylogenet. Evol. 31 (3), 1064–1083. doi: 10.1016/j.ympev.2003.10.003
Monecke, S., Coombs, G., Shore, A. C., Coleman, D. C., Akpaka, P., Borg, M., et al. (2011). A Field Guide to Pandemic, Epidemic and Sporadic Clones of Methicillin-Resistant Staphylococcus aureus. PloS One 6 (4), e17936. doi: 10.1371/journal.pone.0017936
Monecke, S., Jatzwauk, L., Müller, E., Nitschke, H., Pfohl, K., Slickers, P., et al. (2016). Diversity of SCCmec Elements in Staphylococcus aureus as Observed in South-Eastern Germany. PloS One 11 (9), e0162654. doi: 10.1371/journal.pone.0162654
Monecke, S., Kanig, H., Rudolph, W., Müller, E., Coombs, G., Hotzel, H., et al. (2010). Characterisation of Australian MRSA Strains ST75- and ST883-MRSA-IV and Analysis of Their Accessory Gene Regulator Locus. PloS One 5 (11), e14025. doi: 10.1371/journal.pone.0014025
Monecke, S., Müller, E., Braun, S. D., Armengol-Porta, M., Bes, M., Boswihi, S., et al. (2021). Characterisation of S. aureus/MRSA CC1153 and Review of Mobile Genetic Elements Carrying the Fusidic Acid Resistance Gene fusC. Sci. Rep. 11 (1), 8128. doi: 10.1038/s41598-021-86273-4
Monecke, S., Slickers, P., Ehricht, R. (2008). Assignment of Staphylococcus aureus Isolates to Clonal Complexes Based on Microarray Analysis and Pattern Recognition. FEMS Immunol. Med. Microbiol. 53 (2), 237–251. doi: 10.1111/j.1574-695X.2008.00426.x
Monecke, S., Stieber, B., Roberts, R., Akpaka, P. E., Slickers, P., Ehricht, R. (2014). Population Structure of Staphylococcus aureus From Trinidad & Tobago. PloS One 9 (2), e89120. doi: 10.1371/journal.pone.0089120
Ng, J. W., Holt, D. C., Lilliebridge, R. A., Stephens, A. J., Huygens, F., Tong, S. Y., et al. (2009). Phylogenetically Distinct Staphylococcus aureus Lineage Prevalent Among Indigenous Communities in Northern Australia. J. Clin. Microbiol. 47 (7), 2295–2300. doi: 10.1128/JCM.00122-09
Nimmo, G. R., Steen, J. A., Monecke, S., Ehricht, R., Slickers, P., Thomas, J. C., et al. (2015). ST2249-MRSA-III: A Second Major Recombinant Methicillin-Resistant Staphylococcus aureus Clone Causing Healthcare Infection in the 1970s. Clin. Microbiol. Infect. 21 (5), 444–450. doi: 10.1016/j.cmi.2014.12.018
Ohnishi, T., Shinjoh, M., Ohara, H., Kawai, T., Kamimaki, I., Mizushima, R., et al. (2018). Purulent Lymphadenitis Caused by Staphylococcus argenteus, Representing the First Japanese Case of Staphylococcus argenteus (Multilocus Sequence Type 2250) Infection in a 12-Year-Old Boy. J. Infect. Chemother. 24 (11), 925–927. doi: 10.1016/j.jiac.2018.03.018
Okuda, K. V., Toepfner, N., Alabi, A. S., Arnold, B., Belard, S., Falke, U., et al. (2016). Molecular Epidemiology of Staphylococcus aureus From Lambarene, Gabon. Eur. J. Clin. Microbiol. Infect. Dis. 35 (12), 1963–1973. doi: 10.1007/s10096-016-2748-z
Olatimehin, A., Shittu, A. O., Onwugamba, F. C., Mellmann, A., Becker, K., Schaumburg, F. (2018). Staphylococcus aureus Complex in the Straw-Colored Fruit Bat (Eidolon helvum) in Nigeria. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00162
Parks, D. H., Chuvochina, M., Chaumeil, P. A., Rinke, C., Mussig, A. J., Hugenholtz, P. (2020). A Complete Domain-to-Species Taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38 (9), 1079–1086. doi: 10.1038/s41587-020-0501-8
Parks, D. H., Chuvochina, M., Rinke, C., Mussig, A. J., Chaumeil, P.-A., Hugenholtz, P. (2022). GTDB: An Ongoing Census of Bacterial and Archaeal Diversity Through a Phylogenetically Consistent, Rank Normalized and Complete Genome-Based Taxonomy. Nucleic Acids Res. 50 (D1), D785–D794. doi: 10.1093/nar/gkab776
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A Standardized Bacterial Taxonomy Based on Genome Phylogeny Substantially Revises the Tree of Life. Nat. Biotechnol. 36 (10), 996–1004. doi: 10.1038/nbt.4229
Pumipuntu, N. (2019). Staphylococcus argenteus: An Emerging Subclinical Bovine Mastitis Pathogen in Thailand. Vet. World 12 (12), 1940–1944. doi: 10.14202/vetworld.2019.1940-1944
Rigaill, J., Grattard, F., Grange, S., Forest, F., Haddad, E., Carricajo, A., et al. (2018). Community-Acquired Staphylococcus argenteus Sequence Type 2250 Bone and Joint Infection, France, 2017. Emerg. Infect. Dis. 24 (10), 1958–1961. doi: 10.3201/eid2410.180727
Roberts, M. C., Feßler, A. T., Monecke, S., Ehricht, R., No, D., Schwarz, S. (2018). Molecular Analysis of Two Different MRSA Clones ST188 and ST3268 From Primates (Macaca spp.) in a United States Primate Center. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02199
Rossi, B. F., Bonsaglia É, C. R., Castilho, I. G., Dantas, S. T. A., Langoni, H., Pantoja, J. C. F., et al. (2020). First Investigation of Staphylococcus argenteus in a Brazilian Collections of S. aureus Isolated From Bovine Mastitis. BMC Vet. Res. 16 (1), 252. doi: 10.1186/s12917-020-02472-7
Ruimy, R., Armand-Lefevre, L., Barbier, F., Ruppe, E., Cocojaru, R., Mesli, Y., et al. (2009). Comparisons Between Geographically Diverse Samples of Carried Staphylococcus aureus. J. Bacteriol. 191 (18), 5577–5583. doi: 10.1128/JB.00493-09
Schaumburg, F., Alabi, A. S., Kock, R., Mellmann, A., Kremsner, P. G., Boesch, C., et al. (2012). Highly Divergent Staphylococcus aureus Isolates From African non-Human Primates. Environ. Microbiol. Rep. 4 (1), 141–146. doi: 10.1111/j.1758-2229.2011.00316.x
Schaumburg, F., Pauly, M., Anoh, E., Mossoun, A., Wiersma, L., Schubert, G., et al. (2015). Staphylococcus aureus Complex From Animals and Humans in Three Remote African Regions. Clin. Microbiol. Infect. 21 (4), 345 e1–345 e8. doi: 10.1016/j.cmi.2014.12.001
Scholtzek, A. D., Hanke, D., Walther, B., Eichhorn, I., Stöckle, S. D., Klein, K. S., et al. (2019). Molecular Characterization of Equine Staphylococcus aureus Isolates Exhibiting Reduced Oxacillin Susceptibility. Toxins (Basel) 11 (9). doi: 10.3390/toxins11090535
Schuster, D., Rickmeyer, J., Gajdiss, M., Thye, T., Lorenzen, S., Reif, M., et al. (2017). Differentiation of Staphylococcus argenteus (Formerly: Staphylococcus aureus Clonal Complex 75) by Mass Spectrometry From S. aureus Using the First Strain Isolated From a Wild African Great Ape. Int. J. Med. Microbiol. 307 (1), 57–63. doi: 10.1016/j.ijmm.2016.11.003
Schutte, A. H. J., Strepis, N., Zandijk, W. H. A., Bexkens, M. L., Bode, L. G. M., Klaassen, C. H. W. (2021). Characterization of Staphylococcus roterodami SpNov., a New Species Within the Staphylococcus aureus Complex Isolated From a Human Foot Infection. Int. J. Syst. Evol. Microbiol. 71 (9). doi: 10.1099/ijsem.0.004996
Senok, A., Nassar, R., Kaklamanos, E. G., Belhoul, K., Abu Fanas, S., Nassar, M., et al. (2020). Molecular Characterization of Staphylococcus aureus Isolates Associated With Nasal Colonization and Environmental Contamination in Academic Dental Clinics. Microb. Drug Resist. 26 (6), 661–669. doi: 10.1089/mdr.2019.0318
Shittu, A. O., Mellmann, A., Schaumburg, F. (2020). Molecular Characterization of Staphylococcus aureus Complex From Fomites in Nigeria. Infect. Genet. Evol. 85, 104504. doi: 10.1016/j.meegid.2020.104504
Söderquist, B., Wildeman, P., Stenmark, B., Stegger, M. (2020). Staphylococcus argenteus as an Etiological Agent of Prosthetic Hip Joint Infection: A Case Presentation. J. Bone Jt. Infect. 5 (4), 172–175. doi: 10.7150/jbji.44848
Soge, O. O., No, D., Michael, K. E., Dankoff, J., Lane, J., Vogel, K., et al. (2016). Transmission of MDR MRSA Between Primates, Their Environment and Personnel at a United States Primate Centre. J. Antimicrob. Chemother. 71 (10), 2798–2803. doi: 10.1093/jac/dkw236
Suzuki, Y., Kubota, H., Ono, H. K., Kobayashi, M., Murauchi, K., Kato, R., et al. (2017). Food Poisoning Outbreak in Tokyo, Japan Caused by Staphylococcus argenteus. Int. J. Food Microbiol. 262, 31–37. doi: 10.1016/j.ijfoodmicro.2017.09.005
Tång Hallbäck, E., Karami, N., Adlerberth, I., Cardew, S., Ohlén, M., Engström Jakobsson, H., et al. (2018). Methicillin-Resistant Staphylococcus argenteus Misidentified as Methicillin-Resistant Staphylococcus aureus Emerging in Western Sweden. J. Med. Microbiol. 67 (7), 968–971. doi: 10.1099/jmm.0.000760
Tong, S. Y., Schaumburg, F., Ellington, M. J., Corander, J., Pichon, B., Leendertz, F., et al. (2015). Novel Staphylococcal Species That Form Part of a Staphylococcus aureus-Related Complex: The Non-Pigmented Staphylococcus argenteus Sp. Nov. And the Non-Human Primate-Associated Staphylococcus schweitzeri Sp. Nov. Int. J. Syst. Evol. Microbiol. 65 (Pt 1), 15–22. doi: 10.1099/ijs.0.062752-0
van den Beld, M. J., Reubsaet, F. A. (2012). Differentiation Between Shigella, Enteroinvasive Escherichia coli (EIEC) and Noninvasive Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 31 (6), 899–904. doi: 10.1007/s10096-011-1395-7
Wang, T., Tanaka, M., Sato, K. (1998). Detection of grlA and gyrA Mutations in 344 Staphylococcus aureus Strains. Antimicrob. Agents Chemother. 42 (2), 236–240. doi: 10.1128/aac.42.2.236
Keywords: Staphylococcus aureus, Staphylococcus schweitzeri, Staphylococcus argenteus, Staphylococcus singaporensis, Staphylococcus roterodami, whole genome sequencing (WGS), DNA microarray
Citation: Monecke S, Schaumburg F, Shittu AO, Schwarz S, Mühldorfer K, Brandt C, Braun SD, Collatz M, Diezel C, Gawlik D, Hanke D, Hotzel H, Müller E, Reinicke M, Feßler AT and Ehricht R (2022) Description of Staphylococcal Strains from Straw-Coloured Fruit Bat (Eidolon helvum) and Diamond Firetail (Stagonopleura guttata) and a Review of their Phylogenetic Relationships to Other Staphylococci. Front. Cell. Infect. Microbiol. 12:878137. doi: 10.3389/fcimb.2022.878137
Received: 17 February 2022; Accepted: 16 March 2022;
Published: 11 May 2022.
Edited by:
Percy Schröttner, Technische Universität Dresden, GermanyReviewed by:
Selvaraj Anthonymuthu, University of California, Irvine CA, United StatesCopyright © 2022 Monecke, Schaumburg, Shittu, Schwarz, Mühldorfer, Brandt, Braun, Collatz, Diezel, Gawlik, Hanke, Hotzel, Müller, Reinicke, Feßler and Ehricht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Monecke, c3RlZmFuLm1vbmVja2VAbGVpYm5pei1pcGh0LmRl
†These authors share senior authorship
‡Retired
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.