 Kai Xia
Kai Xia Renyuan Gao
Renyuan Gao Xiaocai Wu
Xiaocai Wu Jing Sun1
Jing Sun1 Jian Wan
Jian Wan Tianqi Wu
Tianqi Wu Lu Yin
Lu Yin Chunqiu Chen
Chunqiu Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 13 April 2022
Sec. Microbiome in Health and Disease
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.864944
This article is part of the Research Topic Inflammation and Repair in Inflammatory Bowel Disease: Clinical, Translational and Experimental Studies View all 6 articles
Background: Crohn’s disease (CD) is a chronic nonspecific inflammatory bowel disease (IBD) with an increasing incidence worldwide. The etiology of CD is still obscure, but microbial dysbiosis has been recognized as an essential factor contributing to CD. However, few studies have revealed the microbiome’s signatures and reciprocal correlations between multiple sites in patients with CD over different disease stages. This study investigated the specific microbial architectures of the oral cavity, sputum, and ileum in patients with CD in the active and remission stages.
Methods: Microbial samples from the oral cavity, sputum, and ileum were collected from patients with CD in the active and remission stages and healthy controls. The microbial composition was assessed by 16S ribosomal RNA (rRNA) gene sequencing. In addition, bioinformatics methods were used to demonstrate the microbial signatures, functional changes, and correlations between microbiota and clinical data in CD.
Results: Compared with healthy controls, a distinct microbiota dysbiosis in the oral cavity, sputum, and ileum of patients with CD was identified, characterized by alterations in microbiota biodiversity and composition. The oral cavity and sputum microbiota showed significantly lower microbial diversity in patients with CD than in healthy controls. In terms of microbiota composition, the microbiota changes in the oral cavity of patients with CD were similar to those in the sputum, while they were different from those in the ileum. In the oral cavity and sputum of patients with CD, a lower relative abundance of Firmicutes and Actinobacteria was observed compared to healthy controls, which was most prominent in the active stage. In contrast, an increased relative abundance of Fusobacteria, Porphyromonas, and Haemophilus was observed in patients with CD. The predicted metabolic pathways involved in the oral cavity, sputum, and ileum were similar, predominantly involving metabolism, environmental information processing, and genetic information processing.
Conclusion: The results revealed the alterations of microbiota architecture in the oral cavity, sputum, and ileum of patients with CD, which varied across disease stages. Studying microbiota dysbiosis may bring new insights into the etiology of CD and lead to novel treatments.
Crohn’s disease (CD) is a complex chronic inflammatory bowel disease (IBD) that involves genetic susceptibility and environmental triggers (Kong et al., 2021). Although the underlying cause is unknown, a growing number of studies have revealed that intestinal microbial dysbiosis plays an essential role in the occurrence and development of CD due to its impact on intestinal mucosal immunity and barrier function (Bernstein and Forbes, 2017; Sokol et al., 2017; Dovrolis et al., 2020). Firmicutes and Bacteroidetes synthesize short-chain fatty acids (SCFAs) from carbohydrates, inducing the expression of related proteins required for tight junction formation, contributing to intestinal mechanical barriers (Fesler et al., 2020). In addition, Bacteroidetes can produce polysaccharide A (PSA), which has anti-inflammatory effects, inducing Treg cells to release interleukin (IL)-10 to protect the intestinal barrier (Ramakrishna et al., 2019). However, these microbiotas are found in lower numbers in the colon of patients with CD (Lloyd-Price et al., 2019), destroying the intestinal barrier function and increasing CD susceptibility. However, only a few studies have reported the microbiota changes in the ileum and their role in the relapse of CD (Sokol et al., 2020; Olaisen et al., 2021).
CD also has several extraintestinal manifestations, such as glossitis, angular cheilitis, oral mucosal plaques, oral candidiasis, interstitial lung diseases (ILDs), and the like (Katsanos et al., 2015; Danve, 2019). An earlier study showed that some dominant genera in the oral cavity, including Streptococcus, Prevotella, Neisseria, Haemophilus, Veillonella, and Gemella, contributed to dysbiosis in the salivary microbiota of patients with CD (Said et al., 2014). Furthermore, Rautava et al. (2015) demonstrated microbial dysbiosis of the oral cavity in a mouse colitis model. Read et al. (2021) proposed that specific bacteria could translocate from the oral cavity of patients with CD to the intestine and directly cause gut dysbiosis. Similarly, Ubags et al. (Ubags and Marsland, 2017) reported that there was also a potential association between the intestine and the lung via intestinal microbiota translocation. However, the microbial signatures from the respiratory tract and the sputum of CD patients remain underestimated.
The present study aimed to explore the characteristics of the oral cavity, sputum, and ileum microbiota of patients with CD in the active and remission stages and investigate the stage-specific microbiota profiles. In addition, the functional alterations of microbiota from multi-ecological sites and correlations between the dominant microbiota strains and clinical factors were also explored.
All the study participants were enrolled from the Shanghai Tenth People’s Hospital from July 2021 to August 2021, including patients in the active stage, remission stage, and healthy controls. The diagnosis of CD was based on a comprehensive evaluation of clinical symptoms, laboratory examinations, endoscopic features, radiological findings, and histological features by two experienced physicians (Ding et al., 2020). Patients with CD in the active stage all underwent ileostomy, and the patients in the remission stage came to the hospital for the reversal of ileostoma. The exclusion criteria included severe oral and respiratory diseases diagnosed within 1 month, such as dental caries, periodontitis, oral malignant tumor, oral CD, peri-oral CD, pneumonia, and lung cancer. Meanwhile, a history of probiotics or prebiotic consumption and antibiotic use within 1 month and other acute or chronic gastrointestinal disorders were also excluded. People who came to the hospital for physical examination were recruited as healthy controls. Among them, people with a history of chronic inflammatory conditions including IBD and current gastrointestinal or respiratory symptoms were excluded. The Ethics Committee of Shanghai Tenth People’s Hospital approved this study protocol (Ethical Approval Number: SHSY-IEC-4.1/21-145/01). Informed consent was obtained from all patients. All procedures were performed following the Declaration of Helsinki and its later amendments. This study has been registered in ClinicalTrial.gov (NCT04965584).
The nutrition risk screening score (2002) (Kondrup et al., 2003) and Crohn’s disease activity index (CDAI) (Yoshida, 1999) were recorded for each recruited patient. A CDAI score over 150 was regarded as the active stage, whereas a CDAI score of 150 or below was defined as the remission stage.
Saliva samples from the tongue base of the recruited patients and healthy controls were collected with a sterile cotton swab and immediately placed into a 2-ml sterile container. Before sputum sample collection, the participants were instructed to gargle three times and inhale 5 ml hypertonic sodium chloride solution by atomization for 10 min, then coughed hard to obtain sputum samples. Fecal samples from the ileal stoma were collected with a sterile cotton swab and placed into a 2-ml sterile container. In addition, 2 g of cefmetazole was routinely used intraoperatively, and there was no history of antibiotic use before surgery for those patients (Figure 1A). All samples were transported to the laboratory with an ice pack within 2 h, frozen immediately, and stored at −80°C before analyses.

Figure 1 Human body model diagram and experimental flow diagram. (A) Microbial samples were collected from the tongue base, sputum and ileal stoma. (B) Microbial samples from the oral cavity, sputum, and ileum were collected from patients with CD in the active and remission stages and healthy controls. The microbial composition was assessed by 16S rRNA gene sequencing. Bioinformatics methods were used to demonstrate the alpha and beta diversity of microbiota, the structure of microbiota, correlations between microbiota and clinical factors, and functional characterization of microbiota.
Microbial DNA was extracted from all samples using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s instructions. The final DNA concentration and purification were determined by a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA), and the DNA quality was checked by 1% agarose gel electrophoresis. The V3–V4 hypervariable regions of the bacteria 16S ribosomal RNA (rRNA) gene were amplified with primers 338F (5′- ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) with the thermocycler PCR system (GeneAmp 9700, ABI, USA). The PCR reactions were conducted using the following procedure: 3 min of denaturation at 95°C, 27 cycles of 30 s each at 95°C, 30 s for annealing at 55°C, and 45 s for elongation at 72°C, and a final extension at 72°C for 10 min. PCR reactions were performed in triplicate, with 20 μl mixture containing 4 μl of 5× FastPfu buffer, 2 μl of 2.5 mM deoxyribonucleotide triphosphates (dNTPs), 0.8 μl of each primer (5 μM), 0.4 μl of FastPfu polymerase, and 10 ng of template DNA. The resulting PCR products were extracted from a 2% agarose gel, further purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using QuantiFluor™-ST (Promega, USA) according to the manufacturer’s protocol.
Purified amplicons were pooled in equimolar ratios and paired-end sequenced (2 × 300) on an Illumina MiSeq platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Raw fastq files were demultiplexed, quality filtered by Trimmomatic, and merged by FLASH with the following criteria: (i) the reads were truncated at any site receiving an average quality score <20 over a 50-bp sliding window; (ii) primers were exactly matched, allowing two nucleotide mismatching, and reads containing ambiguous bases were removed; and (iii) sequences whose overlap was longer than 10 bp were merged according to their overlap sequence.
Operational taxonomic units (OTUs) were clustered with 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. The taxonomy of each 16S rRNA gene sequence was analyzed by the RDP Classifier algorithm (http://rdp.cme.msu.edu/) against the Silva (SSU123) 16S rRNA database using a confidence threshold of 70%.
Alpha diversity was analyzed using the Mothur software (version 1.30.2 https://www.mothur.org/wiki/Download_mothur). To evaluate alpha diversity, the Chao index was calculated, representing the abundance of microbiota, while the Shannon index represented microbiota diversity. A Venn diagram was drawn by Venn Diagram of software R (version 3.3.1) to show the similarity and overlap of microbiota composition of each group. In addition, the Community Bar diagram was drawn by software R (version 3.3.1) to show the abundance of microbiota in multi-ecological sites. The principal coordinates analysis (PCoA) is a multivariate statistical analysis method that reduces the dimensionality of multidimensional data to extract the most important elements. The differences between groups are presented in a two-dimensional coordinate graph (Liu et al., 2020). Furthermore, the Wilcoxon rank-sum test (R package “stats” and python package “scipy”) was used to analyze the inter-group differences of microbiota. A correlation heatmap was drawn with the “pheatmap” package of software R (version 3.3.1), visually reflecting the association between clinical factors and dominant microbiotas in the corresponding ecological sites. In addition, the PICRUSt software (version 1.1.0 http://picrust.github.io/picrust/) was used to predict the function of 16S in the amplicon-sequencing results. The Benjamini–Hochberg method was used to correct multiple comparisons, and an false discovery rate (FDR) value <0.05 was considered statistical significant (Gao et al., 2021). p < 0.05 at two sides was considered significant in all statistical analyses Figure 1B.
The 16S rRNA gene sequence data sets have been deposited in the NCBI Sequence Read Archive (SRA) under the study accession number PRJNA773617.
A total of 43 patients (23 patients in the active stage and 20 patients in the remission stage) and 18 healthy controls were recruited in this study (Table 1). Patients with CD were at higher nutritional risk (p < 0.001) than their healthy counterparts. Most patients with CD had a regular diet and had no history of smoking, alcohol, or hematochezia, similar to the healthy controls. Moreover, the CD lesions were mainly located in the ileocolon. The levels of hematological indexes, including C-reactive protein (CRP), white blood cell (WBC), hemoglobin, albumin, prealbumin, and erythrocyte sedimentation rate (ESR), were significantly different between the active and remission stages (all p < 0.05). However, the levels of inflammatory biomarkers, including procalcitonin (PCT), IL-1, IL-6, IL-8, and IL-10, were similar between the active and remission stages of patients with CD (Table 1).
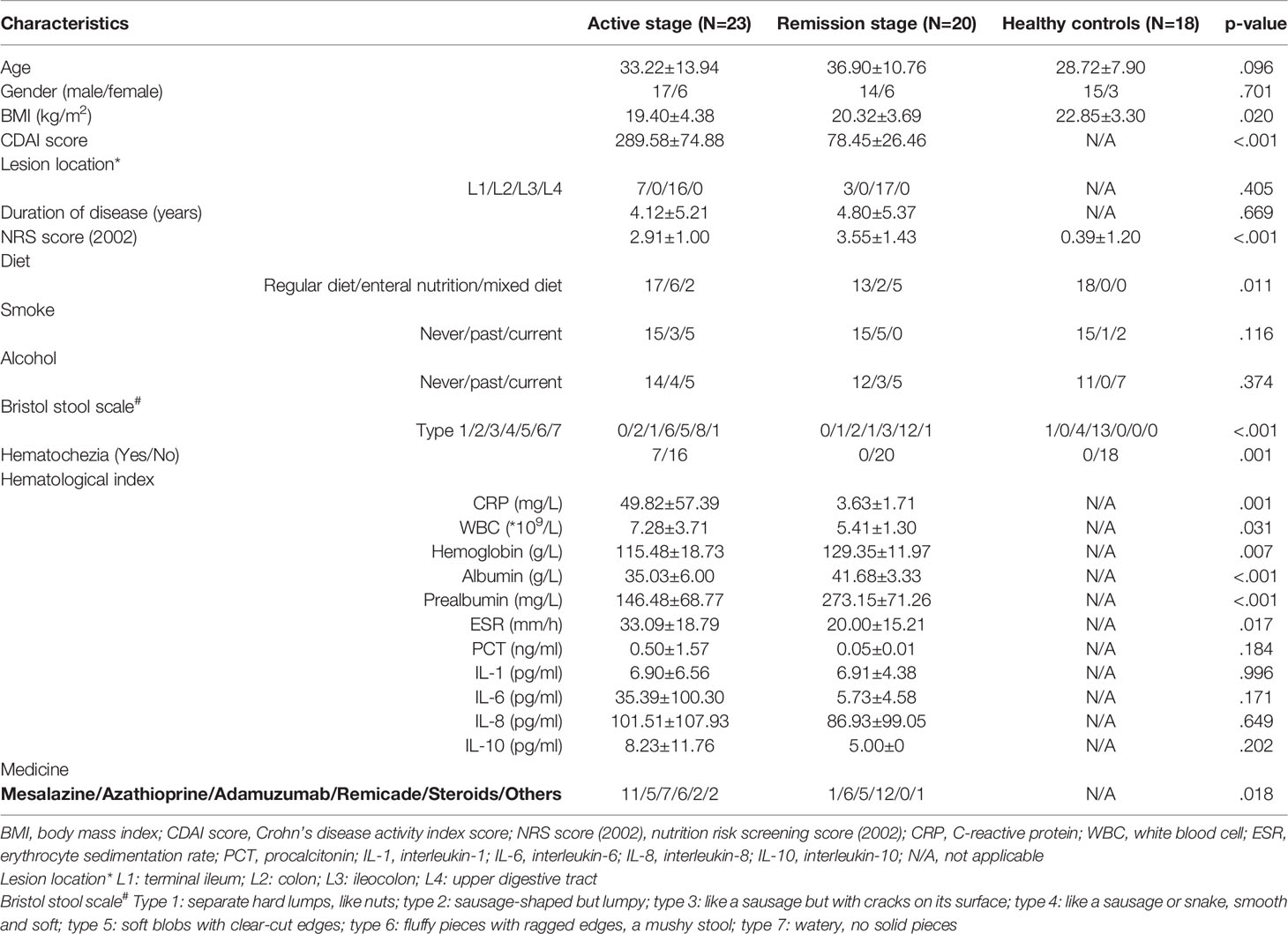
Table 1 Baseline demographic and clinical characteristics of patients with CD and healthy controls.
In this study, the Chao and Shannon indexes were used to analyze the alpha diversity of microbiota. The Chao index was used to reflect the abundance of microbiota in the samples, and the Shannon index was used to indicate microbiota diversity. Patients in the remission stage had a significantly lower Chao index of oral microbiota than healthy controls on both phylum and genus levels (p = 0.006 and p = 0.002) (Figures 2A, B). Similar results were observed in sputum samples (Figures 2C, D). In addition, the Shannon index of the oral microbiota in healthy controls was significantly higher than that in both groups of patients with CD (Figures 2E, F). However, there was no significant difference in sputum microbiota in patients with CD and healthy controls (Figures 2G, H). As for the ileal microbiota, the Chao and Shannon indexes of the active stage were higher than those of the remission stage, but the differences were not statistically significant (Supplementary Figures S1A–D).
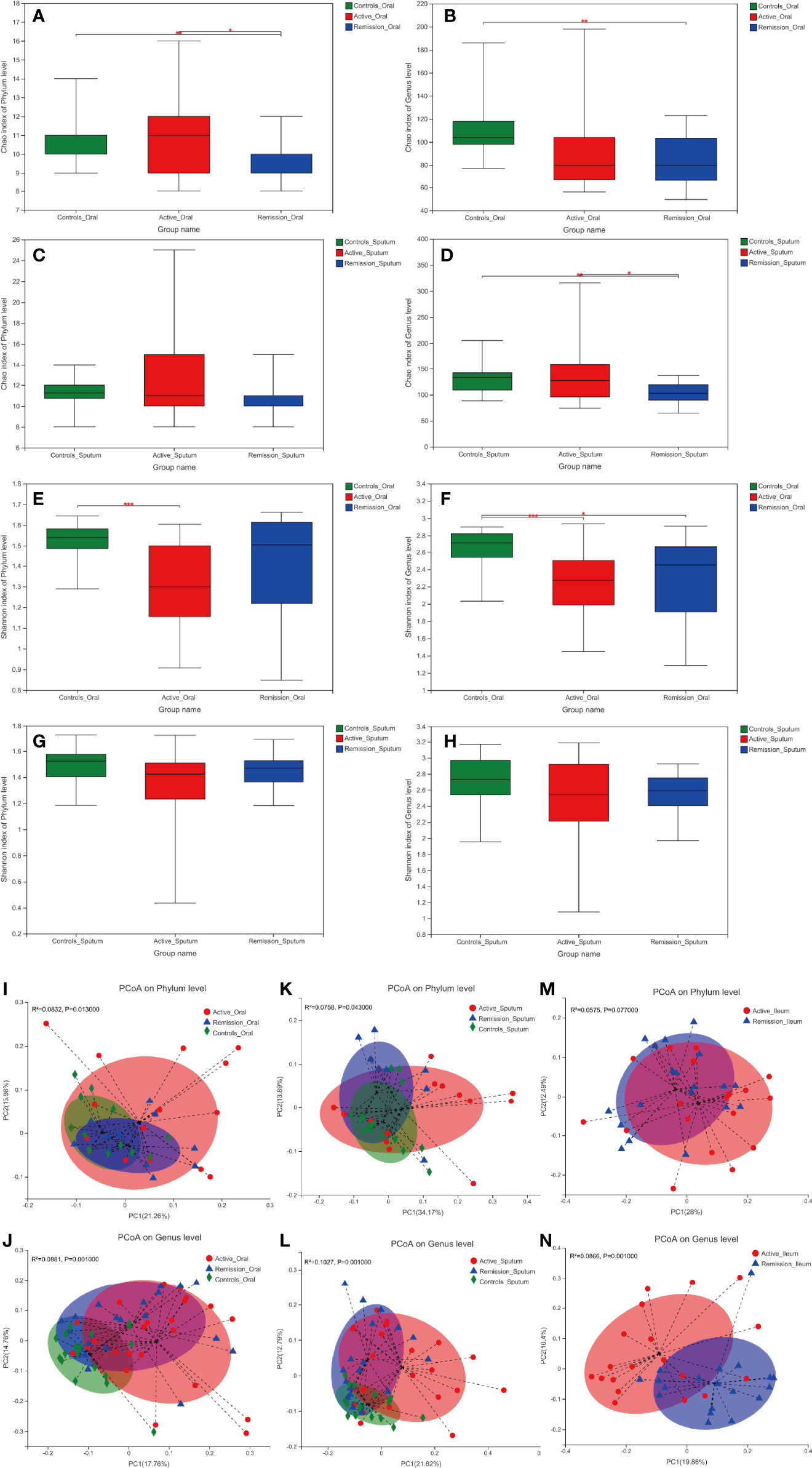
Figure 2 Alpha and beta diversity of microbiota in the oral cavity and sputum of patients with CD and healthy controls. (A–D) Chao diversity index of phylum and genus levels in the samples of oral cavity and sputum. (E–H) Shannon diversity index of phylum and genus levels in the samples of oral cavity and sputum. (I–N) Principle coordinate analysis (PCoA) on the phylum and genus levels using the unweighted UniFrac distance and Adonis test. Red color represents samples from active stage, blue indicates remission stage, while green color indicates healthy controls. *, **, and *** correspond to p-values <0.05, 0.01, and 0.001, respectively.
Principle coordinate analysis (PCoA) using the unweighted UniFrac distances also demonstrated that the microbiota in the oral cavity (p = 0.013 and p = 0.001) and sputum (p = 0.043 and p = 0.001) microbiota differed significantly among different stages of CD and healthy controls on phylum and genus levels, respectively (Figures 2I–L). In terms of ileal microbiota, a notable difference was observed between active and remission stages on the genus level (p = 0.001) but not on the phylum level (Figures 2M, N).
The total number of OTUs across all samples was 65 on the phylum level, among which 29 were identified in the active stage, 19 in the remission stage, and 17 in the healthy controls. Fifteen of these OTUs were common to all three groups (Figure 3A). The relative abundances of Firmicutes and Actinobacteria were the highest in the active stage and were lower in the remission stage of CD and lowest in the healthy controls. On the contrary, Bacteroidetes and Fusobacteria were predominant in the remission stage of CD and healthy controls (Figure 3C).
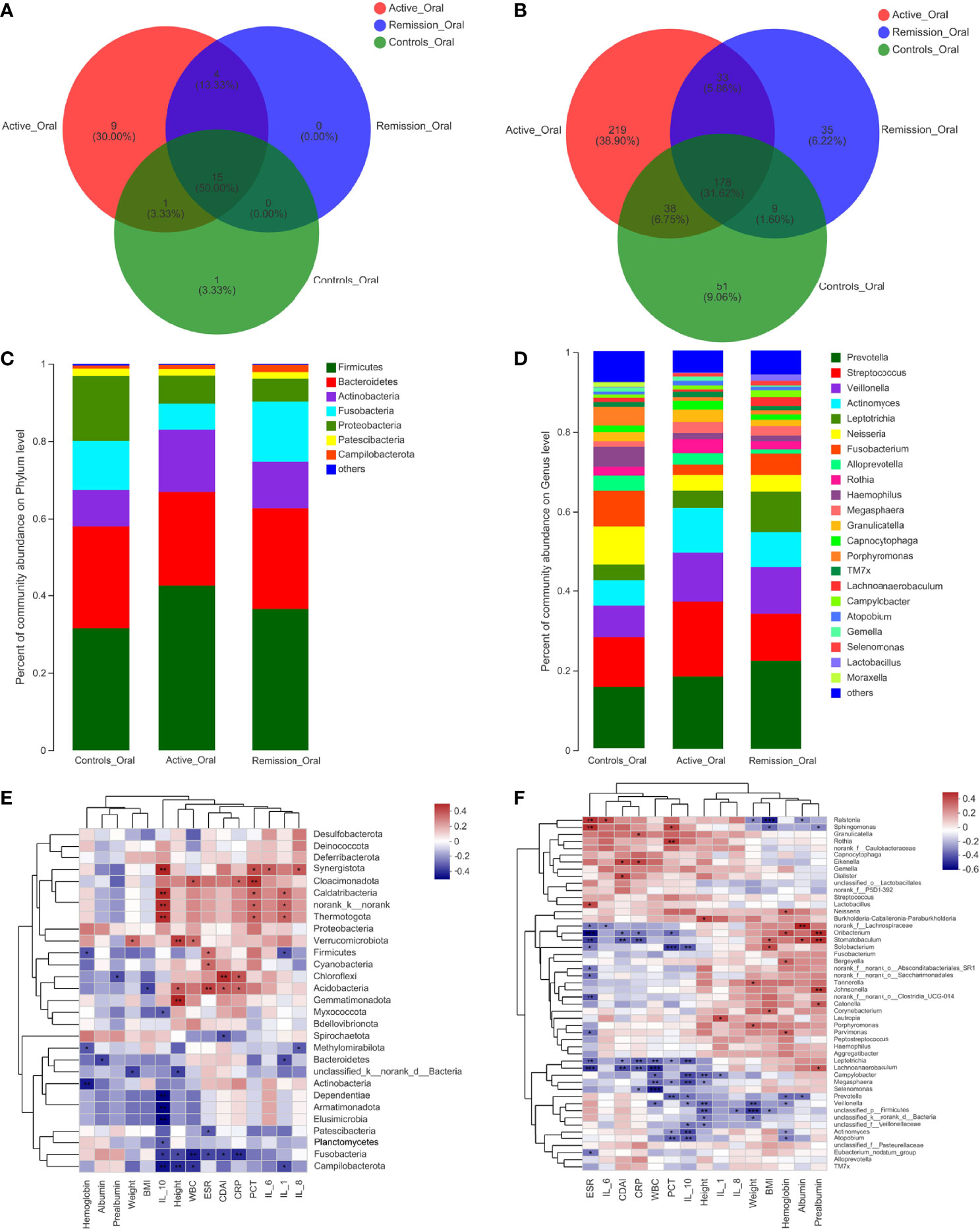
Figure 3 Structure analysis of the oral microbiota in patients with CD. (A, B) Venn diagram indicating the overlap of OTUs in the categories on the phylum and genus levels, respectively. (C, D) Barplot of the relative abundances of different taxa on the phylum and genus levels, respectively. (E, F) Heatmap revealing the association between oral microbiota and clinical factors on the phylum and genus levels, respectively. *, **, and *** correspond to p-values <0.05, 0.01, and 0.001, respectively.
We then aimed to investigate the stage-specific oral microbial profiles. Regarding the oral microbiota, Fusobacteria was more abundant in the remission stage of CD (p = 0.026), while Chloroflexi was more abundant in the active stage on the phylum level (p = 0.032) (Figure 4A). The relative abundances of Actinobacteria and Synergistota were higher in the active stage of CD (p = 0.047 and p = 0.036), while Fusobacteria and Spirochaetota were more abundant in the healthy controls (p = 0.006 and p < 0.001) (Figure 4B). In addition, the relative abundances of Spirochaetota and Synergistota were higher in the healthy controls than in the remission stage of CD (p = 0.036 and p < 0.001, respectively) (Figure 4C).
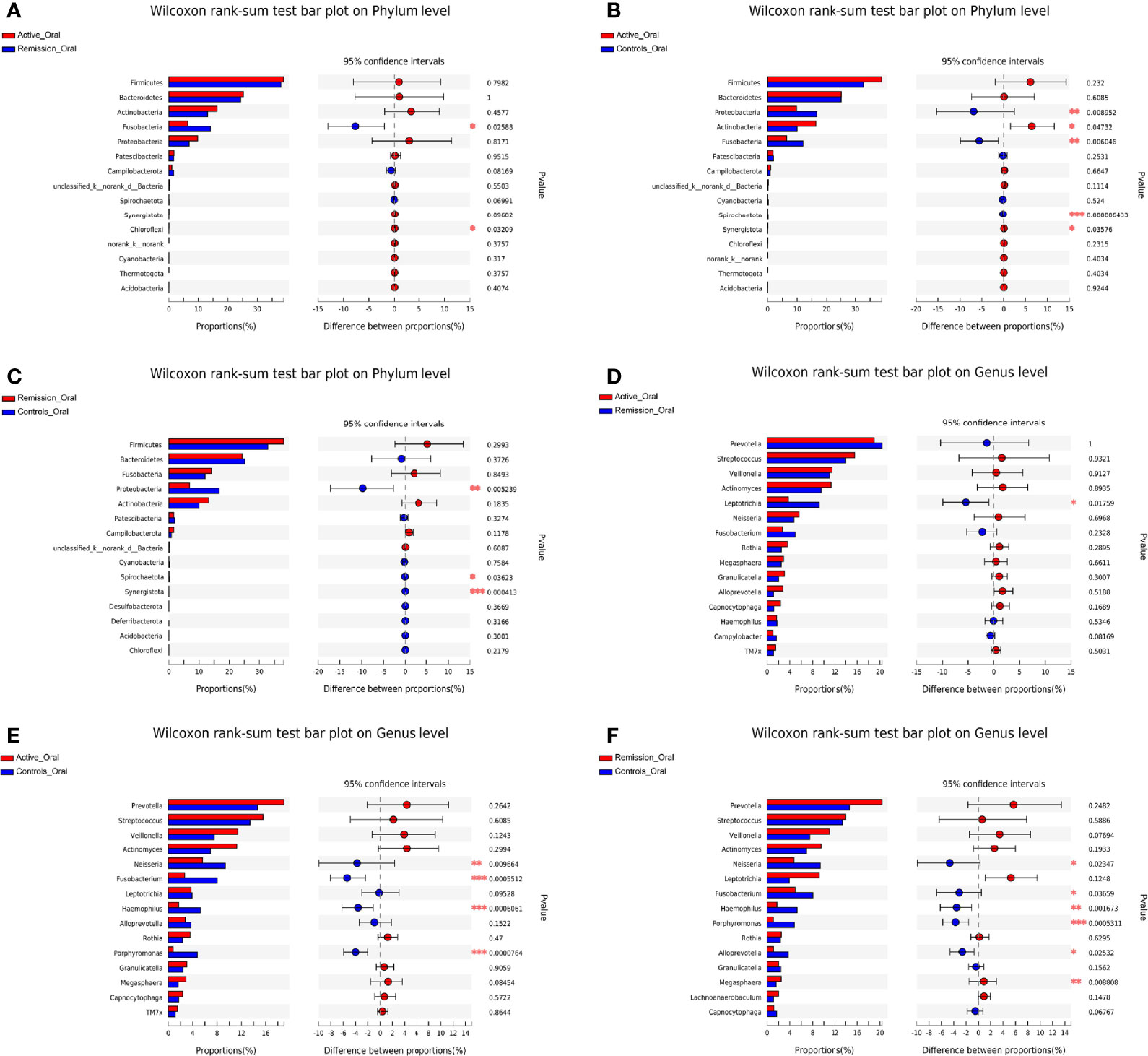
Figure 4 Inter-group difference analysis of microbiota in the oral cavity of patients with CD and healthy controls. (A–C) Wilcoxon rank-sum test bar plot of different taxa on the phylum level. (D–F) Wilcoxon rank-sum test bar plot of different taxa on the genus level. *, **, and *** correspond to p-values <0.05, 0.01, and 0.001, respectively.
On the genus level, 999 OTUs were identified in total, 468 were identified in the active stage, 255 in the remission stage, and 276 in the healthy controls. A total of 178 OTUs were shared across all three groups. The active stage group and the remission stage group had 33 overlapping OTUs, while the active stage group and the control group shared 38 OTUs. In contrast, the remission stage group and the control group had nine OTUs in common (Figure 3B). The relative abundances of Veillonella, Actinomyces, Megasphaera, and Atopobium decreased gradually from the active stage and remission stage of CD to healthy controls. However, the relative abundances of Fusobacterium, Haemophilus, and Porphyromonas increased gradually along the same sequence. Furthermore, Campylobacter was abundant in the oral cavity of patients with CD but less predominant in the healthy controls (Figure 3D). The only oral microbiota with a significant difference between the active and remission stages was Leptotrichia (p = 0.018), which was more abundant in the remission stage (Figure 4D). The relative abundances of Fusobacterium, Haemophilus, Porphyromonas, and Neisseria demonstrated a higher prominence in healthy controls than in the active stage of CD (p < 0.001, p < 0.001, p < 0.001, and p = 0.010) (Figure 4E). Comparing the remission stage to healthy controls, Porphyromonas, Haemophilus, Neisseria, Fusobacterium, and Alloprevotella were more abundant in the healthy controls, whereas Megasphaera was more abundant in the remission stage. These differences in microbiotas were statistically significant (all p < 0.05) (Figure 4F).
The total number of OTUs across all samples was 78 on the phylum level; 34 were identified in the active stage, 22 in the remission stage, and 22 in the healthy controls, respectively. Seventeen OTUs were common to all three groups (Figure 5A). The relative abundances of Firmicutes and Actinobacteria were highest in the active stage of CD and lowest in healthy controls, while the contrary was observed for Fusobacteria (Figure 5C).
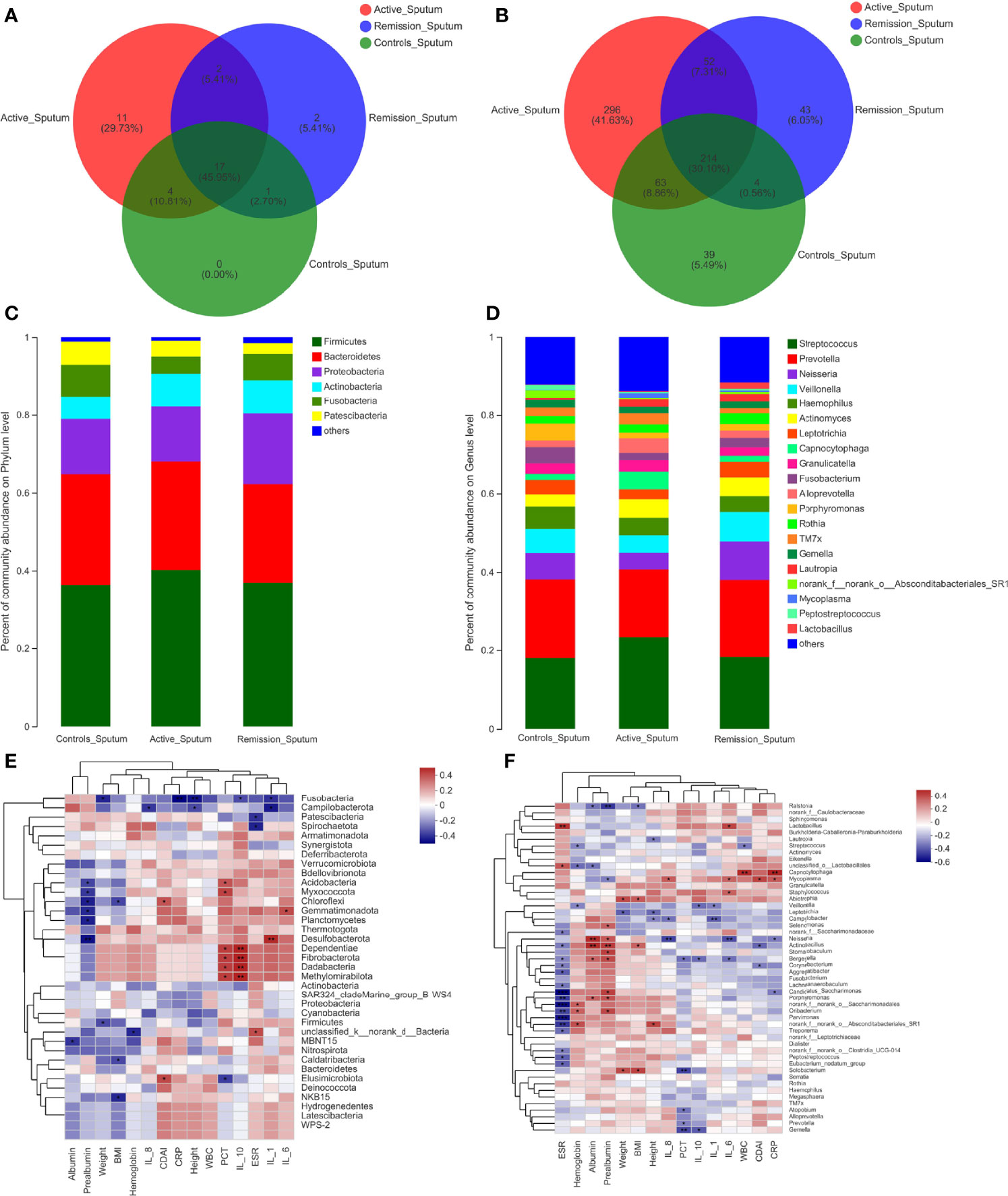
Figure 5 Structure analysis of the sputum microbiota in patients with CD. (A, B) Venn diagram indicating the overlap of OTUs in the categories on the phylum and genus levels, respectively. (C, D) Barplot of the relative abundances of different taxa on the phylum and genus levels, respectively. (E, F) Heatmap revealing the association between sputum microbiota and clinical factors on the phylum and genus levels, respectively. *, **, and *** correspond to p-values <0.05, 0.01, and 0.001, respectively.
Regarding the sputum microbiota, Chloroflexi was the only microbiota that was significantly enriched in the active stage (p = 0.006) (Figure 6A). The relative abundances of Fusobacteria, Spirochaetota, and Synergistota were significantly higher in the healthy controls, especially compared to the active stage of CD (p = 0.022, p = 0.034, and p = 0.017, respectively) (Figure 6B). Furthermore, Actinobacteria was more abundant in the remission stage of CD (p = 0.022), whereas Patescibacteria and Synergistota were more abundant in the healthy controls (p = 0.031 and p = 0.040) (Figure 6C).
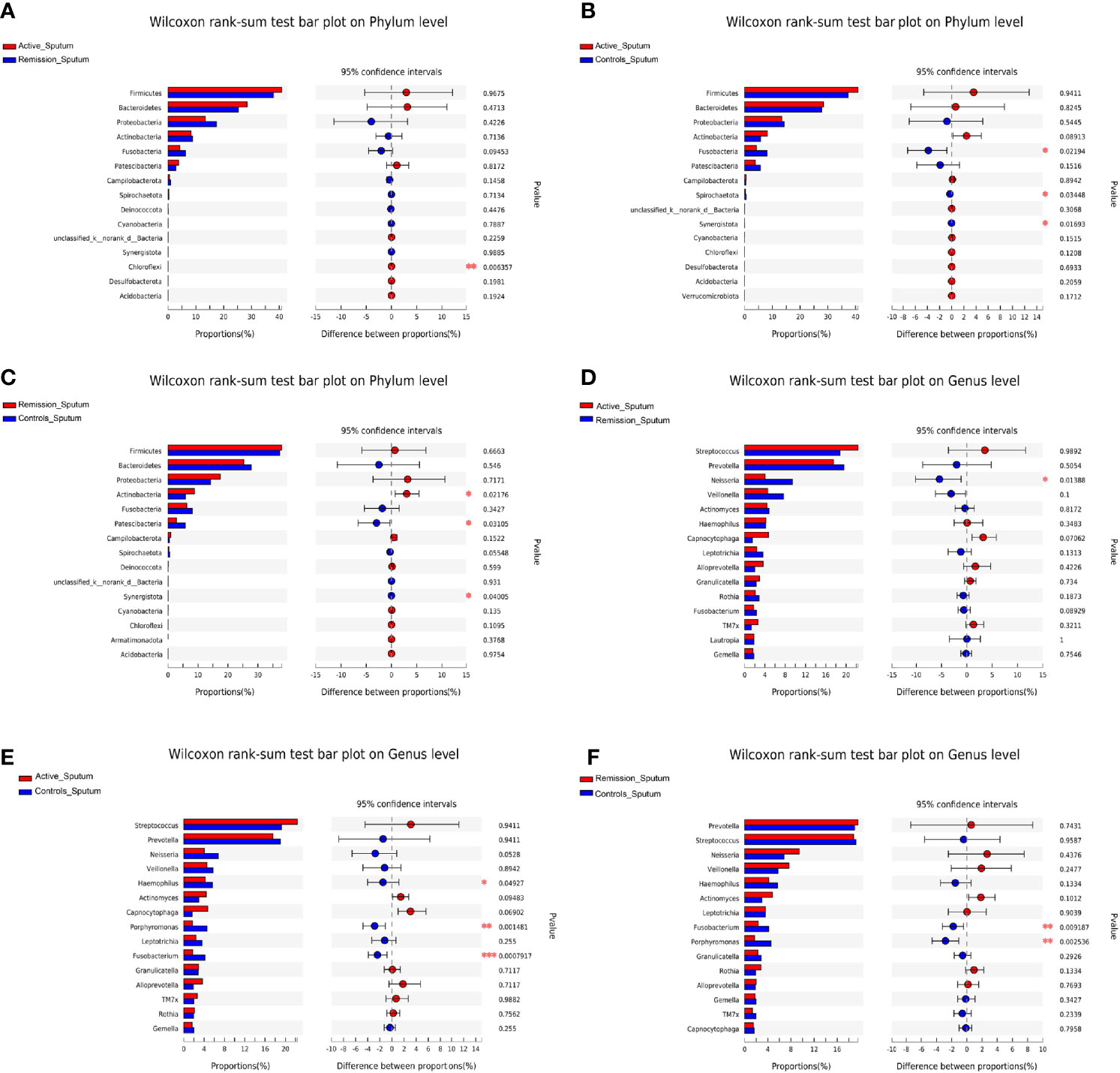
Figure 6 Inter-group difference analysis of sputum microbiota of patients with CD and healthy controls. (A–C) Wilcoxon rank-sum test bar plot of different taxa on the phylum level. (D–F) Wilcoxon rank-sum test bar plot of different taxa on the genus level. *, **, and *** correspond to p-values <0.05, 0.01, and 0.001, respectively.
On the genus level, the total number of OTUs across all samples was 1,258. The three groups shared 214 OTUs, active stage and remission stage groups shared 52 OTUs, the active stage and control groups shared 63 OTUs, and the remission stage and control groups shared 4 OTUs (Figure 5B). The relative abundance of Streptococcus declined gradually from the active stage, remission stage of CD to healthy controls. On the contrary, the relative abundances of Prevotella, Fusobacterium, Porphyromonas, and Haemophilus were lowest in the active stage of CD and highest in the healthy controls (Figure 5D). The only sputum microbiota with a significant difference between the active and remission stages was Neisseria (p = 0.014), as it was more abundant in the remission stage (Figure 6D). In contrast, the relative abundances of Fusobacterium, Haemophilus, and Porphyromonas were much higher in the healthy controls (p < 0.001, p = 0.049, and p = 0.001) (Figure 6E). Furthermore, the relative abundances of Fusobacterium and Porphyromonas were significantly higher in the healthy controls than in the remission stage group (p = 0.009 and p = 0.004) (Figure 6F).
The total number of OTUs across all samples was 56 on the phylum level, 30 were identified in the active stage, and 26 in the remission stage. Twenty-two OTUs were found in common in the active stage and remission stage groups (Supplementary Figure S1E). The amount of OTUs on the genus level was 1,105, and the two groups shared 353 OTUs. 321 were exclusively identified in the active stage and 78 in the remission stage (Supplementary Figure S1F). The relative abundances of Bacteroidetes and Desulfobacterota were much higher in the active stage on the phylum level (all p < 0.001) (Figure 7C). However, the relative abundances of Firmicutes and Patescibacteria were higher in the active stage than in the remission stage of CD, but the differences between these microbiotas were not statistically significant (Figures 7A, C). On the genus level, the relative abundance of Bacteroides was much higher in the active stage of CD (p < 0.001). On the contrary, the relative abundances of Streptococcus, Veillonella, and Lactococcus were higher in the remission stage of CD (p = 0.033, p < 0.001, and p = 0.004) (Figures 7B, D).
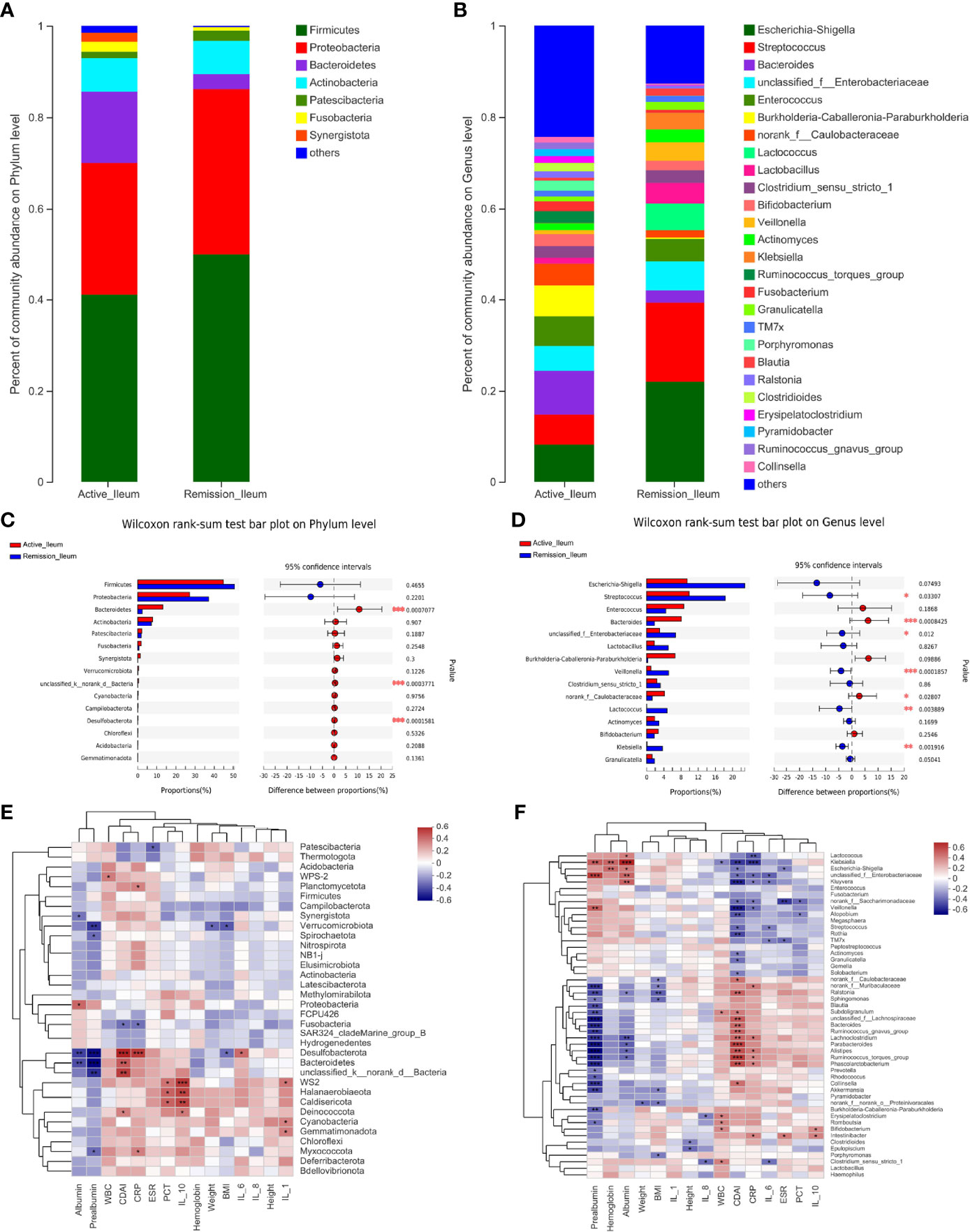
Figure 7 Structure analysis of the ileal microbiota in patients with CD. (A, B) Barplot of the relative abundances of different taxa on the phylum and genus levels, respectively. (C, D) Wilcoxon rank-sum test bar plot of different taxa on the phylum and genus levels, respectively. (E, F) Heatmap revealing the association between ileal microbiota and clinical factors on the phylum and genus levels, respectively. *, **, and *** correspond to p-values <0.05, 0.01, and 0.001, respectively.
To identify the correlations between dominant microbiota and several clinical factors, the Spearman’s rank correlation coefficient was used. Regarding the oral microbiota, ESR levels (R = 0.308, p = 0.045) were positively associated with Firmicutes, whereas IL-1 (R = −0.324, p = 0.034) and hemoglobin levels (R = −0.317, p = 0.038) in the blood of patients with CD showed negative correlations (Figure 3E). Similarly, the relative abundance of Firmicutes in the sputum samples was also positively correlated with ESR levels (R = 0.343, p = 0.030), while it was negatively correlated with hemoglobin levels (R = −0.375, p = 0.017) (Figure 5E). However, no significant correlations between the relative abundance of Firmicutes and clinical factors were observed in the ileal samples (Figure 7E). On the genus level, PCT levels in the blood of patients with CD were found to be negatively correlated with the relative abundance of Prevotella in the oral and sputum samples (R = −0.407, p = 0.007 and R = −0.321, p = 0.043, respectively) (Figures 3F, 5F). Moreover, IL-10 (R = −0.360, p = 0.018), hemoglobin (R = −0.377, p = 0.013), and albumin (R = −0.369, p = 0.015) concentrations were also negatively correlated with the relative abundance of Prevotella in the oral samples (Figure 3F). The relative abundance of Streptococcus in the sputum samples was negatively correlated with white blood cell count (R = −0.334, p = 0.035) and hemoglobin concentration (R = −0.313, p = 0.049), whereas no significant correlations in the oral samples were observed (Figures 3F, 5F). Regarding the ileal microbiota, hemoglobin (R = 0.414, p = 0.010) and albumin (R = 0.367, p = 0.023) concentrations showed positive correlations with the relative abundance of Escherichia–Shigella, whereas CDAI score (R = −0.353, p = 0.030) and ESR levels (R = −0.333, p = 0.041) in the blood were negatively associated with the microbiota. In addition, CDAI score (R = −0.390, p = 0.016) and IL-6 concentration (R = −0.335, p = 0.040) were negatively associated with the relative abundance of Streptococcus (Figure 7F).
To characterize the microbial functions alterations in patients with CD, we analyzed clusters of orthologous group (COG) functional classifications for microbiota and characterization of different metagenomes of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways levels. Oral microbial gene processes, such as translation, ribosomal structure and biogenesis, general function prediction only, amino acid transport and metabolism, replication, recombination and repair, and cell wall/membrane/envelope biogenesis pathways were highly abundant in all samples (Supplementary Figure S2A). In addition, the microbiota of sputum samples showed similar functional characteristics (Supplementary Figure S2B). However, the most active COG functional categories in the ileum were amino acid transport and metabolism, followed by general function prediction only, carbohydrate transport, and metabolism and transcription (Supplementary Figure S2C). Regarding the relative abundance of different metagenomes of KEGG pathways levels, the predicted metabolic pathways in the oral cavity, sputum, and ileum were similar, predominantly involving metabolism, environmental and genetic information processing (Figures 8A–C).
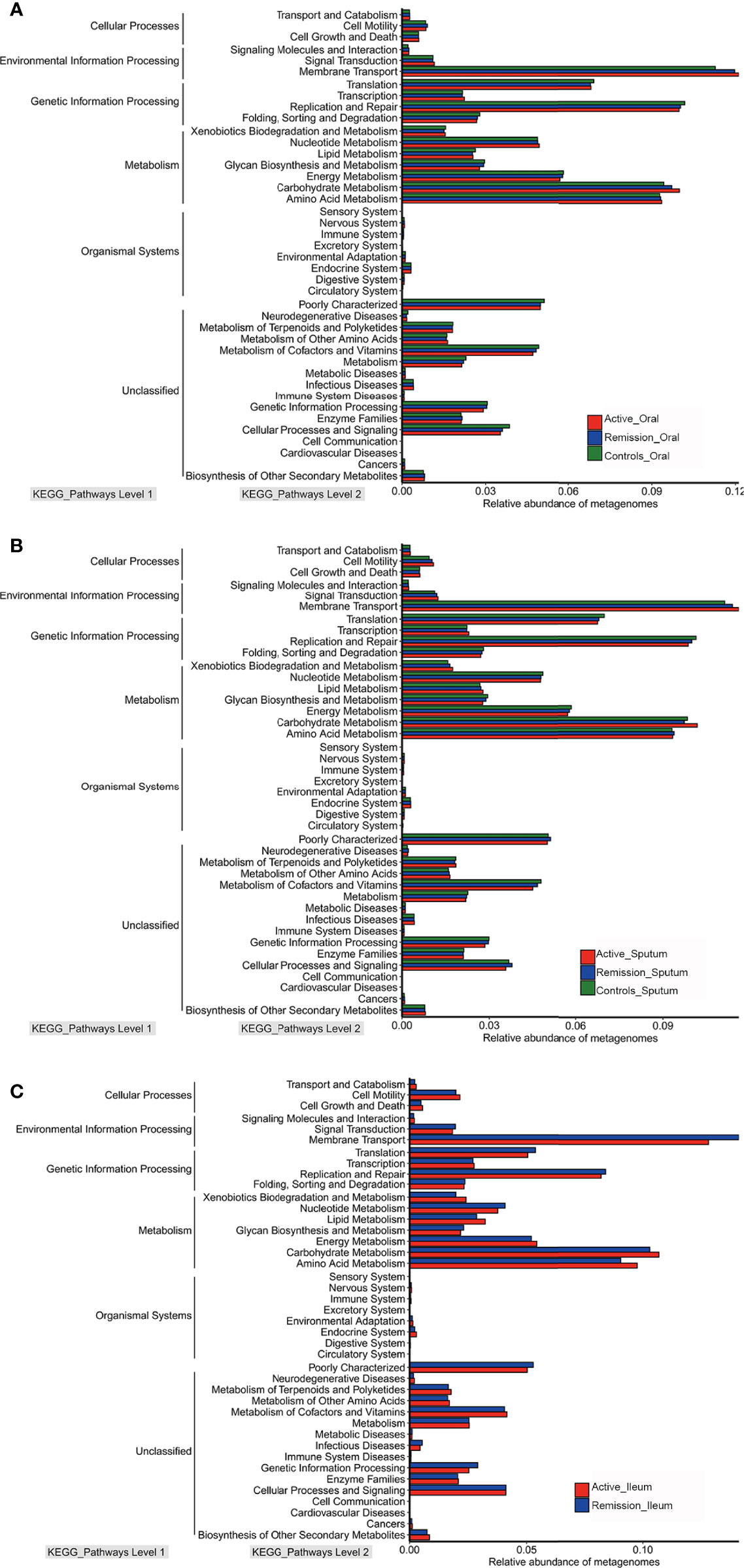
Figure 8 Relative abundances of metagenomes of different KEGG pathways level in microbiota of patients with CD. (A) Relative abundances of metagenomes of different KEGG pathways level in oral microbiota of patients with CD. (B) Relative abundances of metagenomes of different KEGG pathways level in sputum microbiota of patients with CD. (C) Relative abundances of metagenomes of different KEGG pathways level in ileal microbiota of patients with CD. Red color represents samples from active stage, blue indicates remission stage, while green color indicates healthy controls, respectively.
The oral cavity is the entrance to the digestive tract, which is colonized by one of the most diverse microbiotas of the human body. The human oral microbiome database (HOMD) had recorded about 700 prokaryote species in the human oral cavity, some of which are closely related to human health and disease (Dewhirst et al., 2010). Moreover, growing evidence revealed that some gastrointestinal tract diseases were correlated with dysbiosis of the lung microecology, leading to crosstalk between these organs, termed the gut–lung axis (Budden et al., 2017; Budden et al., 2019). An earlier study showed that more than 48% of adult IBD patients had different degrees of abnormal lung function (Ji et al., 2014), and up to 71% of children and adolescents with CD had abnormal bronchial hyperreactivity (Mansi et al., 2000). Thus, there are emerging evidence that indicated the potential associations between IBD and the lungs (Mateer et al., 2015; Cozzi et al., 2018).
The present study identified the stage-specific alterations of microbiota architecture in the oral cavity, sputum, and ileum of patients with CD. The oral microbiota had a statistically significant decrease in alpha diversity compared to healthy controls, which aligned with Xun’s report (Xun et al., 2018). In addition, we found that the oral microbiota of healthy controls clustered with that from the remission stage of CD, whereas most of the oral microbiota from the active stage of CD clustered separately. A previous study by Zhang et al. (2020) described similar findings. Regarding the composition of oral microbiota of patients with CD, the relative abundances of Veillonella, Actinomyces, Megasphaera, and Atopobium were highest in the active stage and lowest in healthy controls. On the contrary, the relative abundances of Fusobacterium, Haemophilus, and Porphyromonas were lowest in the active stage and highest in healthy controls. Said et al. (2014) also reported the depletion of Haemophilus in the salivary microbiota of patients with CD. These evidence demonstrated the complicated microbiota signatures in the oral microbiota of patients with CD.
Furthermore, several studies had confirmed the enteric pathogenic potential of Campylobacter in the oral cavity (Man, 2011; Ismail et al., 2012; Kirk et al., 2018). Nielsen et al. (2011) showed that oral Campylobacter induced impairment of the intestinal barrier function by inducing apoptosis in HT-29/B6 intestinal epithelial cells, providing a plausible explanation to the conclusion reached in this study. Thus, over-growth and colonization of Campylobacter in the gastrointestinal tract may contribute to the destruction of cell structures, impairment of the intestinal absorption function, and inflammatory disorders in CD (Qi et al., 2021).
The present study also investigates the characterization of sputum microbiota during the active and remission stages of patients with CD, as this relationship remains understudied. Comparing the structural changes and functional characterization of sputum microbiota of patients with CD, we found that changes in the sputum were similar to those in the oral cavity. Furthermore, the concept of the gut–lung axis is gradually being accepted. The fragments and metabolites derived from intestinal microbiota are transported to the lung and modulated the lung immune response by translocation across the intestinal barrier and the mesenteric lymphatic system (Piersigilli et al., 2020). For instance, intestinal microbiota-mediated production of SCFAs could exert anti-inflammatory effects by modulating immune cell migration and suppressing the activation of nuclear factor kappa B (NF-κB) pathways in the lung (Anand and Mande, 2018). In addition, SCFAs were found to stimulate Treg cells to prevent airway inflammation by inhibiting histone deacetylases (HDACs) or activating acetate and propionate (Thorburn et al., 2015).
Moreover, SCFAs could enhance the generation of dendritic cell precursors, preventing allergic inflammation of the airway (Trompette et al., 2014). Similarly, the dysbiosis of lung microbiota could also affect the gut microbiota (Barcik et al., 2020; Yang et al., 2020). Clinical evidence showed that influenza infection of the lung induced gut microbiota dysbiosis, such as increased Enterobacteriaceae and decreased lactococci and lactobacilli, in the intestine (Looft and Allen, 2012). This research may be an opportunity to further explore the relationship between the microbiota in the respiratory tract and CD and provide new options for the clinical treatment of CD.
Until now, the fecal microbiota of patients with CD has been extensively studied. However, the use of fecal samples alone in most of these studies indicates that the findings were specific to the colonic content (Zmora et al., 2018). Recently, Ruigrok et al. (2021) revealed the composition and metabolic potential of the human small intestinal microbiota within the context of IBD. However, no studies revealed the characterization of small intestinal microbiota during different stages of CD, indicating another innovative aspect of our research. Hattori et al. (2020) reported that the abundances of Streptococcus and Clostridium were higher in the mucosal healing group, which could predict mucosal healing in the small intestine in patients with CD. Our results revealed that these microbiotas were markedly enriched in the remission stage of CD, which further confirmed the above conclusion. Ruigrok et al. (2021) reported Veillonella, Streptococcus, and Actinomyces were abundant in the small intestine of patients with CD. This finding confirmed that the relative abundances of these microbiotas were higher in the remission stage of CD. Several studies had revealed that the colonic microbiota in patients with CD and showed a decrease in probiotics but an increase in the pathogenic microbiota (Dong et al., 2019). Our study demonstrated that the relative abundances of Lactococcus and Lactobacillus were higher in the remission stage of CD than in the active stage, whereas the contrary was observed for Fusobacterium. This suggested that although the structure of the colon was different from that of the small intestine, the abundance of some microbiota may be relatively similar.
While the results of our study offer a detailed insight into the microbiota in multi-ecological sites of patients with CD, some limitations need to be addressed. Since fecal samples from the ileal stoma were collected during the operation, the patients were administrated with antibiotics routinely since the beginning of surgery in the clinic. Currently, we could not estimate the potential influence of antibiotics on the microbiota structure within such a short time, as no strong evidence elucidates these issues so far. Although the influence of antibiotics on results could not be completely avoided, our experimental protocol has minimized the influence. In addition, most previous studies collected samples from healthy controls during routine endoscopies, from patients undergoing intestinal resections or from sudden death individuals (El Aidy et al., 2015). Therefore, it is challenging to collect microbial ileal samples from healthy controls without potential damage. Regarding the collection of sputum samples, the oral cavity is the only way to obtain sputum samples, which inevitably leads to a small amount of oral microbiota mixed in this process. Weiser et al. (2022) revealed that induced sputum captured a microbiota signature representative of the lower airway in 80% of cases and was a direct, non-invasive intervention. Therefore, induced sputum is an essential and practical approach to studying the respiratory microbiota signatures. Perhaps, the contamination can be minimized if a more convenient collection method becomes available, and bioinformatics techniques could also help eliminate the background contamination in the future. In addition, the conclusions of our analyses are also limited by the small sample size. More samples are needed to confirm the changes in some specific microbiota and their roles in the onset of CD.
In conclusion, our study provided insights into the microbiota composition and their functional characterization in the oral cavity, sputum, and ileum of patients with CD compared to healthy controls. Although we only identified the specific microbiota of patients with CD during different disease stages, not referred to the causality, we still displayed the novel microbiota signatures of various sites and their potential relationships with CD. We believe that multi-omics research, including metagenomics, metabolomics, proteomics, and experimental exploration, will provide a more comprehensive understanding of the roles of microbiota in CD, especially on the causality between microbiota and CD, which will improve the treatment strategy for CD in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA773617.
The studies involving human participants were reviewed and approved by the Ethics Committee of Shanghai Tenth People’s Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was not obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
KX experimental design, implementation research, analysis and interpretation of data, and drafting of papers. RG experimental design, implementation research, analysis and interpretation of data, and revision of papers. XW, JS, JW, and TW experimental design, analysis and interpretation of data, and revision of papers. JF, LY, and CC experimental design, revision of papers, and funding support. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.864944/full#supplementary-material
Supplementary Figure 1 | Structure analysis of the ileal microbiota in patients with CD. (A, B) Chao diversity index of phylum level and genus level in the ileal samples. (C, D) Shannon diversity index of phylum level and genus level in the ileal samples. (E, F) Venn diagram indicating the overlap of OTUs in the categories on the phylum level and genus level, respectively.
Supplementary Figure 2 | COG functional classifications of microbiota in patients with CD. (A–C) COG functional classifications of microbiota in the oral cavity, sputum and ileum, respectively.
Anand, S., Mande, S. S. (2018). Diet, Microbiota and Gut-Lung Connection. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02147
Barcik, W., Boutin, R. C. T., Sokolowska, M., Finlay, B. B. (2020). The Role of Lung and Gut Microbiota in the Pathology of Asthma. Immunity. 52 (2), 241–255. doi: 10.1016/j.immuni.2020.01.007
Bernstein, C. N., Forbes, J. D. (2017). Gut Microbiome in Inflammatory Bowel Disease and Other Chronic Immune-Mediated Inflammatory Diseases. Inflammation Intest Dis. 2 (2), 116–123. doi: 10.1159/000481401
Budden, K. F., Gellatly, S. L., Wood, D. L., Cooper, M. A., Morrison, M., Hugenholtz, P., et al. (2017). Emerging Pathogenic Links Between Microbiota and the Gut-Lung Axis. Nat. Rev. Microbiol. 15 (1), 55–63. doi: 10.1038/nrmicro.2016.142
Budden, K. F., Shukla, S. D., Rehman, S. F., Bowerman, K. L., Keely, S., Hugenholtz, P., et al. (2019). Functional Effects of the Microbiota in Chronic Respiratory Disease. Lancet Respir. Med. 7 (10), 907–920. doi: 10.1016/S2213-2600(18)30510-1
Cozzi, D., Moroni, C., Addeo, G., Danti, G., Lanzetta, M. M., Cavigli, E., et al. (2018). Radiological Patterns of Lung Involvement in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 1–10. doi: 10.1155/2018/5697846
Danve, A. (2019). Thoracic Manifestations of Ankylosing Spondylitis, Inflammatory Bowel Disease, and Relapsing Polychondritis. Clin. Chest Med. 40 (3), 599–608. doi: 10.1016/j.ccm.2019.05.006
Dewhirst, F. E., Chen, T., Izard, J., Paster, B. J., Tanner, A. C., Yu, W. H., et al. (2010). The Human Oral Microbiome. J. Bacteriol. 192 (19), 5002–5017. doi: 10.1128/JB.00542-10
Ding, N. S., McDonald, J. A. K., Perdones–Montero, A., Rees, D. N., Adegbola, S. O., Misra, R., et al (2020). Metabonomics and the Gut Microbiome Associated With Primary Response to Anti-TNF Therapy in Crohn's Disease. J. Crohns. Colitis. 14 (8), 1090–1102. doi: 10.1093/ecco-jcc/jjaa039
Dong, L. N., Wang, M., Guo, J., Wang, J. P. (2019). Role of Intestinal Microbiota and Metabolites in Inflammatory Bowel Disease. Chin. Med. J. (Engl) 132 (13), 1610–1614. doi: 10.1097/CM9.0000000000000290
Dovrolis, N., Michalopoulos, G., Theodoropoulos, G. E., Arvanitidis, K., Kolios, G., Sechi, L. A., et al. (2020). The Interplay Between Mucosal Microbiota Composition and Host Gene-Expression is Linked With Infliximab Response in Inflammatory Bowel Diseases. Microorganisms 8 (3), 438. doi: 10.3390/microorganisms8030438
El Aidy, S., van den Bogert, B., Kleerebezem, M. (2015). The Small Intestine Microbiota, Nutritional Modulation and Relevance for Health. Curr. Opin. Biotechnol. 32, 14–20. doi: 10.1016/j.copbio.2014.09.005
Fesler, Z., Mitova, E., Brubaker, P. L. (2020). GLP-2, EGF, and the Intestinal Epithelial IGF-1 Receptor Interactions in the Regulation of Crypt Cell Proliferation. Endocrinology 161 (4), 1–14. doi: 10.1210/endocr/bqaa040
Gao, R., Zhu, Y., Kong, C., Xia, K., Li, H., Zhu, Y., et al. (2021). Alterations, Interactions, and Diagnostic Potential of Gut Bacteria and Viruses in Colorectal Cancer. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.657867
Hattori, S., Nakamura, M., Yamamura, T., Maeda, K., Sawada, T., Mizutani, Y., et al. (2020). The Microbiome Can Predict Mucosal Healing in Small Intestine in Patients With Crohn’s Disease. J. Gastroenterol. 55 (12), 1138–1149. doi: 10.1007/s00535-020-01728-1
Ismail, Y., Mahendran, V., Octavia, S., Day, A. S., Riordan, S. M., Grimm, M. C., et al. (2012). Investigation of the Enteric Pathogenic Potential of Oral Campylobacter Concisus Strains Isolated From Patients With Inflammatory Bowel Disease. PloS One 7 (5), e38217. doi: 10.1371/journal.pone.0038217
Ji, X., Wang, L., Lu, D. (2014). Pulmonary Manifestations of Inflammatory Bowel Disease. World J. Gastroenterol. 20 (37), 13501–13511. doi: 10.1016/S1579-2129(06)60404-7
Katsanos, K. H., Torres, J., Roda, G., Brygo, A., Delaporte, E., Colombel, J. F. (2015). Review Article: non-Malignant Oral Manifestations in Inflammatory Bowel Diseases. Aliment Pharmacol. Ther. 42 (1), 40–60. doi: 10.1111/apt.13217
Kirk, K. F., Méric, G., Nielsen, H. L., Pascoe, B., Sheppard, S. K., Thorlacius-Ussing, O., et al. (2018). Molecular Epidemiology and Comparative Genomics of Campylobacter Concisus Strains From Saliva, Faeces and Gut Mucosal Biopsies in Inflammatory Bowel Disease. Sci. Rep. 8 (1), 1902. doi: 10.1038/s41598-018-20135-4
Kondrup, J., Rasmussen, H. H., Hamberg, O., Stanga, Z. (2003). Nutritional Risk Screening (NRS 2002): A New Method Based on an Analysis of Controlled Clinical Trials. Clin. Nutr. 22 (3), 321–336. doi: 10.1016/s0261-5614(02)00214-5
Kong, C., Yan, X., Liu, Y., Huang, L., Zhu, Y., He, J., et al. (2021). Ketogenic Diet Alleviates Colitis by Reduction of Colonic Group 3 Innate Lymphoid Cells Through Altering Gut Microbiome. Signal Transduct Target Ther. 6 (1), 154. doi: 10.1038/s41392-021-00549-9
Liu, W., Zhang, R., Shu, R., Yu, J., Li, H., Long, H., et al. (2020). Study of the Relationship Between Microbiome and Colorectal Cancer Susceptibility Using 16srrna Sequencing. BioMed. Res. Int. 2020, 1–17. doi: 10.1155/2020/7828392
Lloyd-Price, J., Arze, C., Ananthakrishnan, A. N., Schirmer, M., Avila-Pacheco, J., Poon, T. W., et al. (2019). Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature. 569 (7758), 655–662. doi: 10.1038/s41586-019-1237-9
Looft, T., Allen, H. K. (2012). Collateral Effects of Antibiotics on Mammalian Gut Microbiomes. Gut Microbes 3 (5), 463–467. doi: 10.4161/gmic.21288
Man, S. M. (2011). The Clinical Importance of Emerging Campylobacter Species. Nat. Rev. Gastroenterol. Hepatol. 8 (12), 669–685. doi: 10.1038/nrgastro.2011.191
Mansi, A., Cucchiara, S., Greco, L., Sarnelli, P., Pisanti, C., Franco, M. T., et al. (2000). Bronchial Hyperresponsiveness in Children and Adolescents With Crohn's Disease. Am. J. Respir. Crit. Care Med. 161 (3 Pt 1), 1051–1054. doi: 10.1164/ajrccm.161.3.9906013
Mateer, S. W., Maltby, S., Marks, E., Foster, P. S., Horvat, J. C., Hansbro, P. M., et al. (2015). Potential Mechanisms Regulating Pulmonary Pathology in Inflammatory Bowel Disease. J. Leukoc. Biol. 98 (5), 727–737. doi: 10.1189/jlb.3RU1114-563R
Nielsen, H. L., Nielsen, H., Ejlertsen, T., Engberg, J., Günzel, D., Zeitz, M., et al. (2011). Oral and Fecal Campylobacter Concisus Strains Perturb Barrier Function by Apoptosis Induction in HT-29/B6 Intestinal Epithelial Cells. PloS One 6 (8), e23858. doi: 10.1371/journal.pone.0023858
Olaisen, M., Flatberg, A., Granlund, A. V. B., Martinsen, T. C., Sandvik, A. K., Fossmark, R. (2021). Bacterial Mucosa-Associated Microbiome in Inflamed and Proximal Noninflamed Ileum of Patients With Crohn's Disease. Inflammation Bowel Dis. 27 (1), 12–24. doi: 10.1093/ibd/izaa107
Piersigilli, F., Van Grambezen, B., Hocq, C., Danhaive, O. (2020). Nutrients and Microbiota in Lung Diseases of Prematurity: The Placenta-Gut-Lung Triangle. Nutrients. 12 (2), 469. doi: 10.3390/nu12020469
Qi, Y., Zang, S., Wei, J., Yu, H., Yang, Z., Wu, H., et al. (2021). High-Throughput Sequencing Provides Insights Into Oral Microbiota Dysbiosis in Association With Inflammatory Bowel Disease. Genomics. 113 (1 Pt 2), 664–676. doi: 10.1016/j.ygeno.2020.09.063
Ramakrishna, C., Kujawski, M., Chu, H., Li, L., Mazmanian, S. K., Cantin, E. M. (2019). Bacteroides Fragilis Polysaccharide A Induces IL-10 Secreting B and T Cells That Prevent Viral Encephalitis. Nat. Commun. 10 (1), 2153. doi: 10.1038/s41467-019-09884-6
Rautava, J., Pinnell, L. J., Vong, L., Akseer, N., Assa, A., Sherman, P. M. (2015). Oral Microbiome Composition Changes in Mouse Models of Colitis. J. Gastroenterol. Hepatol. 30 (3), 521–527. doi: 10.1111/jgh.12713
Read, E., Curtis, M. A., Neves, J. F. (2021). The Role of Oral Bacteria in Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 18 (10), 731–742. doi: 10.1038/s41575-021-00488-4
Ruigrok, R. A. A. A., Collij, V., Sureda, P., Klaassen, M. A. Y., Bolte, L. A., Jansen, B. H., et al. (2021). The Composition and Metabolic Potential of the Human Small Intestinal Microbiota Within the Context of Inflammatory Bowel Disease. J. Crohns Colitis. 15 (8), 1326–1338. doi: 10.1093/ecco-jcc/jjab020
Said, H. S., Suda, W., Nakagome, S., Chinen, H., Oshima, K., Kim, S., et al. (2014). Dysbiosis of Salivary Microbiota in Inflammatory Bowel Disease and Its Association With Oral Immunological Biomarkers. DNA Res. 21 (1), 15–25. doi: 10.1093/dnares/dst037
Sokol, H., Brot, L., Stefanescu, C., Auzolle, C., Barnich, N., Buisson, A., et al. (2020). Prominence of Ileal Mucosa-Associated Microbiota to Predict Postoperative Endoscopic Recurrence in Crohn's Disease. Gut. 69 (3), 462–472. doi: 10.1136/gutjnl-2019-318719
Sokol, H., Leducq, V., Aschard, H., Pham, H. P., Jegou, S., Landman, C., et al. (2017). Fungal Microbiota Dysbiosis in IBD. Gut. 66 (6), 1039–1048. doi: 10.1136/gutjnl-2015-310746
Thorburn, A. N., Mckenzie, C. I., Shen, S., Stanley, D., Macia, L., Mason, L. J., et al. (2015). Evidence That Asthma is a Developmental Origin Disease Influenced by Maternal Diet and Bacterial Metabolites. Nat. Commun. 6, 7320. doi: 10.1038/ncomms8320
Trompette, A., Gollwitzer, E. S., Yadava, K., Sichelstiel, A. K., Sprenger, N., Ngom-Bru, C., et al. (2014). Gut Microbiota Metabolism of Dietary Fiber Influences Allergic Airway Disease and Hematopoiesis. Nat. Med. 20 (2), 159–166. doi: 10.1038/nm.3444
Ubags, N. D. J., Marsland, B. J. (2017). Mechanistic Insight Into the Function of the Microbiome in Lung Diseases. Eur. Respir. J. 50 (3), 1602467. doi: 10.1183/13993003.02467-2016
Weiser, R., Oakley, J., Ronchetti, K., Tame, J. D., Hoehn, S., Jurkowski, T. P., et al. (2022). The Lung Microbiota in Children With Cystic Fibrosis Captured by Induced Sputum Sampling. J. Cyst Fibros. 7, 54. doi: 10.1016/j.jcf.2022.01.006
Xun, Z., Zhang, Q., Xu, T., Chen, N., Chen, F. (2018). Dysbiosis and Ecotypes of the Salivary Microbiome Associated With Inflammatory Bowel Diseases and the Assistance in Diagnosis of Diseases Using Oral Bacterial Profiles. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01136
Yang, D., Xing, Y., Song, X., Qian, Y. (2020). The Impact of Lung Microbiota Dysbiosis on Inflammation. Immunology. 159 (2), 156–166. doi: 10.1111/imm.13139
Yoshida, E. M. (1999). The Crohn's Disease Activity Index, its Derivatives and the Inflammatory Bowel Disease Questionnaire: A Review of Instruments to Assess Crohn's Disease. Can. J. Gastroenterol. 13 (1), 65–73. doi: 10.1155/1999/506915
Zhang, T., Kayani, M. U. R., Hong, L., Zhang, C., Zhong, J., Wang, Z., et al. (2020). Dynamics of the Salivary Microbiome During Different Phases of Crohn's Disease. Front. Cell Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.544704
Keywords: Crohn’s disease, microbiota, oral cavity, sputum, ileum, 16S rRNA gene sequence
Citation: Xia K, Gao R, Wu X, Sun J, Wan J, Wu T, Fichna J, Yin L and Chen C (2022) Characterization of Specific Signatures of the Oral Cavity, Sputum, and Ileum Microbiota in Patients With Crohn’s Disease. Front. Cell. Infect. Microbiol. 12:864944. doi: 10.3389/fcimb.2022.864944
Received: 29 January 2022; Accepted: 17 March 2022;
Published: 13 April 2022.
Edited by:
Manuela Neuman, University of Toronto, CanadaReviewed by:
Traci L Testerman, University of South Carolina, United StatesCopyright © 2022 Xia, Gao, Wu, Sun, Wan, Wu, Fichna, Yin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Xia, OTg3MjM5OTQ1QHFxLmNvbQ==; Lu Yin, eWlubHVtYXN0ZXIwMTA1QDEyNi5jb20=; Chunqiu Chen, Y2hlbmNodW5xaXU2QDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.