 Yuxin Guo
Yuxin Guo Chenxi Zeng
Chenxi Zeng Chenjie Ma1
Chenjie Ma1 Shaowei Zhai
Shaowei Zhai Mao Lin
Mao Lin- 1Fisheries College, Engineering Research Center of the Modern Technology for Eel Industry, Ministry of Education, Jimei University, Xiamen, Fujian, China
- 2Key Laboratory of Healthy Mariculture for the East China Sea, Ministry of Agriculture and Rural Affairs, Xiamen, Fujian, China
In this paper, the whole genome of the multidrug-resistant Aeromonas hydrophila MX16A was comprehensively analyzed and compared after sequencing by PacBio RS II. To shed light on the drug resistance mechanism of A. hydrophila MX16A, a Kirby-Bauer disk diffusion method was used to assess the phenotypic drug susceptibility. Importantly, resistance against β-lactam, sulfonamides, rifamycins, macrolides, tetracyclines and chloramphenicols was largely consistent with the prediction analysis results of drug resistance genes in the CARD database. The varied types of resistance genes identified from A. hydrophila MX16A revealed multiple resistance mechanisms, including enzyme inactivation, gene mutation and active effusion. The publicly available complete genomes of 35 Aeromonas hydrophila strains on NCBI, including MX16A, were downloaded for genomic comparison and analysis. The analysis of 33 genomes with ANI greater than 95% showed that the pan-genome consisted of 9556 genes, and the core genes converged to 3485 genes. In summary, the obtained results showed that A. hydrophila exhibited a great genomic diversity as well as diverse metabolic function and it is believed that frequent exchanges between strains lead to the horizontal transfer of drug resistance genes.
Introduction
Occurrences of multi-drug resistance (MDR) have become increasingly frequent in recent decades, complicating the treatment of certain diseases caused by microorganisms in humans (Williams and Bardsley, 1999; Thomas et al., 2016; Boucher et al., 2017). Multidrug-resistant bacteria (MDRB) could render a large number of antibiotic drugs ineffective, leading to severe public health concerns and affecting virtually all population groups around the world (Lautenbach and Fishman, 1999).The antibiotic-rich environment promotes horizontal gene transfer between bacteria, increasing the drug resistance of bacteria and causing a great risk to the public (Skwor et al., 2020).Furthermore, with constant evolution of MDRB, food production and safety may also be affected.
Aeromonas spp., a rod-shape and facultative anaerobic bacteria species, is widely distributed in natural environments, particularly in aquatic media (Chauret et al., 2001; Scoaris et al., 2008; Araujo et al., 1991; Rafei et al., 2018). The mesophilic species exhibits optimal growth at 35–37 °C and is involved in a variety of human infections whereas sychrophilic species can be grown at lower temperatures of 22–25 °C and frequently result in nonmotile diseases in fish (Janda and Abbott, 2010). This species of gram-negative bacteria, A. hydrophila being one representative, is commonly associated with a variety of diseases including septicaemia, gastroenteritis, and wound infections frequently affecting aquatic animals, terrestrial animals, and humans (Adamski et al., 2006; Parker and Shaw, 2011; Igbinosa et al., 2012; Rutteman, 2017).
From 1977 to 2016, three generations of various types of sequencing technologies have been developed. The second and third generation sequencing technologies referred commonly to as next generation sequencing technology, has evolved significantly with increase in sequencing speed, decrease in sequencing cost, since its inception in 2004. Currently, the third-generation sequencing platforms include Helicos™ Genetic Analysis System by SeqLL, SMRT Sequencing by Pacific Biosciences, Nanopore sequencing by Oxford Nanopore, to name a few (Ambardar et al., 2016). PacBio’s DNA sequencing has been used for detailed genomic investigations of bacterial isolates. Whole genome sequencing of bacteria has become an important tool for understanding and analyzing bacterial evolution. The complete understanding of the antibiotic resistance mechanisms of MDR A. hydrophila is crucial for the further development of therapeutic agents. We therefore conducted PacBio’s single molecule real time (SMRT™) DNA sequencing and the preliminary analysis of the MDRB strain A. hydrophila MX16A. Our results highlight resistance-related genes to aid in the understanding of genetic mechanisms of drug resistance of A. hydrophila and the preliminary prediction of virulence factors present in the genome. We further conducted a comparative genomic analysis of MX16A and reference A. hydrophila strains to generate the pan-core genome in order to understand the exclusive and shared genes and characteristics of the species of A. hydrophila. The results obtained herein will mine and develop the value of the existing reference genomes, and gain complete variation information of the entire species.
Materials and methods
Bacterial strain and antimicrobial susceptibility test
A. hydrophila strain MX16A was isolated from Jiulong River, Fujian, China (Lin et al., 2015), and cultured with Luria-Bertani broth (Hopebio, Qingdao, China) at 30 °C in a shaking incubator for 24 h. In previous experiments, MX16A was found to be insensitive to a variety of antibiotics, and contain various resistance genes and virulence genes detected by PCR.The toxicity tests showed that the median lethal concentration (LD50) of A. hydrophila MX16A to zebrafish was 1.6×106 CFU/fish, indicating that MX16A had pathogenicity to aquatic animals. Antimicrobial susceptibility testing was performed using the Kirby-Bauer disk diffusion method (OXOID, UK). All results were determined by the Clinical and Laboratory Standards Institude (CLSI) guidelines (M100-S23).
DNA extraction and 16S rRNA gene sequencing
Bacterial genomic DNA was extracted using the TIANamp Bacteria DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. The strain identity was confirmed by 16S rRNA gene sequencing using specific primers (27F: 5’-AGAGTTTGATCCTGGCTCA G-3’; 1492R: 5’-GGTTACCTTGTTACGACTT-3’). The 16S rRNA sequence results from A. hydrophila MX16A were deposited into the GenBank Database with accession number KJ806394.1 (https://www.ncbi.nlm.nih.gov/search/all/?term=KJ806394.1).
Library preparation and whole genome sequencing
High-quality DNA was sent to Majorbio Company (Shanghai, China) for sequencing by PacBio RS II, a single molecule real time (SMRT™) DNA sequencing technology (Pacific Biosciences, Menlo Park, USA). Briefly, A. hydrophila MX16A genome DNA was first randomly sheared into 8-10 kb fragments. After repairing the damage and ends of the fragmented DNA, some appropriately sized double-standard DNA fragments called SMRTbell™ (structurally linear and topologically circular), capped by hairpin loops at both ends of the fragmented pieces. Next, a primer and a polymerase are annealed to the adapter, and the annealed templates were bound to DNA polymerase located at the bottom of zero-mode waveguides (ZMWs) in SMRT™ Cell (single molecule real time reaction pore, one SMRT™ Cell included 150 thousand ZMWs) (Ardui et al., 2018). Finally, the MX16A plate was set up for sequencing. This PacBio TGS sequencing procedure has been previously described and is shown in Figure 1.
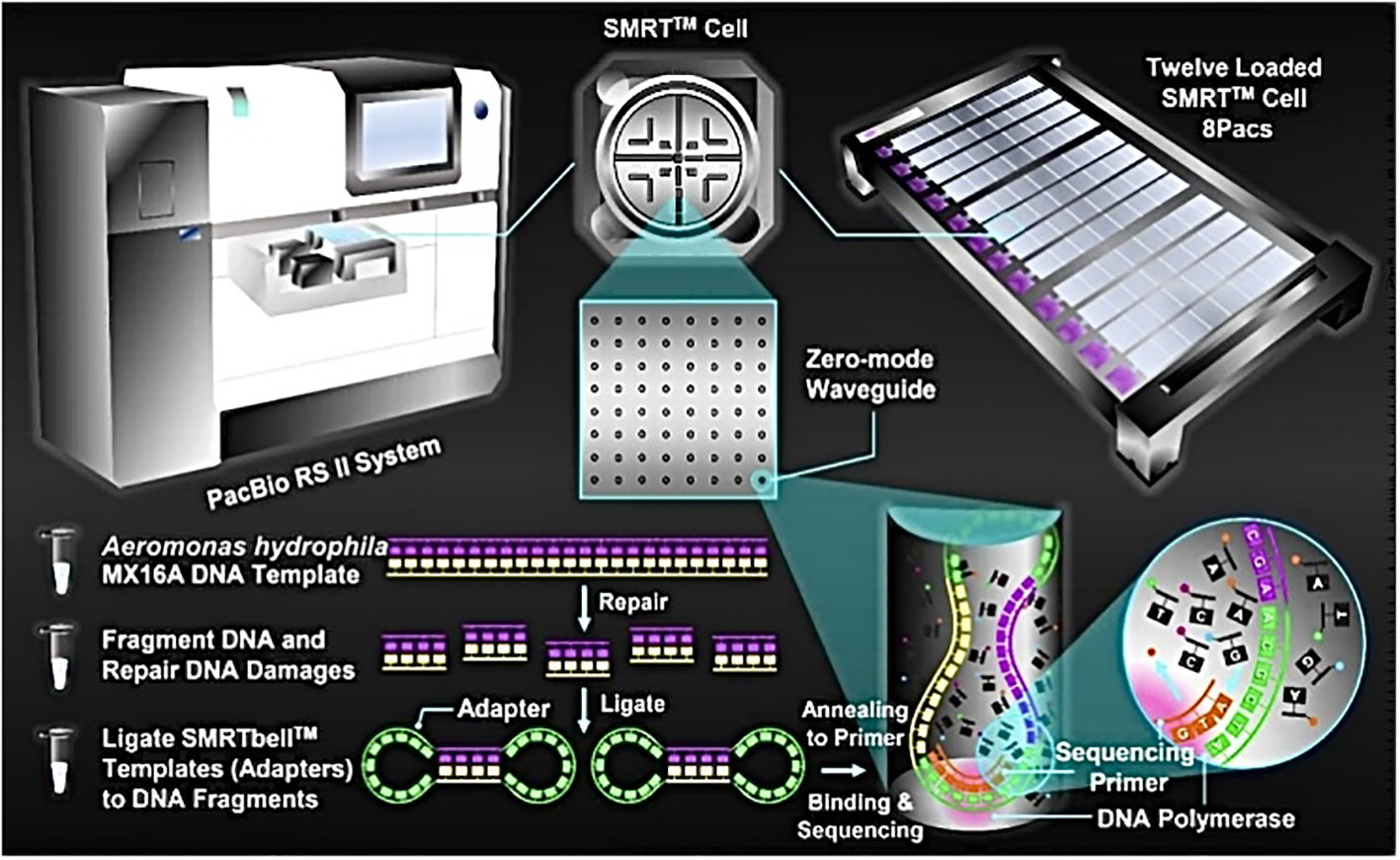
Figure 1 Schematic representation of PacBio’s DNA sequencing. A. hydrophila MX16A DNA sample was extracted by using a TIANamp Bacteria DNA Kit (Tiangen Biotech, Beijing, China) following the manufacturer’s instructions.
Gene prediction, rapid annotation, and data analysis
Gene prediction was carried out using Glimmer v3.02 while rRNA- and tRNA-encoding of genes was predicted using Barrnap v0.4.2 and tRNAscan-SE v1.3.1. Gene annotation and statistics were performed using blastp (BLAST 2.2.28+) and compared to the Non-Redundant (Nr) Protein Sequence Database (https://blast.ncbi.nlm.nih.gov/), Genes Database (https://www.genome.jp/kegg/genes.html/), String Database (http://string-db.org/), and Gene Ontology (GO) Database (http://www.gene-ontology.org/). Analysis of antibiotic-related genes of the A. hydrophila MX16A genome was performed using the Comprehensive Antibiotic Resistance Database (CARD, https://card.mcmaster.ca/). A circular map based on the bacterial chromosomal genome was generated using Circos v0.64 in Perl environment.
Genbank accession number of MX16A’s genome
The genome sequence of A. hydrophila strain MX16A has been deposited in GenBank and may be found using accession number CP018201.1.
Genome data
The genome of A. hydrophila was obtained from the publicly available genome database in the National Center for Biotechnology Information (NCBI). A total of 35 genomic sequences were analyzed in a comparative fashion throughout this study. The sequences were statistically classified by the host and source, including human, fish, or other hosts isolated from China, Asia (except China), South America, North America, Africa or Europe, respectively.
Determination of average nucleotide identity
To avoid any possible genomic misidentification, all genomes were subjected to ANI check using the web based ANI calculator available at http://enve-omics.ce.gatech.edu/ani/index. In the ANI calculation, the strain A. hydrophila MX16A was used as a standard to analyze the genetic relationship between the A. hydrophila MX16A genome and the downloaded reference genome.
Analysis of pan-genome and core-genome
Generated faa files of 33 strains were used from the Prokaryotic Dynamic Programming Genefinding Algorithm (Prodigal). These served as input files for the Bacterial Pan Genome Analysis tool (BPGA) to perform a core-pan-genome analysis. USEARCH clustering algorithm was used as parameters to run BPGA. The identity value was 0.5 and the number of combinations was 500.
Prediction of drug resistance genes and virulence genes
Prediction of drug resistance genes and virulence genes was re-performed for 35 genomic sequences. Gff files of 35 strains were generated by Prokka. Drug resistance genes were predicted by AMRfinder (https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/), and virulence factors were identified based on the core data set of the VFDB (http://www.mgc.ac.cn-/VFs/). The identity threshold used was 90% and the coverage threshold was 60%.
Results
Strain identification and antibiotic susceptibility
A total of 16S rRNA gene sequences clearly identified the isolate as A. hydrophila. Antimicrobial susceptibility testing was carried out based on the Kirby-Bauer method (Table 1). A. hydrophila MX16A was proved to be resistant to most antibiotics tested, including cefotaxime, amoxicilli, trimethoprim-sulfamethoxazole, rifampicin, erythromycin, tetracycline, streptomycin and chloramphenicol which represent β-lactams, macrolides, rifamycins, tetracyclines, sulfonamides, aminoglycosides, and chloramphenicols respectively. Based on the drug sensitivity test, it can be concluded that A. hydrophila MX16A was indeed a multidrug-resistant bacterium.
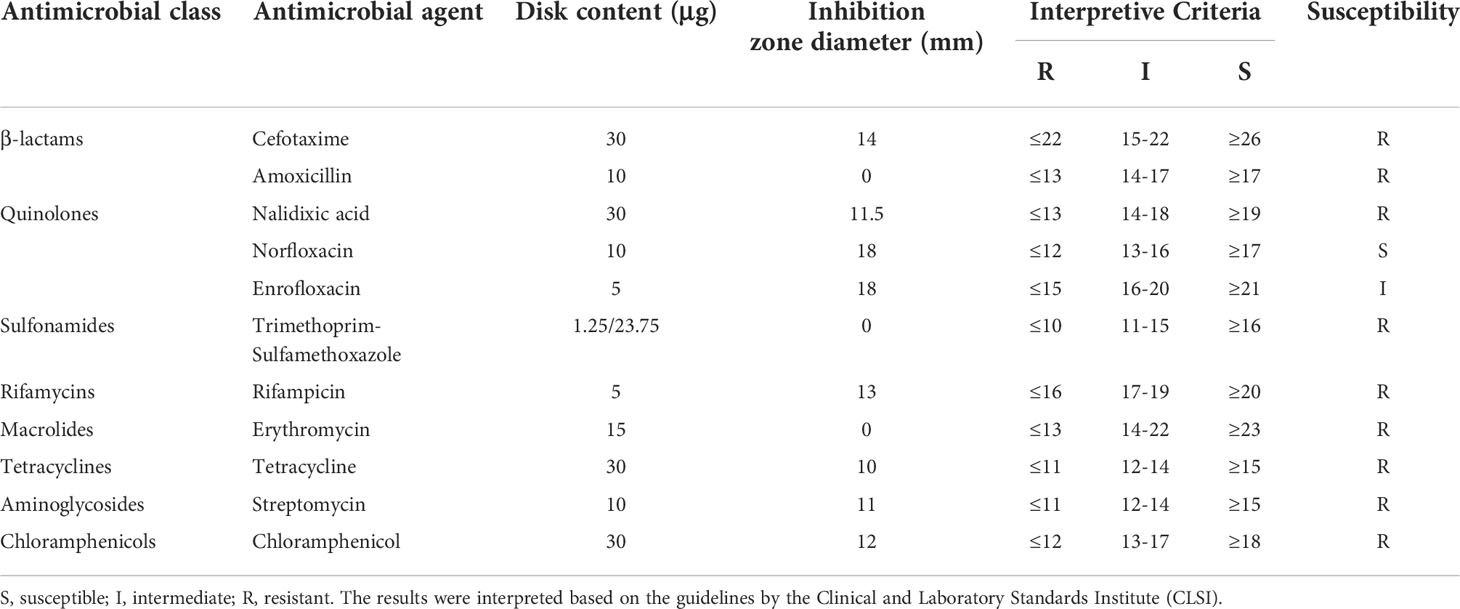
Table 1 Antimicrobial susceptibility profile of A. hydrophila MX16A.
Whole genome analysis
The complete genome of A. hydrophila MX16A consisted of a single circular 4,168,374 bp chromosome with 4418 genes and no plasmid. The general genome features included the genomic GC content, intergeneric region length, and the number of RNA genes (Table 2). The circular genome map was consistent with the characteristics of bacterial genome (Figure 2). Furthermore, we performed a GO analysis to determine the functional classification of differentially expressed genes (DEGs) in the regulation process of MX16A. All DGEs could be combined into three main GO categories, namely biological process, cellular component, and molecular function, along with 43 subcategories as shown in Figure 3.

Table 2 General genome features of A. hydrophila MX16A.
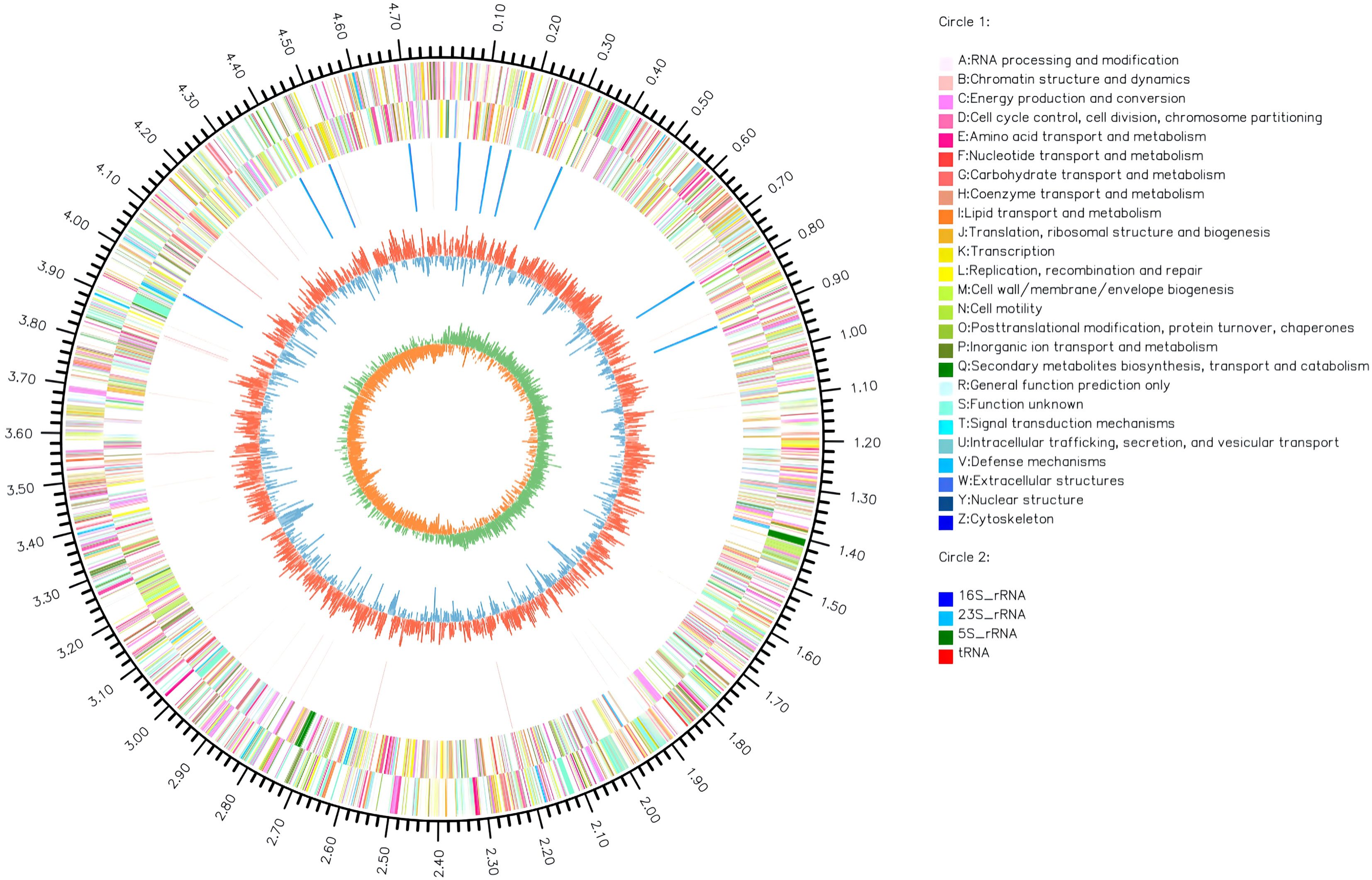
Figure 2 Circos circular map of A. hydrophila MX16A genome. The outer circle indicates the size of the genome, every scale represents 0.5 Mb; The second and third circles represent the CDS from positive-sense and negative-sense strand of genome sequences, different colors represent various functions of COG; The fourth circle represents tRNA and rRNA; The fifth circle shows the (G+C) content, the outer red portion indicates that the GC content of the region is higher than the average GC content of the whole genome; higher peaks indicate a larger difference with the average GC content; the inner blue signals indicate that the GC content of the region was lower than the average GC content of the whole genome; higher peaks indicate a larger difference with the average GC content; the innermost circle shows the GC-Skew value. The innermost circle features the GC-Skew value, the specific algorithm was (G-C)/(G+C), useful to determine the leading strand and lagging strand. In general, the leading strand GC features skew>0, the lagging strand GC features skew<0. This may further help to determine the replication starting point and the cumulative offset may also assist in determining the starting point, i.e. cumulative offset minimum, and the end point, i.e. cumulative offset maximum, of replication, particularly important for cyclic genomes.
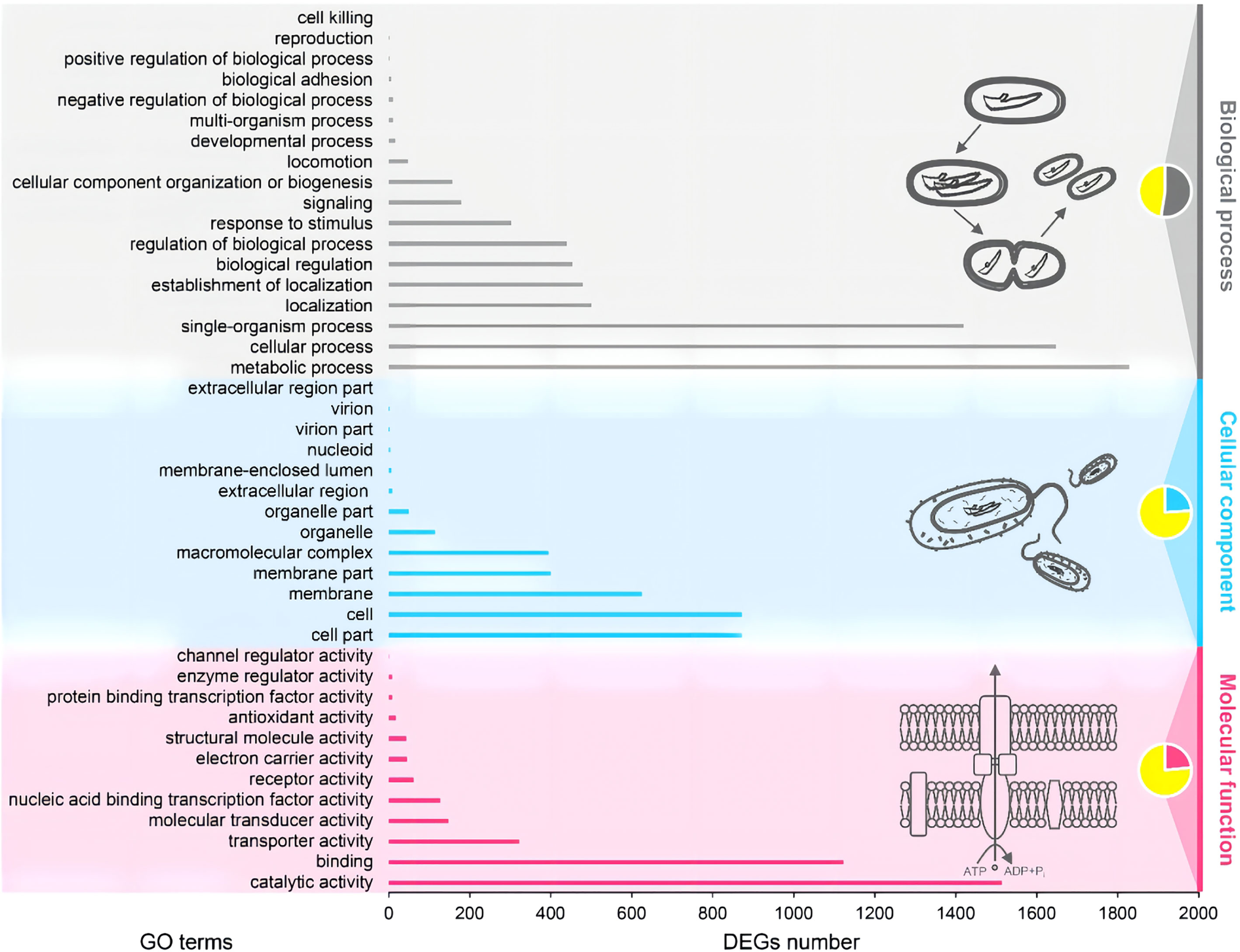
Figure 3 Gene ontology (GO) functional classification of A. hydrophila MX16A. DEGs, Differentially expressed genes.
Types and distribution of resistance-associated genes
Analysis of antibiotic-related genes of A. hydrophila MX16A associated with resistance to β-lactams (ampC, bla2, blaZ), tetracyclines (tetA), quinolones (gyrA, parC), sulfonamides (sulA, folC), aminoglycosides (aphA, aacC, aadA), chloramphenicols (cat), macrolides (msbA), and rifamycins (rpoB) was performed using CARD databases. Some other efflux-associated genes following by acrA, acrB, emrA, emrB, emrD, emrE, mdtK, tolC, bcr, and ABCC10 (gene symbol of efflux protein MRP7) were also identified (Table 3 and Figure 4).

Table 3 Resistance genes of the A. hydrophila MX16A genome extracted by CARD and Nr analysis.
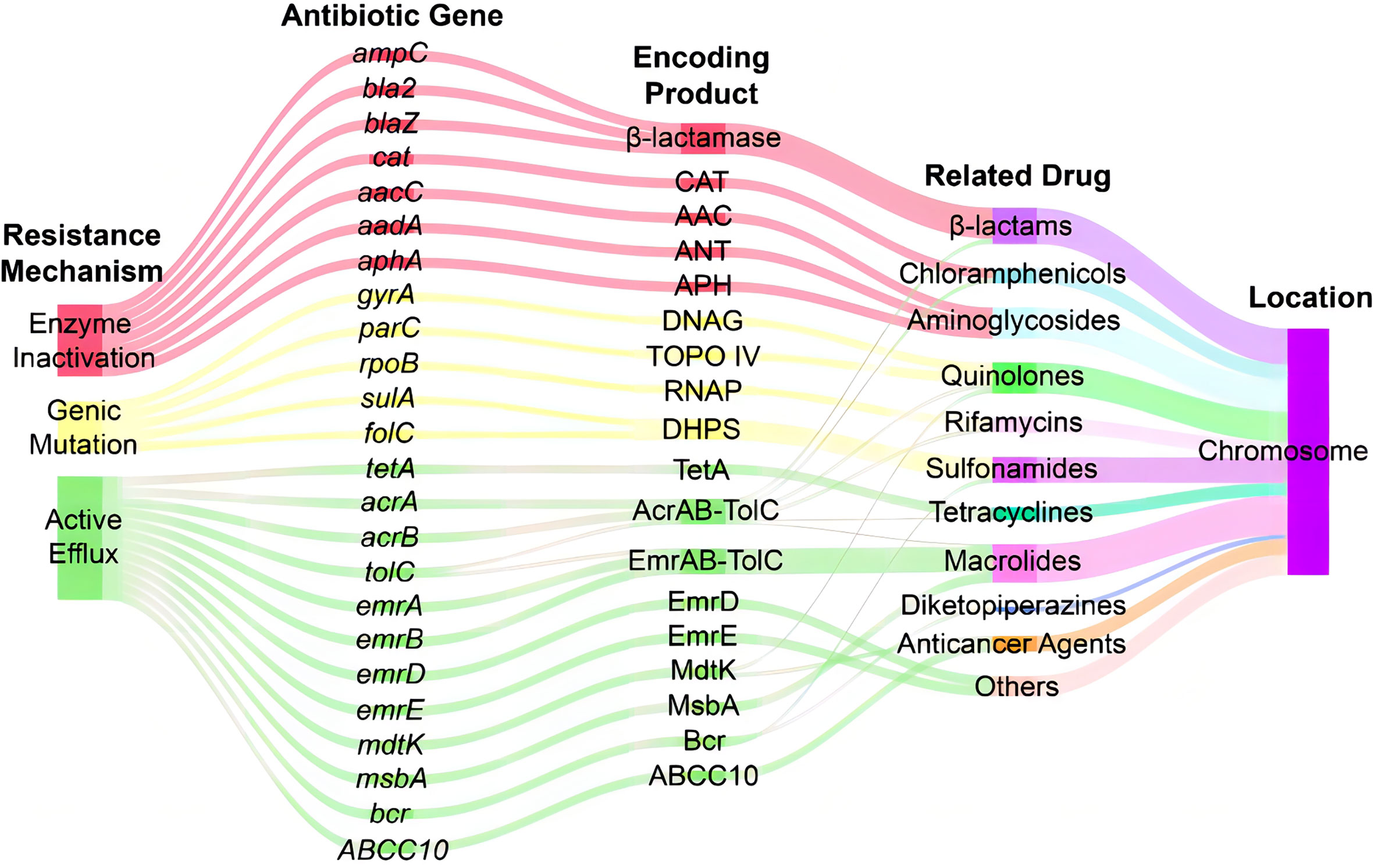
Figure 4 Sankey diagram of distribution of resistance-associated genes in A. hydrophila MX16A. CAT, chloramphenicol acetyltransferase; AAC, acetyltransferase; ANT, adenylyltransferase; APH, phosphotransferase; DNAG, DNA gyrase; TOPO IV, topoisomerase IV; RNAP, RNA polymerase; DHPS, dihydropteroate synthase.
However, antimicrobial susceptibility test (Table 1) revealed that MX16A was not resistant to enrofloxacin and norfloxacin. We further compared the common mutation sites on the quinolone resistance- determining regions (QRDR) of gyrA and parC genes and found that Ser83 on gyrA and Ser87 on parC were both mutated to Ile. For this reason, MX16A was performed additional drug sensitivity tests to nalidixic acid and found to be resistant to it.
Overview of the genomes for comparison
The complete genomes of 35 Aeromonas hydrophila strains isolated from different regions and hosts on NCBI, including MX16A, were downloaded for genomic comparison (Table 4).
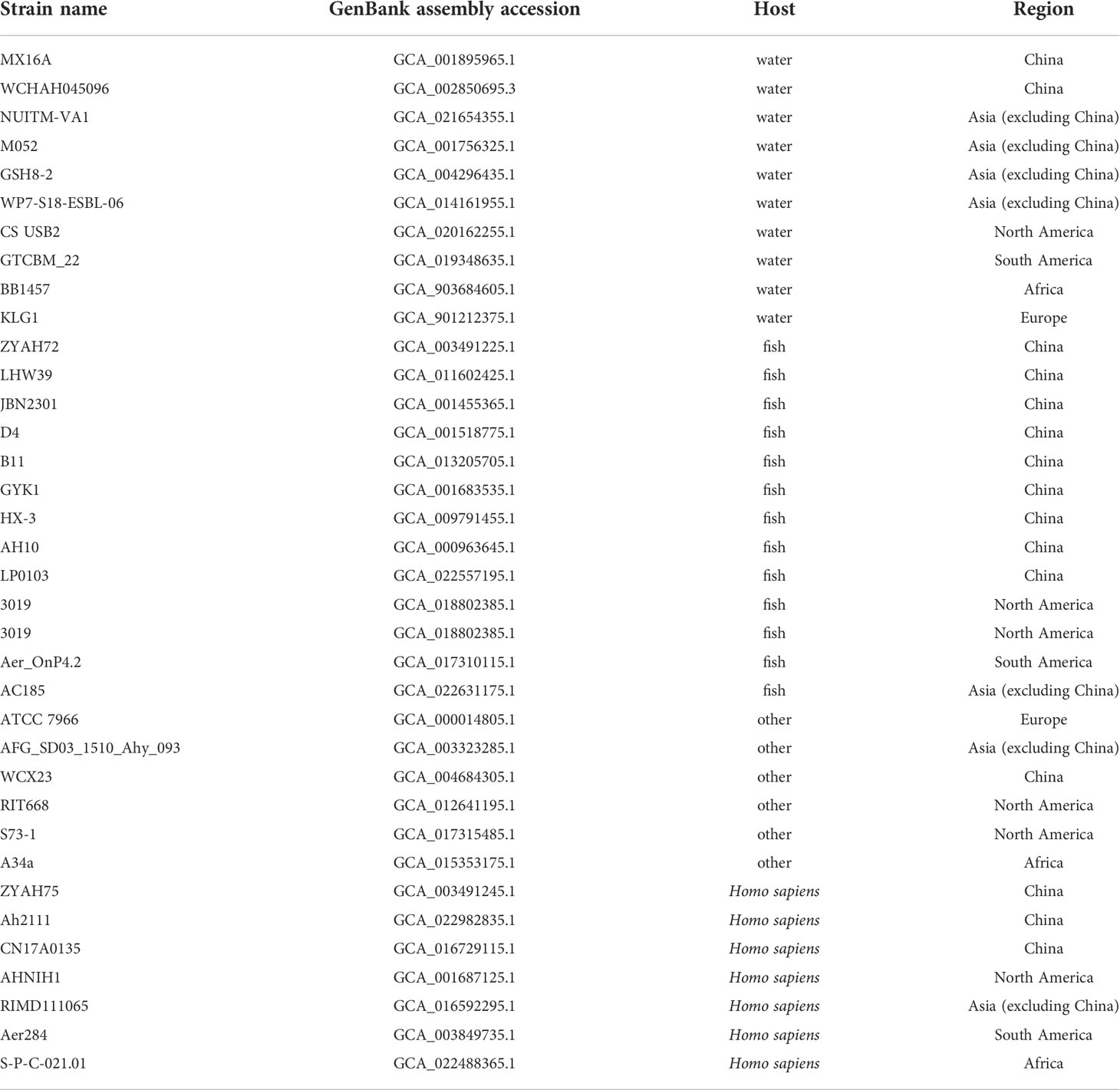
Table 4 Genomes information of 35 srains of A. hydrophila used in the present study.
Comparative analysis of average nucleotide identity
In recent studies, whole-genome average nucleotide identity (ANI) has emerged as a robust method for assessing species boundaries. Typically, organisms belonging to the same species have ≥95% ANI values. ANI represents the average nucleotide identity of all orthologous genes shared between any two genomes and offers robust resolution between strains of the same or closely related species (showing 80–100% ANI) (Jain et al., 2010). We performed ANI analysis on the whole genome of 35 Aeromonas hydrophila strains used in this study (Figure 5; Supplementary Table 1). The figure shows a region with high similarity and a higher genomic diversity. The analysis revealed that strains GCA_014161955.1 and GCA_022982835.1 featured high similarity with A. hydrophila MX16A. The corresponding ANI was 99.6% and 98.6%, respectively. The similarity of strains GCA_013205705.1 and GCA_903684605.1 with MX16A was below 95%. This finding may indicate that these two strains were misclassified as A. hydrophila.

Figure 5 Heat map of ANI pairings between any two genomes of MX16A (in red font) and other A. hydrophila strains downloaded from NCBI database.
Comparative genome analysis
Compared with A. hydrophila MX16A, only GCA_014161955.1 and GCA_022982835.1 featured greater ANI than 98%. Using the genomes of the three strains as inputs, OrthoFinder was used to pally align the protein sequences of the strains and detect the shared protein between the strains. Based on the generated files (Orthogroups. GeneCount. TSV) for processing, the protein contained in each of the three genomes were sorted out. Jvenn was used to draw a comparative analysis diagram of the clustering results of the three strains protein (Figure 6). The results obtained were similar to the ANI prediction and strain MX16A was demonstrated to be more similar to GCA_014161955.1 as these two strains shared most of the protein. Strain GCA_022982835.1 featured a larger number of unique proteins.
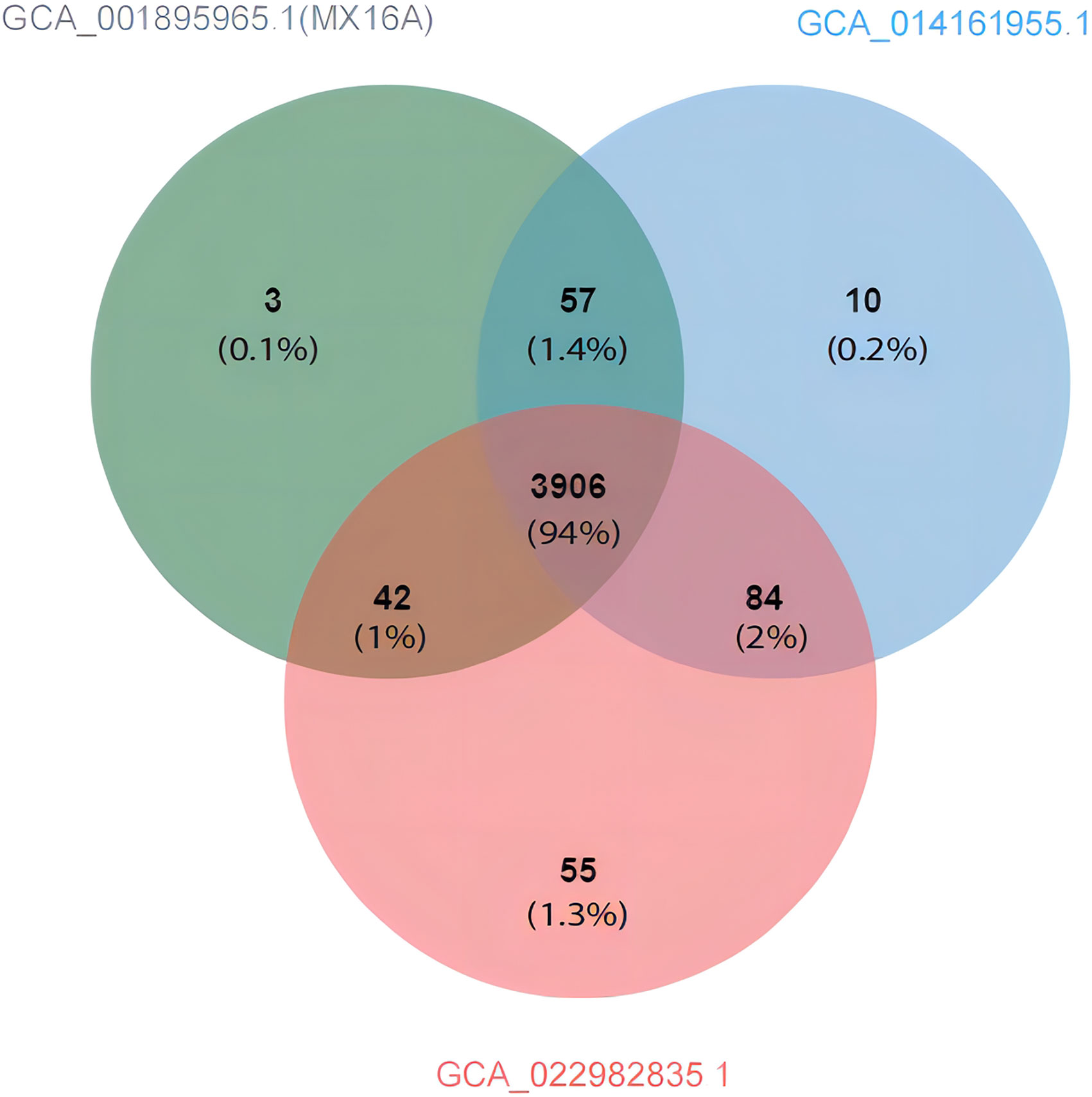
Figure 6 Venn diagram showing the protein shared and exclusive among A. hydrophila GCA_001895965.1 (MX16A), GCA_014161955.1, and GCA_022982835.1.
Pan-core genome analysis
Excluding the two strains with an ANI lower than 95%, the remaining 33 strains were analyzed regarding their pan-core genome. As the number of genes increased, the number of core genes gradually decreased and was then found to plateau while the pan-genome continued to expand. The final core genome size was converged to 3485 genes and the final pan-genome was converged to 9556 genes (Supplementary Tables 2, 3), presenting an open state (Figure 7). It may be speculated in this context that the A. hydrophila genome featured a more stable core gene cluster with high partial genome variability. These highly variable sections of the genome potentially enable different environmental adaptations.
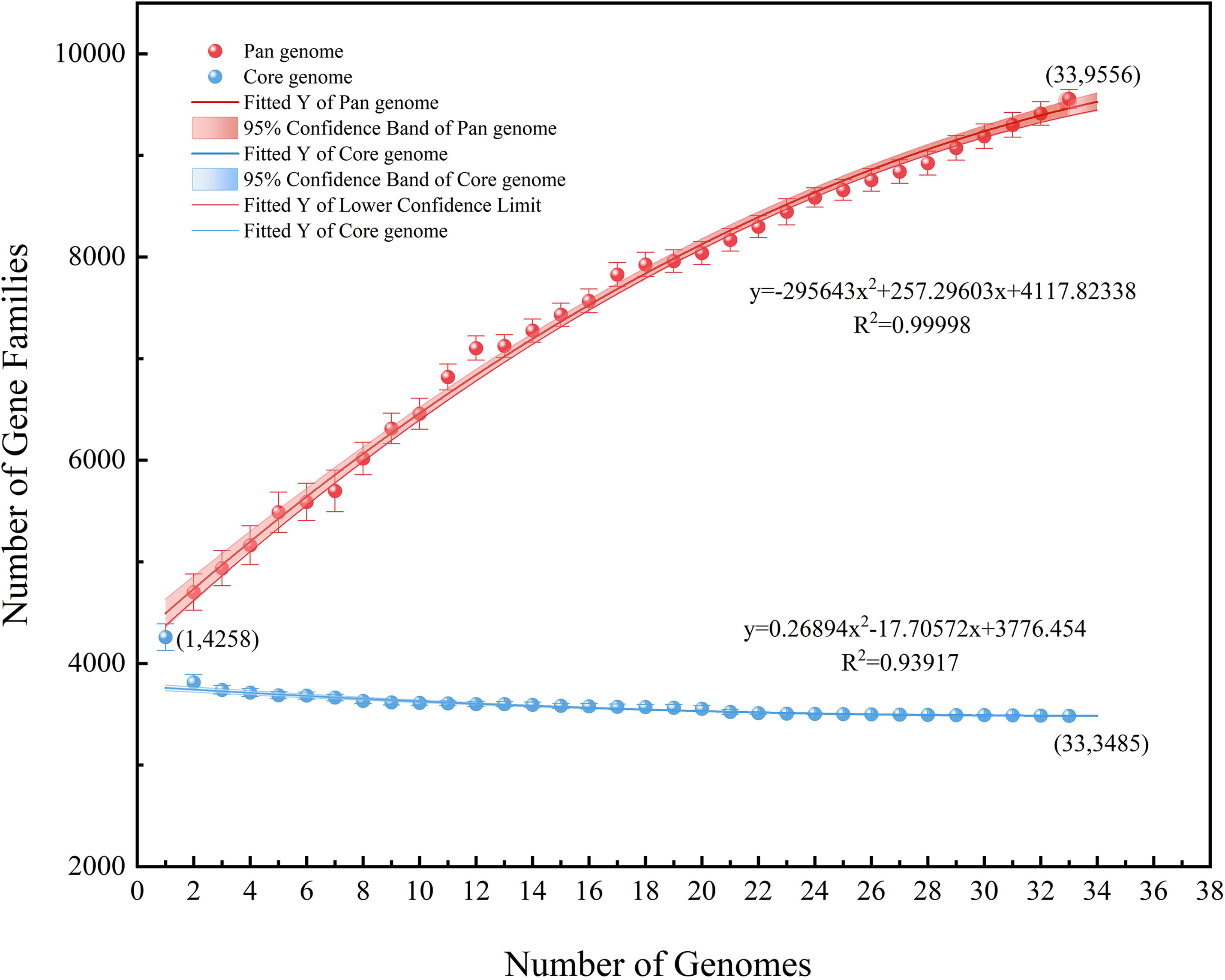
Figure 7 Pan-core genome analysis of 33 strains of A. hydrophila, except the two possibly misclassified strains (ANI<95%).
Analysis of drug resistance and virulence factors
The online tool ITOL (https://itol.embl.de/) was used to generate evolutionary trees and maps of drug resistance genes and virulence factors (Figure 8). A total of 12 classes of resistance genes were predicted in these genomes, of which β-lactam and β-lactam:carbapenem genes were shared by 35 strains. Genes of β-lactam:cephalosporin were also shared by alomost all strains, except the two possibly misclassified strains GCA_013205705.1 and GCA_903684605.1 (ANI<95%). Strain MX16A featured a larger number of resistance genes (Supplementary Table 4). For virulence factors, 6 classes of virulence factors could be determined, common to most strains. The strains GCA_003491225.1, GCA_001455365.1, GCA_001518775.1, GCA_001683535.1 and GCA_011602425.1 exhibited highly similar drug resistance genes and virulence factors, all isolated from China with fish being the host. However, these strains stem from different Chinese provinces such as Guangdong and Hubei, potentially resulting in spreading due to breeding and transportation.
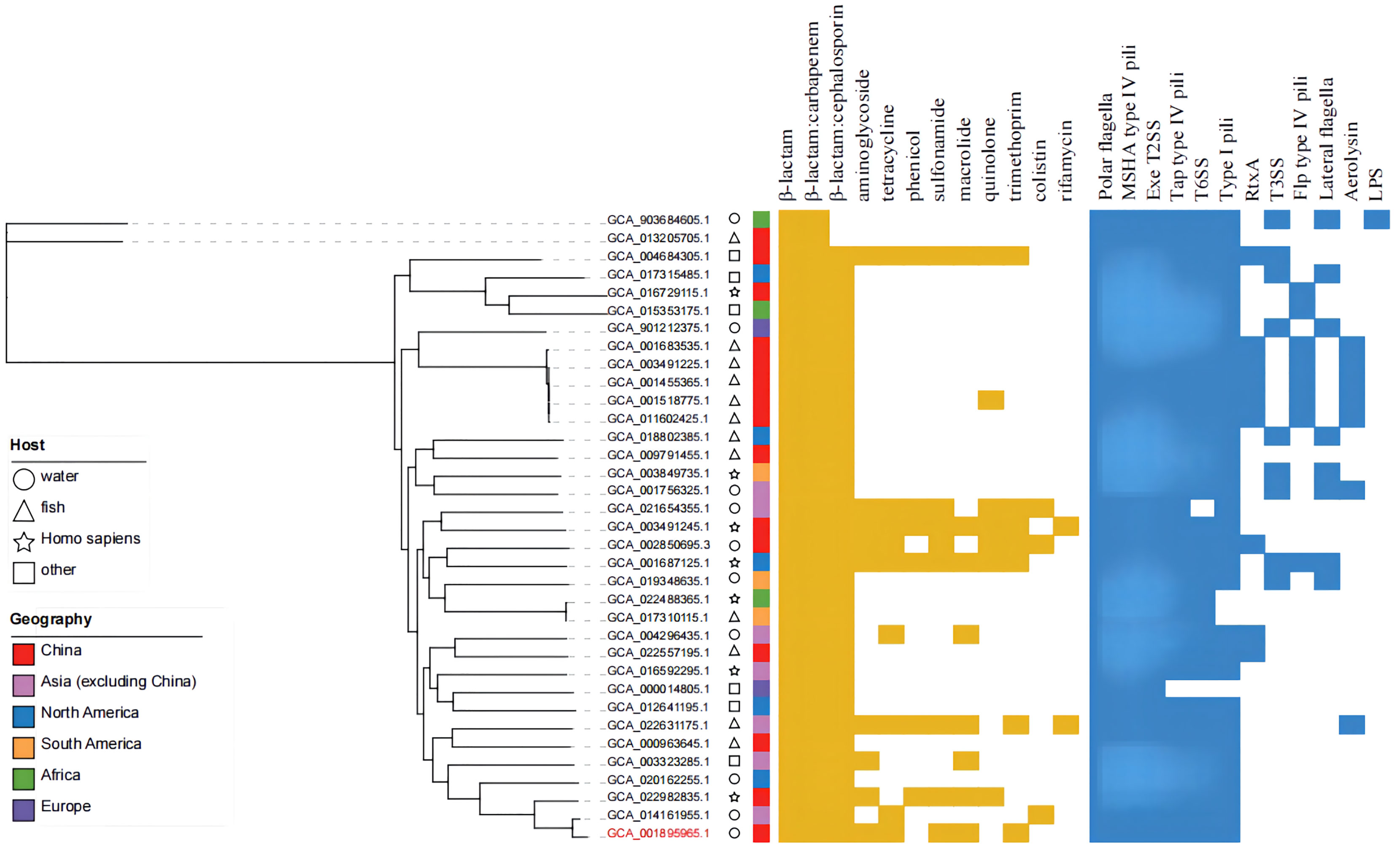
Figure 8 Phylogenetic tree, distribution of drug-resistance genes and virulence factors of 35 strains of A. hydrophila.
Discussion
Drug resistance and resistance-associated genes of A. hydrophila MX16A
From antibiotic sensitivity tests. we could determine that A. hydrophila MX16A was resistant to all clinically used drugs except for some quinolones. This finding was consistent with the results of the majority of drug resistance genes predicted by CARD.
Enzyme mediated drug resistance
β-lactam antibiotics have been extensively used to against gram-negative pathogens due to their broad spectrum of antibacterial activity these drugs exhibit until the appearance of the β-lactam inactivator β-lactamase (Abraham and Chain, 1988). The latter enzymes hydrolyze the β-lactam ring and therefore reduce the overall activity of β-lactam drugs (Davies, 1994). The process of enzymatic hydrolization is regarded as the primary resistance mechanism to β-lactam antibiotics in drug-resistant pathogens. Overexpression of various chromosomally-mediated-β-lactamases in A. hydrophila is generally associated with β-lactam as reported previously (Ko et al., 1998; Saavedra et al., 2004; Balsalobre et al., 2009). As such, the chromosome-borne enzyme AmpC coded as gene ampC has been found to inactivate carbapenem antibiotics in Enterobacteriaceae bacteria (Suh et al., 2010) while the β-lactamase Bla2, coded as chromosomal gene bla2, has been found to be expressed in Escherichia coli, resulting in β-lactam hydrolyzation and drug resistance against antibiotics based on carbapenem, cephalosporinase, and penicillinase (Materon et al., 2003).
Chloramphenicol acetyltransferase (CAT), encoded by the gene cat, represents the main factor for chloramphenicol resistance demonstrated by some bacteria strains. This type of antibiotic inactivating enzyme may catalyze the acetyl-S-CoA-dependent acetylation of chloramphenicol at the 3-hydroxyl group to transfer chloramphenicol to a metabolite without antimicrobial activity (Shaw, 1983). Moreover, in bacteria, resistance to aminoglycosides is usually deemed to enzymatic inactivation by acetyltransferases (encoded by gene aac), adenylyltransferases (encoded by gene aad), and phosphotransferases (encoded by gene aph) (Shaw et al., 1993).
Genetic mutation-mediated drug resistance
Naturally occurring resistance to fluoroquinolones in gram-negative bacteria is usually due to the genetic mutation of DNA gyrase (encoded by gyrA) and topoisomerase IV (encoded by parC), both of which represent the enzyme targets of fluoroquinolones in the bacterial cell and play a key role in DNA replication (Drlica and Zhao, 1997; Hooper, 2000). These enzymatic changes in the target site may block the formation of compounds by these enzymes and fluoroquinolones, usually responsible for bacterial DNA replication inhibition (Hooper, 2000). Furthermore, the region for mutations within gyrA and parC has been described as the quinolone-resistance determining region (QRDR) (Yoshida et al., 1990) and is associated with the mechanism of quinolone-resistance in Aeromonas spp. as reported previously (Alcaide et al., 2010). Moreover, anti-rifampicin strains have been reported with alteration of the rpoB gene encoding the beta subunit of RNA polymerase. A large majority of rifampicin-resistant isolates including Aeromonas strains are related to specific mutations within the 81-bp region (rifampicin-resistance determining region, RRDR) of the rpoB gene as reported in the literature (Herrera et al., 2003; Zhou et al., 2012; Pridgeon et al., 2013).
For living cells, folate derivatives, synthesized by most bacterial species, generally represent the requisite cofactors participating in the biosynthesis of purines, pyrimidines, and amino acids. Thus, enzymes in the pathway of folate biosynthesis represent the paramount targets to folate-inhibitors, including sulfonamides, to antagonize pathogens (Goulding et al., 2005). Earlier reports have revealed that sulfonamide-resistance was closely associated with genetic mutation of the dihydropteroate synthase (DHPS) encoded by the chromosomal gene sul and fol (e.g. sulA and folC) in bacteria (Maskell et al., 1997; Swedberg and Ringertz, 1998; Haasum et al., 2001).
Efflux protein-mediated drug resistance
Efflux pumps are one of the major causes of MDR in almost all bacterial species with gene-encoding proteins found on the chromosomes or plasmids (Webber and Piddock, 2003; Poole, 2007). According to the structural diversity, energy sources, and substrates, efflux pumps of gram-negative bacteria can be divided into five families: ATP-binding cassette (ABC) superfamily, major facilitator (MF) superfamily, resistance nodulation division (RND) family, multi-antimicrobial extrusion (MATE) family, and small multidrug-resistance (SMR) family (Blair et al., 2014).
As members of the ABC superfamily proteins, MRP7/ABCC10 (gene symbol ABCC10) may transport amphipathic anions and cause resistance to several anticancer agents such as docetaxel and vincristine (Hopperborge et al., 2004; Kruh et al., 2007) while MsbA protein (encoded by gene msbA and localized to the inner membrane) was found to cause resistance to erythromycin by means of ATP binding and hydrolytic properties in both gram-positive (Woebking et al., 2005) and gram-negative (Ghanei et al., 2007) microorganisms. The corresponding gene tetA located on the chromosome encoding TetA efflux protein (generally associated with MF superfamily) frequently observed in Aeromonas spp. isolates (Jacobs and Chenia, 2007) has been described to leave the host bacterium unharmed by actively transporting intracellular tetracycline out of cell cytoplasm (Roberts, 2005; Wang et al., 2014). Previous studies have revealed that overproduction of MdtK, a MATE family membrane transport protein, encoded by gene mdtK, could induce resistance to norfloxacin, doxorubicin, and acriflavine in Salmonella enterica (Horiyama et al., 2010).
TolC (encoded by tolC), an outer membrane protein, functions as the outer membrane channel to several MDR efflux systems (Benz et al., 1993), including the AcrAB-TolC (RND-type) and EmrAB-TolC (MF-type). AcrAB-TolC represents the main efflux system commonly found in various gram-negative bacteria (Pradel and Pagès, 2002; Baucheron et al., 2004; de Cristóbal et al., 2006), forming by a tripartite complex comprised of a cytosolic membrane pump (AcrB, encoded by acrB), a periplasmic membrane fusion protein (AcrA, encoded by acrA), and a TolC protein (Du et al., 2014). The EmrAB-TolC efflux system is also composed of three componentes including the membrane protein EmrA (encoded by emrA), transporter protein EmrB (encoded by emrB), and outer membrane protein TolC (Lomovskaya and Lewis, 1992). These two above-mentioned efflux systems, particularly AcrAB-TolC, have been reported to significantly affect the antimicrobial resistance to multiple antibiotic classes such as tetracyclines, chloramphenicols, rifamicins, β-lactams, quinolones, and macrolides in gram-native bacteria (Okusu et al., 1996; Nishino et al., 2003; Baucheron et al., 2004; Chollet et al., 2004; Elkins and Mullis, 2007). Moreover, EmrD (MF superfamily) and EmrE (SMR family) represent two membrane transporter proteins codified by genes emrD and emrE, respectively. Both of these proteins may induce drug resistance by involving adaptation to low energy shock or exporting diverse drug substrates protecting bacteria from antibiotic damage according to various studies reported in the literature (Naroditskaya et al., 1993; Rotem and Schuldiner, 2004). Moreover, gene bcr encoding a drug resistance bacterial transmembrane protein (Bcr), was also identified from the A. hydrophila MX16A genome involved in bicyclomycin and sulfathiazole resistance in E. coli (Bentley et al., 1993; Hayashi et al., 2010).
Comparative genome analysis
As described above, we were able to assess the diversity level of the biological genome to some extent related to the ratio of core genome to pangenome. The pangenome analysis of A. hydrophila described in this paper showed a large genomic diversity and further demonstrated that this species could adapt to and survive in different environmental conditions. In a previous study found in the literature (Ghatak et al., 2016), the pan-genome and core genes of 16 strains of Aeromonas in the genus Aeromonas spp. were analyzed and the results showed that A. hydrophila exhibited a higher genome diversity than A. veronii and A. caviae. Combined with our analytical data, the number of genes in the core genome did not change significantly, further confirming that the core genome of A. hydrophila is indeed stable. Finally, it may also be speculated that the genome of A. hydrophila features rich metabolic functions.
Analysis of virulence and drug resistance factors
Type III secretion systems are considered an important virulence mechanism for aeromonads (Sierra et al., 2010; Krzymińska et al., 2012). In this study, we did not predict genes for the type III virulence system, however, Ghatak et al., reported genes related to the type III secretion systems are present in all genomes of A. hydrophila, presumably due to the use of different databases. Importantly, a different drug resistance gene database for A. hydrophila MX16A was used in this report, resulting in some differences in the forecasted results. Several strains with more and roughly similar resistance genes stemed from different host types and were distributed in different regions. Due to the wide geographical distribution of A. hydrophila and the wide distribution of its hosts, it is hypothesized that frequent gene exchange between strains may lead to the transfer of corresponding drug resistance genes.
Conclusions
In this study, we report preliminary results regarding the environmental MDR Aeromonas hydrophila strain MX16A based on complete genome sequence analysis. The results of resistance gene prediction analysis were mostly consistent with the phenotypic antimicrobial susceptibility test. A variety of antibiotic-related genes identified from the A. hydrophila MX16A genome indicates that this bacterium may pose a potential threat to both human and aquatic animal health with phylogenetic analysis further confirming this conclusion. The diverse types and distributions of antibiotic genes of A. hydrophila MX16A suggested that the bacterial resistance mechanism was variable as follows: enzyme inactivation, genetic mutation, and active efflux. However, it still remains unclear which and how these three variables influence the overall drug resistance mechanism of A. hydrophila MX16A. Thus, future studies may now focus on the genomic characteristics potentially further leading to the development of targeted antibiotics. A. hydrophila MX16A exhibited close similarities to GCA_022982835.1. Due to the finding that humans are the primary host for the GCA_022982835.1 strain, it may be concluded that the pathway of human infection with Aeromonas may be due to the presence of this bacterial species in water and infected aquatic animals. Genome comparison studies carried out throughout this research further revealed that A. hydrophila displays a wide host and geographic distribution pattern, is not conserved, features high genome variability, and horizontal gene transfer may promote the evolution of other strains.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
YG and CZ carried out the experiments and drafted the manuscript. ML directed the research and revised the manuscript. CM isolated the bacterial strain. HC, XJ, SZ, and XX analyzed the genomic data and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Key R&D Program of China (2020YFD0900102), the Earmarked Fund for China Agriculture Research System (No.CARS-46), the Xiamen Ocean and Fishery Development Special Fund (21CZP007HJ07).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.1042350/full#supplementary-material
References
Abraham, E. P., Chain, E. (1988). An enzyme from bacteria able to destroy penicillin. 1940. Rev. Infect. Dis. 10 (4), 677–678.
Adamski, J., Koivuranta, M., Leppänen, E. (2006). Fatal case of myonecrosis and septicaemia caused by aeromonas hydrophila in Finland. Scand. J. Infect. Dis. 38 (11-12), 1117–1119. doi: 10.1080/00365540600684389
Alcaide, E., Blasco, M. D., Esteve, C. (2010). Mechanisms of quinolone resistance in aeromonas species isolated from humans, water and eels. Res. Microbiol. 161 (1), 40–45. doi: 10.1016/j.resmic.2009.10.006
Ambardar, S., Gupta, R., Trakroo., D., Lal., R., Vakhlu., J. (2016). High throughput sequencing: An overview of sequencing chemistry. Indian. J. Microbiol. 56 (4), 394–404. doi: 10.1007/s12088-016-0606-4
Araujo, R. M., Arribas, R. M., Pares, R. (1991). Distribution of aeromonas species in waters with different levels of pollution. J. Appl. Bacteriol. 71 (2), 182–186. doi: 10.1111/j.1365-2672
Ardui, S., Ameur, A., Vermeesch, J. R., Hestand, M. S. (2018). Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic. Acids Res. 46 (5), 2159–2168. doi: 10.1093/nar/gky066
Balsalobre, L. C., Dropa, M., Lincopan, N., Mamizuka, E. M., Matté, G. R., Matté, M. H. (2009). Detection of metallo-β-Lactamases-Encoding genes in environmental isolates of aeromonas hydrophila and aeromonas jandaei. Lett. Appl. Microbiol. 49 (1), 142–145. doi: 10.1111/j.1472-765X.2009.02625.x
Baucheron, S., Tyler, S., Boyd, D., Mulvey, M. R., Chaslus-Dancla, E., Cloeckaert, A. (2004). AcrAB-TolC efflux-mediated multi-drug resistance in salmonella enterica serovar typhimurium DT104. Antimicrob. Agents. Ch. 48 (10), 3729–3735. doi: 10.1128/AAC.48.10
Bentley, J., Hyatt, L. S., Ainley, K., Parish, J. H., Herbert, R. B., White, G. R. (1993). Cloning and sequence analysis of an escherichia coli gene conferring bicyclomycin resistance. Gene. 127 (1), 117–120. doi: 10.1016/0378-1119(93)90625-d
Benz, R., Maier, E., Gentschev, I. (1993). TolC of escherichia coli functions as an outer membrane channel. Zentralbl. Bakteriol. 278 (2-3), 187–196. doi: 10.1016/s0934-8840(11)80836-4
Blair, J. M., Richmond, G. E., Piddock, L. J. (2014). Multidrug efflux pumps in gram-negative bacteria and their role in antibiotic resistance. Future. Microbiol. 9 (10), 1165. doi: 10.2217/fmb.14.66
Boucher, H. W., Cosgrove, S. E., Cox, E., Talbot, G. H. (2017). The fight against multidrug-resistant bacteria. Ann. Intern. Med. 166 (1), 78. doi: 10.7326/L16-0584
Chauret, C., Volk, C., Creason, R., Jarosh, J., Robinson, J., Warnes, C. (2001). Detection of aeromonas hydrophila in a drinking-water distribution system: A field and pilot study. Can. J.Microbiol. 47 (8), 782–786. doi: 10.1139/w01-070
Chollet, R., Chevalier, J., Bryskier, A., Pagès, J. M. (2004). The AcrAB-TolC pump is involved in macrolide resistance but not in telithromycin efflux in enterobacter aerogenes and escherichia coli. Antimicrob. Agents. Ch. 48 (9), 3621–3624. doi: 10.1128/AAC.48.9
Davies, J. (1994). Inactivation of antibiotics and the dissemination of resistance genes. Science. 264 (5157), 375–382. doi: 10.1126/science.8153624
de Cristóbal, R. E., Vincent, P. A., Salomón, R. A. (2006). Multidrug resistance pump acrab-tolc is required for high-level, tet(a)-mediated tetracycline resistance in escherichia coli. J. Antimicrob. Chemother. 58 (1), 31–36. doi: 10.1093/jac/dkl172
Drlica, K., Zhao, X. (1997). DNA Gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61 (3), 377. doi: 10.1128/mmbr.61.3.377-392.1997
Du, D., Wang, Z., James, N. R., Voss, J. E., Klimont, E., Oheneagyei, T., et al. (2014). Structure of the acrab-tolc multidrug efflux pump. Nature. 509 (7501), 512–515. doi: 10.1038/nature13205
Elkins, C. A., Mullis, L. B. (2007). Substrate competition studies using whole-cell accumulation assays with the major tripartite multidrug efflux pumps of escherichia coli. Antimicrob. Agents. Ch. 51 (3), 923–929. doi: 10.1128/AAC.01048-06
Ghanei, H., Abeyrathne, P. D., Lam, J. S. (2007). Biochemical characterization of MsbA from pseudomonas aeruginosa. J. Biol. Chem. 282 (37), 26939. doi: 10.1074/jbc.M702952200
Ghatak, S., Blom, J., Das, S., Sanjukta, R., Puro, K., Mawlong, M., et al. (2016). Pan-genome analysis of aeromonas hydrophila, aeromonas veronii and aeromonas caviae indicates phylogenomic diversity and greater pathogenic potential for aeromonas hydrophila. Antonie. Van. Leeuwenhoek. 109 (7), 945–956. doi: 10.1007/s10482-016-0693-6
Goulding, C. W., Apostol, M. I., Sawaya, M. R., Phillips, M., Parseghian, A., Eisenberg, D. (2005). Regulation by oligomerization in a mycobacterial folate biosynthetic enzyme. J. Mol. Biol. 349 (1), 61–72. doi: 10.1016/j.jmb.2005.03.023
Haasum, Y., Ström, K., Wehelie, R., Luna, V., Roberts, M. C., Maskell, J. P., et al. (2001). Amino acid repetitions in the dihydropteroate synthase of streptococcus pneumoniae lead to sulfonamide resistance with limited effects on substratekm. Antimicrob. Agents. Ch. 45 (3), 805–809. doi: 10.1128/AAC.45.3.805-809.2001
Hayashi, M., Tabata, K., Yagasaki, M., Yonetani, Y. (2010). Effect of multidrug-efflux transporter genes on dipeptide resistance and overproduction in escherichia coli. Fems. Microbiol. Lett. 304 (1), 12–19. doi: 10.1111/j.1574-6968.2009.01879.x
Herrera, L., Jiménez, S., Valverde, A., García-Aranda, M. A., Sáez-Nieto, J. A. (2003). Molecular analysis of rifampicin-resistant mycobacterium tuberculosis isolated in spain, (1996-2001). description of new mutations in the rpob gene and review of the literature. Int. J. Antimicrob. Agents. 21 (5), 403–408. doi: 10.1016/s0924-8579(03)00036-0
Hooper, D. C. (2000). Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis. 31 (Suppl 2), S24–S28. doi: 10.1086/314056
Hopperborge, E., Chen, Z. S., Shchaveleva, I., Belinsky, M. G., Kruh, G. D. (2004). Analysis of the drug resistance profile of multidrug resistance protein 7 (abcc10). Cancer. Res. 64 (14), 4927. doi: 10.1158/0008-5472.CAN-03-3111
Horiyama, T., Yamaguchi, A., Nishino, K. (2010). TolC ddependency of multidrug efflux systems in salmonella enterica serovar typhimurium. J. Antimicrob. Chemother. 65 (7), 1372–1376. doi: 10.1093/jac/dkq160
Igbinosa, I. H., Igumbor, E. U., Aghdasi, F., Tom, M., Okoh, A. I. (2012). Emerging aeromonas species infections and their significance in public health. Scientific. World. J. 2012 (3), 625023. doi: 10.1100/2012/625023
Jacobs, L., Chenia, H. Y. (2007). Characterization of integrons and tetracycline resistance determinants in aeromonas spp. isolated from south African aquaculture systems. Int. J. Food. Microbiol. 114 (3), 295–306. doi: 10.1016/j.ijfoodmicro.2006.09.030
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., Aluru, S. (2010). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9 (1), 5114. doi: 10.1038/s41467-018-07641-9
Janda, J. M., Abbott, S. L. (2010). The genus aeromonas: Taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23 (1), 35–73. doi: 10.1128/CMR.00039-09
Ko, W. C., Wu, H. M., Chang, T. C., Yan, J. J., Wu, J. J. (1998). Inducible β-lactam resistance in aeromonas hydrophila: Therapeutic challenge for antimicrobial therapy. J. Clin. Microbiol. 36 (11), 3188–3192. doi: 10.1128/JCM.36.11.3188-3192.1998
Kruh, G. D., Guo, Y., Hopperborge, E., Belinsky, M. G., Chen, Z. S. (2007). ABCC10, ABCC11, and ABCC12. Pflugers. Arch. 453 (5), 675–684. doi: 10.1007/s00424-006-0114-1
Krzymińska, S., Mokracka, J., Koczura, R., Cwiertnia, A., Kaznowski, A. (2012). Aeromonas spp.-mediated cell-contact cytotoxicity is associated with the presence of type III secretion system. Antonie. Van. Leeuwenhoek. 101 (2), 243–251. doi: 10.1007/s10482-011-9627-5
Lautenbach, E., Fishman, N. O. (1999). Wagging the dog: Antibiotic use and the emergence of resistance. J. Gen. Intern. Med. 14 (10), 643–645. doi: 10.1046/j.1525-1497.1999.t01-1-07029.x
Lin, M., Liang, J., Zhang, X., Wu, X., Yan, Q., Luo, Z. (2015). Genetic diversity of three classes of integrons in antibiotic-resistant bacteria isolated from jiulong river in southern China. Environ. .Sci. pollut. Res. Int. 22 (15), 11930–11939. doi: 10.1007/s11356-015-4480-0
Lomovskaya, O., Lewis, K. (1992). Emr, an escherichia coli locus for multidrug resistance. Proc. Natl. Acad. Sci. U. S. A. 89 (19), 8938–8942. doi: 10.1073/pnas.89.19.8938
Maskell, J. P., Sefton, A. M., Hall, L. M. (1997). Mechanism of sulfonamide resistance in clinical isolates of streptococcus pneumoniae. Antimicrob. Agents. Ch. 41 (10), 2121–2126. doi: 10.1128/AAC.41.10.2121
Materon, I. C., Queenan, A. M., Koehler, T. M., Bush, K., Palzkill, T. (2003). Biochemical characterization of -lactamases Bla1 and Bla2 from bacillus anthracis. Antimicrob. Agents. Ch. 47 (6), 2040–2042. doi: 10.1128/AAC.47.6.2040-2042.2003
Naroditskaya, V., Schlosser, M. J., Fang, N. Y., Lewis, K. (1993). An e. coli, gene EmrD is involved in adaptation to low energy shock. Biochem. Biophys. Res. Commun. 196 (2), 803–809. doi: 10.1006/bbrc.1993.2320
Nishino, K., Yamada, J., Hirakawa, H., Hirata, T., Yamaguchi, A. (2003). Roles of TolC-dependent multidrug transporters of escherichia coli in resistance to β-lactams. Antimicrob. Agents. Ch. 47 (9), 3030–3033. doi: 10.1128/AAC.47.9.3030-3033.2003
Okusu, H., Ma, D., Nikaido, H. (1996). Acrab efflux pump plays a major role in the antibiotic resistance phenotype ofescherichia colimultiple- antibiotic-resistance (mar) mutants. J. Bacteriol. 178 (1), 306–308. doi: 10.1128/jb.178.1.306-308.1996
Parker, J. L., Shaw, J. G. (2011). Aeromonas spp. clinical microbiology and disease. J. Infect. 62 (2), 109–118. doi: 10.1016/j.jinf.2010.12.003
Poole, K. (2007). Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 39 (3), 162–176. doi: 10.1080/07853890701195262
Pradel, E., Pagès, J. M. (2002). The acrab-tolc efflux pump contributes to multidrug resistance in the nosocomial pathogen enterobacter aerogenes. Antimicrob. Agents. Ch. 46 (8), 2640. doi: 10.1128/AAC.46.8.2640-2643.2002
Pridgeon, J. W., Mu, X., Klesius, P. H. (2013). Biochemical and molecular characterization of the novobiocin and rifampicin resistant aeromonas hydrophila vaccine strain Al09-71n+r compared to its virulent parent strain Al09-71. Vet. Microbiol. 165 (3-4), 349–357. doi: 10.1016/j.vetmic.2013
Rafei, R., Kassaa, I. A., Osman, M., Dabboussi, F., Hamze, M. (2018). Distribution and antibiotic susceptibility profiles of aeromonas spp. from different aquatic environments in north Lebanon. J. Infect. Dev. Ctries. 12 (2.1), 6S. doi: 10.3855/jidc.10055
Roberts, M. C. (2005). Update on acquired tetracycline resistance genes. Fems. Microbiol. Lett. 245 (2), 195–203. doi: 10.1016/j.femsle.2005.02.034
Rotem, D., Schuldiner, S. (2004). EmrE, a multidrug transporter from escherichia coli, transports monovalent and divalent substrates with the same stoichiometry. J. Biol. Chem. 279 (47), 48787–48793. doi: 10.1074/jbc.M408187200
Rutteman, B. (2017). Aeromonas wound infection in a healthy boy. Jmm. Case. Rep. 4 (11), e005129. doi: 10.1099/jmmcr.0.005129
Saavedra, M. J., Guedesnovais, S., Alves, A., Rema, P., Tacão, M., Correia, A., et al. (2004). Resistance to beta-lactam antibiotics in aeromonas hydrophila isolated from rainbow trout (oncorhynchus mykiss). Int. Microbiol. 7 (3), 207–211.
Scoaris, D. O., Colacite, J., Nakamura, C. V., Ueda-Nakamura, T., de Abreu Filho, B. A., Dias Filho, B. P. (2008). Virulence and antibiotic susceptibility of aeromonas spp. isolated from drinking water. Antonie. Van. Leeuwenhoek. 93 (1-2), 111–122. doi: 10.1007/s10482-007-9185-z
Shaw, W. V. (1983). Chloramphenicol acetyltransferase: Enzymology and molecular biolog. Crc. Crit. Rev. Biochem. 14 (1), 1–46. doi: 10.3109/10409238309102789
Shaw, K. J., Rather, P. N., Hare, R. S., Miller, G. H. (1993). Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57 (1), 138–163. doi: 10.1128/mr.57.1.138-163.1993
Sierra, J. C., Suarez, G., Sha, J., Baze, W. B., Foltz, S. M., Chopra, A. K. (2010). Unraveling the mechanism of action of a new type III secretion system effector AexU from aeromonas hydrophila. Microb. Pathog. 49 (3), 122–134. doi: 10.1016/j.micpath.2010.05.011
Skwor, T., Stringer, S., Haggerty, J., Johnson, J., Duhr, S., Johnson, M., et al. (2020). Prevalence of potentially pathogenic antibiotic-resistant aeromonas spp. in treated urban wastewater effluents versus recipient riverine populations: a 3-year comparative study. Appl. Environ. Microbiol. 86 (3), e02053–e02019. doi: 10.1128/AEM.02053-19
Suh, B., Bae, I. K., Kim, J., Jeong, S. H., Yong, D., Lee, K. (2010). Outbreak of meropenem- resistant serratia marcescens comediated by chromosomal ampc beta-lactamase overproduction and outer membrane protein loss. Antimicrob. Agents. Ch. 54 (12), 5057–5061. doi: 10.1128/AAC.00768-10
Swedberg, G., Ringertz, S. O. (1998). Sulfonamide resistance in streptococcus pyogenes is associated with differences in the amino acid sequence of its chromosomal dihydropteroate synthase. Antimicrob. Agents. Ch. 42 (5), 1062–1067. doi: 10.1128/AAC.42.5.1062
Thomas, B. E., Shanmugam, P., Malaisamy, M., Ovung, S., Suresh, C., Subbaraman, R., et al. (2016). Psycho-Socio-Economic issues challenging multidrug resistant tuberculosis patients: a systematic review. Plos. One 11 (1), e0147397. doi: 10.1371/journal.pone.0147397
Wang, W., Guo, Q., Xu, X., Sheng, Z. K., Ye, X., Wang, M. (2014). High-level tetracycline resistance mediated by efflux pumps tet(a) and tet(a)-1 with two start codons. J. Med. Microbiol. 63 (Pt 11), 1454–1459. doi: 10.1099/jmm.0.078063-0
Webber, M. A., Piddock, L. J. (2003). The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 51 (1), 9–11. doi: 10.1093/jac/dkg050
Williams, D. H., Bardsley, B. (1999). The vancomycin group of antibiotics and the fight against resistant bacteria. Angew. Chem. Int. Ed. Engl. 38 (9), 1172–1193. doi: 1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C
Woebking, B., Reuter, G., Shilling, R. A., Velamakanni, S., Shahi, S., Venter, H., et al. (2005). Drug-lipid a interactions on the escherichia coli ABC transporter MsbA. J.Bacteriol. 187 (18), 6363–6369. doi: 10.1128/JB.187.18.6363-6369.2005
Yoshida, H., Bogaki, M., Nakamura, M., Nakamura, S. (1990). Quinolone resistance- determining region in the DNA gyrase gyra gene of escherichia coli. Antimicrob. Agents. Ch. 34 (6), 1271–1272. doi: 10.1128/AAC.34.6.1271
Keywords: Aeromonas hydrophila, complete genome sequencing, comparative genomics, multidrug resistance, drug resistance mechanism
Citation: Guo Y, Zeng C, Ma C, Cai H, Jiang X, Zhai S, Xu X and Lin M (2022) Comparative genomics analysis of the multidrug-resistant Aeromonas hydrophila MX16A providing insights into antibiotic resistance genes. Front. Cell. Infect. Microbiol. 12:1042350. doi: 10.3389/fcimb.2022.1042350
Received: 12 September 2022; Accepted: 18 October 2022;
Published: 03 November 2022.
Edited by:
Pengfei Li, Guangxi Academy of Sciences, ChinaReviewed by:
Yuting Deng, Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences, ChinaMingyou Li, Shanghai Ocean University, China
Copyright © 2022 Guo, Zeng, Ma, Cai, Jiang, Zhai, Xu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mao Lin, bGlubWFvQGptdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship