
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 28 November 2022
Sec. Virus and Host
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.1035566
This article is part of the Research TopicHuman Organoid Technology for Virus ResearchView all 5 articles
 Clarissa Clifton1†
Clarissa Clifton1† Brian F. Niemeyer1†
Brian F. Niemeyer1† Richard Novak2†
Richard Novak2† Uryan Isik Can3†
Uryan Isik Can3† Kelly Hainline2
Kelly Hainline2 Kambez H. Benam1,4,5*
Kambez H. Benam1,4,5*In vitro culture and differentiation of human-derived airway basal cells under air-liquid interface (ALI) into a pseudostratified mucociliated mucosal barrier has proven to be a powerful preclinical tool to study pathophysiology of respiratory epithelium. As such, identifying differentiation stage-specific biomarkers can help investigators better characterize, standardize, and validate populations of regenerating epithelial cells prior to experimentation. Here, we applied longitudinal transcriptomic analysis and observed that the pattern and the magnitude of OMG, KRT14, STC1, BPIFA1, PLA2G7, TXNIP, S100A7 expression create a unique biosignature that robustly indicates the stage of epithelial cell differentiation. We then validated our findings by quantitative hemi-nested real-time PCR from in vitro cultures sourced from multiple donors. In addition, we demonstrated that at protein-level secretion of BPIFA1 accurately reflects the gene expression profile, with very low quantities present at the time of ALI induction but escalating levels were detectable as the epithelial cells terminally differentiated. Moreover, we observed that increase in BPIFA1 secretion closely correlates with emergence of secretory cells and an anti-inflammatory phenotype as airway epithelial cells undergo mucociliary differentiation under air-liquid interface in vitro.
The human airway epithelium (bronchial and bronchiolar) is a pseudostratified columnar tissue that is composed of multiple epithelial cell sub-types such as basal cells, mucin-producing (goblet) cells, ciliated cells and Club cells. This mucosal barrier protects us against inhaled environmental stimuli, including pathogens, pollutants and allergens. Moreover, the airway epithelial dysfunction has been shown to be central to pathogenesis of several debilitating respiratory disorders, such as chronic obstructive pulmonary disease (COPD), asthma, primary ciliary dyskinesia (PCD), and cystic fibrosis (CF) (Martinez-Anton et al., 2013; Callejas-Diaz et al., 2020).
Over the past few decades, rigorous efforts by multiple groups led to optimization of culture conditions for ex vivo isolation and selective expansion, if required, of human airway epithelial cells (hAEpCs) from lung biopsies or airway brushings for subsequent in vitro differentiation under air-liquid interface (ALI) (Fulcher et al., 2005; Schmid et al., 2006; Ibricevic et al., 2006; Button et al., 2012; Manzanares et al., 2015; Zhao et al., 2017; Everman et al., 2018; Jiang et al., 2018). In this process, squamous-like undifferentiated monolayers of airway basal stem cells turn into terminally differentiated pseudostratified, polarized, mucociliated epithelia over the course of several weeks (typically 3–4 weeks at ALI). Such in vitro system is a reductionist and minimalist bottom-up approach for emulating human airway function and biology, which to a reasonable degree can reproduce tissue-level complexities of human airway epithelium. Thus, it has been widely adapted for preclinical studies into pathophysiology of lung bronchial and bronchiolar epithelia during homeostasis and when challenged with external stimuli.
Evaluating differential RNA and protein expression profiles as epithelial cells mature and differentiate has been the subject of a number of studies (Martinez-Anton et al., 2013; Callejas-Diaz et al., 2020; Bonser et al., 2021). For instance, Martinez-Anton et al., conducted global miRNA and gene expression profiling and compared day 28 (D28) well-differentiated normal human bronchial epithelial (NHBE) cells against their D0 counterpart – i.e., the undifferentiated confluent NHBE cells at time of ALI induction (Martinez-Anton et al., 2013). They found a total of 55 miRNAs and 1,201 transcript probe sets differentially expressed in D28 vs. D0. However, the authors did not characterize changes in gene expression throughout the differentiation process, as the study objective was not to elucidate biosignatures indicative of stage of hAEpCs in vitro differentiation. Similarly, Callejas‐Díaz and colleagues performed a transcriptome‐wide analysis on differentiating human nasal mucosa-derived basal stem cells in vitro (Callejas-Diaz et al., 2020). While the authors included D14 ALI samples in their studies, the focus of the study was to compare biological processes that differ during differentiation in healthy donor-derived nasal stem cells against those obtained from subjects with chronic rhinosinusitis with nasal polyps. More recently, Bonser et al., leveraging single-cell RNA-sequencing (scRNA-seq) data, devised antibody panels to characterize and isolate the major epithelial cell subsets by flow cytometry from human in vitro cultures (Bonser et al., 2021). Nevertheless, the overarching goal of study was not to identify differentiation stage-specific biosignatures.
Thus, we set to discover gene expression states and trajectories that correlate with various stages of mucociliary differentiation. We applied bulk transcriptomic analysis of in vitro cultured hAEpCs using a longitudinal sampling approach, validated our findings with sensitive hemi-nested real-time qPCR, and further confirmed one of the biomarkers by evaluating corresponding protein levels in the secretome of in vitro cultured hAEpCs.
Primary human small airway and bronchial epithelial cells were either purchased from commercial sources (Promocell, Lonza, LifeLine Cell Technology) or obtained from Drs. Fernando Holguin and Oliver Eickelberg. The donor information is provided in Supplementary Table 1. In total, bronchial (donor n = 5) and bronchiolar (donor n = 6) cells from eleven different donors were used in our studies. Of these, eight were known non-smokers, two had smoking history, and it was unknown if one donor had any prior smoking (Supplementary Table 1). In addition, eight donors (who were non-smokers) were confirmed healthy by appropriate vendor/provider, two donor lungs (from subjects with smoking history) were received as healthy but we observed emphysematous changes in parts of the lung; however, cells for in vitro culture were isolated only from non-diseased normal areas, and for one donor (with unknow smoking history information) the cause of death was head trauma. The cells were differentiated using transwell insert (TWI) culture systems (Corning, Cat. # CLS3401). Briefly, cells were expanded in PneumaCult™ Ex-Plus media (StemCell Cat. # 0540) containing 1x hydrocortisone (StemCell Cat. # 07925) and were seeded in 24-well TWIs (0.4 µm pores; polyester membrane) at a density of 3.3e4 cells per insert (= 100,000 cells per cm2) and cultured until full confluence in Ex-Plus media. Then, cells were allowed to equilibrate to PneumaCult™-air-liquid interface (ALI) media (StemCell Cat. # 05001) containing 1X hydrocortisone (StemCell Cat. # 07925) and 1X heparin (StemCell Cat. # 07980) overnight followed by ALI induction through removal of apical media. Every other day basal media was changed while the cells differentiated over 28 days.
Cells were lysed in situ for total RNA isolation on days 0, 2, 4, 6, 8, 10, 14, 21 and 28 post-ALI. For this, 200 µl of ALI media were added to the apical surface of the epithelia in TWIs and the cells were incubated for 10 minutes at 37°C. Then apical washes were performed by gently pipetting the media up and down then aspirating them to remove residual mucus and cell debris. Next, cells were lysed by adding 150 µl of RLT buffer from RNeasy Mini Kit (Qiagen Cat. # 74104). Cell lysates were then added to microcentrifuge tubes containing an additional 150 µl of the RLT buffer. RNA was isolated on-column and DNase-treated according to manufacturer’s instructions. Total RNA from the sample were then submitted to Molecular Biology Core Facilities of Dana-Farber Cancer Institute at Harvard University for analysis using Affymetrix GeneChip™ Human Gene 2.0 ST Array. The results obtained were robust multi-array average (RMA) data normalized and assessed for quality using Affymetrix Power Tools similar to our earlier microarray gene expression analysis (Benam et al., 2016), and then further processed and analyzed using custom scripts in MATLAB; duplicate genes and data lacking gene IDs were removed prior to analysis. Genes with change in expression of over two-fold over the studied time points (D0 through D28 post-ALI) and a p value < 0.01 (by Kruskal-Wallis test) were used for analysis. Data were deposited at GEO (GSE GSE197142).
RNA isolation was performed in situ at days 0, 2, 8, and 28 post-ALI as above. Following RNA extraction, cDNA was generated using from 5 µl of RNA template using SuperScript™ III Reverse Transcriptase (Thermo Fisher Scientific, Cat. # 18080093) and random primers (Thermo Fisher Scientific, Cat. # 4819011). Quantification gene expression was performed using hemi-nested qRT-PCR with iTaq™ Universal SYBR® Green Supermix (Bio-Rad, Cat. # 1725120) using primers targeting OMG, KRT14, STC1, BPIFA1, PLA2G7, TXNIP, and S100A7 (Supplementary Table 2). Briefly, targeted PCR was performed using an initial set of gene specific primers for 20 cycles. The product from this PCR was then diluted 100-fold prior to a second round of PCR, consisting of 45 cycles, which utilized a new internal primer (Supplementary Table 2). Following this, expression levels were calculated by using the delta-delta-CT method with GAPDH serving as a reference gene, as previously described (Benam et al., 2011).
Secreted protein levels were measured from the basal compartment of the TWIs at indicated days follow ALI. For BPIFA1, analysis was performed using an enzyme-linked immunosorbent assay (ELISA) kit (RayBiotech, Cat. # ELH-PLUNC-1) by following the manufacturer’s protocol. Briefly, samples and standards were loaded onto pre-coated tube strip plates and allowed to incubate at room temperature for 2 hours and 30 minutes while shaking at 150 rpm. After washing the plates, biotinylated antibody was added to the samples and they were incubated for an additional hour at room temperature at 150 rpm. After another round of washing, HRP-streptavidin was added to each well and the plates were incubated for 45 minutes at room temperature while shaking at 150 rpm. The samples were then washed and treated with 3,3,5,5’-tetramethylbenzidine solution for 30 minutes at room temperature while shaking at 150 rpm. To stop the reaction, 0.2 M sulfuric acid was added to each well. Plates were analyzed within 10 minutes of addition of the stop solution at a 450 nm wavelength using a Synergy HTX Multi-Mode Microplate Reader from BioTek on the The Gen5™ Microplate Reader and Imager software, also from BioTek.
For secreted cytokines and chemokines, the basolateral compartment media from TWIs were collected and analyzed for CSF2, IP10, MCP1, RANTES, IL6, and IL8 using custom MilliPlex assay kits (Millipore). Analyte concentrations were determined according to the manufacturer’s instructions using a Luminex FlexMap 3D system coupled with Luminex XPONENT software (Luminex).
hAEpCs at D0, 2, 8 and 35 post-ALI were removed from TWIs by incubating with Accutase (Invitrogen, Cat. #: 00-4555-56) for 20 minutes. Single-cell suspensions were fix in 4% paraformaldehyde for 15 minutes at 37C, permeabilized with 0.2% Triton X-100 for 15 minutes at room temperature, and then stained using the following primary antibodies: anti-β tubulin IV [β-tub IV] (GeneTex, Cat. #: GTX11315), anti-acetylated tubulin [α-tub] (Santa Cruz Biotechnology, Cat. #: SC-23950), anti-Keratin 5 [Krt5] (Biolegend, Cat. #: 905901), anti-nerve growth factor receptor [NGFR] (Biolegend, Cat. #: 345109), anti-mucin 5AC [Muc5AC] (Invitrogen, Cat. #: MA5-12178), anti-mucin 5B [Muc5B] (Santa Cruz Biotechnology, Cat. #: SC-21768), anti-Club cell protein 10 [CC10] (Proteintech, Cat. #: 26909-1-AP). Secondary antibodies used were Goat anti-chicken IgY-FITC (Biolegend, Cat. #: 410802), goat anti-mouse IgG-BV510 (Biolegend, Cat. #: 405331), and donkey anti-rabbit IgG-AF647(Biolegend, Cat. #: 406414). Basal cells were defined as either β-tub IV– Krt5+ or α-tub– NGFR+. Club and mucin-producing goblet cells were defined as CC10+ and Muc5AC+/Muc5B+, respectively. Ciliated cells were defined as either β-tub IV+ Krt5– or α-tub+ NGFR–.
Statistical analysis on BPIFA1 secretion (ELISA) and flow cytometry (on frequency of epithelial sub-types) was performed by non-parametric Mann-Whitney test using GraphPad PRISM. The multiplexed bead-based protein levels (cytokine/chemokine secretion) were analyzed by paired Student’s t-test (comparing each of time points D-2, D2, D4, D6, D8, D10, D12, D14, D16, and D18 post-ALI independently against D0) using GraphPad PRISM. The data were considered statistically significant when p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
We cultured primary human small airway epithelial cells (hSAEpCs) in vitro on porous membrane of transwell inserts and guided them to full differentiation under ALI for 28 days. Then, on days 0 (that is just prior to introducing ALI), 2, 4, 6, 8, 10, 14, 21 and 28 we lysed the cells in situ, collected their RNA and subjected them to whole-transcript expression analysis (Figure 1A). Our objective here was to identify molecular signatures based on trajectories of single genes that correlate with stage of hSAEpCs differentiation. We first classified genes into similar longitudinal profiles based on similarity of their gene expression patterns for all timepoints. We detected 1,953 differentially expressed genes in total with a fold change (FC) > 2 and p value of < 0.01 (Supplementary Table 3). We further reduced the number of genes, limiting ourselves to only those with the largest magnitude change in gene expression (FC > 8 and p value of < 0.01), resulting in 299 identified genes of interest (Supplementary Table 4). Using K-means clustering, we fit all 299 genes into 9 clusters with distinct expression profiles in order to isolate which genes were most indicative of differentiation (Figure 1B). We selected the number of clusters so that the majority, if not all, of genes within each cluster demonstrate a comparable expression pattern. Next, a total of seven genes were selected (given the partial similarity in gene expression in two pairs of the clusters) as potential biomarkers having a large change in gene expression in response to stage of differentiation. These were oligodendrocyte myelin glycoprotein (OMG), keratin 14 (KRT14), stanniocalcin 1 (STC1), BPI fold containing family A member 1 (BPIFA1, also known as SPLUNC1), phospholipase A2 group VII (PLA2G7), thioredoxin interacting protein (TXNIP) and S100 calcium binding protein A7 (S100A7). To validate our findings, we performed hemi-nested real-time qPCR on these genes on four representative time points (days 0, 2, 8 and 28 following ALI). Hemi-nested real-time qPCR, as opposed to conventional qPCR, was selected to ensure that very low transcript numbers over a wide dynamic range can be reliably detected in our studies. Moreover, to address possible inter-individual variability in identified gene expression profiles, we investigated four healthy donors, encompassing bronchial and bronchiolar epithelia. As illustrated in Figure 2A (right panel), 2B, the most consistent pattern of gene expression across all tested donors belonged to KRT14 and BPIFA1, both in agreement with microarray whole-transcript data [Figures 1, 2A (left panel)]. The detailed changes in genes expression analysis by qPCR is shown in Supplementary Table 5. The expression of KRT14 fell as early as D2 post-ALI (after normalization to reference housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and compared against D0) and continued to drop or stayed low until the last time-point on D28. The expression of BPIFA1 showed the exact opposite trend with a considerable increase dateable on D2, which kept on rising or stay stably high until D28. Plotting the average fold change in expression of the seven genes as heatmap (Figure 2C) further confirmed consistency and magnitude of change in KRT14 and BPIFA1 expression. Analysis also showed that expression of STC1, PLA2G7 and S100A7 can differentially mark D0, D2, D8 and D28 post-ALI.
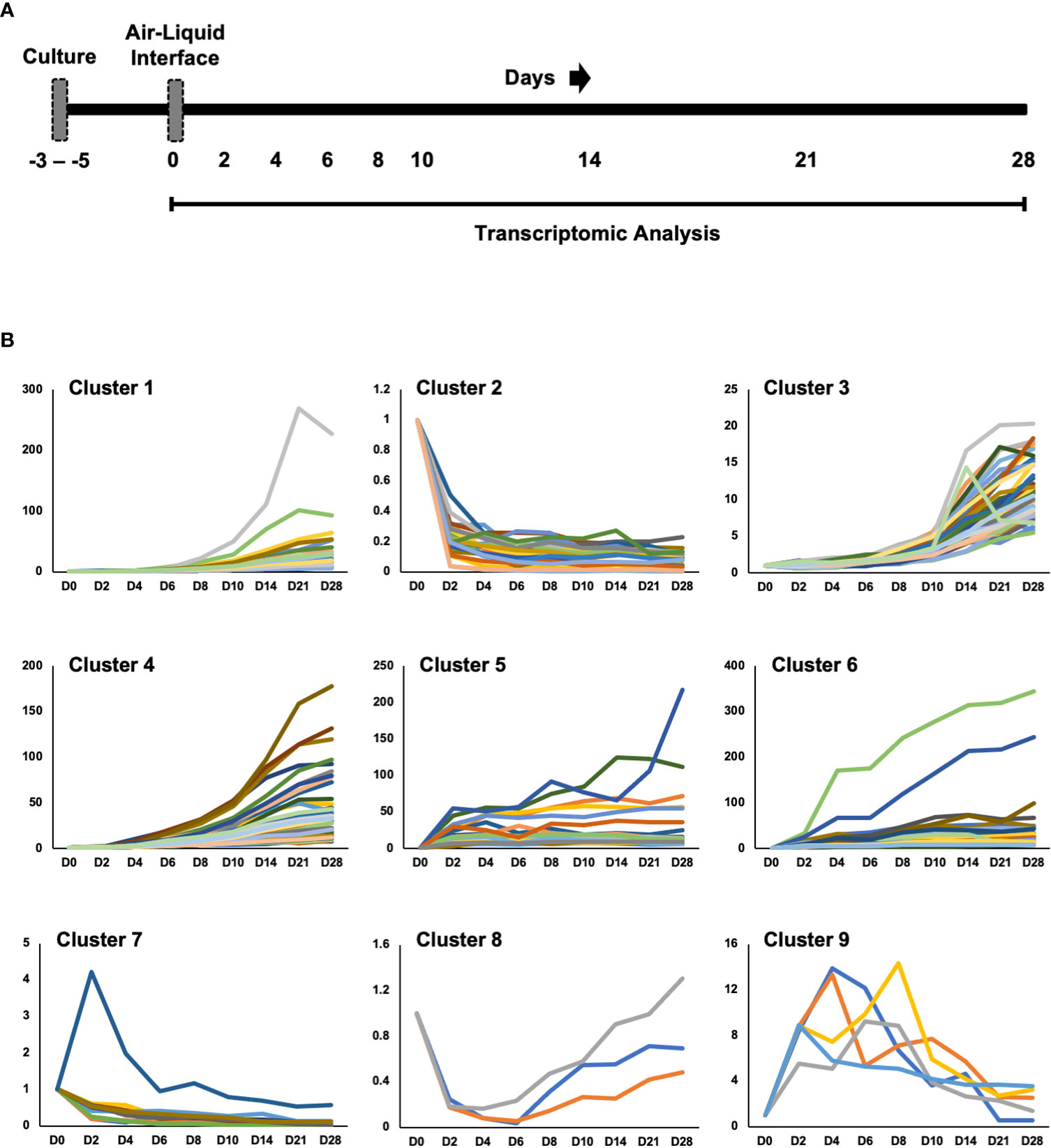
Figure 1 Study Design and Transcriptomic Analysis. (A) Diagram of healthy human small airway epithelial cells (hSAEpCs) (donor #1) guided through differentiation in vitro on transwell insert (TWI) porous membranes. The arrows indicate days following air-liquid interface (ALI) that cells were lysed in situ and used for transcriptomic analysis. (B) K-means clustering of genes differentially expressed during airway epithelial cell differentiation in vitro (these are genes with fold change [FC] > 8 and p < 0.01). D: days post-ALI; n = 3 replicates – i.e., TWI samples, per time point.
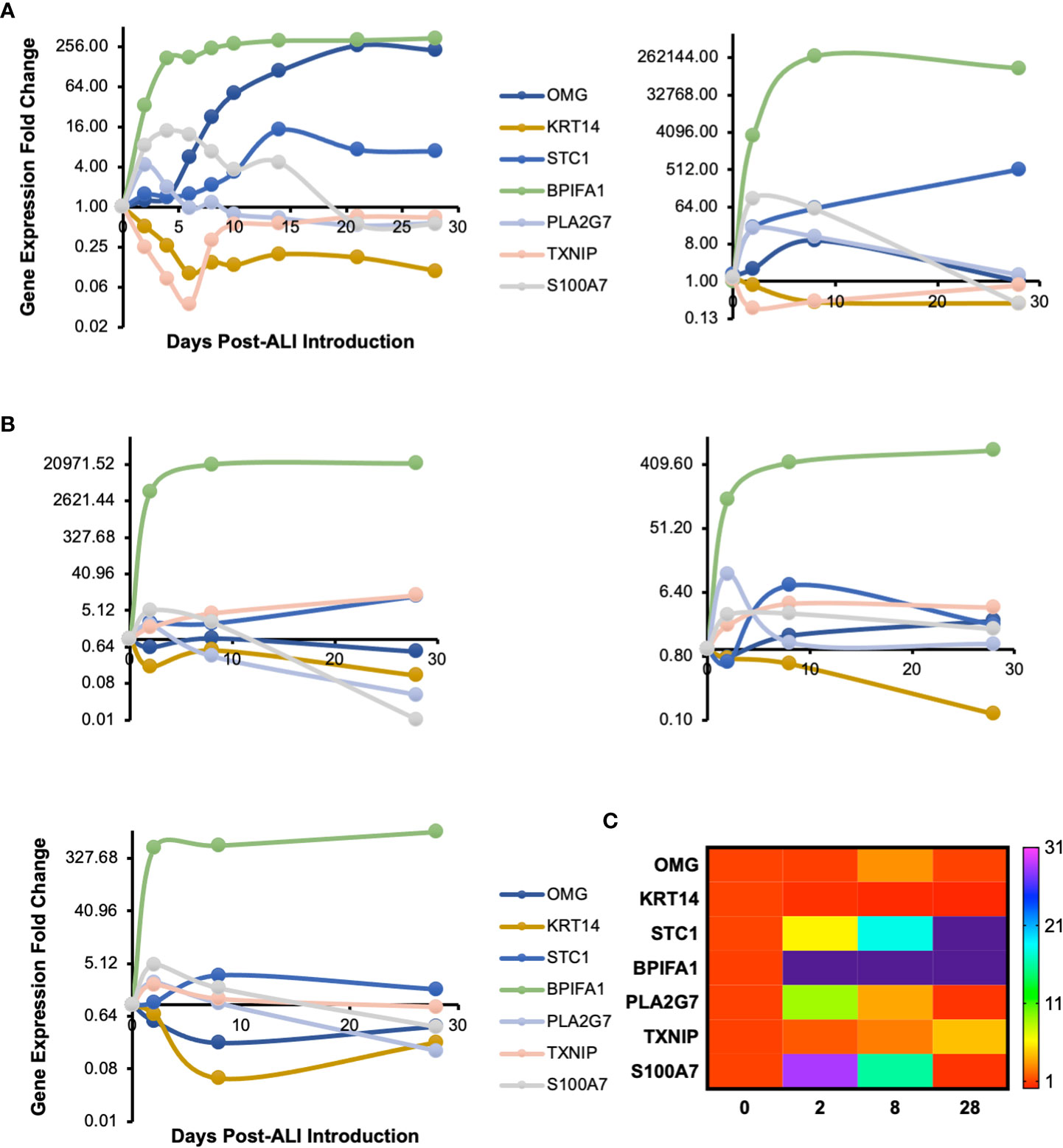
Figure 2 Transcriptomic Signature of In Vitro Differentiating Primary Human Airway Epithelial Cells. (A) Time course profile of expression of select genes from clusters plotted in Figure 1B. Left panel: microarray gene expression; right panel: hemi-nested qPCR validation of microarray data for days 0, 2, 8 and 28 post-ALI (same hSAEpC donor [donor #1]). (B) qPCR plots illustrating comparable patterns in expression of OMG, KRT14, STC1, BPIFA1, PLA2G7, TXNIP and S100A7 in three additional healthy human airway epithelial cell (hAEpC) donors, consistent with (A). Top left (donor #2) and right (donor #3) panels: human bronchial epithelial cells [hBEpCs]: bottom left panel: hSAEpCs (donor #4). (C) Heatmap illustration of change in expression of genes in (A) and (B) across all tested donors (donors #1–4). OMG, oligodendrocyte myelin glycoprotein; KRT14, keratin 14; STC1, stanniocalcin 1; BPIFA1, BPI fold containing family A member 1; PLA2G7, phospholipase A2 group VII; TXNIP, thioredoxin interacting protein; S100A7, S100 calcium binding protein A7.
BPIFA1 is an airway epithelium-derived multi-functional secretory protein that has been shown to have antimicrobial, ion channels-regulatory and airway smooth muscle-relaxing properties (Garcia-Caballero et al., 2009; Britto et al., 2013; Liu et al., 2013; Wu et al., 2017). So, we next evaluated change in secretion of BPIFA1 from basolateral surface of differentiating hAEpCs from multiple donors. We observed (Figure 3) that levels of BPIFA1 reached statistically significant high levels by D8 post-ALI (from undetectable at D0) and remained elevated throughout full differentiation (D28 post-ALI). This trend not only closely mimicked the transcript change in BPIFA1 expression, but also revealed that BPIFA1 can serve as a secreted biomarker of regenerating/differentiating human airway epithelium.
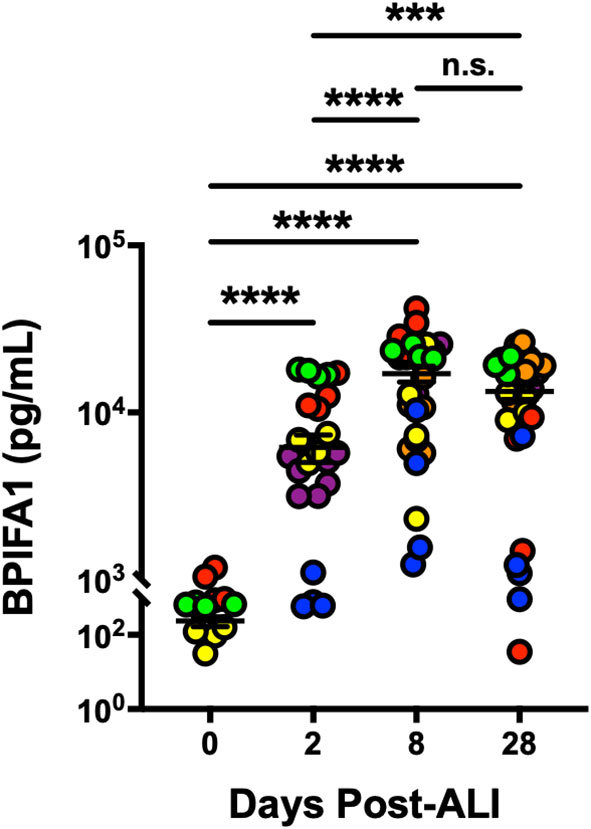
Figure 3 BPIFA1 Is a Secretory Biomarker of Differentiating Human Airway Epithelial Cells. Confirmation of BPIFA1 expression at protein level (secreted basolaterally into culture media) from six hAEpC donors (bronchial epithelial cells derived from two different donors [designated as yellow (donor #5) and red (donor #6)], and small airway epithelial cells derived from four separate donors [designated as purple (donor #7), orange (donor #8), blue (donor #9), and green (donor #10)]) with n = 4-7 replicates per donor. Data were analyzed by non-parametric Mann-Whitney test. ***p < 0.001, ****p < 0.0001, n.s., not significant.
Next, we evaluated abundance of bronchial and bronchiolar epithelial sub-types from multiple donors as they went through ALI differentiation in vitro (Figure 4A). We observed significant increase in frequency of secretory cells (goblet and Club cells) as well as ciliated cells as the epithelial cells differentiated, while the frequency of basal cells showed the opposite trend. Notably, emergence of goblet cells occurred as early as D2 post-ALI with additional increases evident on D8 and D35, whereas marked increase in abundance of Club and ciliated cells was evident only at D35 post-ALI upon full differentiation (Figure 4A). Our data indicated that differentiation into mucin-producing cells shows a similar pattern to BPIFA1 transcriptomic expression and protein secretion. Lastly, we examined baseline secretion of colony-stimulating factor 2 (CSF2), interferon-gamma-inducible protein 10 (IP10), monocyte chemoattractant protein-1 (MCP1), regulated upon activation, normal T cell expressed and presumably secreted (RANTES), interleukin 6 (IL6), and IL8, as representative inflammatory cytokines and chemokines, during the course of epithelial cells differentiation (Figure 4B). We found that the level of all these proteins increases with induction of ALI, peaking at D2-4 post ALI before showing a downward trend to low levels. Interestingly, this pattern inversely correlates with BPIFA1 secretion.
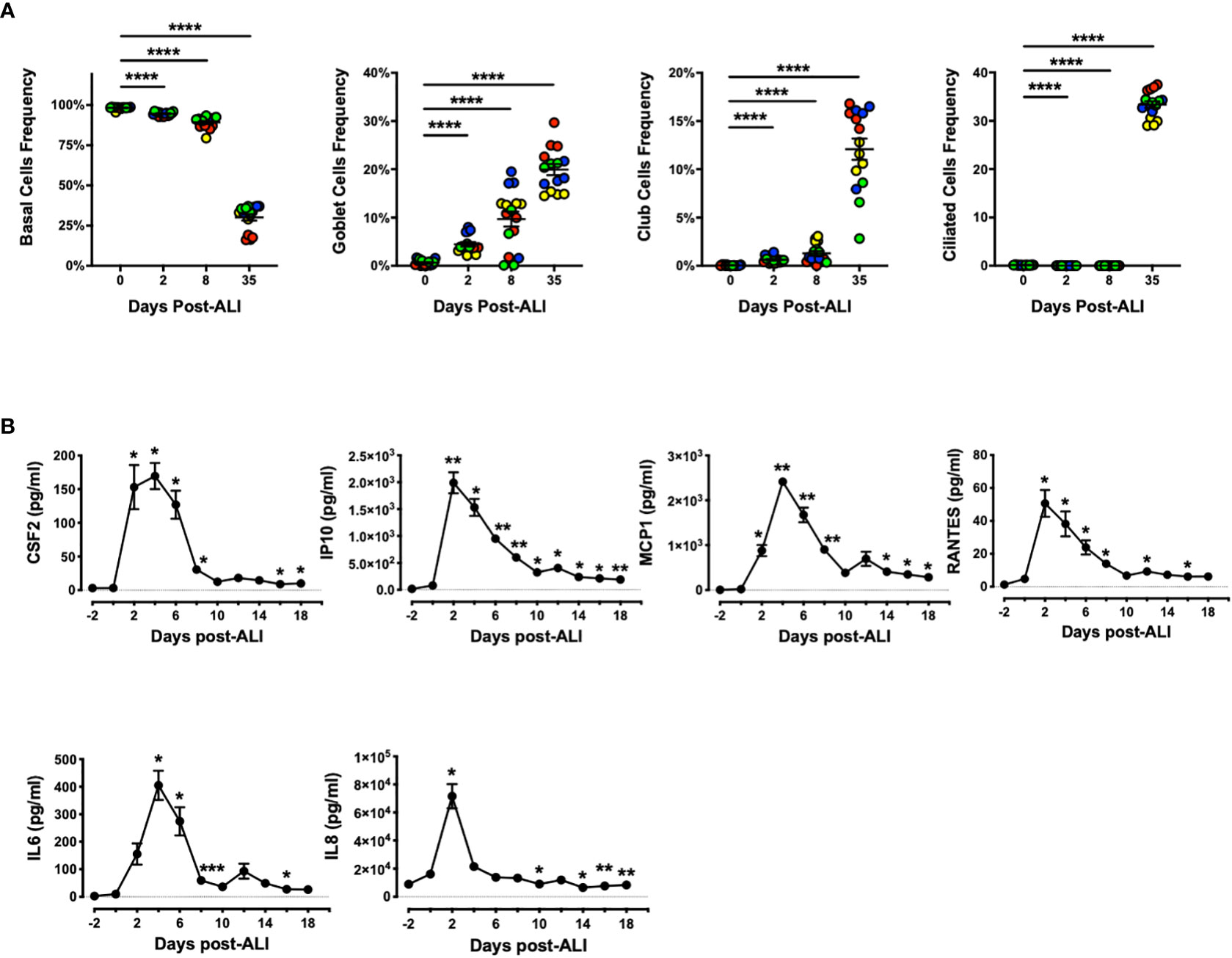
Figure 4 Change in Frequency of Airway Epithelial Sub-types and Secreted Inflammatory Proteins During In Vitro Differentiation. (A) The abundance of basal, goblet, Club, and ciliated cells were analyzed by flow cytometry on D0, 2, 8 and 35 post-ALI from four hAEpC donors (bronchial epithelial cells derived from two different donors [designated as yellow (donor #5) and red (donor #6)], and small airway epithelial cells derived from two separate donors [designated as blue (donor #9) and green (donor # 10)]) with n = 4 replicates per donor. Data were analyzed by non-parametric Mann-Whitney test. (B) Time course secretion analysis of inflammatory cytokines/chemokines for a representative human bronchial epithelial cell (donor #11) with n = 3–6 replicates. Data were analyzed by paired Student’s t-test test. *p < 0.05, **p < 0.01, ****p < 0.0001. CSF2, colony-stimulating factor 2; IP10, interferon-gamma-inducible protein 10; MCP1, monocyte chemoattractant protein-1; RANTES, regulated upon activation; normal T cell expressed and presumably secreted; IL6, interleukin 6; IL8, interleukin 8.
In this study, we cultured and differentiated hAEpCs (bronchial and bronchiolar) in vitro, applied whole-transcript expression analysis and found that the profile of OMG, KRT14, STC1, BPIFA1, PLA2G7, TXNIP, S100A7 expression creates a unique biosignature that reveals the stage of epithelial cells differentiation. In addition, we successfully validated our bulk transcriptomic data in multiple donors using hemi-nested qPCR, and showed that, at protein-level, secretion of epithelium-derived pathophysiologically relevant BPIFA1 well reflects the gene expression profile. Notably the patterns of genes and protein expression were comparable between the bronchial and bronchiolar (small airway) epithelial cells. Lastly, we correlated abundance of epithelial sub-types and secretory baseline inflammatory markers with levels of BPIFA1.
Here, our overarching goal was not to delineate biological processes or pathways that are induced or inhibited upon mucociliary differentiation, rather we wanted to identify individual genes whose expression states at any given timepoint, following ALI induction and after normalization to housekeeping gene, can provide insight into hAEpC differentiation stage. To obtain meaningful results from the relatively large data set comprising of 27 samples (9 timepoints × 3 replicates per timepoint), we used K-means clustering for stratifying our microarray gene expression data into clusters of genes with similar longitudinal expression patterns. Since K-means clustering requires a priori knowledge of the number of clusters, we empirically found that clustering the differentially expressed genes into 9 groups yields optimal clustering to identify patterns of genes that behave similarly while avoiding multiple groups with overly similar patterns.
Our experimental strategy that led to identification of BPIFA1 as a biomarker of differentiating hAEpCs is in line with studies by Campos et al. (Campos et al., 2004), where authors detected BPIFA1 (named ‘palate, lung, nasal epithelium clone protein (PLUNC)’ at the time) as a major protein in secretions of in vitro-cultured hAEpCs. However, while Campos and colleagues reported a dramatic increase in BPIFA1 release into culture media of hAEpCs during the second week of ALI (consistent with our findings), their studies predominantly focused on characterization of BPIFA1 as an epithelium-derived ethanol-soluble protein. In contrast, in our studies we applied a systemic analytical approach where evaluation of whole-transcript expression data led us to identification of BPIFA1 as a biomarker of differentiating epithelial cells under ALI. Moreover, Campos et al. neither examined change in BPIFA1 mRNA expression throughout mucociliary differentiation process, nor reported a transcript signature indicative of differentiation stage.
Regulation of BPIFA1 transcript and protein levels by differentiated state of human airway epithelium provides a new angle into understanding its biological significance. The majority of prior works focused on role of immune and inflammatory signals in driving or being influenced by BPIFA1 expression (Britto et al., 2013; Britto et al., 2019; Wrennall et al., 2021). For instance, Britto and colleagues using in vivo murine models demonstrated that Bpifa1 is key for pulmonary recruitment and transmigration of neutrophils to the airways in response to LPS challenge, and during acute lung inflammation, the most downregulated genes in Bpifa1-/- mice are Cxcl9 and Cxcl10 (Britto et al., 2019). Similarly, Bpifa1 was downregulated in bronchoalveolar lavage fluid (BALF) of C57Bl/6 mice following viral and bacterial pneumonia, and Th1 and Th2 inflammation inhibited its protein expression in vivo (Britto et al., 2013). As such an insight into change in expression of BPIFA1 based on differentiated state of the airway epithelium provides a new window for further characterization of its biological significance and potentially as a prognostic biomarker.
The additional 4 genes validated using hemi-nested qPCR may find further utility in expanding the differentiation biosignature for research and clinical applications by adding additional differentiation state resolution and sensitivity. KRT14 is a biomarker of airway basal stem cells. Therefore, its expression reduction following ALI is in line with observation that the number of basal cells drops and terminally differentiated cells (e.g., ciliated cells, secretory cells) increase in frequency post-ALI induction. STC1 has been for most part studied in the setting of cellular inflammation and carcinogenesis (Yeung et al., 2012; Zhao et al., 2020); however, a recent study found that STC1 is detectable in human blood serum, its level is significantly lower in asthmatics compared with healthy control subjects, and intranasal administration of recombinant human STC1 reduces airway hyperresponsiveness and inflammation in mice (Xu et al., 2020). These findings imply a protective role for STC1 (at least in the context of allergic asthma), and our gene expression data, for the first time, reveal that STC mRNA levels increase as hAEpCs differentiate. In our studies, expression of PLA2G7 and S100A7 peaked on D2 and then started to drop to D0 or lower levels by D28 post-ALI. PLA2G7 is a secreted enzyme which degrades and inactivates platelet-activating factor (PAF) (Karabina and Ninio, 2006), yet its expression in respiratory epithelium had not been reported before. Here, we show the first evidence of PLA2G7 expression and its correlation with stage of differentiation in hAEpCs. Antimicrobial peptide S100A7, whose expression in our studies was comparable to that of PLA2G7, has been shown to be expressed by well-differentiated human airway epithelia (Andresen et al., 2011); however, here we provide the first evidence on correlation of S100A7 gene expression with stage of hAEpCs differentiation in vitro.
The transcriptomic molecular signature we identified in this study may serve as an initial or complementary step for utilization as diagnostic in clinical setting by detecting human airway epithelial regeneration state. This requires thorough validation of our results in large sample numbers (to be obtained, for instance, through airway brushing of healthy and diseased donors). The clinical need for such use should also be clearly laid out upfront, as airway brushing, lung biopsy acquisition or bronchoalveolar lavage fluid collection may not be feasible in all patient settings. In addition, the impact of an underlying co-morbidity must be taken into consideration.
In our studies, we also demonstrated that induction of BPIFA1 expression can likely be attributed to emergence of mucin-producing goblet cells as these cells appeared as early as two days following ALI and their number further increased on days 8 and 35. This is consistent with earlier reports. Using affinity-purified antibodies, Bingle and colleagues showed that BPIFA1 is a secreted product of goblet cells in vitro and in airways of whole human lungs (Bingle et al., 2010; Bingle et al., 2012). Interestingly, De Smet et al., through histochemical and transcriptomic analysis of airway in a large cohort of never-smokers and smokers with and without COPD, found that BPIFA1 is elevated in COPD patients and correlates with disease severity and goblet cell hyperplasia (De Smet et al., 2018). It has been shown that BPIFA1 expression in lungs of laboratory mice occurs in Club cells, co-localizing with secretoglobin family 1A member 1 (SCGB1A1) (Leeming et al., 2015). Thus, it is likely that this epithelial sub-type in our human in vitro cultures may also contribute to BPIFA1 secretion as cells differentiate.
It has been reported that BPIFA1, in addition to its antimicrobial, ion transport-influencing, and smooth muscle-relaxing properties, has an immunomodulatory function (Gaillard et al., 2010; Britto et al., 2013; Britto and Cohn, 2015; Walton et al., 2016; Wu et al., 2017; Akram et al., 2018; Khanal et al., 2021). Chu et al. reported that BPIFA1 significantly suppresses human airway epithelial IL8 production induced by Mycoplasma pneumoniae-derived lipoproteins and Toll-like receptor 2 (TLR2) ligand Pam3CSK4 (Chu et al., 2007). Another study reported that BPIFA1 inhibits airway eosinophilic inflammation in a murine airway allergic inflammation in vivo, and attenuates LPS-induced eotaxin-2 production from alveolar macrophages (Thaikoottathil et al., 2012). These findings are in line with decrease in inflammatory cytokine/chemokine secretion from hAEpCs in our studies while BPIFA1 levels go up. While we attribute the anti-inflammatory effect to BPIFA1 during epithelial cells differentiation, future studies are needed to validate this using blocking antibodies, small molecule inhibitors and/or genetic knockout systems.
Our studies here have a number of drawbacks. We drew our conclusions based on the limited number of donors we tested. In addition, we studied the differentiating epithelia in the absence of other organ-level complexities such as other lung airway cells/tissues (e.g., vascular endothelium, airway smooth muscle, fibroblasts, tissue-resident immune cells), physiologically relevant mechanical cues (e.g., rhythmic airflow associated with breathing) and sub-epithelial extracellular matrix. We also did not investigate the expression profile in diseased subject-derived epithelial cells. Future studies should address these drawbacks. Lastly, we plan in future studies to evaluate apical secretion of BPIFA1 to understand whether its levels correlate with basally secreted pattern, and to what is found in sputum or bronchoalveolar lavage of human subjects at health and disease.
In summary, this study, based on a limited number of primary human airway epithelial cell donors, reveals a unique transcriptomic molecular signature for various stages of mucociliary differentiation. It also shows that protein production of BPIFA1 closely mimics its gene expression profile, pointing to BPIFA1 as a secreted biomarker of regenerating human airway epithelium in vitro.
The datasets presented in this study have been deposited in Gene Expression Omnibus (GEO) under accession number GSE197142, which is available via https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197142.
KHB and RN conceptualized the initial gene expression studies; KHB designed and planned the study direction; KHB, CC, BFN, UC performed qPCR and protein secretion experiments; BFN performed flow cytometry studies, KHB and RN designed and planned hemi-nested qPCR; KH performed qPCR conditions optimization; RN analyzed the microarray gene expression data; KHB wrote up the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Division of Pulmonary, Allergy and Critical Care Medicine at University of Pittsburgh, the Wyss Institute for Biologically Inspired Engineering at Harvard University, the U.S. National Institutes of Health (U01EB029085; R41ES031639), and the U.S. Department of Defense Congressionally Directed Medical Research Programs Discovery Award (W81XWH2010035). The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the U.S. Department of Defense or the U.S. government.
We thank Ms. Eva Zittel for her assistance with qPCR primers testing, staff at Molecular Biology Core Facilities, Dana-Farber Cancer Institute, for their assistance with sample preparation and microarray chip processing for gene expression studies.
KB is founder and holds equity in Pneumax, LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.1035566/full#supplementary-material
Akram, K. M., Moyo, N. A., Leeming, G. H., Bingle, L., Jasim, S., Hussain, S., et al. (2018). An innate defense peptide BPIFA1/SPLUNC1 restricts influenza a virus infection. Mucosal Immunol. 11, 71–81. doi: 10.1038/mi.2017.45
Andresen, E., Lange, C, Strodthoff, D., Goldmann, T., Fischer, N., Sahly, H., et al. (2011). S100A7/psoriasin expression in the human lung: unchanged in patients with COPD, but upregulated upon positive s. aureus detection. BMC Pulm Med. 11, 10. doi: 10.1186/1471-2466-11-10
Benam, K. H., Novak, R., Nawroth, J., Hirano-Kobayashi, M., Ferrante, T. C., Choe, Y., et al. (2016). Matched-comparative modeling of normal and diseased human airway responses using a microengineered breathing lung chip. Cell Syst. 3(5), 456–466.e454. doi: 10.1016/j.cels.2016.10.003
Benam, K. H., Kok, W. L., McMichael, A. J., Ho, L. P. (2011). Alternative spliced CD1d transcripts in human bronchial epithelial cells. PLoS One 6, e22726. doi: 10.1371/journal.pone.0022726
Bingle, C. D., Wilson, K., Lunn, H., Barnes, F. A., High, A. S., Wallace, W. A., et al. (2010). Human LPLUNC1 is a secreted product of goblet cells and minor glands of the respiratory and upper aerodigestive tracts. Histochem Cell Biol. 133, 505–515. doi: 10.1007/s00418-010-0683-0
Bingle, L., Wilson, K., Musa, M., Araujo, B., Rassl, D., Wallace, W. A., et al. (2012). BPIFB1 (LPLUNC1) is upregulated in cystic fibrosis lung disease. Histochem Cell Biol. 138 (5), 749–758. doi: 10.1007/s00418-012-0990-8
Bonser, L. R., Koh, K. D., Johansson, K., Choksi, S. P., Cheng, D., Liu, L., et al. (2021). Flow-cytometric analysis and purification of airway epithelial-cell subsets. Am. J. Respir. Cell Mol. Biol. 64, 308–317. doi: 10.1165/rcmb.2020-0149MA
Britto, C. J., Cohn, L. (2015). Bactericidal/Permeability-increasing protein fold-containing family member A1 in airway host protection and respiratory disease. Am. J. Respir. Cell Mol. Biol. 52, 525–534. doi: 10.1165/rcmb.2014-0297RT
Britto, C. J., Liu, Q., Curran, D. R., Patham, B., Dela Cruz, C. S., Cohn, L.. (2013). Short palate, lung, and nasal epithelial clone-1 is a tightly regulated airway sensor in innate and adaptive immunity. Am. J. Respir. Cell Mol. Biol. 48, 717–724. doi: 10.1165/rcmb.2012-0072OC
Britto, C. J., Niu, N., Khanal, S., Huleihel, L., Herazo-Maya, J. D., Thompson, A., et al. (2019). BPIFA1 regulates lung neutrophil recruitment and interferon signaling during acute inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 316, L321–L333. doi: 10.1152/ajplung.00056.2018
Button, B., Cai, L.-H., Ehre, C., Kesimer, M., Hill, D. B., Sheehan, J. K., et al. (2012). A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337, 937–941. doi: 10.1126/science.1223012
Callejas-Diaz, B., Fernandez, G., Fuentes, M., Martínez-Antón, A., Alobid, I., Roca-Ferrer, J., et al. (2020). Integrated mRNA and microRNA transcriptome profiling during differentiation of human nasal polyp epithelium reveals an altered ciliogenesis. Allergy 75, 2548–2561. doi: 10.1111/all.14307
Campos, M. A., Abreu, A. R., Nlend, M. C., Cobas, M. A., Conner, G. E., Whitney, P. L.. (2004). Purification and characterization of PLUNC from human tracheobronchial secretions. Am. J. Respir. Cell Mol. Biol. 30, 184–192. doi: 10.1165/rcmb.2003-0142OC
Chu, H. W., Thaikoottathil, J., Rino, J. G., Zhang, G., Wu, Q., Moss, T., et al. (2007). Function and regulation of SPLUNC1 protein in mycoplasma infection and allergic inflammation. J. Immunol. 179 (6), 3995–4002. doi: 10.4049/jimmunol.179.6.3995
De Smet, E. G., Leen Seys, L. J., Verhamme, F. M., Vanaudenaerde, B. M., Brusselle, G. G., Bingle, C. D., et al. (2018). Association of innate defense proteins BPIFA1 and BPIFB1 with disease severity in COPD. Int. J. Chron Obstruct Pulmon Dis. 13, 11–27. doi: 10.2147/COPD.S144136
Everman, J. L., Rios, C., Seibold, M. A. (2018). Primary airway epithelial cell gene editing using CRISPR-Cas9. Methods Mol. Biol. 1706, 267–292. doi: 10.1007/978-1-4939-7471-9_15
Fulcher, M. L., Gabriel, S., Burns, K. A., Yankaskas, J. R., Randell, S. H. (2005). Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 107, 183–206. doi: 10.1385/1-59259-861-7:183
Gaillard, E. A., Kota, P., Gentzsch, M., Dokholyan, N. V., Stutts, M. J., Tarran, R.. (2010). Regulation of the epithelial na+ channel and airway surface liquid volume by serine proteases. Pflugers Arch. 460 (1), 1–17. doi: 10.1007/s00424-010-0827-z
Garcia-Caballero, A., Rasmussen, J. E., Gaillard, E., Watson, M. J., Olsen, J. C., Donaldson, S. H., et al. (2009). SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. U.S.A. 106, 11412–11417. doi: 10.1073/pnas.0903609106
Ibricevic, A., Pekosz, A., Walter, M. J., Newby, C., Battaile, J. T., Brown, E. G., et al. (2006). Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 80 (15), 7469–7480. doi: 10.1128/JVI.02677-05
Jiang, D., Schaefer, N., Chu, H. W. (2018). Air-liquid interface culture of human and mouse airway epithelial cells. Methods Mol. Biol. 1809, 91–109. doi: 10.1007/978-1-4939-8570-8_8
Karabina, S. A., Ninio, E. (2006). Plasma PAF-acetylhydrolase: an unfulfilled promise? Biochim. Biophys. Acta 1761, 1351–1358. doi: 10.1016/j.bbalip.2006.05.008
Khanal, S., Webster, M., Niu, N., Zielonka, J., Nunez, M., Chupp, G., et al. (2021). SPLUNC1: A novel marker of cystic fibrosis exacerbations. Eur. Respir. J. 58. doi: 10.1183/13993003.00507-2020
Leeming, G. H., Kipar, A., Hughes, D. J., Bingle, L., Bennett, E., Moyo, N. A., et al. (2015). Gammaherpesvirus infection modulates the temporal and spatial expression of SCGB1A1 (CCSP) and BPIFA1 (SPLUNC1) in the respiratory tract. Lab. Invest. 95, 610–624. doi: 10.1038/labinvest.2014.162
Liu, Y., Di, M. E., Chu, H. W., Liu, X., Wang, L., Wenzel, S., et al. (2013). Increased susceptibility to pulmonary pseudomonas infection in Splunc1 knockout mice. J. Immunol. 191, 4259–4268. doi: 10.4049/jimmunol.1202340
Manzanares, D., Krick, S., Baumlin, N., Dennis, J. S., Tyrrell, J., Tarran, R., et al. (2015). Airway surface dehydration by transforming growth factor beta (TGF-beta) in cystic fibrosis is due to decreased function of a voltage-dependent potassium channel and can be rescued by the drug pirfenidone. J. Biol. Chem. 290, 25710–25716. doi: 10.1074/jbc.M115.670885
Martinez-Anton, A., Sokolowska, M., Kern, S., Davis, A. S., Alsaaty, S., Taubenberger, J. K., et al. (2013). Changes in microRNA and mRNA expression with differentiation of human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 49, 384–395. doi: 10.1165/rcmb.2012-0368OC
Schmid, A., Bai, G., Schmid, N., Zaccolo, M., Ostrowski, L. E., Conner, G. E., et al. (2006). Real-time analysis of cAMP-mediated regulation of ciliary motility in single primary human airway epithelial cells. J. Cell Sci. 119, 4176–4186. doi: 10.1242/jcs.03181
Thaikoottathil, J. V., Martin, R. J., Di, P. Y., Minor, M., Case, S., Zhang, B., et al. (2012). SPLUNC1 deficiency enhances airway eosinophilic inflammation in mice. Am. J. Respir. Cell Mol. Biol. 47, 253–260. doi: 10.1165/rcmb.2012-0064OC
Walton, W. G., Ahmad, S., Little, M. S., Kim, C. S. K., Tyrrell, J., Lin, Q., et al. (2016). Structural features essential to the antimicrobial functions of human SPLUNC1. Biochemistry 55, 2979–2991. doi: 10.1021/acs.biochem.6b00271
Wrennall, J. A., Ahmad, S., Worthington, E. N., Wu, T., Goriounova, A. S., Voeller, A. S., et al. (2021). A SPLUNC1 peptidomimetic inhibits Orai1 and reduces inflammation in a murine allergic asthma model. Am. J. Respir. Cell Mol. Biol. 66(3), 271–282. doi: 10.1165/rcmb.2020-0452OC
Wu, T., Huang, J., Moore, P. J., Little, M. S., Walton, W. G., Fellner, R. C., et al. (2017). Identification of BPIFA1/SPLUNC1 as an epithelium-derived smooth muscle relaxing factor. Nat. Commun. 8, 14118. doi: 10.1038/ncomms14118
Xu, J., Meng, Y., Jia, M., Jiang, J., Yang, Y., Ou, Y., et al. (2020). Epithelial expression and role of secreted STC1 on asthma airway hyperresponsiveness through calcium channel modulation. Allergy. 76 (8), 2475–2487. doi: 10.1111/all.14727
Yeung, B. H., Law, A. Y., Wong, C. K. (2012). Evolution and roles of stanniocalcin. Mol. Cell Endocrinol. 349, 272–280. doi: 10.1016/j.mce.2011.11.007
Zhao, J., Minami, Y., Etling, E., Coleman, J. M., Lauder, S. N., Tyrrell, V., et al. (2017). Preferential generation of 15-HETE-PE induced by IL-13 regulates goblet cell differentiation in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 57, 692–701. doi: 10.1165/rcmb.2017-0031OC
Keywords: BPIFA1, human airway epithelium, biomarker, differentiation, inflammation
Citation: Clifton C, Niemeyer BF, Novak R, Can UI, Hainline K and Benam KH (2022) BPIFA1 is a secreted biomarker of differentiating human airway epithelium. Front. Cell. Infect. Microbiol. 12:1035566. doi: 10.3389/fcimb.2022.1035566
Received: 02 September 2022; Accepted: 07 November 2022;
Published: 28 November 2022.
Edited by:
Veronica Krenn, University of Milano-Bicocca, ItalyReviewed by:
Xiaopeng Qi, Shandong University, ChinaCopyright © 2022 Clifton, Niemeyer, Novak, Can, Hainline and Benam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kambez H. Benam, YmVuYW1rQHBpdHQuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.