 Reshma S. Rudraraju1†
Reshma S. Rudraraju1† Samer S. Daher2†Ricardo Gallardo-Macias2Xin Wang3
Samer S. Daher2†Ricardo Gallardo-Macias2Xin Wang3 Matthew B. Neiditch1*‡
Matthew B. Neiditch1*‡ Joel S. Freundlich2,4*‡
Joel S. Freundlich2,4*‡- 1Department of Microbiology, Biochemistry and Molecular Genetics, New Jersey Medical School, Rutgers University, Newark, NJ, United States
- 2Department of Pharmacology, Physiology, and Neuroscience, New Jersey Medical School, Rutgers University, Newark, NJ, United States
- 3Department of Immunology and Infectious Diseases, Harvard University T.H. Chan School of Public Health, Boston, MA, United States
- 4Department of Medicine, Center for Emerging and Re-emerging Pathogens, New Jersey Medical School, Rutgers University, Newark, NJ, United States
Recent studies have reported the β-ketoacyl-acyl carrier protein KasA as a druggable target for Mycobacterium tuberculosis. This review summarizes the current status of major classes of KasA inhibitors with an emphasis on significant contributions from structure-based design methods leveraging X-ray crystal structures of KasA alone and in complex with inhibitors. The issues addressed within each inhibitor class are discussed while detailing the characterized interactions with KasA and structure-activity relationships. A critical analysis of these findings should lay the foundation for new KasA inhibitors to study the basic biology of M. tuberculosis and to form the basis of new antitubercular molecules of clinical significance with activity against drug-sensitive and drug-resistant infections.
The cell wall of Mycobacterium tuberculosis plays a defining role in terms of its interactions with the host immune system and with antitubercular drugs (Rahlwes et al., 2019; Dulberger et al., 2020). Its cell wall is comprised of peptidoglycan (PG) covalently attached to the heteropolysaccharide arabinogalactan (AG) via phosphoryl-N-acetylglucosaminosyl-rhamnosyl linkage units (P-GlcNAc-Rha). AG is in turn esterified at its non-reducing ends to long α-alkyl β-hydroxy fatty acids known as mycolic acids (MAs). This PG-AG-MA triumvirate is viewed as the inner leaflet of a mycomembrane with the outer leaflet primarily comprised of non-covalently bound mycolic acids, trehalose monomycolate, and trehalose dimycolate (Zuber et al., 2008; Hoffmann et al., 2008; Marrakchi et al., 2014). MAs are synthesized by the fatty acid synthase-I (FAS-I) and fatty acid synthase-II (FAS-II) proteins (Figure 1) (Brennan and Nikaido, 1995; Cole et al., 1998). The FAS-I cycle generates fatty acids (FAs) with shorter chain fragments ranging up to C16-C18 and C24-C26 (Takayama et al., 2005), in which the latter corresponds to the α-branch found in MAs. The FAS-II cycle further elongates the FAs to afford C50-C56 meromycolates (Marrakchi et al., 2014). Pks13 condenses the FAS-I cycle fatty acid product and the FAS-II cycle meromycolic acid product to form the MA (Portevin et al., 2004; Leger et al., 2009; Gavalda et al., 2009). Despite the fact that the two cycles differ in the carrier protein, molecular organization, and substrates, they perform analogous reaction sequences with an iterative series of steps relying on consecutive additions of a two-carbon (acetate) unit ultimately from malonyl-Coenzyme A (CoA) to an acyl moiety. The malonyl group is transferred from malonyl-CoA to the mycobacterial acyl carrier protein (AcpM) by malonyl CoA-ACP transacylase (MtFabD, Rv2243) to form malonyl-AcpM. The condensation of malonyl-AcpM with acyl-CoA is catalyzed by the β-ketoacyl-ACP synthase III (MtFabH, Rv0533c) to form β-ketoacyl-AcpM and, thus, link the FAS-I and FAS-II cycles. Generally, there are four main enzymes involved in each cycle of elongation. The nicotinamide adenine dinucleotide phosphate-dependent 3-ketoacyl-acyl carrier protein reductase (MabA, Rv1483) reduces the β-keto group. Heterodimeric (3R)-hydroxyacyl-ACP dehydratase (HadAB, Rv0635 – Rv0636, and HadBC, Rv0636 – Rv0637) subsequently dehydrates the resulting β-hydroxy intermediate into enoyl-AcpM. Then, the nicotinamide adenine dinucleotide hydrogen (NADH)-dependent trans-2-enoyl-ACP reductase (InhA, Rv1484) reduces enoyl-AcpM into acyl-AcpM. Subsequent cycles of elongation are carried out by KasA (Rv2245) and KasB (Rv2246) (Figure 2) that elongate the acyl-AcpM by two carbons to form the β-ketoacyl-AcpM. KasA and KasB are 67% identical and 86% similar by protein sequence, and they exhibit differential substrate preferences (Bhatt et al., 2007; Bhatt et al., 2007).
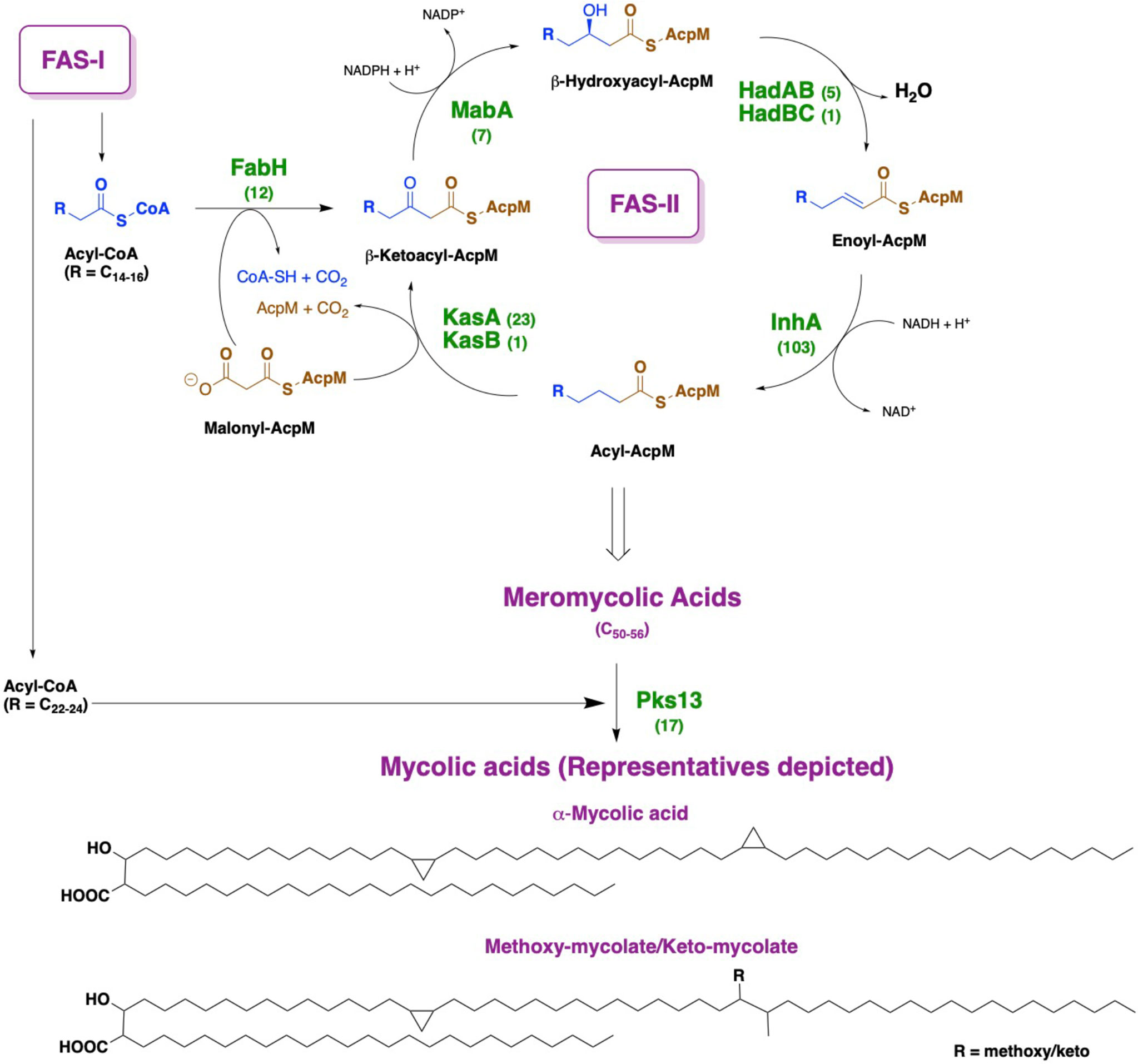
Figure 1 Depiction of mycolic acid biosynthesis through the FAS-I and FAS-II pathways. AcpM: mycobacterial acyl carrier protein. By each protein name, the number of corresponding X-ray crystal structures deposited in the Protein Data Bank is annotated.
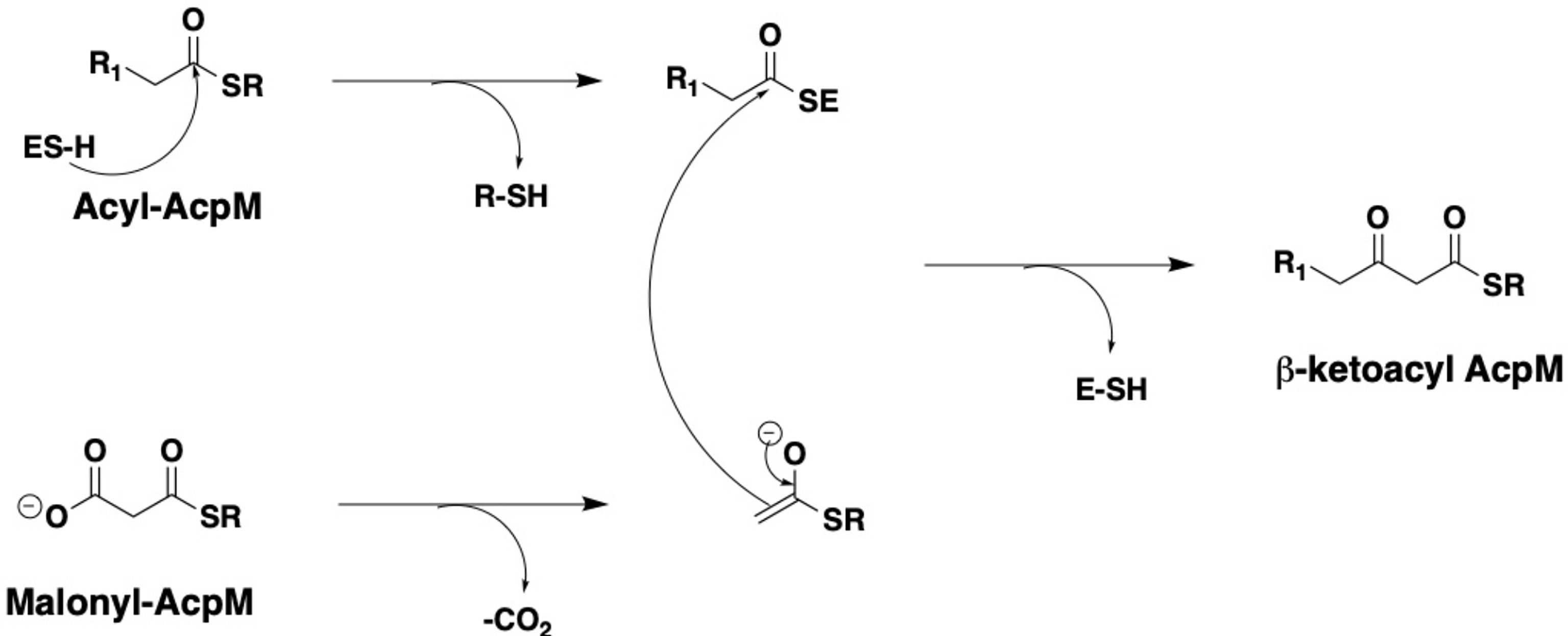
Figure 2 General catalytic mechanism of the KasA and KasB enzymes.
MA synthesis inhibition has represented a fertile ground for antitubercular drug targeting (Abrahams and Besra, 2020; Fernandes et al., 2022; Dartois and Rubin, 2022), and this is undoubtedly due to the availability of X-ray crystal structures of many involved proteins (Figure 1) in the Protein Data Bank (www.rcsb.org). This strategy is highly favorable because developed inhibitors are expected to lack mechanism-based toxicity as mammals rely primarily on a FAS-I system (Bhatt et al., 2007). For instance, isoxyl and thioacetazone are known to target the HadAB/HadBC (3R)-hydroxyacyl-ACP dehydratase complex, whereas isoniazid and ethionamide prevent the reduction of enoyl-AcpM by targeting InhA (Banerjee et al., 1994; Vilcheze et al., 2006). We highlight the fact that an M. tuberculosis β-ketoacyl synthase is not currently the target of a tuberculosis drug. Thus, a therapeutic inhibiting KasA should be clinically useful versus both drug-sensitive and drug-resistant infections.
KasA is an essential and vulnerable drug target in vitro
kasA has been identified as an essential gene for the M. tuberculosis laboratory strain H37Rv under in vitro growth conditions via multiple transposon-based approaches, including the original transposon site hybridization method (TRaSH) (Sassetti et al., 2003), deep sequencing of transposon insertions (TnSeq) (Griffin et al., 2011; Zhang et al., 2012), and a more comprehensive TnSeq analysis (DeJesus et al., 2017) to account for TA insertability with a hidden Markov model. In contrast, the in vitro essentiality of kasB appears to be dependent on the experimental conditions (growth media and nature of genetic disruption) (Minato et al., 2019). Extensive studies show no evidence of kasA as a conditional non-essential gene, indicating that the in vitro requirement for kasA is independent of culture conditions. Consistent with these findings with laboratory strains, kasA was also revealed as essential for in vitro cultures of clinical isolates belonging to the most prevalent lineages: Euro-American, East Asian, and Indo-Oceanic (Carey et al., 2018). A gene vulnerability study based on the mycobacterial CRISPRi system (Rock et al., 2017) identified kasA as a vulnerable gene in both laboratory strain H37Rv and clinical isolate HN878 (Bosch et al., 2021). These studies indicate that M. tuberculosis in vitro growth is highly dependent on kasA and the FAS-II pathway in general. The data supporting the in vitro essentiality and vulnerability of kasA in M. tuberculosis have created a significant impetus to attain in vivo validation of this drug target.
Protein crystallography has provided key insights into KasA catalysis
X-ray crystallographic analyses of KasA alone (or apo) (Luckner et al., 2009; Kumar et al., 2018), KasA-C171Q in complex with fortuitously-bound phospholipid (Luckner et al., 2009; Schiebel et al., 2013), and KasA bound to small molecule inhibitors (Schiebel et al., 2013; Abrahams et al., 2016; Kumar et al., 2018; Cunningham et al., 2020; Inoyama et al., 2020) have provided atomic-level mechanistic insights into the KasA structure. In the crystals, KasA exhibits C2 symmetry and is composed of a five-layer αβαβα structure, a fingerprint of thiolases (Figure 3). Based on our understanding of KasA structure and function, and for the benefit of discussion, it can be divided into two regions – the core and cap domains. Within the core domain lies the catalytic triad residues (Cys171, His311, and His345) and the phoshopantetheine tunnel, which opens into the malonyl binding region. The proposed role of the catalytic triad in KasA acylation, decarboxylation and condensation is described in Figure 4. The acyl channel is a hydrophobic channel in the cap region formed by helices α2, α5, α9 and α5′. It can be accessed either through the malonyl binding site or from the opening of acyl channel in the cap region. It accommodates the growing acyl chain. The acyl chain binding site is traced by the phospholipid bound to KasA in PDB ID 4C6W (Schiebel et al., 2013). The cap domain is disordered in the apo structure but becomes ordered upon acylation of KasA Cys171 (Luckner et al., 2009; Kumar et al., 2018). The cap domain is proposed to be responsible for significant functional differences between KasA and KasB (Schiebel et al., 2013). How the core and cap domains function to condense the acyl donor and malonyl-AcpM substrates to form β-ketoacyl-AcpM products is outlined below.
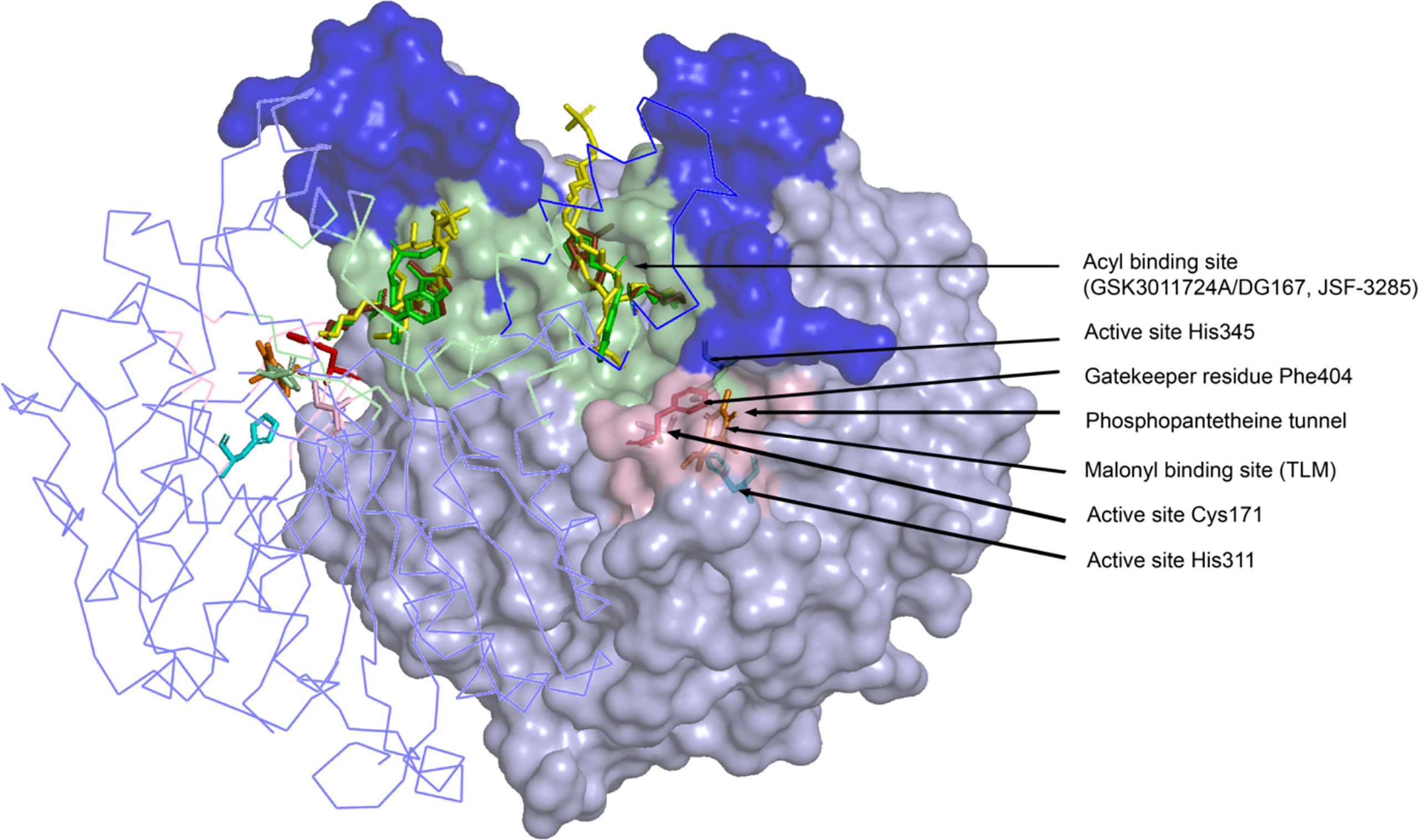
Figure 3 Crystal structure of the KasA dimer with inhibitors bound (GSK3011724A/DG167, JSF-3285, TLM) at different binding sites. The monomer on the left is represented as a ribbon tracing the alpha carbons and the monomer on the right is depicted as a surface. The dark blue surface indicates the cap region of KasA excluding the acyl channel whereas the light blue surface indicates the core domain; Light green surface, acyl binding site; pink surface, malonyl binding site; yellow sticks; phospholipid bound to KasA (PDB ID 4C6W); green sticks; DG167 (PDB ID 5W2P); brown sticks; JSF-3285 (PDB ID 6P9L); orange sticks; TLM (PDB ID 4C6U); pink sticks, Cys171; red sticks, Phe404; cyan sticks, His 311 and His 345. For clarity, only the KasA dimer from PDB ID 6P9L is shown.
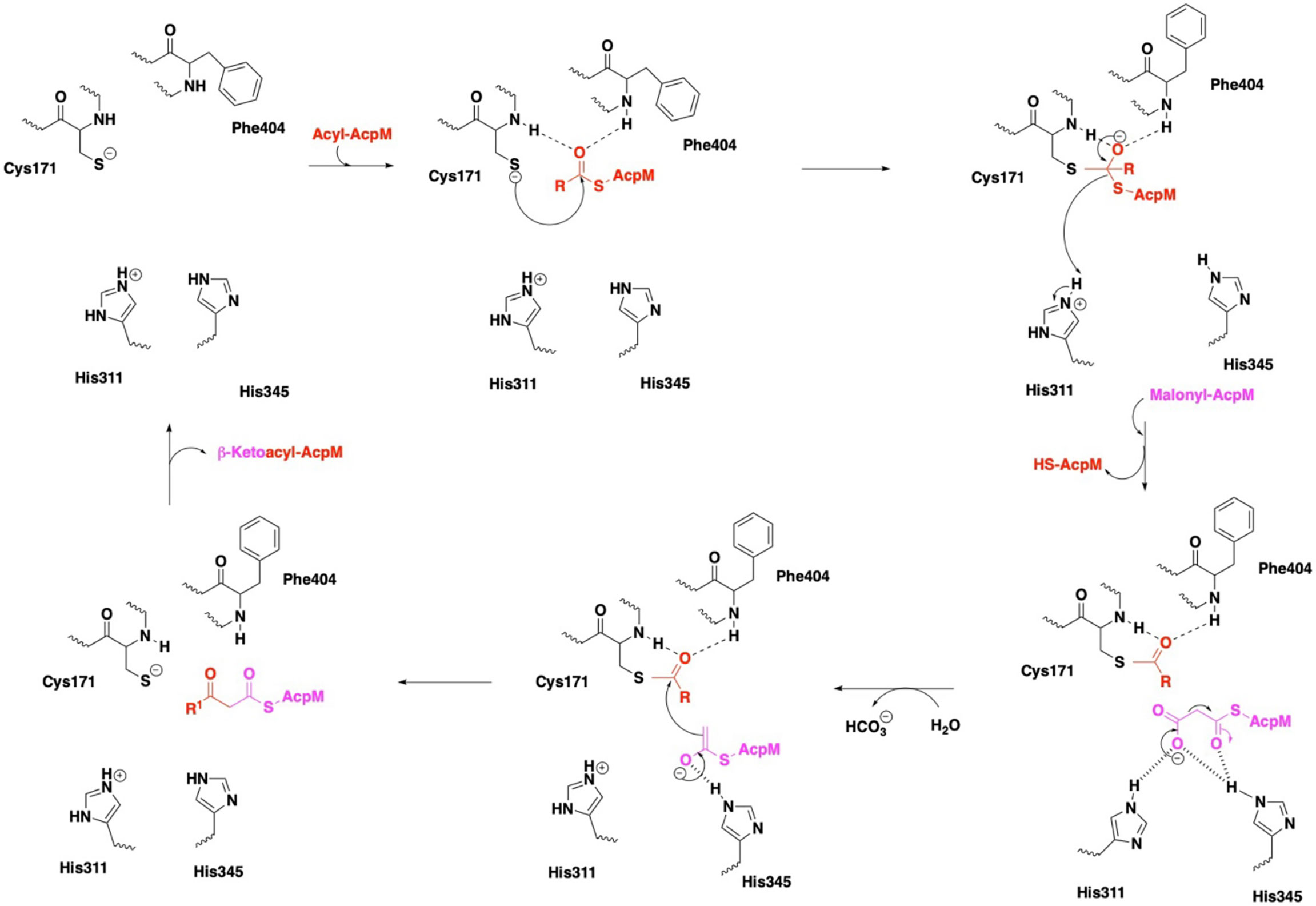
Figure 4 Mechanism of KasA catalysis with emphasis on the catalytic triad. In the KasA resting state, Cys171 and His311 are proposed to be deprotonated and protonated, respectively. Acylation of KasA occurs by nucleophilic attack of Cys171 on the acyl-AcpM. Several theories have been offered to explain the decarboxylation step, and it is unclear as to the protonation state of His311. The ensuing condensation of this enolate with the Cys171 bound acyl moiety occurs to elongate the acyl chain length by two carbons and then release the product.
In the apo state, the KasA active site residues are in a zwitterionic state, i.e., Cys171 is deprotonated and His311 is protonated. In step one, apo KasA binds to an acyl-AcpM, charging Cys171 with an acyl chain that is at least C16-18 and maximally C36-40 (Slayden and Barry, 2002). It is important to note that the acyl chain is covalently attached to AcpM via a phosphopantetheine group. Two models have been proposed to explain how the acyl chain accesses the KasA active site. In model one, it is hypothesized that both the acyl-AcpM and the malonyl-AcpM substrates are delivered through the phosphopantetheine tunnel mediated by the binding of acidic AcpM residues to basic residues on the KasA surface (namely Arg74, Arg 78, Arg79, Arg 135, Arg214 and Lys136) (Lee et al., 2011; Schiebel et al., 2013). In model two, it is proposed that KasA residues 115 – 147 in the flexible cap region of both monomers move in a scissor-like motion to allow direct access of the acyl chain into the hydrophobic cavity (Luckner et al., 2009). A structure of KasA in complex with acyl-AcpM may explain how the complex overcomes a variety of steric factors and the hydrophilic and hydrophobic nature of the acyl and phosphopantetheine channels. Structure-function studies do suggest that acylation of Cys171 likely induces a conformational change of gatekeeper residue Phe404 along with the additional gatekeeper residues Leu116 and Tyr126 (Schiebel et al., 2013). Acylated KasA adopts an open conformation, which facilitates the binding of malonyl-AcpM by widening the phosphopantethiene tunnel entrance and increasing the size of the malonyl binding site. Decarboxylation of malonyl-AcpM is then mediated by His311 and His345. It is important to note that different theories have been proposed to explain the protonation states of the catalytic triad residues during acylation and decarboxylation (Lee et al., 2011; Lee and Engels, 2014). Subsequent Claisen condensation occurs by a nucleophilic attack of the enolate on the thioester intermediate, yielding the product β-ketoacyl-AcpM (Figure 4).
The thiolactomycin chemotype as a malonyl binding site inhibitor
Thiolactomycin (TLM; Figure 5) is a thiolactone natural product, isolated from Nocardia spp., that initially was noted for its modest in vitro growth inhibitory efficacy versus a range of Gram-positive and Gram-negative bacteria (Oishi et al., 1982). TLM has been shown to exhibit low cytotoxicity to mammalian cells. Analysis of TLM serum levels in orally dosed rats evidenced rapid absorption and clearance (Miyakawa et al., 1982). Accordingly, TLM and its analogs have exhibited only modest in vivo efficacy in mouse models of infection with Staphylococcus aureus, Klebsiella pneumoniae, and Serratia marcescens (Miyakawa et al., 1982; Bommineni et al., 2016).
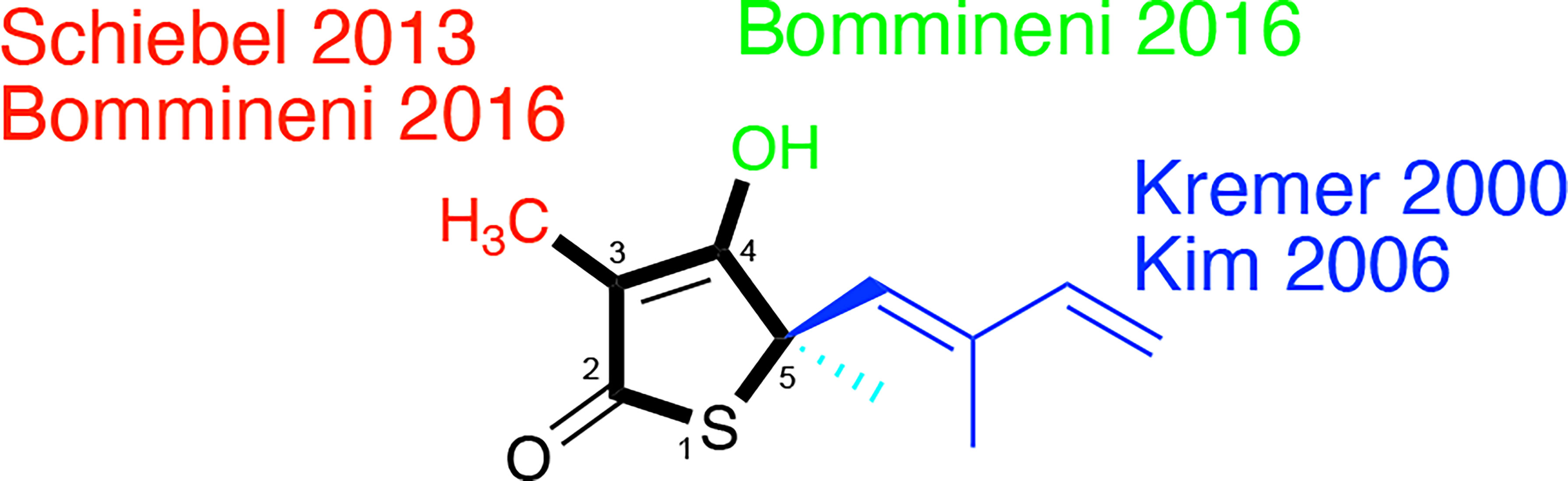
Figure 5 Thiolactomycin and its analogs. Noted are efforts, referenced within, regarding analogs at the specific positions of the thiophen-2(5H)-one ring system which are numbered.
Slayden et al. demonstrated TLM in vitro efficacy versus M. tuberculosis albeit with racemic, synthetic material with a MIC = 120 µM on solid media (Slayden et al., 1996); the active enantiomer features a 5R-stereocenter (Kim et al., 2006). This group also showed that TLM afforded bactericidal efficacy against M. tuberculosis in an infected murine bone marrow-derived macrophage model and inhibition of mycolic biosynthesis within M. smegmatis. Knowledge of its inhibition of E. coli β-ketoacyl synthases (Nishida et al., 1986; Magnuson et al., 1993) hinted at its engagement of KasA and KasB which was evidenced through overexpression studies in M. bovis BCG (Kremer et al., 2000). Studies with the purified M. tuberculosis proteins showed that TLM inhibited all three annotated M. tuberculosis β−ketoacyl synthases in various functional assays. Whereas the metrics of inhibition vary according to the assay, in general, TLM inhibition followed the order of KasA > KasB > FabH (Choi et al., 2000; Schaeffer et al., 2001; Kim et al., 2006; Machutta et al., 2010).
In 2000, Kremer et al. reported a subset of TLM analogs designed without the initial guidance from an X-ray crystal structure (Kremer et al., 2000). Their assay results demonstrated the importance of the TLM isoprenyl group in maintaining whole-cell activity against M. tuberculosis as judged by MIC but showed potential disconnects between MIC and the respective inhibitions of mycolic acid biosynthesis and FAS-II biosynthesis in M. smegmatis. While the authors constructed a KasA homology model from E. coli FabF (PBD ID: 1KAS) (Huang et al., 1998), their analysis was limited to the observation of a hydrophobic pocket that could be engaged by the isoprenyl. This observation would later be supported by a 2009 reported X-ray crystallographic study (Luckner et al., 2009) that illustrated the orientation of the isoprenoid moiety toward an extended pocket where two water molecules were present stabilizing the loop from Asp272 to Pro280. Furthermore, a thorough examination of a range of substitutions for the isoprenyl moiety was performed by Kim and colleagues (Kim et al., 2006). In an assay quantifying KasA catalytic activity, the authors found linear or branched alkyl, cycloalkyl, and aryl moieties failed to afford TLM analogs with inhibition within six-fold of TLM. In fact, all but two compounds demonstrated IC50 values in excess of 100 µM. Importantly, all analogs failed to exhibit significant M. tuberculosis growth inhibitory efficacy versus the H37Rv strain.
The 2009 structural biology report (Luckner et al., 2009) was critical to elucidating the details of how TLM binds to KasA (Figures 3, 6). More specifically, it illustrated how TLM occupies the malonyl binding site. TLM interactions with the active site residues and the binding mode of TLM itself differ when KasA is in the closed (apo) or open (acylated) conformations (PDB IDs 2WGE and 2WGG, respectively). The isoprenoid chain and the double bond in the thiophen-2(5H)-one of TLM are critical for KasA inhibitory activity. In apo KasA, the TLM carbonyl oxygen formed two hydrogen bonds with the sulfhydryl group of Cys171 and sidechain N-H of His345, whereas in the C171Q mutant (that mimics acylated KasA) two hydrogen bonds were formed with the sidechain N-H moieties of His311 and His345. Furthermore, in the apo state, the KasA phenyl ring of gatekeeper residue Phe404 formed a face-to-face interaction with the thiolactone ring. With the KasA C171Q mutant, however, a conformational change shifted the Phe404-TLM interaction to edge-to-face. The orientation of the TLM isoprenoid tail in the lipophilic pocket also varied between the apo KasA and KasA-C171Q structures. Movement of Phe404 from the closed to open conformation led to a shift in residues Leu116, Val142, and Met146, and helix α9. This conformational change increased the size of the malonyl binding site and the phosphopantetheine tunnel. This explains the slow (Machutta et al., 2010) and preferential binding of TLM to acylated KasA as compared to apo KasA.
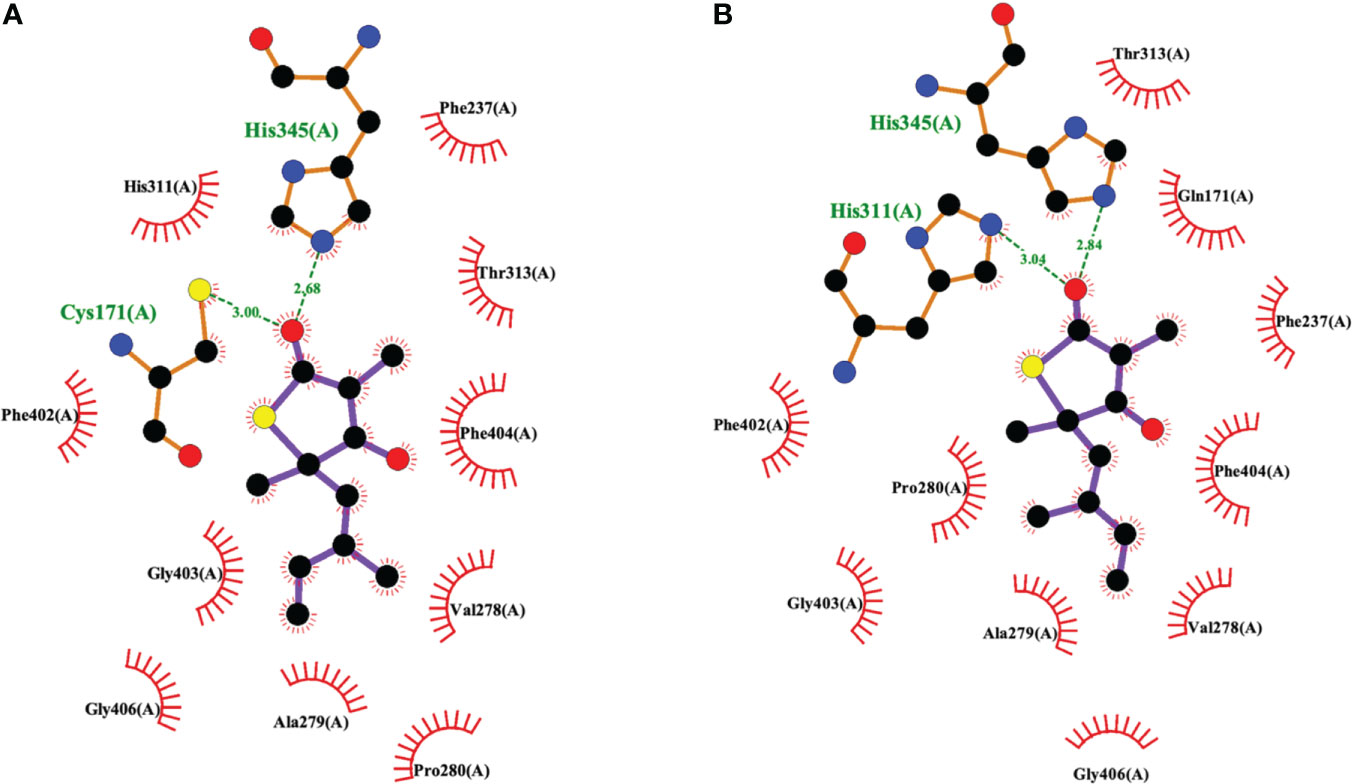
Figure 6 Interaction profiles for TLM bound to (A) wild type KasA (PDB ID 2WGE) and (B) KasA-C171Q (PDB ID 2WGG). Hydrogen bonds are shown as green dashed lines and distances are measured in Angstroms.
Schiebel and colleagues leveraged this structural information in the pursuit of TLM analogs (Schiebel et al., 2013). Attempting to mimic the alkyl portion of the acyl chain of bound acyl-AcpM, linear hydrophobic substituents (e.g, ethyl, n-propyl, and 4-azido-n-butyl) were introduced at the 3-position of the thiolactone ring (Figure 5). The TLM alkyl-AcpM mimics displayed a 4 – 18 fold reduction in KasA binding affinity for the C171Q mutant in comparison to TLM. However, they did maintain the TLM slow onset of binding phenotype. X-ray crystal structures demonstrated how the introduced alkyl chain oriented towards the aromatic cavity formed by Phe210, Phe237, Phe239, and His345 (PDB IDs 4C70, 4C6Z, 4C71; Figure 7). Interestingly, upon comparison to the whole-cell efficacy of TLM versus the M. tuberculosis H37Rv strain, the 3-ethyl analog was approximately equipotent while the 3-n-propyl derivative was about eightfold less active (Bommineni et al., 2016). Kapilashrami continued this effort and their para-substituted phenethyl and phenylbutyl analogs generally offered enhanced binding affinity for wild type KasA and an improved initial equilibrium binding constant for the C171Q mutant as compared to TLM, but failed to exhibit slow binding kinetics (Kapilashrami et al., 2013). Slow onset of binding has been demonstrated to be a key aspect of long residence time inhibitors and to their in vivo efficacy (Lu and Tonge, 2008; Lu and Tonge, 2010). These results serve as a reminder of the intricacies associated with inhibitor binding to KasA and the value of detailed kinetic measurements. Furthermore, an X-ray crystal structure of KasA bound to one of these 3-alkylphenyl analogs would help explain these observations.
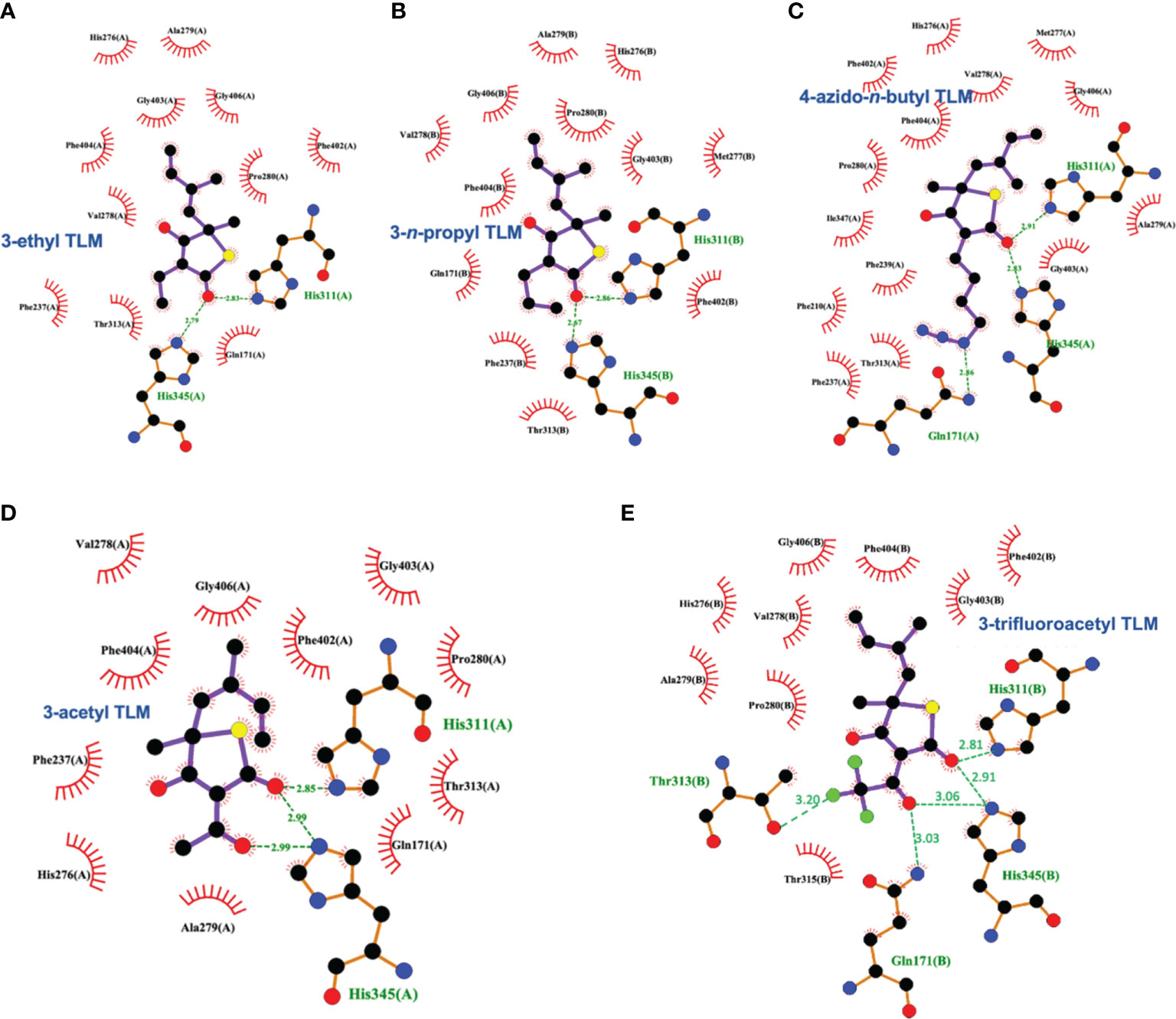
Figure 7 Interaction profiles for KasA C171Q bound to the following TLM analogs: (A) 3-ethyl (PDB ID 4C6Z), (B) 3-n-propyl (PDB ID 4C70), (C) 4-azido-n-butyl (PDB ID 4C71), (D) 3-acetyl (PDB ID 4C72), and (E) 3-trifluoroacetyl (PDB ID 4C73). Hydrogen bonds are shown as green dashed lines and distances are measured in Angstroms.
TLM 3-substituted acyl analogs (e.g., acetyl and trifluoroacetyl) utilized the acyl moiety to mimic the diketo group of malonyl-AcpM. These derivatives offered 2 – 4 fold increased affinity for the C171Q mutant as compared to TLM while still maintaining slow onset kinetics. X-ray crystallographic studies again proved insightful (Figure 7). The structure of either 3-acyl analog is supportive of the increased binding affinity being due to the formation of a new hydrogen-bonding interaction between the acyl carbonyl and His345. A potentially favorable interaction between the trifluoromethyl group in the 3-trifluoroacetyl analog and the sidechain hydroxyl of Thr313 may also be present. In comparison to the whole-cell efficacy of TLM versus the M. tuberculosis H37Rv strain, the 3-acetyl and 3-trifluoroacetyl analogs were 4- and 32-fold less potent, respectively (Bommineni et al., 2016). Kapilashrami reported additional acyl analogs with a longer alkyl chain (i.e., n-Pr, C15H31, (CH2)5(4-PhC6H4)). These were not slow onset binders and they failed to offer improvements in binding affinity to wild type KasA or initial equilibrium binding constant for the C171Q mutant. These results further support the value of an X-ray crystal structure of KasA bound to malonyl-AcpM, or a non-reactive mimic.
Removal of the TLM 3-methyl group was also pursued and led to a decrease in whole-cell potency (MIC > 100 µM) while providing gains in the binding constants for the wild type and C171Q forms of KasA, respectively (Bommineni et al., 2016). The disconnect between enzyme binding and whole-cell efficacy could potentially be further explored by examining the effect of methyl group removal on compound accumulation and/or metabolism within M. tuberculosis (Wang et al., 2019; Wang et al., 2020). Furthermore, alkylation of the 4-OH of this desmethyl TLM abrogated not only growth inhibition of M. tuberculosis but also binding to wild type KasA or KasA-C171Q. An observed decrease in potency via removal of the hydroxy group may be consistent with the loss of a potential water-mediated hydrogen bond with the carbonyl oxygen of Val278. It is also possible that the newly introduced alkyl substituent makes unfavorable interactions with nearby KasA residues. While analysis of the relevant crystal structures does not reveal obvious unfavorable interactions, it is, however, possible that unfavorable interactions could occur when KasA adopts different conformations, e.g., when complexed with AcpM.
The indazole sulfonamide chemotype as an acyl channel inhibitor
GSK3011724A, an indazole sulfonamide (Figure 8), was first identified as an antitubercular by GlaxoSmithKline (GSK) through a high-throughout whole-cell phenotypic screening campaign (Ballell et al., 2013; Rebollo-Lopez et al., 2015). This compound and other screening actives were made available to the research community to explore their mechanism of action and optimization. GSK and our laboratory (renaming the molecule as DG167; a convention that will be used throughout this review) each reported on their respective initial studies which characterized the property profile and mechanism of action of this indazole sulfonamide (Abrahams et al., 2016; Kumar et al., 2018). In our case, we were drawn to DG167 because of its signature as an inhibitor of cell wall biosynthesis through its induction of iniBAC (Wilson et al., 2013) that lacked cross-resistance with current front- and second-line tuberculosis drugs. While the two reports differ in terms of their exact values for different property metrics, in general, DG167 exhibited promising in vitro (i.e., sub-micromolar) potency versus the M. tuberculosis H37Rv strain, a lack of cytotoxicity to model mammalian cell lines, and good aqueous solubility. Both groups determined through the generation of spontaneous resistant mutants that DG167 targets KasA with acceptable frequencies of resistance for an antitubercular drug discovery molecule. The protein target was further confirmed via additional techniques, such as FAS-II thin layer chromatography (TLC) to evidence mycolic acid biosynthesis inhibition, binding constant quantification with respect to purified KasA, functional inhibition of purified KasA, and target pull-down with a bead-bound analog of DG167.
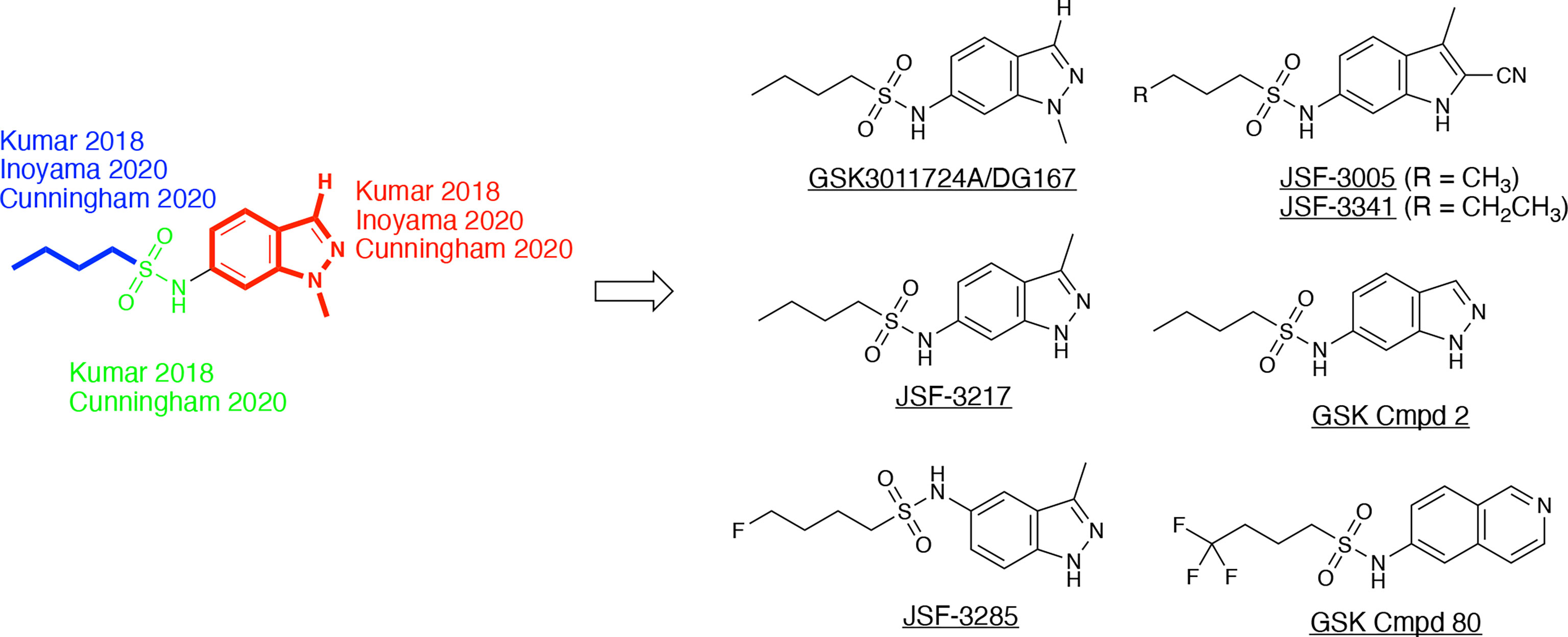
Figure 8 DG167 and its analogs. The derivatives, as referenced herein, are noted with regard to alterations in the heterocyclic core and sulfonamide moiety.
The X-ray crystal structure of this compound has been elucidated bound to KasA (Abrahams et al., 2016; Kumar et al., 2018). The GSK report established the initial crystal structure (PDB ID 5LD8) and our work had significant contributions in confirming and correcting the initial findings (PDB 5W2P). The GSK structure depicted the acyl channel of each KasA subunit to be occupied by one molecule of DG167 and one molecule of polyethylene glycol (PEG). The purification and crystallization conditions, however, excluded PEG. It is, therefore, unclear to us why PEG was modeled in this structure and continued to be modeled in the more recently determined structures of KasA (Cunningham et al., 2020). In contrast, the electron density maps of our published structure of KasA-DG167 enabled us to unambiguously build two molecules of DG167 (DG167A and DG167B) in the acyl binding site (Figures 3, 9). DG167B was in the position modeled as PEG in the GSK publication. KasA-DG167 binding was stabilized by both hydrophobic and hydrogen bond interactions. For instance, the DG167A sulfonamide N-H formed a hydrogen bond with the Glu199 sidechain. An intermolecular hydrogen bond was observed between the DG167A sulfonamide oxygen and the DG167B sulfonamide N-H. Both indazole units of DG167A and DG167B contributed to hydrophobic interactions throughout the acyl channel. We observed the aliphatic moiety of DG167A mirrored the binding of the phospholipid acyl tail reported in previously determined structures of KasA (e.g., PDB ID 4C6W) (Schiebel et al., 2013). More specifically, this n-butyl moiety of the 6-sulfonamide was bound in a narrow hydrophobic channel lined by Gly200, Ile202, Pro206, Phe239, His345, and Ile347. Furthermore, the 1-methyl group fit into a shallow pocket defined by Pro201, Glu203, and Pro206, and engaged these amino acids through hydrophobic interactions. The DG167B indazole nitrogen formed water-mediated hydrogen bonds with Gly115, Asn148, and Ala170. Additionally, the DG167B indazole mediated hydrophobic interactions across the KasA/KasA′ dimer interface. It is important to note that in the KasA-DG167 structure, gatekeeper residue Phe404 was in the closed conformation where acyl chain access to the acyl channel was restricted. This was, to our knowledge, the first time a KasA ligand (such as an inhibitor) was demonstrated to bind KasA in the closed (nonacylated) conformation. It is remarkable that DG167 can access the acyl channel while Phe404 is in the closed conformation, defeating the elaborate mechanisms KasA has evolved to prevent the entry of cellular free fatty acids into the acyl channel.
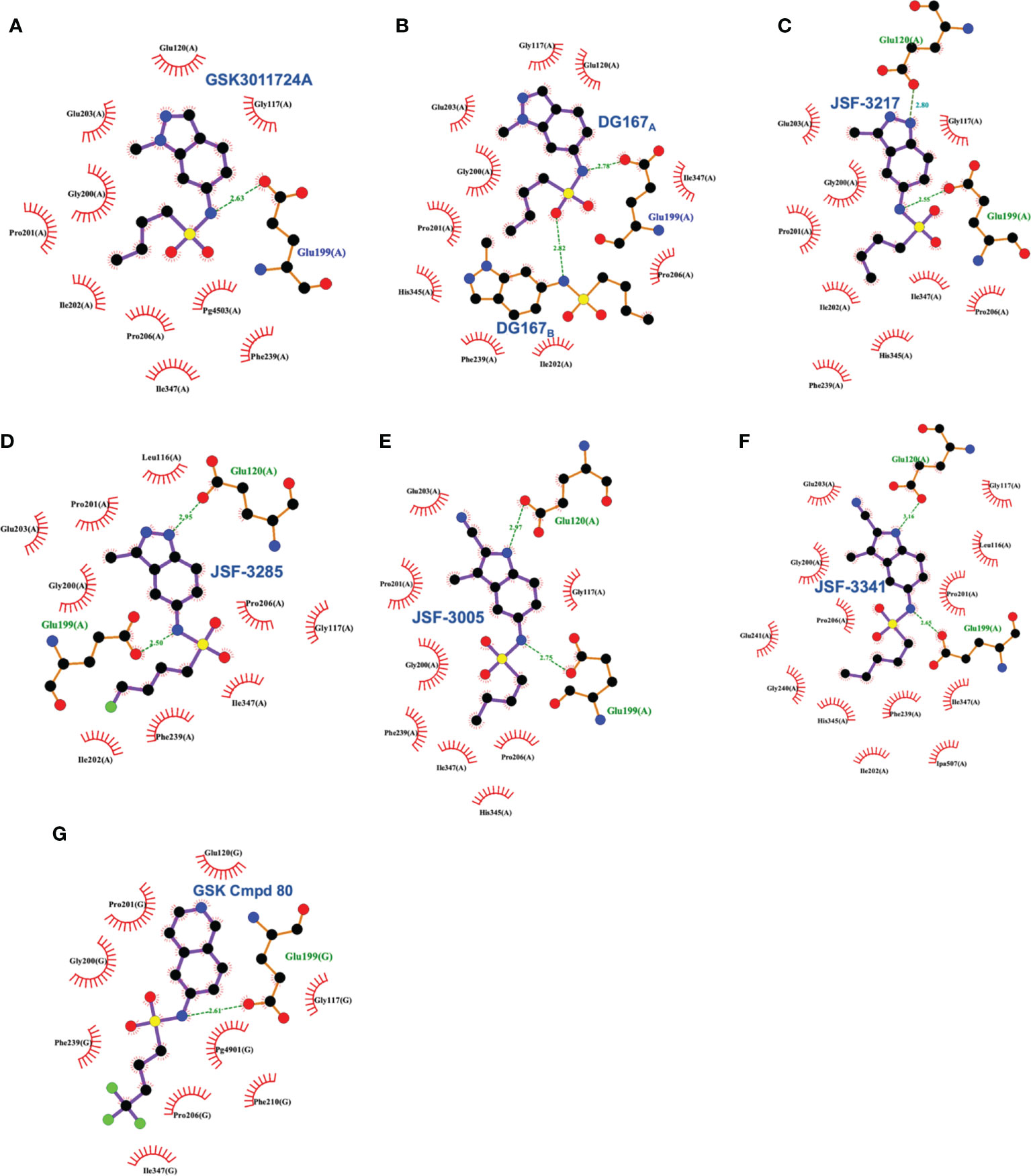
Figure 9 Interaction profiles for wild type KasA bound to the following sulfonamides: (A) GSK3011724A (PDB ID 5LD8), (B) DG167 (PDB ID 5W2P), (C) JSF-3217 (PDB ID 5W2S), (D) JSF-3285 (PDB ID 6P9L), (E) JSF-3005 (PDB ID 6P9K), (F) JSF-3341 (PDB ID 6P9M), and (G) GSK Cmpd 80 (PDB ID 6Y2J). Hydrogen bonds are shown as green dashed lines and distances are measured in Angstroms.
With the target of DG167 firmly established as KasA, the question as to its in vivo profile arose. Studies in both laboratories found the compound to exhibit a modest pharmacokinetic (PK) profile in mice with some evidence of toxicity at doses ≥300 mg/kg once-daily oral (qd po). We proposed that the oral exposure was limited by demethylation of the 1-methyl moiety (half-life t1/2 = 10.1 min) to afford the whole-cell inactive 1H-indazole, as observed through incubation of DG167 in the presence of mouse liver microsomes (MLM). Consistent with its modest PK profile, we did not observe DG167 (100 mg/kg qd po) to exhibit the ability to reduce M. tuberculosis infection in a sub-acute model of infection when quantifying bacterial burden in the lungs of female BALB/c mice dosed for 2 weeks as compared to the lung bacterial burden at the outset of compound treatment. The GSK report first profiled DG167 in a “fast” and less conservative model (Rullas et al., 2010) with 8 d of drug treatment commencing 1 d post infection of C57BL/6 mice. Quantification of the reduction of lung bacterial burden utilized the level in mouse lung for the no-drug control arm at the end of treatment as the comparison. GSK reported cidal activity in this infection model as well as in a more typical chronic model of infection albeit with what we would term a less conversative calculation of bacterial load reduction.
These in vivo results, in our minds, formed the basis of an optimization problem where we hypothesized that DG167 analogs with enhanced metabolic stability and mouse PK profile would demonstrate significant bactericidal efficacy in vivo. In our structure-based optimization reported in 2020, the central design hypothesis was to transpose the indazole 1-nitrogen of DG167 to afford 3-methyl-5-sulfonamide indazoles or indoles (Inoyama et al., 2020). It was hypothesized that this would maintain the hydrophobic interactions of the DG167 1-methyl moiety that our 2018 publication (Kumar et al., 2018) had demonstrated could not be replaced without losses in whole-cell potency and surprisingly also in MLM stability. We entertained a small number of potential alterations to the sulfonamide substituent, having learned from our earlier work (Kumar et al., 2018) that n-alkyl groups larger than pentyl and smaller than butyl afforded poorer MIC values. Furthermore, consistent with the relative narrowness of the hydrophobic channel recognizing the sulfonamide alkyl group, we were cognizant that substitutions on all but the terminal carbon of the n-butyl, or its replacement with carbocyclic or aromatic/heteroaromatic groups, were not tolerated. It should be noted that we had preliminarily reported on one transposed indazole (compound 5g or JSF-3217; Figure 8) in our 2018 publication that exhibited greater whole-cell activity (MIC = 0.2 µM) than DG167 but with only marginally better MLM stability (t1/2 = 11.5 min). Critically, an X-ray structure of JSF-3217 bound to KasA (PDB ID 5W2S) demonstrated the protein-inhibitor interactions that were in our initial designs (Figure 9). Analysis of the X-ray crystal structure led us to postulate that a second molecule of JSF-3217 cannot bind in the DG167B site because it would force the indazole N(1)-H in close proximity to the hydrophobic surface associated with the KasA dimer interface. In addition to the sulfonamide N-H hydrogen bond with Glu199 similar to the one observed in the DG167 structure, the N-H group of the transposed indazole formed a new hydrogen bond with Glu120. Only one molecule of JSF-3217 bound to a KasA monomer and did not contact the other protomer in the KasA dimer. The KasA–JSF-3217 structure also showed that single molecule acyl channel occupancy was sufficient to stabilize the KasA flap (Luckner et al., 2009) (residues 115 – 147). Further optimization efforts with the transposed indazoles culminated in JSF-3285, which replaced the n-butyl of JSF-3217 with a 4-fluorobutyl. The main improvements realized with JSF-3285 as compared to DG167 were its improved mouse PK profile (AUC0-5h = 59323 vs. 1965 h*ng/mL), MLM stability (t1/2 = 28.4 vs. 10.1 min), and kinetic aqueous solubility (S = 483 vs. 324 µM) while slightly enhancing in vitro potency (MIC = 0.20 vs. 0.39 µM). Critically, the in vitro efficacy of JSF-3285 versus the H37Rv laboratory strain was maintained versus a set of 48 drug-sensitive and drug-resistant clinical strains. This observation furthers our confidence that a KasA-targeting therapeutic will be of significant utility versus both drug-sensitive and drug-resistant infections. We determined the X-ray crystal structure of KasA complexed with JSF-3285 (PDB ID: 6P9L) (Figure 9). Overall, the JSF-3285 and JSF-3217 binding modes were similar. The JSF-3285 sulfonamide alkyl moiety, however, reached further into the hydrophobic channel, making different hydrophobic contacts than the JSF-3217 alkyl sulfonamide. Overall, we were gratified to have structural data to support our structure-based design hypothesis; removal of the metabolic instability of DG167 afforded an advanced compound for further study.
In addition to the transposed indazole optimization campaign, our analysis of the X-ray structure of the KasA-DG167 structure supported removal of the N(2) from the transposed indazole design to afford 3-methyl-5-sulfonamide indoles where we could explore additional interactions with KasA through modification of the 2-substituent. Key molecules in this campaign were JSF-3005 and JSF-3341 (Figure 8) which both featured a 2-cyano group and the sulfonamide moiety as n-butyl or n-pentyl, respectively. Their in vitro growth inhibitory potencies versus the Mtb H37Rv strain were 0.78 and 0.20 µM, respectively. Each indole was crystallized bound to the KasA (Figure 9). Both JSF-3005 and JSF-3341 utilized similar hydrogen bonding interactions between the indole N-H and KasA Glu120, as well as the sulfonamide N-H and KasA Glu199. However, a few differences were observed like additional electron density corresponding to an unknown molecule identified in the acyl channel of the KasA–JSF-3005 complex. The alkyl chain in JSF-3341 is longer by one carbon than in JSF-3005, extending 0.9 Å deeper into the hydrophobic channel. The sulfonamide group in JSF-3341 was slightly shifted by 0.5 Å when compared to JSF-3005. The additional methyl group on the sulfonamide of JSF-3341 also mediated additional hydrophobic contacts not observed in the KasA–JSF-3005 complex structure.
JSF-3005, JSF-3341, and JSF-3285 were studied thoroughly in an effort to proceed to in vivo efficacy assessment. Summarily, all three compounds were rigorously confirmed to primarily target KasA through the following methods in addition to the previously described X-ray crystallography with their KasA complex: quantification of KasA binding via microscale thermophoresis, fatty acid TLC, and drug-resistant mutant generation and sequencing. The three molecules were assessed in mouse models of sub-acute and chronic M. tuberculosis infection. In the sub-acute model with four weeks of compound treatment, JSF-3005 dosing (100 mg/kg qd po) led to increases in the lung bacterial burden and JSF-3341 dosing (200 mg qd po) afforded bacteriostatic activity. JSF-3285 was dosed at 100 mg/kg and 200 mg/kg qd po, in accordance with dose proportionality and tolerability studies, in the sub-acute infection model, and we were delighted to observe an ca. 2 log10 reduction in bacterial burden as quantified by the lung bacterial burden post 4 weeks of treatment as compared to the bacterial burden at the beginning of compound treatment. The corresponding chronic infection assessment in BALB/c mice demonstrated JSF-3285 at 100 or 200 mg/kg to reduce the bacterial burden in the lungs >2 log10 colony-forming units (CFUs) after 4 weeks as compared to the bacterial burden at the start of the treatment. The addition of JSF-3285 (200 mg/kg) to INH or RIF (either at 10 mg/kg) improved efficacy of the front-line drug by about 1 log10 CFUs. Furthermore, the bactericidal efficacy of JSF-3285 at doses ranging from 200 – 20 mg/kg qd po was examined. At 20 mg/kg qd po, INH and RIF each afforded just more than a 2 log10 reduction in CFUs. JSF-3285 exhibited an approximately 1.5 – 2.0 log10 reduction in CFUs at doses of 20 – 200 mg/kg. These experiments provided rigorous validation for JSF-3285 as a preclinical tuberculosis drug candidate and critically established in vivo pharmacological validation of KasA with conservative quantification of cidal bacterial efficacy.
Building on their earlier work (Abrahams et al., 2016), a GSK team reported (Cunningham et al., 2020) on the results of their evolution of DG167 in a manuscript submitted four months after the submission of our report; both papers were published in March 2020. They verified the whole-cell potential of the transposed indazole chemotype found by us (Kumar et al., 2018; Inoyama et al., 2020), but mainly focused on their finding of the Ames mutagenicity of the amine metabolite of their initial hit or select transposed indazoles (e.g., GSK Cmpd 2 in Figure 8). Our efforts to address this concern will be published separately. While the amount of the amine metabolite found in the urine of a single Sprague Dawley rat dosed at 300 mg/kg po in each case was not quantified, the in vivo observation of an amine with an Ames positive signal was a significant concern in the absence of further studies.
The GSK team proposed several strategies to prevent the formation of a mutagenic amine, primarily by substituting for the sulfonamide with other moieties that would not suffer hydrolysis. Additionally, the steric and/or electronic environment was modified around the 6-membered ring of the core heterocycle or the 5-membered ring of the indazole was replaced with other ring systems; the goal was to eliminate the mutagenicity of the amine formed. These alterations failed to find afford a coalescence of antitubercular whole-cell efficacy with a lack of Ames mutagenicity associated with the related amine metabolite.
A significant factor in this outcome was the necessity of maintaining the sulfonamide, which, as described earlier, serves as a recognition element providing an appropriate straight-chain alkyl hydrophobe along with the hydrogen bond between its N-H to Glu199. Changes of the central heterocycle from indazole to indole, 2,3-dihydro-1H-indene, and isoquinoline (e.g., GSK Cmpd 80 in Figure 8) were achieved, but they do not appear to have offered sufficient advantages worthy of further exploration. The indole substitution was not surprising given that it involves the removal of the indazole nitrogen from DG167 to afford a 5-substituted indole. The corresponding N-(2,3-dihydro-1H-inden-5-yl)butane-1-sulfonamide exhibited modest efficacy (MIC = 16.3 µM) while lacking the N-H to proposedly hydrogen bond with Glu120. Finally, the GSK Cmpd 80 isoquinoline sulfonamide demonstrated modest potency (MIC = 15 µM) and was structurally characterized bound to KasA (PDB ID 6Y2J; Figure 9). While its n-butyl sulfonamide exhibited similar interactions as DG167, its basic nitrogen was shown to engage a water molecule through hydrogen-bonding; Glu120 did not interact with the inhibitor. These results led the authors to speculate on the need for the planarity of the heterocycle given the proximity of Gly200 and Pro201. However, it is not obvious that this would contribute significantly to the binding energy. We suspect that the loss of hydrogen-bonding to Glu120 is primarily responsible for the reduction in compound activity.
Summary
This review focuses on the promise of KasA as a tuberculosis drug target and, in particular, on the importance of structure-based design approaches to deliver KasA inhibitors of translational significance. Thus, we did not discuss in-depth earlier stage molecules, such as cerulenin (Schaeffer et al., 2001) and platensimycin (Brown et al., 2009), that have not been supported by published X-ray crystal structures in complex with KasA. We also have not described computational approaches (Puhl et al., 2020) involving docking and/or machine learning methods to design KasA binders. Most likely, X-ray crystal structures of these compounds bound to KasA will be necessary to enable their design optimization. Our analysis of the literature supports KasA as a highly valuable target for future study. A panoply of approaches has been utilized to vet KasA with regard to its essentiality and vulnerability. Two binding sites (i.e., the malonyl binding and acyl binding sites) have been targeted with small molecules and the relevant interactions have been discerned through X-ray crystallography. KasA acyl binding channel targeting with drug-like small molecules has been shown to be critical to significantly reduce the M. tuberculosis infection in mice, while efforts with malonyl binding site inhibitors have fallen short thus far of in vivo pharmacologic validation. While the current in vivo active molecules have limitations, we expect that ongoing studies with JSF-3285 and newer series from our laboratories, GSK, and others will find the requisite balancing of efficacy and toxicity profiles. We anticipate that structure-based design efforts, as summarized herein, will play a prominent role in these endeavors. Ultimately, we are optimistic that the sum total of efforts with M. tuberculosis KasA inhibitors will afford one or more molecules for clinical studies that will positively impact drug regimens for both drug-sensitive and drug-resistant infections.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported at Rutgers University – New Jersey Medical School by the US National Institutes of Health grants U19AI142731 (MN and JF).
Acknowledgments
We want to thank Dr. Clifton E. Barry III (NIH/NIAID) and Dr. Alexander L. Perryman (Repare Therapeutics) for helpful discussions.
Conflict of interest
MN and JF are listed as inventors on patent filings pertinent to the indole and indazole compounds mentioned in this manuscript as employees of Rutgers University.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrahams, K. A., Besra, G. S. (2020). Mycobacterial drug discovery. RSC Med. Chem. 11, 1354–1365. doi: 10.1039/D0MD00261E
Abrahams, K. A., Chung, C. W., Ghidelli-Disse, S., Rullas, J., Rebollo-Lopez, M. J., Gurcha, S. S., et al. (2016). Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 7, 12581. doi: 10.1038/ncomms12581
Ballell, L., Bates, R. H., Young, R. J., Alvarez-Gomez, D., Alvarez-Ruiz, E., Barroso, V., et al. (2013). Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 8, 313–321. doi: 10.1002/cmdc.201200428
Banerjee, A., Dubnau, E., Quemard, A., Balasubramanian, V., Um, K. S., Wilson, T., et al. (1994). InhA, a gene encoding a target for isoniazid and ethionamide in M. tuberculosis. Science 263, 227–230. doi: 10.1126/science.8284673
Bhatt, A., Fujiwara, N., Bhatt, K., Gurcha, S. S., Kremer, L., Chen, B., et al. (2007). Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc. Natl. Acad. Sci. U.S.A. 104, 5157–5162. doi: 10.1073/pnas.0608654104
Bhatt, A., Molle, V., Besra, G. S., Jacobs, W. R., Jr., Kremer, L. (2007). The Mycobacterium tuberculosis FAS-II condensing enzymes: Their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 64, 1442–1454. doi: 10.1111/j.1365-2958.2007.05761.x
Bommineni, G. R., Kapilashrami, K., Cummings, J. E., Lu, Y., Knudson, S. E., Gu, C., et al. (2016). Thiolactomycin-based inhibitors of bacterial beta-ketoacyl-ACP synthases with in vivo activity. J. Med. Chem. 59, 5377–5390. doi: 10.1021/acs.jmedchem.6b00236
Bosch, B., DeJesus, M. A., Poulton, N. C., Zhang, W., Engelhart, C. A., Zaveri, A., et al. (2021). Genome-wide gene expression tuning reveals diverse vulnerabilities of m. tuberculosis Cell. 184, 4579–92.e24. doi: 10.1016/j.cell.2021.06.033
Brennan, P. J., Nikaido, H. (1995). The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29–63. doi: 10.1146/annurev.bi.64.070195.000333
Brown, A. K., Taylor, R. C., Bhatt, A., Futterer, K., Besra, G. S. (2009). Platensimycin activity against mycobacterial beta-ketoacyl-ACP synthases. PLoS One 4, e6306. doi: 10.1371/journal.pone.0006306
Carey, A. F., Rock, J. M., Krieger, I. V., Chase, M. R., Fernandez-Suarez, M., Gagneux, S., et al. (2018). TnSeq of Mycobacterium tuberculosis clinical isolates reveals strain-specific antibiotic liabilities. PLoS Pathog. 14, e1006939. doi: 10.1371/journal.ppat.1006939
Choi, K. H., Kremer, L., Besra, G. S., Rock, C. O. (2000). Identification and substrate specificity of beta -ketoacyl (acyl carrier protein) synthase III (mtFabH) from Mycobacterium tuberculosis. J. Biol. Chem. 275, 28201–28207. doi: 10.1074/jbc.M003241200
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Cunningham, F., Esquivias, J., Fernandez-Menendez, R., Perez, A., Guardia, A., Escribano, J., et al. (2020). Exploring the SAR of the beta-Ketoacyl-ACP synthase inhibitor GSK3011724A and optimization around a genotoxic metabolite. ACS Infect. Dis. 6, 1098–1109. doi: 10.1021/acsinfecdis.9b00493
Dartois, V. A., Rubin, E. J. (2022). Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat. Rev. Microbiol. 20. doi: 10.1038/s41579-022-00731-y
DeJesus, M. A., Gerrick, E. R., Xu, W., Park, S. W., Long, J. E., Boutte, C. C., et al. (2017). Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8, e02133–e02116. doi: 10.1128/mBio.02133-16
Dulberger, C. L., Rubin, E. J., Boutte, C. C. (2020). The mycobacterial cell envelope - a moving target. Nat. Rev. Microbiol. 18, 47–59. doi: 10.1038/s41579-019-0273-7
Fernandes, G. F. S., Thompson, A. M., Castagnolo, D., Denny, W. A., Dos Santos, J. L. (2022). Tuberculosis drug discovery: Challenges and new horizons. J. Med. Chem. 65, 7489–7531. doi: 10.1021/acs.jmedchem.2c00227
Gavalda, S., Leger, M., van der Rest, B., Stella, A., Bardou, F., Montrozier, H., et al. (2009). The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem. 284, 19255–19264. doi: 10.1074/jbc.M109.006940
Griffin, J. E., Gawronski, J. D., Dejesus, M. A., Ioerger, T. R., Akerley, B. J., Sassetti, C. M. (2011). High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PloS Pathog. 7, e1002251. doi: 10.1371/journal.ppat.1002251
Hoffmann, C., Leis, A., Niederweis, M., Plitzko, J. M., Engelhardt, H. (2008). Disclosure of the mycobacterial outer membrane: Cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U. S. A. 105, 3963–3967. doi: 10.1073/pnas.0709530105
Huang, W., Jia, J., Edwards, P., Dehesh, K., Schneider, G., Lindqvist, Y. (1998). Crystal structure of beta-ketoacyl-acyl carrier protein synthase II from e.coli reveals the molecular architecture of condensing enzymes. EMBO J. 17, 1183–1191. doi: 10.1093/emboj/17.5.1183
Inoyama, D., Awasthi, D., Capodagli, G. C., Tsotetsi, K., Sukheja, P., Zimmerman, M., et al. (2020). A preclinical candidate targeting Mycobacterium tuberculosis KasA. Cell Chem. Biol. 27, 560–570. doi: 10.1016/j.chembiol.2020.02.007
Kapilashrami, K., Bommineni, G. R., Machutta, C. A., Kim, P., Lai, C. T., Simmerling, C., et al. (2013). Thiolactomycin-based beta-ketoacyl-AcpM synthase a (KasA) inhibitors: Fragment-based inhibitor discovery using transient one-dimensional nuclear overhauser effect NMR spectroscopy. J. Biol. Chem. 288, 6045–6052. doi: 10.1074/jbc.M112.414516
Kim, P., Zhang, Y. M., Shenoy, G., Nguyen, Q. A., Boshoff, H. I., Manjunatha, U. H., et al. (2006). Structure-activity relationships at the 5-position of thiolactomycin: an intact (5R)-isoprene unit is required for activity against the condensing enzymes from Mycobacterium tuberculosis and escherichia coli. J. Med. Chem. 49, 159–171. doi: 10.1021/jm050825p
Kremer, L., Douglas, J. D., Baulard, A. R., Morehouse, C., Guy, M. R., Alland, D., et al. (2000). Thiolactomycin and related analogues as novel anti-mycobacterial agents targeting KasA and KasB condensing enzymes in Mycobacterium tuberculosis. J. Biol. Chem. 275, 16857–16864. doi: 10.1074/jbc.M000569200
Kumar, P., Capodagli, G. C., Awasthi, D., Shrestha, R., Maharaja, K., Sukheja, P., et al. (2018). Synergistic lethality of a binary inhibitor of Mycobacterium tuberculosis KasA. mBio 9, e02101–e02117. doi: 10.1128/mBio.02101-17
Lee, W., Engels, B. (2014). The protonation state of catalytic residues in the resting state of KasA revisited: Detailed mechanism for the activation of KasA by its own substrate. Biochemistry 53, 919–931. doi: 10.1021/bi401308j
Lee, W., Luckner, S. R., Kisker, C., Tonge, P. J., Engels, B. (2011). Elucidation of the protonation states of the catalytic residues in mtKasA: Implications for inhibitor design. Biochemistry 50, 5743–5756. doi: 10.1021/bi200006t
Leger, M., Gavalda, S., Guillet, V., van der Rest, B., Slama, N., Montrozier, H., et al. (2009). The dual function of the Mycobacterium tuberculosis FadD32 required for mycolic acid biosynthesis. Chem. Biol. 16, 510–519. doi: 10.1016/j.chembiol.2009.03.012
Luckner, S. R., Machutta, C. A., Tonge, P. J., Kisker, C. (2009). Crystal structures of mycobacterium tuberculosis KasA show mode of action within cell wall biosynthesis and its inhibition by thiolactomycin. Structure 17, 1004–1013. doi: 10.1016/j.str.2009.04.012
Lu, H., Tonge, P. J. (2008). Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc. Chem. Res. 41, 11–20. doi: 10.1021/ar700156e
Lu, H., Tonge, P. J. (2010). Drug-target residence time: Critical information for lead optimization. Curr. Opin. Chem. Biol. 14, 467–474. doi: 10.1016/j.cbpa.2010.06.176
Machutta, C. A., Bommineni, G. R., Luckner, S. R., Kapilashrami, K., Ruzsicska, B., Simmerling, C., et al. (2010). Slow onset inhibition of bacterial beta-ketoacyl-acyl carrier protein synthases by thiolactomycin. J. Biol. Chem. 285, 6161–6169. doi: 10.1074/jbc.M109.077909
Magnuson, K., Jackowski, S., Rock, C. O., Cronan, J. E., Jr. (1993). Regulation of fatty acid biosynthesis in escherichia coli. Microbiol. Rev. 57, 522–542. doi: 10.1128/mr.57.3.522-542.1993
Marrakchi, H., Laneelle, M. A., Daffe, M. (2014). Mycolic acids: Structures, biosynthesis, and beyond. Chem. Biol. 21, 67–85. doi: 10.1016/j.chembiol.2013.11.011
Minato, Y., Gohl, D. M., Thiede, J. M., Chacon, J. M., Harcombe, W. R., Maruyama, F., et al. (2019). Genomewide assessment of mycobacterium tuberculosis conditionally essential metabolic pathways. mSystems 4, e00070–e00019. doi: 10.1128/mSystems.00070-19
Miyakawa, S., Suzuki, K., Noto, T., Harada, Y., Okazaki, H. (1982). Thiolactomycin, a new antibiotic. IV. biological properties and chemotherapeutic activity in mice. J. Antibiot. (Tokyo). 35, 411–419. doi: 10.7164/antibiotics.35.411
Nishida, I., Kawaguchi, A., Yamada, M. (1986). Effect of thiolactomycin on the individual enzymes of the fatty acid synthase system in escherichia coli. J. Biochem. 99, 1447–1454. doi: 10.1093/oxfordjournals.jbchem.a135614
Oishi, H., Noto, T., Sasaki, H., Suzuki, K., Hayashi, T., Okazaki, H., et al. (1982). Thiolactomycin, a new antibiotic. i. taxonomy of the producing organism, fermentation and biological properties. J. Antibiot. (Tokyo). 35, 391–395. doi: 10.7164/antibiotics.35.391
Portevin, D., De Sousa-D’Auria, C., Houssin, C., Grimaldi, C., Chami, M., Daffe, M., et al. (2004). A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. U.S.A. 101, 314–319. doi: 10.1073/pnas.0305439101
Puhl, A. C., Lane, T. R., Vignaux, P. A., Zorn, K. M., Capodagli, G. C., Neiditch, M. B., et al. (2020). Computational approaches to identify molecules binding to Mycobacterium tuberculosis KasA. ACS Omega. 5, 29935–29942. doi: 10.1021/acsomega.0c04271
Rahlwes, K. C., Sparks, I. L., Morita, Y. S. (2019). Cell walls and membranes of actinobacteria. Subcell. Biochem. 92, 417–469. doi: 10.1007/978-3-030-18768-2_13
Rebollo-Lopez, M. J., Lelievre, J., Alvarez-Gomez, D., Castro-Pichel, J., Martinez-Jimenez, F., Papadatos, G., et al. (2015). Release of 50 new, drug-like compounds and their computational target predictions for open source anti-tubercular drug discovery. PloS One 10, e0142293. doi: 10.1371/journal.pone.0142293
Rock, J. M., Hopkins, F. F., Chavez, A., Diallo, M., Chase, M. R., Gerrick, E. R., et al. (2017). Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2, 16274. doi: 10.1038/nmicrobiol.2016.274
Rullas, J., Garcia, J. I., Beltran, M., Cardona, P. J., Caceres, N., Garcia-Bustos, J. F., et al. (2010). Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob. Agents Chemother. 54, 2262–2264. doi: 10.1128/AAC.01423-09
Sassetti, C. M., Boyd, D. H., Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Schaeffer, M. L., Agnihotri, G., Volker, C., Kallender, H., Brennan, P. J., Lonsdale, J. T. (2001). Purification and biochemical characterization of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthases KasA and KasB. J. Biol. Chem. 276, 47029–47037. doi: 10.1074/jbc.M108903200
Schiebel, J., Kapilashrami, K., Fekete, A., Bommineni, G. R., Schaefer, C. M., Mueller, M. J., et al. (2013). Structural basis for the recognition of mycolic acid precursors by KasA, a condensing enzyme and drug target from Mycobacterium tuberculosis. J. Biol. Chem. 288, 34190–34204. doi: 10.1074/jbc.M113.511436
Slayden, R. A., Barry, C. E., 3rd. (2002). The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis. Tuberculosis (Edinb). 82, 149–160. doi: 10.1054/tube.2002.0333
Slayden, R. A., Lee, R. E., Armour, J. W., Cooper, A. M., Orme, I. M., Brennan, P. J., et al. (1996). Antimycobacterial action of thiolactomycin: An inhibitor of fatty acid and mycolic acid synthesis. Antimicrob. Agents Chemother. 40, 2813–2819. doi: 10.1128/AAC.40.12.2813
Takayama, K., Wang, C., Besra, G. S. (2005). Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18, 81–101. doi: 10.1128/CMR.18.1.81-101.2005
Vilcheze, C., Wang, F., Arai, M., Hazbon, M. H., Colangeli, R., Kremer, L., et al. (2006). Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 12, 1027–1029. doi: 10.1038/nm1466
Wang, X., Inoyama, D., Russo, R., Li, S. G., Jadhav, R., Stratton, T. P., et al. (2020). Antitubercular triazines: Optimization and intrabacterial metabolism. Cell. Chem. Biol. 27, 172–85.e11. doi: 10.1016/j.chembiol.2019.10.010
Wang, X., Perryman, A. L., Li, S. G., Paget, S. D., Stratton, T. P., Lemenze, A., et al. (2019). Intrabacterial metabolism obscures the successful prediction of an InhA inhibitor of Mycobacterium tuberculosis. ACS Infect. Dis. 5, 2148–2163. doi: 10.1021/acsinfecdis.9b00295
Wilson, R., Kumar, P., Parashar, V., Vilcheze, C., Veyron-Churlet, R., Freundlich, J. S., et al. (2013). Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 9, 499–506. doi: 10.1038/nchembio.1277
Zhang, Y. J., Ioerger, T. R., Huttenhower, C., Long, J. E., Sassetti, C. M., Sacchettini, J. C., et al. (2012). Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 8, e1002946. doi: 10.1371/journal.ppat.1002946
Keywords: mycobacterium tuberculosis, KasA, β-ketoacyl synthase, structure-based drug discovery, medicinal chemistry
Citation: Rudraraju RS, Daher SS, Gallardo-Macias R, Wang X, Neiditch MB and Freundlich JS (2022) Mycobacterium tuberculosis KasA as a drug target: Structure-based inhibitor design. Front. Cell. Infect. Microbiol. 12:1008213. doi: 10.3389/fcimb.2022.1008213
Received: 31 July 2022; Accepted: 24 August 2022;
Published: 15 September 2022.
Edited by:
Ben Gold, Weill Cornell Medicine, United StatesReviewed by:
Cynthia Dowd, George Washington University, United StatesWilliam Jacobs, Albert Einstein College of Medicine, United States
Copyright © 2022 Rudraraju, Daher, Gallardo-Macias, Wang, Neiditch and Freundlich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew B. Neiditch, bmVpZGl0bWJAbmptcy5ydXRnZXJzLmVkdQ==; Joel S. Freundlich, ZnJldW5kanNAcnV0Z2Vycy5lZHU=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship