 Yinghu Chen1,2,3
Yinghu Chen1,2,3 Lingfeng Mao3
Lingfeng Mao3 Dengming Lai1,2,3Weize Xu1,2,3Yuebai Zhang1,2
Dengming Lai1,2,3Weize Xu1,2,3Yuebai Zhang1,2 Sihao Wu3Di Yang3Shaobo Zhao1,2Zhicong Liu1,2Yi Xiao1,2
Sihao Wu3Di Yang3Shaobo Zhao1,2Zhicong Liu1,2Yi Xiao1,2 Yi Tang3Xiaofang Meng3Min Wang3Jueliang Shi3
Yi Tang3Xiaofang Meng3Min Wang3Jueliang Shi3 Qixing Chen1,2,3*
Qixing Chen1,2,3* Qiang Shu1,2,3*
Qiang Shu1,2,3*- 1The Children’s Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2National Clinical Research Center for Child Health, Hangzhou, China
- 3Joint Research Center for Molecular Diagnosis of Severe Infection in Children, Binjiang Institute of Zhejiang University, Hangzhou, China
Objectives: To develop a rapid and low-cost method for 16S rDNA nanopore sequencing.
Methods: This was a prospective study on a 16S rDNA nanopore sequencing method. We developed this nanopore barcoding 16S sequencing method by adding barcodes to the 16S primer to reduce the reagent cost and simplify the experimental procedure. Twenty-one common pulmonary bacteria (7 reference strains, 14 clinical isolates) and 94 samples of bronchoalveolar lavage fluid from children with severe pneumonia were tested. Results indicating low-abundance pathogenic bacteria were verified with the polymerase chain reaction (PCR). Further, the results were compared with those of culture or PCR.
Results: The turnaround time was shortened to 6~8 hours and the reagent cost of DNA preparation was reduced by employing a single reaction adding barcodes to the 16S primer in advance. The accuracy rate for the 21 common pulmonary pathogens with an abundance ≥ 99% was 100%. Applying the culture or PCR results as the gold standard, 71 (75.5%) of the 94 patients were positive, including 25 positive cultures (26.6%) and 52 positive quantitative PCRs (55.3%). The median abundance in the positive culture and qPCR samples were 29.9% and 6.7%, respectively. With an abundance threshold increase of 1%, 5%, 10%, 15% and 20%, the test sensitivity decreased gradually to 98.6%, 84.9%, 72.6%, 67.1% and 64.4%, respectively, and the test specificity increased gradually to 33.3%, 71.4%, 81.0%, 90.5% and 100.0%, respectively.
Conclusions: The nanopore barcoding 16S sequencing method can rapidly identify the pathogens causing bacterial pneumonia in children.
Introduction
Next-generation sequencing (NGS) was first reported for the diagnosis of neuroleptospirosis in 2014 (Wilson et al., 2014). Subsequently, the NGS has been tentatively applied in detecting pathogens causing infection, such as the coronavirus disease 2019 (COVID-19) and plague (Zhu et al., 2020; Stefan et al., 2016). However, due to the high cost and long turnaround time, NGS is usually applied to detect pathogens causing infections with treatment failure rather than early-stage severe infections in clinical setting (Consensus et al., 2020; Hutchins et al., 2019). For children with severe bacterial infection, rapid pathogen identification is important for the administration of appropriate antibiotics. Nanopore sequencing technology, with the advantages of long reads, and real-time sequencing and analysis, makes it possible to quickly detect and diagnose local pathogen (Sedlazeck et al., 2018; Shiota et al., 2018). Several studies on the application of nanopore sequencing in pathogen diagnosis have been reported (Charalampous et al., 2019; Leggett et al., 2020; Baldan et al., 2021; Zhao et al., 2021). Besides, nanopore sequencing may be used to screen for drug resistance mutations and virulence genes of pathogens (Zhou et al., 2021; Ferreira et al., 2021).
In this study, we developed a nanopore barcoding 16S rDNA sequencing (NB16S-seq) method, which was a 16S full-length gene amplification with barcoded primers prior to targeted third-generation sequencing (TGS). The amplification of full-length 16S rDNA improves the sensitivity and decreases the limit of detection. In addition, the barcode sequence is added to the primer in advance, and this optimization can detect the amplification of full-length 16S rDNA and the ligation of barcodes in one step. The above features not only reduce the reagents cost of end-repair and dA-tailing for all samples to one pooled sample but also avoid the aerosol contamination from the barcode addition step. This prospective study was carried out to ascertain the significance of the NB16S-seq in the identification of bacterial pneumonia pathogens.
Methods
Study design and participants
This was a prospective study on an improved 16S rDNA nanopore sequencing method. Before testing clinical samples, the 21 common pathogens of bacterial pneumonia (7 reference strains and 14 clinical isolates) were sequenced by the NB16S-seq to establish its accuracy.
Bronchoalveolar lavage fluid (BALF) (5 ml) from patients with pulmonary infection was collected in the respiratory endoscopy room between December 2021 and January 2022, and pulmonary infection was confirmed by either pulmonary X-ray or CT. The BALF was sent for culture, NB16S-seq test and M. pneumonia RNA detection. Pathogens with negative cultures and positive NB16S-seq (abundant ≥ 5.0%) were verified by quantitative polymerase chain reaction (qPCR). If only one pathogen was detected and its abundance was between 1% and 5%, the qPCR verification was also performed.
The gold standard of the BALF testing is either culture or qPCR. We collected the following data: age, sex, symptoms, diagnosis, antibiotics use, imaging, and duration from admission to sequencing, etc. Routine clinical microbiological investigation included the BALF culture and detection of routine respiratory virus (including respiratory syncytial virus, adenovirus, influenza A and B, and parainfluenza A, B and C) antigens from nasopharyngeal swabs with Rapid Antigen Detection Kits (Kaibili, Hangzhou Innovation Biotechnology Co., Ltd., Hangzhou, China).
Definitions
‘Respiratory pathogens’ or ‘pathogens’ were defined in this study as common causes of respiratory infection to differentiate them from commensal organisms. The respiratory pathogens identified in this study were S. pneumoniae, H. influenzae, M. catarrhalis, M. pneumoniae, S. aureus, S. agalactiae, L. gormanii, B. pertussis, P. aeruginosa, S. marcescens, and A. baumannii.
DNA extraction
Samples were processed in the genetic diagnostic center. The BALF was collected and stored at 4 °C before testing. The 7 reference strains and 13 clinically isolated strains were inoculated on Columbia blood plate culture medium for DNA extraction. These strains were identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS; Bruker Daltoniks, Germany) before DNA extraction. The QIAamp® DNA Mini Kit (QIAGEN, Germany) was used for the purification of microbial DNA. BALF (300μL) was first centrifuged at 13,000 rpm for 5 min to remove the supernatant, and then, the precipitate was suspended in normal saline (200 μL). The mixture was added to buffer ATL (180 μL) and proteinase K (20 μL), mixed by vortexing and incubated at 56 °C for 30 min at 500 rpm in a thermomixer (Eppendorf, Germany). For the 21 bacterial strains, 200 μL of sample (1×108 CFU/ml), which was directly added to buffer ATL (180 μL) and proteinase K (20 μL), was incubated at 56 °C for 30 min at 500 rpm in a thermomixer after mixing well. The following experimental steps were carried out according to the manufacturer’s instructions.
16S rDNA full-length gene amplification and nanopore sequencing
The concentration of isolated DNA from samples was measured with a Qubit dsDNA HS Assay Kit and a Qubit 4.0 Fluorometer (Thermo Fisher, USA) according to the instructions of the manufacturer. A total of 5~10 ng of DNA template (10 μL) was used for 16S rDNA full-length sequence amplification (primers: 5’-barcode-27F-AGAGTTTGATCMTGGCTCAG-3’, 5’-barcode-1492R-CGGTTACC TTGTTACGACTT-3’) with 25 μL of Q5 Hot Start High-Fidelity 2X Master Mix (M0494L, New England Biolabs, USA) and 14.5 μL of nuclease-free water (New England Biolabs, USA). PCR amplicons were visualized by 1% agarose gel electrophoresis, followed by purification using 1x AMPure XP beads (Beckman Coulter, USA), and the DNA concentration was measured with a Qubit 4.0 Fluorometer. 1~48 amplified DNA was pooled in equimolar amounts. The pooled DNA was end-repaired and dA-tailed with NEBNext FFPE DNA Repair Mix (New England Biolabs, USA) and NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs, USA), followed by purification with AMPure XP beads (Beckman Coulter, USA). Then, the nanopore sequencing was performed using the ligation sequencing kit SQK-LSK110 (Oxford Nanopore Technologies, UK), and the DNA library was loaded into an R9.4.1 flow cell (Oxford Nanopore Technologies, UK) for sequencing for 1~3 hours in the GridION platform according to the sample number. In every sequencing process run, negative process controls were added to the DNA extraction and amplification steps with sterile nuclease-free water to monitor contamination from the environment.
Bioinformatics and pathogen identification
First, raw reads were demultiplexed with an in-house script to eliminate interference from error splitting. Porechop was used to trim the adapter and barcode on the end of the reads (Wick et al., 2018). Seqkit was used to filter reads, retaining those with a size range of 1.3–1.6 kb and qscore ≧ 12 (Shen et al., 2016). To identify the pathogen, clean reads were mapped to the 16S database collected from NCBI and GTDB using minimap2 v2.17 (Li H, 2018) with the parameter “-ax map-ont”, and the program samtools v1.10 (Danecek et al., 2021) was used to remove unmapped, nonunique mapped and multiple-mapped reads with the parameter “view -F2308”. The species abundance in the sample were incorporated into the biological classification according to the NCBI taxonomy tree. The pathogen identification was determined according to the matching between the species abundance in the sample and the guide for pathogens in the Manual of Clinical Microbiology, 12th edition. The thresholds used in this study for pathogen identification were defined as follows: the valid read number was greater than 5000, and the reliable species abundance for pathogen identification was 1%. To reduce the impact of similar homologous segments of the same species and miscellaneous bacteria in the environment on the analytical results, we tabulated bacterial species that were frequently cultured in the BALF samples into a table (for details, see Supplementary Table 1). If the test bacteria met the following conditions, they were considered “detected bacteria”: ① the abundance of detected bacteria was more than 1% and the species was in the above mentioned high-frequency bacterial table; ② if not in the Supplementary Table 1, at most two species with abundance ≥5% were reported in the same genus; and ③ if there were more than ten species not in the Supplementary Table 1, they were classified according to their abundance, and only the first ten species were reported.
Statistical analysis
Stata 15 software was used for ROC analysis, and the sensitivity, specificity and area under the ROC curve were calculated under different cross-sections. Data are expressed as the median (interquartile range).
Results
Rapid NB16S-seq test method
The NB16S-seq workflow is shown in Figure 1. The report time was 8 hours after receiving the BALF sample and could be shortened to 6 hours. This was significantly shorter than the time of culture and NGS. Rapid diagnosis may enhance the early identification of pathogens and contribute to the successful treatment of severe infection.
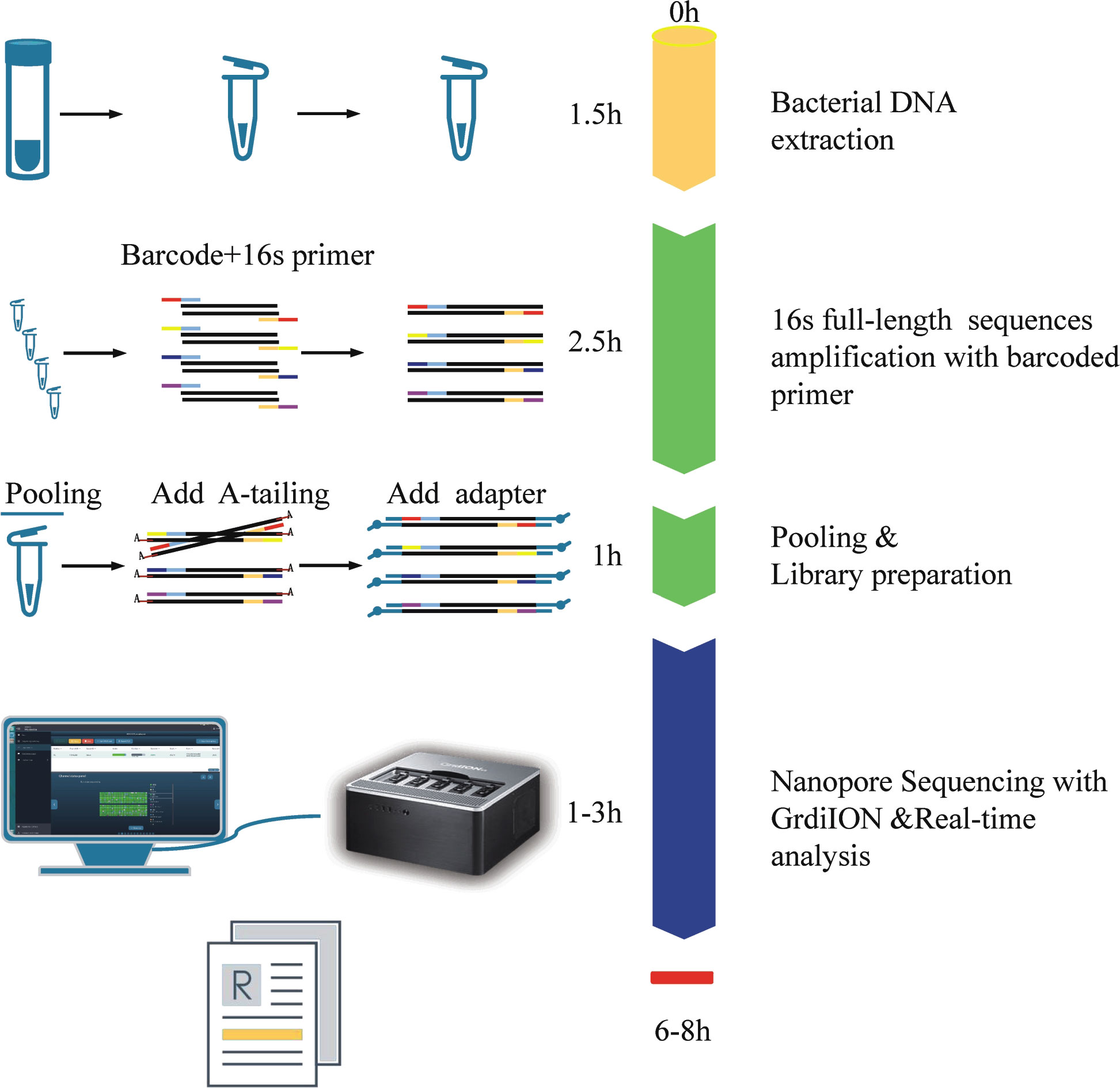
Figure 1 The process and schematic representation of NB16S-seq. The turnaround time was approximately 6~8 h from sample collection to results.
Evaluating the NB16S-seq test with reference strains, clinical isolates and BALF specimens
The NB16S-seq results for these 21 bacterial strains are shown in Table 1. All bacterial species in Table 1 were correctly detected with an abundance of ≥ 99%, suggesting that the NB16S-seq is accurate in the detection of common pathogens of pulmonary infection.
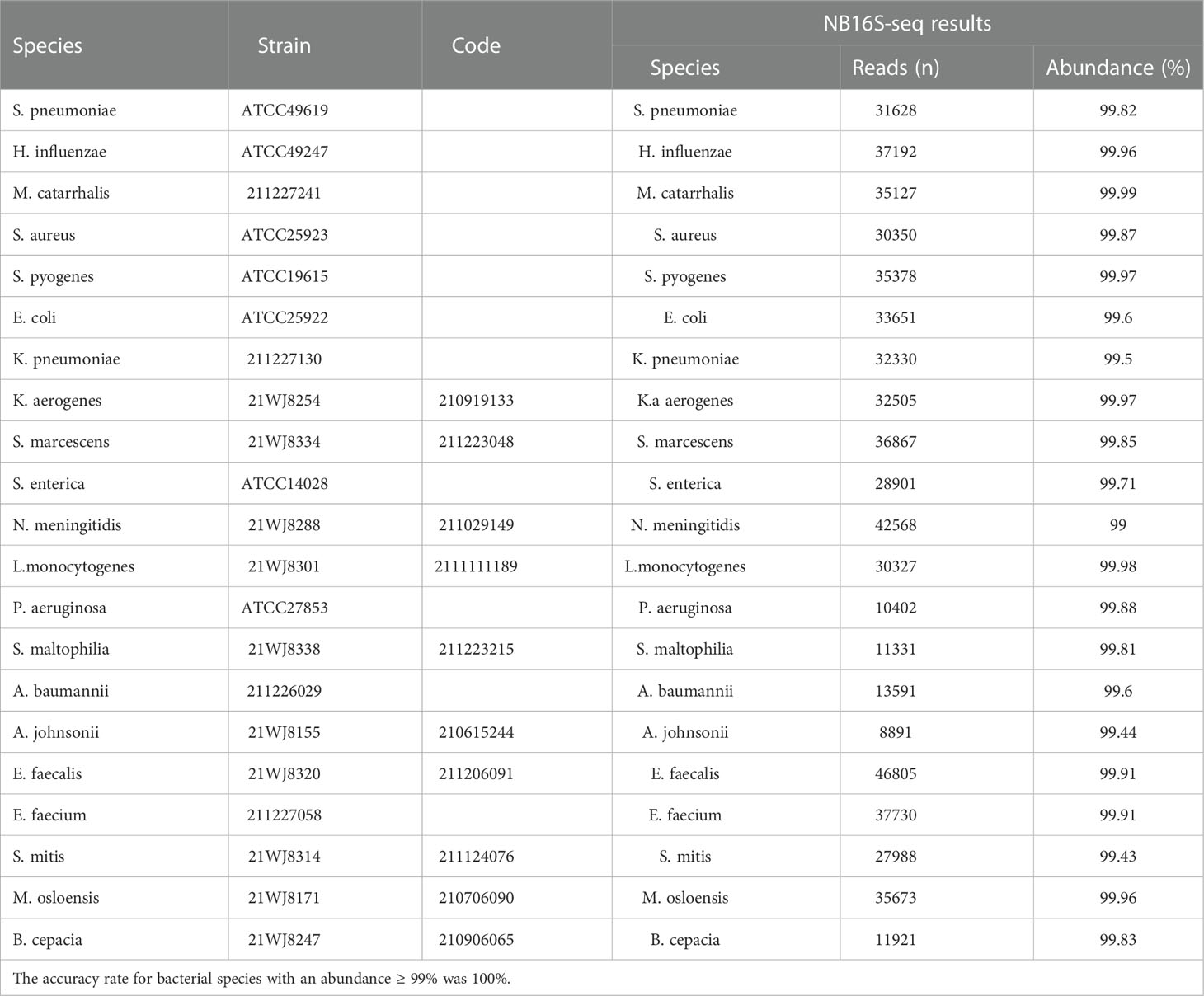
Table 1 Evaluating the NB16S-seq test with 21 common respiratory pathogens. Seven reference strains and 14 clinical isolates were used to verify the detection results of the NB16S-seq.
Among the 110 eligible BALF samples, 94 met the criteria (Figure 2A). The demographic and clinical characteristics of the patients are shown in Table 2, including 88 cases of community acquired pneumonia, 6 cases of hospital acquired pneumonia (HAP), 15 cases of acute respiratory distress syndrome, 20 cases with ICU admission, 1 case of progression, 2 cases of death, and 91 cases of improvement or recovery. The NB16S-seq results regarding bacteria and their abundances are shown in a heatmap (Figure 2B, detail data seen in Supplementary Table 2). The heatmap shows many commensal organisms, fewer opportunistic pathogens, and various abundant common pathogens. This may be related to the following: ① opportunistic pathogens are often isolated in HAP, and only 6 HAP children (6.4%) were enrolled; ② commensal organisms exist in the oropharynx, which might contaminate BALF specimens; and ③ eighty-eight patients (93.6%) received antibiotics before sampling, potentially mitigating pathogen abundance.
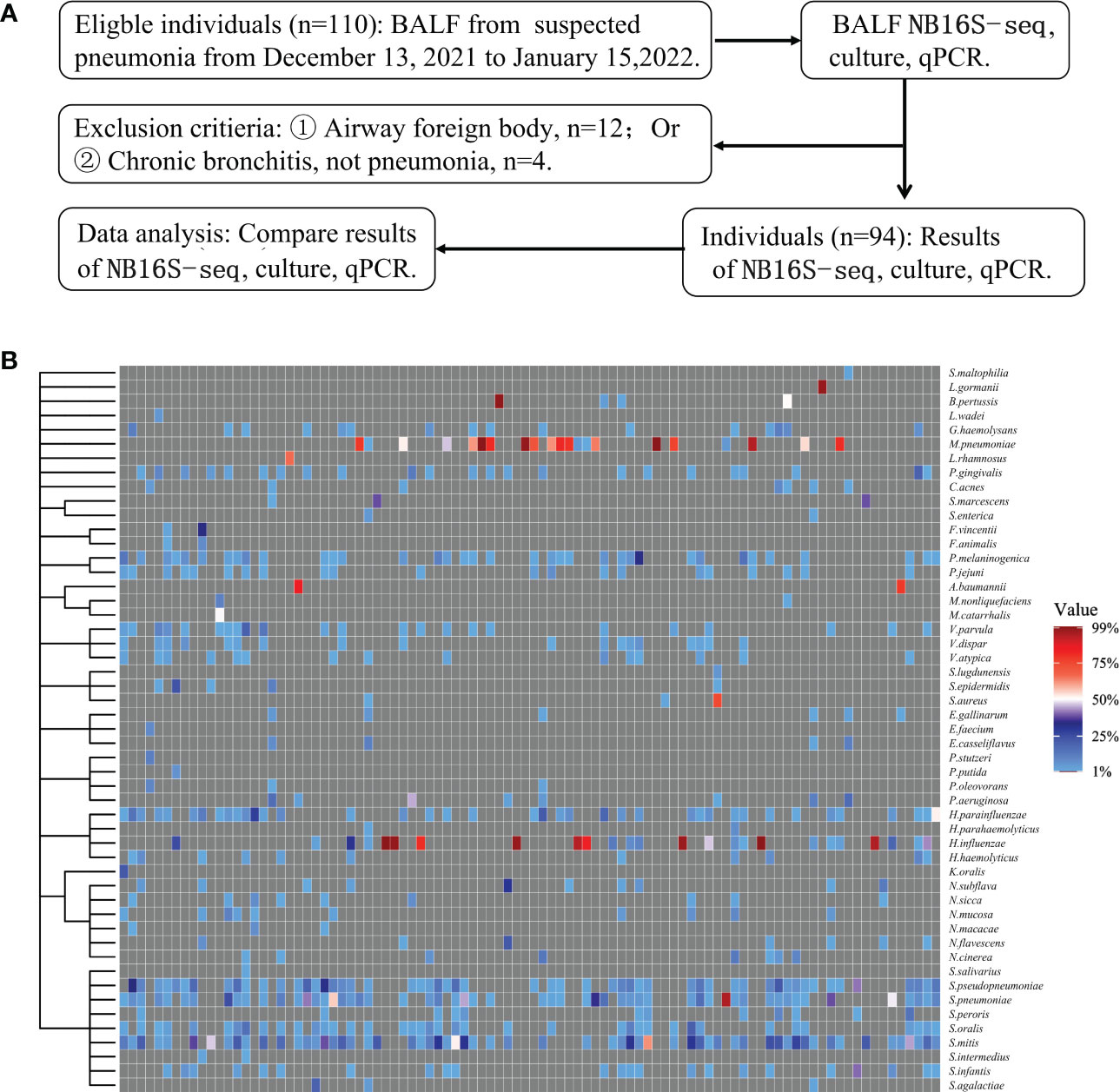
Figure 2 Evaluation of NB16S-seq in the etiological diagnosis of pediatric pneumonia (A): BALF form pediatric pneumonia tested with NB16S-seq, culture or qPCR in our hospital between November 2021 and January 2022. (B). Heatmap of bacteria abundance ≥1.0% in 94 BALF samples tested by NB16S-seq. Each column in the (C) indicates a sample. Formula of abundance = (number of sequences for a species/total number of sequences in the specimen) ×100%. The value indicates abundance, and different colors of the value bar represent the different NB16S-seq abundances.
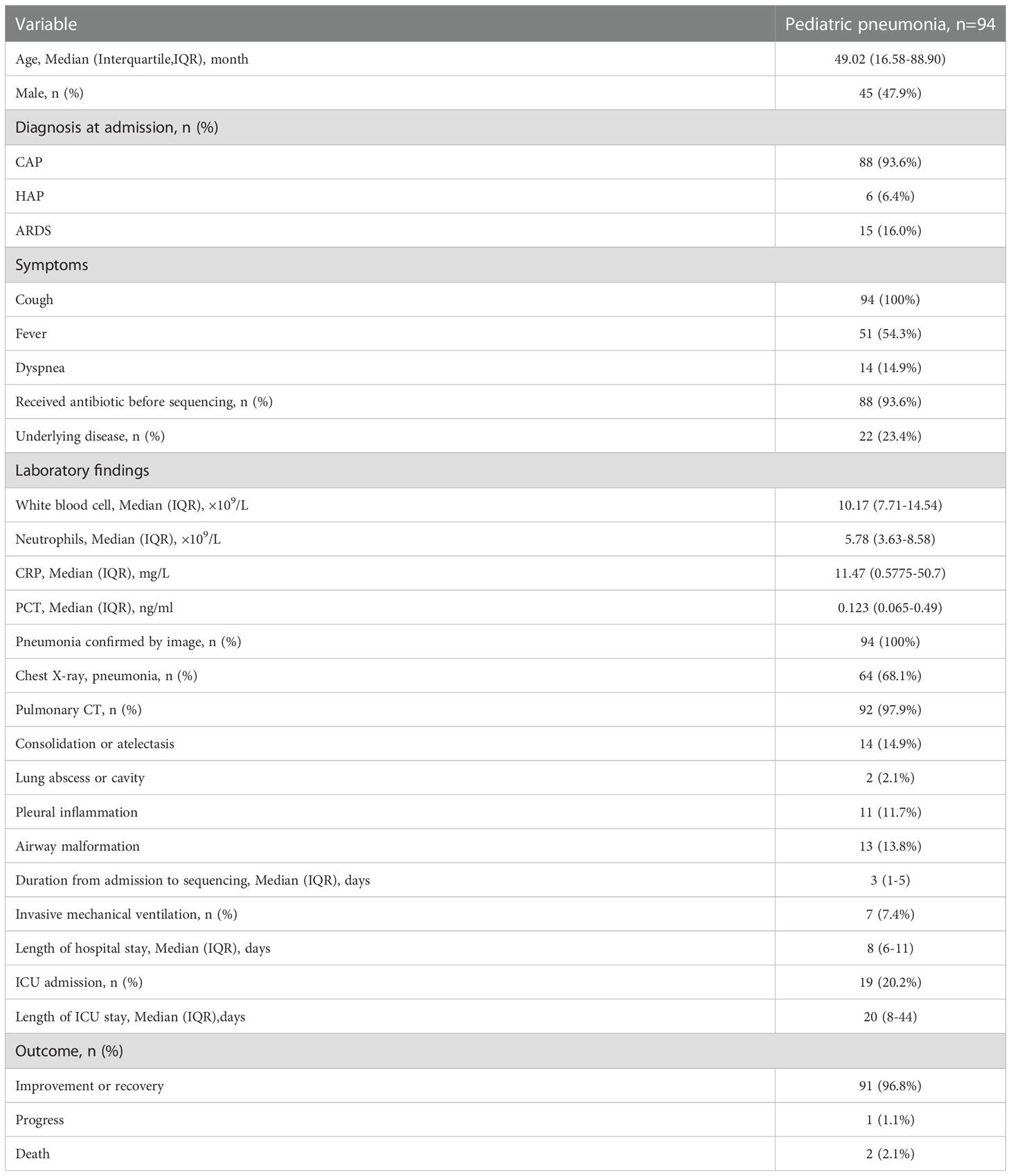
Table 2 Demographic and clinical characteristics of the 94 pediatric pneumonia patients.
Exploring the appropriate NB16S-seq threshold to identify pathogen
Applying the BALF culture or qPCR results as the gold standard, BALF NB16S-seq results were analyzed (details in Supplementary Table 3). Seventy-one (75.5%) of the 94 BALF specimens were positive according to the gold standard, 25 (26.6%) were culture positive, and 52 (55.3%) were qPCR positive. The number of samples containing 0, 1, 2, 3 and 5 pathogenic bacteria with detected abundance ≥ 1% for each bacterium were 7, 54, 28, 4 and 1, respectively. The relationship between NB16S-seq abundance and positive culture, positive PCR and negative PCR is shown in Figure 3A, and the median abundances were 29.9%, 6.7% and 4.2%, respectively. The abundance on bacterial NB16S-seq was closely related to the positive culture and qPCR results. The NB16S-seq abundance of three-quarters of the BALF with positive culture and positive qPCR was ≥5.2% and 2.7%, respectively.
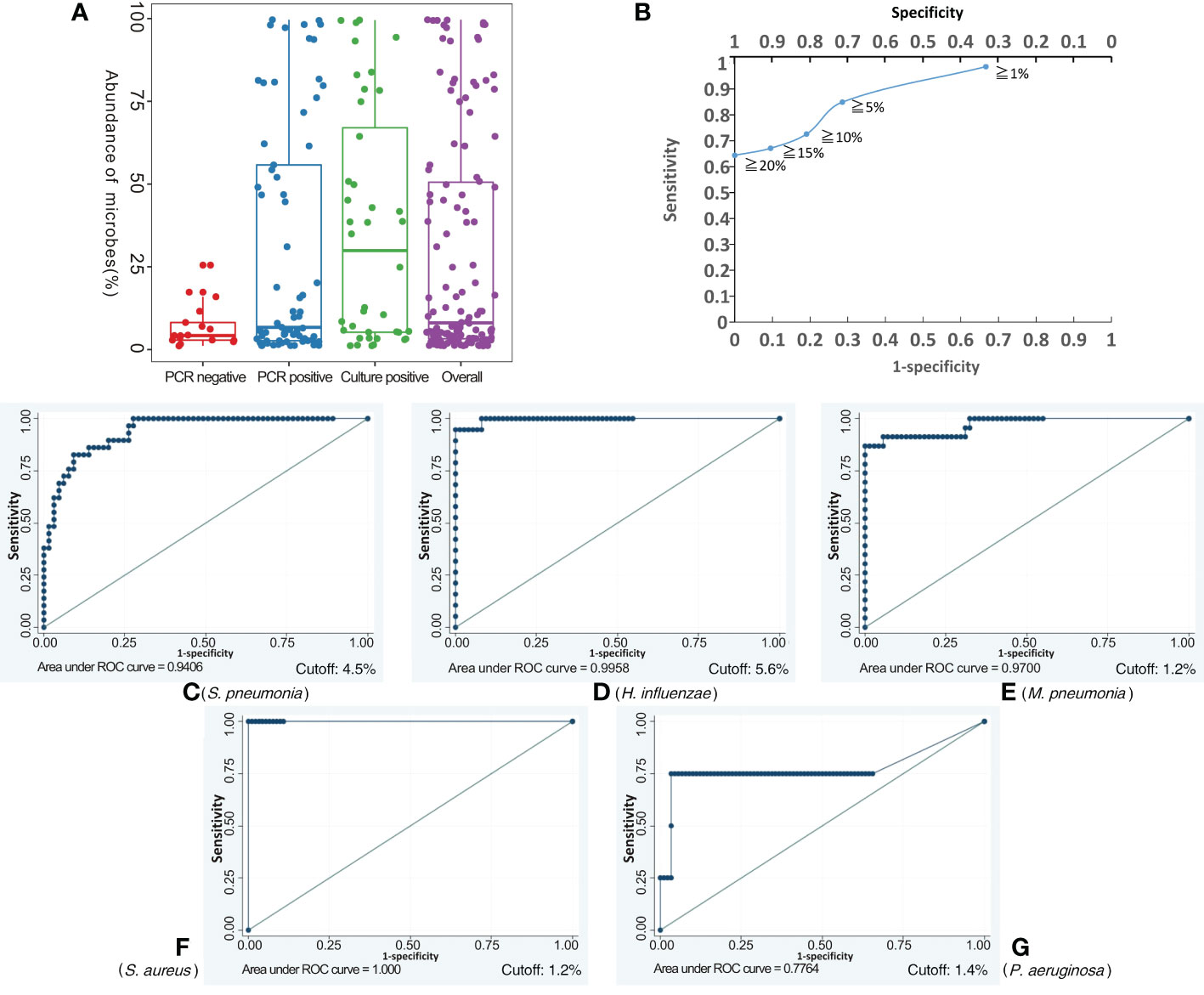
Figure 3 Exploring the appropriate NB16S-seq threshold to identify pathogenic bacteria. The relationship between BALF NB16S-seq abundance and positive culture, positive qPCR and negative qPCR results (A). Correlation between NB16S-seq abundance threshold and sensitivity and specificity of BALF pathogen detection (B). For common respiratory pathogens, an NB16S-seq abundance ≥ 5% is an appropriate criterion to designate a sample positive for a pathogen (C-G). The relationship between the NB16S-seq abundance threshold and the sensitivity and specificity for different strains, S. pneumonia (C), H. influenza (D), M. pneumonia (E), S. aureus (F), and P. aeruginosa (G). A value of 4.5% was an appropriate cutoff for S. pneumonia (C), 5.6% for H. influenzae (D), 1.2% for M. pneumonia (E) and S. aureus (F), and 1.4% for P. aeruginosa (G).
With the increase in the specimens’ NB16S-seq abundance threshold to 1%, 5%, 10%, 15% and 20%, the test sensitivity decreased gradually to 98.6%, 84.9%, 72.6%, 67.1% and 64.4%, respectively, and the test specificity increased progressively to 33.3%, 71.4%, 81.0%, 90.5% and 100.00%, respectively (see Figure 3B). If a low NB16S-seq abundance of common pathogens was detected in the BALF, the relationship between the bacteria and pulmonary infection was further evaluated, as these might have been colonizing bacteria. Therefore, for all common respiratory pathogens with an NB16S-seq abundance ≥ 1%, a cut-off value ≥ 5% was applied because at this cut-off the Youden Index (84.9% + 71.4% - 100% = 56.3%) was the largest.
According to the gold standard, S. pneumonia was detected 29 times, M. pneumoniae 23 times, H. influenzae 18 times, P. aeruginosa 4 times, A. baumannii thrice, both S. aureus and B. pertussis twice, along with M. catarrhalis, S. agalactiae, L. gormanii, and S. marcescens being detected once in 94 BALF specimens (details in Supplementary Table 3). When the cut-off value of specimens’ NB16S-seq abundance was the same, the sensitivity and specificity of each pathogen were different (Figure 3). The relationship between the NB16S-seq abundance threshold, together with the sensitivity and specificity of common strains was analyzed (Table 3). Accordingly, 4.5% was an appropriate cut-off for S. pneumonia (Figure 3C), 5.6% for H. influenzae (Figure 3D), 1.2% for M. pneumonia (Figure 3E) and S. aureus (Figure 3F), and 1.4% for P. aeruginosa (Figure 3G). The appropriate cutoff value of S. pneumonia and H. influenzae was higher than that of M. pneumonia, S. aureus and P.aeruginosa, which may be related to the oropharyngeal colonization of H. influenzae and S. pneumonia, while the possibility of colonization of the other three bacteria was low.
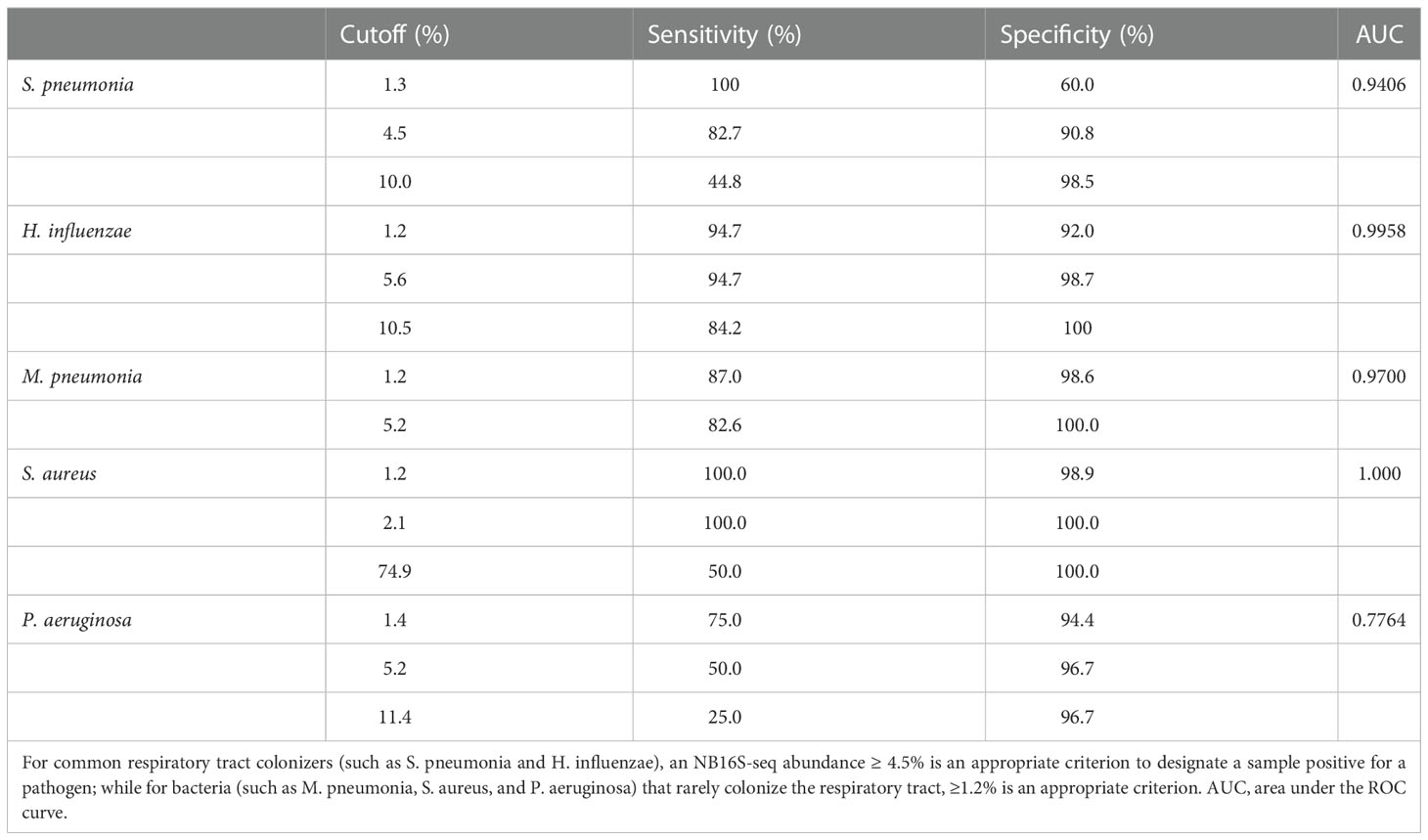
Table 3 The relationship between the NB16S-seq abundance threshold and the sensitivity and specificity for different strains according to the gold standard test (either culture or PCR) results.
To optimize the interpretations of NB16S-seq results, an abundance cut-off of 1.0% might be appropriate for organisms that are not typical colonizers of upper respiratory tract (e.g., M. pneumonia, S. aureus, and P. aeruginosa), and 5.0% for the other eight bacteria that typically colonize the upper respiratory. NB16S-seq abundances were evaluated with these two different cut-off values, the true or false positive, and true or false negative results are shown in Table 4, the sensitivity, specificity, positive and negative predictive values were 85.9%, 73.6%, 83.9% and 76.5%, respectively, and after applying two different abundance cut-off values for two different group of bacteria, Youden Index (85.9%+73.6% -100%=59.5%) was higher than that (56.3%) of single 5.0% cutoff value.
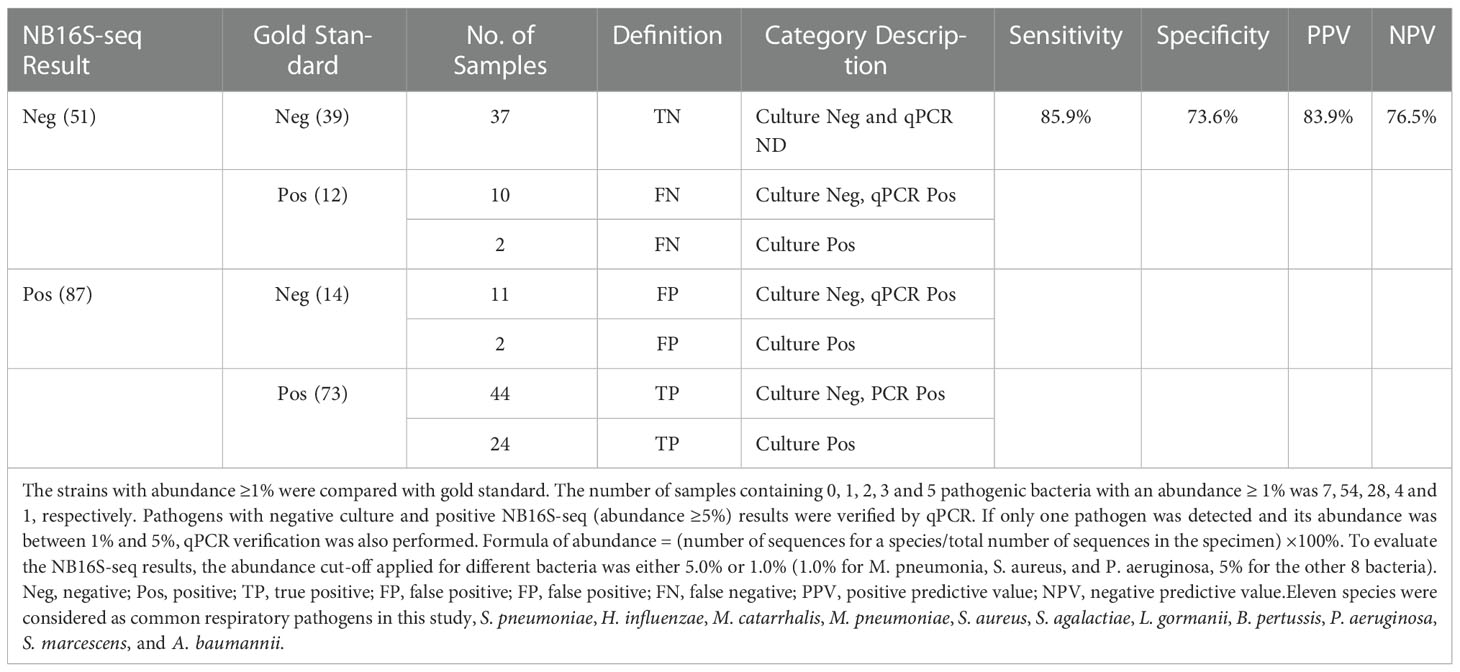
Table 4 Evaluating BALF NB16S-seq with the gold standard test (either culture or qPCR).
Discussion
Pneumonia is one of the main causes of death in children under 5 years old (Liu et al., 2015). Timely and appropriate antibiotic treatment could significantly reduce mortality. However, diversity and antibiotic resistance complicates the pathogenic treatment (Yiang et al., 2021). Rapid and sensitive pathogen identification is important. Routine pathogen detection cannot meet clinical needs due to prolonged time period required or limited detection of single or few kinds of pathogens (Holter et al., 2015; Hassibi et al., 2018; Klein et al., 2021).
In recent years, the application of nanopore sequencing technology in pathogen detection has gained much attention. Compared with the NGS, nanopore sequencing technology has the following advantages (Goldberg et al., 2015; Sedlazeck et al., 2018): ① shorter time from sampling to results, ② simpler sequencing library construction, ③ long sequence reading without assembly, and ④ real-time sequencing and analysis. Compared with the previous 16S rDNA nanopore sequencing method, the NB16S-seq has the following advantages: ① reduced the reagent cost for each sample, and ② optimized the experimental procedure.
The NB16S-seq method includes bacterial DNA extraction, 16S rDNA amplification, pooling and library preparation, nanopore sequencing and real-time analysis. The NB16S-seq was verified for 7 reference strains and 14 clinical isolates. The accuracy rate for species was 100%, and their abundance was ≥ 99%. In the process of 16S rDNA amplification and species database comparison, there was no error, and no other species appeared incorrectly. The 16S rDNA is a commonly used molecular clock in bacterial systematic classification. Bacterial universal primers can amplify the 16S rDNA fragments of all bacteria, and the difference in variable regions can be used to distinguish different bacteria. As an increasing number of bacterial 16S rDNA sequences are being determined and included in the NCBI and GTDB databases, it is faster and more convenient to identify species based on the 16S rDNA (Robeson et al., 2021). The similarity rate of 16S rDNA among some species (such as: S. pneumoniae and S. mitis) is greater than 99%, which intricates the difficulty of species identification (Church et al., 2020; Yatera et al., 2021). Interestingly, the NB16S-seq can effectively distinguish them.
Among the bacteria detected by the NB16S-seq, oropharyngeal-colonizing bacteria were the most common. In particular, the BALF culture reported them as colonizing bacteria, and the NB16S-seq listed them in the appendix results as detected bacteria. These bacteria can cause infective endocarditis, meningitis, and local tissue abscesses. The NGS showed that colonizing anaerobic bacteria in the oropharynx are closely related to lung abscesses (Mu et al., 2021).
Some pathogenic bacteria, such as S. pneumoniae and H. influenzae may colonize the oropharynx. Furthermore, differentiating commensal from pathogens is complex. However, our results showed the NB16S-seq abundance to be beneficial in distinguishing pathogens from commensals. When the NB16S-seq abundance was increased from 1% to 5%, the specificity also increased from 33.3% to 71.5%, and significantly reduced the false-positive rate. When the abundance extended to 20%, the specificity was 100%. Some pathogenic bacteria, such as M. pneumoniae, S. aureus and P. aeruginosa rarely colonize the oropharynx. Interestingly, they had a high specificity when the abundance was ≥ 1%, implying that the bacteria exist in the BALF.
Conclusions
We report a rapid pathogen diagnostic method, the NB16S-seq, for use in bacterial pneumonia with an optimized experimental procedure and lower cost. Pathogens can be identified in 6~8 h. For oropharyngeal-colonizing pathogens, an NB16S-seq abundance of 5% is the best cutoff point, and 1% is the best cutoff for pathogens that rarely colonize.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: We have uploaded the sequencing to the GSA, Project Number is PRJCA011155, which can be visited in https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA011155.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics committee of The Children’s Hospital of Zhejiang University of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
YC and LM contributed equally to this work and share first authorship. YC, DL, and WX collected and analyzed medical data of the patients, wrote and revised the manuscript. YZ, ZL, and SZ participated in the treatment of the patients and data collection. LM, SW, DY, YX, YT, XM, MW, and JS participated in the Improved targeting 16S rDNA nanopore sequencing and data analysis. QC and QS contributed to the treatment plan and made a critical revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (+226-2022-00060), “Pioneer” and “Leading Goose” R&D Program of Zhejiang Province (2023C03028), and the National Natural Science Foundation of China (82071812).
Acknowledgments
We thank Professor Kangchen Zhao from Jiangsu Provincial Center for Disease Control and Prevention for providing primers of the 7 common pathogenic bacteria.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.1001607/full#supplementary-material
References
Baldan, R., Cliff, P. R., Burns, S., Medina, A., Smith, G. C., Batra, R., et al. (2021). Development and evaluation of a nanopore 16S rRNA gene sequencing service for same day targeted treatment of bacterial respiratory infection in the intensive care unit. J. Infect. 83 (2), 167–174. doi: 10.1016/j.jinf.2021.06.014
Charalampous, T., Kay, G. L., Richardson, H., Aydin, A., Baldan, R., Jeanes, C., et al. (2019). Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 37 (7), 783–792. doi: 10.1038/s41587-019-0156-5
Church, D. L., Cerutti, L., Gürtler, A., Griener, T., Zelazny, A., Emler, S. (2020). Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin. Microbiol. Rev. 33 (4), e00053–e00019. doi: 10.1128/CMR.00053-19
Consensus Group Of Experts On Application Of Metagenomic Next Generation Sequencing In The Pathogen Diagnosis In Clinical Moderate And Severe Infections, Professional Committee Of Sepsis And Shock Chinese Research Hospital Association, Professional Committee Of Microbial Toxins Chinese Society For Microbiology, Professional Committee Of Critical Care Medicine Shenzhen Medical Association (2020). Expert consensus for the application of metagenomic next generation sequencing in the pathogen diagnosis in clinical moderate and severe infections. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 32 (5), 531–536. doi: 10.3760/cma.j.cn121430-20200228-00095
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10 (2), giab008. doi: 10.1093/gigascience/giab008
Ferreira, F. A., Helmersen, K., Visnovska, T., Jørgensen, S. B., Aamot, H. V. (2021). Rapid nanopore-based DNA sequencing protocol of antibiotic-resistant bacteria for use in surveillance and outbreak investigation. Microb. Genom. 7 (4), 557. doi: 10.1099/mgen.0.000557
Goldberg, B., Sichtig, H., Geyer, C., Ledeboer, N., Weinstock, G. M. (2015). Making the leap from research laboratory to clinic: Challenges and opportunities for next-generation sequencing in infectious disease diagnostics. mBio 6 (6), e01888–e01815. doi: 10.1128/mBio.01888-15
Hassibi, A., Manickam, A., Singh, R., Bolouki, S., Sinha, R., Jirage, K. B., et al. (2018). Multiplexed identification, quantification and genotyping of infectious agents using a semiconductor biochip. Nat. Biotechnol. 36 (8), 738–745. doi: 10.1038/nbt.4179
Holter, J. C., Müller, F., Bjørang, O., Samdal, H. H., Marthinsen, J. B., Jenum, P. A., et al. (2015). Etiology of community-acquired pneumonia and diagnostic yields of microbiological methods: a 3-year prospective study in Norway. BMC Infect. Dis. 15, 64. doi: 10.1186/s12879-015-0803-5
Hutchins, R. J., Phan, K. L., Saboor, A., Miller, J. D., Muehlenbachs, A., CDC NGS Quality Workgroup (2019). Practical guidance to implementing quality management systems in public health laboratories performing next-generation sequencing: Personnel, equipment, and process management (Phase 1). J. Clin. Microbiol. 57 (8), e00261–e00219. doi: 10.1128/JCM.00261-19
Klein, M., Bacher, J., Barth, S., Atrzadeh, F., Siebenhaller, K., Ferreira, I., et al. (2021). Multicenter evaluation of the unyvero platform for testing bronchoalveolar lavage fluid. J. Clin. Microbiol. 59 (3), e02497–e02420. doi: 10.1128/JCM.02497-20
Leggett, R. M., Alcon-Giner, C., Heavens, D., Caim, S., Brook, T. C., Kujawska, M., et al. (2020). Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat. Microbiol. 5 (3), 430–442. doi: 10.1038/s41564-019-0626-z
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34 (18), 3094–3100. doi: 10.1093/bioinformatics/bty191
Liu, L., Oza, S., Hogan, D., Perin, J., Rudan, I., Lawn, J. E., et al. (2015). Global, regional, and national causes of child mortality in 2000-13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet 385 (9966), 430–440. doi: 10.1016/S0140-6736(14)61698-6
Mu, S., Hu, L., Zhang, Y., Liu, Y., Cui, X., Zou, X., et al. (2021). Prospective evaluation of a rapid clinical metagenomics test for bacterial pneumonia. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.684965
Robeson, M. S., 2nd, O'Rourke, D. R., Kaehler, B. D., Ziemski, M., Dillon, M. R., Foster, J. T., et al. (2021). RESCRIPt: Reproducible sequence taxonomy reference database management. PloS Comput. Biol. 17 (11), e1009581. doi: 10.1371/journal.pcbi.1009581
Sedlazeck, F. J., Rescheneder, P., Smolka, M., Fang, H., Nattestad, M., von Haeseler, A., et al. (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15 (6), 461–468. doi: 10.1038/s41592-018-0001-7
Shen, W., Le, S., Li, Y., Hu, F. (2016). SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS One 11 (10), e0163962. doi: 10.1371/journal.pone.0163962
Shiota, H., Barral, S., Buchou, T., Tan, M., Couté, Y., Charbonnier, G., et al. (2018). Nut directs p300-dependent, genome-wide H4 hyperacetylation in Male germ cells. Cell Rep. 24 (13), 3477–3487.e6. doi: 10.1016/j.celrep.2018.08.069
Stefan, C. P., Koehler, J. W., Minogue, T. D. (2016). Targeted next-generation sequencing for the detection of ciprofloxacin resistance markers using molecular inversion probes. Sci. Rep. 6, 25904. doi: 10.1038/srep25904
Wick, R. R., Volkening, J. J., Loman, N. (2018). Porechop is a tool for finding and removing adapters from Oxford nanpore reads. Available at: https://github.com/rrwick/Porechop.
Wilson, M. R., Naccache, S. N., Samayoa, E., Biagtan, M., Bashir, H., Yu, G., et al. (2014). Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl. J. Med. 370 (25), 2408–2417. doi: 10.1056/NEJMoa1401268
Yatera, K., Noguchi, S., Mukae, H. (2021). Perspective on the clone library method for infectious diseases. Respir. Investig. 59 (6), 741–747. doi: 10.1016/j.resinv.2021.07.003
Yiang, G. T., Tzeng, I. S., Shui, H. A., Wu, M. Y., Peng, M. Y., Chan, C. Y., et al. (2021). Early screening of risk for multidrug-resistant organisms in the emergency department in patients with pneumonia and early septic shock: Single-center, retrospective cohort study. Shock 55 (2), 198–209. doi: 10.1097/SHK.0000000000001599
Zhao, N., Cao, J., Xu, J., Liu, B., Liu, B., Chen, D., et al. (2021). Targeting RNA with next- and third-generation sequencing improves pathogen identification in clinical samples. Adv. Sci. (Weinh) 8 (23), e2102593. doi: 10.1002/advs.202102593
Zhou, M., Wu, Y., Kudinha, T., Jia, P., Wang, L., Xu, Y., et al. (2021). Comprehensive pathogen identification, antibiotic resistance, and virulence genes prediction directly from simulated blood samples and positive blood cultures by nanopore metagenomic sequencing. Front. Genet. 12. doi: 10.3389/fgene.2021.620009
Keywords: nanopore sequencing, 16S rDNA, bacterial pneumonia, BALF, prospective study
Citation: Chen Y, Mao L, Lai D, Xu W, Zhang Y, Wu S, Yang D, Zhao S, Liu Z, Xiao Y, Tang Y, Meng X, Wang M, Shi J, Chen Q and Shu Q (2023) Improved targeting of the 16S rDNA nanopore sequencing method enables rapid pathogen identification in bacterial pneumonia in children. Front. Cell. Infect. Microbiol. 12:1001607. doi: 10.3389/fcimb.2022.1001607
Received: 23 July 2022; Accepted: 19 December 2022;
Published: 09 January 2023.
Edited by:
Jeyaprakash Rajendhran, Madurai Kamaraj University, IndiaReviewed by:
Rusung Tan, University of British Columbia, CanadaRubayet Hasan, Liflabs Ontario, Canada
Copyright © 2023 Chen, Mao, Lai, Xu, Zhang, Wu, Yang, Zhao, Liu, Xiao, Tang, Meng, Wang, Shi, Chen and Shu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qixing Chen, cWl4aW5nY2hlbkB6anUuZWR1LmNu; Qiang Shu, c2h1cWlhbmdAemp1LmVkdS5jbg==