
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 13 January 2022
Sec. Parasite and Host
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.810398
This article is part of the Research Topic Identification and Characterization of Novel Antigens of Malarial Parasites View all 7 articles
 Md Atique Ahmed1*
Md Atique Ahmed1* Gauspasha Yusuf Deshmukh2
Gauspasha Yusuf Deshmukh2 Rehan Haider Zaidi2
Rehan Haider Zaidi2 Ahmed Saif3
Ahmed Saif3 Mohammed Abdulrahman Alshahrani3Syeda Wasfeea Wazid4
Mohammed Abdulrahman Alshahrani3Syeda Wasfeea Wazid4 Saurav Jyoti Patgiri1Fu-Shi Quan5,6*
Saurav Jyoti Patgiri1Fu-Shi Quan5,6*Malaria is a major public health concern, and any tangible intervention during the pre-elimination phase can result in a significant reduction in infection rates. Recent studies have reported that antigens producing cross-protective immunity can play an important role as vaccines and halt malaria transmission in different endemic regions. In this study, we studied the genetic diversity, natural selection, and discovered novel conserved epitopes of a high molecular weight rhoptry protein 2 (RhopH2) in clinical samples of Plasmodium knowlesi and Plasmodium vivax cross-protective domains, which has been proven to produce cross-protective immunity in both species. We found low levels of nucleotide diversity (P. knowlesi; π ~ 0.0093, SNPs = 49 and P. vivax π ~ 0.0014, SNPs = 23) in P. knowlesi (n = 40) and P. vivax (n = 65) samples in the PkRhopH2 cross-protective domain. Strong purifying selection was observed for both species (P. knowlesi; dS - dN = 2.41, p < 0.009, P. vivax; dS - dN = 1.58, p < 0.050). In silico epitope prediction in P. knowlesi identified 10 potential epitopes, of which 7 epitopes were 100% conserved within clinical samples. Of these epitopes, an epitope with 10 amino acids (QNSKHFKKEK) was found to be fully conserved within all P. knowlesi and P. vivax clinical samples and 80%–90% conservation within simian malaria ortholog species, i.e., P. coatneyi and P. cynomolgi. Phylogenetic analysis of the PkRhopH2 cross-protective domain showed geographical clustering, and three subpopulations of P. knowlesi were identified of which two subpopulations originated from Sarawak, Malaysian Borneo, and one comprised only the laboratory lines from Peninsular Malaysia. This study suggests that RhopH2 could be an excellent target for cross-protective vaccine development with potential for outwitting strain as well as species-specific immunity. However, more detailed studies on genetic diversity using more clinical samples from both species as well as the functional role of antibodies specific to the novel conserved epitope identified in this study can be explored for protection against infection.
Malaria is a vector-borne disease which is prevalent in more than a hundred countries with 228 million malaria cases and an incidence of 405,000 deaths in 2019, the majority of which are due to Plasmodium falciparum infections (World Health Organisation, 2019). Controlling the spread of malarial infection can result in changes in species distribution patterns; for example, in one area, the spread of P. falciparum and P. vivax has decreased dramatically, but the spread of the zoonotic malaria, P. knowlesi, has increased significantly in Southeast Asian countries (Ahmed and Cox-Singh, 2015; Yusof et al., 2016). P. knowlesi, for example, caused 5% of malaria cases in Sabah in 2004, but 98% in 2017 (William et al., 2013; Cooper et al., 2020). This simian malaria parasite, P. knowlesi, was reported as a major cause of human malaria in Sarawak, Malaysia, in a paper published 16 years ago (Singh et al., 2004). Since then, several Southeast Asian countries have reported zoonotic malaria cases due to P. knowlesi. The whole-genome and genetic studies on P. knowlesi identified that there are at least three subpopulations in clinical samples from Malaysia, and out of these, two of them are linked to the primary monkey hosts, Macaca nemestrina and Macaca fascicularis (Ahmed et al., 2014; Assefa et al., 2015; Pinheiro et al., 2015; Ahmed et al., 2016)
The first clinical manifestation due to malaria starts during the asexual stages of the parasite when merozoites are released from RBCs. The invasion of the parasite into red blood cells (RBC) is a complex process which engages proteins on the merozoite surface and sequentially releases them from the apical organelles (micronemes and rhoptries) (Quintana et al., 2018). During merozoite egress and host cell invasion, invasive malaria merozoites have a typical apical complex set of secretory organelles that are discharged in a tightly controlled and highly regulated order (Sherling et al., 2019). Among the prominent organelles, the rhoptries are club-shaped, twinned structures which have a bulbous body that narrows to a narrow neck as it approaches the merozoite’s apical prominence (Sherling et al., 2019). Rhoptry proteins are essential for the Plasmodium parasite’s ability to enter and replicate in human red blood cells (RBCs). These proteins are also involved in the invasion of target cells by sporozoites, such as mosquito salivary glands and mammalian hepatocytes (Ishino et al., 2019). PkRhopH2, a high molecular mass protein in the rhoptries (161 kDa), was found to be highly immunogenic (with cross-protective immunity) with growth inhibitory activities (Muh et al., 2020). Host cell attachment and tight-junction formation are mediated by rhoptry neck proteins; however, the function of rhoptry bulb proteins is unclear due to a lack of functional studies (Ghosh et al., 2017). More than 30 rhoptry proteins have been identified in P. falciparum to date (Counihan et al., 2013). RhopH2 localizes to the bulb region and interacts with RhopH3, RhopH1, the erythrocyte cytoskeleton, and exported proteins that are involved in the remodeling of the host cell leading to increase in permeability in RBCs (Counihan et al., 2013).
P. knowlesi and P. vivax have a close phylogenetic relationship with 89% gene orthologs between them (Tachibana et al., 2012). Thus, these ortholog genes with roles in red blood cell invasion are proposed as attractive cross-species vaccine candidates (Cornejo and Escalante, 2006; Carlton et al., 2008). The cross-reactivity between P. falciparum and P. vivax is due to the presence of common or similar shared B and T-cell epitopes and homology between the plasmodial proteins (Diggs and Sadun, 1965; Maitland et al., 1997; Woodberry et al., 2008; Kawai et al., 2009; Muh et al., 2018; Muh et al., 2020). A recent study showed highly efficient cross-reactive RhopH2 antibodies against P. vivax to P. knowlesi which inhibit parasite growth in vitro and cross-immunogenicity in clinical samples, thereby highlighting its potential use for cross-protective immunity against both parasites in endemic areas (Muh et al., 2020). The same researchers have shown that in both P. vivax and P. knowlesi, the apical asparagine (Asn)-rich protein (AARP) antigen has been linked to long-lasting cross-species protective immunity (Muh et al., 2018). Previous studies have also found that P. falciparum antigens with structural similarities, such as erythrocyte membrane protein 1 variations and variant surface antigen 2-CSA, P. vivax AMA1, and P. falciparum AMA1, demonstrated cross-reactivity via conserved epitopes (Klein et al., 2008; Drew et al., 2018; Gnidehou et al., 2019).
In this study, we determined the genetic diversity and natural selection acting at the RhopH2 cross-protective domain (Muh et al., 2020) from P. knowlesi as well as P. vivax samples: for P. knowlesi, 40 samples [37 clinical samples and 3 laboratory lines (along with the H-strain)] from Malaysia, and for P. vivax, 65 PvRhopH2 gene sequences retrieved from clinical samples from 10 countries. We also predicted the cross-species epitopes in P. vivax and P. knowlesi using bioinformatics tools. Phylogenetic analysis was conducted to understand the relationships between clinical samples and other ortholog species of Plasmodium and determine conserved epitope regions. Since this is the first study on RhopH2 sequences obtained from clinical samples of both species, the results of this study will be helpful in understanding the level of polymorphism within the functional domains in field samples for future functional and strain-transcending vaccine development studies. This will be beneficial for the rational design and formulation of a blood-stage vaccine against P. knowlesi and P. vivax.
Thirty-seven PkRhopH2 gene sequences were retrieved from a public database (https://www.ebi.ac.uk/ena/browser/home) from clinical samples originating from Malaysian Borneo and 3 previously isolated lines from Peninsular Malaysia (along with the H-strain PKNH_0727900) (Supplementary Table 1) (Assefa et al., 2015). Sixty-five PvRhopH2 gene sequences were retrieved from clinical samples from 10 countries from PlasmoDB (https://plasmodb.org) (Supplementary Table 2) along with 3 reference strains of P. vivax (Sal-1; PVX_099930, P01; PVP01_072900 and P. vivax-like Pvl01; PVL_000087200). Sequences were aligned using the CLUSTAL-W program in MegAlign Lasergene v 7.0 (DNASTAR), and polymorphism and phylogenetic analyses were conducted in MEGA 5.0 software. In order to determine the relationship between PkRhopH2 sequences (laboratory lines and clinical samples from Sarawak, Malaysian Borneo), phylogenetic analyses were conducted using deduced amino acid sequences using the maximum likelihood (ML) method based on the Poisson correction model as described in MEGA 5.0 with 1,000 bootstrap replicates to test the robustness of the trees. The interspecies phylogenetic analysis was also performed by using the same method in P. falciparum (PF3D7_0929400), P. cynomolgi (PCYB_073680), P. coatneyi (PCOAH_00016180), P. knowlesi (PKNH_0727900), and P. vivax Sal-1 (PVX_099930). Phylogenetic analysis was also conducted using 65 PvRhopH2 deduced amino acid sequences and its ortholog species using the same method as used for P. knowlesi sequences.
B cell epitopes are antigenic determinant, portion of foreign protein, or antigen that can be used for developing a peptide vaccine (Saha and Raghava, 2006). In this study, in order to find cross-reactive epitopes between P. vivax and P. knowlesi, B cell epitopes were predicted in silico in RhopH2 amino acid sequences (domain previously studied (Muh et al., 2020) by using the Bcpred server http://www.imtech.res.in/raghava/bcepred/bcepred_team.html (Saha and Raghava, 2006) and the antibody epitope prediction server at the IEDB Analysis resource, by using the Emini Surface Accessibility Prediction model http://tools.immuneepitope.org/bcell (Emini et al., 1985). The Bcpred software predicts B cell epitopes based on amino acid properties, i.e., hydrophilicity, flexibility, polarity, and exposed surface, and a threshold score of 2.38 is considered for epitope prediction. The potential conservation of epitopes between P. knowlesi, P. vivax, and other primate malaria species was investigated.
Sequence diversity (π) was determined by DnaSP v5.10 software (Librado and Rozas, 2009). Number of parsimony informative sites, polymorphic sites, synonymous (silent mutations) and non-synonymous substitution (replacement changes), singletons, number of haplotypes (H), and haplotype and nucleotide diversity within PkRhopH2 and PvRhopH2 gene exon 1 (from 64 to 1,161 nt) were also determined by DnaSP software. Nucleotide diversity was also graphically represented using the window length of 100 and step size of 25 bp. The rate of non-synonymous substitution per non-synonymous site (dS) and the rate of synonymous substitution per synonymous site (dN) which determine the natural selection were determined using the method of Nei and Gojobori (1986). Additionally, more analyses were performed to determine natural selection, such as Tajima’s D, Fu and Li’s D*, and F* neutrality tests, which were implemented in DnaSP v5.10 software. Under neutrality, Tajima’s D value should be zero. The negative value of Tajima’s D is indicative of population expansion, and the positive as well as significant value indicates positive selection/balancing. Tajima’s D values were also represented graphically using DnaSP software. Fu and Li’s D* and F* positive and significant values indicate population contraction; singleton excess and negative values indicate population expansion.
The amino acid sequence identity of the RhopH2 region (64 to 1,161 nt, Exon I) which exhibited high cross-reactivity (Muh et al., 2020) with the P. knowlesi H-strain and P. vivax Sal I was found to be 74.44%. A schematic diagram of the full-length 10-exon structure of the RhopH2 gene of P. knowlesi in comparison to P. vivax sal-1 is shown in Figure 1A. A conserved 10-exon structure was observed within both species with length variations in Exons II, V, VII, and IX in P. vivax (Figure 1A). The phylogenetic analysis performed using deduced amino acid sequences in the ML method showed that PkRhopH2 is more closely related with P. coatneyi in comparison to its other orthologs in P. vivax, P. cynomolgi, and P. falciparum (Figure 1B). However, no geographical clustering was noted for P. vivax samples originating from 10 countries.
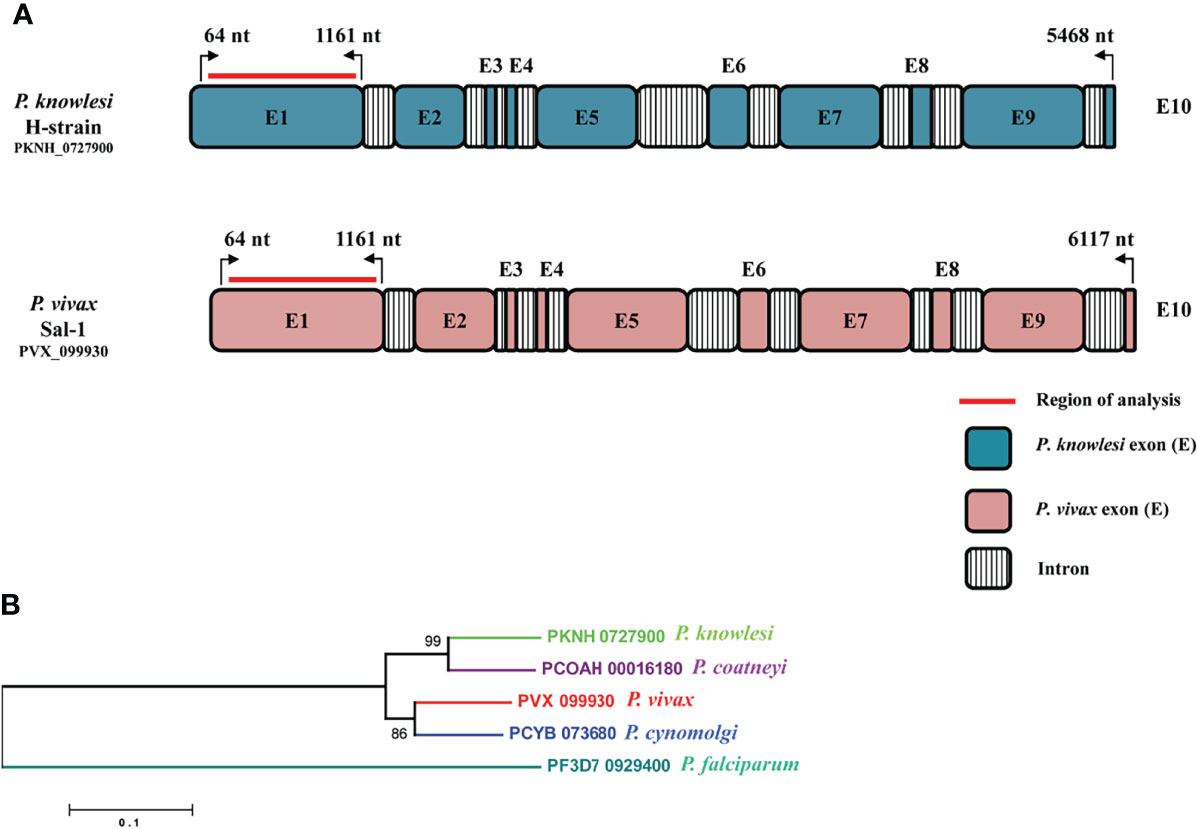
Figure 1 (A) Schematic representation of the P. knowlesi full-length RhopH2 gene in H-strain (PKNH_0727900, 5,468 bp) and P. vivax sal-1 strain (PVX_099930, 6,117 bp). The introns and exons were determined as described in PlasmoDB (www.plasmodb.org). The number on top with bend arrows represents position of the nucleotides in P. knowlesi and in P. vivax. The bold red line indicates the region of analysis for this study. (B) Interspecies phylogenetic relationships between RhopH2 full-length amino acid sequences of orthologs i.e., P. knowlesi (PKNH_0727900), P. coatneyi (PCOAH_00016180), P. vivax (PVX_099930), P. cynomolgi (PCYB_073680), and P. falciparum (PF3D7_0929400).
The nucleotide alignment of 40 PkRhopH2 sequences revealed that there were 49 single-nucleotide polymorphisms (SNPs) (Figure S1), of which 24 were synonymous substitutions and 22 non-synonymous substitutions. The overall nucleotide diversity was found to be π = 0.00936 which was higher compared to PvRhopH2; π = 0.00147 (Table 1). Analysis of 65 PvRhopH2 sequences revealed 23 SNPs (14 were synonymous substitutions and 9 non-synonymous substitutions). Twenty-three PvRhopH2 SNPs observed within 65 sequences are shown in Figure S5. PkRhopH2 had 38 parsimony informative sites out of which three were tri-variants, 8 singleton variable sites, 34 haplotypes with the haplotype diversity of Hd = 0.988 (Table 1). PvRhopH2 sequences revealed 23 singleton sites, 6 parsimony informative sites, and 9 haplotypes with haplotype diversity of Hd = 0.548 (Table 1). The graphical representation of the nucleotide diversity for both species is shown in Figures 2A, B, respectively. Graphical representations of Tajima’ D values are shown in Figures S4A, B. The amino acid sequence alignment of 40 PkRhopH2 sequences identified 3 sites with triple variants (T225S/N, Q281R/K, V302S/A) (Figure S2). The amino acid sequence alignment of 65 PvRhopH2 sequences with 9 non-synonymous substitutions is shown in Figure S6.
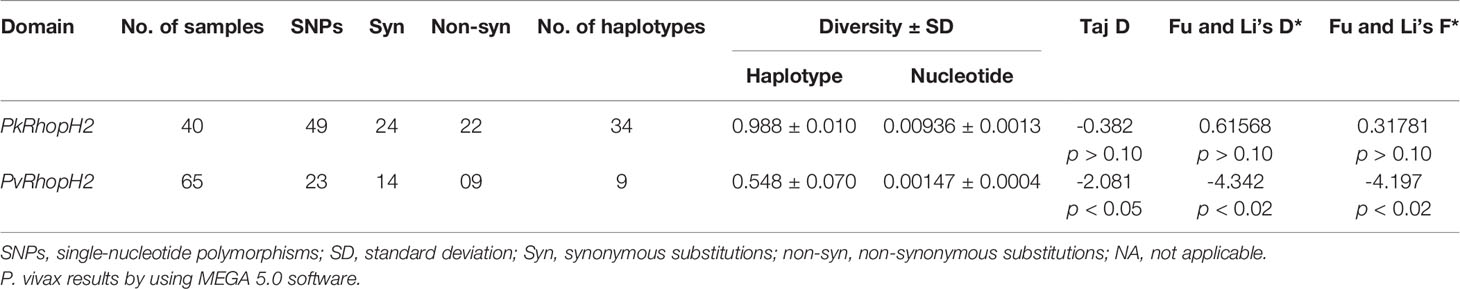
Table 1 Estimates of nucleotide diversity, haplotype diversity, and neutrality indices of P knowlesi and P. vivax RhopH2 genes.
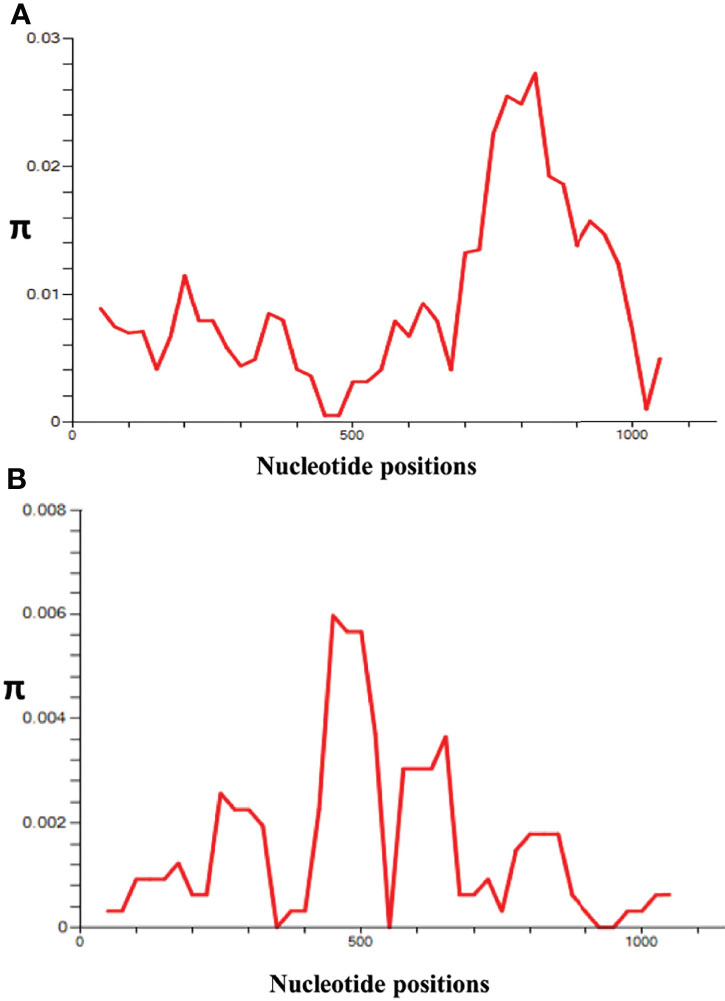
Figure 2 l (A) Graphical representation of nucleotide diversity (π) within the PkRhopH2 gene showing high diversity in the region from 700 to 900 nt. (B) Graphical representation of nucleotide diversity (π) within the PvRhopH2 gene showing high diversity in the region from 400 to 550 nt. The window length and step size of the π graph are 100 and 25, respectively, as implemented in DnaSP software v5.0.
The schematic representation of 22 non-synonymous substitutions observed within 40 samples with reference to P. knowlesi reference H strain is shown in Figure 3A. Similarly, the P. vivax non-synonymous substitutions within 65 samples with reference to the Sal-1 strain are shown in Figure 3B.
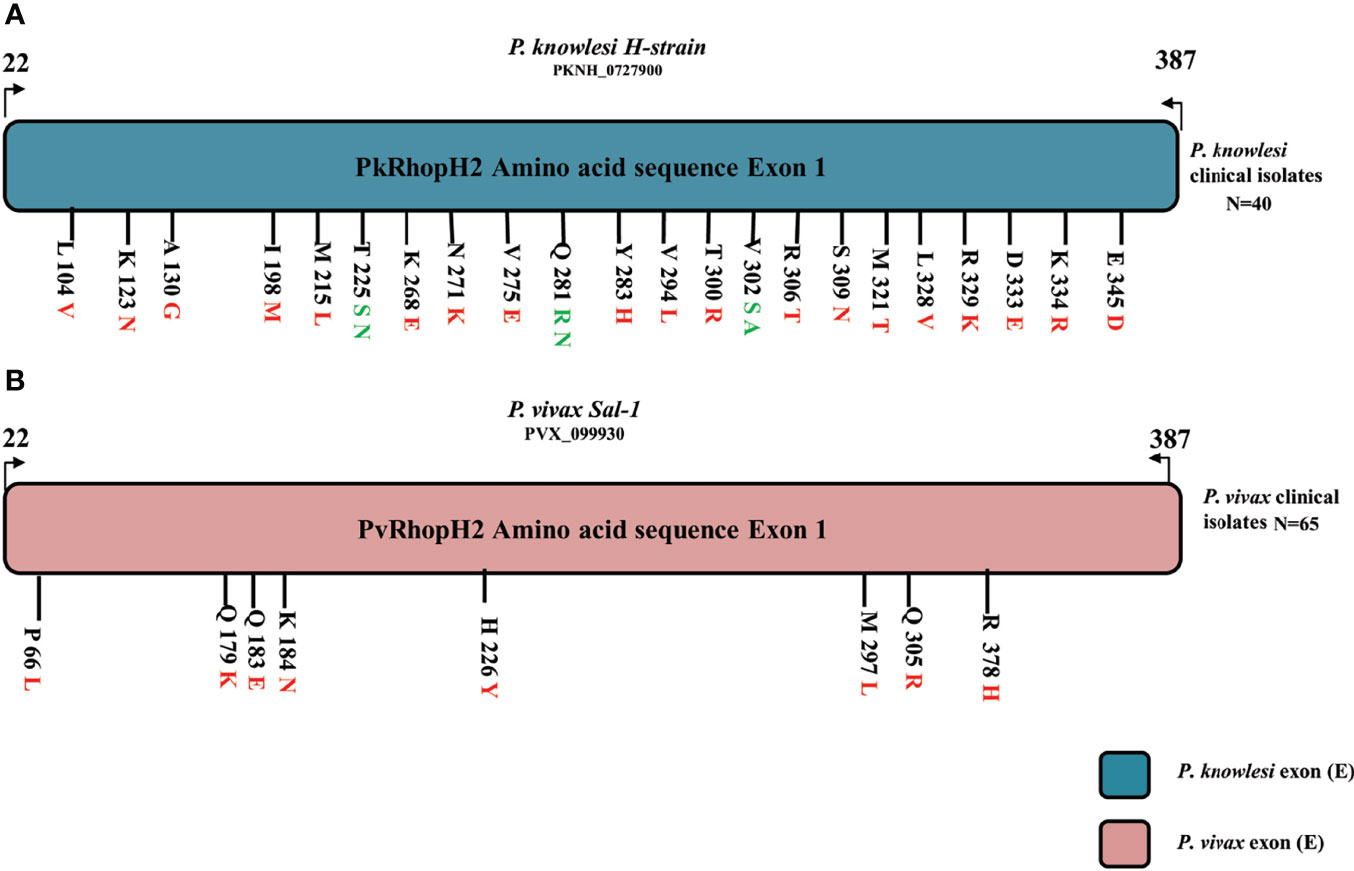
Figure 3 (A) Schematic representation of amino acid polymorphism in clinical isolates in P. knowlesi RhopH2 protein. Amino acid polymorphism is represented by black lines with letter and numbers. The letter before number represents the reference H-strain amino and polymorphism is represented in red (two variants) and green color (three variants). Similarly in P. vivax clinical samples in (B). The alphabet letter represents the amino acid one-letter code, and numbers represent amino acid position.
The natural selection analysis of the RhopH2 gene from 40 sequences indicated that the gene is under negative or purifying selection (dS-dN = 2.41, p < 0.009) probably due to functional constraints (Table 1). We found a similar strong negative selection acting at the PvRhopH2 domain (dS-dN = 1.59, p < 0.05). The overall Tajima’s D value was negative for both species (PkRhopH2; D = -0.38, p > 0.05 and PvRhopH2; D = -2.08, p < 0.05), which indicates purifying selection and population expansion. Fu and Li’s D* and F* values were positive (0.618 and 0.317) but not significant for PkRhopH2. Significant values were obtained for PvhopH2 -4.32 and -4.17, respectively (Table 1).
The phylogenetic analysis of the 40 samples of PkRhopH2 amino acid sequences with its ortholog species in Plasmodium by using the maximum likelihood method identified three different population clusters or subpopulation (Cluster 1, Cluster 2, and Cluster 3) (Figure 4). Out of these three clusters, two clusters originated from Malaysian Borneo and cluster 3 belonged to laboratory lines containing the H-strain. These clusters were linked to the primary hosts of P. knowlesi which are Macaca nemestrina (cluster 1) and Macaca fascicularis (cluster 2) as previously reported (Divis et al., 2015; Pinheiro et al., 2015; Ahmed et al., 2016; Ahmed et al., 2018a). The phylogenetic analysis of 65 PvRhopH2 amino acid sequences with its ortholog species in Plasmodium using the ML method revealed that there was no geographical clustering (Figure S7).
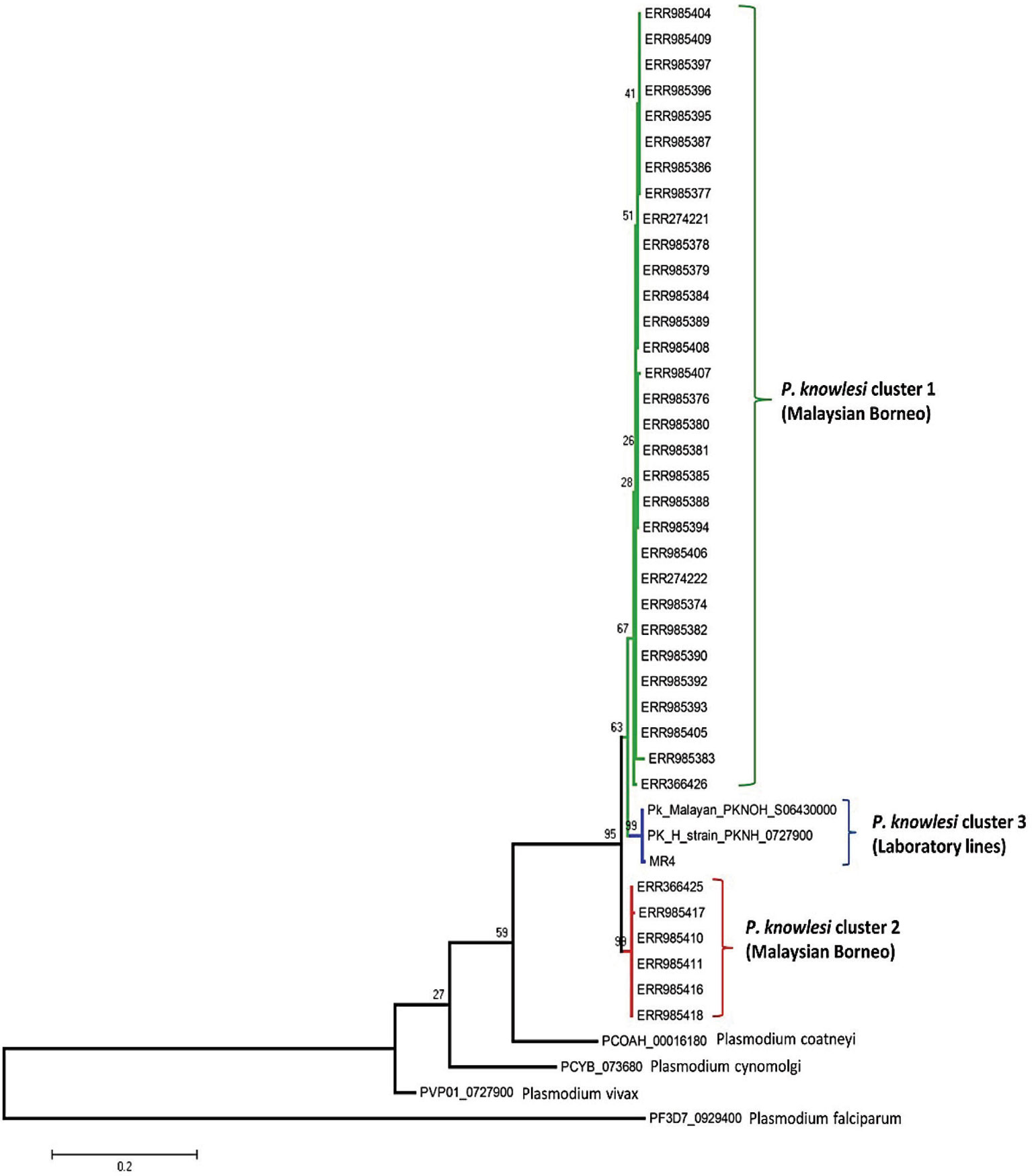
Figure 4 Phylogenetic tree of PkRhopH2 proteins (cross-protective domain, amino acid positions 22–387) from clinical samples of Malaysia and its orthologs in other Plasmodium species is constructed based on the maximum likelihood method. Cluster 1 and cluster 2 represent the two subpopulations of clinical samples from Malaysian Borneo, and the 3rd cluster contains lab strains of P. knowlesi. Bootstrap values are indicated by numbers at nodes.
Epitope prediction using Bcpred and IEDB servers identified 11 epitopes in P. knowlesi RhopH2 (Table 2A and Figure 5A). In P. vivax, Bcpred and IEDB servers identified 11 and 9 RhopH2 epitopes, respectively (Table 2B and Figure 5B). The comparison of epitope outputs from both servers revealed a total of 10 P. knowlesi RhopH2 epitopes ranging in length from 6 to 15 amino acids (Table 2A), while 9 P. vivax RhopH2 epitopes were identified by both servers (Table 2B). The interspecies comparison of epitopes identified a highly conserved epitope comprising of 10 amino acid (256QNSKHFKKEK265) (Tables 2A, B). This conservation of epitope was also observed in all 3 P. knowlesi clusters which were identified by phylogenetic analysis as well as 62 clinical samples of P. vivax from 10 countries. However, in comparison with the conserved epitope region found in P. knowlesi and P. vivax, there was a difference of 2 amino acids (256ENSKHFKKDK265) in its simian orthologs, i.e., P. coatneyi and P. cynomolgi and 1 amino acid difference (256QNSKHFKKDK265), respectively (Figure S3). Analysis of diversity and prevalence of the 10 PkRhopH2 epitopes (common epitopes identified by both software) in clinical samples indicated that 7 epitopes (70%) were 100% conserved (Table 3). The remaining 3 epitopes had at least 2–4 variants (Table 3). Analysis of diversity and prevalence of 9 PvRHopH2 epitopes (common epitopes identified by both software) from 65 samples from 10 countries indicated that 7 epitopes (77%) were 100% conserved (Table 4).
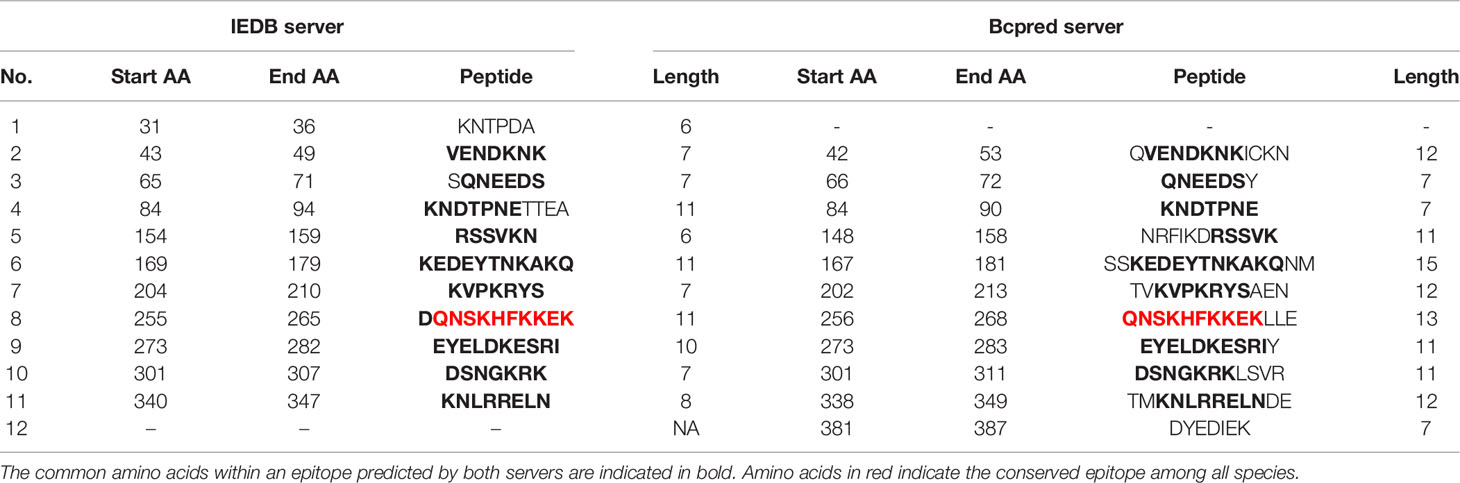
TABLE 2(A) The possible epitope predicted by using the IEDB server and Bcpred server in P. knowlesi shown in the above table.
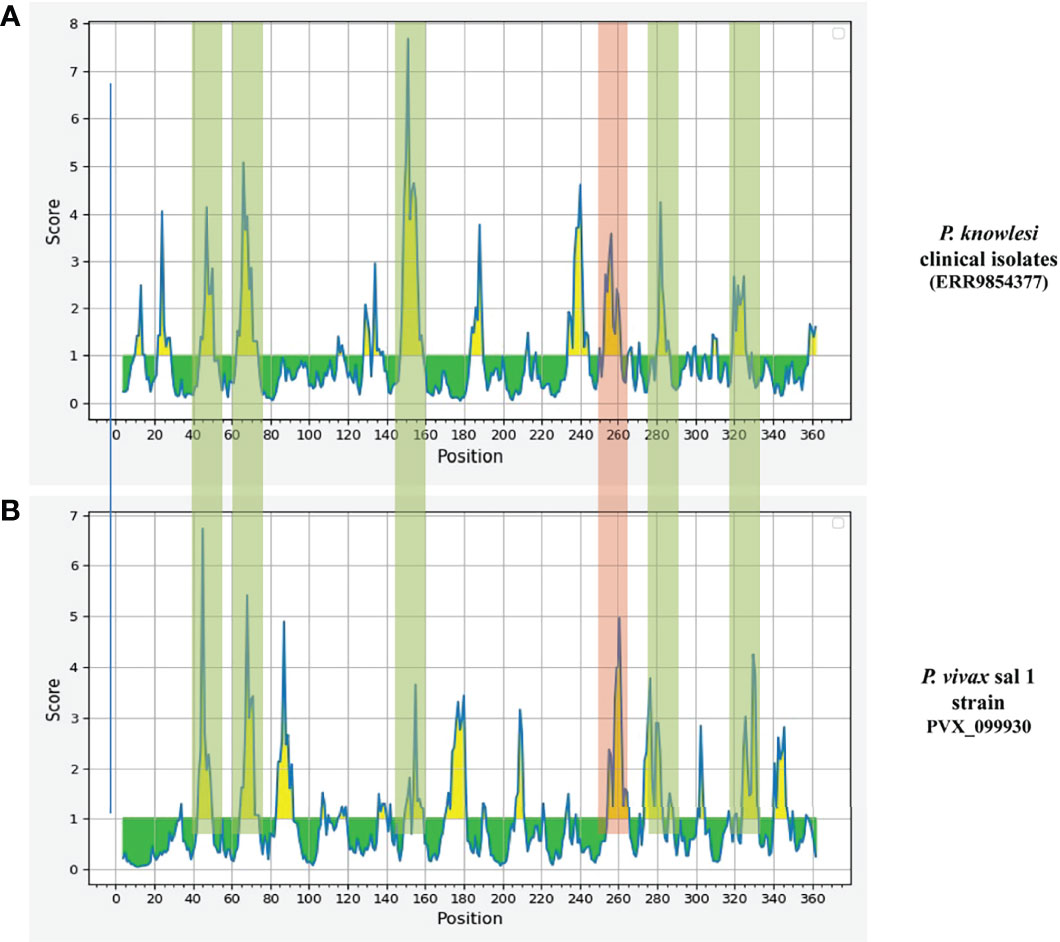
Figure 5 The graphs (A P. knowlesi and B P. vivax) are obtained from epitope prediction software IEDB. The graph represents the possible epitope region in RhopH2 protein. The peaks in graph show the possible epitope region in RhopH2 protein. The highlighted part represents the overlapping of the peaks in P. knowlesi and P. vivax. This indicates there is possibility of epitopes which have one or more common amino acids. The peak highlighted in red regions shows a 100% conserved amino acid sequence in all three clusters of P. knowlesi clinical isolate and P. vivax.
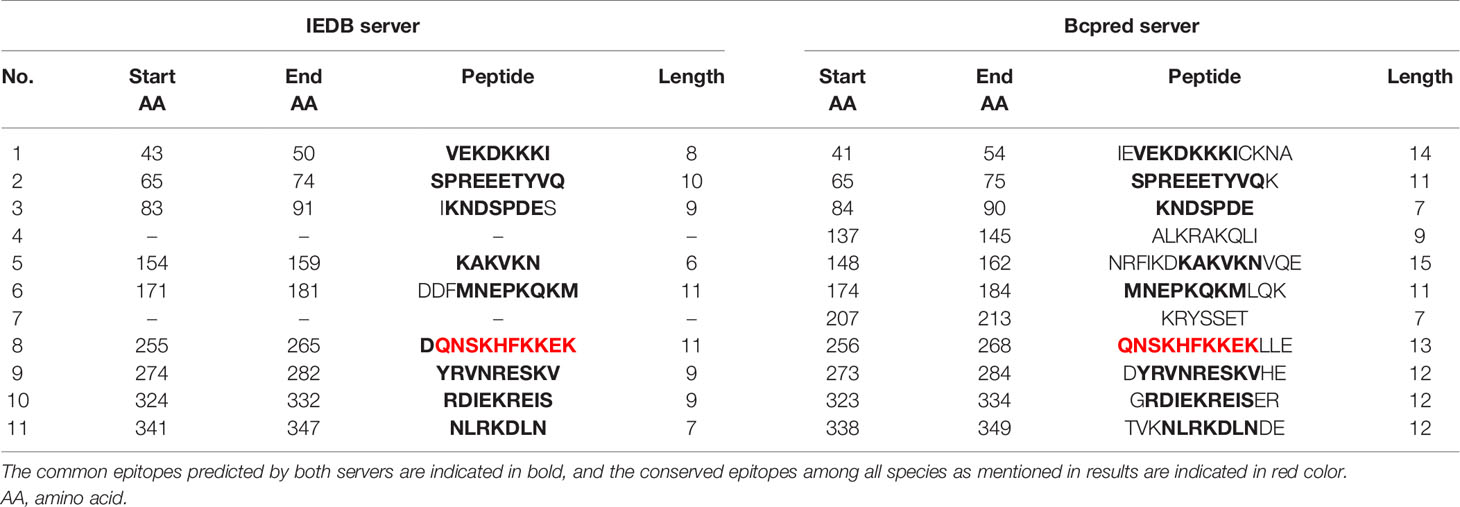
Table 2(B) The epitope predicted by using the IEDB server and Bcpred server in P. vivax shown in the above table.
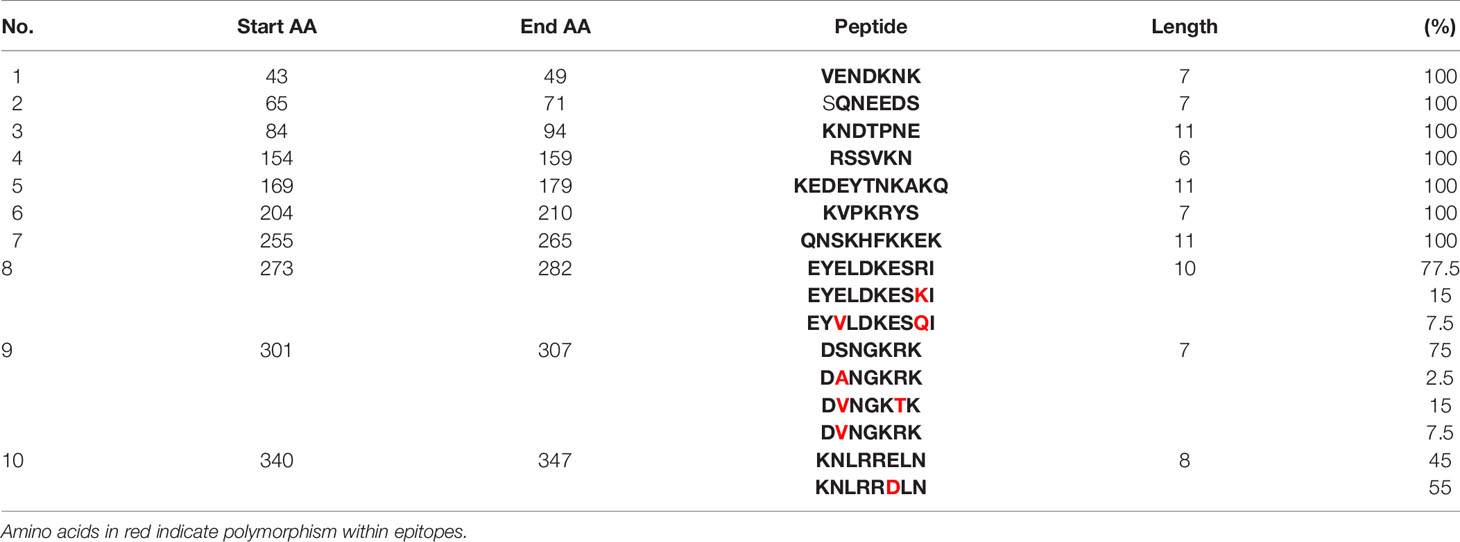
Table 3 Diversity of the common epitopes predicted by both servers and their prevalence in clinical isolates of P. knowlesi.
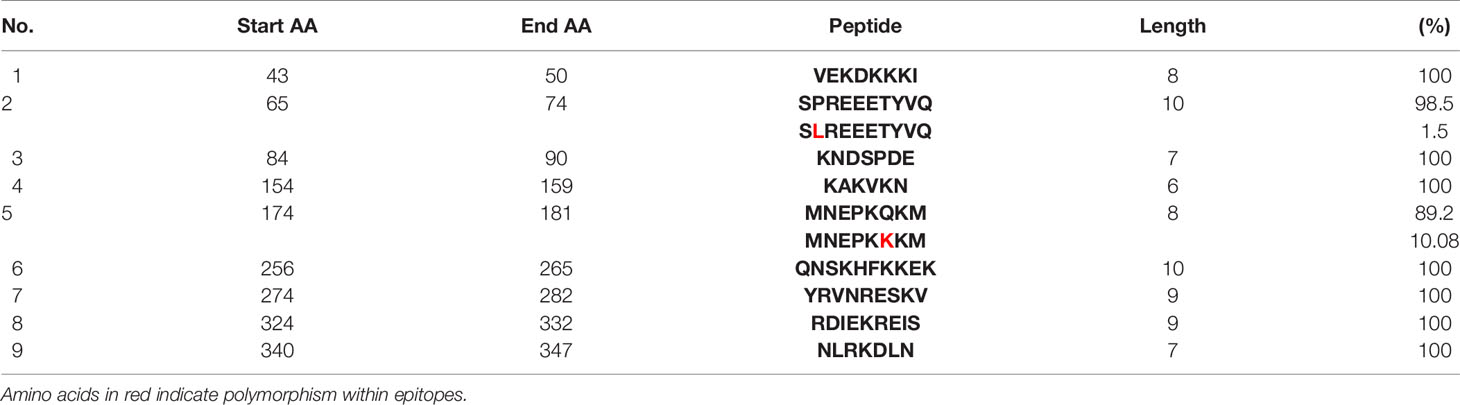
Table 4 Diversity of the common epitopes predicted by both servers and their prevalence in 65 clinical isolates of P. vivax RhopH2.
Antigens which are expressed during blood stages of the malaria parasite’s life cycle, specifically during the merozoite invasion process, e.g., micronemes and rhoptries, are excellent candidates for vaccine development as they are exposed to host immune response. An ideal vaccine candidate is expected to possess low levels of polymorphism but high and long-lasting antigenicity, along with strain-transcending efficacy across different geographical locations. We studied the genetic diversity and natural selection and predicted the B cell epitopes of a RhopH2 domain in P. knowlesi and P. vivax which has previously shown cross-species immunity (Muh et al., 2020).
Recently, the role of cross-species protective immunity has been reported using apical asparagine (Asn)-rich protein (AARP) in P. vivax and P. knowlesi (Muh et al., 2018). In this study, the overall nucleotide diversity of PvRhopH2 and PkRhopH2 was found to be low (π ~ 0.0014 and 0.009, respectively). These diversity values were lower than a previously reported cross-species candidate AARP, indicating that RhopH2 can be an excellent vaccine candidate as the antigen also showed growth inhibitory as well as cross-species reactive immunity (Muh et al., 2020). The amino acid sequence identity of RhopH2 between the P. knowlesi H-strain and P. vivax Sal I was found to be 74.44% which was similar to findings of AARP (Muh et al., 2018). Tests of natural selection for both species indicated strong purifying selection probably due to functional constrains in the cross-protective domain studied here; however, Taj’s D and Li and Fu’s D* and F* values were positive for P. knowlesi but not significant. This is probably due to existence of P. knowlesi subpopulations (Emini et al., 1985; Assefa et al., 2015; Ahmed et al., 2016; Ahmed et al., 2018b). The graphical representation of the nucleotide diversity was high from nucleotide positions 765 to 810. The overall average Tajima’s D value was found out to be negative, but it was positive in the regions of high diversity indicating probable epitope regions.
The ML phylogenetic tree identified 3 subpopulations of PkRhopH2, cluster 1 and cluster 2 from Malaysian Borneo and cluster 3 comprising only the laboratory lines as observed in other invasion genes and population genomic studies (Assefa et al., 2015; Pinheiro et al., 2015; Ahmed et al., 2016; Yusof et al., 2016; Divis et al., 2017; Ahmed et al., 2018a; Ahmed et al., 2018b; Ahmed et al., 2018c; Ahmed et al., 2019).
In this study, we also investigated whether shared epitopes are present in both species in the RhopH2 domain where high cross reactivity and immunogenicity have been observed by Muh et al. (2020). Among multiple epitopes identified by both the software (Bcpred server and IEDB server), a conserved epitope comprising 10 amino acids (QNSKHFKKEK) was found in both P. knowlesi and P. vivax clinical samples, indicating the possible reason for high cross-species reactivity and immunogenicity as observed by Muh et al. (2020). Interestingly, Taj’s D values around the epitope region gave a positive peak which may further confirm the prediction. This conserved epitope region was also present in all the three clusters of P. knowlesi obtained through phylogenetic analysis, which could indicate high antigenicity in clinical samples. This Rhoph2 epitope was also found in other simian malaria parasites, i.e., P. cynomolgi and P. coatneyi, which showed 80%–90% conservation indicating the possibility of cross-species reactivity and immunogenicity; however, further studies need to be conducted to understand the functional aspect of these epitopes. P. falciparum antigens with structural similarities, such as erythrocyte membrane protein 1 variations and variant surface antigen 2-CSA, P. vivax AMA1, and P. falciparum AMA1, demonstrated cross-reactivity via conserved epitopes (Klein et al., 2008; Gnidehou et al., 2019). The results obtained in this study thus provide further supportive evidence for the existence of cross-protective immunity between P. vivax and P. knowlesi conferred through a shared common epitope. This could serve as a vaccination strategy to protect Southeast Asian residents from P. knowlesi infections. To our knowledge, this is the first study to identify novel RhopH2 epitopes and genetic characterization in both species, i.e. P. knowlesi and P. vivax, thereby contributing significantly toward new knowledge and understanding of the cross-species epitopes for vaccine development. Through our study, the functional role of antibodies specific to the novel conserved epitope identified in this study can be explored for protection against malaria infection.
Publicly available datasets were analyzed in this study. The repository is ENA (European nucleotide archive https://www.ebi.ac.uk/ena/browser/home), and accession numbers can be found in the Supplementary Material.
MAAh participated in the conception, design of the study, data collection, analysis, interpretation, and manuscript preparation. GD, RZ, AS, MAAl, SP, and SW participated in the laboratory procedures, data collection, and analysis and manuscript preparation. All authors contributed to the article and approved the submitted version.
This study was funded by the Department of Biotechnology, Govt. of India, No.BT/RLF/Re-entry/09/2017, Deanship of Scientific Research at Najran University, Najran, Saudi Arabia NU/IFC/ENT/01/007, Ministry of Health & Welfare, Republic of Korea (HV20C0142) and the National Research Foundation of Korea (NRF) (2018R1A6A1A03025124).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.810398/full#supplementary-material
Ahmed, M. A., Chu, K. B., Quan, F. S. (2018a). The Plasmodium knowlesi Pk41 Surface Protein Diversity, Natural Selection, Sub Population and Geographical Clustering: A 6-Cysteine Protein Family Member. PeerJ 6, e6141. doi: 10.7717/peerj.6141
Ahmed, M. A., Cox-Singh, J. (2015). Plasmodium knowlesi - an Emerging Pathogen. ISBT Sci. Ser. 10, 134–140. doi: 10.1111/voxs.12115
Ahmed, M. A., Fauzi, M., Han, E. T. (2018b). Genetic Diversity and Natural Selection of Plasmodium knowlesi Merozoite Surface Protein 1 Paralog Gene in Malaysia. Malar. J. 17, 115. doi: 10.1186/s12936-018-2256-y
Ahmed, M. A., Fong, M. Y., Lau, Y. L., Yusof, R. (2016). Clustering and Genetic Differentiation of the Normocyte Binding Protein (Nbpxa) of Plasmodium knowlesi Clinical Isolates From Peninsular Malaysia and Malaysia Borneo. Malar. J. 15, 241. doi: 10.1186/s12936-016-1294-6
Ahmed, M. A., Lau, Y. L., Quan, F. S. (2018c). Diversity and Natural Selection on the Thrombospondin-Related Adhesive Protein (TRAP) Gene of Plasmodium knowlesi in Malaysia. Malar. J. 17, 274. doi: 10.1186/s12936-018-2423-1
Ahmed, A. M., Pinheiro, M. M., Divis, P. C., Siner, A., Zainudin, R., Wong, I. T., et al. (2014). Disease Progression in Plasmodium knowlesi Malaria is Linked to Variation in Invasion Gene Family Members. PloS Negl. Trop. Dis. 8, e3086. doi: 10.1371/journal.pntd.0003086
Ahmed, M. A., Saif, A., Quan, F. S. (2019). Diversity Pattern of Plasmodium knowlesi Merozoite Surface Protein 4 (MSP4) in Natural Population of Malaysia. PloS One 14, e0224743. doi: 10.1371/journal.pone.0224743
Assefa, S., Lim, C., Preston, M. D., Duffy, C. W., Nair, M. B., Adroub, S. A., et al. (2015). Population Genomic Structure and Adaptation in the Zoonotic Malaria Parasite Plasmodium knowlesi. Proc. Natl. Acad. Sci. U. S. A. 112, 13027–13032. doi: 10.1073/pnas.1509534112
Carlton, J. M., Adams, J. H., Silva, J. C., Bidwell, S. L., Lorenzi, H., Caler, E., et al. (2008). Comparative Genomics of the Neglected Human Malaria Parasite Plasmodium Vivax. Nature 455, 757–763. doi: 10.1038/nature07327
Cooper, D. J., Rajahram, G. S., William, T., Jelip, J., Mohammad, R., Benedict, J., et al. (2020). Plasmodium knowlesi Malaria in Sabah, Malaysi-2017: Ongoing Increase in Incidence Despite Near-Elimination of the Human-Only Plasmodium Species. Clin. Infect. Dis. 70, 361–367. doi: 10.1093/cid/ciz237
Cornejo, O. E., Escalante, A. A. (2006). The Origin and Age of Plasmodium Vivax. Trends Parasitol. 22, 558–563. doi: 10.1016/j.pt.2006.09.007
Counihan, N. A., Kalanon, M., Coppel, R. L., De Koning-Ward, T. F. (2013). Plasmodium Rhoptry Proteins: Why Order Is Important. Trends Parasitol. 29, 228–236. doi: 10.1016/j.pt.2013.03.003
Diggs, C. L., Sadun, E. H. (1965). Serological Cross Reactivity Between Plasmodium Vivax and Plasmodium Falciparum as Determined by a Modified Fluorescent Antibody Test. Exp. Parasitol. 16, 217–223. doi: 10.1016/0014-4894(65)90046-9
Divis, P. C., Lin, L. C., Rovie-Ryan, J. J., Kadir, K. A., Anderios, F., Hisam, S., et al. (2017). Three Divergent Subpopulations of the Malaria Parasite Plasmodium knowlesi. Emerg. Infect. Dis. 23, 616–624. doi: 10.3201/eid2304.161738
Divis, P. C., Singh, B., Anderios, F., Hisam, S., Matusop, A., Kocken, C. H., et al. (2015). Admixture in Humans of Two Divergent Plasmodium knowlesi Populations Associated With Different Macaque Host Species. PloS Pathog. 11, e1004888. doi: 10.1371/journal.ppat.1004888
Drew, D. R., Sanders, P. R., Weiss, G., Gilson, P. R., Crabb, B. S., Beeson, J. G. (2018). Functional Conservation of the AMA1 Host-Cell Invasion Ligand Between P. Falciparum and P. Vivax: A Novel Platform to Accelerate Vaccine and Drug Development. J. Infect. Dis. 217, 498–507. doi: 10.1093/infdis/jix583
Emini, E. A., Hughes, J. V., Perlow, D. S., Boger, J. (1985). Induction of Hepatitis A Virus-Neutralizing Antibody by a Virus-Specific Synthetic Peptide. J. Virol. 55, 836–839. doi: 10.1128/jvi.55.3.836-839.1985
Ghosh, S., Kennedy, K., Sanders, P., Matthews, K., Ralph, S. A., Counihan, N. A., et al. (2017). The Plasmodium Rhoptry Associated Protein Complex is Important for Parasitophorous Vacuole Membrane Structure and Intraerythrocytic Parasite Growth. Cell Microbiol. 19. doi: 10.1111/cmi.12733
Gnidehou, S., Mitran, C. J., Arango, E., Banman, S., Mena, A., Medawar, E., et al. (2019). Cross-Species Immune Recognition Between Plasmodium Vivax Duffy Binding Protein Antibodies and the Plasmodium Falciparum Surface Antigen VAR2CSA. J. Infect. Dis. 219, 110–120. doi: 10.1093/oxfordjournals.molbev.a040410
Ishino, T., Murata, E., Tokunaga, N., Baba, M., Tachibana, M., Thongkukiatkul, A., et al. (2019). Rhoptry Neck Protein 2 Expressed in Plasmodium Sporozoites Plays a Crucial Role During Invasion of Mosquito Salivary Glands. Cell Microbiol. 21, e12964. doi: 10.1111/cmi.12964
Kawai, S., Hirai, M., Haruki, K., Tanabe, K., Chigusa, Y. (2009). Cross-Reactivity in Rapid Diagnostic Tests Between Human Malaria and Zoonotic Simian Malaria Parasite Plasmodium knowlesi Infections. Parasitol. Int. 58, 300–302. doi: 10.1016/j.parint.2009.06.004
Klein, M. M., Gittis, A. G., Su, H. P., Makobongo, M. O., Moore, J. M., Singh, S., et al. (2008). The Cysteine-Rich Interdomain Region From the Highly Variable Plasmodium Falciparum Erythrocyte Membrane Protein-1 Exhibits a Conserved Structure. PloS Pathog. 4, e1000147. doi: 10.1371/journal.ppat.1000147
Librado, P., Rozas, J. (2009). DnaSP V5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Maitland, K., Williams, T. N., Newbold, C. I. (1997). Plasmodium Vevax and P. Falciparum: Biological Interactions and the Possibility of Cross-Species Immunity. Parasitol. Today 13, 227–231. doi: 10.1016/S0169-4758(97)01061-2
Muh, F., Ahmed, M. A., Han, J. H., Nyunt, M. H., Lee, S. K., Lau, Y. L., et al. (2018). Cross-Species Analysis of Apical Asparagine-Rich Protein of Plasmodium Vivax and Plasmodium knowlesi. Sci. Rep. 8, 5781. doi: 10.1038/s41598-018-23728-1
Muh, F., Kim, N., Nyunt, M. H., Firdaus, E. R., Han, J. H., Hoque, M. R., et al. (2020). Cross-Species Reactivity of Antibodies Against Plasmodium Vivax Blood-Stage Antigens to Plasmodium knowlesi. PloS Negl. Trop. Dis. 14, e0008323. doi: 10.1371/journal.pntd.0008323
Nei, M., Gojobori, T. (1986). Simple Methods for Estimating the Numbers of Synonymous and Nonsynonymous Nucleotide Substitutions. Mol. Biol. Evol. 3, 418–426. doi: 10.1093/infdis/jiy467
Pinheiro, M. M., Ahmed, M. A., Millar, S. B., Sanderson, T., Otto, T. D., Lu, W. C., et al. (2015). Plasmodium knowlesi Genome Sequences From Clinical Isolates Reveal Extensive Genomic Dimorphism. PloS One 10, e0121303. doi: 10.1371/journal.pone.0121303
Quintana, M. D. P., Ch’ng, J. H., Zandian, A., Imam, M., Hultenby, K., Theisen, M., et al. (2018). SURGE Complex of Plasmodium Falciparum in the Rhoptry-Neck (SURFIN4.2-RON4-GLURP) Contributes to Merozoite Invasion. PloS One 13, e0201669. doi: 10.1371/journal.pone.0201669
Saha, S., Raghava, G. P. (2006). Prediction of Continuous B-Cell Epitopes in an Antigen Using Recurrent Neural Network. Proteins 65, 40–48. doi: 10.1002/prot.21078
Sherling, E. S., Perrin, A. J., Knuepfer, E., Russell, M. R. G., Collinson, L. M., Miller, L. H., et al. (2019). The Plasmodium Falciparum Rhoptry Bulb Protein RAMA Plays an Essential Role in Rhoptry Neck Morphogenesis and Host Red Blood Cell Invasion. PloS Pathog. 15, e1008049. doi: 10.1371/journal.ppat.1008049
Singh, B., Kim Sung, L., Matusop, A., Radhakrishnan, A., Shamsul, S. S., Cox-Singh, J., et al. (2004). A Large Focus of Naturally Acquired Plasmodium knowlesi Infections in Human Beings. Lancet 363, 1017–1024. doi: 10.1016/S0140-6736(04)15836-4
Tachibana, S., Sullivan, S. A., Kawai, S., Nakamura, S., Kim, H. R., Goto, N., et al. (2012). Plasmodium Cynomolgi Genome Sequences Provide Insight Into Plasmodium Vivax and the Monkey Malaria Clade. Nat. Genet. 44, 1051–1055. doi: 10.1038/ng.2375
William, T., Rahman, H. A., Jelip, J., Ibrahim, M. Y., Menon, J., Grigg, M. J., et al. (2013). Increasing Incidence of Plasmodium knowlesi Malaria Following Control of P. Falciparum and P. Vivax Malaria in Sabah, Malaysia. PloS Negl. Trop. Dis. 7, e2026. doi: 10.1371/journal.pntd.0002026
Woodberry, T., Minigo, G., Piera, K. A., Hanley, J. C., De Silva, H. D., Salwati, E., et al. (2008). Antibodies to Plasmodium Falciparum and Plasmodium Vivax Merozoite Surface Protein 5 in Indonesia: Species-Specific and Cross-Reactive Responses. J. Infect. Dis. 198, 134–142. doi: 10.1086/588711
Keywords: Plasmodium knowlesi, Plasmodium vivax, conserved cross-species, polymorphism, rhopH2, vaccine
Citation: Ahmed MA, Deshmukh YG, Zaidi RH, Saif A, Alshahrani MA, Wazid SW, Patgiri SJ and Quan F-S (2022) Identification, Mapping, and Genetic Diversity of Novel Conserved Cross-Species Epitopes of RhopH2 in Plasmodium knowlesi With Plasmodium vivax. Front. Cell. Infect. Microbiol. 11:810398. doi: 10.3389/fcimb.2021.810398
Received: 06 November 2021; Accepted: 22 December 2021;
Published: 13 January 2022.
Edited by:
Tania F. De Koning-Ward, Deakin University, AustraliaReviewed by:
Surendra Kumar Prajapati, Henry M Jackson Foundation for the Advancement of Military Medicine (HJF), United StatesCopyright © 2022 Ahmed, Deshmukh, Zaidi, Saif, Alshahrani, Wazid, Patgiri and Quan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Md Atique Ahmed, YXRpcWJpb3RlY2hAZ21haWwuY29t; Fu-Shi Quan, ZnF1YW4wMUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.