
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 14 January 2022
Sec. Bacteria and Host
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.798246
This article is part of the Research Topic Otitis Media View all 18 articles
 Christina L. Elling1,2
Christina L. Elling1,2 Melissa A. Scholes1,3Sven-Olrik Streubel1,3
Melissa A. Scholes1,3Sven-Olrik Streubel1,3 Eric D. Larson1Todd M. Wine1,3Tori C. Bootpetch1Patricia J. Yoon1,3
Eric D. Larson1Todd M. Wine1,3Tori C. Bootpetch1Patricia J. Yoon1,3 Jennifer M. Kofonow4Samuel P. Gubbels1Stephen P. Cass1Charles E. Robertson4
Jennifer M. Kofonow4Samuel P. Gubbels1Stephen P. Cass1Charles E. Robertson4 Herman A. Jenkins1
Herman A. Jenkins1 Jeremy D. Prager1,3
Jeremy D. Prager1,3 Daniel N. Frank4
Daniel N. Frank4 Kenny H. Chan1,3Norman R. Friedman1,3Allen F. Ryan5
Kenny H. Chan1,3Norman R. Friedman1,3Allen F. Ryan5 Regie Lyn P. Santos-Cortez1,2,6*
Regie Lyn P. Santos-Cortez1,2,6*Otitis media (OM) is a leading cause of childhood hearing loss. Variants in FUT2, which encodes alpha-(1,2)-fucosyltransferase, were identified to increase susceptibility to OM, potentially through shifts in the middle ear (ME) or nasopharyngeal (NP) microbiotas as mediated by transcriptional changes. Greater knowledge of differences in relative abundance of otopathogens in carriers of pathogenic variants can help determine risk for OM in patients. In order to determine the downstream effects of FUT2 variation, we examined gene expression in relation to carriage of a common pathogenic FUT2 c.461G>A (p.Trp154*) variant using RNA-sequence data from saliva samples from 28 patients with OM. Differential gene expression was also examined in bulk mRNA and single-cell RNA-sequence data from wildtype mouse ME mucosa after inoculation with non-typeable Haemophilus influenzae (NTHi). In addition, microbiotas were profiled from ME and NP samples of 65 OM patients using 16S rRNA gene sequencing. In human carriers of the FUT2 variant, FN1, KMT2D, MUC16 and NBPF20 were downregulated while MTAP was upregulated. Post-infectious expression in the mouse ME recapitulated these transcriptional differences, with the exception of Fn1 upregulation after NTHi-inoculation. In the NP, Candidate Division TM7 was associated with wildtype genotype (FDR-adj-p=0.009). Overall, the FUT2 c.461G>A variant was associated with transcriptional changes in processes related to response to infection and with increased load of potential otopathogens in the ME and decreased commensals in the NP. These findings provide increased understanding of how FUT2 variants influence gene transcription and the mucosal microbiota, and thus contribute to the pathology of OM.
Infection and inflammation of the middle ear (ME), known as otitis media (OM), is the most frequently diagnosed disease in infants and young children in the United States and is globally a leading cause of hearing loss (Monasta et al., 2012; GBD, 2021). In children, an estimated 60% of hearing loss is due to preventable causes, and infections and chronic OM account for around 31% of pediatric hearing loss (Schilder et al., 2016; GBD, 2021). In the United States, treatment of OM costs over $5 billion annually and typically includes antibiotics and surgery such as tympanostomy tube insertion (Schilder et al., 2016; Suaya et al., 2018). OM risk and pathology are influenced by many factors including environmental factors such as age, sex, daycare attendance and breastfeeding as well as genetic factors (Zhang et al., 2014; Brennan-Jones et al., 2015). Heritability of OM is estimated to be as high as 74%; furthermore, genes related to OM predisposition are known to function in pathways that include innate immune response, cell-mediated immune dysfunction and pathogen-host-environment interactions (Casselbrant et al., 1999; Mittal et al., 2014).
OM is often bacterial or viral in origin, wherein pathogens in the nasopharynx (NP) migrate via the Eustachian tube to the ME. This creates an inflammatory cycle in the ME with an accumulation of mucus and fluid which can lead to permanent damage and hearing loss (Rosenfeld et al., 2013). It is important to note that prior to infection, the ME is essentially sterile as it is generally separated from the external environment by the tympanic membrane, whereas the NP has an established microbiota that can vary based on microbial exposure and host genetics, but these microbes in the NP do not become resident in the ME if the Eustachian tube is functioning well (Jervis-Bardy et al., 2019). Some NP commensals are potential opportunistic otopathogens of the ME (Yatsyshina et al., 2016). It is well-known that increased abundance of potential otopathogens in the NP is associated with higher risk for OM (Jervis-Bardy et al., 2017; Browne et al., 2021; Xu et al., 2021).
FUT2 (MIM 182100) encodes alpha-(1,2)-fucosyltransferase which is responsible for secretion and expression of ABO(H) antigens on mucosal epithelia (Kelly et al., 1995). Secretory status directly influences pathogen binding in mucosal epithelia in multiple organ systems. The FUT2 stop variant c.461G>A (p.Trp154*; rs601338) has been associated with multiple mucosal phenotypes and is in strong linkage disequilibrium (LD) with a synonymous FUT2 variant rs681343 that was previously associated with childhood ear infections in genome-wide association studies (GWAS) (Pickrell et al., 2016; Tian et al., 2017). This variant has also been confirmed to confer familial OM risk in multiple cohorts (Santos-Cortez et al., 2018). Additionally, Fut2 expression transiently increased in the mouse ME after infection with non-typeable Haemophilus influenzae (NTHi), which is a common otopathogen in humans (Santos-Cortez et al., 2018).
Non-secretors, i.e., homozygous for FUT2 c.461G>A, show higher rates of bacterial infections [e.g. with Streptococcus pneumoniae, NTHi in different organ systems], but decreased susceptibility to viral infection (i.e. viral diarrhea or HIV-1), possibly due to the effects of the glycan on the mucus barrier (Magalhaes et al., 2016; Azad et al., 2018; Santos-Cortez et al., 2018). Though FUT2 is well-studied, to our knowledge there are no previous studies of transcriptome-wide differences in host gene expression based on carriage of the FUT2 c.461G>A variant in humans. Furthermore, to date only seven studies investigated changes in the host microbiota that were associated with carriage of this variant (Rausch et al., 2011; Wacklin et al., 2011; Wacklin et al., 2014; Kumar et al., 2015; Davenport et al., 2016; Kumbhare et al., 2017; Turpin et al., 2018; Chen et al., 2021). These studies were limited to assessment of the gut microbiota according to variant carriage and identified associations seemed to be environment- or disease- specific. While some studies observed no associations between gut microbiome and FUT2 c.461G>A genotype, others noted that Bifidobacterium levels, among other taxa, were significantly different between variant carriers and wildtype (Wacklin et al., 2011; Wacklin et al., 2014; Davenport et al., 2016; Turpin et al., 2018). Furthermore, in Crohn’s Disease and throughout pregnancy, the FUT2 c.461G>A variant was associated with differences in the gut microbiota diversity and abundance of individual taxa (Rausch et al., 2011; Kumar et al., 2015).
In order to further elucidate the role of FUT2 in OM pathogenesis, the goal of this study was to investigate the potential downstream effects of the FUT2 c.461G>A (p.Trp154*) variant on gene expression and site-specific colonization by commensals and known otopathogens. Characterization of this common variant and its role in the interplay between host genetics, host immune response, and mucosal microbiotas not only expands our general understanding of these complex relationships but also, within the context of OM, provides clinically relevant insight that can be used to better determine individual risk and inform treatment. In this study, we performed differential expression (DE) analysis on RNA-sequence data from saliva of OM-affected individuals and identified multiple differentially expressed genes based on carriage of the FUT2 c.461G>A variant. These DE genes were replicated using genome-wide expression data from infected mouse ME. We also performed microbiota analysis using 16S rRNA sequence data from ME and NP samples of OM-affected individuals and identified bacterial taxa that were different in relative abundance according to genotype.
Ethical approval was obtained from the COMIRB prior to the start of the study. Informed consent was obtained from study participants, including parents of children enrolled in the study. The IACUC of the Veterans Affairs Medical Center, San Diego, California granted approval for mouse studies.
Clinical data were obtained from 91 pediatric patients undergoing surgery for OM, with information on age, sex, self-reported ethnicity, family history, breastfeeding history, history of exposure to smoking, OM diagnoses and surgical technique (Table 1). We also had clinical information and samples from 15 adult patients with OM, but these samples were removed from further analyses because of marked differences in expression and microbiota profiles due to age (Figure 1). DNA samples were collected from the 91 pediatric patients with OM using the Oragene-DNA OGR-500 or OGR-575 kits (DNA Genotek, Ottawa, Ontario, Canada).
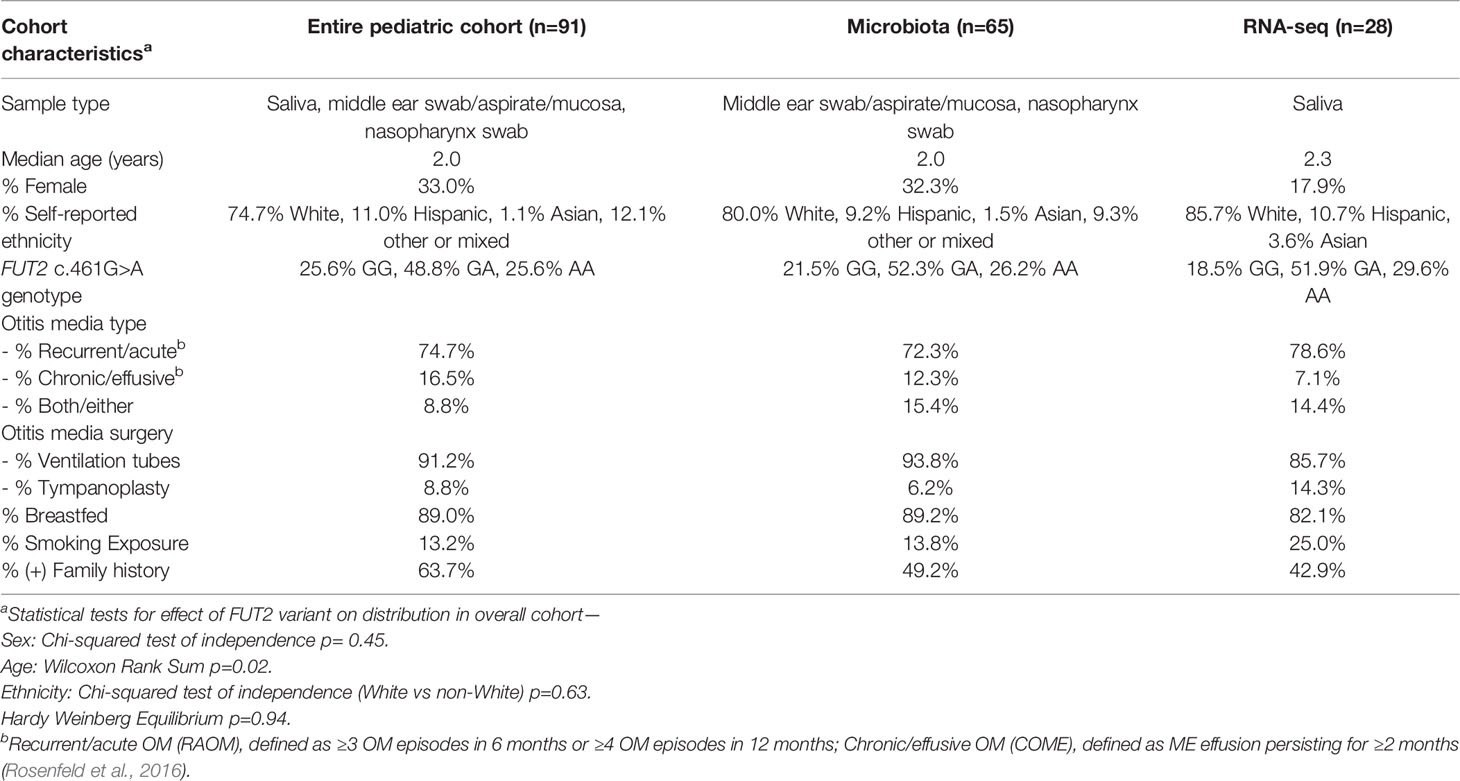
Table 1 Characteristics of OM patients by dataset.
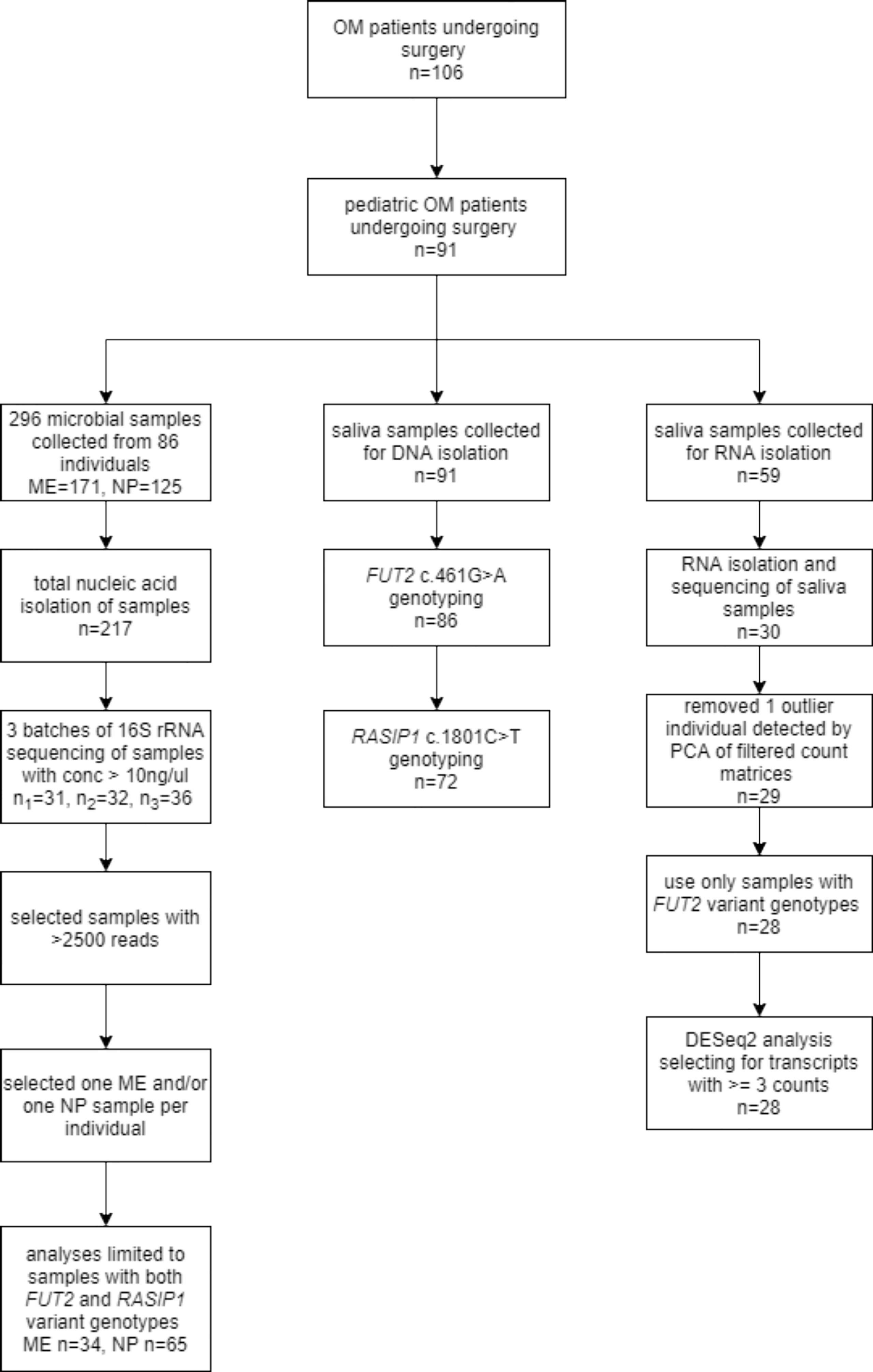
Figure 1 Study flowchart. The flowchart shows the number of saliva and microbial samples included for genotyping, RNA-seq and microbiota analyses.
Saliva samples were also collected from pediatric patients with OM using Oragene-RNA RE-100 kits and sufficient RNA was isolated from 30 samples using the manufacturer’s protocol (Figure 1). A total of 296 microbial samples were obtained from the ME (n=171) and NP (n=125) of 86 individuals, including 74 ME swabs, 86 ME aspirates, and seven ME mucosal tissue samples. Four ME cholesteatoma/granuloma tissue samples and 125 NP swabs were also collected (Figure 1). Microbial DNA was isolated from 217 (73%) samples using the Epicentre Masterpure Complete DNA Purification Kit (Lucigen, Middleton, WI, USA); the rest of the samples from which no microbial DNA was isolated were excluded from further study (Figure 1).
A variant in RASIP1 c.1801C>T (p.Arg601Cys; rs2287922) is in moderate LD with the FUT2 variant c.461G>A (r2 = 0.82) and with the rs681343 variant (r2 = 0.65) that was associated with childhood ear infections (Pickrell et al., 2016; Buniello et al., 2019). Sanger sequencing was performed for the FUT2 NM_000511.6:c.461G>A and RASIP1 NM_017805.3:c.1801C>T variants using DNA from saliva samples of pediatric patients with OM. Both variants were in Hardy-Weinberg equilibrium within the entire cohort and in each cohort used for RNA-seq and microbiota analyses (Table 1).
Thirty salivary RNA samples (median RIN=7.1) were submitted for RNA-sequencing at the University of Colorado Denver Genomics and Microarray Core, as previously described (Larson et al., 2019). In summary, RNA samples were processed using the Nugen Trio RNA-Seq Kit (Tecan, Redwood City, CA, USA). Sequencing was performed on an Illumina HiSeq 4000 with an average of 31 million reads per sample. Reads were trimmed using the FASTX-Toolkit v0.0.13 and aligned using STAR v2.5.3a (Dobin et al., 2013). Principal components analysis (PCA) was performed on this dataset and one outlier sample was removed from further analyses due to not clustering with other samples (Supplementary Figure 1). Transcript counts were summarized at the gene level and analyses included genes with an average read count >3. DE analysis was performed on 28 samples (Figure 1) according to carriage of the FUT2 c.461G>A variant using the DESeq2 package in R (Love et al., 2014), with correction for age, sex and batch effects (Supplementary Figure 1). Results were considered significant for genes with log2-transformed fold change > ± 2 and false discovery rate (FDR)-adjusted p-value <0.05 using the Benjamini-Hochberg method.
FUT2, RASIP1 and DE genes were used as input in NetworkAnalyst for construction of a protein-protein interaction network using the IMEx interactome database (Xia et al., 2014; Xia et al., 2015). Pathway enrichment analysis was performed on the resulting network using the KEGG and PANTHER GO-slim BP databases in NetworkAnalyst (Kanehisa and Goto, 2000; Kanehisa, 2019; Mi et al., 2019; Kanehisa et al., 2021). Pathways with an FDR-adjusted p<0.05 were deemed significantly enriched.
A total of 171 ME and 125 NP samples were obtained from 86 Coloradan pediatric patients with OM and submitted for 16S rRNA sequencing. Microbial DNA isolation was performed using the Epicentre MasterPure™ Kit. In order to test for contaminating bacterial DNA in reagents or plastics, every batch of samples that was submitted for 16S rRNA gene PCR and sequencing included ≥3 negative process controls. Bacterial profiles were determined by broad-range PCR amplification and sequence analysis of the 16S rRNA gene V1V2 regions, as previously described (Santos-Cortez et al., 2018; Frank et al., 2021). Illumina paired-end sequencing was performed on MiSeq using the 600 cycle version 3 kit. Assembled and quality-filtered sequences were aligned and classified with SINA (1.3.0-r23838) using the 418,497 bacterial sequences in Silva 115NR99 (Pruesse et al., 2012; Quast et al., 2013). Operational taxonomic units (OTUs) were produced by clustering sequences with identical taxonomic assignments (median: 115,176 sequences/sample; interquartile range: 46,274.5 – 170,300.0). Goods coverage scores were ≥99.7% for all samples, indicating adequate depth of sequence coverage for all samples. Of the 296 microbial samples submitted for sequencing, 79 did not pass quality control (DNA concentration ≥10 ng/ul; 2500 reads after sequencing; Figure 1). Because it was not possible to determine whether the lack of microbial DNA is due to a relatively sterile ME or from a sample collection issue, these 79 samples were excluded. Bacterial alpha-diversity indices (richness, diversity, and evenness; Robertson et al., 2013) were tested for association with carriage of each of the FUT2 c.461G>A or RASIP1 c.1801C>T variants independently via Wilcoxon test and adjusted for ethnicity (Robertson et al., 2013). Associations of individual OTUs with FUT2 c.461G>A and RASIP1 c.1801C>T variants were assessed using linear regression with sample batch as a covariate. To minimize multiple-comparisons, only taxa with a prevalence >10% and relative abundance >1% were included in the analysis. Beta-diversity was determined via PERMANOVA using the Morisita-Horn dissimilarity index and adjusted for age, sex and batch effects. R software was used for data analyses and figure generation.
All animal experiments were performed according to the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and carried out in strict accordance with an approved Institutional Animal Care and Use Committee (IACUC) protocol (A13-022) of the Veteran Affairs Medical Center (San Diego, CA). All animal experiments employed the best efforts for minimizing animal suffering under general anesthesia according to the NIH guidelines.
For gene array studies, wild-type (WT) C57/WB F1 hybrid mice were purchased from the Jackson Laboratory (Bar Harbor, ME USA). NTHi strain 3655 (non-typeable, biotype II, originally isolated from the ME of a child with OM in St Louis, MO USA) was cultured in defined liquid media (Coleman et al., 2003). To induce ME infection, mice were deeply anesthetized with an intraperitoneal injection of rodent cocktail (13.3 mg/ml ketamine hydrochloride, 1.3 mg/ml xylazine, 0.25 mg/ml acepromazine; at 0.1-0.2 ml per 25-30 g body weight of the mouse). The bullae were bilaterally exposed through soft tissue dissection via a ventral approach. A hole was made in the bulla with a 23 gauge needle, allowing approximately 5 μl of NTHi inoculum (~5x104 CFU/mL) to be injected using a Hamilton syringe with a 30-gauge needle. After the injection of NTHi inoculum, the tympanic membranes were visually inspected and confirmed to be intact. The incision was then stapled and the mice were given normal saline and analgesics (buprenorphine at 0.05mg/Kg) subcutaneously while recovering on the heated mat. Following recovery from anesthesia the mice appeared healthy, with a clinical activity index ≤ 3 throughout the duration of OM.
Gene array data were generated as previously described (Hernandez et al., 2015). In summary, forty mice per time point were inoculated bilaterally with NTHi. Mucosal tissue and exudate were harvested from 20 mice at each of the following intervals – 0 hours (0h, no treatment), 3h, 6h, 1 day (1d), 2d, 3d, 5d and 7d after inoculation – then pooled. The tissue was homogenized in TRIzol (Life Technologies, Carlsbad, CA) and total RNA extracted, reverse transcribed and re-transcribed in vitro to generate biotinylated cRNA probes that were hybridized to 2 Affymetrix MU430 2.0 microarrays. Hybridization intensity data were median-normalized and differences in gene transcript expression levels evaluated using variance-modeled posterior inference (VAMPIRE) (Hsiao et al., 2005). Bonferroni multiple testing correction (αBonf < 0.05) was applied to identify only those genes with the most robust changes. The data were duplicated at each time point to obtain a second, independent biological replicate. Thus each data point represents 2 separate samples consisting of 20 mice each, and 4 Affymetrix arrays. A total of 3,605 genes, approximately 14.4% of the mouse genome, defined the signature of acute, NTHi-induced OM across time. Hybridization of RNA to specific gene probes was assessed at individual time points by comparison to uninfected MEs, after Bonferroni correction for multiple tests, using Genespring GX 7.3 (Agilent Technologies, Santa Clara, CA).
For single-cell RNASeq, the same ME inoculation protocol was followed, except that C57BL/6J mice (Jackson Labs) were employed. Single-cell samples for RNA-sequencing were generated from the entire contents of the mouse ME (Ryan et al., 2020). For each of three independent samples, tissue was harvested from both ears of six young adult C57-BL6 mice 6 hours after inoculation of the ME with NTHi. Single-cell libraries were generated using the 10X Genomics (Pleasanton, CA, USA) Chromium Single Cell 3’ Reagent Kit V2. cDNA synthesis, barcoding, and library preparation were then carried out on a 10X Genomics Chromium Controller according to the manufacturers’ instructions. After validating quality of cDNA library, sequencing was performed on an Illumina HiSeq 2500 (Illumina, San Diego, CA USA). Reads were demultiplexed and aligned to the murine reference genome (mm10 with annotations from Ensembl, release 84). 10X Genomics Cellranger aggr and Seurat were used to generate PCA clustering (Satija et al., 2015). The expression of well-recognized marker genes identified 24 distinct cell types (Ryan et al., 2020). Linearized relative expression levels of each gene examined in this study were log-transformed from single-cell mRNA copy numbers, normalized, and scaled for each cell type. Data were visualized in 10X Genomics cLoupe, with UMI numbers expressed colorimetrically for each cell.
Samples were collected from 91 pediatric patients with OM with ages ranging from 8.7 months to 14.9 years old (median 2.0 years; Table 1). Of these, 86 had sufficient DNA sample for Sanger sequencing of FUT2 c.461G>A (Figure 1) and 83.3% are homozygous or heterozygous for the FUT2 variant. Carriage of the FUT2 variant was not associated with age, sex, ethnicity or OM diagnosis among children with OM (Table 1). In the entire cohort and in each subset analyses, males were predominant (≥81%), which is a known phenomenon for OM (Paradise et al., 1997).
RNA-seq data from 28 pediatric patients (0.8 to 14.8 years old; Table 1) passed QC and were available for analysis according to FUT2 genotype. DE analysis was performed using FUT2 c.461G>A variant carriage as the classifier (5 wildtype and 23 variant carriers) and with adjustment for age, sex and batch effects (Table 1 and Supplementary Figure 1). Five DE genes were significant, namely: FN1 (log-fold change = -3.7, FDR-adj-p=0.006); KMT2D/MLL2 (log-fold change = -3.8, FDR-adj-p=0.04); MUC16 (log-fold change = -4.3, FDR-adj-p=0.04); MTAP (log-fold change = +5.4, FDR-adj-p=0.006); and NBPF20 (log-fold change = -3.5, FDR-adj-p=0.04). In carriers of the FUT2 c.461G>A variant, FN1, KMT2D/MLL2, MUC16 and NBPF20 were downregulated whereas MTAP was upregulated (Figure 2).
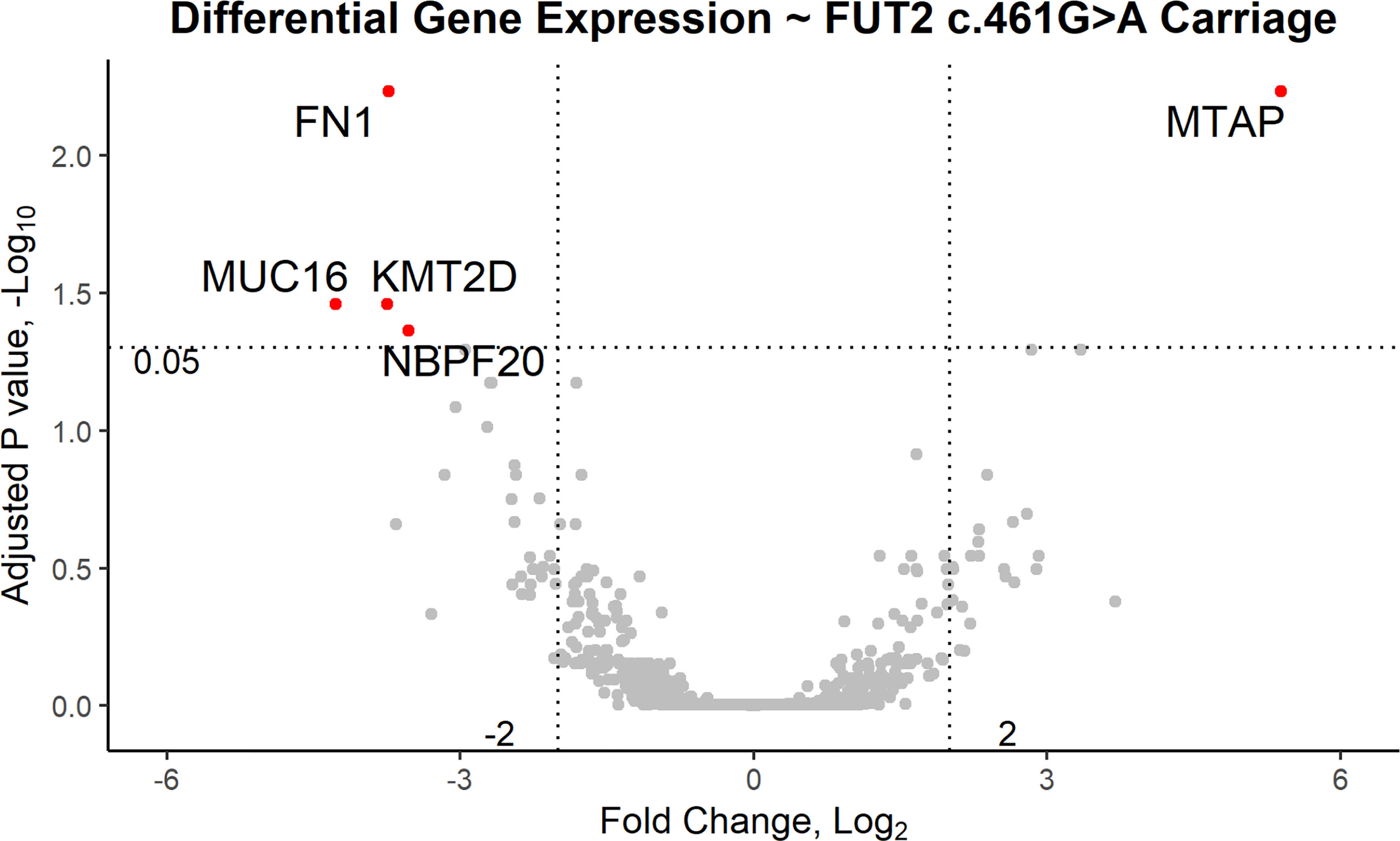
Figure 2 Volcano plot of differentially expressed genes based on carriage of the FUT2 c.461G>A variant in patients with OM. In variant carriers, KMT2D/MLL2, MUC16, NBPF20 and FN1 were downregulated (FDR-adjusted p < 0.05, log2 fold change < -2) and MTAP was upregulated (FDR-adjusted p < 0.05, log2 fold change > 2).
To further investigate how FUT2, FN1, KMT2D/MLL2, MUC16, MTAP, NBPF20 and RASIP1 are related, these genes were used as input for network analysis. RASIP1, FN1, KMT2D/MLL2 and MTAP were connected in a single protein-protein interaction network (Figure 3A). Pathway enrichment analysis of this network revealed 27 significant pathways in KEGG and 21 significant processes in PANTHER GO-slim BP, many of which overlap (Figures 3B, C and Table 2). Among these are processes pertaining to viral and bacterial infection, cell cycle regulation, apoptosis, and endocytosis (Table 2).
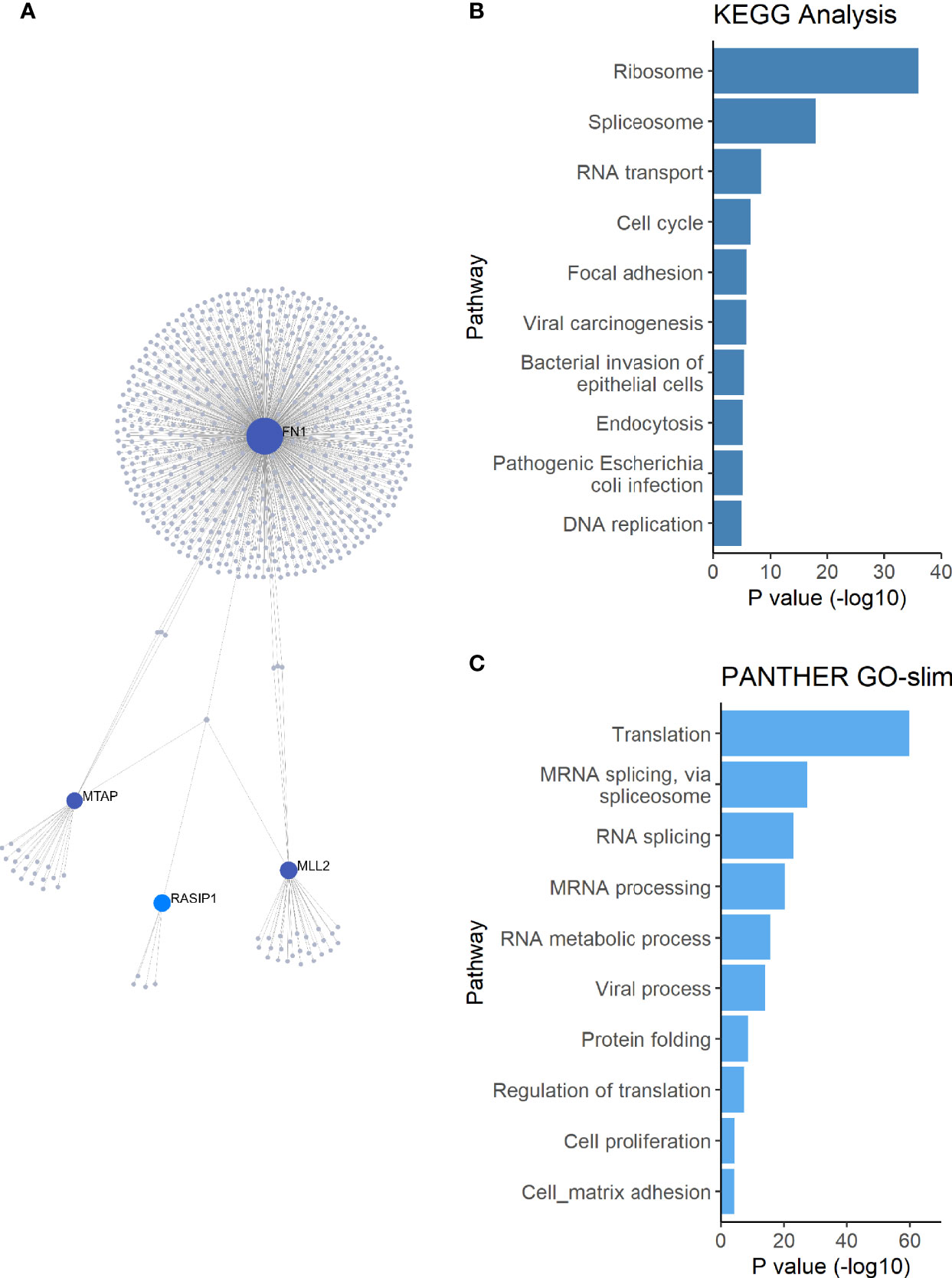
Figure 3 Network and pathway enrichment analysis of differentially expressed genes. (A) A single PPI network was constructed using the FUT2, RASIP1 and the DE genes as input. (B) KEGG and (C) PANTHER GO-slim:BP pathway enrichment analysis results, showing the top 10 pathways with the smallest p-values. MUC16 and FUT2 are not connected to this network, suggesting a different mechanism for the interaction of these two genes in relation to OM.
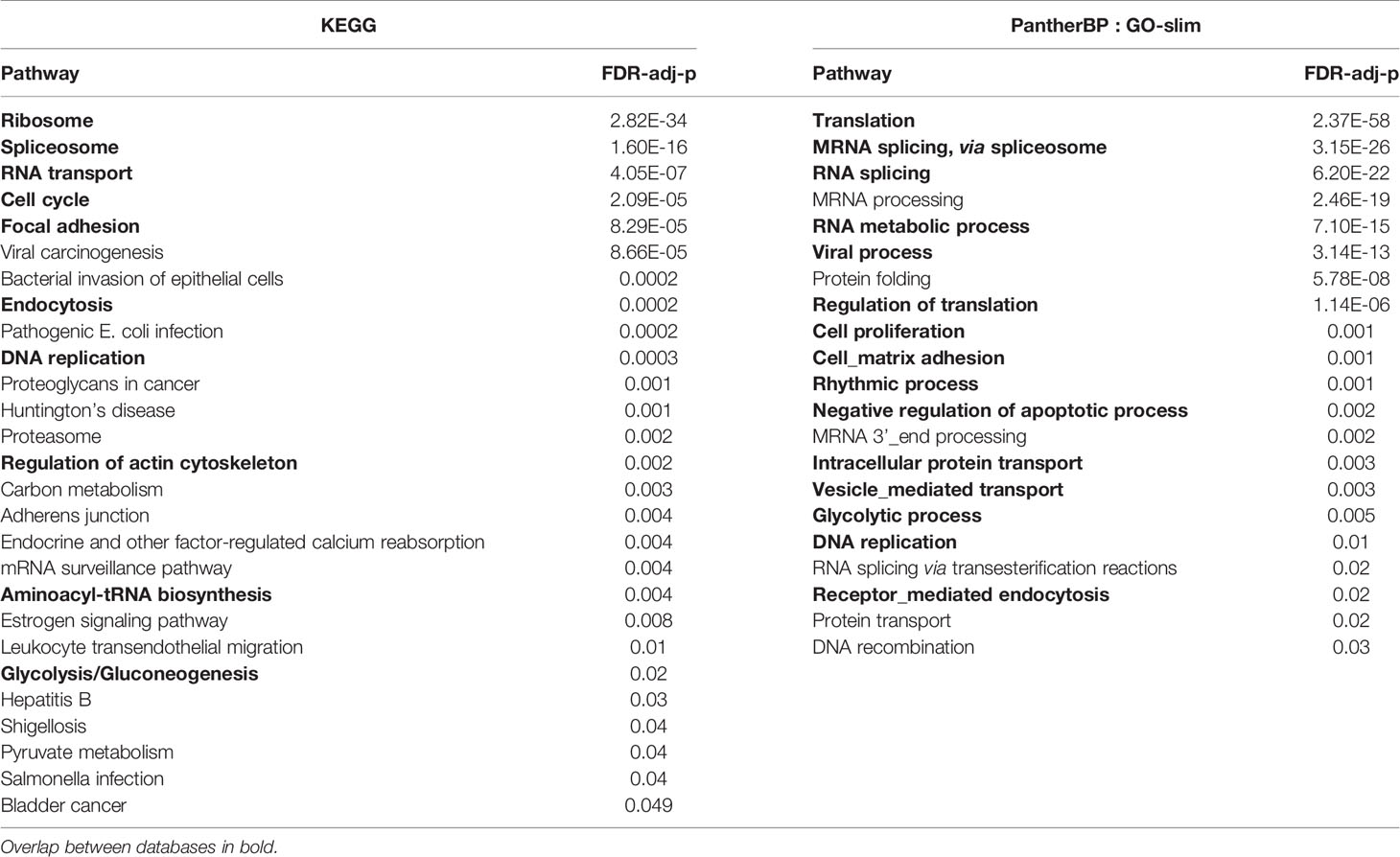
Table 2 Significant pathways within network connecting DE genes.
To further understand the role and interactions between FUT2, RASIP1 and DE genes, expression of orthologs Fut2, Fn1, Kmt2d, Muc16, Mtap and Rasip1 were measured by gene array in ME of wildtype mice at multiple time points (from 3 hours to 7 days) post-infection with NTHi (Figure 4 and Tables 3, 4). The NBPF gene family results from segmental duplication events in primate, thus an ortholog for NBPF20 is not present in mice (Vandepoele et al., 2005). Expression of Fut2, Rasip1 and Mtap were significantly increased after inoculation, with Fut2 and Mtap peaking around one day post-inoculation, and Rasip1 and Fn1 at 3 hours post-inoculation (Figure 4). Additionally, expression of Muc16 was significantly decreased one day post-inoculation. For Fn1, Mtap and Muc16, DE was sustained through days 2-7 post-inoculation, including when OM is supposedly in recovery phase (Hernandez et al., 2015). Kmt2d showed no significant changes in ME expression at any point during the 7 days when compared to control mice (Figure 4).
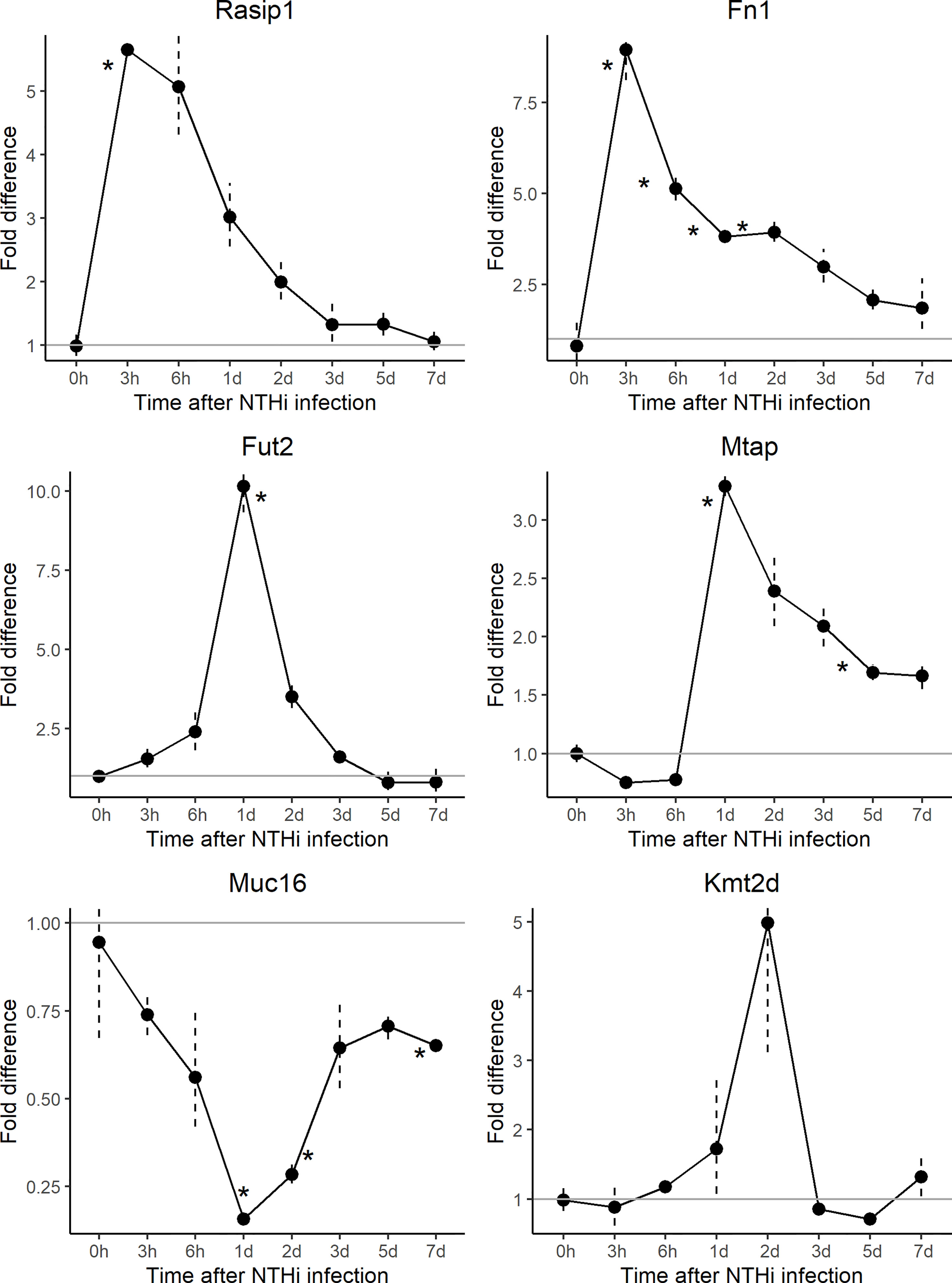
Figure 4 Gene array expression data for select genes post-inoculation with non-typeable Haemophilus influenzae (NTHi). Mouse middle ear expression of select genes across different time points, shown as fold change in middle ears inoculated with NTHi as compared to placebo. Fut2, Muc16 and Mtap reached peak change in expression at 24 hours post-inoculation while Muc16 demonstrated sustained downregulation. On the other hand, Rasip1 and Fn1 reached peak upregulation at 3 hours post-inoculation. In this experiment, time point 0h represents uninfected middle ear. *p < 0.05; see Table 3 for gene expression values by time point and gene.
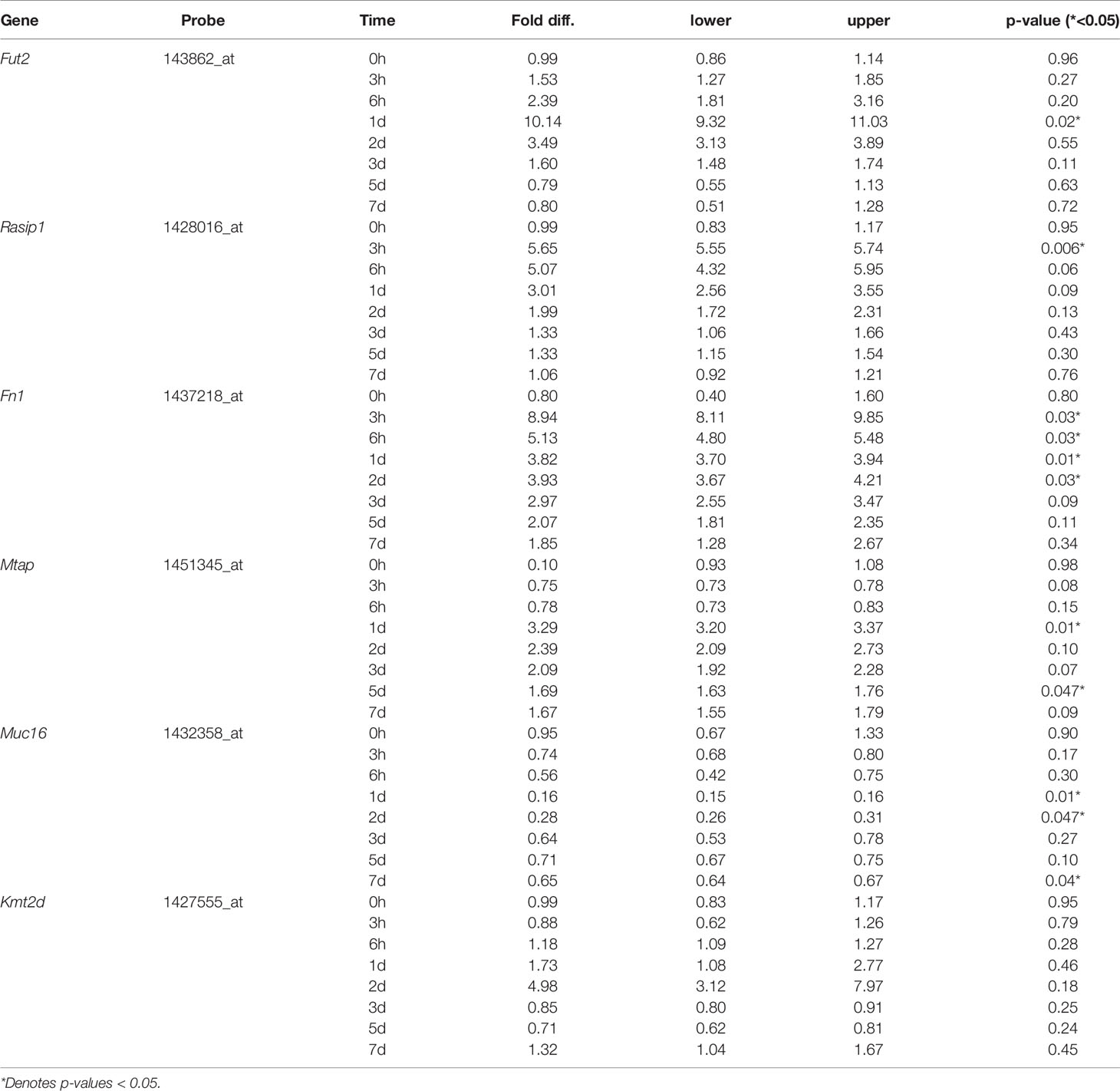
Table 3 Mouse ME gene expression values by time point.

Table 4 Comparison of DE gene regulation in human saliva of FUT2 c.461G>A variant carriers vs non-carriers and NTHi- vs placebo-inoculated mouse middle ear (ME).
Single-cell RNA-sequence (scRNA-Seq) data were derived from the MEs of NTHi-infected mice six hours after inoculation (Figure 5 and Table 5). In uninfected ME (time point 0h), Fut2 was expressed primarily in ciliated epithelial cells (Hydin+). Muc16 was expressed in most epithelial cells except basal epithelial cells (Krt14+). Rasip1 was expressed in most endothelial cells (Egfl7+) and Fn1 mostly in stromal cells (Col1a2+) and melanocytes (Mlana+). Mtap and Kmt2d were modestly expressed in all ME cell types. Six hours after ME inoculation with NTHi, when overall expression data was strongest (Figure 5), Fut2 had increased expression in non-ciliated epithelial cells (Krt18/19+) and Muc16 in all epithelial cell types. Rasip1 continued to be expressed in endothelial cells, but was also observed in polymorphonuclear cells (PMNs) and monocytes (Csf1r+). Fn1 increased expression in stromal cells and monocytes and some endothelial cells (Figure 5). Mtap and Kmt2d remained moderately expressed in all ME cell types except infiltrating PMNs and red blood cells. Level of gene expression per cell peaked at 1 day, and then declined (Table 5). Taken together, the mouse ME expression profiles for Fut2, Rasip1 and DE genes support the findings of DE genes in OM patients using RNA-seq data from saliva (Table 4), and also the overall expression of these genes in other human mucosal tissues (Table 6).
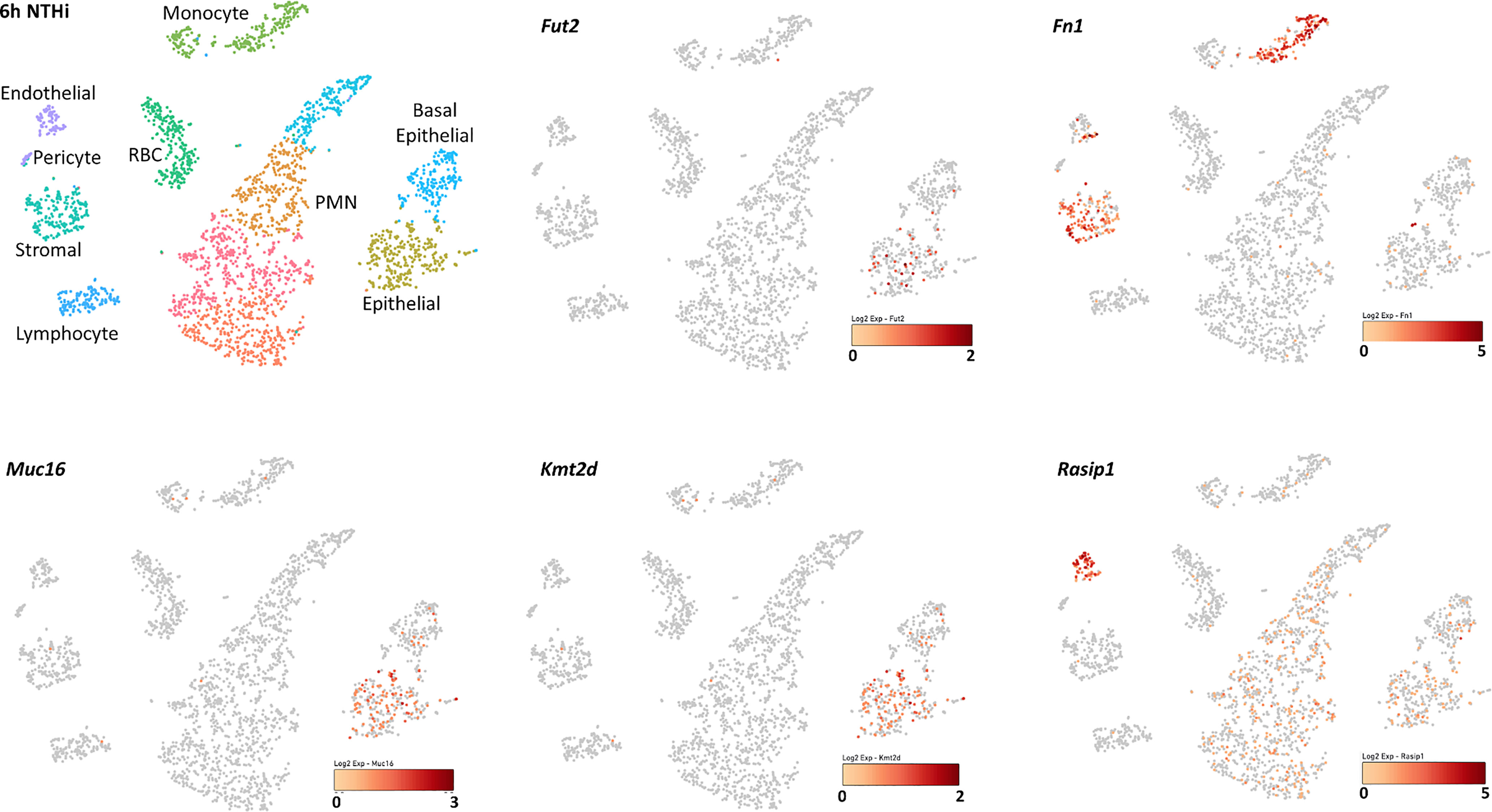
Figure 5 Single-cell RNA-seq expression data from mouse middle ear 6 hours after inoculation with NTHi. Expression of Fut2, Fn1, Muc16, Kmt2d and Rasip1 in wildtype mouse middle ear, 6 hours post-infection. Cell types of the various PCA clusters were identified by the expression of unique marker genes. Darkness of color (red for each gene) indicates level of UMI expression by each cell, according to the associated log2 scale. Epithelial cell clusters were identified based on the expression of Krt18 and/or Krt19. Basal epithelial cells also express Krt14, while ciliated epithelial cells express Hydin. Stromal cells are identified by Col1a2. Vascular endothelial cells express Egfl7 and Flt4, lymphatic endothelial cells Egfl7 and Flt1. Melanocytes express Mlana and pericytes Rgs5. Monocytes express Csf1r, lymphocytes Ptprcap, polymorphonuclear cells Il1f9 and Stfa2l1, and red blood cells Hba-a1.
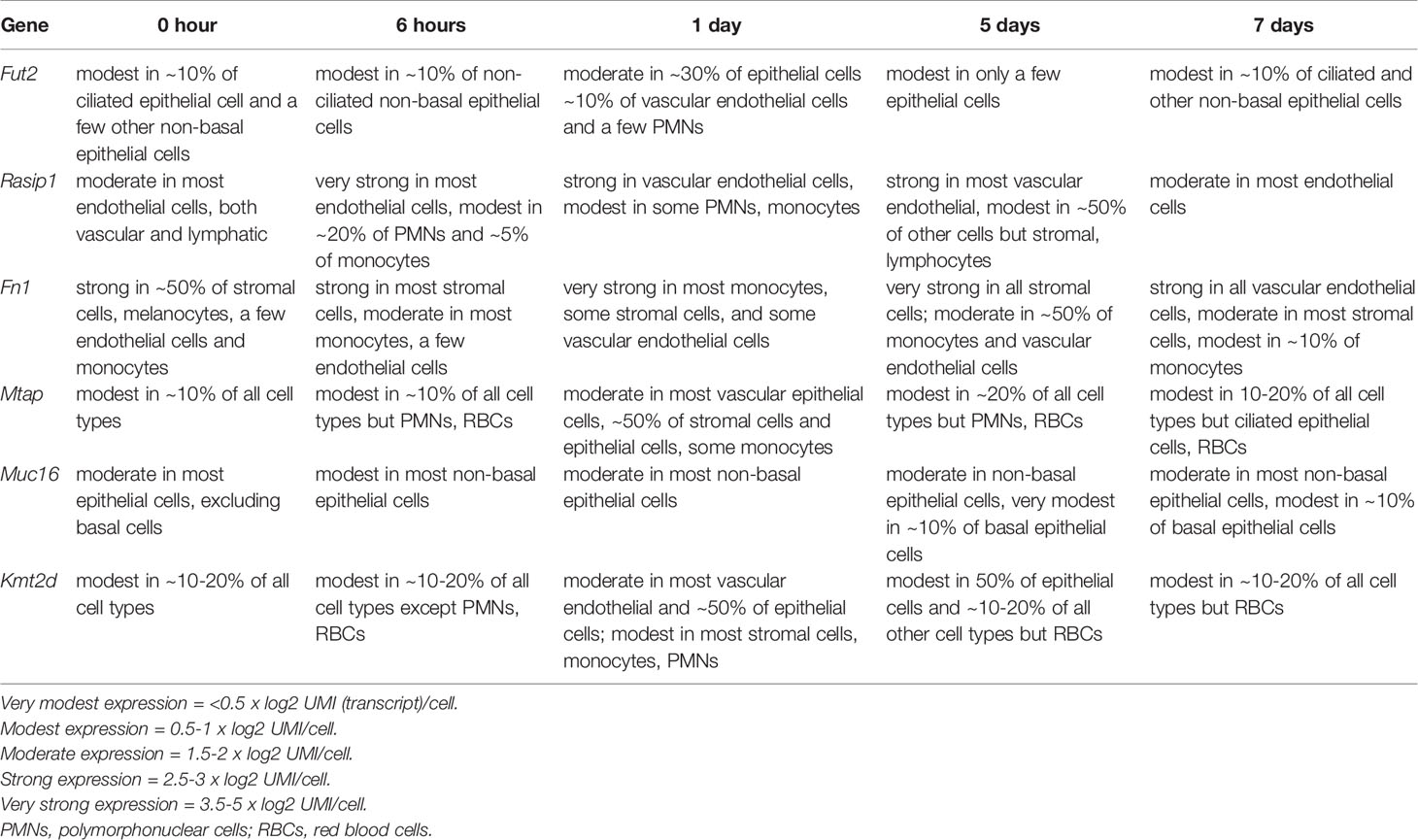
Table 5 Single-cell RNA-seq expression levels in mouse ME by time point after NTHi inoculation.
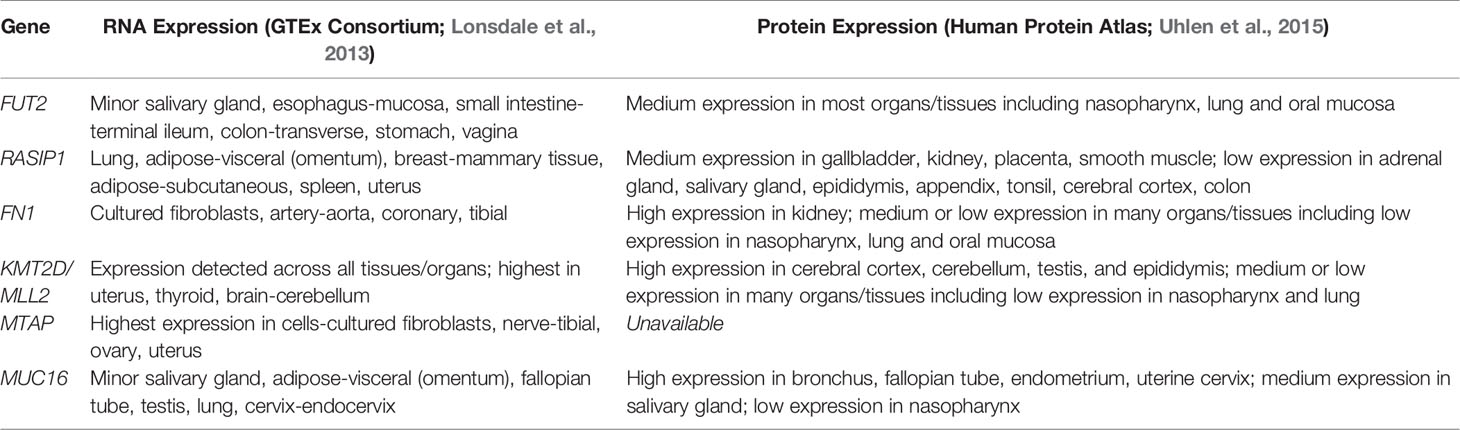
Table 6 Known RNA and protein expression profiles of FUT2, RASIP1 and DE genes in human tissues.
A total of 296 microbial samples were collected from the NP and ME of 86 children (Figure 1). For microbiota analyses, samples were filtered for: (1) those with >2500 16S rRNA sequencing reads; (2) one ME and one NP sample per individual where bilateral samples were collected (if bilateral, right-sided sample was used); and (3) available genotypes for FUT2 and RASIP1 variants (Figure 1). No differences were identified between right and left NP or ME samples from the same individuals in PCA and PERMANOVA analyses (data not shown). After filtering, 16S rRNA sequence data from 34 ME and 65 NP samples were analyzed according to carriage of the FUT2 c.461G>A (p.Trp154*) variant.
In the ME, based on carriage of the FUT2 variant, Chao1 which denotes bacterial richness was significant when all ethnic groups were included (p=0.03); however, all alpha-diversity indices were not significant when only individuals of European descent were included in analyses (Supplementary Table 1). Overall microbiota composition (i.e., beta-diversity) did not differ significantly by FUT2 variant according to PERMANOVA analysis with adjustment for age, sex, or batch effects (Figure 6A). Additionally, the relative abundances of Haemophilus (nominal p=0.03) and Moraxella (nominal p=0.02) were increased with wildtype FUT2 genotype, whereas increased Propionibacterium (nominal p=0.04) and Anoxybacillus (nominal p=0.02) were associated with presence (homozygous or heterozygous genotypes combined) of the variant (Figure 6C and Supplementary Table 2). Performing these analyses by genotype had no overall effect on results (Supplementary Figure 2).

Figure 6 Relative abundance of individual taxa in middle ears (ME) and nasopharynges (NP) of carriers and non-carriers of the FUT2 c.461G>A variant. (A) Cumulative relative abundance profiles in the ME of wildtype (n=8) and carriers (n=26) of FUT2 c.461G>A. (B) Cumulative relative abundance profiles in the NP of wildtype (n=14) and carriers (n=51) of FUT2 c.461G>A. Plots showing p-values for relative abundance of individual bacterial taxa in the (C) ME and (D) NP of wildtype versus variant carriers after adjusting for batch. Blue lines indicate taxa that were increased in wildtype, red lines for carriers. Dashed lines indicate significance thresholds where the red line is unadjusted-p=0.1 (non-significant) and green lines indicate unadjusted-p=0.05 and unadjusted-p=0.01. (C) In the ME, Haemophilus (1) and Moraxella (2) were nominally associated with wildtype, whereas Propionibacterium (3) and Anoxybacillus (4) were nominally associated with variant carriage. (D) In the NP, Candidate Division TM7 (1) was significantly associated with wildtype (FDR-adj-p=0.009). Additionally, Selenomonas (2) and Actinobacillus (3) were nominally associated with wildtype whereas Propionibacterium (4) was nominally associated with variant carriage.
In the NP, there were also no significant differences in alpha- or beta-diversity (Supplementary Table 1 and Figure 6B). Similar to ME, Propionibacterium had increased relative abundance in the NP (nominal p=0.01) among carriers of the FUT2 variant. In addition, the relative abundances of Actinobacillus (nominal p=0.03), Selenomonas (nominal p=0.03) and Candidate Division TM7 (Saccharibacteria; nominal p=0.0002) were increased in wildtype individuals (Figure 6D, Supplementary Table 2). When individual taxa were tested for association by genotype, no taxa were significant (Supplementary Figure 2). Note however that these FUT2-microbiota associations were nominal and were non-significant after FDR correction, with the exception of Candidate Division TM7 in the NP (FDR-adjusted p=0.009).
Sanger sequencing of DNA samples confirmed that the RASIP1 c.1801C>T and FUT2 c.461G>A variants are in moderate LD in our cohort as the genotypes for 57 of 71 (80.3%) individuals were identical. In the ME, similar to findings with the FUT2 variant, an increased relative abundance of Haemophilus (nominal p=0.04) was associated with wildtype genotype whereas increased Propionibacterium (nominal p=0.04) was associated with the RASIP1 variant (Figure 7C and Supplementary Table 3). When analyzed by genotype, Haemophilus remained nominally associated with wildtype (Supplementary Figure 3). In the NP, increased abundance of Propionibacterium (nominal p=0.006), chloroplast (FDR-adjusted p=0.05), Escherichia-Shigella (nominal p=0.04) and Staphylococcus (nominal p=0.04) was associated with carriage of the RASIP1 variant, whereas increased abundance of Candidate Division SR1 (FDR-adjusted p=0.05), Candidate Division TM7 (FDR-adjusted p=0.05), and Actinobacillus (nominal p=0.01) was associated with wildtype genotype (Figure 7D and Supplementary Table 3).

Figure 7 Relative abundance of individual taxa in the middle ears (ME) and nasopharynges (NP) of carriers and non-carriers of the RASIP1 c.1801C>T variant. (A) Cumulative relative abundance profiles in the ME of wildtype (n=8) and carriers (n=26) of RASIP1 c.1801C>T. (B) Cumulative relative abundance profiles in the NP of wildtype (n=15) and carriers (n=50) of RASIP1 c.1801C>T. Plots showing p-values for relative abundance of individual bacterial taxa in the (C) ME and (D) NP of wildtype versus carriers after adjusting for batch. Blue lines indicate taxa that were increased in wildtype, red lines for carriers. Dashed lines indicate significance thresholds where the red line is unadjusted-p=0.1 (non-significant) and green lines indicate unadjusted-p=0.05 and unadjusted-p=0.01. (C) In the ME, Gammaproteobacteria (1) was nominally associated with wildtype, whereas Propionibacterium (2) was nominally associated with variant carriage. (D) In the NP, Candidate Division SR1 (1) and Candidate Division TM7 (2) were significantly associated with wildtype, and Chloroplast (5) with variant carriage (FDR-adj-p=0.05). Additionally, Actinobacillus (3) was nominally associated with wildtype, whereas Propionibacterium (4), Staphylococcus (6) and Escherichia-Shigella (7) were nominally associated with variant carriage.
Variants in FUT2, including the c.461G>A (p.Trp154*) variant investigated here, have been associated with increased susceptibility to OM but their functional role in OM pathology has not been fully elucidated. Although FUT2 has been studied by many groups, to our knowledge this is the first study in which RNA-seq data combined with ME and NP microbiotas have been examined in relation to carriage of the FUT2 c.461G>A variant. Our results suggest that the FUT2 variant confers OM susceptibility through its modulation of MUC16 expression and downstream induction of FN1 and MTAP after microbe binding and pathogen colonization (Figures 2, 3 and Table 2). These DE findings were supported by similar regulation of expression in NTHi-infected ME of wildtype mice, whether by bulk mRNA-seq or single-cell RNA-seq data (Figure 4 and Tables 3, 4). Because these genes were differentially regulated in response to OM in the infected wildtype mouse ME, the results of this DE analysis suggest that the FUT2 c.461G>A variant magnifies the downstream response to infection (for example, downregulated MUC16, upregulated MTAP), and/or reverses the direction of regulation (e.g. downregulation of FN1 in carriers of the FUT2 variant; Figures 2, 4 and Table 4). Alternatively, DE genes may vary depending on the predominant otopathogen during infection: in other words, whether commensal or otopathogenic bacteria bind to ME mucosal epithelium via A antigen, the expression of which is affected by heterozygous or homozygous genotype for the FUT2 c.461G>A variant (Figures 6, 7; Santos-Cortez et al., 2018).
RASIP1 is expressed in ME endothelial cells and provides another avenue for investigation in relation to FUT2 c.461G>A variant carriage. RASIP1 c.1801G>T, previously identified by GWAS to be in LD with FUT2 c.461G>A (Pickrell et al., 2016), is also in moderate LD with FUT2 c.461G>A in the sample set. RASIP1 is part of the PPI immune network including MTAP, KMT2D and FN1 (Figure 3A and Table 2), which led us to question whether the expression and microbiota effects we observed were being driven by the RASIP1 missense variant rather than the FUT2 stop variant. When examining the changes in the expression of these genes in wildtype mice after NTHi inoculation, Rasip1 and Fn1 expression peaked at 3 hours post-inoculation, whereas Fut2 peaked at one day post-inoculation, in concordance with Muc16 and Mtap expression (Figure 4 and Table 3). Additionally, in the single-cell RNA-seq data from mouse ME, we observed Rasip1 and Fn1 expression in endothelial cells versus epithelial expression of Fut2 and Muc16 (Figure 5 and Table 5). When examined together, these expression profiles strongly support FUT2 as mediating OM susceptibility within the ME mucosal epithelium. In particular, the downregulation of MUC16 in OM patients with the FUT2 stop variant might indicate a prolonged recovery phase when MUC16 is expected to return to normal levels as part of the normal response to acute OM. MUC16 downregulation is therefore a potential avenue for future research, for example, whether this effect of FUT2 knockdown is a mechanism for an acute infection to proceed to recurrence or chronicity (Kerschner et al., 2013).
Dysbiosis of the NP and ME mucosal microbiotas is supported by our data here and in our previous studies in which the ME of FUT2 c.461G>A variant carriers were enriched in potentially otopathogenic taxa such as Propionibacterium, and decreased for established otopathogens Haemophilus and Moraxella, although these associations were nominal (Santos-Cortez et al., 2018). This could be attributed to the effect of FUT2 c.461G>A on pathogen binding, wherein those homozygous for the FUT2 variant are non-secretors of ABO(H) antigens on the epithelia surface (Table 6); these antigens can serve as ligands to which some bacteria may bind and thus affect the commensal and pathogen loads of the NP and ME. Interestingly the only bacterial taxon that has a significant association with the FUT2 variant after correction for multiple testing is Candidate Division TM7, which is also known as Saccharibacteria (Figure 6). Little is known about Saccharibacteria and its reported associations with human mucosal disease have been variable, though there is some evidence that it parasitizes other bacteria and can kill its host bacterium, thereby modulating the overall microbiota (Bor et al., 2019).
The change in relative abundance of chloroplast in the NP corresponding to RASIP1 variant carriage is an unusual result. This is potentially due to a sequence misclassification of cyanobacteria in the reference database rather than systematic contamination during isolation from the kit or reagents. Though general contamination is a possible explanation, if this were the case its presence would be detected among all samples or the effect would be eliminated by the adjustment for batch during analyses. Furthermore, chloroplast contamination would be negatively correlated with number of reads per sample as contamination would be less prominent in samples with higher bacterial loads. However, we did not observe these in our samples and during analyses. Thus, it is unlikely that the identification of chloroplast as being differentially abundant in carriers of the RASIP1 variant is due to general contamination, though random, non-systematic contamination cannot be ruled out. Note that the main findings in this work are more likely explained by carriage of the FUT2 variant and not the RASIP1 variant.
In addition to the impact on pathogen and commensal binding to epithelia, the DE and network analyses suggest that the FUT2 c.461G>A variant also has a downstream effect on basic cellular pathways (Figures 2, 3 and Tables 4, 7). For example, FN1 is a modulator of ME anti-inflammatory response (Song et al., 2013) as well as a binding site for otopathogen Staphylococcus aureus (Fowler et al., 2000) and group A Streptococcus (McNitt et al., 2018). FN1 protein expression was also previously demonstrated to be dysregulated by viral infection (Simon et al., 2015; Qiao et al., 2021); however, viruses are not included in this study due to sample collection methods. Notably we only observed a nominal increase in Staphylococcus abundance in the NP (but not ME) of carriers of the RASIP1 variant (Figure 7), but not in carriers of the FUT2 variant (Figure 6). In addition, NTHi inoculation of mouse ME resulted in upregulation of Fn1 (Figure 4 and Table 3). In contrast, in our OM patients with the FUT2 stop variant, FN1 was downregulated (Figure 2 and Table 4), indicating that non-functional FUT2 might also affect the direction of regulation of the immune network that includes FN1 and also RASIP1, MTAP and MLL2/KMT2D (Figure 3). It should be noted that KMT2D variants are responsible for Kabuki Syndrome which is characterized by increased rates of OM as well as other immunological abnormalities (Hoffman et al., 2005; Ng et al., 2010; Yap et al., 2020; Boniel et al., 2021).
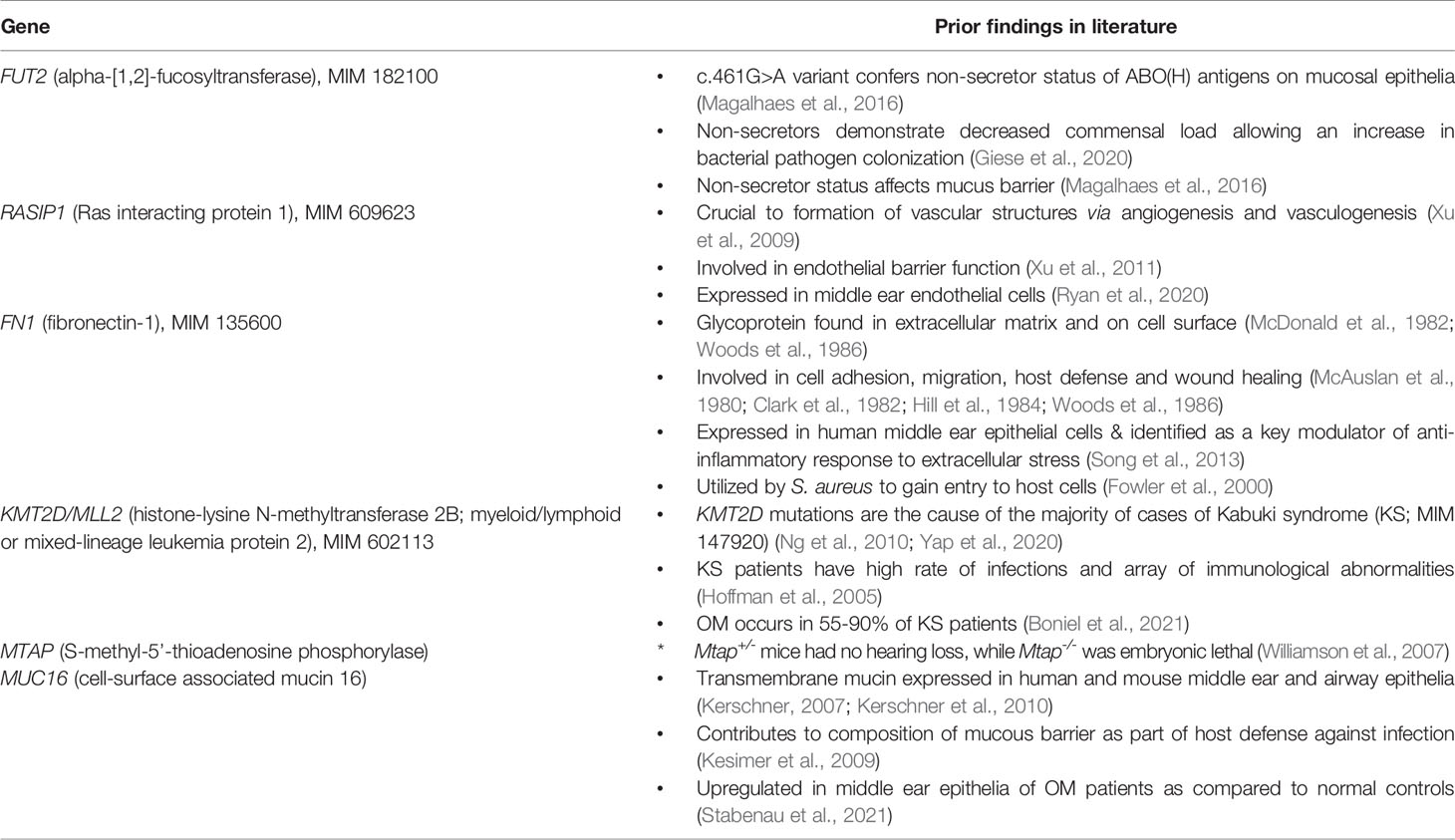
Table 7 Summary of relevant knowledge of FUT2, RASIP1 and DE genes.
In conclusion, we propose that the mechanistic effects of the FUT2 c.461G>A variant on OM susceptibility are two-fold: (1) Non-secretor status conferred by this FUT2 stop variant alters the profiles of bacterial taxa that bind to ME and NP mucosal epithelia and thereby increases susceptibility to bacterial infection in mucosal epithelia; and (2) FUT2 variants affect expression of genes including downregulation of MUC16 and those connected to an immune network, which leads to further susceptibility to infection as well as impaired immune responses (Figure 3) and basic cellular processes (Table 2) within the ME mucosal epithelium. Through increased understanding of the effects of pathogenic variants on dysbiosis and gene regulation in OM, the ability to determine risk for patients due to specific genetic variants may be improved, and thereafter enhance prevention and treatment protocols for OM using more targeted antibiotics for otopathogens associated with these variants.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/gap/, phs001941.v1.p1 https://www.ncbi.nlm.nih.gov/sra/, BioProject ID PRJNA748418.
The studies involving human participants were reviewed and approved by Colorado Multiple Institutional Review Board. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by Institutional Animal Care and Use Committee Veterans Affairs Medical Center, San Diego, California.
RS-C conceptualized the study. MS, S-OS, TW, PY, SG, SC, HJ, JP, KC, and NF recruited patients and collected samples. TB performed isolation of human DNA and RNA and microbial DNA samples and submitted them for sequencing. CE and EL performed RNA sequence analyses. CE performed network analyses. JK, CR, and DF performed 16S rRNA sequencing. CE and DF performed analyses of microbiota data. AR performed mouse expression studies. CE, AR, and RS-C wrote the manuscript. All authors read and approved the manuscript.
This work was supported by the National Institutes of Health (NIH) - National Institute on Deafness and Other Communication Disorders (NIDCD) via grant R01 DC015004 (to RS-C). Mouse studies were supported by grant R01 DC000129 from NIH-NIDCD (to AR). CE was supported by the T32 DC012280 grant from NIH-NIDCD (to Sue C. Kinnamon and HJ). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
AR is a cofounder of Otonomy, Inc., serves as a consultant and member of the Scientific Advisory Board, and holds an equity position in the company. The UCSD and San Diego VA Committees on Conflict of Interest have approved this relationship. Otonomy, Inc. had no role in the conduct of this study and the writing of the manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the patients and families for their participation in the study. We also thank the department’s Clinical Research Coordinator core for their assistance with subject enrollment.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.798246/full#supplementary-material
Azad, M. B., Wade, K. H., Timpson, N. J. (2018). FUT2 Secretor Genotype and Susceptibility to Infections and Chronic Conditions in the ALSPAC Cohort. Wellcome. Open Res. 3, 65. doi: 10.12688/wellcomeopenres.14636.2
Boniel, S., Szymanska, K., Smigiel, R., Szczaluba, K. (2021). Kabuki Syndrome-Clinical Review With Molecular Aspects. Genes (Basel) 12 (4), 468. doi: 10.3390/genes12040468
Bor, B., Bedree, J. K., Shi, W., McLean, J. S., He, X. (2019). Saccharibacteria (TM7) in the Human Oral Microbiome. J. Dent. Res. 98 (5), 500–509. doi: 10.1177/0022034519831671
Brennan-Jones, C. G., Whitehouse, A. J., Park, J., Hegarty, M., Jacques, A., Eikelboom, R. H., et al. (2015). Prevalence and Risk Factors for Parent-Reported Recurrent Otitis Media During Early Childhood in the Western Australian Pregnancy Cohort (Raine) Study. J. Paediatr. Child Health 51 (4), 403–409. doi: 10.1111/jpc.12741
Browne, J. J., Matthews, E. H., Taylor-Robinson, A. W., Kyd, J. M. (2021). Otopathogen Interactions in the Nasopharynx of Children, and the Predictive Value of Nasopharyngeal Aspirate Culture for the Aetiology of Upper Respiratory Infections. J. Paediatr. Child Health 57 (7), 1016–1022. doi: 10.1111/jpc.15370
Buniello, A., MacArthur, J. A. L., Cerezo, M., Harris, L. W., Hayhurst, J., Malangone, C., et al. (2019). The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 47 (D1), D1005–D1012. doi: 10.1093/nar/gky1120
Casselbrant, M. L., Mandel, E. M., Fall, P. A., Rockette, H. E., Kurs-Lasky, M., Bluestone, C. D., et al. (1999). The Heritability of Otitis Media: A Twin and Triplet Study. JAMA 282 (22), 2125–2130. doi: 10.1001/jama.282.22.2125
Chen, Y., Shen, L., Chen, B., Han, X., Yu, Y., Yuan, X., et al. (2021). The Predictive Prognostic Values of CBFA2T3, STX3, DENR, EGLN1, FUT4, and PCDH7 in Lung Cancer. Ann. Transl. Med. 9 (10), 843. doi: 10.21037/atm-21-1392
Clark, R. A., Lanigan, J. M., DellaPelle, P., Manseau, E., Dvorak, H. F., Colvin, R. B. (1982). Fibronectin and Fibrin Provide a Provisional Matrix for Epidermal Cell Migration During Wound Reepithelialization. J. Invest. Dermatol. 79 (5), 264–269. doi: 10.1111/1523-1747.ep12500075
Coleman, H. N., Daines, D. A., Jarisch, J., Smith, A. L (2003). Chemically Defined Media for Growth of Haemophilus Influenzae Strains. J. Clin. Microbiol. 41 (9), 44408–4410. doi: 10.1111/1523-1747.ep12500075
Davenport, E. R., Goodrich, J. K., Bell, J. T., Spector, T. D., Ley, R. E., Clark, A. G. (2016). ABO Antigen and Secretor Statuses Are Not Associated With Gut Microbiota Composition in 1,500 Twins. BMC Genomics 17 (1), 941. doi: 10.1186/s12864-016-3290-1
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 29 (1), 15–21. doi: 10.1093/bioinformatics/bts635
Fowler, T., Wann, E. R., Joh, D., Johansson, S., Foster, T. J., Hook, M. (2000). Cellular Invasion by Staphylococcus Aureus Involves a Fibronectin Bridge Between the Bacterial Fibronectin-Binding MSCRAMMs and Host Cell Beta1 Integrins. Eur. J. Cell Biol. 79 (10), 672–679. doi: 10.1078/0171-9335-00104
Frank, D. N., Giese, A. P. J., Hafren, L., Bootpetch, T. C., Yarza, T. K. L., Steritz, M. J., et al. (2021). Otitis Media Susceptibility and Shifts in the Head and Neck Microbiome Due to SPINK5 Variants. J. Med. Genet. 58 (7), 442–452. doi: 10.1136/jmedgenet-2020-106844
GBD 2019 Hearing Loss Collaborators (2021). Hearing Loss Prevalence and Years Lived With Disability 1990-2019: Findings From the Global Burden of Disease Study 2019. Lancet 397 (10278), 996–1009. doi: 10.1016/S0140-6736(21)00516-X
Giese, A. P. J., Ali, S., Isaiah, A., Aziz, I., Riazuddin, S., Ahmed, Z. M. (2020). Genomics of Otitis Media (OM): Molecular Genetics Approaches to Characterize Disease Pathophysiology. Front. Genet. 11, 313. doi: 10.3389/fgene.2020.00313
Hernandez, M., Leichtle, A., Pak, K., Webster, N. J., Wasserman, S. I., Ryan, A. F. (2015). The Transcriptome of a Complete Episode of Acute Otitis Media. BMC Genomics 16, 259. doi: 10.1186/s12864-015-1475-7
Hill, H. R., Shigeoka, A. O., Augustine, N. H., Pritchard, D., Lundblad, J. L., Schwartz, R. S. (1984). Fibronectin Enhances the Opsonic and Protective Activity of Monoclonal and Polyclonal Antibody Against Group B Streptococci. J. Exp. Med. 159 (6), 1618–1628. doi: 10.1084/jem.159.6.1618
Hoffman, J. D., Ciprero, K. L., Sullivan, K. E., Kaplan, P. B., McDonald-McGinn, D. M., Zackai, E. H., et al. (2005). Immune Abnormalities are a Frequent Manifestation of Kabuki Syndrome. Am. J. Med. Genet. A. 135 (3), 278–281. doi: 10.1002/ajmg.a.30722
Hsiao, A., Ideker, T., Olefsky, J. M., Subramaniam, S. (2005). VAMPIRE Microarray Suite: A Web-Based Platform for the Interpretation of Gene Expression Data. Nucleic Acids Res. 33 (Web Server issue), W627–W632. doi: 10.1093/nar/gki443
Jervis-Bardy, J., Carney, A. S., Duguid, R., Leach, A. J. (2017). Microbiology of Otitis Media in Indigenous Australian Children: Review. J. Laryngol. Otol. 131 (S2), S2–S11. doi: 10.1017/S0022215116009294
Jervis-Bardy, J., Leong, L. E. X., Papanicolas, L. E., Ivey, K. L., Chawla, S., Woods, et al. (2019). Examining the Evidence for an Adult Healthy Middle Ear Microbiome. mSphere 4 (5), e00456–19. doi: 10.1128/mSphere.00456-19
Kanehisa, M. (2019). Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 28 (11), 1947–1951. doi: 10.1002/pro.3715
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., Tanabe, M. (2021). KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 49 (D1), D545–D551. doi: 10.1093/nar/gkaa970
Kanehisa, M., Goto, S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28 (1), 27–30. doi: 10.1093/nar/28.1.27
Kelly, R. J., Rouquier, S., Giorgi, D., Lennon, G. G., Lowe, J. B. (1995). Sequence and Expression of a Candidate for the Human Secretor Blood Group Alpha(1,2)Fucosyltransferase Gene (FUT2). Homozygosity for an Enzyme-Inactivating Nonsense Mutation Commonly Correlates With the non-Secretor Phenotype. J. Biol. Chem. 270 (9), 4640–4649. doi: 10.1074/jbc.270.9.4640
Kerschner, J. E. (2007). Mucin Gene Expression in Human Middle Ear Epithelium. Laryngoscope 117 (9), 1666–1676. doi: 10.1097/MLG.0b013e31806db531
Kerschner, J. E., Hong, W., Taylor, S. R., Kerschner, J. A., Khampang, P., Wrege, K. C., et al. (2013). A Novel Model of Spontaneous Otitis Media With Effusion (OME) in the Oxgr1 Knock-Out Mouse. Int. J. Pediatr. Otorhinolaryngol. 77 (1), 79–84. doi: 10.1016/j.ijporl.2012.09.037
Kerschner, J. E., Li, J., Tsushiya, K., Khampang, P. (2010). Mucin Gene Expression and Mouse Middle Ear Epithelium. Int. J. Pediatr. Otorhinolaryngol. 74 (8), 864–868. doi: 10.1016/j.ijporl.2010.04.014
Kesimer, M., Scull, M., Brighton, B., DeMaria, G., Burns, K., O’Neal, W., et al. (2009). Characterization of Exosome-Like Vesicles Released From Human Tracheobronchial Ciliated Epithelium: A Possible Role in Innate Defense. FASEB J. 23 (6), 1858–1868. doi: 10.1096/fj.08-119131
Kumar, H., Wacklin, P., Nakphaichit, M., Loyttyniemi, E., Chowdhury, S., Shouche, Y., et al. (2015). Secretor Status Is Strongly Associated With Microbial Alterations Observed During Pregnancy. PLoS One 10 (7), e0134623. doi: 10.1371/journal.pone.0134623
Kumbhare, S. V., Kumar, H., Chowdhury, S. P., Dhotre, D. P., Endo, A., Matto, J., et al. (2017). A Cross-Sectional Comparative Study of Gut Bacterial Community of Indian and Finnish Children. Sci. Rep. 7 (1), 10555. doi: 10.1038/s41598-017-11215-y
Larson, E. D., Magno, J. P. M., Steritz, M. J., Llanes, E., Cardwell, J., Pedro, M., et al. (2019). A2ML1 and Otitis Media: Novel Variants, Differential Expression, and Relevant Pathways. Hum. Mutat. 40 (8), 1156–1171. doi: 10.1002/humu.23769
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., et al. (2013). The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 45 (6), 580–585. doi: 10.1038/ng.2653
Love, M. I., Huber, W., Anders, S. (2014). Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data With Deseq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Magalhaes, A., Rossez, Y., Robbe-Masselot, C., Maes, E., Gomes, J., Shevtsova, A., et al. (2016). Muc5ac Gastric Mucin Glycosylation is Shaped by FUT2 Activity and Functionally Impacts Helicobacter Pylori Binding. Sci. Rep. 6, 25575. doi: 10.1038/srep25575
McAuslan, B. R., Hannan, G. N., Reilly, W., Stewart, F. H. (1980). Variant Endothelial Cells. Fibronectin as a Transducer of Signals for Migration and Neovascularisation. J. Cell Physiol. 104 (2), 177–186. doi: 10.1002/jcp.1041040207
McDonald, J. A., Kelley, D. G., Broekelmann, T. J. (1982). Role of Fibronectin in Collagen Deposition: Fab’ to the Gelatin-Binding Domain of Fibronectin Inhibits Both Fibronectin and Collagen Organization in Fibroblast Extracellular Matrix. J. Cell Biol. 92 (2), 485–492. doi: 10.1083/jcb.92.2.485
McNitt, D. H., Choi, S. J., Keene, D. R., Van De Water, L., Squeglia, F., Berisio, R., et al. (2018). Surface-Exposed Loops and an Acidic Patch in the Scl1 Protein of Group A Streptococcus Enable Scl1 Binding to Wound-Associated Fibronectin. J. Biol. Chem. 293 (20), 7796–7810. doi: 10.1074/jbc.RA118.002250
Mi, H., Muruganujan, A., Ebert, D., Huang, X., Thomas, P. D. (2019). PANTHER Version 14: More Genomes, a New PANTHER GO-Slim and Improvements in Enrichment Analysis Tools. Nucleic Acids Res. 47 (D1), D419–D426. doi: 10.1093/nar/gky1038
Mittal, R., Robalino, G., Gerring, R., Chan, B., Yan, D., Grati, M., et al. (2014). Immunity Genes and Susceptibility to Otitis Media: A Comprehensive Review. J. Genet. Genomics 41 (11), 567–581. doi: 10.1016/j.jgg.2014.10.003
Monasta, L., Ronfani, L., Marchetti, F., Montico, M., Vecchi Brumatti, L., Bavcar, A., et al. (2012). Burden of Disease Caused by Otitis Media: Systematic Review and Global Estimates. PLoS One 7 (4), e36226. doi: 10.1371/journal.pone.0036226
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., McMillin, M. J., Gildersleeve, H. I., et al. (2010). Exome Sequencing Identifies MLL2 Mutations as a Cause of Kabuki Syndrome. Nat. Genet. 42 (9), 790–793. doi: 10.1038/ng.646
Paradise, J. L., Rockette, H. E., Colborn, D. K., Bernard, B. S., Smith, C. G., Kurs-Lasky, M., et al. (1997). Otitis Media in 2253 Pittsburgh-Area Infants: Prevalence and Risk Factors During the First Two Years of Life. Pediatrics 99 (3), 318–333. doi: 10.1542/peds.99.3.318
Pickrell, J. K., Berisa, T., Liu, J. Z., Segurel, L., Tung, J. Y., Hinds, D. A. (2016). Detection and Interpretation of Shared Genetic Influences on 42 Human Traits. Nat. Genet. 48 (7), 709–717. doi: 10.1038/ng.3570
Pruesse, E., Peplies, J., Glockner, F. O. (2012). SINA: Accurate High-Throughput Multiple Sequence Alignment of Ribosomal RNA Genes. Bioinformatics 28 (14), 1823–1829. doi: 10.1093/bioinformatics/bts252
Qiao, D., Skibba, M., Xu, X., Garofalo, R. P., Zhao, Y., Brasier, A. R. (2021). Paramyxovirus Replication Induces the Hexosamine Biosynthetic Pathway and Mesenchymal Transition via the IRE1alpha-XBP1s Arm of the Unfolded Protein Response. Am. J. Physiol. Lung Cell Mol. Physiol. 321 (3), L576–L594. doi: 10.1152/ajplung.00127.2021
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi: 10.1093/nar/gks1219
Rausch, P., Rehman, A., Kunzel, S., Hasler, R., Ott, S. J., Schreiber, et al. (2011). Colonic Mucosa-Associated Microbiota is Influenced by an Interaction of Crohn Disease and FUT2 (Secretor) Genotype. Proc. Natl. Acad. Sci. U. S. A. 108 (47), 19030–19035. doi: 10.1073/pnas.1106408108
Robertson, C. E., Harris, J. K., Wagner, B. D., Granger, D., Browne, K., Tatem, B., et al. (2013). Explicet: Graphical User Interface Software for Metadata-Driven Management, Analysis and Visualization of Microbiome Data. Bioinformatics 29 (23), 3100–3101. doi: 10.1093/bioinformatics/btt526
Rosenfeld, R. M., Schwartz, S. R., Pynnonen, M. A., Tunkel, D. E., Hussey, H. M., Fichera, J. S., et al. (2013). Clinical Practice Guideline: Tympanostomy Tubes in Children. Otolaryngol. Head Neck Surg. 149 (1 Suppl), S1–35. doi: 10.1177/0194599813487302
Rosenfeld, R. M., Shin, J. J., Schwartz, S. R., Coggins, R., Gagnon, L., Hackell, J. M., et al. (2016). Clinical Practice Guideline: Otitis Media With Effusion (Update). Otolaryngol. Head Neck Surg. 154 (1 Suppl), S1–S41. doi: 10.1177/0194599815623467
Ryan, A. F., Nasamran, C. A., Pak, K., Draf, C., Fisch, K. M., Webster, N., et al. (2020). Single-Cell Transcriptomes Reveal a Complex Cellular Landscape in the Middle Ear and Differential Capacities for Acute Response to Infection. Front. Genet. 11, 358. doi: 10.3389/fgene.2020.00358
Santos-Cortez, R. L. P., Chiong, C. M., Frank, D. N., Ryan, A. F., Giese, A. P. J., Bootpetch Roberts, T., et al. (2018). FUT2 Variants Confer Susceptibility to Familial Otitis Media. Am. J. Hum. Genet. 103 (5), 679–690. doi: 10.1016/j.ajhg.2018.09.010
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F., Regev, A. (2015). Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 33 (5), 495–502. doi: 10.1038/nbt.3192
Schilder, A. G., Chonmaitree, T., Cripps, A. W., Rosenfeld, R. M., Casselbrant, M. L., Haggard, M. P., et al. (2016). Otitis Media. Nat. Rev. Dis. Primers 2, 16063. doi: 10.1038/nrdp.2016.63
Simon, P. F., McCorrister, S., Hu, P., Chong, P., Silaghi, A., Westmacott, G., et al. (2015). Highly Pathogenic H5N1 and Novel H7N9 Influenza A Viruses Induce More Profound Proteomic Host Responses Than Seasonal and Pandemic H1N1 Strains. J. Proteome Res. 14 (11), 4511–4523. doi: 10.1021/acs.jproteome.5b00196
Song, J. J., Kwon, J. Y., Park, M. K., Seo, Y. R. (2013). Microarray Analysis of Gene Expression Alteration in Human Middle Ear Epithelial Cells Induced by Micro Particle. Int. J. Pediatr. Otorhinolaryngol. 77 (10), 1760–1764. doi: 10.1016/j.ijporl.2013.08.012
Stabenau, K. A., Zimmermann, M. T., Mathison, A., Zeighami, A., Samuels, T. L., Chun, R. H., et al. (2021). RNA Sequencing and Pathways Analyses of Middle Ear Epithelia From Patients With Otitis Media. Laryngoscope 131 (11), 2590–2597. doi: 10.1002/lary.29551
Suaya, J. A., Gessner, B. D., Fung, S., Vuocolo, S., Scaife, J., Swerdlow, D. L., et al. (2018). Acute Otitis Media, Antimicrobial Prescriptions, and Medical Expenses Among Children in the United States During 2011-2016. Vaccine 36 (49), 7479–7486. doi: 10.1016/j.vaccine.2018.10.060
Tian, C., Hromatka, B. S., Kiefer, A. K., Eriksson, N., Noble, S. M., Tung, J. Y., et al. (2017). Genome-Wide Association and HLA Region Fine-Mapping Studies Identify Susceptibility Loci for Multiple Common Infections. Nat. Commun. 8 (1), 599. doi: 10.1038/s41467-017-00257-5
Turpin, W., Bedrani, L., Espin-Garcia, O., Xu, W., Silverberg, M. S., Smith, M. I., et al. (2018). FUT2 Genotype and Secretory Status are Not Associated With Fecal Microbial Composition and Inferred Function in Healthy Subjects. Gut. Microbes 9 (4), 357–368. doi: 10.1080/19490976.2018.1445956
Uhlen, M., Fagerberg, L., Hallstrom, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-Based Map of the Human Proteome. Science 347 (6220), 1260419. doi: 10.1126/science.1260419
Vandepoele, K., Van Roy, N., Staes, K., Speleman, F., van Roy, F. (2005). A Novel Gene Family NBPF: Intricate Structure Generated by Gene Duplications During Primate Evolution. Mol. Biol. Evol. 22 (11), 2265–2274. doi: 10.1093/molbev/msi222
Wacklin, P., Makivuokko, H., Alakulppi, N., Nikkila, J., Tenkanen, H., Rabina, J., et al. (2011). Secretor Genotype (FUT2 Gene) is Strongly Associated With the Composition of Bifidobacteria in the Human Intestine. PLoS One 6 (5), e20113. doi: 10.1371/journal.pone.0020113
Wacklin, P., Tuimala, J., Nikkila, J., Sebastian, T., Makivuokko, H., Alakulppi, N., et al. (2014). Faecal Microbiota Composition in Adults is Associated With the FUT2 Gene Determining the Secretor Status. PLoS One 9 (4), e94863. doi: 10.1371/journal.pone.0094863
Williamson, R. E., Darrow, K. N., Michaud, S., Jacobs, J. S., Jones, M. C., Eberl, D. F., et al. (2007). Methylthioadenosine Phosphorylase (MTAP) in Hearing: Gene Disruption by Chromosomal Rearrangement in a Hearing Impaired Individual and Model Organism Analysis. Am. J. Med. Genet. A. 143A (14), 1630–1639. doi: 10.1002/ajmg.a.31724
Woods, A., Couchman, J. R., Johansson, S., Hook, M. (1986). Adhesion and Cytoskeletal Organisation of Fibroblasts in Response to Fibronectin Fragments. EMBO J. 5 (4), 665–670. doi: 10.1002/j.1460-2075.1986.tb04265.x
Xia, J., Benner, M. J., Hancock, R. E. (2014). NetworkAnalyst–integrative Approaches for Protein-Protein Interaction Network Analysis and Visual Exploration. Nucleic Acids Res. 42 (Web Server issue), W167–W174. doi: 10.1093/nar/gku443
Xia, J., Gill, E. E., Hancock, R. E. (2015). NetworkAnalyst for Statistical, Visual and Network-Based Meta-Analysis of Gene Expression Data. Nat. Protoc. 10 (6), 823–844. doi: 10.1038/nprot.2015.052
Xu, K., Chong, D. C., Rankin, S. A., Zorn, A. M., Cleaver, O. (2009). Rasip1 is Required for Endothelial Cell Motility, Angiogenesis and Vessel Formation. Dev. Biol. 329 (2), 269–279. doi: 10.1016/j.ydbio.2009.02.033
Xu, L., Earl, J., Bajorski, P., Gonzalez, E., Pichichero, M. E. (2021). Nasopharyngeal Microbiome Analyses in Otitis-Prone and Otitis-Free Children. Int. J. Pediatr. Otorhinolaryngol. 143, 110629. doi: 10.1016/j.ijporl.2021.110629
Xu, K., Sacharidou, A., Fu, S., Chong, D. C., Skaug, B., Chen, Z. J., et al. (2011). Blood Vessel Tubulogenesis Requires Rasip1 Regulation of GTPase Signaling. Dev. Cell 20 (4), 526–539. doi: 10.1016/j.devcel.2011.02.010
Yap, C. S., Jamuar, S. S., Lai, A. H. M., Tan, E. S., Ng, I., Ting, T. W., et al. (2020). Identification of KMT2D and KDM6A Variants by Targeted Sequencing From Patients With Kabuki Syndrome and Other Congenital Disorders. Gene 731, 144360. doi: 10.1016/j.gene.2020.144360
Yatsyshina, S., Mayanskiy, N., Shipulina, O., Kulichenko, T., Alyabieva, N., Katosova, L., et al. (2016). Detection of Respiratory Pathogens in Pediatric Acute Otitis Media by PCR and Comparison of Findings in the Middle Ear and Nasopharynx. Diagn. Microbiol. Infect. Dis. 85 (1), 125–130. doi: 10.1016/j.diagmicrobio.2016.02.010
Keywords: FUT2, microbiota, otitis media, p.Trp154*, RNA-sequencing, rs601338
Citation: Elling CL, Scholes MA, Streubel S-O, Larson ED, Wine TM, Bootpetch TC, Yoon PJ, Kofonow JM, Gubbels SP, Cass SP, Robertson CE, Jenkins HA, Prager JD, Frank DN, Chan KH, Friedman NR, Ryan AF and Santos-Cortez RLP (2022) The FUT2 Variant c.461G>A (p.Trp154*) Is Associated With Differentially Expressed Genes and Nasopharyngeal Microbiota Shifts in Patients With Otitis Media. Front. Cell. Infect. Microbiol. 11:798246. doi: 10.3389/fcimb.2021.798246
Received: 19 October 2021; Accepted: 20 December 2021;
Published: 14 January 2022.
Edited by:
Kevin Mason, Nationwide Children’s Hospital, United StatesReviewed by:
Kenneth L. Brockman, Medical College of Wisconsin, United StatesCopyright © 2022 Elling, Scholes, Streubel, Larson, Wine, Bootpetch, Yoon, Kofonow, Gubbels, Cass, Robertson, Jenkins, Prager, Frank, Chan, Friedman, Ryan and Santos-Cortez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Regie Lyn P. Santos-Cortez, cmVnaWUuc2FudG9zLWNvcnRlekBjdWFuc2NodXR6LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.