 Xiaoyao Peng
Xiaoyao Peng Zhixuan Luo1
Zhixuan Luo1 Luhua Zhang
Luhua Zhang Ying Li
Ying Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 04 November 2021
Sec. Molecular Bacterial Pathogenesis
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.768108
As a complex multicellular structure of the vascular system at the central nervous system (CNS), the blood-brain barrier (BBB) separates the CNS from the system circulation and regulates the influx and efflux of substances to maintain the steady-state environment of the CNS. Lipopolysaccharide (LPS), the cell wall component of Gram-negative bacteria, can damage the barrier function of BBB and further promote the occurrence and development of sepsis-associated encephalopathy (SAE). Here, we conduct a literature review of the direct and indirect damage mechanisms of LPS to BBB and the relationship between these processes and SAE. We believe that after LPS destroys BBB, a large number of inflammatory factors and neurotoxins will enter and damage the brain tissue, which will activate brain immune cells to mediate inflammatory response and in turn further destroys BBB. This vicious circle will ultimately lead to the progression of SAE. Finally, we present a succinct overview of the treatment of SAE by restoring the BBB barrier function and summarize novel opportunities in controlling the progression of SAE by targeting the BBB.
All organisms with a well-developed central nervous system (CNS) have a blood-brain barrier (BBB). The CNS has always been in a dynamic equilibrium environment that is significantly different from the peripheral environment and this is mainly determined by the barrier function of the BBB (Obermeier et al., 2013). The BBB is mainly composed of vascular endothelial cells (ECs) and basement membrane (BM), and there are also pericytes and astrocyte end-feet covering them, which together constitute the neurovascular unit (NVU) to maintain the function of BBB (Armulik et al., 2010; Yao et al., 2014; Kutuzov et al., 2018; Nikolakopoulou et al., 2019; Huang et al., 2020b; Kaplan et al., 2020; Yue et al., 2020).
In addition, neurons and microglial cells, as components of the NVU, can also directly or indirectly affect the development and functions of the BBB (da Fonseca et al., 2014; Xu et al., 2017; Haruwaka et al., 2019; Su et al., 2020; Zhang et al., 2020). As a protective barrier at the interface of the brain and the peripheral environment, BBB successfully shields the CNS from toxic substances circulating in the blood by strictly controlling the material exchange between blood and brain tissue (Liebner et al., 2018). And, as a dynamic interface, BBB’s structure is often damaged in pathological conditions, leading to the disruption of barrier function, which is mainly manifested in the increased permeability of both transcellular and paracellular pathways. Lipopolysaccharide (LPS), also known as endotoxin, which is the cell wall component of Gram-negative bacteria, is the main cause of sepsis (Opal, 2010; Yan et al., 2016; Berger et al., 2017; Hao et al., 2017; Skirecki and Cavaillon, 2019). Purified LPS is widely used to construct pathological models of sepsis and its complications (Kingsley and Bhat, 2016; Varatharaj and Galea, 2017). Intensive researches have demonstrated that LPS could destroy the BBB through a variety of mechanisms, and the damaged BBB would promote the development of multiple brain diseases, including the sepsis-associated encephalopathy (SAE) (Sweeney et al., 2019). In recent years, SAE caused by the disrupted BBB has attracted extensive attention, in which a CNS often has no obvious infection, but patients have significant cognitive dysfunction, memory decline, and other symptoms of serious complications (Iwashyna et al., 2010; Golzari and Mahmoodpoor, 2014; Varatharaj and Galea, 2017; Kikuchi et al., 2019).
In view of the fact that LPS can damage the BBB to cause serious diseases, here we review the relevant literature in recent years to elaborate the specific mechanism of LPS on BBB damage from two aspects of direct and indirect effects, and to discuss the links between BBB damaging by LPS and SAE, and the possible means to treat SAE by targeting the BBB.
Controlling substances and cells into and out of CNS is the most important function of BBB, especially preventing toxins and pathogens from entering the brain. It guarantees the relative stability of CNS under different conditions, which is conferred by the low permeability of transcellular pathway and paracellular pathway of BBB. The low permeability of paracellular pathways is mainly determined by tight junctions (TJs), which mainly consist of densely distributed transmembrane protein claudin (especially claudin-5), occluding, tricellulins, junctional adhesion molecules, and intracellular support proteins such as zona occludens (ZO) (Haseloff et al., 2015; Tietz and Engelhardt, 2015). TJs are mostly located between brain ECs, sealing the paracellular pathway of molecules, greatly reducing the permeation of polar solutes from plasma to extracellular fluid of the brain, which play an important role in maintaining the integrity of BBB (Sivandzade et al., 2020; Yu et al., 2020; Winkler et al., 2021). In addition, adherens junctions (AJs) formed by intercellular cadherin (e.g., vascular-endothelial (VE-) cadherin) and intracellular catenin (e.g., epithelial (E-)cadherin), which connect cells together, are the basis of TJs and are also involved in maintaining the barrier function of BBB (Blecharz-Lang et al., 2018; Yang et al., 2018).
LPS has been demonstrated in several studies to enhance BBB permeability by killing TJs and AJs, decreasing their expression, and altering their distribution, allowing hazardous chemicals to pass across the BBB and induce CNS dysfunction (Cheng et al., 2018). Seok et al. have confirmed that LPS can reduce the amount of TJs proteins, ZO-1 and claudin-5, and change their cellular localization by transferring them from the cell membrane to the cytoplasm after 14 days’ treatment (Veszelka et al., 2007; Seok et al., 2013; Banks et al., 2015). LPS produced by Bacteroides fragilis has been found to damage the BBB by disrupting intercellular adhesion proteins and cleaving cadherin (Alexandrov et al., 2020). Besides, LPS of Escherichia coli O111:B4 treatment directly cause the formation of the paracellular gap by increasing the expression of C-X-C Motif Chemokine Receptor 2 (CXCR2) on brain ECs in a time-dependent manner (4-24 h post LPS injection), which was involved in endothelial actin polymerization and actin stress fiber formation (Wu et al., 2020). Another important factor involved in LPS-mediated damage to TJs is matrix metalloproteinase (MMP), especially MMP-9 (Dal-Pizzol et al., 2013), which participates in the degradation of TJs and thereby elevates BBB permeability (Ren et al., 2015). MMP is regulated by a variety of cytokines on the mitogen-activated protein kinase (MAPK) pathway without overexpression in normal brain tissues (Hong et al., 2005; Malinsky et al., 2013; Yi et al., 2020). Qin et al. found that LPS could increase phosphorylation of p38MAPK and inhibition of p38MAPK could significantly inhibit LPS-induced MMP-2 overexpression at both the mRNA and protein levels, thereby attenuating the effect of LPS on occluding (Qin et al., 2015) (Figure 1). It is worth mentioning that c-Jun amino-terminal kinase (JNK) in the MAPK family plays a similar function to p38MAPK (Qin et al., 2015). This suggests that the MAPK family may be involved in the process that LPS disrupts TJs (Qin et al., 2015).
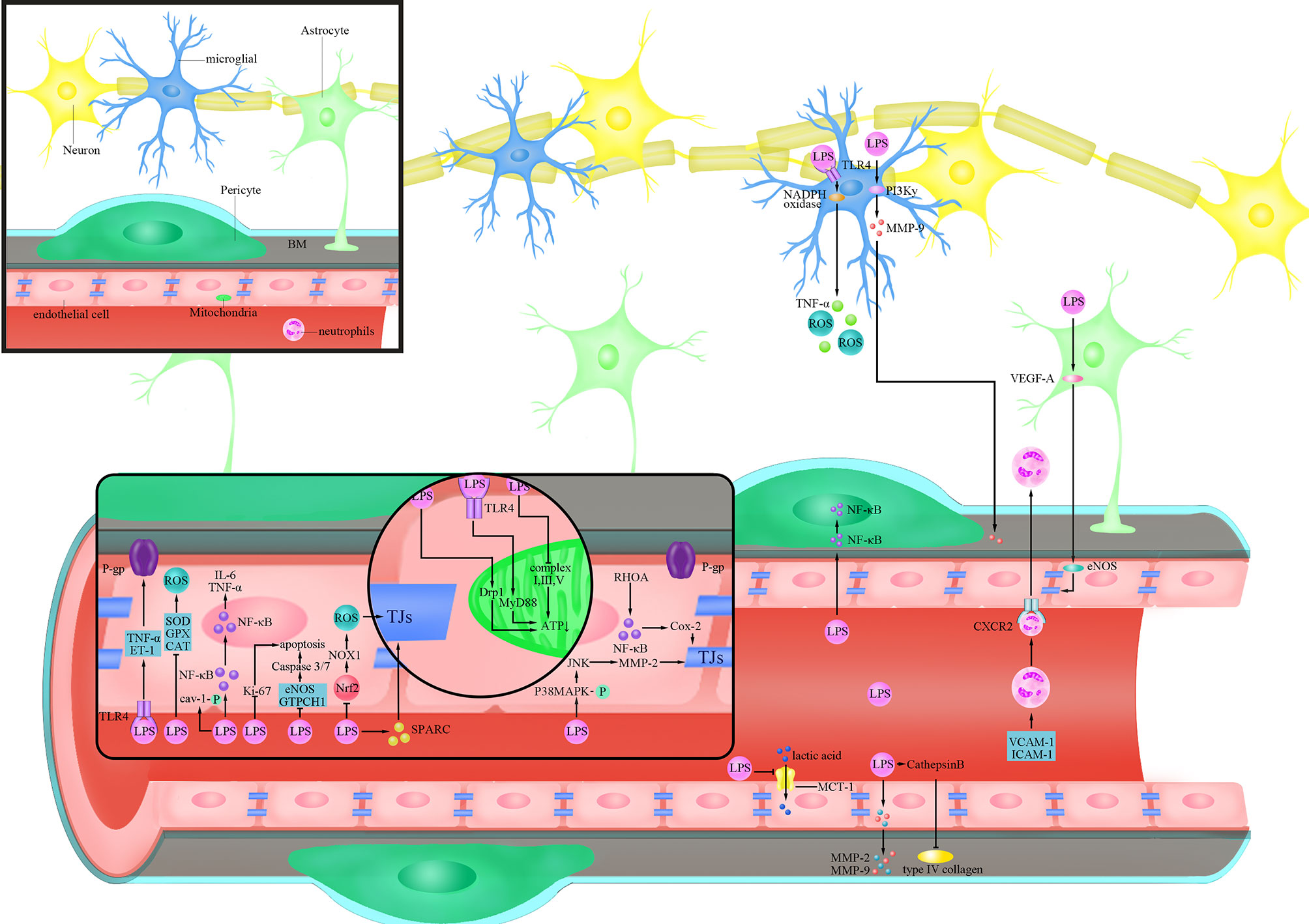
Figure 1 Cellular responses of BBB evoked by LPS. lipopolysaccharide (LPS) activates Ras homolog family member A (RHOA) and Nuclear factor-k-gene binding (NF-κB), and destroys the tight junctions (TJs) protein by triggering the production of cyclooxygenase-2 (Cox-2); LPS inhibits the expression of Nuclear factor-erythroid 2-related factor 2 (Nrf2), thereby increasing the expression of Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)1 and destroying the TJs protein;LPS increases the phosphorylation of p38 mitogen-activated protein kinase (MAPK), activates c-Jun amino-terminal kinase (JNK), triggers the expression of matrix metalloproteinase (MMP)-2, and destroys the TJs protein; LPS reduces the expression of TJs protein by inducing secreted protein acidic and rich in cysteine (SPARC); LPS activates Tumour Necrosis Factor-α (TNF-α) and endothelin (ET)-1 after binding to Toll-like receptor 4 (TLR4), and reduces the activity of P-glycoprotein(P-gp); LPS increases the phosphorylation of caveolin-1(cav-1); LPS inhibits endothelial nitric oxide synthase (eNOS) and Guanosine triposphate cyclohydrolase 1 (GTPCH1) and increases the activity of Caspase3/7 to trigger cell apoptosis; LPS trigger cell apoptosis by inhibiting the expression of Ki-67; LPS activates dynamin-related protein-1(Drp1) or activates myeloid differentiation factor 88 (MyD88) after binding to TLR4 to reduce ATP produced by mitochondria; LPS inhibits the production of ATP by inhibiting complex I, III and IV LPS inhibits the expression of Superoxide Dismutase (SOD), glutathione peroxidase (GPX) and catalase (CAT) to increase the level of reactive oxygen species (ROS); LPS trigger the translocation of NF-κB into the nucleus to promote the secretion of inflammatory factors such as Interleukin(IL)-6 and TNF-α; LPS inhibits the expression of Monocarboxylate transporter-1(MCT-1) and inhibits lactic acid influx; LPS trigger the secretion of MMP-2/9 in endothelial cells and directly degrades extracellular matrix (ECM) and digest basement membrane (BM); LPS increases the activity of Cathepsin B and cleave type IV collagen; LPS triggers the translocation of NF-κB to the nucleus in pericytes; LPS trigger the increase of the levels of Vascular cell adhesion molecule-1(VCAM-1) and Intercellular adhesion molecule-1(ICAM-1) in blood vessels, as well as the combination of neutrophils and C-X-C Motif Chemokine Receptor 2 (CXCR2) to trigger the infiltration of the brain; LPS trigger the secretion of vascular endothelial growth factor A (VEGF-A) in astrocytes and destroys TJs protein by activating eNOS; LPS binds to TLR4 on microglia and trigger the production of TNF-α and ROS through the NOX pathway; LPS produces MMP-9 through the phosphoinositide 3-kinase (PI3K)γ pathway and acts on the BM.
Nuclear factor-k-gene binding (NF-κB) is a key regulator of inflammation-related gene expression in human cells, and LPS can stimulate NF-κB expression by binding to Toll-like receptor 4 (TLR4). (Tiruppathi et al., 2008; Verstrepen et al., 2008; Xu et al., 2021). Hu et al. found that LPS can destroy BBB by reducing the level of occludin through TLR4/NF-κB pathway (Hu et al., 2017). Ras homolog family member A(RHOA), which induces intracellular inflammatory signal transduction and acts as a regulator of the integrity of intercellular junctions, has been found to be involved in promoting disassembly of TJs (Forster, 2008; Yamamoto et al., 2008). Studies have found that LPS of E. coli O55:B5 can activate RHOA as well as downstream NF-κB, and lead to increased phosphorylation of myosin light chain (MLC), which ultimately causes a decrease in the expression of TJs proteins, claudin-5 and ZO-1 (He et al., 2011) (Figure 1). In addition, studies found that the NF-κβ/cyclooxygenase-2 (Cox-2) pathway is involved in the process of BBB damage by LPS of E. coli O111:B4, and inhibition of Cox-2 prevented the LPS-induced decrease in Transendothelial electrical resistance (TEER) and improved the amount of ZO-1, showing a decrease in BBB permeability (Kikuchi et al., 2019) (Figure 1). Therefore, NF-κB and its associated molecules play an important role in the process of TJs damage in inflammatory responses mediated by LPS, ultimately disrupting BBB function.
Oxidative stress, which is mainly manifested by excessive production of reactive oxygen species (ROS), occurs widely when the balance between oxidation and antioxidation is broken (Tarafdar and Pula, 2018). Treatment of human brain microvascular endothelial cells (BMECs) with LPS of E. coli O111:B4 leads to an abnormal increase in ROS, which further caused a significant decrease in occludin and claudin-5 after 8 hours’ treatment, and a time-dependent decrease in TEER, indicating the damage of BBB (Zhao et al., 2014). In addition to causing direct damage to TJs, ROS induced by LPS of E. coli O111:B4 can also activate other molecular pathways such as RHOA/phosphoinositide 3-kinase (PI3K)/protein kinase B(AKT) to reduce the expression of occludin and claudin-5, and cause the reduction of TEER (Schreibelt et al., 2007). Studies have proved that the phosphorylation of MAPK can be inhibited when the ROS production induced by LPS of E. coli O111:B4 is inhibited, thus decreasing the phosphorylation of TJs and protecting the integrity of BBB (Deli et al., 2005; Cardoso et al., 2010; Seok et al., 2013). Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is an important source of ROS and is demonstrated to participate in the process of BBB dysfunction (Chrissobolis and Faraci, 2008; Ma et al., 2017). In the study of Zhao et al., LPS of E. coli O111:B4 treatment increased the activity of NADPH oxidase (NOX) in a time-dependent manner (0-24 hours post LPS treatment), and the accumulation of ROS induced by LPS was significantly reduced after inhibiting the activity of NOX (Zhao et al., 2014). Exogenous antioxidants such as Superoxide Dismutase(SOD) and 4,4’-Diaminodiphenylsulfone, Dapsone (DDS) were found to reduce the damage of LPS to BBB by preventing LPS-induced ROS accumulation, thereby restoring the expression of TJ proteins to maintain the low permeability of BBB (Miller et al., 2009; Cho et al., 2010; Zhou et al., 2014).In view of this, activating endogenous antioxidant molecules inhibited by LPS may be a better solution to attenuate the damage of LPS to BBB. As a transcription factor that regulates the expression of multiple antioxidant genes, Nuclear factor-erythroid 2-related factor 2 (Nrf2) deficiency was found to cause an increase in intracellular ROS levels (Hyeon et al., 2013). Li et al. found that LPS treatment led to an increase production of ROS in both in vivo and in vitro experiments by inhibiting the expression of Nrf2, which was accompanied by decreased TEER and ZO-1, two indicators for a breakdown of the BBB (Li et al., 2018) (Figure 1). After artificially silencing the expression of Nrf2, a significant increase in ROS related to the enhanced expression of NOX1 can be observed, and this effect was amplified by the presence of LPS (Li et al., 2018) (Figure 1). Researchers have attempted to activate AMPK in both genetic and non-genetic ways since long-term exposure to E. coli O111:B4 LPS was shown to cause a reduction in its activity (Zhao et al., 2014; Taile et al., 2020). The results showed that activation of AMPK significantly reduced LPS-induced NOX and ROS production after 24 hours’ treatment, ultimately improving the expression of occludin and claudin-5 (Zhao et al., 2014). Activating AMPK with metformin by Takata et al. showed reduced permeability of BBB, which manifests itself specifically as an increase in TEER and a decrease in Evans blue albumin permeability (Takata et al., 2013). Therefore, LPS can dysregulate the oxidative and antioxidant responses in ECs by inducing the expression of ROS, which ultimately leads to damage of TJs and disruption of BBB.
As a cysteine rich acidic secreted protein, secreted protein acidic and rich in cysteine (SPARC) is highly expressed not only at the early stage of cerebrovascular development, but also at sites such as injury and tumors, and it is related to the angiogenesis and the integrity of BBB (Vincent et al., 2008). Addition of exogenous LPS caused an increase in SPARC, which in turn led to a decrease in the expression of ZO-1 and occluding, and finally a rise in BBB permeability at 24 h in a concentration-dependent manner (Alkabie et al., 2016). Researchers believe that SPARC’s primary function after stimulated by LPS is to promote ECs proliferation at the damaged site and to protect BBB, but SPARC also reduces the expression of ZO-1 and occluding, and eventually increases BBB permeability (Bradshaw and Sage, 2001; Endl et al., 2001; Chlenski and Cohn, 2010; Alkabie et al., 2016) (Figure 1).
Concerning the transcellular transport of substances by the BBB, it is generally accepted that there are four pathways to transport ions and molecules between the blood and the brain (Daneman and Prat, 2015; Langen et al., 2019): i) Free diffusion of lipid-soluble substances and gas small molecules; ii) polar molecules that cannot diffuse freely are transported by solute carriers (Winkler et al., 2015; Chien et al., 2018); iii) A variety of lipid-soluble substances can be selectively transported by ATP-binding cassette transporters (ABCs) (Bauer et al., 2019); iv) Vesicular transport of macromolecules, also known as transcytosis. Notably, vesicular transport in ECs at the BBB was slower than that in most peripheral ECs, which may be due to inhibition of caveolae-mediated transcytosis (Andreone et al., 2017; Sadeghian et al., 2018). Since the first two pathways is mainly related to the concentration difference of intracellular and extracellular solutes rather than a regulatory role of the cell itself, the low permeability of BBB is achieved primarily by the efflux effects of ABCs and the inefficient vesicle transport.
P-glycoprotein (P-gp), an important efflux transporter belonging to ABCs (Linville et al., 2019). Reduced expression and activity of P-gp may cause an accumulation of toxic metabolites in the CNS, leading to an increased risk of cerebral diseases. Studies showed that LPS of E. coli O111:B4 can disrupt BBB permeability by affecting the activity of P-gp, causing various neurotoxins and pathogenic microorganisms to fail to drain out of the brain (Veszelka et al., 2007; Dohgu and Banks, 2008; Cardoso et al., 2012). In the study of Hartz et al., [N-ϵ(4-nitrobenzofurazan-7-yl)-d-Lys8]-cyclosporin A, a substance that accurately and specifically reflects the activity of P-gp in the vascular lumen, was declined in a concentration-dependent way after brain capillaries were treated with LPS, indicating a decrease in the activity of P-gp upon LPS treatment (Hartz et al., 2006). Further studies showed that LPS activates molecules such as tumor necrosis factor-α (TNF-α), endothelin (ET)-1 by binding to TLR4 on the EC membrane, which ultimately causes a rapid and reversible decline in P-gp activity (Bauer et al., 2004; Hartz et al., 2006) (Figure 1). In the study of Salkeni et al., they excluded the roles of blood circulating factors in the actions of LPS of Salmonella typhimurium on the activity of P-gp by intraperitoneal perfusion of CD-1 mice, and their results also showed that LPS inhibited the activity of P-gp independent of prostaglandins or nitric oxide (NO) (Salkeni et al., 2009). It is worth noting that LPS reduced P-gp activity by directly activating nitric oxide synthase (NOS) by Hartz et al., which is inconsistent with the study by Salkeni et al., and the possible reason may be due to the involvement of microglia in the latter study (Hartz et al., 2006; Salkeni et al., 2009). Pan et al. showed that LPS of S. typhimurium regulated NF-κB to elevate P-gp expression after 48 hours’ treatment, while the efflux activity of P-gp decreased, so the regulation of P-gp by LPS was most likely to be achieved by post-translational modifications (Hartz et al., 2006; Pan et al., 2010).
In addition to ABCs, caveolae-associated transcytosis also plays an important role in maintaining low permeability in the transcellular pathway of the BBB. Caveolin-1 (cav-1) is a major component of caveolae and its expression level or activity would have a significant impact on transcellular permeability (Keaney and Campbell, 2015; Liu et al., 2015; Linden et al., 2019). Wang et al. found that LPS had no obvious effect on the expression of cav-1, but would increase the phosphorylation degree of cav-1 after treatment for 4 and 24 hours, and inhibition of cav-1 phosphorylation with drugs was accompanied by a decrease in BBB permeability (Wang et al., 2019a) (Figure 1). Increased phosphorylation of cav-1upon LPS treatment may have led to structural changes of caveolae, ultimately elevating the permeability of BBB. Collectively, LPS affects the permeability of the BBB transcellular pathway mainly through regulating of P-gp and cav-1.
Vascular ECs constitute the main cellular components of the BBB, and structural damage to the endothelium or a decrease in the number of ECs may elevate the permeability of the BBB (Tam and Watts, 2010; Banks et al., 2015). Studies showed that the apoptosis of ECs is closely related to BBB damage and cerebral microcirculation disturbance, so the effect of LPS on the apoptosis of ECs of BBB may refine the mechanism of damage to the BBB by LPS (Yamazaki et al., 2019). Cardoso et al. found that LPS of E. coli O111:B4 treatment causes time-dependent apoptosis of mouse BMECs (Cardoso et al., 2012). A similar result was also reported by Li et al., who found that LPS could increase the apoptosis of bend.3 cells and decrease their activity after 24 hours’ treatment (Li et al., 2020). To further clarify the underlying mechanism of cell apoptosis induced by LPS, apoptosis analysis of human microvascular endothelial cell(HCMEC/D3) treated by LPS was performed by Liu et al (Liu et al., 2020). They showed that as the concentration of LPS increased, the activity of Caspase3/7 rose, the expression of the pro-oncogene Bax increased and the tumor suppressor gene Bcl-2 decreased (Liu et al., 2020) (Figure 1). They further found that the effects of LPS on HCMECs could be reversed by enhancing the expression of endothelial nitric oxide synthase (eNOS) and Guanosine triphosphate cyclohydrolase 1 (GTPCH1), two proteins that are involved in ECs protection, indicating that LPS may damage the function of BBB by promoting EC apoptosis via inhibiting eNOS and GTPCH1 (Zheng et al., 2018; Liu et al., 2020) (Figure 1). In addition, Boitsova et al. found that the expression of Ki-67 protein, which is associated with cell proliferation, significantly decreased in rat BMECs after treated the LPS of E. coli O111:B4 for 72 hours (Boitsova et al., 2018) (Figure 1). These results indicated that LPS can promote apoptosis and reduce the proliferation of EC, and ultimately destroy the structure and disrupt the function of BBB.
Mitochondria are the center of cellular energy metabolism. Abnormal energy metabolism of mitochondria in ECs of the BBB is usually followed by bioenergetics disruption of ECs, which may finally cause the BBB dysfunction (Alelwani et al., 2020). Haileselassie et al. found that mitochondrial damage in ECs and subsequent disruption of the BBB were caused by reduced mitochondrial membrane potential and increased oxidative stress after LPS treatment (Haileselassie et al., 2020). They also showed that the permeability of microvascular ECs/astrocytes co-culture system increased upon LPS treatment for 24 hours, and this phenomenon resulted from pathological activation of dynamin-related protein-1 (Drp1) (Haileselassie et al., 2020)(Figure 1). Inhibition of Drp1 with P110, an inhibitor of Drp1- Fission 1 (Fis1) interaction, was found to attenuate LPS induced oxidative stress as well as elevate the membrane potential of mitochondria, which consequently improved the integrity of BBB (Haileselassie et al., 2020). Besides, the injection of LPS resulted in a decline in the mitochondrial ATP as well as a rise in ROS levels in the brain, which might be attributed to LPS of E. coli O55:B5 interfering with mitochondrial transcription as well as oxidative phosphorylation (Suliman et al., 2003). Monocarboxylate transporter-1 (MCT-1), as a lactate receptor expressed at luminal and abluminal membranes, carries on the influx and efflux of lactate between cells and contributes to the effective metabolic coupling of NVU cells (Lottes et al., 2015). Boitsova et al. found that LPS of E. coli O111:B4 treatment inhibited the expression of MCT-1 in BMECs after 24 hours’ treatment, which consequently leads to insufficient uptake of lactate by cells (Boitsova et al., 2018) (Figure 1). Thereby, the mitochondrial dynamic effects mediated by lactate are later suppressed, and then the metabolic state of mitochondria, especially energy production, was disrupted. As lactate transport is closely associated with proliferation activity of BMECs and highly selected permeability of the BBB, it was found that cell proliferation activity decreased while BBB barrier function was impaired in the presence of LPS (Stapor et al., 2014; Tang et al., 2014).
To explore the roles of LPS in oxidative phosphorylation and mitochondrial volume in ECs, cultured cerebrovascular endothelial cells (cCVECs) were used as an in vitro BBB model by Doll et al. (2015). They found that the expression of myeloid differentiation factor 88 (MyD88), an adaptor protein involved in TLR4 signaling, increased in cCVECs upon LPS of E. coli O55:B5 stimulation, and it was clearly colocalized with mitochondria, suggesting that LPS could affect mitochondrial function directly through TLR4 mediated signaling (Doll et al., 2015; Fritz et al., 2018) (Figure 1). They further analyzed the effect of LPS of E. coli O55:B5 on mitochondrial energy metabolism by detecting cellular energy consumption oxygen. Results showed that no significant change in basal oxygen consumption was observed, but the maximal respiration and spare capacity in cCVECs decreased significantly, which were not related to the decrease of cell number or cell viability (Doll et al., 2015; Lou et al., 2016). These results demonstrate that the impairment of mitochondrial energy metabolism in ECs occurs after LPS treatment (Doll et al., 2015; Lou et al., 2016). Considering that it is the respiratory chain complex proteins that play a decisive role in mitochondrial respiration, the expression of the respiratory chain complexes was examined after LPS treatment by Doll et al (Doll et al., 2015; Chen et al., 2018; Tsai et al., 2020). They demonstrated that the expression of complex I, complex III, and complex IV decreased after LPS of E. coli O55:B5 treatment, suggesting that LPS inhibits oxidative phosphorylation of CVECs (Doll et al., 2015) (Figure 1). Specifically inhibiting complex I or V caused a rapid rise in the permeability of cCVEC and a disruption of linear cell-cell junctions in both in vivo and in vitro experiments (Doll et al., 2015). Therefore, mitochondria in ECs plays a key role in maintaining BBB integrity, and LPS can damage the barrier function of the BBB by inhibiting mitochondria.
The oxidative stress and inflammation in ECs induced by LPS can damage BBB (Zhang et al., 2010; Zhao et al., 2015). Li et al. found that the expression of three antioxidant enzymes, glutathione peroxidase (GPX), catalase (CAT), and SOD were inhibited in Bend.3 cells after LPS treatment for 24 hours, which resulted in an increased level of ROS (Ceretta et al., 2012; Li et al., 2020) (Figure 1). In addition, reduced glutathione/oxidized glutathione (GSH/GSSG), an indicator of cellular antioxidant capacity, was similarly inhibited in Bend.3 cells by LPS after 24 hours’ treatment (Buendia et al., 2016; Lu et al., 2016; Ugun-Klusek et al., 2017; Zheng et al., 2021). Li et al. also showed that the activation of Nrf2 elevated the expression of antioxidant enzymes GPX, CAT, SOD as well as GSH/GSSG, which alleviated the BBB dysfunction induced by LPS (Li et al., 2020). These results suggest that inhibition of the antioxidant capacity of ECs leading to a rise in the level of ROS is an important mechanism that LPS causes BBB damage. In addition, mouse BMECs were also found to produce inflammatory responses upon LPS stimulation, specifically by directly producing inflammatory factors such as interleukin (IL)-1β, IL-18, IL-6, and TNF-α, and these factors are able to mediate the damaging effects of LPS on the BBB (Smyth et al., 2018; Li et al., 2020). As oxidative stress and the inflammatory response are frequently inextricably linked, occurring concurrently in response to LPS stimulation and interacting with each other, both aspects need to be taken into consideration to protect ECs and BBB (Zhao et al., 2011; Li et al., 2020).
Based on the above studies we believe that LPS can disturb the functional status of the BBB by directly affecting processes such as proliferation or apoptosis of ECs, mitochondrial energy metabolism, inflammatory responses, as well as oxidative stress.
The composition of BBB, including vascular ECs, pericytes, and the foot processes of astrocytes, are all attached to the BM, which is composed of extracellular matrix (ECM) (Cardoso et al., 2010). Once the BM is disrupted, the function of the BBB will also be impaired. MMP can directly degrade the ECM as well as digest the BM (Cardoso et al., 2010). Studies found an increased expression of MMP upon LPS treatment, which was accompanied by increased permeability of BBB (Tilling et al., 1998; Yang et al., 2007; Vanlaere and Libert, 2009; Cardoso et al., 2010; Coisne and Engelhardt, 2011; Lee et al., 2011; Zhao et al., 2011; Qin et al., 2015).
Dal-Pizzol et al. used cecal ligation and puncture (CLP, a widely used animal model of sepsis without direct injection of LPS), which allows intestinal bacteria to invade the body cavity to establish a sepsis model and found that the increased level of LPS in the blood was accompanied by elevated BBB permeability (Dal-Pizzol et al., 2013). They further showed that the elevated permeability of BBB was most likely to be related to the increased contents and activity of MMP-2 and MMP-9 in the presence of LPS, as the injection of inhibitors of MMP-2 and MMP-9 reduced BBB permeability in model mice (Dal-Pizzol et al., 2013). Cardoso et al. also confirmed that the expression of MMP-2 and MMP-9 in rat BMECs was significantly increased after LPS of E. coli O111:B4 treatment for 4 hours, and results of sodium fluorescein and TEER assays showed obviously disrupted barrier function of BMECs (Cardoso et al., 2012) (Figure 1). Inhibition of MMP-9 was also found to protect the microvascular BM in the presence of LPS in a study by Wang et al (Wang et al., 2019a). In addition, they discovered that in response to LPS, the activity of cathepsin B, which can cleave type IV collagen, an important component of BM, was significantly increased in plasma and brain tissue, which is consistent with the finding that the distribution of type IV collagen in brain ECs became discontinuous after LPS stimulation by confocal microscopy (Shoji et al., 2014; Daneman and Prat, 2015; Wang et al., 2019a)(Figure 1). Pericytes, an essential component of the NVU, wrap around the surface of ECs and embed in BM (Sweeney et al., 2016). Nishioku et al. found that rupture of BM occurred after LPS of E. coli O55:B5 treatment and that pericytes are detached from the rupture of BM, which were accompanied by an increase of the permeability of BBB at 6 and 24 hours (Nishioku et al., 2009). Collectively, the integrity of BM is essential for maintaining the barrier function of BBB and the damage to the BM by LPS would lead to the destruction of BBB.
As an integral part of the NVU in the BBB, microglia play an important role in maintaining the integrity of BBB (da Fonseca et al., 2014; Liebner et al., 2018). Activating microglia surround blood vessels is a protective response that prevents more pathogens from entering the brain tissue (Bowyer et al., 2020). Sumi et al. found that a small dose of LPS of E. coli O55:B5 could activate most of microglia in the rat brain microvascular endothelial cell (RBEC)/microglial co-culture system at 6 hours, which appeared as a morphological change from round to bipolar rod shape with enlarged cytoplasm and cell bodies, and an enhanced ionized calcium binding adapter molecule 1 (Iba1) staining (Nishioku et al., 2009; Sumi et al., 2010). The activation of microglia by LPS of E. coli O55:B5 resulted in reduced RBEC permeability, and the degree of this effect was closely related to the amount as well as the degree of activation of microglia (Nayak et al., 2010). Further immunofluorescence staining showed that due to the activation of microglia, the immunostaining activity of ZO-1, claudin-5, occludin, and other TJ proteins in the intercellular borders became fragmented, and the above dysfunction could be alleviated by NOX inhibitor (Sumi et al., 2010). Considering that NOX is an important source of ROS, and LPS can promote the production of inflammatory factors by binding to TLR4, researchers believe that LPS in microglia destroys the structure and function of BBB by generating ROS through the TLR4-NOX signaling pathway (Chow et al., 1999; Akira and Takeda, 2004; Sumi et al., 2010; Anthoney et al., 2018) (Figure 1). Also, Nishioku et al. demonstrated the disruption of BBB by TNF-α only in the presence of microglia (Nishioku et al., 2010) (Figure 1). Bowyer et al. found that the microglia can be activated by LPS, which were particularly manifested as a mass migration to blood vessels (Bowyer et al., 2020). This study revealed that LPS activates microglia not only by directly binding to receptors on them, but also by released cytokines by damaging blood vessels.
We previously described the mechanisms that LPS enhance BBB permeability by altering P-gp activity in ECs, and some researchers believe that vascular ECs and microglia are also involved in these processes. Matsumoto et al. used the accumulation of rhodamine 123 in cells to assess the function and activity of P-gp, and discovered that LPS of E. coli O55:B5 may decrease P-gp activity in brain ECs in a microglia-dependent way after 6 hours’ treatment (Matsumoto et al., 2012). They further proved that LPS can reduce the activity of P-gp by activating NOX in microglia to produce inflammatory factors such as TNF-α (Matsumoto et al., 2012; Carey et al., 2020). PI3K-γ is a key mediator of LPS induced activation and phagocytosis of microglia. Frister et al. found that in microglia, LPS of E. coli O55:B5 increased the expression of MMP in a PI3K-γ-dependent pathway, thereby increasing the permeability of the BBB (Frister et al., 2014) (Figure 1). Therefore, like ECs, microglia can also produce MMP to destroy the BM and open BBB (Yadollah et al., 2003). Taken together, microglia participate in damaging the barrier function of BBB in response to LPS through inflammatory cascade amplification and oxidative stress (Desai et al., 2007; Gao and Hong, 2008).
As an integral part of the NVU of the BBB, astrocytes attach to capillaries of the CNS as end feet, and secrete various trophic factors and ECM proteins (Kaplan et al., 2020). In response to stimulation with LPS, astrocytes are activated to secrete cytokines, which are followed by activation of microglia, triggering a more drastic inflammatory response (Tarassishin et al., 2014; Vutukuri et al., 2018; Bowyer et al., 2020; Silva et al., 2020). Park et al. found that after male Sprague-Dawley rats were injected with LPS, the filaments and terminal feet of astrocytes were significantly sparse at 12 hours and 7 days, and these changes were accompanied by increased vascular permeability, which was assessed by infusing the vessel and then visualizing the concentration of extravasated vessels, suggesting that the integrity of BBB was disrupted (Park et al., 2015). Studies have further shown that LPS elevates the expression of vascular endothelial growth factor A (VEGF-A) by astrocytes, followed by activating eNOS, inhibiting the expression of claudin-5 and occluding, and ultimately disrupting the barrier function of the BBB (Argaw et al., 2009; Argaw et al., 2012; Park et al., 2015) (Figure 1). Therefore, maintaining the normal morphology and function of astrocytes is an important way to preserve BBB function.
It is now widely recognized that pericytes are spatially the closest cells to brain ECs, and the roles of pericytes in the process of LPS damaging BBB have attracted much attention (Bonkowski et al., 2011; Mae et al., 2011). Vutukuri et al. found that the activation of pericytes was observed in whole male C57BL/6 mice brain tissue after LPS of E. coli O55:B5 treatment for 4 hours, which was accompanied by an increase in the levels of TNF-α and IL-1β, and in the permeability of BBB (Vutukuri et al., 2018). Smyth et al. found translocation of NF-κB into the nucleus in pericytes after LPS of E. coli O26:B6 treatment for 1 hour, indicating an activated inflammatory response in pericytes in the presence of LPS (Smyth et al., 2018) (Figure 1). Further studies have found that the inflammatory response mediated by signal transducer and activator of transcription 1(STAT1), and similar to others against dodecapentaplegic-2/3(SMAD2/3) is also very similar in pericytes and ECs (Smyth et al., 2018). These results suggest that inflammatory responses mediated by pericytes and ECs have a concerted response (Smyth et al., 2018). Kovac et al. found that LPS of S. typhimurium can stimulate pericytes to produce many chemokines and pro-inflammatory factors (Kovac et al., 2011). Notably, IL-9 generated by pericytes in response to LPS of S. typhimurium can further stimulate astrocytes to produce chemokine-ligand-20 (CCL-20), which promotes T lymphocyte infiltration into the CNS and amplifies the inflammatory response in vivo (Zhou et al., 2011). Pericytes may therefore play a role in the inflammatory response in the brain tissue upon LPS stimulation to disrupt the barrier function of BBB.
The BBB prevents peripheral immune cells from entering the brain tissue, but when the BBB barrier function is broken, they can penetrate the brain tissue, highlighting that the systemic inflammatory response may compromise the BBB’s barrier function (Obermeier et al., 2013; Prinz and Priller, 2017). Park et al. found that neutrophils were infiltrated in brain tissue in male Sprague–Dawley rats after treated with LPS for 12 hours and 7 days, and BBB destruction occurred simultaneously (Park et al., 2015). Haileselassie et al. showed that vascular leukocyte adhesion factors such as vascular cell adhesion molecule-1(VCAM-1) and intercellular adhesion molecule-1(ICAM-1) increased in LPS model of sepsis after 24 hours’ treatment, indicating that inflammatory cells have adhered and transmigrated to vessels (Haileselassie et al., 2020) (Figure 1). The CXCR2, a G protein coupled receptor, is involved in a series of leukocyte cellular responses, such as cell recruitment and migration (Figure 1) (Jones et al., 1997; Casilli et al., 2005). Wu et al. found that neutrophils were significantly infiltrated in the cerebral cortex of mice after LPS of E. coli O111:B4 injection for 24 hours, while blocking CXCR2 significantly alleviated the effects (Wu et al., 2020) (Figure 1). Silwedel et al. also observed that LPS of E. coli O55:B5 caused an increased expression of C-X-C motif chemokine ligand (CXCL)11 and a decreased level of CXCL12 after 4 hours’ treatment, which resulted from the increase of leukocyte migration (Silwedel et al., 2018). In summary, LPS can induce the migration of neutrophils to the brain tissue. However, studies found that LPS of E. coli O111:B4 elevates CXCR2 on brain ECs but reduces CXCR2 on neutrophils, suggesting that ECs are important in inducing neutrophil infiltration under the stimulation of LPS (Rios-Santos et al., 2007; Wu et al., 2020). As an important part of systemic inflammation, the response of neutrophils is specifically manifested as mediating and amplifying inflammation in the brain tissue, thereby destroying the structure of the BBB.
Sepsis, a pathological process causing dysfunction of one’s own organs due to bacterial infection and its resulting dysregulated inflammatory response, is the leading cause of death in critically ill patients with significant morbidity and mortality, representing a high proportion of readmissions after treatment discharge (Fleischmann et al., 2016a; Fleischmann et al., 2016b; Chen et al., 2019; Gadre et al., 2019). Sepsis often leads to multiple organ dysfunctions, such as the heart, liver as well as kidneys, and causes severe complications (Savio et al., 2017; Sun et al., 2018; Poston and Koyner, 2019). The brain dysfunction caused by sepsis, namely SAE, is one of the most common and serious complications (Annane and Sharshar, 2015; Savio et al., 2017; Sun et al., 2018; Ziesmann and Marshall, 2018; Poston and Koyner, 2019; Loscher and Gericke, 2020). The CNS of patients with SAE often does not have obvious infections, instead neuroinflammation and oxidative stress appear in brain tissue, and patients have significant symptoms such as cognitive dysfunction, memory decline, and clinical coma or confusion (Iwashyna et al., 2010; Gofton and Young, 2012; Golzari and Mahmoodpoor, 2014; Catalao et al., 2017; Jesus et al., 2020). Given that disruption of the BBB is an important mechanism underlying the development of SAE and would confer a poor prognosis and long-term impact on quality of life, we will focus on the relationship between BBB destruction and SAE.
It is commonly assumed that neuroinflammation produced by oxidative stress and mitochondrial dysfunction are the primary pathophysiological processes implicated in the development of SAE. (Cornelius et al., 2013; Sonneville et al., 2013). In normal circumstances, only a small number of inflammatory factors such as TNF-α, IL-1, and IL-6, from the systemic circulation may pass through the BBB to enter the brain (Tauber et al., 2017). However, the disruption of BBB will make a wide range of inflammatory cytokines, neurotoxins, complement as well as pathogens to enter the brain tissue when systemic inflammation occurs (Dhaya et al., 2018; Nwafor et al., 2019; Ren et al., 2020). Not only that, the NVU that makes up the BBB also mediates partial inflammatory responses, oxidative stress, and other reactions, for example, activating microglia to release NO, cytokines, and ROS, to aggravate BBB dysfunction in turn, so BBB disruption can be not only a cause but also a consequence of SAE (da Fonseca et al., 2014; Danielski et al., 2018).
Alexander et al. found that after intraperitoneal injection of LPS of E. coli O55:B55, the concentration of TNF-α, a key metabolic mediator in sepsis in the blood, increased significantly after 1 hour’s treatment, which resulted in a decrease in the integrity of BBB, infiltration of neutrophils into the brain and the subsequent SAE symptoms (Alexander et al., 2008). An increased activation of glial cells, as well as neuronal apoptosis, was observed over time by using LPS of E. coli O127:B8 to induce peripheral inflammatory responses by Semmler et al (Semmler et al., 2005). In a sepsis model with CLP, Imamura et al. found that the LPS level in the blood increased, the BBB was destroyed, the expression of IL-1β and its receptors in the brain tissue increased, and brain inflammation occurred (Imamura et al., 2011). They also showed that IL-1β was one of the first increased inflammatory factors in sepsis and the dysfunctional BBB, in turn, allowed other inflammatory mediators including IL-1β to enter the brain as well as the cerebrospinal fluid, consequently leading to the appearance of cognitive impairment (Imamura et al., 2011; Mina et al., 2014). Mina et al. used IL-1β receptor antagonist to antagonize IL-1β, which reversed BBB damage and cognitive impairment after brain oxidative damage, confirming that IL-1β is an important factor mediating cognitive impairment in sepsis (Mina et al., 2014).
It is known that during the early stage of sepsis, there is an activation of glial cells at the brain vessels as well as the secretion of large amounts of cytokines, which leads to the disrupted BBB and symptoms of brain inflammation (Comim et al., 2011). Microglia are brain resident macrophages that are normally in a resting state and can immediately go into an activated state upon stimulation by injury or factors like LPS, after which they rapidly undergo proliferation, migration, engulf and secrete cytokines (Greenhalgh et al., 2020; Zengeler and Lukens, 2021). Normal microglial responses in brain tissue are obligatory for phagocytic clearance of damaged neural cells and tissue debris, but persistent or vigorous microglial responses can damage the CNS (Greenhalgh et al., 2020; Zengeler and Lukens, 2021). When BBB permeability rises, especially in the hippocampus, a large number of cytokines, such as TNF-α and IL-6, enter the brain tissue to cause oxidative stress and inflammatory responses in microglia, which plays a major role in the development of cognitive impairment in SAE (Michels et al., 2015b). Moreover, the oxidative stress response mediated by NOX in microglia manifesting as excessive ROS production, is also involved in the process of LPS damage to TJ and AJ proteins and SAE occurrence (Hernandes et al., 2014). Collectively, microglia can be activated by LPS or cytokines such as TNF-α and IL-6, and destroy the BBB during the initial phase of sepsis by directly mediating neuroinflammation in the brain and participating in the direct spread of neuroinflammation in the course of SAE (Hawkins and Davis, 2005; Ronaldson and Davis, 2020).
Sepsis is a dysregulated systemic inflammatory response that is initially aimed at eradicating pathogens or pathogenic factors, but the abnormal inflammatory response would cause BBB disruption and neuronal damage, accompanied by the appearance of cognitive dysfunction, mental disorder, and other clinical symptoms. Clinical studies demonstrated BBB damage in patients who died of sepsis, and a significant decrease of TJ proteins (Sharshar et al., 2007; Erikson et al., 2020). Sharshar et al. demonstrated that brain damage caused by sepsis was specifically manifested as an elevated permeability of BBB by magnetic resonance imaging, which is strongly associated with poor prognosis (Sharshar et al., 2007). Disrupted BBB with a rise in permeability was also observed in a pathological model of sepsis through CLP (Zhao et al., 2015; Silva et al., 2020). These results indicated that impaired BBB may serve as an initiating link as well as a driver of SAE, which leads to an infiltration of various cytokines and leukocytes in brain tissue and causes neuronal apoptosis and dysfunction, ending up with a series of neuropsychiatric symptoms with a high mortality.
Sepsis is an abnormal or dysregulated host response to stimuli from external factors as well as internal organ dysfunction (Lelubre and Vincent, 2018). Current studies have redefined sepsis as ‘life-threatening organ dysfunction caused by a dysregulated host response to infection’ (Singer et al., 2016).To date, sepsis has been a major public health problem worldwide and a leading cause of the death of critical illness (Vincent et al., 2014; Fleischmann et al., 2016a). Survivors of sepsis usually have physical and cognitive dysfunctions, giving rise to a large burden on society and patients’ families (Iwashyna et al., 2010; Iwashyna et al., 2012). SAE, as one of the most serious and frequent complications of sepsis, leads to an extremely poor prognosis for patients as well as a substantial reduction patient’s life quality. Here, we will discuss the possibility of BBB as an effective target for treating SAE. As LPS can disrupt the structure and function of the BBB, and the damaged BBB is closely related to the development of SAE, we believe that targeting the BBB may be an effective strategy to prevent and treat SAE.
Glial cells play an important role in both LPS disruption of the BBB and the development of SAE after BBB disruption (Haruwaka et al., 2019; Spampinato et al., 2019). Monique et al. showed that inhibition of the activity of microglia could decrease the permeability of BBB that were elevated by the actions of systemic cytokines, and reverse the long-term cognitive impairment (Michels et al., 2015b). They further found that the protective effect by inhibiting microglia on BBB is partly through the cluster of differentiation (CD)40-CD40 ligand pathway, and anti-CD40 treatment ameliorated long-term cognitive impairment caused by sepsis (Michels et al., 2015a) (Figure 2). Studies in recent years have found that dexmedetomidine, a clinical drug commonly used for sedation, plays a neuroprotective role through an anti-inflammatory mechanism (Liu et al., 2017; Wang et al., 2019b). Mei et al. found that dexmedetomidine could activate 2A adrenoceptors on astrocytes but not microglia to decrease proinflammatory factors in the brain, preserve BBB integrity, and enhance learning and memory performance by activating 2A adrenoceptors on astrocytes in septic mice (Mei et al., 2021) (Figure 2). It was shown that the high mobility group box protein 1 (HMGB1) released from astrocytes and microglia is an important factor in regulating BBB permeability in inflammatory response (Festoff et al., 2016) (Figure 2). Dexmedetomidine can improve the integrity of BBB as well as cognitive impairment by inhibiting HMGB1 (Hu et al., 2018; Mei et al., 2021). Therefore, HMGB1 may represent a new target for the treatment of sepsis and SAE (Wang et al., 2014). Several studies have demonstrated the pro-inflammatory effects of hyperglycemia (Amorim et al., 2019). Huang et al. found that the activity of astrocytes and microglia, as well as that of NF-κB and MAPKs was inhibited after using insulin to control blood glucose in Sprague-Dawley rats, which accompanied by reduced permeability of BBB and alleviated SAE, showing full recovery of the mechanical withdrawal threshold and thermal withdrawal latency as well as an increased electroencephalography frequency (Huang et al., 2020a) (Figure 2). Besides, studies have also confirmed that insulin can directly inhibit brain tissue inflammatory response and oxidative stress injury, which consequently improves brain tissue damage (Chen et al., 2014). Omarigliptin is a novel once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor developed for the treatment of type 2 diabetes (Biftu et al., 2014). Studies have found that Omarigliptin can not only maintain the integrity of the BBB by inhibiting neuroinflammation, but also reduce the expression of MMP-2 and MMP-9 to protect TJs protein (Du and Wang, 2020). These results suggest that, for the management of sepsis patients in intensive care unit (ICU), attentions should also be paid to the blood glucose changes and the application of insulin, to reduce the likelihood of SAE (Huang et al., 2020a). Overall, protecting the BBB by inhibiting the activity of glial cells may be helpful for the treatment of SAE. But studies also have found that in the early stage of SAE, the morphology of glial cells begins to change without obvious disruption of the BBB (Griton et al., 2020). Therefore, introducing prophylactic drugs that inhibit neuroinflammation would be helpful to avoid the rapid deterioration of SAE after BBB disruption.
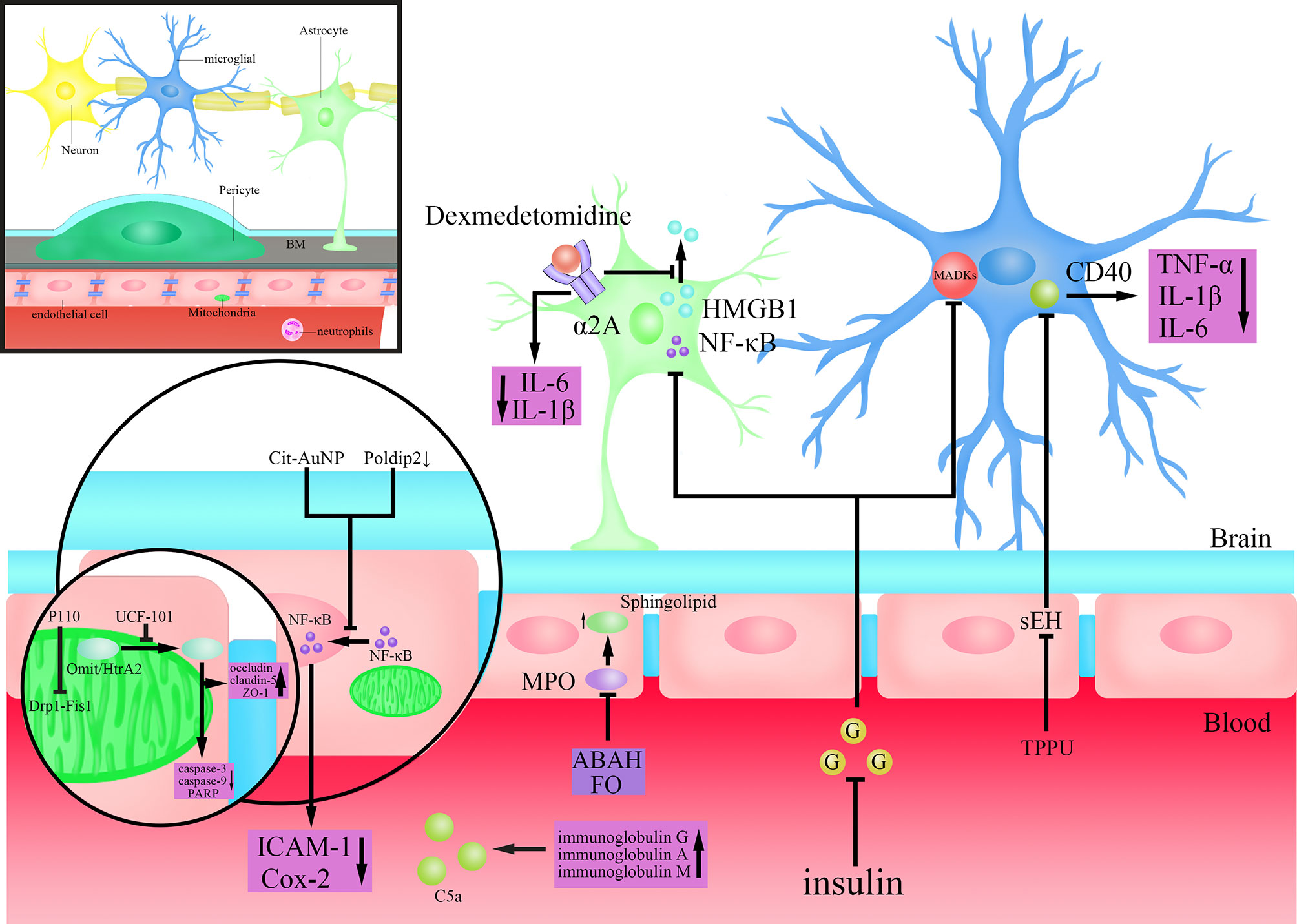
Figure 2 Mechanisms of action of drugs that may target the BBB to treat SAE. Dexmedetomidine activates α2A adrenoceptors on astrocytes to inhibit the extracellular release of high mobility group box protein 1 (HMGB1) and reduce the levels of Interleukin(IL)-6 and IL-1β in brain tissue; Insulin inhibits the activity of Nuclear factor-k-gene binding (NF-κB) in astrocytes and mitogen-activated protein kinase (MAPK)s in microglia after lowering blood sugar; Citrate-covered gold nanoparticles (Cit-AuNP) or targeted inhibition of Poldip2 inhibits the nuclear translocation process of Nuclear factor-k-gene binding (NF-κB) in endothelial cells to reduce the secretion of Intercellular adhesion molecule-1(ICAM-1) and cyclooxygenase-2 (Cox-2); TPPU(N-[1-(1-oxopropyl)-4-piperidinyl]-N’-[4-(trifluoromethoxy)phenyl)-urea) inhibits Cluster of differentiation (CD)40 by inhibiting soluble epoxide hydrolase (SEH), and reduces the secretion of Tumour Necrosis Factor-α (TNF-α), IL-1β and IL-6; 4-aminobenzoic acid hydrazide (ABAH) or Fish oil (FO) maintain the steady state of Sphingolipid by inhibiting Myeloperoxidase (MPO); P110(an inhibitor of dynamin-related protein-1(Drp1)-Fission 1(Fis1) interaction) inhibits the Drp1-Fis1 interaction; UCF-101(an inhibitor of Omi/HtrA2) inhibits the translocation of Omi/high-temperature requirement serine protease A2 (HtrA2) from mitochondria to cytoplasm, antagonizes the Caspase-dependent apoptosis pathway and reduces poly (ADP-ribose) polymerase (PARP) levels, while also protecting the tight junctions (TJs) protein; Injection of immunoglobulin G or a combination of immunoglobulin A and M inhibits complement(C)5a.
NF-κB, as a proinflammatory cell transcription factor that exists in almost all cells, was found to participate in the BBB disruption by LPS (Xu et al., 2021). Nod-like receptor protein 3 (NLRP3) plays an important role in the inflammation cascade by amplifying the inflammation response (Islam et al., 2020). Chen et al. found a suppression of the NF-kB/NLRP3 inflammatory pathway by directly binding to the promoter region of NLRP3, which resulted in a decreased cell apoptosis and inflammation, and an alleviation of BBB disruption and SAE (Chen et al., 2020). Kikuchi et al. showed that inhibition of poldip2 could attenuate BBB disruption caused by sepsis by regulating the nuclear translocation process of NF-κB as well as Cox-2 expression, preventing the occurrence and development of SAE (Kikuchi et al., 2019) (Figure 2). Di Bella et al. found that citrate-covered gold nanoparticles (Cit-AuNP) could prevent the process of NF-κB translocation to the nucleus by decreasing the phosphorylation of inhibitor of kappa B alpha (IκBα), which was followed by decreased expression of pro-inflammatory factor ICAM-1 on leukocytes and reduced adhesion of leukocyte to the endothelium of intracerebral blood vessels, ultimately alleviating the inflammatory response in the brain by BBB protection and reducing the occurrence of SAE during sepsis (Di Bella et al., 2021) (Figure 2). Wang et al. found that by inhibiting soluble epoxide hydrolase (sEH) using TPPU(N-[1-(1-oxopropyl)-4-piperidinyl]-N’-[4-(trifluoromethoxy)phenyl)-urea), which is required for the activity of epoxyeicosatrienoic acids (EETs), a substance involved in maintaining the intact structure of cerebral blood vessels, the barrier function of BBB was improved, followed by an alleviation of long-term encephalopathy in CLP model mice (Iliff et al., 2010; Wang et al., 2020) (Figure 2). Myeloperoxidase (MPO) in microglia and neutrophils not only regulates inflammation-related signaling pathways but also promotes ROS production to mediate oxidative stress (Patel, 2016). Sphingolipid, as the biologically active family of lipids found, plays a key role in maintaining the structure and function of the BBB, and is also closely related to the pathological state of the CNS (Kuperberg and Wadgaonkar, 2017). Ullen et al. found that 4-aminobenzoic acid hydrazide (ABAH), an inhibitor of MPO, alleviated BBB dysfunction induced by LPS by maintaining the homeostasis of sphingolipid, thereby improving the function of brain (Ullen et al., 2013; Goeritzer et al., 2020) (Figure 2). Fish oil (FO) is an important source of long-chain n-3 polyunsaturated fatty acids (PUFAs) with immunomodulatory and anti-inflammatory effects (Li et al., 2014). Recent studies have found that FO enriched lipid emulsion is able to prevent long-term cognitive impairment caused by sepsis, by protecting the BBB as well as inhibiting the activity of MPO (Della Giustina et al., 2020) (Figure 2). Interestingly, Margotti et al. found that neutrophils infiltration into brain tissue due to BBB rupture caused an increased production of MPO, accompanied by lipid oxidative damage in the hippocampus and prefrontal cortex, suggesting a vicious circle between BBB destruction and MPO production (Margotti et al., 2020). These results suggested that MPO may become a potential drug target for the treatment of brain dysfunction caused by sepsis (Margotti et al., 2020). Besides, anti-inflammatory and antioxidant drugs, such as metformin, platonin have been shown to exhibit neuroprotective effects, and can improve the decreased expression of TJ proteins induced by sepsis, thereby improving BBB function and attenuating sepsis-induced brain injury (Yeh et al., 2015; Ismail Hassan et al., 2020). Therefore, anti-inflammation and anti-oxidation represent important strategies to protect BBB and brain tissues.
Mitochondria are vital to cell metabolism because they provide ATP for living activities via oxidative phosphorylation, and play an important part in the cell apoptosis and programmed cell death processes (Bock and Tait, 2020). Impairment of mitochondrial energy metabolism or abnormal apoptosis in BMECs both affect the barrier function of BBB (Chen et al., 2017). Therefore, mitochondria may represent a potential strategy in the treatment of SAE. Haileselassie et al. found that the mitochondrial dysfunction mediated by the interaction of Drp1-Fis1 in response to LPS leads to increased permeability of the BBB and a direct toxic effect on primary cortical ECs, but the above results can all be eliminated by Drp1-Fis1 interaction inhibitor P110 (Haileselassie et al., 2020) (Figure 2). So, inhibiting mitochondrial dysfunction to protect both BBB and neurons could be a potentially new therapeutic strategy for treating SAE. Omi/high-temperature requirement serine protease A2 (HtrA2), a proapoptotic serine protease mainly found in and released from mitochondria, is found to be involved in the development of BBB dysfunction and SAE induced by sepsis through a caspase-dependent mitochondrial pathway (Wang et al., 2018; Hu et al., 2019). After inhibiting Omi/HtrA2 translocation from mitochondria to the cytosol by UCF-101(an inhibitor of Omi/HtrA2), oxidative damage in the brain tissue and cognitive dysfunction induced by sepsis were significantly improved (Hu et al., 2013) (Figure 2). Further studies found that inhibiting Omi/HtrA2 not only protects neurons, but also alleviates the apoptosis of brain ECs and the decreased TJs protein induced by LPS (Wang et al., 2018; Hu et al., 2019) (Figure 2). In addition, Xu et al. alleviated BBB disruption induced by sepsis and protected synaptic plasticity by inhibiting caspase-1-mediated apoptosis, thus improving the cognitive impairment in a mouse model of sepsis (Xu et al., 2019). Therefore, improving mitochondrial dysfunction and related apoptosis pathways are helpful to protect neurons and maintain the barrier function of BBB, which represent a new potential treatment strategy for SAE.
Pathogenic factors in sepsis such as LPS, after binding to TLR4, result in dysregulated inflammatory responses through complex immune activation, so targeting immune activating responses that inhibit neuroinflammation may be protective against BBB disruption by sepsis (Hartz et al., 2006; Miao et al., 2021). Intravenous immunoglobulin is a therapeutic treatment with anti-infective, anti-inflammatory, and immunomodulatory effects (Hamano et al., 2013; Di Rosa et al., 2014). Studies showed that intravenous immunoglobulin can serve as an important part of the adjuvant treatment of sepsis, but for 40 years the method is still controversial (Hamano et al., 2013; Di Rosa et al., 2014; Szakmany, 2019; Jarczak et al., 2020). Esen et al. found that the injection of immunoglobulin G or a combination of immunoglobulin A and M both exerted neuroprotective effects, which improved the integrity of the BBB, and alleviated the symptoms of sepsis in rats (Esen et al., 2012) (Figure 2). Considering the regulatory effects of immunoglobulins on the complement system and the finding that inhibition of complement C5a could protect the BBB during sepsis, the protective effect of immunoglobulins on the BBB is most likely achieved through the regulation of the complement system (Basta, 2008; Flierl et al., 2009; Esen et al., 2017) (Figure 2). Overall, as the dysregulated inflammatory response in sepsis is coupled with the initial immunomodulatory response, more studies could be performed to refine novel strategies to treat SAE by suppressing the immune activation response of excessive neuroinflammation.
The BBB is essential for maintaining homeostasis within the brain’s biochemical environment (Liebner et al., 2018). As the barrier of CNS, the low permeability of BBB separates neural tissue from circulating blood, and the infiltration of leukocytes (such as neutrophils, lymphocytes) in CNS is much less than that in peripheral tissues, CNS was often considered as an immune privileged site in the past (Liebner et al., 2018). However, BBB is often disturbed in response to various pathogenic agents, causing the entry of cytokines, pathogens as well as immune cells from blood into the CNS through BBB, which is closely associated with the occurrence and development of CNS diseases such as SAE (Sweeney et al., 2019).
This review discussed the mechanisms of the BBB damage by LPS, a major pathogenic agent of sepsis, through direct and indirect actions. The direct damage on the BBB by LPS is by disrupting the BBB’s structure. Among the mechanisms, the paracellular pathways maintained by TJs and AJs, the transcellular pathway, P-gp, and vesicle trafficking are the fundamental forms by which LPS raises the permeability of the BBB and disrupts its barrier function. The paracellular and transcellular pathways can be regarded as central links in future studies on how to protect the BBB from being damaged by toxic substances like LPS. As the basic components of BBB, the endothelium and BM, are the targets of LPS. Once their energy metabolism status is abnormally altered, a direct disruption of the BBB’s structure would occur and thus its barrier function cannot be maintained. Besides, LPS also damages the BBB indirectly by acting on microglia, astrocytes, pericytes, and neutrophils. Notably, the roles performed by these cells in the disruption of the BBB by LPS may be interpreted as a succession or amplification, implying that the effects on the structure and function of the BBB by LPS are largely or even entirely through the mechanisms detailed in the direct actions section. Recent studies have found that the communication between cells that make up the NVU and the basic structure of the BBB was more complex and diverse than previously realized, and cells of the NVU were involved in dynamically regulating the barrier function of the BBB (Kaplan et al., 2020). Therefore, more researches are needed to explore how to raise the infiltration of drugs into the brain to act on these cells, and thus to protect the BBB.
Sepsis attracts the attention of clinicians and researchers due to the poor prognosis of patients in critical care medicine (Fleischmann et al., 2016a; Fleischmann et al., 2016b; Gadre et al., 2019). In this review, we discussed the link between the BBB disruption and the occurrence of SAE, aiming at finding novel strategies to treat SAE by maintaining the barrier function of BBB. Systemic injection of LPS has been shown to cause damage to the BBB of recipient animals, resulting in the subsequent entry of peripheral cytokines into the brain (Vuuren et al., 2020). It is believed that the main contribution of the damaged BBB to the occurrence and development of SAE is to cause a large number of inflammatory factors and neurotoxins to enter the brain tissue, accompanied by immune cells in the brain tissue such as microglia being activated during systemic inflammation, thereby mediating neuroinflammation and oxidative stress. Subsequently, inflammation and oxidative stress in the brain tissue, in turn, amplify the extent of BBB disruption, which creates a vicious cycle that would fuel the generation and progression of SAE. Therefore, restoring the barrier function of BBB may serve as an effective approach to treat SAE.
According to current research, neuroinflammation and oxidative stress are key factors in the development of SAE, and inhibiting inflammatory reactions has a significant impact on SAE therapy. (Yeh et al., 2015; Kikuchi et al., 2019; Ismail Hassan et al., 2020; Wang et al., 2020; Di Bella et al., 2021). Therefore, elucidating mechanisms of the inflammation and oxidative stress that are closely related to the occurrence of SAE will be important for the development of drugs to treat SAE. More importantly, it was found that drugs that are generally recognized to have the ability to inhibit inflammatory and oxidative stress responses also have neuroprotective effect (Yeh et al., 2015; Ismail Hassan et al., 2020). It is important to note that the efficacy of intravenous immunoglobulin therapy for sepsis has been controversial, but targeting the pathway that inhibits the immune activation response to neuroinflammation is hoped to lead the way from the very beginning to the development of SAE and is likely turn out to be a brand-new strategy in the treatment of SAE. We believe that researchers may try to block the damage on the BBB by LPS to prevent and treat SAE in the further studies.
XP and YL conceived and wrote the manuscript. SH and ZL conceived the figure. YL and LZ reviewed the original draft. All authors contributed to the article and approved the submitted version.
This study was funded by Innovation and Entrepreneurship Training Program of Sichuan province (S202010632272).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akira, S., Takeda, K. (2004). Toll-Like Receptor Signalling. Nat. Rev. Immunol. 4, 499–511. doi: 10.1038/nri1391
Alelwani, W., Elmorsy, E., Kattan, S. W., Babteen, N. A., Alnajeebi, A. M., Al-Ghafari, A., et al. (2020). Carbamazepine Induces a Bioenergetics Disruption to Microvascular Endothelial Cells From the Blood-Brain Barrier. Toxicol. Lett. 333, 184–191. doi: 10.1016/j.toxlet.2020.08.006
Alexander, J. J., Jacob, A., Cunningham, P., Hensley, L., Quigg, R. J. (2008). TNF Is a Key Mediator of Septic Encephalopathy Acting Through Its Receptor, TNF Receptor-1. Neurochem. Int. 52, 447–456. doi: 10.1016/j.neuint.2007.08.006
Alexandrov, P. N., Hill, J. M., Zhao, Y., Bond, T., Taylor, C. M., Percy, M. E., et al. (2020). Aluminum-Induced Generation of Lipopolysaccharide (LPS) From the Human Gastrointestinal (GI)-Tract Microbiome-Resident Bacteroides Fragilis. J. Inorg Biochem. 203, 110886. doi: 10.1016/j.jinorgbio.2019.110886
Alkabie, S., Basivireddy, J., Zhou, L., Roskams, J., Rieckmann, P., Quandt, J. A. (2016). SPARC Expression by Cerebral Microvascular Endothelial Cells In Vitro and Its Influence on Blood-Brain Barrier Properties. J. Neuroinflammation. 13, 225. doi: 10.1186/s12974-016-0657-9
Amorim, R. G., Guedes, G. D. S., Vasconcelos, S. M. L., Santos, J. C. F. (2019). Kidney Disease in Diabetes Mellitus: Cross-Linking Between Hyperglycemia, Redox Imbalance and Inflammation. Arq Bras. Cardiol. 112, 577–587. doi: 10.5935/abc.20190077
Andreone, B. J., Chow, B. W., Tata, A., Lacoste, B., Ben-Zvi, A., Bullock, K., et al. (2017). Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 94, 581–594.e585. doi: 10.1016/j.neuron.2017.03.043
Annane, D., Sharshar, T. (2015). Cognitive Decline After Sepsis. Lancet Respir. Med. 3, 61–69. doi: 10.1016/S2213-2600(14)70246-2
Anthoney, N., Foldi, I., Hidalgo, A. (2018). Toll and Toll-Like Receptor Signalling in Development. Development 145, dev156018. doi: 10.1242/dev.156018
Argaw, A. T., Asp, L., Zhang, J., Navrazhina, K., Pham, T., Mariani, J. N., et al. (2012). Astrocyte-Derived VEGF-A Drives Blood-Brain Barrier Disruption in CNS Inflammatory Disease. J. Clin. Invest. 122, 2454–2468. doi: 10.1172/JCI60842
Argaw, A. T., Gurfein, B. T., Zhang, Y., Zameer, A., John, G. R. (2009). VEGF-Mediated Disruption of Endothelial CLN-5 Promotes Blood-Brain Barrier Breakdown. Proc. Natl. Acad. Sci. U. S. A. 106, 1977–1982. doi: 10.1073/pnas.0808698106
Armulik, A., Genove, G., Mae, M., Nisancioglu, M. H., Wallgard, E., Niaudet, C., et al. (2010). Pericytes Regulate the Blood-Brain Barrier. Nature 468, 557–561. doi: 10.1038/nature09522
Banks, W. A., Gray, A. M., Erickson, M. A., Salameh, T. S., Damodarasamy, M., Sheibani, N., et al. (2015). Lipopolysaccharide-Induced Blood-Brain Barrier Disruption: Roles of Cyclooxygenase, Oxidative Stress, Neuroinflammation, and Elements of the Neurovascular Unit. J. Neuroinflammation. 12, 223. doi: 10.1186/s12974-015-0434-1
Basta, M. (2008). Ambivalent Effect of Immunoglobulins on the Complement System: Activation Versus Inhibition. Mol. Immunol. 45, 4073–4079. doi: 10.1016/j.molimm.2008.07.012
Bauer, B., Hartz, A., Fricker, G., Miller, D. (2004). Pregnane X Receptor Up-Regulation of P-Glycoprotein Expression and Transport Function at the Blood-Brain Barrier. Mol. Pharmacol. 66, 413–419. doi: 10.1124/mol.66.3
Bauer, M., Karch, R., Wulkersdorfer, B., Philippe, C., Nics, L., Klebermass, E. M., et al. (2019). A Proof-Of-Concept Study to Inhibit ABCG2- and ABCB1-Mediated Efflux Transport at the Human Blood-Brain Barrier. J. Nucl. Med. 60, 486–491. doi: 10.2967/jnumed.118.216432
Berger, R. E., Rivers, E., Levy, M. M. (2017). Management of Septic Shock. N. Engl. J. Med. 376, 2282–2285. doi: 10.1056/NEJMclde1705277
Biftu, T., Sinha-Roy, R., Chen, P., Qian, X., Feng, D., Kuethe, J. T., et al. (2014). Omarigliptin (MK-3102): A Novel Long-Acting DPP-4 Inhibitor for Once-Weekly Treatment of Type 2 Diabetes. J. Med. Chem. 57, 3205–3212. doi: 10.1021/jm401992e
Blecharz-Lang, K. G., Wagner, J., Fries, A., Nieminen-Kelha, M., Rosner, J., Schneider, U. C., et al. (2018). Interleukin 6-Mediated Endothelial Barrier Disturbances Can Be Attenuated by Blockade of the IL6 Receptor Expressed in Brain Microvascular Endothelial Cells. Transl. Stroke Res. 9, 631–642. doi: 10.1007/s12975-018-0614-2
Bock, F. J., Tait, S. W. G. (2020). Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 21, 85–100. doi: 10.1038/s41580-019-0173-8
Boitsova, E. B., Morgun, A. V., Osipova, E. D., Pozhilenkova, E. A., Martinova, G. P., Frolova, O. V., et al. (2018). The Inhibitory Effect of LPS on the Expression of GPR81 Lactate Receptor in Blood-Brain Barrier Model In Vitro. J. Neuroinflammation. 15, 196. doi: 10.1186/s12974-018-1233-2
Bonkowski, D., Katyshev, V., Balabanov, R. D., Borisov, A., Dore-Duffy, P. (2011). The CNS Microvascular Pericyte: Pericyte-Astrocyte Crosstalk in the Regulation of Tissue Survival. Fluids Barriers CNS 8, 8. doi: 10.1186/2045-8118-8-8
Bowyer, J. F., Sarkar, S., Burks, S. M., Hess, J. N., Tolani, S., O’Callaghan, J. P., et al. (2020). Microglial Activation and Responses to Vasculature That Result From an Acute LPS Exposure. Neurotoxicology 77, 181–192. doi: 10.1016/j.neuro.2020.01.014
Bradshaw, A. D., Sage, E. H. (2001). SPARC, A Matricellular Protein That Functions in Cellular Differentiation and Tissue Response to Injury. J. Clin. Invest. 107, 1049–1054. doi: 10.1172/JCI12939
Buendia, I., Michalska, P., Navarro, E., Gameiro, I., Egea, J., Leon, R. (2016). Nrf2-ARE Pathway: An Emerging Target Against Oxidative Stress and Neuroinflammation in Neurodegenerative Diseases. Pharmacol. Ther. 157, 84–104. doi: 10.1016/j.pharmthera.2015.11.003
Cardoso, F. L., Brites, D., Brito, M. A. (2010). Looking at the Blood-Brain Barrier: Molecular Anatomy and Possible Investigation Approaches. Brain Res. Rev. 64, 328–363. doi: 10.1016/j.brainresrev.2010.05.003
Cardoso, F. L., Kittel, A., Veszelka, S., Palmela, I., Toth, A., Brites, D., et al. (2012). Exposure to Lipopolysaccharide and/or Unconjugated Bilirubin Impair the Integrity and Function of Brain Microvascular Endothelial Cells. PloS One 7, e35919. doi: 10.1371/journal.pone.0035919
Carey, A. N., Fisher, D. R., Bielinski, D. F., Cahoon, D. S., Shukitt-Hale, B. (2020). Walnut-Associated Fatty Acids Inhibit LPS-Induced Activation of BV-2 Microglia. Inflammation 43, 241–250. doi: 10.1007/s10753-019-01113-y
Casilli, F., Bianchini, A., Gloaguen, I., Biordi, L., Alesse, E., Festuccia, C., et al. (2005). Inhibition of Interleukin-8 (CXCL8/IL-8) Responses by Repertaxin, a New Inhibitor of the Chemokine Receptors CXCR1 and CXCR2. Biochem. Pharmacol. 69, 385–394. doi: 10.1016/j.bcp.2004.10.007
Catalao, C. H. R., Santos-Junior, N. N., da Costa, L. H. A., Souza, A. O., Alberici, L. C., Rocha, M. J. A. (2017). Brain Oxidative Stress During Experimental Sepsis Is Attenuated by Simvastatin Administration. Mol. Neurobiol. 54, 7008–7018. doi: 10.1007/s12035-016-0218-3
Ceretta, L. B., Reus, G. Z., Abelaira, H. M., Ribeiro, K. F., Zappellini, G., Felisbino, F. F., et al. (2012). Increased Oxidative Stress and Imbalance in Antioxidant Enzymes in the Brains of Alloxan-Induced Diabetic Rats. Exp. Diabetes Res. 2012, 302682. doi: 10.1155/2012/302682
Chen, Y. J., Chen, F. L., Chen, J. H., Wu, M. M., Chen, Y. L., Chien, D. S., et al. (2019). Epidemiology of Sepsis in Taiwan. Med. (Baltimore) 98, e15725. doi: 10.1097/MD.0000000000015725
Cheng, X., Yang, Y. L., Yang, H., Wang, Y. H., Du, G. H. (2018). Kaempferol Alleviates LPS-Induced Neuroinflammation and BBB Dysfunction in Mice via Inhibiting HMGB1 Release and Down-Regulating TLR4/MyD88 Pathway. Int. Immunopharmacol. 56, 29–35. doi: 10.1016/j.intimp.2018.01.002
Chen, D., Li, X., Zhang, L., Zhu, M., Gao, L. (2018). A High-Fat Diet Impairs Mitochondrial Biogenesis, Mitochondrial Dynamics, and the Respiratory Chain Complex in Rat Myocardial Tissues. J. Cell Biochem. 119, 9602. doi: 10.1002/jcb.27068
Chen, S., Tang, C., Ding, H., Wang, Z., Liu, X., Chai, Y., et al. (2020). Maf1 Ameliorates Sepsis-Associated Encephalopathy by Suppressing the NF-Kb/NLRP3 Inflammasome Signaling Pathway. Front. Immunol. 11, 594071. doi: 10.3389/fimmu.2020.594071
Chen, K., Wang, N., Diao, Y., Dong, W., Sun, Y., Liu, L., et al. (2017). Hydrogen-Rich Saline Attenuates Brain Injury Induced by Cardiopulmonary Bypass and Inhibits Microvascular Endothelial Cell Apoptosis Via the PI3K/Akt/GSK3beta Signaling Pathway in Rats. Cell Physiol. Biochem. 43, 1634–1647. doi: 10.1159/000484024
Chen, Q., Yu, W., Shi, J., Shen, J., Gao, T., Zhang, J., et al. (2014). Insulin Alleviates the Inflammatory Response and Oxidative Stress Injury in Cerebral Tissues in Septic Rats. J. Inflammation (Lond). 11, 18. doi: 10.1186/1476-9255-11-18
Chien, H. C., Colas, C., Finke, K., Springer, S., Stoner, L., Zur, A. A., et al. (2018). Reevaluating the Substrate Specificity of the L-Type Amino Acid Transporter (Lat1). J. Med. Chem. 61, 7358–7373. doi: 10.1021/acs.jmedchem.8b01007
Chlenski, A., Cohn, S. L. (2010). Modulation of Matrix Remodeling by SPARC in Neoplastic Progression. Semin. Cell Dev. Biol. 21, 55–65. doi: 10.1016/j.semcdb.2009.11.018
Cho, S. C., Park, M. C., Keam, B., Choi, J. M., Cho, Y., Hyun, S., et al. (2010). DDS, 4,4’-Diaminodiphenylsulfone, Extends Organismic Lifespan. Proc. Natl. Acad. Sci. U. S. A. 107, 19326–19331. doi: 10.1073/pnas.1005078107
Chow, J. C., Young, D. W., Golenbock, D. T., Christ, W. J., Gusovsky, F. (1999). Toll-Like Receptor-4 Mediates Lipopolysaccharide-Induced Signal Transduction. J. Biol. Chem. 274, 10689–10692. doi: 10.1074/jbc.274.16.10689
Chrissobolis, S., Faraci, F. M. (2008). The Role of Oxidative Stress and NADPH Oxidase in Cerebrovascular Disease. Trends Mol. Med. 14, 495–502. doi: 10.1016/j.molmed.2008.09.003
Coisne, C., Engelhardt, B. (2011). Tight Junctions in Brain Barriers During Central Nervous System Inflammation. Antioxid. Redox. Signal. 15, 1285–1303. doi: 10.1089/ars.2011.3929
Comim, C. M., Vilela, M. C., Constantino, L. S., Petronilho, F., Vuolo, F., Lacerda-Queiroz, N., et al. (2011). Traffic of Leukocytes and Cytokine Up-Regulation in the Central Nervous System in Sepsis. Intensive Care Med. 37, 711–718. doi: 10.1007/s00134-011-2151-2
Cornelius, C., Crupi, R., Calabrese, V., Graziano, A., Milone, P., Pennisi, G., et al. (2013). Traumatic Brain Injury: Oxidative Stress and Neuroprotection. Antioxid. Redox. Signal. 19, 836–853. doi: 10.1089/ars.2012.4981
da Fonseca, A. C., Matias, D., Garcia, C., Amaral, R., Geraldo, L. H., Freitas, C., et al. (2014). The Impact of Microglial Activation on Blood-Brain Barrier in Brain Diseases. Front. Cell Neurosci. 8, 362. doi: 10.3389/fncel.2014.00362
Dal-Pizzol, F., Rojas, H. A., dos Santos, E. M., Vuolo, F., Constantino, L., Feier, G., et al. (2013). Matrix Metalloproteinase-2 and Metalloproteinase-9 Activities Are Associated With Blood-Brain Barrier Dysfunction in an Animal Model of Severe Sepsis. Mol. Neurobiol. 48, 62–70. doi: 10.1007/s12035-013-8433-7
Daneman, R., Prat, A. (2015). The Blood-Brain Barrier. Cold Spring Harb. Perspect. Biol. 7, a020412. doi: 10.1101/cshperspect.a020412
Danielski, L. G., Giustina, A. D., Badawy, M., Barichello, T., Quevedo, J., Dal-Pizzol, F., et al. (2018). Brain Barrier Breakdown as a Cause and Consequence of Neuroinflammation in Sepsis. Mol. Neurobiol. 55, 1045–1053. doi: 10.1007/s12035-016-0356-7
Deli, M. A., Abraham, C. S., Kataoka, Y., Niwa, M. (2005). Permeability Studies on In Vitro Blood-Brain Barrier Models: Physiology, Pathology, and Pharmacology. Cell Mol. Neurobiol. 25, 59–127. doi: 10.1007/s10571-004-1377-8
Della Giustina, A., Goldim, M. P., Danielski, L. G., Florentino, D., Garbossa, L., Joaquim, L., et al. (2020). Fish Oil-Rich Lipid Emulsion Modulates Neuroinflammation and Prevents Long-Term Cognitive Dysfunction After Sepsis. Nutrition 70, 110417. doi: 10.1016/j.nut.2018.12.003
Desai, B. S., Monahan, A. J., Carvey, P. M., Hendey, B. (2007). Blood-Brain Barrier Pathology in Alzheimer’s and Parkinson’s Disease: Implications for Drug Therapy. Cell Transplant. 16, 285–299. doi: 10.3727/000000007783464731
Dhaya, I., Griton, M., Raffard, G., Amri, M., Hiba, B., Konsman, J. P. (2018). Bacterial Lipopolysaccharide-Induced Systemic Inflammation Alters Perfusion of White Matter-Rich Regions Without Altering Flow in Brain-Irrigating Arteries: Relationship to Blood-Brain Barrier Breakdown? J. Neuroimmunol. 314, 67–80. doi: 10.1016/j.jneuroim.2017.11.009
Di Bella, D., Ferreira, J. P. S., Silva, R. N. O., Echem, C., Milan, A., Akamine, E. H., et al. (2021). Gold Nanoparticles Reduce Inflammation in Cerebral Microvessels of Mice With Sepsis. J. Nanobiotechnology. 19, 52. doi: 10.1186/s12951-021-00796-6
Di Rosa, R., Pietrosanti, M., Luzi, G., Salemi, S., D’Amelio, R. (2014). Polyclonal Intravenous Immunoglobulin: An Important Additional Strategy in Sepsis? Eur. J. Intern. Med. 25, 511–516. doi: 10.1016/j.ejim.2014.05.002
Dohgu, S., Banks, W. A. (2008). Lipopolysaccharide-Enhanced Transcellular Transport of HIV-1 Across the Blood-Brain Barrier Is Mediated by the P38 Mitogen-Activated Protein Kinase Pathway. Exp. Neurol. 210, 740–749. doi: 10.1016/j.expneurol.2007.12.028
Doll, D. N., Hu, H., Sun, J., Lewis, S. E., Simpkins, J. W., Ren, X. (2015). Mitochondrial Crisis in Cerebrovascular Endothelial Cells Opens the Blood-Brain Barrier. Stroke 46, 1681–1689. doi: 10.1161/STROKEAHA.115.009099
Du, H., Wang, S. (2020). Omarigliptin Mitigates Lipopolysaccharide-Induced Neuroinflammation and Dysfunction of the Integrity of the Blood-Brain Barrier. ACS Chem. Neurosci. 11, 4262–4269. doi: 10.1021/acschemneuro.0c00537
Endl, E., Hollmann, C., Gerdes, J. (2001). Antibodies Against the Ki-67 Protein: Assessment of the Growth Fraction and Tools for Cell Cycle Analysis. Methods Cell Biol. 63, 399–418. doi: 10.1016/s0091-679x(01)63022-x
Erikson, K., Tuominen, H., Vakkala, M., Liisanantti, J. H., Karttunen, T., Syrjala, H., et al. (2020). Brain Tight Junction Protein Expression in Sepsis in an Autopsy Series. Crit. Care 24, 385. doi: 10.1186/s13054-020-03101-3
Esen, F., Orhun, G., Ozcan, P. E., Senturk, E., Kucukerden, M., Giris, M., et al. (2017). Neuroprotective Effects of Intravenous Immunoglobulin Are Mediated Through Inhibition of Complement Activation and Apoptosis in a Rat Model of Sepsis. Intensive Care Med. Exp. 5, 1. doi: 10.1186/s40635-016-0114-1
Esen, F., Senturk, E., Ozcan, P. E., Ahishali, B., Arican, N., Orhan, N., et al. (2012). Intravenous Immunoglobulins Prevent the Breakdown of the Blood-Brain Barrier in Experimentally Induced Sepsis. Crit. Care Med. 40, 1214–1220. doi: 10.1097/CCM.0b013e31823779ca
Festoff, B. W., Sajja, R. K., van Dreden, P., Cucullo, L. (2016). HMGB1 and Thrombin Mediate the Blood-Brain Barrier Dysfunction Acting as Biomarkers of Neuroinflammation and Progression to Neurodegeneration in Alzheimer’s Disease. J. Neuroinflamm. 13, 194. doi: 10.1186/s12974-016-0670-z
Fleischmann, C., Scherag, A., Adhikari, N. K., Hartog, C. S., Tsaganos, T., Schlattmann, P., et al. (2016a). Assessment of Global Incidence and Mortality of Hospital-Treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 193, 259–272. doi: 10.1164/rccm.201504-0781OC
Fleischmann, C., Thomas-Rueddel, D. O., Hartmann, M., Hartog, C. S., Welte, T., Heublein, S., et al. (2016b). Hospital Incidence and Mortality Rates of Sepsis. Dtsch Arztebl Int. 113, 159–166. doi: 10.3238/arztebl.2016.0159
Flierl, M. A., Stahel, P. F., Rittirsch, D., Huber-Lang, M., Niederbichler, A. D., Hoesel, L. M., et al. (2009). Inhibition of Complement C5a Prevents Breakdown of the Blood-Brain Barrier and Pituitary Dysfunction in Experimental Sepsis. Crit. Care 13, R12. doi: 10.1186/cc7710
Forster, C. (2008). Tight Junctions and the Modulation of Barrier Function in Disease. Histochem Cell Biol. 130, 55–70. doi: 10.1007/s00418-008-0424-9
Frister, A., Schmidt, C., Schneble, N., Brodhun, M., Gonnert, F. A., Bauer, M., et al. (2014). Phosphoinositide 3-Kinase Gamma Affects LPS-Induced Disturbance of Blood-Brain Barrier via Lipid Kinase-Independent Control of cAMP in Microglial Cells. Neuromolecular Med. 16, 704–713. doi: 10.1007/s12017-014-8320-z
Fritz, M., Klawonn, A. M., Jaarola, M., Engblom, D. (2018). Interferon- Mediated Signaling in the Brain Endothelium Is Critical for Inflammation-Induced Aversion. Brain Behav. Immun. 67, 54–58. doi: 10.1016/j.bbi.2017.08.020
Gadre, S. K., Shah, M., Mireles-Cabodevila, E., Patel, B., Duggal, A. (2019). Epidemiology and Predictors of 30-Day Readmission in Patients With Sepsis. Chest 155, 483–490. doi: 10.1016/j.chest.2018.12.008
Gao, H. M., Hong, J. S. (2008). Why Neurodegenerative Diseases Are Progressive: Uncontrolled Inflammation Drives Disease Progression. Trends Immunol. 29, 357–365. doi: 10.1016/j.it.2008.05.002
Goeritzer, M., Bernhart, E., Plastira, I., Reicher, H., Leopold, C., Eichmann, T. O., et al. (2020). Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro. Int. J. Mol. Sci. 21, 1143. doi: 10.3390/ijms21031143
Gofton, T. E., Young, G. B. (2012). Sepsis-Associated Encephalopathy. Nat. Rev. Neurol. 8, 557–566. doi: 10.1038/nrneurol.2012.183
Golzari, S. E., Mahmoodpoor, A. (2014). Sepsis-Associated Encephalopathy Versus Sepsis-Induced Encephalopathy. Lancet Neurol. 13, 967–968. doi: 10.1016/S1474-4422(14)70205-4
Greenhalgh, A. D., David, S., Bennett, F. C. (2020). Immune Cell Regulation of Glia During CNS Injury and Disease. Nat. Rev. Neurosci. 21, 139–152. doi: 10.1038/s41583-020-0263-9
Griton, M., Dhaya, I., Nicolas, R., Raffard, G., Periot, O., Hiba, B., et al. (2020). Experimental Sepsis-Associated Encephalopathy Is Accompanied by Altered Cerebral Blood Perfusion and Water Diffusion and Related to Changes in Cyclooxygenase-2 Expression and Glial Cell Morphology But Not to Blood-Brain Barrier Breakdown. Brain Behav. Immun. 83, 200–213. doi: 10.1016/j.bbi.2019.10.012
Haileselassie, B., Joshi, A. U., Minhas, P. S., Mukherjee, R., Andreasson, K. I., Mochly-Rosen, D. (2020). Mitochondrial Dysfunction Mediated Through Dynamin-Related Protein 1 (Drp1) Propagates Impairment in Blood Brain Barrier in Septic Encephalopathy. J. Neuroinflammation. 17, 36. doi: 10.1186/s12974-019-1689-8
Hamano, N., Nishi, K., Onose, A., Okamoto, A., Umegaki, T., Yamazaki, E., et al. (2013). Efficacy of Single-Dose Intravenous Immunoglobulin Administration for Severe Sepsis and Septic Shock. J. Intensive Care 1, 4. doi: 10.1186/2052-0492-1-4
Hao, H., Cao, L., Jiang, C., Che, Y., Zhang, S., Takahashi, S., et al. (2017). Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 25, 856–867.e855. doi: 10.1016/j.cmet.2017.03.007
Hartz, A. M., Bauer, B., Fricker, G., Miller, D. S. (2006). Rapid Modulation of P-Glycoprotein-Mediated Transport at the Blood-Brain Barrier by Tumor Necrosis Factor-Alpha and Lipopolysaccharide. Mol. Pharmacol. 69, 462–470. doi: 10.1124/mol.105.017954
Haruwaka, K., Ikegami, A., Tachibana, Y., Ohno, N., Konishi, H., Hashimoto, A., et al. (2019). Dual Microglia Effects on Blood Brain Barrier Permeability Induced by Systemic Inflammation. Nat. Commun. 10, 5816. doi: 10.1038/s41467-019-13812-z
Haseloff, R. F., Dithmer, S., Winkler, L., Wolburg, H., Blasig, I. E. (2015). Transmembrane Proteins of the Tight Junctions at the Blood-Brain Barrier: Structural and Functional Aspects. Semin. Cell Dev. Biol. 38, 16–25. doi: 10.1016/j.semcdb.2014.11.004
Hawkins, B. T., Davis, T. P. (2005). The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 57, 173–185. doi: 10.1124/pr.57.2.4
He, F., Peng, J., Deng, X. L., Yang, L. F., Wu, L. W., Zhang, C. L., et al. (2011). RhoA and NF-kappaB Are Involved in Lipopolysaccharide-Induced Brain Microvascular Cell Line Hyperpermeability. Neuroscience 188, 35–47. doi: 10.1016/j.neuroscience.2011.04.025
Hernandes, M. S., D’Avila, J. C., Trevelin, S. C., Reis, P. A., Kinjo, E. R., Lopes, L. R., et al. (2014). The Role of Nox2-Derived ROS in the Development of Cognitive Impairment After Sepsis. J. Neuroinflamm. 11, 36. doi: 10.1186/1742-2094-11-36
Hong, S., Park, K. K., Magae, J., Ando, K., Lee, T. S., Kwon, T. K., et al. (2005). Ascochlorin Inhibits Matrix Metalloproteinase-9 Expression by Suppressing Activator Protein-1-Mediated Gene Expression Through the ERK1/2 Signaling Pathway: Inhibitory Effects of Ascochlorin on the Invasion of Renal Carcinoma Cells. J. Biol. Chem. 280, 25202–25209. doi: 10.1074/jbc.M413985200
Huang, C. T., Lue, J. H., Cheng, T. H., Tsai, Y. J. (2020a). Glycemic Control With Insulin Attenuates Sepsis-Associated Encephalopathy by Inhibiting Glial Activation via the Suppression of the Nuclear Factor Kappa B and Mitogen-Activated Protein Kinase Signaling Pathways in Septic Rats. Brain Res. 1738, 146822. doi: 10.1016/j.brainres.2020.146822
Huang, S. F., Othman, A., Koshkin, A., Fischer, S., Fischer, D., Zamboni, N., et al. (2020b). Astrocyte Glutathione Maintains Endothelial Barrier Stability. Redox Biol. 34, 101576. doi: 10.1016/j.redox.2020.101576
Hu, Y., Bi, Y., Yao, D., Wang, P., Li, Y. (2019). Omi/HtrA2 Protease Associated Cell Apoptosis Participates in Blood-Brain Barrier Dysfunction. Front. Mol. Neurosci. 12, 48. doi: 10.3389/fnmol.2019.00048
Hu, Y., Huang, M., Wang, P., Xu, Q., Zhang, B. (2013). Ucf-101 Protects Against Cerebral Oxidative Injury and Cognitive Impairment in Septic Rat. Int. Immunopharmacol. 16, 108–113. doi: 10.1016/j.intimp.2013.03.019
Hu, J., Vacas, S., Feng, X., Lutrin, D., Uchida, Y., Lai, I. K., et al. (2018). Dexmedetomidine Prevents Cognitive Decline by Enhancing Resolution of High Mobility Group Box 1 Protein-Induced Inflammation Through a Vagomimetic Action in Mice. Anesthesiology 128, 921–931. doi: 10.1097/ALN.0000000000002038
Hu, Y., Wang, Z., Pan, S., Zhang, H., Fang, M., Jiang, H., et al. (2017). Melatonin Protects Against Blood-Brain Barrier Damage by Inhibiting the TLR4/ NF-kappaB Signaling Pathway After LPS Treatment in Neonatal Rats. Oncotarget 8, 31638–31654. doi: 10.18632/oncotarget.15780
Hyeon, S., Lee, H., Yang, Y., Jeong, W. (2013). Nrf2 Deficiency Induces Oxidative Stress and Promotes RANKL-Induced Osteoclast Differentiation. Free Radic. Biol. Med. 65, 789–799. doi: 10.1016/j.freeradbiomed.2013.08.005
Iliff, J. J., Jia, J., Nelson, J., Goyagi, T., Klaus, J., Alkayed, N. J. (2010). Epoxyeicosanoid Signaling in CNS Function and Disease. Prostaglandins Other Lipid Mediat. 91, 68–84. doi: 10.1016/j.prostaglandins.2009.06.004
Imamura, Y., Wang, H., Matsumoto, N., Muroya, T., Shimazaki, J., Ogura, H., et al. (2011). Interleukin-1beta Causes Long-Term Potentiation Deficiency in a Mouse Model of Septic Encephalopathy. Neuroscience 187, 63–69. doi: 10.1016/j.neuroscience.2011.04.063
Islam, M. T., Bardaweel, S. K., Mubarak, M. S., Koch, W., Gawel-Beben, K., Antosiewicz, B., et al. (2020). Immunomodulatory Effects of Diterpenes and Their Derivatives Through NLRP3 Inflammasome Pathway: A Review. Front. Immunol. 11, 572136. doi: 10.3389/fimmu.2020.572136
Ismail Hassan, F., Didari, T., Baeeri, M., Gholami, M., Haghi-Aminjan, H., Khalid, M., et al. (2020). Metformin Attenuates Brain Injury by Inhibiting Inflammation and Regulating Tight Junction Proteins in Septic Rats. Cell J. 22, 29–37. doi: 10.22074/cellj.2020.7046
Iwashyna, T. J., Cooke, C. R., Wunsch, H., Kahn, J. M. (2012). Population Burden of Long-Term Survivorship After Severe Sepsis in Older Americans. J. Am. Geriatr. Soc 60, 1070–1077. doi: 10.1111/j.1532-5415.2012.03989.x
Iwashyna, T. J., Ely, E. W., Smith, D. M., Langa, K. M. (2010). Long-Term Cognitive Impairment and Functional Disability Among Survivors of Severe Sepsis. JAMA 304, 1787–1794. doi: 10.1001/jama.2010.1553
Jarczak, D., Kluge, S., Nierhaus, A. (2020). Use of Intravenous Immunoglobulins in Sepsis Therapy-A Clinical View. Int. J. Mol. Sci. 21, 5543. doi: 10.3390/ijms21155543
Jesus, A. A., Passaglia, P., Santos, B. M., Rodrigues-Santos, I., Flores, R. A., Batalhao, M. E., et al. (2020). Chronic Molecular Hydrogen Inhalation Mitigates Short and Long-Term Memory Loss in Polymicrobial Sepsis. Brain Res. 1739, 146857. doi: 10.1016/j.brainres.2020.146857
Jones, S. A., Dewald, B., Clark-Lewis, I., Baggiolini, M. (1997). Chemokine Antagonists That Discriminate Between Interleukin-8 Receptors. Selective Blockers of CXCR2. J. Biol. Chem. 272, 16166–16169. doi: 10.1074/jbc.272.26.16166
Kaplan, L., Chow, B. W., Gu, C. (2020). Neuronal Regulation of the Blood-Brain Barrier and Neurovascular Coupling. Nat. Rev. Neurosci. 21, 416–432. doi: 10.1038/s41583-020-0322-2
Keaney, J., Campbell, M. (2015). The Dynamic Blood-Brain Barrier. FEBS J. 282, 4067–4079. doi: 10.1111/febs.13412
Kikuchi, D. S., Campos, A. C. P., Qu, H., Forrester, S. J., Pagano, R. L., Lassegue, B., et al. (2019). Poldip2 Mediates Blood-Brain Barrier Disruption in a Model of Sepsis-Associated Encephalopathy. J. Neuroinflammation. 16, 241. doi: 10.1186/s12974-019-1575-4
Kingsley, S. M., Bhat, B. V. (2016). Differential Paradigms in Animal Models of Sepsis. Curr. Infect. Dis. Rep. 18, 26. doi: 10.1007/s11908-016-0535-8
Kovac, A., Erickson, M. A., Banks, W. A. (2011). Brain Microvascular Pericytes Are Immunoactive in Culture: Cytokine, Chemokine, Nitric Oxide, and LRP-1 Expression in Response to Lipopolysaccharide. J. Neuroinflamm. 8, 139. doi: 10.1186/1742-2094-8-139
Kuperberg, S. J., Wadgaonkar, R. (2017). Sepsis-Associated Encephalopathy: The Blood-Brain Barrier and the Sphingolipid Rheostat. Front. Immunol. 8, 597. doi: 10.3389/fimmu.2017.00597
Kutuzov, N., Flyvbjerg, H., Lauritzen, M. (2018). Contributions of the Glycocalyx, Endothelium, and Extravascular Compartment to the Blood-Brain Barrier. Proc. Natl. Acad. Sci. U. S. A. 115, E9429–E9438. doi: 10.1073/pnas.1802155115
Langen, U. H., Ayloo, S., Gu, C. (2019). Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 35, 591–613. doi: 10.1146/annurev-cellbio-100617-062608
Lee, S. W., Jung, K. H., Jeong, C. H., Seo, J. H., Yoon, D. K., Suh, J. K., et al. (2011). Inhibition of Endothelial Cell Migration Through the Downregulation of MMP-9 by A-Kinase Anchoring Protein 12. Mol. Med. Rep. 4, 145–149. doi: 10.3892/mmr.2010.389
Lelubre, C., Vincent, J. L. (2018). Mechanisms and Treatment of Organ Failure in Sepsis. Nat. Rev. Nephrol. 14, 417–427. doi: 10.1038/s41581-018-0005-7
Liebner, S., Dijkhuizen, R. M., Reiss, Y., Plate, K. H., Agalliu, D., Constantin, G. (2018). Functional Morphology of the Blood-Brain Barrier in Health and Disease. Acta Neuropathol. 135, 311–336. doi: 10.1007/s00401-018-1815-1
Linden, J. R., Flores, C., Schmidt, E. F., Uzal, F. A., Michel, A. O., Valenzuela, M., et al. (2019). Clostridium Perfringens Epsilon Toxin Induces Blood Brain Barrier Permeability via Caveolae-Dependent Transcytosis and Requires Expression of MAL. PloS Pathog. 15, e1008014. doi: 10.1371/journal.ppat.1008014
Linville, R. M., DeStefano, J. G., Sklar, M. B., Xu, Z., Farrell, A. M., Bogorad, M. I., et al. (2019). Human iPSC-Derived Blood-Brain Barrier Microvessels: Validation of Barrier Function and Endothelial Cell Behavior. Biomaterials 190–191, 24–37. doi: 10.1016/j.biomaterials.2018.10.023
Liu, H., Davis, J. R., Wu, Z. L., Faez Abdelgawad, A. (2017). Dexmedetomidine Attenuates Lipopolysaccharide Induced MCP-1 Expression in Primary Astrocyte. BioMed. Res. Int. 2017, 6352159. doi: 10.1155/2017/6352159
Liu, W. Y., Wang, Z. B., Wang, Y., Tong, L. C., Li, Y., Wei, X., et al. (2015). Increasing the Permeability of the Blood-Brain Barrier in Three Different Models In Vivo. CNS Neurosci. Ther. 21, 568–574. doi: 10.1111/cns.12405
Liu, L., Zhang, H., Shi, Y., Pan, L. (2020). Prostaglandin E1 Improves Cerebral Microcirculation Through Activation of Endothelial NOS and GRPCH1. J. Mol. Neurosci. 70, 2041–2048. doi: 10.1007/s12031-020-01610-y
Li, H., Wang, P., Huang, F., Jin, J., Wu, H., Zhang, B., et al. (2018). Astragaloside IV Protects Blood-Brain Barrier Integrity From LPS-Induced Disruption via Activating Nrf2 Antioxidant Signaling Pathway in Mice. Toxicol. Appl. Pharmacol. 340, 58–66. doi: 10.1016/j.taap.2017.12.019
Li, C. C., Yang, H. T., Hou, Y. C., Chiu, Y. S., Chiu, W. C. (2014). Dietary Fish Oil Reduces Systemic Inflammation and Ameliorates Sepsis-Induced Liver Injury by Up-Regulating the Peroxisome Proliferator-Activated Receptor Gamma-Mediated Pathway in Septic Mice. J. Nutr. Biochem. 25, 19–25. doi: 10.1016/j.jnutbio.2013.08.010
Li, T., Zheng, L. N., Han, X. H. (2020). Fenretinide Attenuates Lipopolysaccharide (LPS)-Induced Blood-Brain Barrier (BBB) and Depressive-Like Behavior in Mice by Targeting Nrf-2 Signaling. BioMed. Pharmacother. 125, 109680. doi: 10.1016/j.biopha.2019.109680
Loscher, W., Gericke, B. (2020). Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain? Pharmaceutics 12, 966. doi: 10.3390/pharmaceutics12100966
Lottes, R. G., Newton, D. A., Spyropoulos, D. D., Baatz, J. E. (2015). Lactate as Substrate for Mitochondrial Respiration in Alveolar Epithelial Type II Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L953–L961. doi: 10.1152/ajplung.00335.2014
Lou, S., Lepak, V. C., Eberly, L. E., Roth, B., Cui, W., Zhu, X. H., et al. (2016). Oxygen Consumption Deficit in Huntington Disease Mouse Brain Under Metabolic Stress. Hum. Mol. Genet. 25, 2813–2826. doi: 10.1093/hmg/ddw138
Lu, M. C., Ji, J. A., Jiang, Z. Y., You, Q. D. (2016). The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 36, 924–963. doi: 10.1002/med.21396
Mae, M., Armulik, A., Betsholtz, C. (2011). Getting to Know the Cast - Cellular Interactions and Signaling at the Neurovascular Unit. Curr. Pharm. Des. 17, 2750–2754. doi: 10.2174/138161211797440113
Malinsky, R. R., Valera, F. C., Cavallari, F. E., Kupper, D. S., Milaneze, C., Silva, J. S., et al. (2013). Matrix Metalloproteinases and Their Impact on Sinusal Extension in Chronic Rhinosinusitis With Nasal Polyps. Eur. Arch. Otorhinolaryngol. 270, 1345–1348. doi: 10.1007/s00405-012-2219-9
Margotti, W., Giustina, A. D., de Souza Goldim, M. P., Hubner, M., Cidreira, T., Denicol, T. L., et al. (2020). Aging Influences in the Blood-Brain Barrier Permeability and Cerebral Oxidative Stress in Sepsis. Exp. Gerontol. 140, 111063. doi: 10.1016/j.exger.2020.111063
Matsumoto, J., Dohgu, S., Takata, F., Nishioku, T., Sumi, N., Machida, T., et al. (2012). Lipopolysaccharide-Activated Microglia Lower P-Glycoprotein Function in Brain Microvascular Endothelial Cells. Neurosci. Lett. 524, 45–48. doi: 10.1016/j.neulet.2012.07.004
Ma, M. W., Wang, J., Zhang, Q., Wang, R., Dhandapani, K. M., Vadlamudi, R. K., et al. (2017). NADPH Oxidase in Brain Injury and Neurodegenerative Disorders. Mol. Neurodegener. 12, 7. doi: 10.1186/s13024-017-0150-7
Mei, B., Li, J., Zuo, Z. (2021). Dexmedetomidine Attenuates Sepsis-Associated Inflammation and Encephalopathy via Central Alpha2a Adrenoceptor. Brain Behav. Immun. 91, 296–314. doi: 10.1016/j.bbi.2020.10.008
Miao, F., Shan, C., Shah, S. A. H., Akhtar, R. W., Geng, S., Ning, D., et al. (2021). The Protective Effect of Walnut Oil on Lipopolysaccharide-Induced Acute Intestinal Injury in Mice. Food Sci. Nutr. 9, 711–718. doi: 10.1002/fsn3.2035
Michels, M., Danieslki, L. G., Vieira, A., Florentino, D., Dall’Igna, D., Galant, L., et al. (2015a). CD40-CD40 Ligand Pathway Is a Major Component of Acute Neuroinflammation and Contributes to Long-Term Cognitive Dysfunction After Sepsis. Mol. Med. 21, 219–226. doi: 10.2119/molmed.2015.00070
Michels, M., Vieira, A. S., Vuolo, F., Zapelini, H. G., Mendonca, B., Mina, F., et al. (2015b). The Role of Microglia Activation in the Development of Sepsis-Induced Long-Term Cognitive Impairment. Brain Behav. Immun. 43, 54–59. doi: 10.1016/j.bbi.2014.07.002
Miller, A. A., Drummond, G. R., De Silva, T. M., Mast, A. E., Hickey, H., Williams, J. P., et al. (2009). NADPH Oxidase Activity Is Higher in Cerebral Versus Systemic Arteries of Four Animal Species: Role of Nox2. Am. J. Physiol. Heart Circ. Physiol. 296, H220–H225. doi: 10.1152/ajpheart.00987.2008
Mina, F., Comim, C. M., Dominguini, D., Cassol, O. J., Jr., Dall Igna, D. M., Ferreira, G. K., et al. (2014). Il1-Beta Involvement in Cognitive Impairment After Sepsis. Mol. Neurobiol. 49, 1069–1076. doi: 10.1007/s12035-013-8581-9
Nayak, D., Huo, Y., Kwang, W. X., Pushparaj, P. N., Kumar, S. D., Ling, E. A., et al. (2010). Sphingosine Kinase 1 Regulates the Expression of Proinflammatory Cytokines and Nitric Oxide in Activated Microglia. Neuroscience 166, 132–144. doi: 10.1016/j.neuroscience.2009.12.020
Nikolakopoulou, A. M., Montagne, A., Kisler, K., Dai, Z., Wang, Y., Huuskonen, M. T., et al. (2019). Pericyte Loss Leads to Circulatory Failure and Pleiotrophin Depletion Causing Neuron Loss. Nat. Neurosci. 22, 1089–1098. doi: 10.1038/s41593-019-0434-z
Nishioku, T., Dohgu, S., Takata, F., Eto, T., Ishikawa, N., Kodama, K. B., et al. (2009). Detachment of Brain Pericytes From the Basal Lamina Is Involved in Disruption of the Blood-Brain Barrier Caused by Lipopolysaccharide-Induced Sepsis in Mice. Cell Mol. Neurobiol. 29, 309–316. doi: 10.1007/s10571-008-9322-x
Nishioku, T., Matsumoto, J., Dohgu, S., Sumi, N., Miyao, K., Takata, F., et al. (2010). Tumor Necrosis Factor-Alpha Mediates the Blood-Brain Barrier Dysfunction Induced by Activated Microglia in Mouse Brain Microvascular Endothelial Cells. J. Pharmacol. Sci. 112, 251–254. doi: 10.1254/jphs.09292sc
Nwafor, D. C., Brichacek, A. L., Mohammad, A. S., Griffith, J., Lucke-Wold, B. P., Benkovic, S. A., et al. (2019). Targeting the Blood-Brain Barrier to Prevent Sepsis-Associated Cognitive Impairment. J. Cent Nerv Syst. Dis. 11, 1179573519840652. doi: 10.1177/1179573519840652
Obermeier, B., Daneman, R., Ransohoff, R. M. (2013). Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat. Med. 19, 1584–1596. doi: 10.1038/nm.3407
Opal, S. M. (2010). Endotoxins and Other Sepsis Triggers. Contrib Nephrol. 167, 14–24. doi: 10.1159/000315915
Pan, W., Yu, C., Hsuchou, H., Kastin, A. J. (2010). The Role of Cerebral Vascular NFkappaB in LPS-Induced Inflammation: Differential Regulation of Efflux Transporter and Transporting Cytokine Receptors. Cell Physiol. Biochem. 25, 623–630. doi: 10.1159/000315081
Park, H. J., Shin, J. Y., Kim, H. N., Oh, S. H., Song, S. K., Lee, P. H. (2015). Mesenchymal Stem Cells Stabilize the Blood-Brain Barrier Through Regulation of Astrocytes. Stem Cell Res. Ther. 6, 187. doi: 10.1186/s13287-015-0180-4
Patel, M. (2016). Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 37, 768–778. doi: 10.1016/j.tips.2016.06.007
Poston, J. T., Koyner, J. L. (2019). Sepsis Associated Acute Kidney Injury. BMJ 364, k4891. doi: 10.1136/bmj.k4891
Prinz, M., Priller, J. (2017). The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 20, 136–144. doi: 10.1038/nn.4475
Qin, L. H., Huang, W., Mo, X. A., Chen, Y. L., Wu, X. H. (2015). LPS Induces Occludin Dysregulation in Cerebral Microvascular Endothelial Cells via MAPK Signaling and Augmenting MMP-2 Levels. Oxid. Med. Cell Longev. 2015, 120641. doi: 10.1155/2015/120641
Ren, C., Li, N., Wang, B., Yang, Y., Gao, J., Li, S., et al. (2015). Limb Ischemic Perconditioning Attenuates Blood-Brain Barrier Disruption by Inhibiting Activity of MMP-9 and Occludin Degradation After Focal Cerebral Ischemia. Aging Dis. 6, 406–417. doi: 10.14336/AD.2015.0812
Ren, C., Yao, R. Q., Zhang, H., Feng, Y. W., Yao, Y. M. (2020). Sepsis-Associated Encephalopathy: A Vicious Cycle of Immunosuppression. J. Neuroinflammation. 17, 14. doi: 10.1186/s12974-020-1701-3
Rios-Santos, F., Alves-Filho, J. C., Souto, F. O., Spiller, F., Freitas, A., Lotufo, C. M., et al. (2007). Down-Regulation of CXCR2 on Neutrophils in Severe Sepsis Is Mediated by Inducible Nitric Oxide Synthase-Derived Nitric Oxide. Am. J. Respir. Crit. Care Med. 175, 490–497. doi: 10.1164/rccm.200601-103OC
Ronaldson, P. T., Davis, T. P. (2020). Regulation of Blood-Brain Barrier Integrity by Microglia in Health and Disease: A Therapeutic Opportunity. J. Cereb Blood Flow Metab. 40, S6–S24. doi: 10.1177/0271678X20951995
Sadeghian, H., Lacoste, B., Qin, T., Toussay, X., Rosa, R., Oka, F., et al. (2018). Spreading Depolarizations Trigger Caveolin-1-Dependent Endothelial Transcytosis. Ann. Neurol. 84, 409–423. doi: 10.1002/ana.25298
Salkeni, M. A., Lynch, J. L., Otamis-Price, T., Banks, W. A. (2009). Lipopolysaccharide Impairs Blood-Brain Barrier P-Glycoprotein Function in Mice Through Prostaglandin- and Nitric Oxide-Independent Pathways. J. Neuroimmune Pharmacol. 4, 276–282. doi: 10.1007/s11481-008-9138-y
Savio, L. E. B., de Andrade Mello, P., Figliuolo, V. R., de Avelar Almeida, T. F., Santana, P. T., Oliveira, S. D. S., et al. (2017). CD39 Limits P2X7 Receptor Inflammatory Signaling and Attenuates Sepsis-Induced Liver Injury. J. Hepatol. 67, 716–726. doi: 10.1016/j.jhep.2017.05.021
Schreibelt, G., Kooij, G., Reijerkerk, A., van Doorn, R., Gringhuis, S. I., van der Pol, S., et al. (2007). Reactive Oxygen Species Alter Brain Endothelial Tight Junction Dynamics via RhoA, PI3 Kinase, and PKB Signaling. FASEB J. 21, 3666–3676. doi: 10.1096/fj.07-8329com
Semmler, A., Okulla, T., Sastre, M., Dumitrescu-Ozimek, L., Heneka, M. T. (2005). Systemic Inflammation Induces Apoptosis With Variable Vulnerability of Different Brain Regions. J. Chem. Neuroanat. 30, 144–157. doi: 10.1016/j.jchemneu.2005.07.003
Seok, S. M., Kim, J. M., Park, T. Y., Baik, E. J., Lee, S. H. (2013). Fructose-1,6-Bisphosphate Ameliorates Lipopolysaccharide-Induced Dysfunction of Blood-Brain Barrier. Arch. Pharm. Res. 36, 1149–1159. doi: 10.1007/s12272-013-0129-z
Sharshar, T., Carlier, R., Bernard, F., Guidoux, C., Brouland, J. P., Nardi, O., et al. (2007). Brain Lesions in Septic Shock: A Magnetic Resonance Imaging Study. Intensive Care Med. 33, 798–806. doi: 10.1007/s00134-007-0598-y
Shoji, A., Kabeya, M., Ishida, Y., Yanagida, A., Shibusawa, Y., Sugawara, M. (2014). Evaluation of cathepsin B activity for degrading collagen IV using a surface plasmon resonance method and circular dichroism spectroscopy. J. Pharm. BioMed. Anal. 95, 47–53. doi: 10.1016/j.jpba.2014.02.009
Silva, A. Y. O., Amorim, E. A., Barbosa-Silva, M. C., Lima, M. N., Oliveira, H. A., Granja, M. G., et al. (2020). Mesenchymal Stromal Cells Protect the Blood-Brain Barrier, Reduce Astrogliosis, and Prevent Cognitive and Behavioral Alterations in Surviving Septic Mice. Crit. Care Med. 48, e290–e298. doi: 10.1097/CCM.0000000000004219
Silwedel, C., Speer, C. P., Haarmann, A., Fehrholz, M., Claus, H., Buttmann, M., et al. (2018). Novel Insights Into Neuroinflammation: Bacterial Lipopolysaccharide, Tumor Necrosis Factor Alpha, and Ureaplasma Species Differentially Modulate Atypical Chemokine Receptor 3 Responses in Human Brain Microvascular Endothelial Cells. J. Neuroinflammation. 15, 156. doi: 10.1186/s12974-018-1170-0
Singer, M., Deutschman, C. S., Seymour, C. W., Shankar-Hari, M., Annane, D., Bauer, M., et al. (2016). The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315, 801–810. doi: 10.1001/jama.2016.0287
Sivandzade, F., Alqahtani, F., Sifat, A., Cucullo, L. (2020). The Cerebrovascular and Neurological Impact of Chronic Smoking on Post-Traumatic Brain Injury Outcome and Recovery: An In Vivo Study. J. Neuroinflamm. 17, 133. doi: 10.1186/s12974-020-01818-0
Skirecki, T., Cavaillon, J. M. (2019). Inner Sensors of Endotoxin - Implications for Sepsis Research and Therapy. FEMS Microbiol. Rev. 43, 239–256. doi: 10.1093/femsre/fuz004
Smyth, L. C. D., Rustenhoven, J., Park, T. I., Schweder, P., Jansson, D., Heppner, P. A., et al. (2018). Unique and Shared Inflammatory Profiles of Human Brain Endothelia and Pericytes. J. Neuroinflammation. 15, 138. doi: 10.1186/s12974-018-1167-8
Sonneville, R., Verdonk, F., Rauturier, C., Klein, I. F., Wolff, M., Annane, D., et al. (2013). Understanding Brain Dysfunction in Sepsis. Ann. Intensive Care 3, 15. doi: 10.1186/2110-5820-3-15
Spampinato, S. F., Bortolotto, V., Canonico, P. L., Sortino, M. A., Grilli, M. (2019). Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood-Brain Barrier Integrity. Front. Pharmacol. 10, 1346. doi: 10.3389/fphar.2019.01346
Stapor, P., Wang, X., Goveia, J., Moens, S., Carmeliet, P. (2014). Angiogenesis Revisited - Role and Therapeutic Potential of Targeting Endothelial Metabolism. J. Cell Sci. 127, 4331–4341. doi: 10.1242/jcs.153908
Su, L., Lei, X., Ma, H., Feng, C., Jiang, J., Jiao, J. (2020). PRDM16 Orchestrates Angiogenesis via Neural Differentiation in the Developing Brain. Cell Death Differ. 27, 2313–2329. doi: 10.1038/s41418-020-0504-5
Suliman, H. B., Carraway, M. S., Welty-Wolf, K. E., Whorton, A. R., Piantadosi, C. A. (2003). Lipopolysaccharide Stimulates Mitochondrial Biogenesis via Activation of Nuclear Respiratory Factor-1. J. Biol. Chem. 278, 41510–41518. doi: 10.1074/jbc.M304719200
Sumi, N., Nishioku, T., Takata, F., Matsumoto, J., Watanabe, T., Shuto, H., et al. (2010). Lipopolysaccharide-Activated Microglia Induce Dysfunction of the Blood-Brain Barrier in Rat Microvascular Endothelial Cells Co-Cultured With Microglia. Cell Mol. Neurobiol. 30, 247–253. doi: 10.1007/s10571-009-9446-7
Sun, Y., Yao, X., Zhang, Q. J., Zhu, M., Liu, Z. P., Ci, B., et al. (2018). Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation 138, 2247–2262. doi: 10.1161/CIRCULATIONAHA.117.032821
Sweeney, M. D., Ayyadurai, S., Zlokovic, B. V. (2016). Pericytes of the Neurovascular Unit: Key Functions and Signaling Pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R., Zlokovic, B. V. (2019). Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 99, 21–78. doi: 10.1152/physrev.00050.2017
Szakmany, T. (2019). Intravenous Immunoglobulin in Sepsis: Can We Find the Right Dose? Minerva Anestesiol. 85, 115–117. doi: 10.23736/S0375-9393.18.13349-9
Taile, J., Arcambal, A., Clerc, P., Gauvin-Bialecki, A., Gonthier, M. P. (2020). Medicinal Plant Polyphenols Attenuate Oxidative Stress and Improve Inflammatory and Vasoactive Markers in Cerebral Endothelial Cells During Hyperglycemic Condition. Antioxidants (Basel). 9, 573. doi: 10.3390/antiox9070573
Takata, F., Dohgu, S., Matsumoto, J., Machida, T., Kaneshima, S., Matsuo, M., et al. (2013). Metformin Induces Up-Regulation of Blood-Brain Barrier Functions by Activating AMP-Activated Protein Kinase in Rat Brain Microvascular Endothelial Cells. Biochem. Biophys. Res. Commun. 433, 586–590. doi: 10.1016/j.bbrc.2013.03.036
Tam, S. J., Watts, R. J. (2010). Connecting Vascular and Nervous System Development: Angiogenesis and the Blood-Brain Barrier. Annu. Rev. Neurosci. 33, 379–408. doi: 10.1146/annurev-neuro-060909-152829
Tang, X., Luo, Y. X., Chen, H. Z., Liu, D. P. (2014). Mitochondria, Endothelial Cell Function, and Vascular Diseases. Front. Physiol. 5, 175. doi: 10.3389/fphys.2014.00175
Tarafdar, A., Pula, G. (2018). The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 19, 3824. doi: 10.3390/ijms19123824
Tarassishin, L., Suh, H. S., Lee, S. C. (2014). LPS and IL-1 Differentially Activate Mouse and Human Astrocytes: Role of CD14. Glia 62, 999–1013. doi: 10.1002/glia.22657
Tauber, S. C., Eiffert, H., Bruck, W., Nau, R. (2017). Septic Encephalopathy and Septic Encephalitis. Expert Rev. Anti Infect. Ther. 15, 121–132. doi: 10.1080/14787210.2017.1265448
Tietz, S., Engelhardt, B. (2015). Brain Barriers: Crosstalk Between Complex Tight Junctions and Adherens Junctions. J. Cell Biol. 209, 493–506. doi: 10.1083/jcb.201412147
Tilling, T., Korte, D., Hoheisel, D., Galla, H. J. (1998). Basement Membrane Proteins Influence Brain Capillary Endothelial Barrier Function In Vitro. J. Neurochem. 71, 1151–1157. doi: 10.1046/j.1471-4159.1998.71031151.x
Tiruppathi, C., Shimizu, J., Miyawaki-Shimizu, K., Vogel, S. M., Bair, A. M., Minshall, R. D., et al. (2008). Role of NF-kappaB-Dependent Caveolin-1 Expression in the Mechanism of Increased Endothelial Permeability Induced by Lipopolysaccharide. J. Biol. Chem. 283, 4210–4218. doi: 10.1074/jbc.M703153200
Tsai, Y. L., Coady, T. H., Lu, L., Zheng, D., Alland, I., Tian, B., et al. (2020). ALS/FTD-Associated Protein FUS Induces Mitochondrial Dysfunction by Preferentially Sequestering Respiratory Chain Complex mRNAs. Genes Dev. 34, 785–805. doi: 10.1101/gad.335836.119
Ugun-Klusek, A., Tatham, M. H., Elkharaz, J., Constantin-Teodosiu, D., Lawler, K., Mohamed, H., et al. (2017). Continued 26S Proteasome Dysfunction in Mouse Brain Cortical Neurons Impairs Autophagy and the Keap1-Nrf2 Oxidative Defence Pathway. Cell Death Dis. 8, e2531. doi: 10.1038/cddis.2016.443
Ullen, A., Singewald, E., Konya, V., Fauler, G., Reicher, H., Nusshold, C., et al. (2013). Myeloperoxidase-Derived Oxidants Induce Blood-Brain Barrier Dysfunction In Vitro and In Vivo. PloS One 8, e64034. doi: 10.1371/journal.pone.0064034
Vanlaere, I., Libert, C. (2009). Matrix Metalloproteinases as Drug Targets in Infections Caused by Gram-Negative Bacteria and in Septic Shock. Clin. Microbiol. Rev. 22, 224–239. doi: 10.1128/CMR.00047-08
Varatharaj, A., Galea, I. (2017). The Blood-Brain Barrier in Systemic Inflammation. Brain Behav. Immun. 60, 1–12. doi: 10.1016/j.bbi.2016.03.010
Verstrepen, L., Bekaert, T., Chau, T. L., Tavernier, J., Chariot, A., Beyaert, R. (2008). TLR-4, IL-1R and TNF-R Signaling to NF-Kappab: Variations on a Common Theme. Cell Mol. Life Sci. 65, 2964–2978. doi: 10.1007/s00018-008-8064-8
Veszelka, S., Pasztoi, M., Farkas, A. E., Krizbai, I., Ngo, T. K., Niwa, M., et al. (2007). Pentosan Polysulfate Protects Brain Endothelial Cells Against Bacterial Lipopolysaccharide-Induced Damages. Neurochem. Int. 50, 219–228. doi: 10.1016/j.neuint.2006.08.006
Vincent, A. J., Lau, P. W., Roskams, A. J. (2008). SPARC Is Expressed by Macroglia and Microglia in the Developing and Mature Nervous System. Dev. Dyn. 237, 1449–1462. doi: 10.1002/dvdy.21495
Vincent, J. L., Marshall, J. C., Namendys-Silva, S. A., Francois, B., Martin-Loeches, I., Lipman, J., et al. (2014). Assessment of the Worldwide Burden of Critical Illness: The Intensive Care Over Nations (ICON) Audit. Lancet Respir. Med. 2, 380–386. doi: 10.1016/S2213-2600(14)70061-X
Vutukuri, R., Brunkhorst, R., Kestner, R. I., Hansen, L., Bouzas, N. F., Pfeilschifter, J., et al. (2018). Alteration of Sphingolipid Metabolism as a Putative Mechanism Underlying LPS-Induced BBB Disruption. J. Neurochem. 144, 172–185. doi: 10.1111/jnc.14236
Vuuren, M. J. V., Nell, T. A., Carr, J. A., Kell, D. B., Pretorius, E. (2020). Iron Dysregulation and Inflammagens Related to Oral and Gut Health Are Central to the Development of Parkinson’s Disease. Biomolecules 11, 30. doi: 10.3390/biom11010030
Wang, H. M., Huang, P., Li, Q., Yan, L. L., Sun, K., Yan, L., et al. (2019a). Post-Treatment With Qing-Ying-Tang, a Compound Chinese Medicine Relives Lipopolysaccharide-Induced Cerebral Microcirculation Disturbance in Mice. Front. Physiol. 10, 1320. doi: 10.3389/fphys.2019.01320
Wang, P., Hu, Y., Yao, D., Li, Y. (2018). Omi/HtrA2 Regulates a Mitochondria-Dependent Apoptotic Pathway in a Murine Model of Septic Encephalopathy. Cell Physiol. Biochem. 49, 2163–2173. doi: 10.1159/000493819
Wang, P., Wang, W., Hu, Y., Li, Y. (2020). Prolonged Soluble Epoxide Hydrolase Reactivity in Brain Endothelial Cells Is Associated With Long Cognitive Deficits in Sepsis. Mol. Neurobiol. 57, 2846–2855. doi: 10.1007/s12035-020-01925-2
Wang, H., Ward, M. F., Sama, A. E. (2014). Targeting HMGB1 in the Treatment of Sepsis. Expert Opin. Ther. Targets. 18, 257–268. doi: 10.1517/14728222.2014.863876
Wang, K., Wu, M., Xu, J., Wu, C., Zhang, B., Wang, G., et al. (2019b). Effects of Dexmedetomidine on Perioperative Stress, Inflammation, and Immune Function: Systematic Review and Meta-Analysis. Br. J. Anaesth 123, 777–794. doi: 10.1016/j.bja.2019.07.027
Winkler, L., Blasig, R., Breitkreuz-Korff, O., Berndt, P., Dithmer, S., Helms, H. C., et al. (2021). Tight Junctions in the Blood-Brain Barrier Promote Edema Formation and Infarct Size in Stroke - Ambivalent Effects of Sealing Proteins. J. Cereb Blood Flow Metab. 41, 132–145. doi: 10.1177/0271678X20904687
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 Reductions Exacerbate Alzheimer’s Disease Vasculo-Neuronal Dysfunction and Degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Wu, F., Chen, X., Zhai, L., Wang, H., Sun, M., Song, C., et al. (2020). CXCR2 Antagonist Attenuates Neutrophil Transmigration Into Brain in a Murine Model of LPS Induced Neuroinflammation. Biochem. Biophys. Res. Commun. 529, 839–845. doi: 10.1016/j.bbrc.2020.05.124
Xu, Z., Liu, C., Wang, R., Gao, X., Hao, C., Liu, C. (2021). A Combination of Lycopene and Human Amniotic Epithelial Cells can Ameliorate Cognitive Deficits and Suppress Neuroinflammatory Signaling by Choroid Plexus in Alzheimer’s Disease Rat. J. Nutr. Biochem. 88, 108558. doi: 10.1016/j.jnutbio.2020.108558
Xu, X. E., Liu, L., Wang, Y. C., Wang, C. T., Zheng, Q., Liu, Q. X., et al. (2019). Caspase-1 Inhibitor Exerts Brain-Protective Effects Against Sepsis-Associated Encephalopathy and Cognitive Impairments in a Mouse Model of Sepsis. Brain Behav. Immun. 80, 859–870. doi: 10.1016/j.bbi.2019.05.038
Xu, B., Zhang, Y., Du, X. F., Li, J., Zi, H. X., Bu, J. W., et al. (2017). Neurons Secrete miR-132-Containing Exosomes to Regulate Brain Vascular Integrity. Cell Res. 27, 882–897. doi: 10.1038/cr.2017.62
Yadollah, O., Lee, C., Jaleh, B., David, C., Saeed, A., Mark, G. (2003). Evaluation of the Immortalised Mouse Brain Capillary Endothelial Cell Line, B.End3, as an In Vitro Blood–Brain Barrier Model for Drug Uptake and Transport Studies. Brain Res. 990, 95–112. doi: 10.1016/S0006-8993(03)03443-7
Yamamoto, M., Ramirez, S. H., Sato, S., Kiyota, T., Cerny, R. L., Kaibuchi, K., et al. (2008). Phosphorylation of Claudin-5 and Occludin by Rho Kinase in Brain Endothelial Cells. Am. J. Pathol. 172, 521–533. doi: 10.2353/ajpath.2008.070076
Yamazaki, Y., Shinohara, M., Shinohara, M., Yamazaki, A., Murray, M. E., Liesinger, A. M., et al. (2019). Selective Loss of Cortical Endothelial Tight Junction Proteins During Alzheimer’s Disease Progression. Brain 142, 1077–1092. doi: 10.1093/brain/awz011
Yang, Y., Estrada, E. Y., Thompson, J. F., Liu, W., Rosenberg, G. A. (2007). Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels Is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb Blood Flow Metab. 27, 697–709. doi: 10.1038/sj.jcbfm.9600375
Yang, T., Sun, Y., Mao, L., Zhang, M., Li, Q., Zhang, L., et al. (2018). Brain Ischemic Preconditioning Protects Against Ischemic Injury and Preserves the Blood-Brain Barrier via Oxidative Signaling and Nrf2 Activation. Redox Biol. 17, 323–337. doi: 10.1016/j.redox.2018.05.001
Yan, J., Mitra, A., Hu, J., Cutrera, J. J., Xia, X., Doetschman, T., et al. (2016). Interleukin-30 (IL27p28) Alleviates Experimental Sepsis by Modulating Cytokine Profile in NKT Cells. J. Hepatol. 64, 1128–1136. doi: 10.1016/j.jhep.2015.12.020
Yao, Y., Chen, Z. L., Norris, E. H., Strickland, S. (2014). Astrocytic Laminin Regulates Pericyte Differentiation and Maintains Blood Brain Barrier Integrity. Nat. Commun. 5, 3413. doi: 10.1038/ncomms4413
Yeh, C. T., Kao, M. C., Chen, C. H., Huang, C. J. (2015). Platonin Preserves Blood-Brain Barrier Integrity in Septic Rats. Acta Anaesthesiol Taiwan. 53, 12–15. doi: 10.1016/j.aat.2015.02.001
Yi, X., Xu, C., Huang, P., Zhang, L., Qing, T., Li, J., et al. (2020). 1-Trifluoromethoxyphenyl-3-(1-Propionylpiperidin-4-Yl) Urea Protects the Blood-Brain Barrier Against Ischemic Injury by Upregulating Tight Junction Protein Expression, Mitigating Apoptosis and Inflammation In Vivo and In Vitro Model. Front. Pharmacol. 11, 1197. doi: 10.3389/fphar.2020.01197
Yue, H., Xie, K., Ji, X., Xu, B., Wang, C., Shi, P. (2020). Vascularized Neural Constructs for Ex-Vivo Reconstitution of Blood-Brain Barrier Function. Biomaterials 245, 119980. doi: 10.1016/j.biomaterials.2020.119980
Yu, Y., Wang, C., Zhang, X., Zhu, J., Wang, L., Ji, M., et al. (2020). Perfluorooctane Sulfonate Disrupts the Blood Brain Barrier Through the Crosstalk Between Endothelial Cells and Astrocytes in Mice. Environ. Pollut. 256, 113429. doi: 10.1016/j.envpol.2019.113429
Zengeler, K. E., Lukens, J. R. (2021). Innate Immunity at the Crossroads of Healthy Brain Maturation and Neurodevelopmental Disorders. Nat. Rev. Immunol. 21, 454–468. doi: 10.1038/s41577-020-00487-7
Zhang, Y., Lu, W., Wang, Z., Zhang, R., Xie, Y., Guo, S., et al. (2020). Reduced Neuronal cAMP in the Nucleus Accumbens Damages Blood-Brain Barrier Integrity and Promotes Stress Vulnerability. Biol. Psychiatry 87, 526–537. doi: 10.1016/j.biopsych.2019.09.027
Zhang, M., Mao, Y., Ramirez, S. H., Tuma, R. F., Chabrashvili, T. (2010). Angiotensin II Induced Cerebral Microvascular Inflammation and Increased Blood-Brain Barrier Permeability via Oxidative Stress. Neuroscience 171, 852–858. doi: 10.1016/j.neuroscience.2010.09.029
Zhao, L., An, R., Yang, Y., Yang, X., Liu, H., Yue, L., et al. (2015). Melatonin Alleviates Brain Injury in Mice Subjected to Cecal Ligation and Puncture via Attenuating Inflammation, Apoptosis, and Oxidative Stress: The Role of SIRT1 Signaling. J. Pineal Res. 59, 230–239. doi: 10.1111/jpi.12254
Zhao, Z., Hu, J., Gao, X., Liang, H., Liu, Z. (2014). Activation of AMPK Attenuates Lipopolysaccharide-Impaired Integrity and Function of Blood-Brain Barrier in Human Brain Microvascular Endothelial Cells. Exp. Mol. Pathol. 97, 386–392. doi: 10.1016/j.yexmp.2014.09.006
Zhao, S., Zhang, L., Lian, G., Wang, X., Zhang, H., Yao, X., et al. (2011). Sildenafil Attenuates LPS-Induced Pro-Inflammatory Responses Through Down-Regulation of Intracellular ROS-Related MAPK/NF-kappaB Signaling Pathways in N9 Microglia. Int. Immunopharmacol. 11, 468–474. doi: 10.1016/j.intimp.2010.12.017
Zheng, Y., Wu, Z., Yi, F., Orange, M., Yao, M., Yang, B., et al. (2018). By Activating Akt/eNOS Bilobalide B Inhibits Autophagy and Promotes Angiogenesis Following Focal Cerebral Ischemia Reperfusion. Cell Physiol. Biochem. 47, 604–616. doi: 10.1159/000490016
Zheng, Q., Zhang, Y., Zhao, Z., Shen, H., Zhao, H., Zhao, M. (2021). Isorhynchophylline Ameliorates Paraquat-Induced Acute Kidney Injury by Attenuating Oxidative Stress and Mitochondrial Damage via Regulating Toll-Interacting Expression. Toxicol. Appl. Pharmacol. 420, 115521. doi: 10.1016/j.taap.2021.115521
Zhou, Y., Sonobe, Y., Akahori, T., Jin, S., Kawanokuchi, J., Noda, M., et al. (2011). IL-9 Promotes Th17 Cell Migration Into the Central Nervous System via CC Chemokine Ligand-20 Produced by Astrocytes. J. Immunol. 186, 4415–4421. doi: 10.4049/jimmunol.1003307
Zhou, T., Zhao, L., Zhan, R., He, Q., Tong, Y., Tian, X., et al. (2014). Blood-Brain Barrier Dysfunction in Mice Induced by Lipopolysaccharide Is Attenuated by Dapsone. Biochem. Biophys. Res. Commun. 453, 419–424. doi: 10.1016/j.bbrc.2014.09.093
Keywords: blood-brain barrier, lipopolysaccharide, sepsis-associated encephalopathy, central nervous system, inflammation, oxidative stress
Citation: Peng X, Luo Z, He S, Zhang L and Li Y (2021) Blood-Brain Barrier Disruption by Lipopolysaccharide and Sepsis-Associated Encephalopathy. Front. Cell. Infect. Microbiol. 11:768108. doi: 10.3389/fcimb.2021.768108
Received: 31 August 2021; Accepted: 14 October 2021;
Published: 04 November 2021.
Edited by:
Monique Michels, Universidade do Extremo Sul Catarinense, BrazilReviewed by:
Jaqueline Generoso, Universidade do Extremo Sul Catarinense, BrazilCopyright © 2021 Peng, Luo, He, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Li, THlpbmcxMDE5QHN3bXUuZWR1LmNu; Luhua Zhang, WmhsdWh1YUBzd211LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.